

Abstract
Summary
Recently, an increasing number of methodological approaches have been proposed to tackle the complexity of metagenomics and microbiome data. In this scenario, reproducibility and replicability have become two critical issues, and the development of computational frameworks for the comparative evaluations of such methods is of utmost importance. Here, we present benchdamic, a Bioconductor package to benchmark methods for the identification of differentially abundant taxa.
Availability and implementation
benchdamic is available as an open-source R package through the Bioconductor project at https://bioconductor.org/packages/benchdamic/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Differential abundance (DA) analysis identifies significant differences in the microbial community composition between groups of samples, providing new insights into the composition of microbial communities and on their associations with the environment. Although many approaches have been proposed for DA analysis, it is widely recognized that the best method (i.e. a method with performances uniformly better than all the others) does not exist and that a careful exploratory data analysis is necessary to address methodological choices (Calgaro et al., 2020; Hawinkel et al., 2019; Nearing et al., 2022; Thorsen et al., 2016; Weiss et al., 2017).
Building on our previous work (Calgaro et al., 2020), we present the benchdamic R/Bioconductor package, which provides a computational framework to guide researchers in the selection of the method that best fits their data.
The structure of benchdamic can be summarized into four main parts (Fig. 1). Each section is developed to answer specific questions when comparing samples from different experimental groups, namely (i) the ability for a given statistical distribution to successfully fit the input data, with particular focus on sparsity and their count nature; (ii) the ability of the DA methods to control the type I error; (iii) the concordance among methods; and (iv) the accuracy of the findings based on a priori biological knowledge. Altogether, benchdamic is a flexible and customizable framework that can be used for the benchmarking of new and existing DA methods.
Fig. 1.
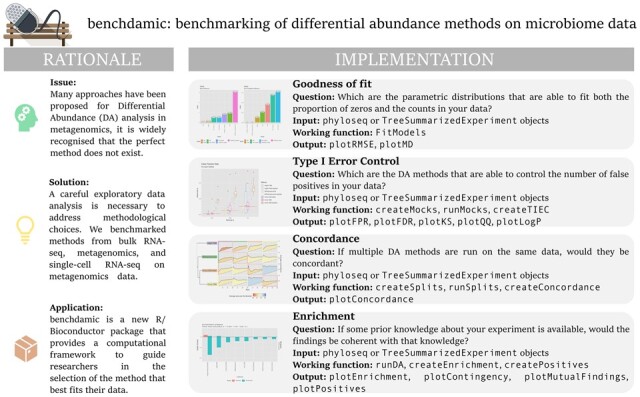
Graphical abstract. Each box on the right represents a step of the analysis where information about the research question, type of input data, working functions and outputs are reported
2 Implementation
benchdamic builds on existing R/Bioconductor infrastructure packages: the primary input of benchdamic’s main functions is a phyloseq or a TreeSummarizedExperiment object (Huang et al., 2021; McMurdie and Holmes, 2013). Ready-to-use normalization and DA methods included in benchdamic are based on the edgeR (Robinson et al., 2010), DESeq2 (Love et al., 2014), limma-voom (Law et al., 2014; Phipson et al., 2016; Ritchie et al., 2015), metagenomeSeq (Paulson et al., 2013), ALDEx2 (Fernandes et al., 2014, 2013), corncob (Martin et al., 2020), MAST (Finak et al., 2015), Seurat (Butler et al., 2018), dearseq (Gauthier et al., 2020), NOISeq (Tarazona et al., 2015), ANCOMBC (Lin and Peddada, 2020; Mandal et al., 2015) and zinbwave (Risso et al., 2018; Van den Berge et al., 2018) packages. Combinations of parameters are possible as well as the inclusion of custom methods (see Supplementary material Section S3).
In the following sections, we briefly outline the main functionality of the package. See Calgaro et al. (2020) for technical details on how these metrics are computed.
2.1 Goodness of fit
DA statistical models are based on different statistical distributions. Five different distributions are available in benchdamic for testing the goodness of fit on user-provided data: negative binomial, zero-inflated negative binomial, zero-inflated Gaussian, truncated Gaussian and Dirichlet-multinomial (see Supplementary material Section S2). Goodness of fit is measured by the ability of each method to correctly estimate the average counts and the probability of observing a zero.
2.2 Type I error control
To investigate the Type I error rate control of each DA method (i.e. the probability of the statistical test to call a feature DA when it is not) mock datasets with no true DA are generated starting from the user-provided data (see Supplementary material Section S4).
Briefly, the dataset is split into two random subsets and DA analysis, based on a chosen list of methods, is performed. The process is repeated N times (N ≥ 1000 suggested). The performances of each method are then summarized and graphically represented considering the false positive rate, false discovery rate, and departure from uniformity for the P-values distribution.
2.3 Concordance
benchdamic can be used to measure the between-method concordance (BMC), in which a DA method is compared to other methods in the same dataset, and the within-method concordance (WMC), in which a method is compared to itself in two random subsets of the same dataset (Supplementary material Section S5). Firstly, the dataset is randomly divided in half to obtain two subsets (Subset1 and Subset2) with samples from two or more biological groups, then DA analysis is performed between two groups, independently on each subset. The process is repeated N times (N ≥ 100 suggested) and average WMC and BMC metrics are computed and summarized using a heatmap representation.
2.4 Enrichment
Enrichment analysis can provide an alternative way of ranking methods in terms of their ability to identify, as significantly different, taxa that are known to be differentially abundant between two groups. DA analysis needs to be performed on a dataset where some a priori knowledge is available (Supplementary material Section S6). Given the direction of the DA features (over- or under-abundant) and the expected group in which the features should be differentially abundant according to the prior knowledge, several contingency tables are created for each DA method. A Fisher exact test is then performed to test the enrichment and the DA features identified by more than one method are highlighted. Additionally, the users will be able to rank the methods based on the difference between the total number of true positives and false positives for several thresholds (based on P-values, adjusted P-values or other statistics). The same approach can also be used to perform power analysis using simulated data (Supplementary material Section S6.8).
3 Conclusions
The benchdamic R/Bioconductor package aims to be a support tool for the identification of DA microbial taxa and the benchmarking of new methods. We envision two main uses of our package: (i) for practitioners interested in performing DA analysis on a new dataset, benchdamic can be used to identify the best DA methods among those already in the literature; (ii) for method developers interested in benchmarking their new approach, benchdamic can be used as an impartial tool to evaluate the relative merits of the new method compared to what is already available.
benchdamic is available as an open-source package through the Bioconductor project. The package includes a vignette with a detailed tutorial.
The future of benchdamic is oriented to the addition of new aspects of analysis e.g. new normalization methods and new DA approaches.
Supplementary Material
Acknowledgements
The authors are grateful to Prof. Levi Waldron for his helpful discussion on the design of the package and feedback.
Funding
This work was supported by the PhD Program in Biotechnology of the University of Verona; Italian Association for Cancer Research [21837]; National Cancer Institute of the National Institutes of Health [2U24CA180996].
Conflict of Interest: none declared.
Contributor Information
Matteo Calgaro, Department of Biotechnology, University of Verona, Verona 37134, Italy.
Chiara Romualdi, Department of Biology, University of Padova, Padova 35131, Italy.
Davide Risso, Department of Statistical Sciences, University of Padova, Padova 35121, Italy.
Nicola Vitulo, Department of Biotechnology, University of Verona, Verona 37134, Italy.
References
- Butler A. et al. (2018) Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol., 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calgaro M. et al. (2020) Assessment of statistical methods from single cell, bulk RNA-seq, and metagenomics applied to microbiome data. Genome Biol., 21, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes A.D. et al. (2013) ANOVA-Like differential expression (ALDEx) analysis for mixed population RNA-Seq. PLoS One, 8, e67019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes A.D. et al. (2014) Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome, 2, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G. et al. (2015) MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol., 16, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier M. et al. (2020) Dearseq: a variance component score test for RNA-seq differential analysis that effectively controls the false discovery rate. NAR Genom. Bioinform., 2, lqaa093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawinkel S. et al. (2019) A broken promise: microbiome differential abundance methods do not control the false discovery rate. Brief Bioinform., 20, 210–221. [DOI] [PubMed] [Google Scholar]
- Huang R. et al. (2021) TreeSummarizedExperiment: a S4 class for data with hierarchical structure. F1000Research, 9, 1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law C.W. et al. (2014) Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol., 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H., Peddada S.D. (2020) Analysis of compositions of microbiomes with bias correction. Nat. Commun., 11, 3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I. et al. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal S. et al. (2015) Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb. Ecol. Health Dis., 26, 27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin B.D. et al. (2020) Modeling microbial abundances and dysbiosis with beta-binomial regression. Ann. Appl. Stat., 14, 94–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie P.J., Holmes S. (2013) Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One, 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nearing J.T. et al. (2022) Microbiome differential abundance methods produce different results across 38 datasets. Nat. Commun., 13, 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson J.N. et al. (2013) Differential abundance analysis for microbial marker-gene surveys. Nat. Methods, 10, 1200–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipson B. et al. (2016) Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann. Appl. Stat., 10, 946–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D. et al. (2018) A general and flexible method for signal extraction from single-cell RNA-seq data. Nat. Commun., 9, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M.E. et al. (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res., 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D. et al. (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarazona S. et al. (2015) Data quality aware analysis of differential expression in RNA-seq with NOISeq R/bioc package. Nucleic Acids Res., 43, e140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsen J. et al. (2016) Large-scale benchmarking reveals false discoveries and count transformation sensitivity in 16S rRNA gene amplicon data analysis methods used in microbiome studies. Microbiome, 4, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berge K. et al. (2018) Observation weights unlock bulk RNA-seq tools for zero inflation and single-cell applications. Genome Biol., 19, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S. et al. (2017) Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome, 5, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.