

Abstract
Hyperinflammation characterized by elevated proinflammatory cytokines known as ‘cytokine storms’ is the major cause of high severity and mortality seen in COVID‐19 patients. The pathology behind the cytokine storms is currently unknown. Increased HMGB1 levels in serum/plasma of COVID‐19 patients were reported by many studies, which positively correlated with the level of proinflammatory cytokines. Dead cells following SARS‐CoV‐2 infection might release a large amount of HMGB1 and RNA of SARS‐CoV‐2 into extracellular space. HMGB1 is a well‐known inflammatory mediator. Additionally, extracellular HMGB1 might interact with SARS‐CoV‐2 RNA because of its high capability to bind with a wide variety of molecules including nucleic acids and could trigger massive proinflammatory immune responses. This review aimed to critically explore the many possible pathways by which HMGB1‐SARS‐CoV‐2 RNA complexes mediate proinflammatory responses in COVID‐19. The contribution of these pathways to impair host immune responses against SARS‐CoV‐2 infection leading to a cytokine storm was also evaluated. Moreover, since blocking the HMGB1‐SARS‐CoV‐2 RNA interaction might have therapeutic value, some of the HMGB1 antagonists have been reviewed. The HMGB1‐ SARS‐CoV‐2 RNA complexes might trigger endocytosis via RAGE which is linked to lysosomal rupture, PRRs activation, and pyroptotic death. High levels of the proinflammatory cytokines produced might suppress many immune cells leading to uncontrolled viral infection and cell damage with more HMGB1 released. Altogether these mechanisms might initiate a proinflammatory cycle leading to a cytokine storm. HMGB1 antagonists could be considered to give benefit in alleviating cytokine storms and serve as a potential candidate for COVID‐19 therapy.
Keywords: COVID‐19, cytokine storm, HMGB1, SARS‐CoV‐2 RNA
Extracellular High Mobility Group Box 1 (HMGB1), which could be a crucial factor behind the COVID‐19‐induced cytokine storm, is long‐known as a potent activator of TLR 4‐mediated proinflammatory cytokines release. HMGB1‐RNA SARS‐CoV‐2 complexes via RAGE might trigger inflammasome activation, pyroptotic cell death, and the initiation of coagulation, followed by a massive production of proinflammatory cytokines which suppressed many immune cells leading to uncontrolled viral infection and cells damage, causing more HMGB1 release. This vicious cycle potentially induced a cytokine storm.
INTRODUCTION
Severe Acute Respiratory Syndrome‐Coronavirus‐2 (SARS‐CoV‐2) belongs to the genus β‐coronavirus, which are enveloped viruses containing a positive single‐stranded ribonucleic acid (RNA). The virus binds to the receptor Angiotensin Converting Enzyme 2 (ACE2) which facilitates its entry into the host cell [1]. The illness caused by SARS‐CoV‐2 is termed Coronavirus Disease‐2019 (COVID‐19) which continues to be a global public health threat. Confirmed cases have risen to over 630 million cases with more than 6.5 million deaths worldwide by November 2022 [2]. Hyperinflammation characterized by elevated proinflammatory cytokines known as ‘cytokine storms’ is the major cause of high severity and mortality seen in COVID‐19. Increased levels of Interleukin‐2 (IL‐2), IL‐4, IL‐6, IL‐8, IL‐10, and tumour necrosis factor (TNF) have been observed in patients with severe COVID‐19 [3, 4]. This condition becomes exacerbated by decreased levels of interferon (IFN) and CD4+ T cells, CD8+ T cells, and B cells as well as natural killer (NK) cell counts [5].
The host immune response to SARS‐CoV‐2 infection causes clinical manifestations of COVID‐19 that vary from mild to severe. In most cases, the immune response works properly to resolve the infection. However, in severe conditions, an uncontrolled host immune response creates vicious cycles between cytokine storms, coagulopathy, and acute respiratory distress syndrome (ARDS) which could rapidly progress to disease worsening and fatalities [6]. Patients with severe COVID‐19 showed high viral load in their blood, lymphocytopenia, and an increase in monocyte‐derived macrophages in the patient's bronchoalveolar fluid. There was also a widespread inflammatory reaction in the form of increased innate immune response and hypercytokinemia. Higher levels of IL‐6 and TNF were detected in critically ill patients compared to mild–moderate patients [7, 8].
Pathogen‐associated molecular patterns (PAMPs) and damage‐associated molecular patterns (DAMPs) have crucial roles in mounting an exaggerated SARS‐CoV‐2 immune response. Lipids, proteins, and viral RNA produced throughout the virus life cycle are referred to as PAMPs and activate their receptors, pattern recognition receptors (PRRs) in membranes, endosomes, and cell cytoplasm. PRRs activate downstream signalling pathways that lead to the secretion of ILs, TNF, and IFN [9]. Other substances capable of interacting with PRRs and triggering an inflammatory response are DAMPs, which are endogenous molecules released after cellular damage or stress. The release of excessive amounts of DAMPs leads to dysregulated life‐threatening hyperinflammatory responses as seen in patients with severe COVID‐19 [10].
One of the most known DAMPs is high mobility group box 1 (HMGB1) [11]. Accumulation of extracellular HMGB1 affects the progression of various respiratory diseases [12]. HMGB1 has been shown to induce inflammatory pathways by triggering the release of cytokines or chemokines in viral respiratory infections. Serum HMGB1 levels were associated with viral replication and the degree of lung pathology. HMGB1 is a potential biomarker to predict the severity of viral infections in the respiratory tract [13, 14]. In line with this finding, HMGB1 is predicted to have an important role in triggering the inflammatory response and is a potential therapeutic target for SARS‐CoV‐2 infection [15, 16, 17, 18]. In this review, we hypothesized that HMGB1 plays a crucial role in SARS‐CoV‐2 infection through its possible interaction with RNA of SARS‐CoV‐2 in triggering the cytokine storm in COVID‐19. This review also discusses the potential for HMGB1 antagonists as therapeutic candidates for COVID‐19.
HIGH MOBILITY GROUP BOX 1 (HMGB1)
HMGB1 is a non‐histone protein involved in the condensation and packing of intranuclear DNA through its non‐specific interactions with DNA. HMGB1 has been reported to be involved in transcription, replication, repair, and recombination due to its binding to DNA [19]. HMGB1, a highly conserved protein, is composed of 215 amino acids and has a molecular weight of ⁓25 kDa. This protein consists of a DNA binding domain and a C‐terminal region. Two homologous N‐terminal boxes in the DNA binding domain, namely A box and B box, are linked by a short basic domain. The A box is located at 1–79 loci and the B box is located at 86–162 loci of the HMGB1 amino acid sequence [20, 21]. The B box can bind Toll‐like receptor 4 (TLR4) and receptor for advanced glycation end products (RAGE) which regulate the production of proinflammatory cytokines. TLR4 binds to the B box at residues 89–108 while RAGE binds to residues 150–183 [22]. Another RAGE binding site identified at residues 23–50 is responsible for reversing apoptosis‐induced tolerance [23]. The acidic C tail composed of 30 amino acid residues of aspartates and glutamates stabilizes HMGB1 under various conditions and enhances its ability to bend DNA [19]. This acidic C‐terminal tail also plays a role in amplifying the anti‐inflammatory effect induced by the A box [24]. The HMGB1 molecule has two nuclear localization sequences located at residues 28–44 in the A box and residues 179–185 located between the B box and the C‐terminal tail. NLS sites are critical for HMGB1 translocation from the nucleus to the cytoplasm. In addition, three cysteine residues at amino acid positions 23 and 45 (A box) and 106 (B box) determine the reduction status and influethe nce biological activity of the extracellular HMGB1 [19, 25, 26] (Figure 1 ).
FIGURE 1.
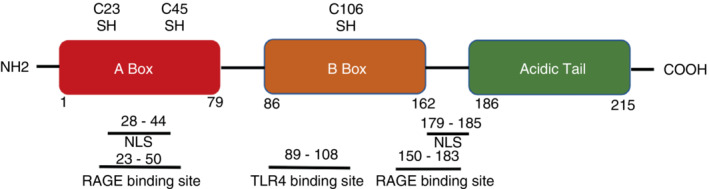
HMGB1 structure. HMGB1 molecule is composed of 215 amino acids, and consists of two boxes for DNA binding domain (A box: aa 1–79 and B box: aa 86–162) and acidic tail (aa 186–215). Two nuclear localization sequences (NLS) sites (aa 28–44 and aa 179–185) are responsible for HMGB1 translocation. Three cysteine residues at positions 23, 45, and 106 determine HMGB1 redox state. HMGB1 triggers inflammation by binding to its receptor. Aa 89–108 and aa 150–183 were considered as HMGB1's binding sites to TLR4 and RAGE. Aa 23–50 is required for RAGE‐dependent reversal of apoptosis‐induced tolerance.
Extracellular HMGB1 is secreted under certain conditions and acts as a cytokine that triggers an immune response, thereby acting as a DAMPs. HMGB1 is expressed by almost all cell types, including all haematopoietic cells. Monocytes, macrophages, mature dendritic cells (DCs), and NK cells induce active secretion through mechanisms triggered by certain factors such as lipopolysaccharides (LPS), IFN‐γ, TNF, and tumour growth factor‐beta (TGF‐β) [25]. HMGB1 can also be released passively by dead cells [11]. The process of releasing HMGB1 requires two steps. In the first step, DAMPs or PAMPs trigger post‐translational modification within nuclear localization signal (NLS) sites which translocate HMGB1 from the nucleus to the cytoplasm. Subsequently, HMGB1 is released into extracellular fluid facilitated by the secretory lysosomes formation or programmed cell death such as pyroptosis or necroptosis [26]. HMGB1 cytoplasm translocation can be induced by type I and II IFN [27, 28].
The biological function of extracellular HMGB1 is influenced by its receptor type, binding complex, and redox state. HMGB1 capacity to regulate inflammation depends on the three cysteine residues redox state: totally reduced (all thiol), partially oxidized (disulfide), and completely oxidized (sulfonyl). All thiol HMGB1 is formed if all cysteine residues (C23, C45, and C106) reside in the fully reduced state with thiol residues. Disulfide bonds between C23 and C45 can be easily formed under mild oxidative conditions. Various cell damage models, such as necrosis, necroptosis, pyroptosis, and apoptosis will influence the isoform of HMGB1 released into the extracellular environment. Necrotic cells secrete HMGB1 in a fully reduced state without acetylation. However, during apoptosis, HMGB1 will be retained in apoptotic bodies, tightly bound to DNA. If phagocytic clearance is not effective, apoptotic bodies undergo secondary necrosis and HMGB1 will be secreted in sulfonyl and disulfide isoforms. Necroptosis cells release HMGB1 in hyperacetylated and fully reduced forms. Pyroptosis is a major pathway for the release of the dangerous disulfide HMGB1. Different redox states of HMGB1 influence its functions. All thiol forms a complex with stromal cell‐derived factor 1 (CXCL12) and triggers immune cells migration via C‐X‐C chemokine receptor type 4 (CXCR4) and RAGE. Only disulfide HMGB1 can interact with TLR4 which exhibits a cytokine‐induced inflammatory response. Totally oxidized HMGB1 molecules have no ability to activate chemokines or cytokines [29].
CORRELATION OF HMGB1 WITH PROINFLAMMATORY CYTOKINES IN COVID‐19: THE PRELIMINARY RESEARCH
The contribution of HMGB1 in the inflammatory response has drawn attention to the role of this protein in SARS‐CoV‐2 infection. SARS‐CoV‐2 infection was shown to trigger HMGB1 secretion which increased over time in cell culture supernatants of Vero‐E6 and Huh7.5. HMGB1 inhibition significantly decreased the cell mortality rate. HMGB1 also has another role in facilitating the SARS‐CoV‐2 infection process, in addition to its role in inflammation. HMGB1 has been reported to induce ACE2 receptor expression [30]. Regulation of ACE2 is maintained by extracellular HMGB1 via RAGE [31]. Effective viral infection is a major cause of cell death which in turn induces a vicious cycle of HMGB1 and massive releases of proinflammatory cytokines, leading to a cytokine storm [10].
Increased HMGB1 levels in serum/plasma of COVID‐19 patients were reported by many studies [31, 32, 33, 34]. HMGB1 levels were significantly increased in COVID‐19 patients compared to healthy controls [33, 34]. The increased HMGB1 levels were positively correlated with the severity of the disease. The average serum level of HMGB1 was significantly elevated in severe compared to moderate COVID‐19 patients. Patients admitted to the intensive care unit (ICU) compared to non‐ICU patients also had higher levels of HMGB1 [31, 32]. Patients' clinical improvement was associated with a decrease in HMGB1 level [31, 34].
The levels of proinflammatory cytokines such as IL‐8, MCP‐3, MCP‐1, IL‐1ra, β‐NGF, IL‐7, IL‐10, RANTES, G‐CSF, IL‐1α, CTACK, and IL‐17A were elevated in COVID‐19 patients and positively correlated with the HMGB1 levels. These data indicated that the overproduction of HMGB1 in COVID‐19 patients was associated with an increase in specific cytokines that characterize a cytokine storm [31, 32]. HMGB1 can be used to determine the prognosis of severe COVID‐19 patients. HMGB1 and IL‐6 levels in COVID‐19 patients were positively correlated with high Sequential Organ Failure Assessment (SOFA) scores, septic shock, acute renal failure, poor oxygenation status, and longer duration of ventilation [34]. Although all of the above studies had limited participants, these studies consistently showed that HMGB1 played a critical role in the outcome of SARS‐CoV‐2 infection, possibly by triggering a cytokine storm. The pathophysiology of how HMGB1 induces cytokine storms needs further exploration.
DYSREGULATION OF THE INNATE AND ADAPTIVE IMMUNE SYSTEMS TRIGGERS PAMPS AND DAMPS SECRETION: THE VIRAL IMMUNE RESPONSE
Cytokines such as IL‐6, TNF, and IFN‐I/III released from SARS‐CoV‐2 infected pulmonary epithelial cells activate the inflammatory response in the resident macrophages and recruit other immune cells such as monocytes, granulocytes, and lymphocytes. NK cells travel to the lungs led by chemokines CCL2, CCL3, and CXCL9/10/11 [35]. DCs, NK cells, and macrophages are the main cells of the innate immune response that act as the first line of defence against SARS‐CoV‐2 infection [36]. Except for tissue macrophages, immune cells express low levels of ACE2 receptors [37]. Although SARS‐CoV might have the ability to infect immune cells, the infection could be abortive [38, 39]. IFN‐α influences the inability of viral replication in the immune cells. Monocyte/macrophages isolated from blood donors expressing IFN‐α did not show the presence of SARS‐CoV antigen. When an anti‐IFN antibody was added to the culture, minimal viral replication was observed [40].
The antiviral activity of IFN plays an important role in modulating the immune response against the SARS virus. IFN blocks viral spreading by inhibiting viral replication and inducing apoptosis of the infected cells [41]. In addition, type I IFN has an immunomodulatory role by promoting upregulation of Major Histocompatibility Complex‐I (MHC‐I) expression in various cells, which is required to optimize the elimination of infected cells by T cells. IFN‐I signalling enhances the cytolytic capacity and survival of NK cells [42]. However, SARS‐CoV‐2 may likely have developed several mechanisms to inhibit IFN production and signalling [35]. It was reported that COVID‐19 patients had a lower level of serum IFN compared to healthy controls. A significant increase in blood IFN levels was correlated with patient clinical improvement and survival [43].
Inhibition of IFN signalling functionally impairs the activity of T cells and NK cells. Evidence showed that SARS‐CoV‐2 acute infection suppressed T and NK cells and caused a broad immune cell reduction including DCs and classic monocytes from peripheral blood of COVID‐19 patients [44]. These DCs have an important role in host defence against SARS‐CoV‐2 infection. Plasmacytoid DCs are the main type I IFN‐producing cells. IL‐12 produced by DC mobilizes NK cells. These DCs are antigen‐presenting cells that present viral antigens in association with MHC‐I and II molecules [45]. Immature DCs reach their maturity after processing viral antigens. The antigens bound to MHC‐I and II are recognized by CD8+ and CD4+ T cells. CD8+ cells proliferate after antigen recognition via MHC‐I on DCs and initiate their cytotoxic activity to kill virus‐infected cells. The introduction of CD4+ cells to viral antigens bound to MHC‐II trigger the activation of CD8+ cells and B cells. Active B cells proliferate into producing antibody plasma cells and memory cells [46]. Antibody production in COVID‐19 patients was predicted to be ineffective due to DCs inhibition. Inadequate antibodies, both in terms of quality and quantity, could mediate the viral entry into host cells. SARS‐CoV‐2 might have the ability to infect cells by binding to antibodies, known as antibody‐dependent enhancement (ADE) [47]. Evidence suggested that ADE response most likely occurred in severe COVID‐19. Increased ADE response induced cytokine production and exacerbated disease progression [48].
DCs isolated from COVID‐19 patients exhibit impaired maturity [44]. These immature DCs express low levels of MHC‐I, MHC‐II, CD80/86, and IFN. The defective DCs could not properly activate CD4+ T cells, CD8+ T cells, and NK cells. In addition, NK cells might induce apoptosis in DCs for their low ability to express MHC‐I [46]. NK cell activity is inhibited in COVID‐19 patients, possibly even undergoing apoptosis [49]. Dysregulation of the immune system, low levels of IFN, and inadequate antibodies could trigger uncontrolled viral infections. High levels of cellular damage induce the release of PAMPs and DAMPs.
The release of PAMPs and DAMPs acts as a ‘signal 0 s’ to initiate an immune response. One of the major PAMPs is derived from microbial nucleic acids, such as viral RNA [50]. The impact of SARS‐CoV‐2 infection goes well beyond the lungs. Viral components such as RNA and protein were identified in multiple organs and body fluids of COVID‐19 patients. The abundance of viral RNA and protein does not always indicate active infection [51]. However, the viral component remains potentially dangerous for its ability to stimulate immune responses [50].
HIGH MOBILITY GROUP BOX 1 TRIGGERS A CYTOKINE STORM IN COVID‐19: THE PATHOPHYSIOLOGY
Extracellular HMGB1 as DAMPs will bind to its receptor, the PRR. TLR4 and RAGE are functional PRRs for HMGB1 [52]. As HMGB1 interacts with TLR4, it stimulates downstream signalling leading to the activation of Nuclear factor kappa B (NF‐KB) and interferon regulator factor 3 (IRF3) [53]. TLR4 is the only TLR capable of activating signalling pathways via two main adapter molecules, Myeloid differentiation primary response 88 (MyD88) and TIR‐domain‐containing adapter‐inducing interferon‐β (TRIF), which trigger NF‐KB and IRF3 activation resulting in the production of inflammatory cytokines and IFNs [54]. HMGB1 interaction with RAGE triggers the activation of Ras which also stimulates signal transduction to the NF‐KB pathway. This pathway involves extracellular signal‐regulated kinases 1 and 2 and p38 mitogen‐activated protein kinase, which stimulates NF‐KB translocation from cytoplasm to nucleus to trigger transcription of proinflammatory cytokines [53].
HMGB1 could also interact with several molecules including nucleic acids. Binding these molecules increases signalling at the HMGB1 receptor (56). Exposure to HMGB1 in the presence of LPS, which is a component of the wall of Gram‐negative bacteria, significantly increases the proinflammatory cytokines production compared to HMGB1 or LPS exposure alone. The underlying mechanism is the ability of HMGB1 to facilitate the internalization of the partner molecules. Several studies have shown that HMGB1 and its molecular partners were internalized via RAGE [55, 56, 57]. HMGB1 facilitated extracellular LPS to gain access to the cytosol via RAGE which was subsequently followed by caspase 11 activation in a murine model. This step is critical for caspase 11‐dependent lethality in endotoxemia and bacterial sepsis [58]. Caspase 11 is homologous to caspase 4 and 5 in humans [59]. All HMGB1 isoforms could be internalized [56]. Endocytosis of the HMGB1 complex would be followed by a transport process to the endo‐lysosomal system [55, 56, 57]. Under normal physiological HMGB1 level, this mechanism is required to eliminate foreign particles. However, in pathological conditions when HMGB1 levels are too high, it acts like a detergent that stimulates lysosomal rupture. It triggers the release of partner molecules into the cytosol [52]. It was demonstrated that macrophage's lysosomes underwent swelling after endocytosis of HMGB1, followed by lysosomal rupture and leakage of its content into the cytosol [55].
HMGB1 has been shown to be able to bind to viral RNA which could be a precondition for the recognition and activation of cytosolic receptors, such as Retinoic acid‐inducible gene (RIG)‐I‐like receptor (RLR) and TLR [60]. The RNA of SARS CoV‐2 is predicted to be able to gain access to the cytosolic receptors mediated by HMGB1 [55]. Endosomal receptors, TLR7 and 8, recognize single‐stranded RNA (ssRNA) of SARS‐CoV‐2 when it accumulates in the endosome. Downstream signal of this interaction is the production of proinflammatory cytokines via the MyD88 pathway [61]. When the RNA of SARS‐CoV‐2 escapes into the cytosol, it could interact with the cytosolic receptors. Retinoic acid‐inducible gene (RIG)‐I‐like receptor (RLR) is a cytoplasmic PRR capable of detecting ssRNA. The RLRs include retinoic acid‐inducible gene (RIG)‐I, melanoma differentiation‐associated gene 5 (MDA5), and laboratory of genetics and physiology 2. Exposure to the ssRNA of SARS CoV‐2 might activate RLR, especially RIG‐1 and MDA5. RLR interacts with a mitochondrial activator of virus signalling (MAVS) which leads to the recruitment and activation of protein kinases TANK‐binding kinase 1 (TBK1)/IKKɛ and IKKα/IKKβ. These protein kinases play a role in the production of IFNs and proinflammatory cytokines through the upregulation of IRF3, IRF7, and NF‐KB [62]. NF‐KB activates the transcription of several pro‐inflammatory genes such as the Nod‐like receptor family, and pyrin domain containing 3 (NLRP3). This pathway induces the formation of the inflammasome and the production of pro‐IL‐1β, pro‐IL‐18, IL‐6, and TNF‐α [63].
The upregulation of cytoplasmic sensors such as NLRP3 triggers inflammasome formation. The inflammasome is a multiprotein complex in the cytoplasm playing a role in the host's reaction to pathogens or tissue damage. NLRP3 inflammasome was activated in severe COVID‐19 patients [16]. Inflammasome formation via NLRP3 triggers the recruitment of complex molecules including adapter protein apoptosis‐associated speck‐like protein containing a CARD (ASC) which in turn activates caspase 1. Caspase 1 cleaves inactive form pro‐IL‐1β and pro‐IL‐18 to mature IL‐1β and IL‐18. Caspase 1 also cleaves gasdermin‐D (GSDMD) which triggers pyroptosis through pore formation at the cell membrane. Later, IL‐1β and IL‐18 are released into the extracellular space [64]. GSDMD is dispensable for the maturation of another IL‐1 family cytokine, IL‐1α. GSDMD pore formation triggers Ca2+ influx which mediates calpains activation. Pro‐IL‐1α is processed into the mature form, IL‐1α, by this protease [65]. It was confirmed that IL‐1α contributed to the deterioration and adverse outcomes of COVID‐19 [66, 67]. GSDMD pore formation‐mediated Ca2+ influx plays a role in coagulation by inducing phosphatidylserine exposure through transmembrane protein 16F which leads to markedly enhanced activation of tissue factor (TF), an initiator of coagulation [68].
Pyroptotic cell death also induces the release of more HMGB1. HMGB1 secretion during pyroptosis is triggered by inflammasome assembly and caspase 1 activation [69]. GSDMD, which inserts the membrane, causes water leakage and cell lysis. HMGB1 is released into the extracellular fluid [70]. The SARS‐CoV viral protein, viroporin protein 3a, can directly activate NLRP3 triggering pyroptotic cell death. SARS‐CoV‐2 genome also contains the sequence which encodes for this protein. It was suspected that SARS‐CoV‐2 also induces a similar response (16). COVID‐19 patients showed an increase in caspase 1 activity accompanied with elevated levels of IL‐1β which could be promoted by the activation of inflammasomes during SARS‐CoV‐2 infection [71]. The interaction between HMGB1 and SARS‐CoV‐2 RNA might trigger an alternative pathway of pyroptosis without the need to activate NLRP3. The leakage of lysosomes after HMGB1 endocytosis led to the release of endosomal enzyme, cathepsin B, into cytosol. Cathepsin B triggers cleavage of caspase 1, by inducing pyroptosome, ASC. Furthermore, ASC activates caspase 1 triggering macrophage pyroptosis. This pathway induces NLRP3 inflammasome‐independent pyroptosis [55] which needs further exploration in COVID‐19.
Many factors might influence the mechanism involving HMGB1 in the pathogenesis of COVID‐19. The HMGB1 redox state might change in the extracellular environment under certain conditions. All thiol HMGB1 could become shifted to disulfide or sulfonyl HMGB1 when exposed to large amounts of reactive oxygen species [22]. In addition, the expression and function of TLRs are regulated by cellular processes (e.g., cell cycle and migration, apoptosis), air pollution, depression, certain drugs (e.g., glucocorticoids, antibiotics), stress, depression, polymorphisms, aging, nutrients, and micronutrients (vitamins and minerals) [72]. Gender affects inflammasome activation. Polymorphonuclear cells in males compared to women showed significantly higher levels of mRNA molecules involved in inflammasome activity such as AIM2, NLRP3, ASC, Casp1, Casp5, and IL‐1β [73]. The mortality rate of confirmed COVID‐19 male patients is higher than that of women in Central Java, Indonesia [74]. Similar data were obtained from many regions worldwide. Male patients are predicted to have a higher risk than women for COVID‐19 severity and admission to the ICU [75]. Aging is associated with low‐grade subclinical inflammation characterized by increased DAMPs and proinflammatory cytokines, and the inflammasome activation, especially NLRP3 [76]. Impairment of the cholinergic system which is known to modulate anti‐inflammatory responses via the vagus nerve is thought to be associated with older age [77]. Sirtuin 1 (SIRT1), a nicotinamide adenine dinucleotide (NAD+) dependent deacetylase, delays cellular senescence and ameliorates the inflammatory effects of HMGB1 [78, 79]. SIRT1 expressions showed a significant decline in aging [80]. This factor might help explain the high mortality rate of elderly patients with COVID‐19 [74]. Excess nutrients could form DAMPs molecules which in turn activate PRRs [81]. PRR mutations are associated with an increase of proinflammatory cytokines [82].
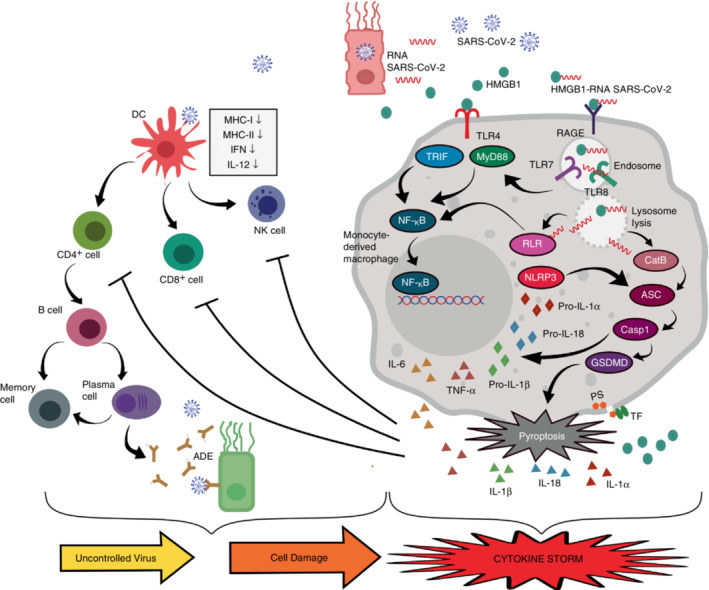
The increased production of proinflammatory cytokines triggered by the HMGB1‐SARS‐CoV‐2 RNA complexes in COVID‐19 patients creates a vicious cycle that compromises the host condition. IL‐6 is responsible for modulating the host immune response during viral infection and suppressing viral replication, which means it has an antiviral role. However, increased systemic level of IL‐6 might have a pro‐viral effect. Imbalanced IL‐6 production after virus infection could induce viral survival and disease expansion [83]. COVID‐19 patients experienced a decrease in both CD4+ and CD8+ T cells, as well as NK cells both in terms of number and function. Inhibition of T cells and NK cells was correlated with high levels of IL‐6 and TNF‐α. High levels of IL‐6 resulted in suppression of the cytolytic activity of NK cells and CD8+ T cells. Both of these cells play a critical role in the lysing of the infected cells. Inhibition of NK cell and CD8+ T cell activity amplifies the proinflammatory cytokine cascade leading to disease worsening [84, 85]. The IL‐1 family including IL‐1β is capable of inducing the secretion of other proinflammatory cytokines such as IL‐6. IL‐1 is also able to induce itself thereby creating a proinflammatory cycle [86]. Another IL‐1 family, IL‐18, is known as a factor that triggers IFN‐γ production. Together with other cytokines, IL‐18 stimulates T cells, CD4+ NKT, mast cells, and basophils [87]. Elevated levels of IL‐18 were correlated with acute respiratory syndrome in patients with severe COVID‐19 [88, 89]. Cytokine storms trigger macrophage activation known as macrophage activation syndrome (MAS) contributing to multi‐organ dysfunction and increasing the rate of mortality [90]. MAS is thought to be involved in the pathogenesis of ARDS due to SARS‐CoV‐2 infection [36, 91] (Figure 2 ).
FIGURE 2.
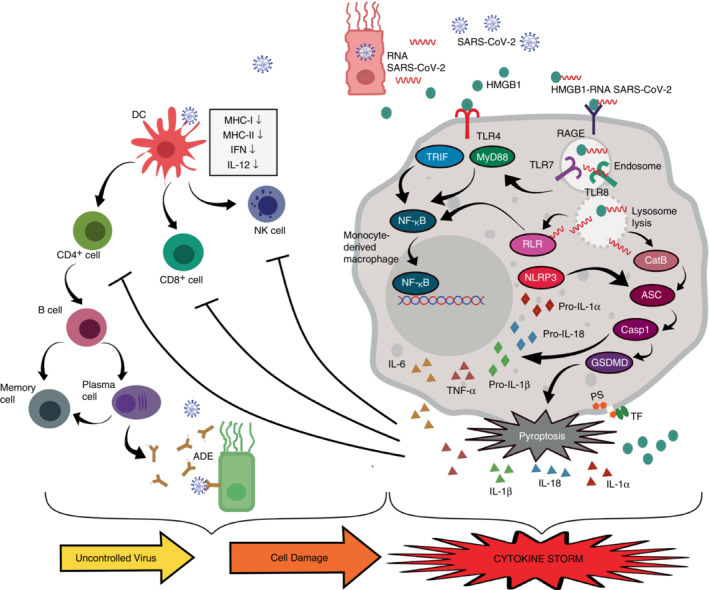
The role of HMGB1 in the cytokine storm in SARS‐CoV‐2 infection. Cell death due to SARS‐CoV‐2 infection releases many DAMPs and PAMPs including HMGB1 and RNA of SARS‐CoV‐2 into the extracellular space. HMGB1 activates TLR4 which triggers an inflammatory response via TRIF and MyD88. HMGB1 binds to viral RNA and triggers endocytosis via RAGE. The HMGB1‐RNA complex induces endo‐lysosomal rupture due to the high acidity of HMGB1. While in the endosome, viral RNA can activate TLR7/8 which in turn triggers an inflammatory response via the MyD88 pathway. The rupture of lysosomes induces viral RNA release into the cytosol. RLR detects this viral RNA and triggers the transcription of inflammasome NLRP3 and pro‐inflammatory cytokine genes such as IL‐6, TNF‐α, pro‐IL‐1β, and pro‐IL‐18 via the NF‐KB pathway. Lysosomal lysis also induces the release of Cathepsin B to mediate pyroptosome ASC which subsequently activates caspase 1. Caspase 1 cleaves GSDMD to trigger pyroptosis. Active caspase 1 cleaves pro‐IL‐1β and pro‐IL‐18 into mature IL‐1β and IL‐18. GSDMD pores mediate calpain activation, resulting in IL‐1α maturation and induce phosphatidylserine (PS) exposure‐mediated tissue factor (TF) activation. Pyroptosis triggers extracellular secretion of IL‐6, TNF, IL‐1α, IL‐1β, IL‐18, and HMGB1. High levels of IL‐6 and TNF‐α inhibit NK cells and T cells. On the other hand, SARS‐CoV‐2 infection inhibits DCs which further suppressed NK cells, T cells, and B cells. Inadequate antibodies production might trigger antibody‐dependent enhancement (ADE) phenomenon. This condition leads to an uncontrolled viral infection and cell damage with more HMGB1 released. Altogether these mechanisms trigger a proinflammatory cycle leading to a cytokine storm.
DAMPs such as proteins, nucleic acids, and extracellular matrix are harmless until they engage with specific PRRs to initiate innate immune responses [10]. Severely ill COVID‐19 patients displayed abundant DAMPs and PAMPs present in their blood and lungs [92]. HMGB1 interaction with other DAMPs or PAMPs could facilitate them to engage with proinflammatory cytosolic receptors via RAGE. This interaction might also affect HMGB1 quantification. Plasma samples pretreatment with perchloric acid capable of dissociating HMGB1‐molecules‐bound complexes revealed higher HMGB1 concentration compared to untreated samples. It seems that HMGB1‐DAMPs/PAMPs complexes might interfere with antibody recognition during ELISA assays which could lead to an underestimated HMGB1 concentration [93].
HMGB1 ANTAGONISTS AS POTENTIAL CANDIDATES FOR COVID‐19 THERAPY
The COVID‐19 treatment guidelines recommend using SARS‐CoV‐2 monoclonal antibodies, antivirals, and corticosteroids or their combination according to certain conditions. Management guidelines are under periodic revisions because of the lack of specific treatment for COVID‐19 [94]. This situation opens an opportunity for COVID‐19 candidate therapy exploration. HMGB1 is known as a candidate for therapeutic targets in inflammatory diseases [95]. Administrations of anti‐HMGB1 were reported to be able to modulate the cytokine profile which was associated with clinical improvement in the in vitro and in vivo sepsis model [56, 96]. Some of the HMGB1 antagonists are potential candidates for COVID‐19 therapy.
Glycyrrhizin.
Glycyrrhizin (GL) was isolated from the root of the plant Glycyrrhiza glabra/licorice. Glycyrrhetinic acid (GA) is a major metabolite of GL [97]. Several reviews highlight the strong potential of GL and its derivatives as candidates for COVID‐19 therapy [98, 99, 100]. Bioinformatics analysis predicts antiviral, antioxidant, and anti‐inflammatory activities of GA could inhibit SARS‐CoV‐2 infection [101].
GL inhibits HMGB1 by direct binding. Molecular docking visualization demonstrated the binding of GL to the HMGB1 surface receptor at residues R23, K42, R109, and K126 in the A and B boxes [102, 103]. GA can also bind to HMGB1 domain although it is less stable than GL, which binds to HMGB1 extracellularly and intracellularly due to its ability to penetrate membrane phospholipids [98]. In addition, both GL and GA are able to inhibit other intracellular and extracellular inflammatory mediators, such as ILs, chemokines, NF‐KB, and mitogen‐activated protein kinase (MAPK) [104]. In addition to its direct binding to HMGB1, GL inhibits HMGB1 translocation from the nucleus to the cytoplasm, thereby blocking its extracellular secretion, both in vitro [105] and in vivo [106]. The inhibition of HMGB1 translocation was done by increasing the expression of the SIRT6 protein [107]. The SARS‐CoV‐2S protein in the receptor binding domain and Orf3a causes cell death through pyroptosis. IL‐1β and HMGB1 are secreted in pyroptosis. GL inhibits this mechanism by decreasing HMGB1 secretion [108].
GL binds to extracellular HMGB1 thereby inhibiting its interaction with TLR4 and RAGE which in turn decreases the expressions of JNK, p38, ERK and IkB. This effect indicated the inhibition of the NF‐KB/MAPK pathway that triggered proinflammatory cytokines. This mechanism was confirmed by the low expression of proinflammatory cytokines in GL therapy. GL had a protective role against acute lung injury (ALI) [105, 109, 110].
In addition to its potential as an HMGB1 antagonist, GL has other mechanisms to inhibit SARS‐CoV‐2 infection. The highly conserved N‐terminal domain of the SARS‐CoV‐2S protein at residues 111–158 enhances viral binding to lipid rafts and facilitates contact with the ACE2 receptors [111]. GL interacts with cholesterol in the membrane and lipid rafts thereby modulating their permeability and interfering with viral attachment and release from host cells [98]. GL's antiviral activity was demonstrated by its ability to inhibit the main protease SARS‐CoV‐2, MPRO. This protease plays a vital role in processing viral polyproteins. Furthermore, MPRO inhibition blocks viral replication [112].
Epigallocatechin‐3‐Gallate (EGCG)
Epigallocatechin‐3‐Gallate inhibits HMGB1 secretion in LPS‐induced macrophage culture even when administered 2–6 h after LPS stimulation. Systemic inhibition of HMGB1 secretion occurred when EGCG was administered intraperitoneally in a septic rat model. Administration of EGCG reduced the mortality rate. The mechanism of how EGCG inhibits HMGB1 secretion is unclear [113]. Epigallocatechin‐3‐Gallate in green tea also triggers the degradation of HMGB1 thereby inhibiting HMGB1 secretion into extracellular space. EGCG binds to HMGB1 in the cytoplasm and forms the EGCG‐HMGB1 complex. This complex triggers the formation of autophagosomes. The fusion of autophagosomes and lysosomes formed auto‐phagolysosomes that degraded HMGB1 [102]. EGCG binds to HMGB1 at residues close to Cy106 and acts as a glue to the A box and B box together. This binding induced a conformational change of the protein, with an increase in polarity and a unique surface electrostatic potential. This change led to the aggregation of HMGB1 which triggered its degradation via autophagy [114]. Molecular docking analysis confirmed by in vitro studies indicated that EGCG also had the ability to inhibit SARS‐CoV‐2 through its activity as an inhibitor to MPRO. This protease is also known as 3‐chymotrypsin‐like protease (3CLPRO) [115, 116].
Cholinergic agonists
Cholinergic agonists such as nicotine and acetylcholine interacted with nicotinic acetylcholine receptor (α7nAChR) to further activate anti‐inflammatory mechanisms. Downstream signalling of this mechanism was the inhibition of HMGB1 secretion and its interaction with TLR4 and RAGE thereby blocking the inflammatory response. Overall, activation of α7nAChR had the potential to attenuate ALI and ARDS [117]. Activation of α7nAChR enhanced NAD+‐SIRT1 pathways [118]. SIRT1 deacetylated specific lysine site of HMGB1 which subsequently prevented HMGB1 translocation from nucleus to cytoplasm and extracellular secretion [79]. Nicotine stimulated α7nAChR resulted in the inhibition of NF‐KB pathway and HMGB1 secretion from human macrophages. This mechanism was protective against sepsis and increased survival in vivo [119]. Choline is a precursor of acetylcholine which is the main neurotransmitter in the cholinergic system. Administration of choline to RAW264.7 macrophage cell culture induced by endotoxin resulted in a decrease in HMGB1 secretion. Similar result was observed in an in vivo study [120]. Acetylcholine and GTS‐21 inhibited HMGB1 endocytosis via RAGE. This inhibitory potential was dose‐dependent and able to suppress TNF release in human macrophage and RAW264.7 culture. The mechanism of how acetylcholine and GTS‐21 inhibit HMGB1 endocytosis is unknown [56]. In silico analysis showed the SARS‐CoV‐2 spike protein binds to α7nAChR and induces dysregulation of the nicotinic cholinergic system which was thought to trigger the inflammatory response in COVID‐19. Cholinergic agonists interfere with this interaction to further activate the anti‐inflammatory response [121].
Haptoglobin
Haptoglobin is a plasma protein that plays a role in the binding of free haemoglobin (Hb). The haptoglobin‐Hb complex binds to the CD163 receptors to further trigger the complex endocytosis process. This mechanism leads to the degradation of free Hb [122]. The role of haptoglobin in triggering Hb degradation was a potential therapeutic candidate for sickle cell anaemia, sepsis, blood transfusion, and subarachnoid haemorrhage [123]. Haptoglobin subunits β bind to HMGB1 A box at residues F18, T22, R24, E25, K28, H31, A54 and K55. Only all thiol and disulfide HMGB1 could bind with the full‐length haptoglobin. Haptoglobin binds to HMGB1 and interacts with CD163 to trigger endocytosis of the haptoglobin‐HMGB1 complex. This process induces the polarization of M2 macrophages which further triggers the production of IL‐10 and an increase in the production of Heme oxygenase‐1 (HO‐1). Haptoglobin‐HMGB1 binding via CD163 triggered anti‐inflammatory activity which was strengthened by a decrease in TNF and IL‐6 levels. These mechanisms protected the host against sepsis [124]. The inhibitory potential of haptoglobin to HMGB1 in COVID‐19 needs to be further explored.
Thrombomodulin
Thrombomodulin (TM) is a thrombin‐binding anticoagulant cofactor expressed on the surface of endothelial cells. The TM structure consists of five domains, with domain D1 binding to HMGB1 while D2 binds to thrombin. Thrombomodulin had anti‐coagulation and anti‐inflammatory activities. Thrombin forms a complex with TM to activate protein C which causes inactivation of factors Va and VIIIa which further blocks subsequent thrombin formation. Thus, TM triggered the anti‐coagulation activity [125]. The anti‐inflammatory activity of TM was triggered by several mechanisms. The activation of protein C triggered the endothelial protein C receptor (EPCR) to activate the protease‐activated receptor 1 (PAR‐1) system. PAR‐1 was induced by thrombin triggering the inflammatory process, but protein C via ECPR induced the anti‐inflammatory activity. TM also inhibited the interaction of TLR4 with its ligands such as HMGB1, histones, and endotoxins thereby inhibiting its proinflammatory activity [126].
The lectin‐like domain on TM could bind to HMGB1 and stimulate its degradation by thrombin [127]. TM induced all thiol and disulfide HMGB1 degradation. Suppression of HMGB1 by this mechanism inhibited allodynia in mice [128]. Administration of recombinant human soluble TM led to a decrease in the expressions of HMGB1, RAGE protein, and mRNA in the cerebrospinal sinus thrombosis model. Thus, TM protective role was not limited to decreasing the level of HMGB1 but also by inhibiting the HMGB1–RAGE interaction. Inhibition of RAGE prevented the production of proinflammatory cytokines characterized by the decreased levels of protein and mRNA of IL‐6, TNF‐α, and IL‐1β [129]. Thrombomodulin had been shown to reduce mortality in septic patients with relatively no side effects [130].
Heparin
Heparin binds directly to HMGB1 thereby blocking its interaction with the receptors on the surface of macrophages. As a result, p38 and ERK1/2 phosphorylation were suppressed characterized by decreased secretion of TNF‐α. Heparin also reduced lethality in mice exposed to LPS and HMGB1 [131]. Inhibition of p38 stimulated a decrease in pulmonary endothelial permeability induced by HMGB1. This inhibitory effect was exerted by unfractionated heparin (UFH) [132]. UFH is a branched glycosaminoglycan. The use of heparin has the potential to cause thrombocytopenia (Heparin‐induced thrombocytopenia/HIT). Low molecular weight heparin (LMWH) has a lower risk of developing HIT [133]. Dociparstat (DSTAT) is a derivative of heparin (UFH) with decreased anticoagulant activity. It can be administered in higher dosages than heparin. DSTAT could inhibit the interaction of HMGB1 with RAGE [134]. DSTAT has undergone phase 2 and 3 clinical trials for COVID‐19 therapy, but unfortunately, it was discontinued due to the low number of participants (ClinicalTrials.gov Identifier: NCT04389840).
Metformin
Metformin is an antidiabetic drug that also has an anti‐inflammatory property. Metformin was able to inhibit HMGB1 activity on LPS stimulation. In vitro study on rabbit annular stem cell culture showed that metformin administration in LPS stimulation led to a decrease in HMGB1 secretion, which accumulated the HMGB1 in the nucleus [135]. Metformin increased Adenosine monophosphate‐activated protein kinase (AMPK) activity. Metformin activated AMPK indirectly through the inhibition of the reaction of ADP to ATP. When the ADP:ATP ratio increased, the AMP:ATP ratio would also increase and trigger AMPK activity [136]. AMPK plays a role in activating the elimination of dead cells or efferocytosis. HMGB1 induces a decrease in efferocytosis. The mechanism of metformin in inhibiting HMGB1 and activating AMPK played a role in improving lung function in patients with ARDS [137]. Metformin binds directly to HMGB1 on the acidic C‐terminal tail, inhibiting p38 phosphorylation in macrophage cells and TNF‐α secretion in mouse serum [138].
Although the above‐mentioned compounds successfully inhibited HMGB1 in the in vitro and in vivo studies, none of the HMGB1 antagonists have been recommended as a standard regiment for COVID‐19 patients in the clinical setting. There is a concern that HMGB1–PAMPs/DAMPs complexes might interfere with HMGB1 antagonist binding causing reduced effectiveness of these compounds which could be misinterpreted as if HMGB1 was not involved in the disease pathogenesis. This challenging issue urgently needs to be resolved in the future.
CONCLUSIONS
DAMPs and PAMPs interaction might impair the host immune response and trigger unintended negative outcomes. This field has not been explored extensively in COVID‐19. In this review, we proposed the pathophysiology of HMGB1‐RNA of SARS‐CoV‐2 complexes induces cytokine storms in COVID‐19. The downstream signals triggered by this interaction also have been described in detailed and comprehensive ways. This pathway needs to be explored further in in vitro and in vivo studies. Elderly, male, and excessive nutrition could exaggerate downstream signalling of this mechanism. When extracellular HMGB1 and SARS‐CoV‐2 RNA accumulations are present in these high‐risk patients, special precautions should be taken. HMGB1 has been shown as a potential molecular target of COVID‐19 therapy. Several identified potential antagonist compounds have been described. Some of these compounds have a double action, such as HMGB1 and SARS‐CoV‐2 downstream signal inhibitors. Glycyrrhizin might have more potential than other compounds for its ability to inhibit intracellularly as well as extracellular HMGB1 and antiviral activity. These compounds should be considered to give benefit in alleviating cytokine storms and may serve as potential candidates for COVID‐19 therapy.
FUNDING STATEMENT
There is no financial support to this work.
AUTHOR CONTRIBUTIONS
Sri Wulandari contributed to the development of the review and authored the initial manuscript. Hartono contributed to the development of the review and curated the review. Tri Wibawa contributed substantially to the contents of the review and supervised the whole process.
CONFLICT OF INTEREST
The authors declare that they have no actual or potential conflict of interest.
ACKNOWLEDGEMENT
None.
Wulandari S, Hartono, Wibawa T. The role of HMGB1 in COVID‐19‐induced cytokine storm and its potential therapeutic targets: A review. Immunology. 2023. 10.1111/imm.13623
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during current study.
REFERENCES
- 1. Naqvi AAT, Fatima K, Mohammad T, Fatima U, Singh IK, Singh A, et al. Insights into SARS‐CoV‐2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. BBA‐Mol Basis Disease. 2020;1866(10):165878. 10.1016/j.bbadis.2020.165878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. World Health Organization WHO coronavirus (COVID‐19) dashboard with vaccination data. Available at https://covid19.who.int/. Accessed 13 Nov 2022
- 3. Del Valle DM, Kim‐Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, et al. An inflammatory cytokine signature predicts COVID‐19 severity and survival. Nat Med. 2020;26(10):1636–43. 10.1038/s41591-020-1051-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu QQ, Cheng A, Wang Y, Li H, Hu L, Zhao X, et al. Cytokines and their relationship with the severity and prognosis of coronavirus disease 2019 (COVID‐19): a retrospective cohort study. BMJ Open. 2020;10(11):1–10. 10.1136/bmjopen-2020-041471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tang Y, Liu J, Zhang D, Xu Z, Ji J, Wen C. Cytokine storm in COVID‐19: the current evidence and treatment strategies. Front Immunol. 2020;11:1–13. 10.3389/fimmu.2020.01708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lega S, Naviglio S, Volpi S, Tommasini A. Recent insight into SARS‐COV2 immunopathology and rationale for potential treatment and preventive strategies in COVID‐19. Vaccine. 2020;8(2):1–30. 10.3390/vaccines8020224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single‐cell landscape of bronchoalveolar immune cells in patients with COVID‐19. Nat Med. 2020;26(6):842–4. 10.1038/s41591-020-0901-9 [DOI] [PubMed] [Google Scholar]
- 8. Martinez FO, Combes TW, Orsenigo F, Gordon S. Monocyte activation in systemic COVID‐19 infection: assay and rationale. EBioMedicine. 2020;59:102964. 10.1016/j.ebiom.2020.102964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID‐19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20(6):363–74. 10.1038/s41577-020-0311-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Land WG. Role of DAMPs in respiratory virus‐induced acute respiratory distress syndrome—with a preliminary reference to SARS‐CoV‐2 pneumonia. Genes Immune. 2021;22(3):141–60. 10.1038/s41435-021-00140-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang H, Wang H, Chavan SS, Andersson U. High mobility group box protein 1 (HMGB1): the prototypical endogenous danger molecule. Mol Med. 2015;21:S6–12. 10.2119/molmed.2015.00087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang M, Gauthier A, Daley LA, Dial K, Wu J, Woo J, et al. The role of HMGB1, a nuclear damage‐associated molecular pattern molecule, in the pathogenesis of lung diseases. Antioxid Redox Signal. 2019;31(13):954–93. 10.1089/ars.2019.7818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Patel MC, Shirey KA, Boukhvalova MS, Vogel SN, Blanco JCG. Serum high‐mobility‐group box 1 as a biomarker and a therapeutic target during respiratory virus infections. MBio. 2018;9(2):1–13. 10.1128/mBio.00246-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rayavara K, Kurosky A, Stafford SJ, Garg NJ, Brasier AR, Garofalo RP, et al. Proinflammatory effects of respiratory syncytial virus‐induced epithelial HMGB1 on human innate immune cell activation. J Immunol. 2018;201(9):2753–66. 10.4049/jimmunol.1800558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cicco S, Cicco G, Racanelli V, Vacca A. Neutrophil extracellular traps (NETs) and damage‐associated molecular patterns (DAMPs): two potential targets for COVID‐ 19 treatment. Mediators Inflamm. 2020;2020:1–25. 10.1155/2020/7527953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van den Berg DF, te Velde AA. Severe COVID‐19: NLRP3 inflammasome dysregulated. Front Immunol. 2020;11:1–6. 10.3389/fimmu.2020.01580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Colavita L, Ciprandi G, Salpietro A, Cuppari C. HMGB1: a pleiotropic activity. Pediatr Allergy Immunol. 2020;31(S26):63–5. 10.1111/pai.13358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Street ME. HMGB1: a possible crucial therapeutic target for COVID‐19? Horm Res Paediatr. 2020;93(2):73–5. 10.1159/000508291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mandke P, Vasquez KM. Interactions of high mobility group box protein 1 (HMGB1) with nucleic acids: implications in DNA repair and immune responses. DNA Repair. 2019;83:102701. 10.1016/j.dnarep.2019.102701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anggayasti WL, Mancera RL, Bottomley S, Helmerhorst E. The self‐association of HMGB1 and its possible role in the binding to DNA and cell membrane receptors. FEBS Lett. 2017;591(2):282–94. 10.1002/1873-3468.12545 [DOI] [PubMed] [Google Scholar]
- 21. Mu SW, Dang Y, Sen WS, Gu JJ. The role of high mobility group box 1 protein in acute cerebrovascular diseases (review). Biomed Reports. 2018;9(3):191–7. 10.3892/br.2018.1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yuan S, Liu Z, Xu Z, Liu J, Zhang J. High mobility group box 1 (HMGB1): a pivotal regulator of hematopoietic malignancies. J Hematol Oncol. 2020;13(1):1–19. 10.1186/s13045-020-00920-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leblanc PM, Doggett TA, Choi J, Hancock MA, Durocher Y, Frank F, et al. An immunogenic peptide in the A‐box of HMGB1 protein reverses apoptosis‐induced tolerance through RAGE receptor. J Biol Chem. 2014;289(11):7777–86. 10.1074/jbc.M113.541474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gong W, Zheng Y, Chao F, Li Y, Xu Z, Huang G, et al. The anti‐inflammatory activity of HMGB1 a box is enhanced when fused with C‐terminal acidic tail. J Biomed Biotechnol. 2010;2010:915234. 10.1155/2010/915234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lotze MT, Tracey KJ. High‐mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5(4):331–42. 10.1038/nri1594 [DOI] [PubMed] [Google Scholar]
- 26. Tang Y, Zhao X, Antoine D, Xiao X, Wang H, Andersson U, et al. Regulation of posttranslational modifications of HMGB1 during immune responses. Antioxidants Redox Signal. 2016;24(12):620–34. 10.1089/ars.2015.6409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kalinina N, Agrotis A, Antropova Y, DiVitto G, Kanellakis P, Kostolias G, et al. Increased expression of the DNA‐binding cytokine HMGB1 in human atherosclerotic lesions: role of activated macrophages and cytokines. Arterioscler Thromb Vasc Biol. 2004;24:2320–5. 10.1161/01.ATV.0000145573.36113.8a [DOI] [PubMed] [Google Scholar]
- 28. Lu B, Antoine DJ, Kwan K, Lundbäck P, Wähämaa H, Schierbeck H, et al. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc Natl Acad Sci USA. 2014;111(8):3068–73. 10.1073/pnas.1316925111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Andersson U, Yang H, Harris H. High‐mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin Immunol. 2018;38:40–8. 10.1016/j.smim.2018.02.011 [DOI] [PubMed] [Google Scholar]
- 30. Wei J, Alfajaro MM, DeWeirdt PC, Hanna RE, Lu‐Culligan WJ, Cai WL, et al. Genome‐wide CRISPR screens reveal host factors critical for SARS‐CoV‐2 infection. Cell. 2021;184:76–91. 10.1016/j.cell.2020.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen R, Huang Y, Quan J, Liu J, Wang H, Billiar TR, et al. HMGB1 as a potential biomarker and therapeutic target for severe COVID‐19. Heliyon. 2020;6:e05672. 10.1016/j.heliyon.2020.e05672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen L, Long X, Xu Q, Tan J, Wang G, Cao Y, et al. Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID‐19 patients. Cell Mol Immunol. 2020;17:992–4. 10.1038/s41423-020-0492-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fan X, Song J‐W, Wang S‐Y, Cao W‐J, Wang X‐W, Zhou M‐J, et al. Changes of damage associated molecular patterns in COVID‐19 patients. Infect Dis Immun. 2021;1(1):20–7. 10.1097/01.ID9.0000733572.40970.6c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sivakorn C, Dechsanga J, Jamjumrus L, Boonnak K, Schultz MJ, Dorndorp AM, et al. High mobility group box 1 and interleukin 6 at intensive care unit admission as biomarkers in critically ill COVID‐19 patients. Am J Trop Med Hyg. 2021;105(1):73–80. 10.4269/ajtmh.21-0165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vabret N, Britton GJ, Gruber C, Hegde S, Kim J, Kuksin M, et al. Immunology of COVID‐19: current state of the science. Immunity. 2020;52:910–41. 10.1016/j.immuni.2020.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Quan C, Li C, Ma H, Li Y. Immunopathogenesis of coronavirus‐induced acute respiratory distress syndrome (ARDS): potential infection‐associated hemophagocytic lymphohistiocytosis. Clin Microbiol Rev. 2021;34(1):e00074–20. 10.1128/CMR.00074-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Song X, Hu W, Yu H, Zhao L, Zhao Y, Zhao X, et al. Little to no expression of angiotensin‐converting enzyme‐2 on most human peripheral blood immune cells but highly expressed on tissue macrophages. Cytometry. 2020;2020:1–10. 10.1002/cyto.a.24285 [DOI] [PubMed] [Google Scholar]
- 38. Wang C, Xie J, Zhao L, Fei X, Zhang H, Tan Y, et al. Alveolar macrophage dysfunction and cytokine storm in the pathogenesis of two severe COVID‐19 patients. EBioMedicine. 2020;57:102833. 10.1016/j.ebiom.2020.102833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Boumaza A, Gay L, Mezouar S, Bestion E, Diallo AB, Michel M, et al. Monocytes and macrophages, targets of severe acute respiratory syndrome coronavirus 2: the clue for coronavirus disease 2019 immunoparalysis. J Infect Dis. 2021;224(3):395–406. 10.1093/infdis/jiab044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yilla M, Harcourt BH, Hickman CJ, McGrew M, Tamin A, Goldsmith CS, et al. SARS‐coronavirus replication in human peripheral monocytes/macrophages. Virus Res. 2005;107(1):93–101. 10.1016/j.virusres.2004.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cinatl J, Michaelis M, Scholz M, Doerr HW. Role of interferons in the treatment of severe acute respiratory syndrome. Expert Opin Biol Ther. 2004;4(6):827–36. 10.1517/14712598.4.6.827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Teijaro JR. Type I interferons in viral control and immune regulation. Curr Opin Virol. 2016;16:31–40. 10.1016/j.coviro.2016.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Contoli M, Papi A, Tomassetti L, Rizzo P, Sega FVD, Fortini F, et al. Blood interferon‐α levels and severity, outcomes, and inflammatory profiles in hospitalized COVID‐19 patients. Front Immunol. 2021;12:1–10. 10.3389/fimmu.2021.648004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou R, To KKW , Wong YC, Liu L, Zhou B, Li X, et al. Acute SARS‐CoV‐2 infection impairs dendritic cell and T cell responses. Immunity. 2020;53:864–77. 10.1016/j.immuni.2020.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Borges RC, Hohmann MS, Borghi SM. Dendritic cells in COVID‐19 immunopathogenesis: insights for a possible role in determining disease outcome. Int Rev Immunol. 2020;40:1–18. 10.1080/08830185.2020.1844195 [DOI] [PubMed] [Google Scholar]
- 46. Han J, Sun J, Zhang G, Chen H. DCs‐based therapies: potential strategies in severe SARS‐CoV‐2 infection. Int J Med Sci. 2021;18:406–18. 10.7150/ijms.47706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Iwasaki A, Yang Y. The potential danger of suboptimal antibody responses in COVID‐19. Nat Rev Immunol. 2020;20(6):339–41. 10.1038/s41577-020-0321-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Khoshkam Z, Aftabi Y, Stenvinkel P, Paige Lawrence B, Rezaei MH, Ichihara G, et al. Recovery scenario and immunity in COVID‐19 disease: a new strategy to predict the potential of reinfection. J Adv Res. 2021;31:49–60. 10.1016/j.jare.2020.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ahmed F, Jo DH, Lee SH. Can natural killer cells be a principal player in anti‐SARS‐CoV‐2 immunity? Front Immunol. 2020;11:5886765. 10.3389/fimmu.2020.586765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Daolin T, Rui K, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0 s that spur autophagy and immunity. Immunol Rev. 2012;249:158–75. 10.1111/j.1600-065X.2012.01146.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Trypsteen W, Van Cleemput J, van Snippenberg W, Gerlo S, Vandekerckhove L. On the whereabouts of SARS‐CoV‐2 in the human body: a systematic review. PLoS Pathog. 2020;16(10):e1009037. 10.1371/journal.ppat.1009037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Andersson U, Ottestad W, Tracey KJ. Extracellular HMGB1: a therapeutic target in severe pulmonary inflammation including COVID‐19? Mol Med. 2020;26:42. 10.1186/s10020-020-00172-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee SA, Kwak MS, Kim S, Shin JS. The role of high mobility group box 1 in innate immunity. Yonsei Med J. 2014;55(5):1165–76. 10.3349/ymj.2014.55.5.1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kawasaki T, Kawai T. Toll‐like receptor signaling pathways. Front Immunol. 2014;5:461. 10.3389/fimmu.2014.00461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xu J, Jiang Y, Wang J, Shi X, Liu Q, Liu Z, et al. Macrophage endocytosis of high‐mobility group box 1 triggers pyroptosis. Cell Death Differ. 2014;21(8):1229–39. 10.1038/cdd.2014.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang H, Liu H, Zeng Q, Imperato GH, Addorisio ME, Li J, et al. Inhibition of HMGB1/RAGE‐mediated endocytosis by HMGB1 antagonist box a, anti‐HMGB1 antibodies, and cholinergic agonists suppresses inflammation. Mol Med. 2019;25:13. 10.1186/s10020-019-0081-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lan J, Luo H, Wu R, Wang J, Zhou B, Zhang Y, et al. Internalization of HMGB1 (high mobility group box 1) promotes angiogenesis in endothelial cells. Arterioscler Thromb Vasc Biol. 2020;40:2922–40. 10.1161/ATVBAHA.120.315151 [DOI] [PubMed] [Google Scholar]
- 58. Deng M, Tang Y, Li W, Tracey KJ, Billiar TR, Lu B. The endotoxin delivery protein HMGB1 mediates caspase‐11‐dependent lethality in sepsis. Immunity. 2018;49:740–3. 10.1016/j.immuni.2018.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–92. 10.1038/nature13683 [DOI] [PubMed] [Google Scholar]
- 60. Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, et al. HMGB proteins function as universal sentinels for nucleic‐acid‐mediated innate immune responses. Nature. 2009;462(7269):99–103. 10.1038/nature08512 [DOI] [PubMed] [Google Scholar]
- 61. Tatematsu M, Funami K, Seya T, Matsumoto M. Extracellular RNA sensing by pattern recognition receptors. J Innate Immun. 2018;10:398–406. 10.1159/000494034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mdkhana B, Sharif‐Askari NS, Ramakrishnan RK, Goel S, Hamid Q, Halwani R. Nucleic acid‐sensing pathways during SARS‐CoV‐2 infection: expectations versus reality. J Inflamm Res. 2021;14:199–216. 10.2147/JIR.S277716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu T, Zhang L, Joo D, Su S‐C. NF‐κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:e17023. 10.1038/sigtrans.2017.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov. 2020;6:36. 10.1038/s41421-020-0167-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tsuchiya K, Hosojima S, Hara H, Kushiyama H, Mahib MR. Gasdermin D mediates the maturation and release of IL‐1α downstream of inflammasomes. Cell Rep. 2021;34:108887. 10.1016/j.celrep.2021.108887 [DOI] [PubMed] [Google Scholar]
- 66. Tamayo‐Velasco Á, Martínez‐Paz P, Peñarrubia‐Ponce MJ, de la Fuente I, Pérez‐González S, Fernández I, et al. HGF, IL‐1α, and IL‐27 are robust biomarkers in early severity stratification of COVID‐19 patients. J Clin Med. 2021;10:2017. 10.3390/jcm10092017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Al‐Muhana IR, Ahmed MM, Al‐Muhana IR, AL‐Hasan BA. Impact of proinflammatory cytokines (interleukin 6, interleukin 1α, and interleukin 1β) on biochemical parameters in severe acute respiratory syndrome coronavirus 2 patients in Iraq. Biomed Biotechnol Res J. 2022;6:170–4. 10.4103/bbrj.bbrj_249_21 [DOI] [Google Scholar]
- 68. Yang X, Cheng X, Tang Y, Qiu X, Wang Y, Kang H, et al. Bacterial endotoxin activates the coagulation cascade through gasdermin D‐dependent phosphatidylserine exposure. Immunity. 2019;51:983–96. 10.1016/j.immuni.2019.11.005 [DOI] [PubMed] [Google Scholar]
- 69. Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, et al. Inflammasome‐dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185:4385–92. 10.4049/jimmunol.1000803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Volchuk A, Ye A, Chi L, Steinberg BE, Goldenberg NM. Indirect regulation of HMGB1 release by gasdermin D. Nat Commun. 2020;11:4561. 10.1038/s41467-020-18443-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ferreira AC, Soares VC, de Azevedo‐Quintanilha IG, da Dias SSG, Fintelman‐Rodrigues N, Sacramento CQ, et al. SARS‐CoV‐2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov. 2021;7:43. 10.1038/s41420-021-00428-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. El‐Zayat SR, Sibaii H, Mannaa FA. Micronutrients and many important factors that affect the physiological functions of toll‐like receptors. Bull Natl Res Cent. 2019;43:123. 10.1186/s42269-019-0165-z [DOI] [Google Scholar]
- 73. Wu X, Cakmak S, Wortmann M, Hakimi M, Zhang J, Böckler D, et al. Sex‐and disease‐specific inflammasome signatures in circulating blood leukocytes of patients with abdominal aortic aneurysm. Mol Med. 2016;22:508–18. 10.2119/molmed.2016.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sutiningsih D, Rahatina VEF, Prabowo Y, Haryanto A, Wibowo MA. Epidemiologic and clinical characteristics of patients with COVID‐19 in Central Java, Indonesia. E3S Web Conf. 2020;202:12014. 10.1051/e3sconf/202020212014 [DOI] [Google Scholar]
- 75. Lakbar I, Luque‐Paz D, Mege JL, Einav S, Leone M. COVID‐19 gender susceptibility and outcomes: a systematic review. PLoS One. 2020;15(11):e0241827. 10.1371/journal.pone.0241827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Latz E, Duewell P. NLRP3 inflammasome activation in inflammaging. Semin Immunol. 2018;40:61–73. 10.1016/j.smim.2018.09.001 [DOI] [PubMed] [Google Scholar]
- 77. Nunzio P, Imbimbo BP. Impairment of the cholinergic anti‐inflammatory pathway in older subjects with severe COVID‐19. Med Hypotheses. 2020;144:110274. 10.1016/j.mehy.2020.110274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lee SH, Lee JH, Lee HY, Min KJ. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019;52(1):24–34. 10.5483/BMBRep.2019.52.1.290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wei S, Gao Y, Dai X, Fu W, Cai S, Fang H, et al. SIRT1‐mediated HMGB1 deacetylation suppresses sepsis‐associated acute kidney injury. Am J Physiol Physiol. 2019;316(1):F20–31. 10.1152/ajprenal.00119.2018 [DOI] [PubMed] [Google Scholar]
- 80. Stamatovic SM, Martinez‐Revollar G, Hu A, Choi J, Keep RF, Andjelkovic AV. Decline in sirtuin‐1 expression and activity plays a critical role in blood‐brain barrier permeability in aging. Neurobiol Dis. 2019;126:105–16. 10.1016/j.nbd.2018.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rheinheimer J, de Souza BM, Cardoso NS, Bauer AC, Crispim D. Current role of the NLRP3 inflammasome on obesity and insulin resistance: a systematic review. Metabolism. 2017;74:1–9. 10.1016/j.metabol.2017.06.002 [DOI] [PubMed] [Google Scholar]
- 82. Verma D, Lerm M, Julinder RB, Eriksson P, Söderkvist P, Särndahl E. Gene polymorphisms in the NALP3 inflammasome are associated with interleukin‐1 production and severe inflammation relation to common inflammatory diseases? Arthritis Rheum. 2008;58:888–94. 10.1002/art.23286 [DOI] [PubMed] [Google Scholar]
- 83. Velazquez‐Salinas L, Verdugo‐Rodriguez A, Rodriguez LL, Borca MV. The role of interleukin 6 during viral infections. Front Microbiol. 2019;10:1057. 10.3389/fmicb.2019.01057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mazzoni A, Salvati L, Maggi L, Capone M, Vanni A, Spinicci M, et al. Impaired immune cell cytotoxicity in severe COVID‐19 is IL‐6 dependent. J Clin Invest. 2020;130(9):4694–703. 10.1172/JCI138554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID‐19). Front Immunol. 2020;11:827. 10.3389/fimmu.2020.00827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cavalli G, Dinarello CA. Anakinra therapy for non‐cancer inflammatory diseases. Front Pharmacol. 2018;9:1157. 10.3389/fphar.2018.01157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yasuda K, Nakanishi K, Tsutsui H. Interleukin‐18 in health and disease. Int J Mol Sci. 2019;20:649. 10.3390/ijms20030649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Satış H, Özger HS, Aysert Yıldız P, Hızel K, Gulbahar Ö, Erbaş G, et al. Prognostic value of interleukin‐18 and its association with other inflammatory markers and disease severity in COVID‐19. Cytokine. 2021;137:155302. 10.1016/j.cyto.2020.155302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tjan LH, Furukawa K, Nagano T, Kiriu T, Nishimura M, Arii J, et al. Early differences in cytokine production by severity of coronavirus disease 2019. J Infect Dis. 2021;223:1145–9. 10.1093/infdis/jiab005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Crayne CB, Albeituni S, Nichols KE, Cron RQ. The immunology of macrophage activation syndrome. Front Immunol. 2019;10:119. 10.3389/fimmu.2019.00119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Jamilloux Y, Henry T, Belot A, Viel S, Fauter M, El T, et al. Should we suppress or stimulate immune responses for COVID‐19? Cytokine and anti‐cytokine interventions. Autoimmun Rev. 2020;19:102567. 10.1016/j.autrev.2020.102567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Naqvi I, Giroux N, Olson L, Morrison SA, Llanga T, Akinade TO , et al. DAMPs/PAMPs induce monocytic TLR activation and tolerance in COVID‐19 patients; nucleic acid binding scavengers can counteract such TLR agonists. Biomaterials. 2022;283:121393. 10.1016/j.biomaterials.2022.121393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Andersson U, Tracey KJ, Yang H. Post‐translational modification of HMGB1 disulfide bonds in stimulating and inhibiting inflammation. Cell. 2021;10:3323. 10.3390/cells10123323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. COVID‐19 Treatment Guidelines Panel . Coronavirus disease 2019 (COVID‐19) treatment guidelines. Bethesda, MD: National Institutes of Health; 2022. Avalaible at https://www.covid19treatmentguidelines.nih.gov/. Accessed 10 Nov 2022. [Google Scholar]
- 95. Vanpatten S, Al‐Abed Y. High mobility group box‐1 (HMGB1): current wisdom and advancement as a potential drug target. J Med Chem. 2018;61:5093–107. 10.1021/acs.jmedchem.7b01136 [DOI] [PubMed] [Google Scholar]
- 96. Stevens NE, Chapman MJ, Fraser CK, Kuchel TR, Hayball JD, Diener KR. Therapeutic targeting of HMGB1 during experimental sepsis modulates the inflammatory cytokine profile to one associated with improved clinical outcomes. Sci Rep. 2017;7:5850. 10.1038/s41598-017-06205-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lim TK. Glycyrrhiza glabra. Edible medicinal and non‐medicinal plants. Volume 10. Springer; 2016. p. 1–659. 10.1007/978-94-017-7276-1 [DOI] [Google Scholar]
- 98. Bailly C, Vergoten G. Glycyrrhizin: an alternative drug for the treatment of COVID‐19 infection and the associated respiratory syndrome? Pharmacol Ther. 2020;214:107618. 10.1016/j.pharmthera.2020.107618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Gomaa AA, Abdel‐Wadood YA. The potential of glycyrrhizin and licorice extract in combating COVID‐19 and associated conditions. Phytomed Plus. 2021;1(3):100043. 10.1016/j.phyplu.2021.100043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Chrzanowski J, Chrzanowska A, Graboń W. Glycyrrhizin: an old weapon against a novel coronavirus. Phyther Res. 2021;35:629–36. 10.1002/ptr.6852 [DOI] [PubMed] [Google Scholar]
- 101. Zheng W, Huang X, Lai Y, Liu X, Jiang Y, Zhan S. Glycyrrhizic acid for COVID‐19: findings of targeting pivotal inflammatory pathways triggered by SARS‐CoV‐2. Front Pharmacol. 2021;12:631206. 10.3389/fphar.2021.631206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Wu AH, He L, Long W, Zhou Q, Zhu S, Wang P, et al. Novel mechanisms of herbal therapies for inhibiting HMGB1 secretion or action evidence‐based complement. Altern Med. 2015;2015:456305. 10.1155/2015/456305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Mollica L, De Marchis F, Spitaleri A, Dallacosta C, Pennacchini D, Zamai M, et al. Glycyrrhizin binds to high‐mobility group box 1 protein and inhibits its cytokine activities. Chem Biol. 2007;14:431–41. 10.1016/j.chembiol.2007.03.007 [DOI] [PubMed] [Google Scholar]
- 104. Richard SA. Exploring the pivotal immunomodulatory and anti‐inflammatory potentials of glycyrrhizic and glycyrrhetinic acids. Mediators Inflamm. 2021;2021:6699560. 10.1155/2021/6699560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zhao F, Fang Y, Deng S, Li X, Zhou Y, Gong Y, et al. Glycyrrhizin protects rats from sepsis by blocking HMGB1 signaling. Biomed Res Int. 2017;2017:9719647. 10.1155/2017/9719647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Li Y, Wang L, Zhang B, Gao F, Yang CM. Glycyrrhizin, an HMGB1 inhibitor, exhibits neuroprotective effects in rats after lithium‐pilocarpine‐induced status epilepticus. J Pharm Pharmacol. 2019;71:390–9. 10.1111/jphp.13040 [DOI] [PubMed] [Google Scholar]
- 107. Chen D, Bellussi LM, Cocca S, Wang J, Passali GC, Hao X, et al. Glycyrrhetinic acid suppressed HMGB1 release by up‐regulation of SIRT6 in nasal inflammation. J Biol Regul Homeost Agents. 2017;31(2):269–77. [PubMed] [Google Scholar]
- 108. Gowda P, Patrick S, Joshi SD, Kumawat RK, Sen E. Glycyrrhizin prevents SARS‐CoV‐2 S1 and Orf3a induced high mobility group box 1 (HMGB1) release and inhibits viral replication. Cytokine. 2021;142:155496. 10.1016/j.cyto.2021.155496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Lee SA, Lee SH, Kim JY, Lee WS. Effects of glycyrrhizin on lipopolysaccharide‐induced acute lung injury in a mouse model. J Thorac Dis. 2019;11(4):1287–302. 10.21037/jtd.2019.04.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Shi X, Yu L, Zhang Y, Liu Z, Zhang H, Zhang Y, et al. Glycyrrhetinic acid alleviates hepatic inflammation injury in viral hepatitis disease via a HMGB1‐TLR4 signaling pathway. Int Immunopharmacol. 2020;84:106578. 10.1016/j.intimp.2020.106578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Fantini J, Di C, Chahinian H, Yahi N. Structural and molecular modelling studies reveal a new mechanism of action of chloroquine and hydroxychloroquine against SARS‐CoV‐2 infection. Int J Antimicrob Agents. 2020;55:105960. 10.1016/j.ijantimicag.2020.105960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. van de Sand L, Bormann M, Alt M, Schipper L, Heilingloh CS, Steinmann E, et al. Glycyrrhizin effectively inhibits SARS‐CoV‐2 replication by inhibiting the viral main protease. Viruses. 2021;13:609. 10.3390/v13040609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Li W, Ashok M, Li J, Yang H, Sama AE, Wang H. A major ingredient of green tea rescues mice from lethal sepsis partly by inhibiting HMGB1. PLoS One. 2007;2(11):e1153. 10.1371/journal.pone.0001153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Meng XY, Li B, Liu S, Kang H, Zhao L, Zhou R. EGCG in green tea induces aggregation of HMGB1 protein through large conformational changes with polarized charge redistribution. Sci Rep. 2016;6:22128. 10.1038/srep22128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Du A, Zheng R, Disoma C, Li S, Chen Z, Li S, et al. Epigallocatechin‐3‐gallate, an active ingredient of traditional Chinese medicines, inhibits the 3CLpro activity of SARS‐CoV‐2. Int J Biol Macromol. 2021;176:1–12. 10.1016/j.ijbiomac.2021.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Jang M, Park YI, Cha YE, Park R, Namkoong S, Lee JI, et al. Tea polyphenols EGCG and theaflavin inhibit the activity of SARS‐CoV‐2 3CL‐protease in vitro evidence‐based complement. Altern Med. 2020;2020:5630838. 10.1155/2020/5630838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Andersson U. The cholinergic anti‐inflammatory pathway alleviates acute lung injury. Mol Med. 2020;26:64. 10.1186/s10020-020-00184-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Li D, Huang F, Ni M, Fu H, Zhang L, Shen FM. α7 nicotinic acetylcholine receptor relieves angiotensin II–induced senescence in vascular smooth muscle cells by raising nicotinamide adenine dinucleotide‐dependent SIRT1 activity. Arterioscler Thromb Vasc Biol. 2016;36(8):1566–76. 10.1161/ATVBAHA.116.307157 [DOI] [PubMed] [Google Scholar]
- 119. Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med. 2004;10(11):1216–21. 10.1038/nm1124 [DOI] [PubMed] [Google Scholar]
- 120. Parrish W, Rosas‐Ballina M, Gallowitsch‐Puerta M, Ochani M, Ochani K, Yang L, et al. Modulation of TNF release by choline requires α7 subunit nAChR mediated signalling. Mol Med. 2008;14:567–74. 10.2119/2008-00079.Parrish [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Alexandris N, Lagoumintzis G, Chasapis CT, Leonidas DD, Papadopoulos GE, Tzartos SJ, et al. Nicotinic cholinergic system and COVID‐19: in silico evaluation of nicotinic acetylcholine receptor agonists as potential therapeutic interventions. Toxicol Reports. 2021;8:73–83. 10.1016/j.toxrep.2020.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Polticelli F, Bocedi A, Minervini G, Ascenzi P. Human haptoglobin structure and function: a molecular modelling study. FEBS J. 2008;275:5648–56. 10.1111/j.1742-4658.2008.06690.x [DOI] [PubMed] [Google Scholar]
- 123. Buehler PW, Humar R, Schaer DJ. Haptoglobin therapeutics and compartmentalization of cell‐free hemoglobin toxicity. Trends Mol Med. 2020;26(7):683–97. 10.1016/j.molmed.2020.02.004 [DOI] [PubMed] [Google Scholar]
- 124. Yang H, Wang H, Levine YA, Gunasekaran MK, Wang Y, Addorisio M, et al. Identification of CD163 as an antiinflammatory receptor for HMGB1‐haptoglobin complexes. JCI Insight. 2018;3(24):1. 10.1172/jci.insight.126617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ito T, Maruyama I. Thrombomodulin: protectorate god of the vasculature in thrombosis and inflammation. J Thromb Haemost. 2011;9(Suppl. 1):168–73. 10.1111/j.1538-7836.2011.04319.x [DOI] [PubMed] [Google Scholar]
- 126. Ito T, Thachil J, Asakura H, Levy JH, Iba T. Thrombomodulin in disseminated intravascular coagulation and other critical conditions: a multi‐faceted anticoagulant protein with therapeutic potential. Crit Care. 2019;23:280. 10.1186/s13054-019-2552-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Bongoni AK, Klymiuk N, Wolf E, Ayares D, Rieben R, Cowan PJ. Transgenic expression of human thrombomodulin inhibits HMGB1‐induced porcine aortic endothelial cell activation. Transplantation. 2016;100(9):1871–9. 10.1097/TP.0000000000001188 [DOI] [PubMed] [Google Scholar]
- 128. Tsujita R, Tsubota M, Hayashi Y, Saeki H, Sekiguchi F, Kawabata A. Role of thrombin in soluble thrombomodulin‐induced suppression of peripheral HMGB1‐mediated allodynia in mice. J Neuroimmune Pharmacol. 2018;13:179–88. 10.1007/s11481-017-9773-2 [DOI] [PubMed] [Google Scholar]
- 129. Gu JJ, Bin CJ, Zhang JH, Zhang H, Sen WS. Recombinant human soluble thrombomodulin protects against brain injury in a CVST rat model, via downregulation of the HMGB1‐RAGE axis. Mol Med Rep. 2016;14:5217–22. 10.3892/mmr.2016.5891 [DOI] [PubMed] [Google Scholar]
- 130. Yoshihiro S, Sakuraya M, Hayakawa M, Ono K, Hirata A, Takaba A, et al. Recombinant human‐soluble thrombomodulin contributes to reduced mortality in sepsis patients with severe respiratory failure: a retrospective observational study using a multicenter dataset. Shock. 2019;51(2):174–9. 10.1097/SHK.0000000000001148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Li L, Ling Y, Huang M, Yin T, Gou SM, Zhan NY, et al. Heparin inhibits the inflammatory response induced by LPS and HMGB1 by blocking the binding of HMGB1 to the surface of macrophages. Cytokine. 2015;72:36–42. 10.1016/j.cyto.2014.12.010 [DOI] [PubMed] [Google Scholar]
- 132. Luan Z, Hu B, Wu L, Jin S, Ma X, Zhang J, et al. Unfractionated heparin alleviates human lung endothelial barrier dysfunction induced by high mobility group box 1 through regulation of P38‐GSK3β‐snail signaling pathway. Cell Physiol Biochem. 2018;46:1907–18. 10.1159/000489375 [DOI] [PubMed] [Google Scholar]
- 133. Junqueira DR, Zorzela LM, Perini E. Unfractionated heparin versus low molecular weight heparins for avoiding heparin‐induced thrombocytopenia in postoperative patients. Cochrane Database Syst Rev. 2017;2017:4. 10.1002/14651858.CD007557.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lasky JA, Fuloria J, Morrison ME, Lanier R, Naderer O, Brundage T, et al. Design and rationale of a randomized, double‐blind, placebo‐controlled, phase 2/3 study evaluating dociparstat in acute lung injury associated with severe COVID‐19. Adv Ther. 2020;38:782–91. 10.1007/s12325-020-01539-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Han Y, Yuan F, Deng C, He F, Zhang Y, Shen H, et al. Metformin decreases LPS‐induced inflammatory response in rabbit annulus fibrosus stem/progenitor cells by blocking HMGB1 release. Aging. 2019;11(22):10252–65. 10.18632/aging.102453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60:1577–85. 10.1007/s00125-017-4342-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Grégoire M, Uhel F, Lesouhaitier M, Gacouin A, Guirriec M, Mourcin F, et al. Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur Respir J. 2018;52:1702590. 10.1183/13993003.02590-2017 [DOI] [PubMed] [Google Scholar]
- 138. Horiuchi T, Sakata N, Narumi Y, Kimura T, Hayashi T, Nagano K, et al. Metformin directly binds the alarmin HMGB1 and inhibits its proinflammatory activity. J Biol Chem. 2017;292(20):8436–46. 10.1074/jbc.M116.769380 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during current study.