

Abstract
Tumors exhibit notable metabolic alterations compared with their corresponding normal tissue counterparts. These metabolic alterations can support anabolic growth, enable survival in hostile environments, and regulate gene expression programs that promote malignant progression. Whether these metabolic changes are selected for during malignant transformation, or can themselves be drivers of tumor initiation, is poorly understood. Suggestively, many of the major bottlenecks for tumor initiation—control of cell fate, proliferation, and survival—are all amenable to metabolic regulation. Here, we will review recent evidence demonstrating a critical role for metabolic pathways in processes that support the earliest stages of tumor development. We will discuss how cell-intrinsic factors, such as cell of origin or transforming oncogene, and cell-extrinsic factors, such as local nutrient availability, promote or restrain tumor initiation. Deeper insight into how metabolic pathways control tumor initiation will improve our ability to design metabolic interventions to limit tumor incidence.
Introduction
Cancer is a subversion of normal tissue homeostasis in which cells escape from, or fail to achieve, their proper identity in space and time, unleashing uncontrolled proliferation. Advances in genetic sequencing have enabled identification of recurrent genomic alterations capable of transforming normal cells and driving tumor growth. Mutations in an ever-lengthening list of oncogenes and tumor suppressors can be sufficient to impair normal developmental pathways and fuel aberrant proliferation. Surprisingly, such mutations are not exclusive to tumors: normal human tissue can also harbor cancer-associated mutations, and individual cells can sustain multiple oncogenic mutations and clonally expand without giving rise to any obvious pathology1,2. Moreover, oncogenic lesions have different outcomes depending on the tissue or cell type in which they are expressed3. These observations indicate that oncogenic mutations are not necessarily sufficient to initiate tumorigenesis and factors beyond genomic alterations—factors such as cell state and environmental pressure—may help determine tumor initiating potential.
Metabolism is emerging as a potential force shaping cancer initiation and tumor progression. To initiate and form a tumor, cancer cells must expand, divide and survive (FIG. 1). Meeting each of these challenges ultimately requires metabolic rearrangements, either to generate biomass required for proliferation or to support pathways enabling survival in hostile or foreign microenvironments. Metabolites can also modulate signaling pathways and gene expression programs that regulate cell state4, thereby potentially altering how cells respond to oncogenic mutations. The challenges inherent in identifying or capturing tumor initiating cells has largely circumscribed our understanding of when metabolism changes during tumor initiation and how metabolic alterations support tumor progression. Nevertheless, work in established tumors is revealing how metabolic networks enable cancer cells to thrive in the face of selective pressures5,6, providing a framework for understanding how metabolism may contribute to tumor initiation. The goal of this review is to leverage work studying metabolic determinants of cancer growth alongside recent studies of metabolic drivers of tumor initiation to provide insight into mechanisms by which metabolites may dictate the earliest stages of tumor initiation and to explore the potential for metabolic interventions to control cancer incidence and progression. Specifically, we will first discuss major barriers to tumorigenesis that may be susceptible to metabolic control. We next provide examples demonstrating the role of cancer cell-intrinsic metabolic networks in tumor progression. Finally, we highlight representative examples of ways that environmental factors can affect tumor incidence.
Fig 1. Challenges in tumor initiation.
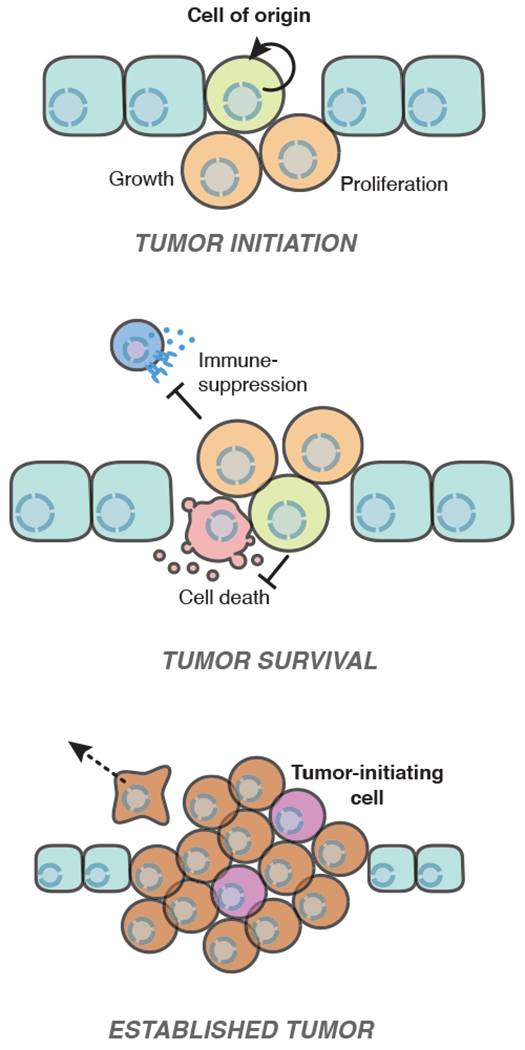
Cells must surmount multiple hurdles to form a tumor. While the primary cell responsible for initiating a tumor can vary between and even within tumor types, the cancer cell of origin is often a stem/progenitor cell or a cell that has acquired the ability to re-enter the cell cycle and undergo aberrant proliferation. To form a tumor, these cancer-initiating cells must evade differentiation and senescence to undergo successive population doublings. Cancer cells must also resist challenges such as immune recognition or cell death induced by changes in tissue architecture or local metabolism. Within an established tumor, a sub-population of “tumor-initiating cells” with stem-like features have the potential to re-initiate tumors; these cells are often therapy-resistant and responsible for cancer relapse after treatment.
Metabolic challenges during tumor initiation
To date, few studies have directly assessed the role of metabolism in tumor initiation. Tumor initiating cells are rare, and therefore difficult to capture and study7. Moreover, modeling initiation is also complicated by unclear cell of origin for many tumor types8,9. Work in this area is also limited by the inherent difficulty uncoupling initiation from progression, as many common assays rely on experimentally detectable tumors that have, by definition, progressed and grown past a few initiating cells. Nevertheless, several key processes required for tumor initiation—continuous self-renewal, proliferation and survival—are known to be under metabolic control. The goal of this section is to highlight how metabolism influences these key processes to illustrate the potential mechanisms through which metabolism may shape tumor initiation.
Supporting aberrant cell proliferation
To unleash hyperplasia, malignant cells must meet the biosynthetic demands of cell proliferation (FIG. 2). To date, much of the work determining how cancer cells rewire metabolism to enable aberrant proliferation has been performed in cells in culture, often with unphysiological—and potentially unlimiting—concentrations of available nutrients. Therefore, a major avenue of current research is elucidating how metabolic demands of cells growing in tumors differs from cells growing in tissue culture. Certainly, isotope tracing studies reveal major differences in metabolic preferences of cancer cells growing in vitro compared to tumors in vivo10,11. Likewise, simply changing medium composition is sufficient to induce large-scale metabolic alterations in cultured cells, implying that nutrient availability is a major factor dictating metabolic phenotypes12,13. These context-specific preferences can translate into altered metabolic demands: a subset of metabolic genes exhibit conditional essentiality depending on exogenous nutrient availability14,15. Somewhat surprisingly, then, recent genetic screens revealed that pancreatic cancer cells growing in 2-dimensions in vitro or as tumors in mice exhibited overall strikingly similar requirements for metabolic genes16,17. Some of these similarities may be because cells were selected in culture—thereby enforcing specific constraints on cell growth—prior to injection into mice for tumor formation assays. Nevertheless, some key differences emerged—specifically, genes involved in heme synthesis were more essential in vivo than in vivo, presumably because of increased heme degradation in vivo16,17. Moreover, 3-dimensional cell cultures preserved in vivo dependencies better than 2-dimensional cell cultures17. These findings suggest that while many of the basic principles of cell growth are similar across a range of environmental conditions, local growth conditions can modulate metabolic strategies that support growth.
Fig 2. Metabolic pathways support biomass accumulation.
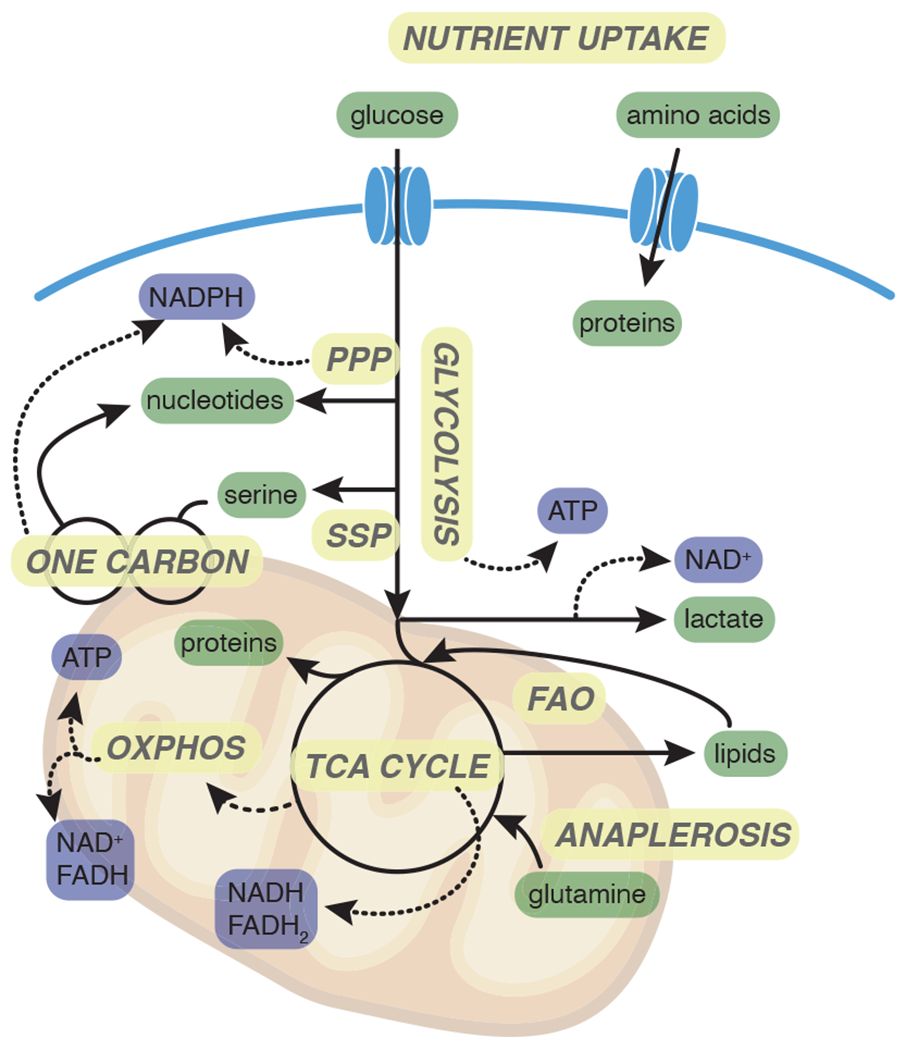
To meet the metabolic demands of proliferation, cells import nutrients including glucose and glutamine that fuel glycolysis and the TCA cycle. Intermediates of glycolysis and the TCA cycle are important precursors for the synthesis of nucleotides, proteins and lipids. Flux through glycolysis and the TCA cycle generates reducing equivalents NADH and FADH2, that are oxidized in the electron transport chain, driving ATP generation through oxidative phosphorylation. PPP: Pentose phosphate pathway; SSP: Serine synthesis pathway; FAO: Fatty acid oxidation; OXPHOS: Oxidative phosphorylation.
Indeed, broadly speaking, the metabolic demands of biomass accumulation are inherently invariant. For example, increasing mass necessarily requires enhanced nutrient uptake. Abundant circulating nutrients such as glucose and amino acids—most notably, glutamine—provide the raw material for the synthesis of membranes, proteins and nucleic acids that are required for cell division. Nutrient uptake is so fundamental to tumor growth that imaging uptake of a radioactive glucose analog, 18F-fluorodeoxyglucose (18F-FDG), via positron emission tomography (PET) is a linchpin of clinical cancer diagnosis, staging and treatment monitoring18. Similarly, imaging 18F-labeled glutamine analogs is emerging as a useful approach for monitoring tumor growth in tissues such as the brain where constitutively high glucose uptake precludes successful application of 18F-FDG imaging19.
Glucose and amino acids, once acquired by proliferating cells, are funneled into central metabolic networks that yield the energy, reducing equivalents and metabolic building blocks required for macromolecule synthesis. Glucose catabolism via glycolysis generates ATP, and glycolytic intermediates additionally serve as initiating metabolites for the pentose phosphate pathway (PPP) and serine synthesis pathway, among others. The PPP and pathways downstream of serine production provide the ribose sugar backbone and other critical intermediates required for nucleotide biosynthesis. Additionally, both pathways can generate NADPH, a carrier of reducing equivalents that is critical for biosynthetic reactions and antioxidant defenses. Amino acids and the glycolytic product pyruvate can be oxidized in the tricarboxylic acid (TCA) cycle, thereby reducing the electron carriers NAD+ and FAD to NADH and FADH220.
Like intermediates of glycolysis, intermediates of the TCA cycle are also precursors for critical biosynthetic pathways. Acetyl-CoA, generated by mitochondrial catabolism of nutrients such as glucose, fatty acids and some amino acids, is required for fatty acid synthesis in the cytosol. As acetyl-CoA cannot cross the inner mitochondrial membrane, cytosolic acetyl-CoA is instead provided by the TCA cycle intermediate citrate, which can transit from the mitochondrion to the cytosol to yield acetyl-CoA through the action of ATP citrate lyase (ACL)21. Furthermore, succinyl-CoA is the precursor for heme production, and oxaloacetate is transaminated to aspartate, an amino acid that in turn is required for both nucleic acid and protein biosynthesis. Such cataplerosis is balanced by anaplerosis—or TCA cycle refilling—mediated most often by catabolism of glutamine22. To keep the TCA cycle running and continually generate biosynthetic precursors, cells must regenerate oxidized electron carriers. As a result, a major benefit accrued by deposition of high-energy electrons into the mitochondrial electron transport chain is not only efficient production of ATP through oxidative phosphorylation (OXPHOS), but also regeneration of NAD+/FAD to facilitate continued substrate oxidation. Analogously in the cytosol, reduction of pyruvate to lactate and concomitant regeneration of NAD+ facilitates continued flux through NAD+-dependent metabolic pathways including glycolysis and serine synthesis23.
While all of these core metabolic processes are critical for proliferation, the relative activity and choice of fuels for each pathway will likely exhibit significant heterogeneity. The precise composition of available nutrients and specific macromolecular requirements will vary depending on the lineage, identity, and environment of any given cell. As a result, while the metabolic challenges imposed by cell proliferation may be relatively constant, the manner in which cells acquire and metabolize nutrients will likely be context-specific. Indeed, increasing evidence demonstrates that tumors display a great degree of metabolic heterogeneity, influenced by cell or tissue of origin and both cell intrinsic and extrinsic factors24,25. In particular, tissue of origin seems to greatly influence tumor metabolic profiles. Mouse models of non-small cell lung cancer (NSCLC) and pancreatic ductal adenocarcinoma (PDAC) revealed that NSCLC tumors are dependent on branched chain amino acids as fuel for metabolic pathways, despite both NSCLC and PDAC tumors being driven by the same initiating lesions26. Importantly, this altered dependence remained even when cancer lines were implanted subcutaneously, indicating that at least some metabolic preferences imposed by cell of origin can dominate even over anatomical location26. Metabolism is not immutable, however: metastatic clones often rewire metabolism when settling into a new tissue niche27,28. The transforming oncogene can also contribute to metabolic diversity: liver tumors induced by MYC expression exhibited elevated glucose and glutamine catabolism compared to tumors induced by MET expression29.
The role of cancer cell lineage and oncogene expression in establishing tumor metabolic profiles suggests that metabolic phenotypes may be established early in tumorigenesis. In support of this model, a recent study found that leukemias arising from different progenitor cells exhibited metabolic profiles consistent with their normal progenitor cell of origin30. Whereas hematopoietic stem cells exhibited little oxidation of glucose-derived pyruvate in the TCA cycle, normal thymic T cells exhibited significant pyruvate oxidation via pyruvate dehydrogenase (PDH); consequently, PDH inhibition impaired development of T cell leukemia but not myeloid neoplasms30. Similarly, studies of normal mammary populations revealed that inherently differential preferences for oxidative phosphorylation and glycolysis appear to be retained in tumors31. These metabolic preferences wired in the tumor cell of origin can impose lasting metabolic vulnerabilities, highlighting the importance of understanding the metabolic profiles of tumor initiating cells31.
The factors that control tumor metabolism likely extend beyond lineage and oncogenic profile. For example, a study of over 80 NSCLC cell lines demonstrated that despite identical culture conditions, cell lines exhibited variety in magnitude and routes of glucose and glutamine usage32. While no one specific feature emerged as a determinant of metabolic preferences, factors such as proteomic, transcriptional and epigenetic profiles—even more than oncogenotype—correlated with a subset of metabolic phenotypes32.
Thus, despite a general dependence on nutrient uptake and catabolism, cancer cells have both hard-wired and context-specific preferences for which nutrients are consumed and how they are catabolized. Consequently, metabolite availability may serve as a restraint on tumor initiation and growth, but specific metabolic environments may exert different selective pressure depending on the identity and demands of the cancer cells themselves. For example, physiological serine levels can be limiting for tumor growth, but upregulation of the serine synthesis machinery allows cancer cells to evade this selective pressure when serine is limiting33. In NSCLC lines, oncogenic activation of the NRF2 transcription factor increases expression of serine synthesis genes, thereby inducing resistance to serine starvation and dependence upon PHGDH, the initiating step in de novo serine synthesis34. Successful tumor initiation therefore seems to depend on cancer cells acquiring mutations that, working alongside their preexisting metabolic preferences, allow cells to leverage available metabolites to meet the metabolic demands of survival and proliferation.
Enabling cancer cell survival
Cell death—whether controlled intrinsically by cancer cells or mediated by immune system—is a potent mechanism of tumor suppression35. Metabolism is critical for cell survival and dysregulated metabolism can trigger a number of forms of cell death. One central mechanism connecting metabolism to cell death is production of reactive oxygen species (ROS), which include hydrogen peroxide (H2O2), hydroxyl radical (·OH), superoxide radical anion (O2−), and lipid hydroperoxides (LOOH). Electrons leaking from the mitochondrial electron transport chain (ETC) represent a major source of cellular ROS, and this leakage is exacerbated by metabolic conditions that favor deposition of electrons into the ETC at a rate beyond the demands for oxidative phosphorylation36. In low doses, ROS can alter cell signaling cascades; higher doses can damage macromolecules including proteins and DNA. Such modest damage can favor tumor initiation, and antioxidants can therefore protect against ROS-induced cancer in several mouse models37 (FIG. 3). At high levels, particularly of LOOH, ROS can hamper cell fitness and even initiate a ROS and iron-dependent form of cell death known as ferroptosis, which is characterized by catastrophic lipid peroxidation in cell membranes38. While suppression of ferroptosis has not been directly linked to tumor initiation, cancer cells may exhibit heightened sensitivity to ferroptotic cell death and some tumors upregulate pathways that guard against ferroptosis39. In this scenario, enhancing antioxidant defenses to counteract the damaging effects of ROS provides an advantage to cancer cells.
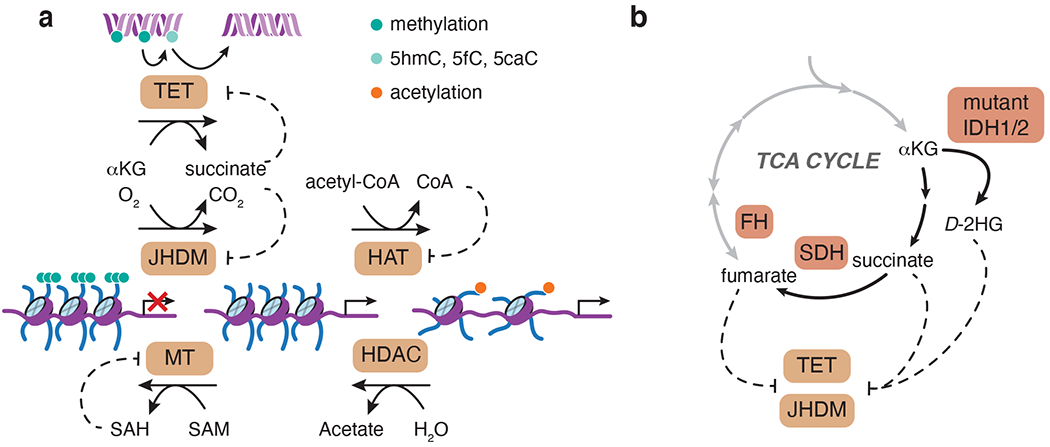
Fig 3. Metabolites have the potential to regulate chromatin-modifying enzymes.
a, S-Adenosyl methionine (SAM) is an obligate methyl donor for DNA and histone methyltransferases (MT). This reaction yields SAH, which can act as a competitive inhibitor of MTs. Acetyl-CoA is used by histone acetyltransferases (HAT) to deposit acetylation marks on histones, a reaction inhibited by CoA. Histone deacetylases (HDAC) can deacetylate histones by hydrolysis. Histone demethylation is mediated by Jumonji-domain-containing (JHDM) αKG-dependent dioxygenases, while αKG-dependent ten-eleven translocation (TET) enzymes facilitate DNA demethylation by iterative oxidation (5mC to 5hmC, 5fC, and 5caC). b, “Oncometabolites” succinate, fumarate and 2-hydroxyglutarate (2HG) share structural similarity with αKG and act as competitive inhibitors of αKG-dependent enzymes. Gain-of-function mutations in IDH1/2 or loss of function of succinate dehydrogenase (SDH) and fumarate hydratase (FH) lead to accumulation of 2HG, succinate and fumarate, respectively.
Indeed, ROS appears to be a major stress early in tumorigenesis: in epithelial cells, matrix detachment is associated with enhanced ROS that can induce a form of cell death known as anoikis40. Mechanistically, reduced glucose uptake following matrix detachment compromises glucose-dependent NADPH production, which is important to sustain endogenous antioxidant programs. Exogenous antioxidants prevent anoikis, allowing epithelial cells to sustain anchorage-independent cell growth—a hallmark of early tumorigenesis40. Antioxidants may also promote later stages of tumor progression, such as metastasis, by facilitating survival in highly oxidative environments41,42. Consistently, somatic mutations favoring constitutive activation of the antioxidant transcription factor NRF2 are common in human cancer, and genetic studies have confirmed that NRF2 promotes incidence and progression of multiple tumor types37. Furthermore, ROS are implicated in differentiation of many adult stem cell lineages, and thus counteracting ROS may favor maintenance of pro-tumorigenic programs of self-renewal43. All together, ROS provide a powerful link between metabolism and cell survival, but the precise role of ROS in tumor initiation is likely to be complex and context-specific.
Metabolism and cell death are also intricately linked through mitochondria, which are required for both energy generation and the induction of a programmed form of cell death known as apoptosis44. Select metabolic triggers including nutrient starvation, hypoxia and oxidative stress can each activate apoptosis, which is characterized by release of cytochrome c from the mitochondrial inner membrane space into the cytosol45. Cytochrome c usually serves as electron shuttle in the mitochondrial electron transport chain. Upon release to the cytosol, cytochrome c activates death-mediating proteases called caspases that ultimately execute apoptotic cell death46. While suppression of apoptosis alone is not sufficient to induce carcinogensis, many common oncogenes and tumor suppressors regulate susceptibility to apoptosis, suggesting that apoptosis cooperates with additional cues to control tumor progression47.
Nutrient starvation can also induce cell death via autophagy, a process in which macromolecules are delivered to the lysosome for degradation and recycling. While short-term autophagy can help cells cope with transient disruptions in nutrient supply, prolonged nutrient restriction can lead to autophagic cell death48. Thus, unlike apoptosis, which is generally accepted as a tumor suppressive mechanism49, autophagy can have both pro- and anti-tumorigenic functions depending on the specific context50. Other nutrient acquisition strategies such as macropinocytosis and receptor-mediated endocytosis may also facilitate cancer cell survival in diverse microenvironments. Macropinocytosis, the bulk uptake of extracellular material including proteins, is prominent in many cancer types—especially the notoriously poorly-perfused pancreatic cancer, and mutations that favor the activation of macropinocytosis allow cells to increase uptake of serum albumin and cellular debris during nutrient limitation51–53. Similarly, large cell lymphoma cells acquire cholesterol via low-density lipoprotein receptor (LDLR)-mediated endocytosis, and the subsequent changes in cellular lipid composition enable cancer cells to resist ferroptotic cell death54. A deepened understanding of how cancer cell type and mutational signature shape the preferred route of cancer cell nutrient acquisition could therefore expose new targetable vulnerabilities to induce lethal starvation in cancer cells.
Non-cell autonomous forms of cell death represent an additional mechanism of tumor suppression. In particular, the immune system provides a potent brake on tumor initiation. Consistent with the notion that malignant cells are continuously detected and cleared as they emerge, immunodeficiency and immunosuppression are associated with enhanced tumor susceptibility in humans55,56. Screening approaches to uncover genetic drivers of immune evasion in cancer cells revealed that an intact immune system selected for emergence of tumors harboring mutations in classic tumor suppressor genes57. More broadly, the mutations that favored tumor growth varied greatly depending on the presence of the immune system, even within a single cancer cell line57. These results highlight the complex alignment of extrinsic and intrinsic factors that is required for successful tumor initiation.
In established tumors, metabolic interplay between cancer and immune cells is increasingly recognized to play a critical role in anti-tumor immunity. Established tumors use two primary mechanisms to withstand elimination: preventing immune recognition through downregulation of antigen presentation and interfering with an ongoing immune reaction through the establishment of an immune-suppressive tumor microenvironment (TME)58,59. Tumor antigen presentation may respond to metabolic alterations in cancer cells: for example, urea cycle dysregulation increases pyrimidine synthesis which in turn triggers nucleotide imbalance. Ultimately, this imbalance induces genetic mutations that increase tumor antigens, rendering tumors susceptible to immune checkpoint therapy60. Similarly, metabolism of both cancer and stromal cells is increasingly implicated in determining immune responses in the TME. The TME is often acidic, hypoxic, and sometimes nutrient-poor, raising the possibility that cancer and stromal cells may compete for nutrients59. Indeed, recent evidence describes immune cells as large consumers of glucose and glutamine: in the TME, myeloid cells consume even more glucose than cancer cells61. Anti-tumor effector T cells may also compete with cancer cells for glucose in the TME: enhancing glycolytic capacity of cancer cells dampens T cell function, thereby allowing otherwise immunogenic tumors to escape immune clearance62. Metabolites likely play roles beyond fuel for expanding T cells. For example, the glycolytic intermediate phosphoenolpyruvate alters Ca2+ signaling to bolster T cell effector function63.
Cancer cells also shape the TME by releasing immune-dampening or -modulating metabolites including D-2-hydroxyglutarate, lactate and ROS64. Impairing cancer cell lactate release by inhibiting lactate dehydrogenase potentiated the anti-tumor immune response in mouse models of melanoma. Mechanistically, high lactate suppresses nuclear factor of activated T cells (NFAT) in T and NK cells, thereby blunting release of pro-inflammatory cytokine interferon-γ65. High lactate levels can also inhibit effector T cell functions while promoting the emergence of immune-suppressive cells including regulatory T cells, whose cellular metabolism appears to adapt to the harsh environment of the TME66. Thus, cancer cells can promote their own survival by interfering with an anti-tumor immune response through nutrient and oxygen depletion or through the release of metabolites that may promote the emergence of immune-suppressive cells59.
Little is known about how cancer cells avoid being cleared in the early stages of initiation before the establishment of the immune-suppressive tumor microenvironment. Suggestively, “latency competent” metastatic cancer cells, which are stem-like cells competent for tumor initiation, evade early detection by innate immunity by entering a slow-cycling, quiescent state67. Quiescent adult stem cells can also suppress MHC-I expression to avoid CD8+ T cell surveillance68. Whether the unique metabolic state of quiescence contributes to immune evasion, and whether such a metabolic state can be coopted to promote survival of cancer initiating cells, remains to be further explored.
Regulating cancer cell fate
Tumor initiation is facilitated by changes in gene expression programs that regulate cellular identity; generally, programs that favor that favor differentiation over self-renewal will antagonize tumor initiation69–72. Studies in both cancer cells and stem cells increasingly implicate metabolism in the control of cell fate programs and this topic has been extensively reviewed elsewhere43,73. Frequently, the links between metabolism and cell fate control center around the potential of metabolites to regulate chemical modifications on DNA and histones. Although transcription factors are ultimate guardians of transcriptional networks, chromatin organization also plays a critical role controlling DNA accessibility and gene expression74. Methylation of DNA and certain histone lysine residues is often associated with gene repression; reciprocally, histone acetylation often co-occurs with an environment permissive for gene expression74,75. Consistent with aberrant regulation of gene expression programs, cancer cells frequently exhibit perturbed chromatin organization, and genetic mutations in genes encoding chromatin regulatory proteins are found in approximately 50% of human tumors76.
Intriguingly, the deposition and removal of these chemical adducts is intimately entwined with cellular metabolism: metabolites are critical substrates and products of many enzymes that control chromatin modifications. Both DNA and histone methyltransferases use S-adenosylmethionine as a methyl donor, producing S-adenosylhomocysteine that in turn can serve as a competitive inhibitor of methyltransferases. Analogously, histone acetyltransferases use acetyl-CoA (or other acyl-CoA species) as a co-substrate and are inhibited by CoA in turn77,78. A family of α-ketoglutarate (αKG) dependent dioxygenases, which includes enzymes responsible for histone demethylation and iterative oxidation of methylated cytosine, consume αKG and molecular oxygen as obligate co-substrates and are inhibited by their product, succinate, as well as structurally similar metabolites such as fumarate and 2-hydroxyglutarate (2HG)79,80. Accordingly, fluctuations in levels of these metabolites have the potential to alter the chromatin landscape and shape gene expression programs that control cell identity77,81–84 (FIG. 4). Most of the above metabolites are central intermediates of the TCA cycle or derived from TCA cycle intermediates, raising the possibility that alterations in TCA cycle dynamics may signal to the nucleus to control gene expression programs85. The ability of these metabolites to control the chromatin landscape depends on enzyme affinity for a given metabolite relative to the concentration of that metabolite and other, similar metabolites that may compete for enzyme active sites. For a full discussion of the biochemical considerations relevant to metabolic control of chromatin modifying enzymes, we direct the reader to several excellent reviews dedicated to this topic75,84,86,87.
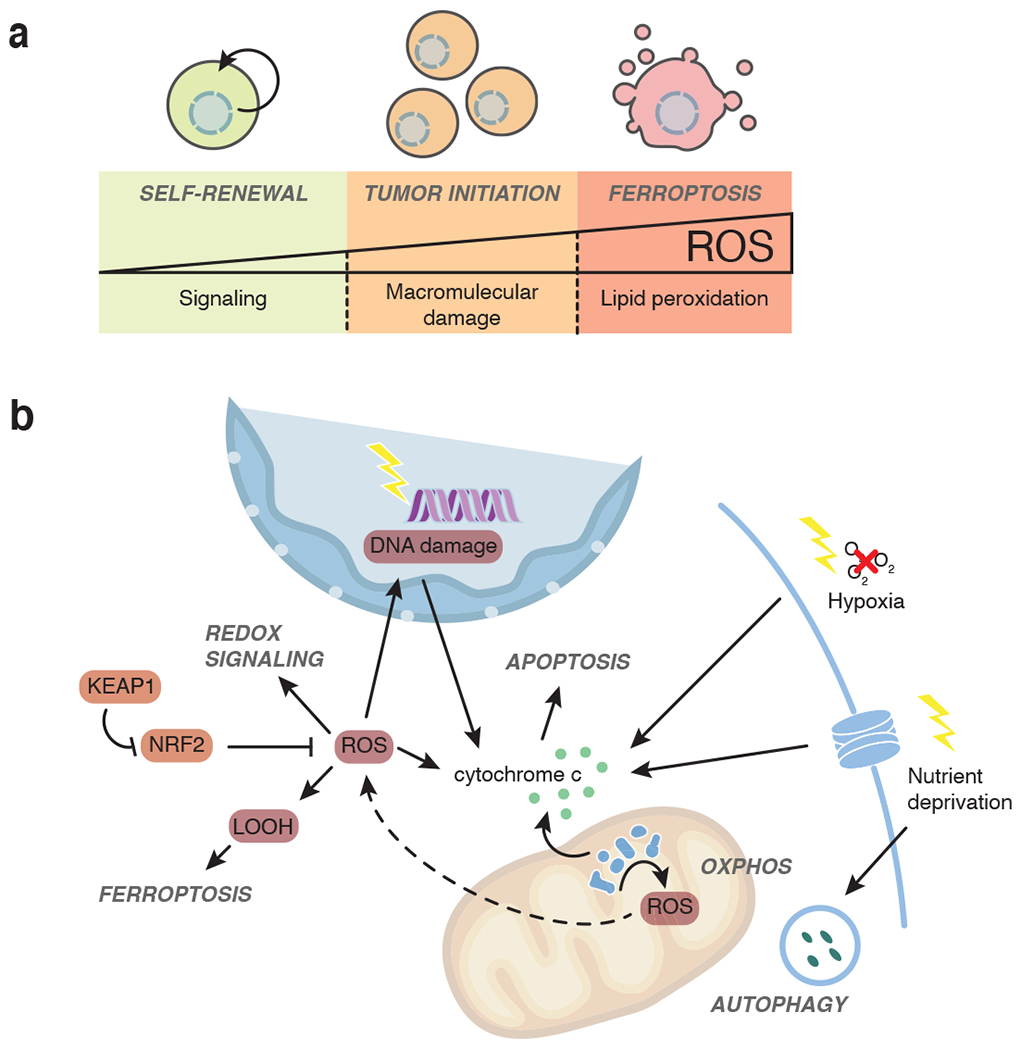
Fig 4. ROS control processes related to tumor evolution.
a, Low levels of ROS are required for stem cells to maintain their self-renewal capacity. Increased levels of ROS can induce macromolecular damage of DNA and proteins and lead to tumor initiation. Excessive ROS accumulation in the form of free radicals can trigger lipid peroxidation and ultimately lead to cell death in the form of ferroptosis. b, ROS arise in part as byproduct of electron flow through the electron transport chain. ROS leaves the mitochondria in the form of hydrogen peroxide (H2O2) or O2− that is converted to H2O2 by superoxide dismutases. H2O2 alters cellular signaling in part via oxidation of cysteine residues of proteins. H2O2 can also be converted to hydroxyl radicals (·OH) that are involved in the generation of lipid peroxides (LOOH), ultimately triggering cell death by ferroptosis. ROS can further damage DNA and proteins, which may lead to apoptotic cell death. Extracellular stresses including hypoxia and nutrient starvation can also trigger apoptosis.
Whether metabolites indeed control chromatin programs that affect tumor initiation remains largely unknown, although initial evidence supporting this notion is emerging and is discussed below. The most compelling evidence linking metabolism to tumor formation is the identification of frequent, recurrent point mutations in genes encoding the metabolic enzymes isocitrate dehydrogenase 1 and 2 (IDH1/2) in a variety of human malignancies88. These mutant versions of IDH1/2 catalyze the reduction of αKG to D-2HG, a metabolite normally present in trace amounts89,90. Accumulation of high levels of D-2HG in IDH1/2 mutant tumors is associated with inhibition of αKG-dependent dioxygenases, histone and DNA hypermethylation, and aberrant self-renewal90–92. Importantly, regulation of chromatin and cell fate is not limited to conditions where mutations drive pathological metabolite accumulation. For example, endogenous metabolic networks support production of αKG, which promotes differentiation and antagonizes tumor formation in multiple lineages93,94. These findings are consistent the notion that metabolic alterations that tip the balance between substrates and inhibitors of αKG-dependent dioxygenases directly modulate chromatin accessibility and gene expression programs related to cell fate and tumorigenesis93.
More broadly, the connection between metabolism and regulation of the chromatin landscape raises the possibility that metabolic wiring can facilitate or antagonize establishment of a chromatin environment that is permissive for tumor initiation. Intriguingly, not all cells are equally amenable to undergo a change in fate. For example, ‘plastic’ cells such as stem and progenitor cells are often the cells responsible for cancer emergence95. However, certain conditions allow differentiated cells to revert to either a progenitor-like state (de-differentiation) or enter a high-plasticity state that favors neoplastic growth. Consequently, the chromatin landscapes that favor tumor initiation may vary depending on the cell of origin or even the transforming oncogene. In this manner, the effect of a given metabolic state on tumor initiation may be highly context specific. Ongoing studies on cell-type specific metabolic profiles will help unravel the degree to which endogenous metabolic networks shape the chromatin landscape to reinforce—or undermine—control of cell identity.
While we focused on chromatin regulation as a suggestive link between metabolism and cell fate control in cancer, there are many other ways in which metabolites could alter gene expression programs that ultimately influence tumor initiation. Indeed, myriad signaling pathways can modulate gene expression programs that control cell fate, and many of these pathways are themselves susceptible to regulation by intracellular metabolites. Most prominently, mammalian target of rapamycin (mTOR) or AMP-activated protein kinase (AMPK)—which are key integrators of nutrient availability and energy status—exert direct or indirect control over signaling networks that ultimately modulate gene expression. More directly, ROS induce differentiation in a variety of cell types43,96. For example, in the skin, mitochondrially-derived ROS are required both for Notch-dependent transcription and β-catenin activation, which control epidermal differentiation and hair follicle specification, respectively97. Major outstanding questions for these potential links between metabolism and cancer cell fate center around physiological relevance and specificity. What degree of metabolic fluctuation is required to exert a given outcome? How can a specific outcome be achieved if a metabolite regulates many distinct enzymes or pathways? Testing the degree to which physiologic shifts in metabolites controls tumor cell fate via specific effectors will likely require combining in vivo metabolite quantification and single-cell sequencing analyses with temporal genomic editing techniques.
Metabolic changes promote tumor initiation
There are many ways that metabolism can support growth and/or progression of established tumors, and this topic has been extensively reviewed elsewhere5,6,22,98. Here, we discuss examples in which cell-intrinsic metabolic features are associated specifically with tumor incidence or with processes known to be involved in tumor initiation.
Hereditary mutations in metabolic enzymes
Direct evidence that metabolic pathways can influence tumor initiation comes from genetic studies of familial cancer predisposition99. Loss of function mutations in genes encoding subunits of the succinate dehydrogenase (SDH) complex and fumarate hydratase (FH), each of which catalyze a portion of the TCA cycle, can result in familial phaeochromocytomas and paragangliomas, closely related neuroendocrine tumors of the adrenal medulla or the autonomic nervous tissue, respectively100–102. Germline SDH mutations also occur in other cancer types including renal carcinoma103 and gastrointestinal stromal tumors104, while germline FH mutations predispose to renal cell carcinoma and hereditary leiomyomatosis of the uterus and skin105. Tumors arising in patients with germline mutations in SDH or FH genes are usually associated with loss of heterozygosity of the remaining wild-type allele, thereby driving complete TCA cycle truncation105,106. The notable tissue-restricted pattern of tumors arising in patients with germline mutations in SDH/FH raises the intriguing possibility that these lineages comprise cells that are uniquely capable of coping with the metabolic disruption imposed by TCA cycle dysfunction. Alternatively, these lineages may be exceptionally susceptible to transformation by the pathways triggered following SDH/FH loss. Perhaps both scenarios are true: recently it has been shown that SDHB deletion and concomitant αKG-dependent dioxygenase inhibition is insufficient to drive tumor development in a murine model of pheochromocytoma but SDHB loss accelerates tumorigenesis when combined with deletion of a tumor suppressor gene107.
Why does SDH or FH mutation predispose to tumor formation? Most likely, the tumor promoting effects of these mutations are due to pathological buildup of their respective substrates, succinate and fumarate. High accumulation of these “oncometabolites” is sufficient to inhibit a subset of αKG-dependent dioxygenases including TET methylcytosine dioxygenases, Jumonji domain containing histone demethylases and the prolyl hydroxylases (PHD) that target the labile α subunits of the hypoxia inducible factor (HIF) complex for degradation (FIG. 5). Indeed, HIFα stabilization even in normoxia is commonly observed in following SDH or FH loss108,109, and tumors with SDH/FH mutations frequently exhibit chromatin hypermethylation99. Other dioxygenases beyond PHDs likely contribute to oncogenic effects of succinate and fumarate. For example, fumarate induces the epithelial-mesenchymal transition in renal cancer cells at least in part by silencing the miR-200 microRNA cluster. These effects are phenocopied by TET inhibition and reversed by αKG supplementation, indicating that TET-mediated control of the miR-200 locus may contribute to malignant phenotypes in FH-deficient renal cancer110. Moreover, succinate, fumarate and 2HG have been implicated in suppressing DNA damage repair through inhibition of KDM4A/B histone demethylases111,112.
Fig 5. Metabolic regulation of hypoxic responses.
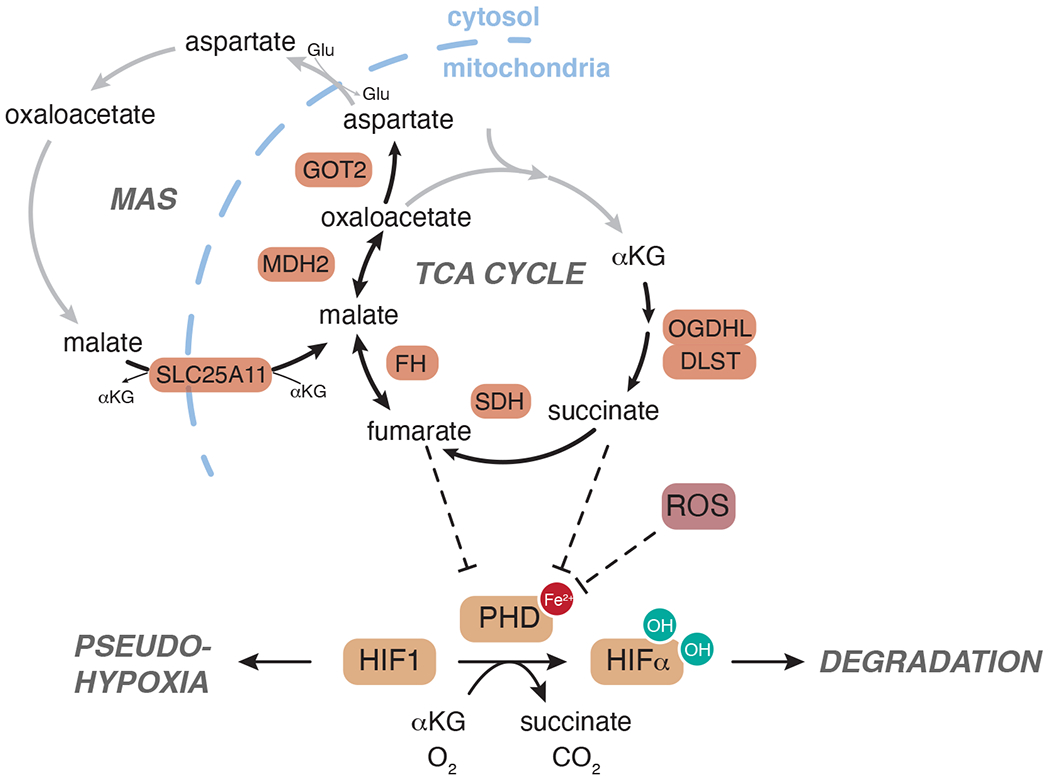
In normoxia, hypoxia inducible factor (HIF) alpha subunits are targeted for degradation by prolyl hydroxylases (PHD), αKG-dependent dioxygenases that hydroxylate select proline residues on HIFα thereby enabling ubiquitination and degradation by the proteasome. Inhibition of PHDs by low substrate availability, as during hypoxia, triggers HIFα stabilization. Stabilized HIFα can dimerize with HIF1β/ARNT to induce transcriptional responses to hypoxia. Accumulation of ROS or of oncometabolites succinate and fumarate can block PHD activity irrespective of hypoxia and induce a pseudo-hypoxic state. Germline mutations in genes encoding subunits of the TCA cycle (OGDHL, DLST, SDH, FH, MDH2) and Malate-Aspartate Shuttle (MAS) (SLC25A11, GOT2) are also linked to development of hereditary cancer syndromes.
However, both SDH and FH inhibition have additional metabolic consequences beyond oncometabolite-mediated dioxygenase inhibition. For example, high levels of ROS following SDHB loss can also trigger HIFα stabilization113. Notably, while depletion of both SDHA and SDHB induce succinate accumulation, SDHA is not commonly mutated in human tumors. Suggestively, in some models only SDHB loss induces HIF-dependent increases in proliferation, arguing that in some contexts ROS may be dominant over oncometabolite accumulation as a driver of tumor growth113. Similarly, the electrophilic fumarate can directly modify reactive cysteine residues that modulate protein function114. Covalent modification of KEAP1 by fumarate releases NRF2, thereby inducing an antioxidant gene program in renal cysts and tumors115,116. Consistently, genetic studies in mice confirmed that renal cyst formation driven by FH loss is independent of HIF116.
Thus, the extent to which hypermethylation and/or HIF stabilization contribute to the pro-tumorigenic effects of SDH/FH loss remains to be elucidated. It is likely that succinate and fumarate accumulation exert context-specific effects, which will only be unraveled by combined genetic and biochemical approaches across multiple tissue types. Even if SDH or FH mutations are insufficient for tumor initiation, HIF activation driven by SDH or FH loss might drive metabolic rewiring that favors adaption to hypoxia, even before low oxygen levels are encountered during tumor growth117. Such pro-hypoxic adaptations may be particularly relevant in hereditary paragangliomas, which often arise in the carotid body that orchestrates the body’s response to low oxygen levels. Paragangliomas are as well characterized by carotid body hyperplasia that mimics the normal cellular expansion of a chronically hypoxic carotid body102,118,119. These cells may therefore represent a cellular lineage uniquely metabolically equipped to withstand and react to the pseudo-hypoxic state driven by the loss of SDH.
While germline mutations causing SDH and FH disruption are by far the dominant examples of metabolic involvement in cancer predisposition syndromes, a handful of additional examples have begun to emerge in recent years, powered by whole-exome sequencing of patients with unexplained hereditary disease. Germline mutations in MDH2, OGDHL, and DLST—all components of the TCA cycle— as well as GOT and SLC25A11—components of the malate-aspartate shuttle that transfers cytosolic reducing equivalents into the TCA cycle—have been identified in patients with pheochromocytoma/paraganglioma (FIG. 5)99. The degree to which these mutations facilitate tumorigenesis and whether these mutations share common mechanisms of pathogenesis remains to be elucidated, and future work may continue to reveal novel roles for germline mutations in metabolic control of cancer susceptibility.
Somatic mutations in metabolic enzymes
The above familial cancer predisposition syndromes support the notion that metabolism can be directly coopted to support tumor growth. Nevertheless, human tumors exhibit only a handful of frequent, recurrent mutations in metabolic enzymes, perhaps because genetic mutations in metabolic enzymes are likely to reduce cell fitness or block metabolic plasticity required for tumor evolution. Consistent with this notion, the most common metabolic mutation in human cancer is not loss-of-function but rather gain-of-function: recurrent IDH1/2 mutations found in a variety of human malignancies including AML120 and gliomas121 occur at specific arginine residues in the catalytic side of the enzyme. These mutations confer neomorphic enzyme activity enabling IDH1/2 to produce the oncometabolite D-2-hydroxyglutarate (D-2HG) instead of the structurally similar αKG89,122 (FIG. 4b). Consistent with the biochemical observation that D-2HG is a competitive inhibitor of several αKG-dependent dioxygenases, IDH-mutant tumors commonly exhibit chromatin hypermethylation and impaired differentiation88.
In gliomas, IDH1 mutations appear to be early events preceding mutations in p53, suggesting that these mutations may contribute to tumor initiation123. Consistently, the introduction of mutant IDH2 is sufficient to reduce differentiation capacity of murine bone marrow cells92 and to enable mesenchymal progenitors to form sarcomas in vivo124. Here, robust accumulation of 2HG induced DNA hypermethylation associated with impaired differentiation into adipocyte or chondrocyte lineages124. In IDH2-mutant leukemia, DNA hypermethylation and impaired differentiation is reversed by pharmacologic inhibition of mutant IDH2, suggesting that continued 2HG production can be required to sustain tumor growth125. Mechanistically, D-2HG-mediated inhibition of the αKG-dependent dioxygenases TET and JHDM appear to play important roles in the tumor-promoting effects of IDH mutations90,92. In support of this model, IDH1/2 and TET2 mutations are mutually exclusive in AML patients92. However, IDH2 and TET2 mutations co-occur in a subset of angioimmunoblastic T-cell lymphomas, indicating that the dioxygenases responsible for the phenotypes downstream of D-2HG accumulation are likely to be context specific126,127. Moreover, while TET2 mutations are frequently found in clonal hematopoiesis—a benign precursor of leukemia—IDH1/2 mutations are not, suggesting that IDH1/2 mutation is not a common initiating event in AML128. The different temporal patterns of IDH1/2 mutation across human tumor types suggest that 2HG production is not a uniform force for tumor initiation but rather may require appropriate cellular contexts to provide a selective advantage to tumor progression.
Changes in copy number may provide another route for metabolic genes to influence tumor progression: PHGDH, which encodes the first step in de novo serine synthesis, is frequently amplified in human cancer and overexpression of PHGDH induces phenotypic alterations associated with transformation in breast epithelial cells129. As more and more cancer genomes are sequenced, the number of enzymes with recurrent—albeit infrequent—mutations is rising. In some cases, there is a clear logical link to tumor progression. For example, D2HGDH, which encodes the gene that converts D-2HG to αKG, is mutated in diffuse large B-cell lymphoma where it controls the ratio of D-2HG to αKG and influences chromatin methylation130. A systematic assessment of how somatic mutation rates in metabolic genes compare to other genes, and whether these mutations appear to arise early or late in tumor evolution, will provide enormous insight into potential routes for metabolic networks to control tumor initiation. Strikingly, recurrent mutations in mitochondrial DNA can occur at a rate similar to those of common cancer drivers, raising the possibility that disruption of oxidative phosphorylation may provide a selective advantage in certain tumor types131.
Metabolic regulation by oncogenes and tumor suppressors
Many common oncogenic drivers directly regulate metabolic pathways, and the impact of major cancer-associated alterations in PI3K-AKT-mTOR, MYC or p53 on metabolic networks have been extensively reviewed elsewhere22,132,133. Nevertheless, it remains unclear whether metabolic deregulation by oncogenes facilitates tumor initiation or merely enables tumor growth5. Uncoupling the role of metabolism from other cancer-promoting effects of oncogenic mutations is complicated, but evidence is beginning to emerge that metabolites can be bona fide effectors of cell fate downstream of common cancer drivers. For example, oncogenic Kras induces pancreatic acinar cells to revert to more duct-like lineages (a process termed acinar-to-ductal metaplasia or ADM) that can then form pancreatic intraepithelial neoplasia (PanIN) lesions and ultimately pancreatic adenocarcinoma (PDAC)134,135. Studying the effect of mutant Kras in primary pancreatic acinar cells revealed that Kras enables accumulation of acetyl-CoA that promotes both sterol biosynthesis and histone acetylation136. Importantly, blocking acetyl-CoA synthesis impaired ADM, showing that these Kras-mediated metabolic changes are required for the early stages of tumor development136. Similarly, metabolic changes driven by loss of p53 may facilitate the transition from pre-malignant PanIN to PDAC. Restoring wild-type p53 function in PDAC cells induced accumulation of αKG and triggered differentiation to a PanIN-like state93. This tumor-suppressive differentiation could be recapitulated even in the absence of p53 by orthogonal methods of inducing αKG and inhibited by enforced succinate accumulation, implicating metabolic regulation of αKG-dependent dioxygenases as a critical component of fate regulation in PDAC cells93. Thus, metabolic alterations favoring αKG-dependent dioxygenase inhibition may be a common feature of cancer-associated mutations beyond SDH/FH familial cancer syndromes. Together, these studies support the notion that metabolism can be an integral component of cell fate transitions in cancer, and it will therefore be of great interest to determine the extent to which specific metabolic alterations downstream of oncogenic mutations control gene expression programs that determine tumor progression.
Intrinsic metabolic phenotypes that affect propensity for transformation
Absent oncogenic mutations, whether endogenous metabolic networks support or antagonize tumor initiation remains largely an open question. Emerging evidence suggests that in some cases, specific metabolic features may serve as a brake on tumor initiation. For example, HSCs have high levels of ascorbate that decrease with differentiation, but ascorbate is a cofactor that potentiates activity of the tumor suppressor TET2137. Accordingly, ascorbate depletion accelerated tumor progression in mouse models of leukemia, suggesting that cell-intrinsic ascorbate pools restrain aberrant self-renewal programs in HSCs137. In drosophila, initiation of brain tumors from neural stem cells requires transition to a highly oxidative state that enables immortalization of tumor-initiating cells138. Thus, context-specific metabolic bottlenecks may limit tumor initiation.
Tissue stem cells often serve as the cell of origin for malignancies, and some evidence suggests that metabolic programs that support stem cell self-renewal may also support tumor initiation and progression73. In the intestine, expression of the mitochondrial pyruvate carrier (MPC) is low in stem cells and increases with differentiation139. MPC inhibition prevents oxidation of glucose-derived pyruvate in the mitochondria and promotes expansion of intestinal stem cells. Consistently, MPC deletion in mice accelerates signs of hyperplasia in response to chemical carcinogenesis and increases the number of tumors formed in both chemical and genetic models of colon cancer140. In humans, MPC expression is reduced at the earliest stages of tumor formation, suggesting that mitochondrial pyruvate oxidation restrains tumor initiation in the colon140. These data support a model wherein the metabolic state enforced by low MPC expression favors self-renewal and tumor initiation in the intestine.
Despite increasing evidence that tissue stem cells often have distinct metabolic profiles compared to their more differentiated counterparts, the difficulty of studying metabolism in rare cell populations has hampered efforts to determine the extent to which metabolic programs of homeostatic stem cells are preserved in cancer141. Studying tumor-associated stem cells has proven more fruitful, and an emerging theme is that these tumor-propagating cells often rely on mitochondrial oxidation to support the signaling and bioenergetic networks that sustain self-renewal. Indeed, inhibition or disruption of mitochondrial respiration targets stem-like cancer cell pools in pancreatic cancer142, glioblastoma143, leukemia144, melanoma145 and lung cancer146. Whether mitochondrial oxidation is permissive for transformation or is rather a requirement for maintenance of tumor-associated stem cells remains to be determined. More nuanced metabolic networks may also play context specific roles. For example, AML-associated stem cells enhance transamination of branched-chain amino acids (BCAA) via BCAT1, which transfers α-amino groups from BCAA to αKG. The subsequent lowered αKG availability and DNA hypermethylation supported leukemia stem-cell function and leukemia initiation147.
Environmental factors favoring tumor initiation
The observation that the metabolic profile of a tumor seems to be derived from its cell of origin raises the question whether certain metabolic phenotypes support the initiation of transformation more than others. While there is little data to date supporting the notion that the metabolic profiles of specific lineages may influence their propensity for transformation, there is increasingly abundant evidence that within a specific lineage, environmental factors can alter susceptibility to tumor initiation and/or progression (FIG. 6).
Fig 6. Environmental factors influencing tumor initiation.
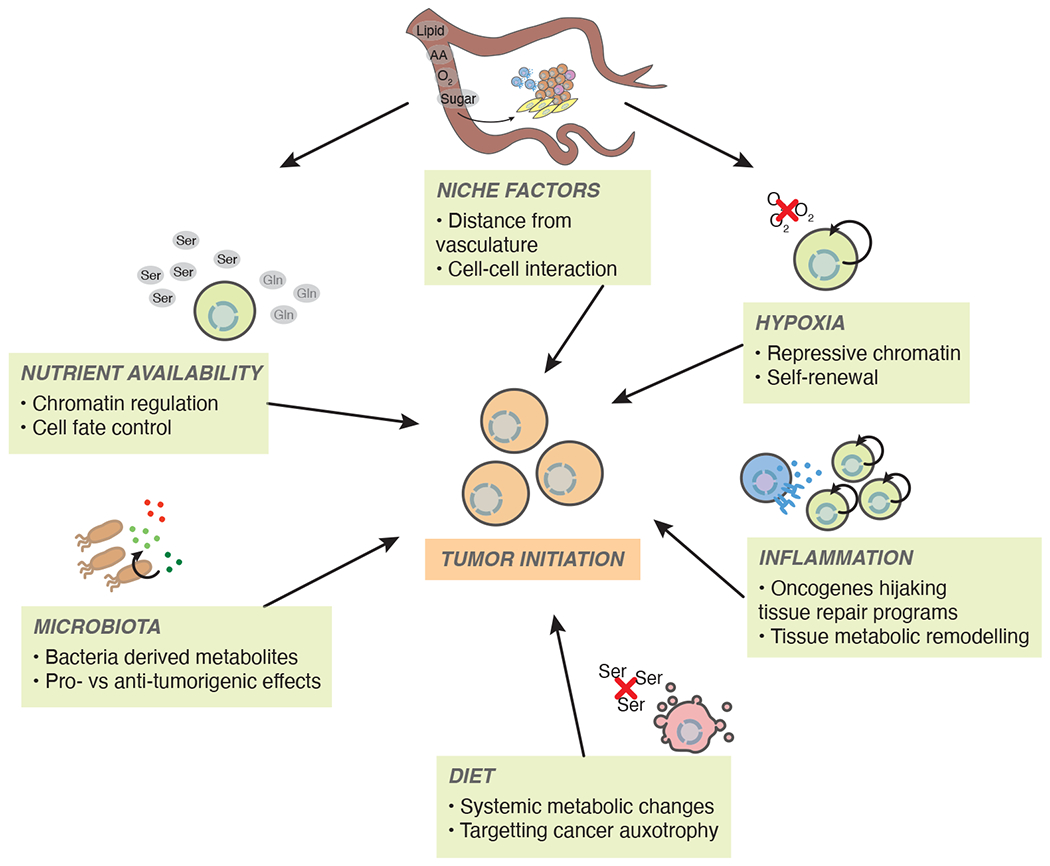
Many systemic and local factors related to metabolism have the potential to alter process related to tumor initiation. Hypoxia and inflammation likely favor tumor growth. Diet, nutrient availability, and microbial homeostasis may potentiate or antagonize tumor initiation and progression. Importantly, these processes are all interconnected and likely to affect many cell types with the potential to alter tumor progression.
Hypoxia
Solid tumors frequently harbor hypoxic regions, in part due to disordered and immature de novo vascularization148. Hypoxia activates transcriptional programs regulating survival, angiogenesis and metabolism149 and can select for aggressive cancer clones, thereby promoting invasion and metastasis150. Several lines of evidence suggest that hypoxia may also be able to contribute to tumor initiation. First, many stem cell populations reside in hypoxic niches, and hypoxia is directly implicated in maintenance of self-renewal programs151. Intriguingly, chromatin modifiers can directly respond to changes in oxygen availability: the histone demethylase KDM6A/UTX that removes trimethylation on histone 3 lysine 27 (H3K27me3) has a relatively low affinity for oxygen that renders it susceptible to inhibition in physiological hypoxia152. In muscle progenitor cells, hypoxia inhibits myogenic differentiation and this effect is reversed by expressing a KDM6A mutant with higher oxygen affinity, thus directly linking oxygen availability to self-renewal of myogenic precursors152. Hypoxia also induces a metabolic state favoring reduction of αKG to L-2HG by LDH and MDH1/2153,154. L-2HG is a more potent inhibitor of many αKG-dependent dioxygenases than its enantiomer D-2HG91,153,154 and hypoxia-induced L-2HG production is associated with a repressive chromatin landscape marked by decreased 5hmC and/or increased histone methylation153,155. Suggestively, inborn errors of metabolism that drive L-2HG accumulation are associated with development of several tumor types156. These findings raise the possibility that L-2HG production induced by hypoxic microenvironments may facilitate maintenance of chromatin landscapes that favor self-renewal and tumor development.
Recent evidence that hypoxia may indeed play an important role in tumor initiation comes from several studies on posterior fossa A (PFA) ependymomas, a subtype of pediatric cerebellar tumors that originate from a highly hypoxic environment157. Hypoxia is required for establishment and culture of patient derived PFA ependymoma cell lines; even transient exposure to ambient oxygen triggered irreversible lethality158. Mechanistically, hypoxia decreases SAM and increases the αKG/succinate ratio, thereby lowering H3K27me3158. Supporting the notion that a metabolic environment favoring low H3K27me3 is important for PFA initiation, genomic studies of PFA tumors demonstrated that PFAs only rarely harbor somatic mutations; rather, transformation appears to be driven by aberrant histone and DNA methylation159. Targeting metabolism and/or chromatin regulators slows PFA growth in vitro and in vivo, consistent with the model that these tumors arise in part because of a metabolic environment that favors hypomethylation157,158,160.
Local nutrient availability
Beyond oxygen, availability of other key nutrients—determined by a combination of local nutrient supply and cellular demand—also has the capacity to shape cell fate programs and tumor progression. Tumors are not uniformly nutrient restricted; rather tumors often harbor increased levels of key nutrients161. Nevertheless, heterogeneous nutrient availability is increasingly recognized as a potential driver of tumor phenotypes. For example, local decreases in glutamine availability—whether from poor vascularization or increased tissue consumption—contributes to intratumoral heterogeneity in chromatin modifications and cancer cell differentiation states162. Mechanistically, glutamine depletion and subsequent αKG restriction limit histone demethylation, thereby favoring a de-differentiated state that can be reversed by supplementing mice with extra glutamine or αKG in mouse models of melanoma and colorectal cancer94,163. The link between regional glutamine depletion and poor differentiation in tumors162 serves as an example how hyper-local variation in nutrient supply might directly influence cell fate control and tumor development.
Other nutrients beyond oxygen and glutamine have the potential to control αKG-dependent dioxygenases. For example, in the skin, high de novo serine synthesis directly yields cytosolic αKG–a byproduct of the transamination step catalyzed by PSAT1—which in turn induces precocious epidermal differentiation164. Pre-malignant stem cells preferentially acquire serine from their environment rather than de novo synthesis, thereby protecting themselves from tumor-suppressive differentiation164. Starving mice of serine reduced the number of tumors formed in response to chemical carcinogenesis and induced H3K27me3 loss and differentiation in established tumors in a manner reversible by enforced succinate accumulation164. Nutrients such as iron and ascorbate—a cofactor that likely potentiates maintenance of iron in the reduced state required for α-ketoglutarate-dependent dioxygenase activity86,165—may also play key roles in determining regional capacity for chromatin regulation. Certainly, providing exogenous ascorbate is sufficient to control TET2 activity and leukemic progression in mice137,166. Advances in spatial metabolomics167 are likely to nominate additional metabolites whose regional variability accompanies or even drives tumor heterogeneity. As nutrient consumption and release profiles vary across organs, it is possible that organ-specific metabolic preferences promote or constrain tumor initiation by altering local metabolite abundance168.
Microbiota
Gut microbiota consume and produce myriad metabolites and have significant potential to affect cancer incidence through both direct and indirect mechanisms169. Changes in gut flora composition can trigger chronic inflammation, release of bacterial toxins and production of damaging bacterial metabolites that can each facilitate tumor initiation169. These processes are most clearly relevant in the context of colorectal cancer (CRC), whose incidence is linked to dietary habits and antibiotic exposure170. For example, non-digestible fiber is fermented by bacteria to yield short-chain fatty acids such as acetate, propionate and butyrate that by and large exert tumor-suppressive effects171. In immune cells and colonocytes, propionate and butyrate activate surface G protein-coupled receptors or enter the cell where they can inhibit histone deacetylases, thereby affecting signaling pathways and gene expression programs171–173. Many metabolites produced by commensal microbes have potential to influence processes related to tumor development171. For example, bacterially-derived reuterin induces oxidative stress that selectively inhibits growth of CRC cells174. Hyper-local production of metabolites may explain regional susceptibility to cancer along the intestinal tract: gallic acid produced by bacteria residing in the distal gut activates WNT signaling to potentiate tumor progression driven by mutant p53175. Supplementing gallic acid enabled high-grade dysplasia in the jejunum, which was otherwise refractory to tumor-promoting effects of mutant p53175. Gut-derived metabolites can also enter the circulation and affect distal organs, with the potential to control signaling and gene expression in tissues beyond the intestinal tract. Elucidating the many interactions between gut-derived metabolites, tumor-initiating cells and stromal cells is likely to provide important insight into fundamental pathways shaping tumor progression and reveal a wide scope for environmental control of cancer susceptibility170.
Inflammation
Chronic inflammation and resulting inflammatory diseases—which are often associated with environmental and lifestyle factors—pose a major burden to public health176. Unlike acute inflammation, which can combat a growing tumor, chronic inflammation is a major driver of all cancer stages, including cancer initiation177. The ongoing immune response that propagates inflammation potently shapes the TME via immune cell infiltration and inflammatory cytokine secretion. As the goal of an ongoing immune response is the resolution of inflammation and the return to tissue homeostasis, released cytokines often activate stem cell expansion and epithelial regeneration to facilitate tissue repair178. In the presence of oncogenic mutations, such stimuli can be potent inducers of tumor initiation177,179. For example, oncogenic Kras only weakly induces PDAC unless mice are subject to pancreatitis180,181. Mechanistically, the combined effects of mutant Kras and local inflammation induce a unique chromatin landscape that supports transcriptional programs antagonizing appropriate tissue repair and promoting tumor progression181,182. Similarly, in the colon, NF-κB activation—a hallmark of pro-inflammatory cytokine signaling—cooperates with β-catenin to favor de-differentiation of villus cells into stem-like cells that promote tumor initiation183. The powerful role of inflammation on tumor initiation highlights the importance of non-genetic factors in tumor progression. Given the links between metabolism, immune responses and cell fate, it is tempting to speculate that metabolism will emerge as a powerful player in both the induction and response to local inflammation. Suggestively, loss of the gluconeogenic enzyme fructose 1,6-bisphosphatase (FBP1)—a common event in hepatocellular carcinoma—induces hepatic stellate cells to adopt a pro-inflammatory senescent state that favors tumor progression184. Likely, metabolic regulation of inflammation and the metabolic consequences of persistent immune responses will collectively influence tumor evolution.
Diet
The notion that diet affects cancer incidence originates in part from epidemiological studies showing a robust association between obesity and cancer185,186. The relationship between obesity and cancer is complex, not least because obesity can be accompanied by dietary alterations such as low fiber intake or high fat and fructose consumption, which by themselves may affect cancer formation187. Increased fat consumption can also trigger microbial dysbiosis that is sufficient to increase intestinal cancer development in genetically susceptible mice, independent of weight gain188,189. Nevertheless, studies in mouse models show that high dietary fat intake and genetically induced obesity each can accelerate initiation and progression of multiple tumor types including liver190, pancreas191 and intestine192. Changes both in circulating hormones and nutrients may contribute to the pro-tumorigenic effects of obesity. For example, cancer cells growing in mice fed a high fat diet increase catabolism of free fatty acids, paradoxically decreasing local availability of this critical nutrient source required for effective CD8+ T cell function193. Reversing fatty acid metabolism in cancer cells restored CD8+ T cell function and blunted the tumor-promoting effect of a high-fat diet193. Similarly, high fat diets increase intestinal stem cell self-renewal and tumorigenicity in mice, and this effect can be recapitulated ex vivo by supply of dietary lipids or pharmacological activation of PPARδ transcriptional networks that induce fatty acid oxidation192,194. Circulating ketones that accumulate during high-fat ketogenic diets may also alter tissue stem cell self-renewal by inhibiting histone deacetylases, although the effects of ketones on gene expression and tumorigenesis appears context-specific195,196. Thus, diet can directly and indirectly remodel nutrient abundance in the TME, thereby affecting many processes that affect tumor growth.
Fructose, abundant in highly processed foods and sweetened beverages, is another component of modern diets that is frequently linked to cancer incidence and may contribute to systemic metabolic disease in humans in part by activating hepatic de novo lipogenesis197,198. High-fructose corn syrup consumption increases villus length and nutrient absorption in the mouse intestine, triggering weight gain independent of caloric intake199. In a mouse model of intestinal cancer driven by Apc deletion, high-fructose corn syrup increases tumor incidence and severity by inducing cancer-cell autonomous metabolic rewiring that favors tumor growth200. Nutrients such as fructose that are linked to cancer incidence likely affect multiple cell and tissue types across the body, and systems biology approaches may be required to deconvolute the mechanisms linking dietary behavior to tumor susceptibility.
Diets do not necessarily have to be pro-tumorigenic; in fact, tailored dietary restrictions may be used to target tumors that rely on exogenous supplies of specific nutrients. For example, many tumor types upregulate enzymes involved in de novo serine synthesis to ensure continuous supply of serine, which is critical for redox homeostasis and protein, nucleotide and phospholipid biosynthesis pathways129,201,202. Transformed cells that do not increase expression of serine synthesis genes thus are highly dependent on extracellular serine; accordingly dietary restriction of serine and its related metabolite glycine specifically slows growth of these serine-dependent tumors203. In the epidermis, serine starvation induces precocious differentiation—presumably by activating de novo serine synthesis which results in stoichiometric production of αKG that in turn promotes epidermal differentiation164. In this manner, endogenous availability of amino acids such as serine may affect stem cell propensity for differentiation and thus antagonize tumor initiation. Similarly, dietary restriction of the essential amino acid methionine also limits tumor growth and enhances the effects of several anti-cancer therapies in mice, showing the power of specific metabolic interventions to control tumor progression204.
The downside of systemic metabolic interventions is that they are inherently non-specific. Metabolic perturbations that affect cancer cells will also affect non-transformed cells in the tumor and even cells in surrounding normal tissues. Crucially, dietary interventions that have the potential to deprive the TME of nutrients and slow tumor growth also can interfere with the functionality of tumor combatting T cells205,206. Taken together, dietary approaches tailored to target cancer cells’ metabolic preferences have the potential to enhance tumor therapy, but effective deployment will require careful consideration of off-target and systemic effects, including effects on the microbiome. Future endeavors to implement diet as complementary cancer therapy will therefore strongly rely on detailed metabolic profiling and on a deepened understanding of the interplay between cancer cells and their niche.
Conclusions
Increasing evidence demonstrates many ways that cell metabolism can directly control survival, proliferation, and differentiation state—all processes intimately entwined with tumor initiation. Specific metabolic alterations never occur in isolation: metabolic pathways are deeply interconnected, and neighboring cells compete for, and share, metabolites both locally and systemically. Therefore, there is an almost staggering number of possibilities for how metabolic networks may contribute to tumor evolution. Nevertheless, unifying principles are beginning to emerge—such as the importance of central metabolic networks controlling biomass accumulation and redox homeostasis for cancer cell proliferation and the ability of select TCA cycle-related metabolites to control gene expression programs that dictate cell state. However, even these central principles have notable exceptions—for example, while most tumors oxidize glucose in the TCA cycle, renal cell carcinomas appear to prefer to secrete glucose as lactate207. A major open question is whether these defining metabolic programs are a byproduct of or requirement for transformation. Given the often tissue-restricted pattern of oncogenic mutations208,209, it is tempting to speculate that the metabolic programs inherent in specific lineages render certain cells more or less amenable to transformation by specific oncogenes. Even within a tissue, the stochastic and heterogenous nature of cancer development210,211 suggests that non-genetic factors such as metabolism likely play a critical role in tumor evolution. Continued insights into the role of metabolism in tumor initiation will require monitoring metabolism with increased temporal and spatial resolution, ideally enabling capture of rare or transient metabolic states that may be stabilized and expanded following oncogene expression. Additional focus on non-transformed or stromal cells that may cooperate or compete with putative cancer cells will also help to reveal how local metabolic networks can control tumor formation. A deeper understanding of the factors that control cancer initiation will provide important insight into both new therapies to target putative cancer-initiating cells and preventative approaches to reduce cancer incidence in susceptible populations.
Key points box.
Metabolism is linked to many key processes required for tumor initiation: cell fate control, biomass accumulation, proliferation and survival.
Through their role as co-substrates for chromatin modifying-enzymes, metabolites have the potential to influence cell fate programs that control tumor initiation.
Hereditary cancer syndromes illustrate how mutations in metabolic enzymes may predispose for cancer through accumulation of “oncometabolites” succinate, fumarate and 2HG.
Intrinsic metabolic configurations of tissues or cells might facilitate or antagonize oncogenic transformation.
Environmental factors including diet, inflammation, hypoxia and nutrient availability interact to control many processes related to tumor initiation and progression.
Acknowledgements
We thank Sanjeethan Baksh for critical reading of the manuscript and members of the Finley lab for helpful discussion. L.W.S.F. is a Searle Scholar. This work was additionally by a Human Frontier Science Program Fellowship LT000200/2021-L (to J.S.B.), grants to L.W.S.F. from the Pershing Square Sohn Prize for Cancer Research and the NIH/NCI (R37CA252305), and the Memorial Sloan Kettering Cancer Center Support Grant P30CA008748.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Martincorena I et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886, doi: 10.1126/science.aaa6806 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frede J, Adams DJ & Jones PH Mutation, clonal fitness and field change in epithelial carcinogenesis. The Journal of Pathology 234, 296–301, doi: 10.1002/path.4409 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Haigis KM, Cichowski K & Elledge SJ Tissue-specificity in cancer: The rule, not the exception. Science 363, 1150–1151, doi: 10.1126/science.aaw3472 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Li X, Egervari G, Wang Y, Berger SL & Lu Z Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nature Reviews Molecular Cell Biology 19, 563–578, doi: 10.1038/s41580-018-0029-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vander Heiden MG & DeBerardinis RJ Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669, doi: 10.1016/j.cell.2016.12.039 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martínez-Reyes I & Chandel NS Cancer metabolism: looking forward. Nature Reviews Cancer 21, 669–680, doi: 10.1038/s41568-021-00378-6 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Ishizawa K et al. Tumor-Initiating Cells Are Rare in Many Human Tumors. Cell Stem Cell 7, 279–282, doi: 10.1016/j.stem.2010.08.009 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Visvader JE Cells of origin in cancer. Nature 469, 314–322, doi: 10.1038/nature09781 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Blanpain C Tracing the cellular origin of cancer. Nature Cell Biology 15, 126–134, doi: 10.1038/ncb2657 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Davidson Shawn M. et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metabolism 23, 517–528, doi: 10.1016/j.cmet.2016.01.007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muir A et al. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. eLife 6, doi: 10.7554/eLife.27713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cantor JR et al. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 169, 258–272.e217, doi: 10.1016/j.cell.2017.03.023 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vande Voorde J et al. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Science Advances 5, doi: 10.1126/sciadv.aau7314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossiter NJ et al. CRISPR screens in physiologic medium reveal conditionally essential genes in human cells. Cell Metabolism 33, 1248–1263.e1249, doi: 10.1016/j.cmet.2021.02.005 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi X et al. Combinatorial GxGxE CRISPR screen identifies SLC25A39 in mitochondrial glutathione transport linking iron homeostasis to OXPHOS. Nature Communications 13, doi: 10.1038/s41467-022-30126-9 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu XG et al. Functional Genomics In Vivo Reveal Metabolic Dependencies of Pancreatic Cancer Cells. Cell Metabolism 33, 211–221.e216, doi: 10.1016/j.cmet.2020.10.017 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biancur DE et al. Functional Genomics Identifies Metabolic Vulnerabilities in Pancreatic Cancer. Cell Metabolism 33, 199–210.e198, doi: 10.1016/j.cmet.2020.10.018 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pavlova Natalya N. & Thompson Craig B. The Emerging Hallmarks of Cancer Metabolism. Cell Metabolism 23, 27–47, doi: 10.1016/j.cmet.2015.12.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Venneti S et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Science Translational Medicine 7, doi: 10.1126/scitranslmed.aaa1009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson DL, Cox MM & Hoskins AA Lehninger principles of biochemistry. Eighth edition. edn, (Macmillan Learning, 2021). [Google Scholar]
- 21.Hatzivassiliou G et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 8, 311–321, doi: 10.1016/j.ccr.2005.09.008 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Pavlova NN, Zhu J & Thompson CB The hallmarks of cancer metabolism: Still emerging. Cell Metabolism 34, 355–377, doi: 10.1016/j.cmet.2022.01.007 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hosios AM & Vander Heiden MG The redox requirements of proliferating mammalian cells. Journal of Biological Chemistry 293, 7490–7498, doi: 10.1074/jbc.TM117.000239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayers JR & Vander Heiden MG Nature and Nurture: What Determines Tumor Metabolic Phenotypes? Cancer Research 77, 3131–3134, doi: 10.1158/0008-5472.Can-17-0165 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Kim J & DeBerardinis RJ Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metabolism 30, 434–446, doi: 10.1016/j.cmet.2019.08.013 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mayers JR et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 353, 1161–1165, doi: 10.1126/science.aaf5171 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rinaldi G et al. In Vivo Evidence for Serine Biosynthesis-Defined Sensitivity of Lung Metastasis, but Not of Primary Breast Tumors, to mTORC1 Inhibition. Molecular Cell 81, 386–397.e387, doi: 10.1016/j.molcel.2020.11.027 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Basnet H et al. Flura-seq identifies organ-specific metabolic adaptations during early metastatic colonization. eLife 8, doi: 10.7554/eLife.43627 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuneva MO et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. 15, 157–170, doi: 10.1016/j.cmet.2011.12.015 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jun S et al. The requirement for pyruvate dehydrogenase in leukemogenesis depends on cell lineage. Cell Metabolism, doi: 10.1016/j.cmet.2021.07.016 (2021). [DOI] [PubMed] [Google Scholar]
- 31.Mahendralingam MJ et al. Mammary epithelial cells have lineage-rooted metabolic identities. Nature Metabolism 3, 665–681, doi: 10.1038/s42255-021-00388-6 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Chen P-H et al. Metabolic Diversity in Human Non-Small Cell Lung Cancer Cells. Molecular Cell 76, 838–851.e835, doi: 10.1016/j.molcel.2019.08.028 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sullivan MR et al. Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metabolism 29, 1410–1421.e1414, doi: 10.1016/j.cmet.2019.02.015 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeNicola GM et al. NRF2 regulates serine biosynthesis in non–small cell lung cancer. Nature Genetics 47, 1475–1481, doi: 10.1038/ng.3421 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strasser A & Vaux DL Cell Death in the Origin and Treatment of Cancer. Molecular Cell 78, 1045–1054, doi: 10.1016/j.molcel.2020.05.014 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Cheung EC & Vousden KH The role of ROS in tumour development and progression. Nature Reviews Cancer, doi: 10.1038/s41568-021-00435-0 (2022). [DOI] [PubMed] [Google Scholar]
- 37.Harris IS & DeNicola GM The Complex Interplay between Antioxidants and ROS in Cancer. Trends in Cell Biology 30, 440–451, doi: 10.1016/j.tcb.2020.03.002 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Stockwell BR & Jiang X The Chemistry and Biology of Ferroptosis. Cell Chemical Biology 27, 365–375, doi: 10.1016/j.chembiol.2020.03.013 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lei G, Zhuang L & Gan B Targeting ferroptosis as a vulnerability in cancer. Nature Reviews Cancer 22, 381–396, doi: 10.1038/s41568-022-00459-0 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schafer ZT et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113, doi: 10.1038/nature08268 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piskounova E et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191, doi: 10.1038/nature15726 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ubellacker JM et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585, 113–118, doi: 10.1038/s41586-020-2623-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chakrabarty RP & Chandel NS Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell Stem Cell 28, 394–408, doi: 10.1016/j.stem.2021.02.011 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Green DR, Galluzzi L & Kroemer G Metabolic control of cell death. Science 345, doi: 10.1126/science.1250256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singh R, Letai A & Sarosiek K Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nature Reviews Molecular Cell Biology 20, 175–193, doi: 10.1038/s41580-018-0089-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andersen Joshua L. & Kornbluth S The Tangled Circuitry of Metabolism and Apoptosis. Molecular Cell 49, 399–410, doi: 10.1016/j.molcel.2012.12.026 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delbridge ARD, Valente LJ & Strasser A The Role of the Apoptotic Machinery in Tumor Suppression. Cold Spring Harbor Perspectives in Biology 4, a008789–a008789, doi: 10.1101/cshperspect.a008789 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang X, Overholtzer M & Thompson CB Autophagy in cellular metabolism and cancer. Journal of Clinical Investigation 125, 47–54, doi: 10.1172/jci73942 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carneiro BA & El-Deiry WS Targeting apoptosis in cancer therapy. Nature Reviews Clinical Oncology 17, 395–417, doi: 10.1038/s41571-020-0341-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Santana-Codina N, Mancias JD & Kimmelman AC The Role of Autophagy in Cancer. Annual Review of Cancer Biology 1, 19–39, doi: 10.1146/annurev-cancerbio-041816-122338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim SM et al. PTEN Deficiency and AMPK Activation Promote Nutrient Scavenging and Anabolism in Prostate Cancer Cells. Cancer Discovery 8, 866–883, doi: 10.1158/2159-8290.Cd-17-1215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Commisso C et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637, doi: 10.1038/nature12138 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kamphorst JJ et al. Human Pancreatic Cancer Tumors Are Nutrient Poor and Tumor Cells Actively Scavenge Extracellular Protein. Cancer Research 75, 544–553, doi: 10.1158/0008-5472.Can-14-2211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garcia-Bermudez J et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 567, 118–122, doi: 10.1038/s41586-019-0945-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zitvogel L, Tesniere A & Kroemer G Cancer despite immunosurveillance: immunoselection and immunosubversion. Nature Reviews Immunology 6, 715–727, doi: 10.1038/nri1936 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Au E, Wong G & Chapman JR Cancer in kidney transplant recipients. Nature Reviews Nephrology 14, 508–520, doi: 10.1038/s41581-018-0022-6 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Martin TD et al. The adaptive immune system is a major driver of selection for tumor suppressor gene inactivation. Science 373, 1327–1335, doi: 10.1126/science.abg5784 (2021). [DOI] [PubMed] [Google Scholar]
- 58.Jhunjhunwala S, Hammer C & Delamarre L Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nature Reviews Cancer 21, 298–312, doi: 10.1038/s41568-021-00339-z (2021). [DOI] [PubMed] [Google Scholar]
- 59.Buck MD, Sowell RT, Kaech SM & Pearce EL Metabolic Instruction of Immunity. Cell 169, 570–586, doi: 10.1016/j.cell.2017.04.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee JS et al. Urea Cycle Dysregulation Generates Clinically Relevant Genomic and Biochemical Signatures. Cell 174, 1559–1570.e1522, doi: 10.1016/j.cell.2018.07.019 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reinfeld BI et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 593, 282–288, doi: 10.1038/s41586-021-03442-1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang C-H et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162, 1229–1241, doi: 10.1016/j.cell.2015.08.016 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ho P-C et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 162, 1217–1228, doi: 10.1016/j.cell.2015.08.012 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leone RD & Powell JD Metabolism of immune cells in cancer. Nature Reviews Cancer 20, 516–531, doi: 10.1038/s41568-020-0273-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brand A et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metabolism 24, 657–671, doi: 10.1016/j.cmet.2016.08.011 (2016). [DOI] [PubMed] [Google Scholar]
- 66.Angelin A et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metabolism 25, 1282–1293.e1287, doi: 10.1016/j.cmet.2016.12.018 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Malladi S et al. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 165, 45–60, doi: 10.1016/j.cell.2016.02.025 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Agudo J et al. Quiescent Tissue Stem Cells Evade Immune Surveillance. Immunity 48, 271–285.e275, doi: 10.1016/j.immuni.2018.02.001 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brown S et al. Correction of aberrant growth preserves tissue homeostasis. Nature 548, 334–337, doi: 10.1038/nature23304 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dow Lukas E. et al. Apc Restoration Promotes Cellular Differentiation and Reestablishes Crypt Homeostasis in Colorectal Cancer. Cell 161, 1539–1552, doi: 10.1016/j.cell.2015.05.033 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ying Z, Sandoval M & Beronja S Oncogenic activation of PI3K induces progenitor cell differentiation to suppress epidermal growth. Nature Cell Biology 20, 1256–1266, doi: 10.1038/s41556-018-0218-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Warrell RP et al. Differentiation Therapy of Acute Promyelocytic Leukemia with Tretinoin (All-trans-Retinoic Acid). New England Journal of Medicine 324, 1385–1393, doi: 10.1056/nejm199105163242002 (1991). [DOI] [PubMed] [Google Scholar]
- 73.Intlekofer AM & Finley LWS Metabolic signatures of cancer cells and stem cells. Nat Metab 1, 177–188, doi: 10.1038/s42255-019-0032-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Laugesen A & Helin K Chromatin Repressive Complexes in Stem Cells, Development, and Cancer. Cell Stem Cell 14, 735–751, doi: 10.1016/j.stem.2014.05.006 (2014). [DOI] [PubMed] [Google Scholar]
- 75.Reid MA, Dai Z & Locasale JW The impact of cellular metabolism on chromatin dynamics and epigenetics. Nature Cell Biology 19, 1298–1306, doi: 10.1038/ncb3629 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Flavahan WA, Gaskell E & Bernstein BE Epigenetic plasticity and the hallmarks of cancer. Science 357, doi: 10.1126/science.aal2380 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee Joyce V. et al. Akt-Dependent Metabolic Reprogramming Regulates Tumor Cell Histone Acetylation. Cell Metabolism 20, 306–319, doi: 10.1016/j.cmet.2014.06.004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fan J, Krautkramer KA, Feldman JL & Denu JM Metabolic Regulation of Histone Post-Translational Modifications. ACS Chemical Biology 10, 95–108, doi: 10.1021/cb500846u (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xiao M et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes & Development 26, 1326–1338, doi: 10.1101/gad.191056.112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xu W et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 19, 17–30, doi: 10.1016/j.ccr.2010.12.014 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schvartzman JM, Thompson CB & Finley LWS Metabolic regulation of chromatin modifications and gene expression. Journal of Cell Biology 217, 2247–2259, doi: 10.1083/jcb.201803061 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Carey BW, Finley LWS, Cross JR, Allis CD & Thompson CB Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416, doi: 10.1038/nature13981 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mentch Samantha J. et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metabolism 22, 861–873, doi: 10.1016/j.cmet.2015.08.024 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Su X, Wellen KE & Rabinowitz JD Metabolic control of methylation and acetylation. Current Opinion in Chemical Biology 30, 52–60, doi: 10.1016/j.cbpa.2015.10.030 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martínez-Reyes I & Chandel NS Mitochondrial TCA cycle metabolites control physiology and disease. Nature Communications 11, doi: 10.1038/s41467-019-13668-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Baksh SC & Finley LWS Metabolic Coordination of Cell Fate by α-Ketoglutarate-Dependent Dioxygenases. Trends in Cell Biology 31, 24–36, doi: 10.1016/j.tcb.2020.09.010 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Suganuma T & Workman JL Chromatin and Metabolism. Annu Rev Biochem 87, 27–49, doi: 10.1146/annurev-biochem-062917-012634 (2018). [DOI] [PubMed] [Google Scholar]
- 88.Pirozzi CJ & Yan H The implications of IDH mutations for cancer development and therapy. Nature Reviews Clinical Oncology 18, 645–661, doi: 10.1038/s41571-021-00521-0 (2021). [DOI] [PubMed] [Google Scholar]
- 89.Dang L et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744, doi: 10.1038/nature08617 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lu C et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478, doi: 10.1038/nature10860 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chowdhury R et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO reports 12, 463–469, doi: 10.1038/embor.2011.43 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Figueroa ME et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 18, 553–567, doi: 10.1016/j.ccr.2010.11.015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morris J. P. t. et al. alpha-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 573, 595–599, doi: 10.1038/s41586-019-1577-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tran TQ et al. α-Ketoglutarate attenuates Wnt signaling and drives differentiation in colorectal cancer. Nature Cancer 1, 345–358, doi: 10.1038/s43018-020-0035-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gupta PB, Pastushenko I, Skibinski A, Blanpain C & Kuperwasser C Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 24, 65–78, doi: 10.1016/j.stem.2018.11.011 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Khacho M et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell 19, 232–247, doi: 10.1016/j.stem.2016.04.015 (2016). [DOI] [PubMed] [Google Scholar]
- 97.Hamanaka RB et al. Mitochondrial reactive oxygen species promote epidermal differentiation and hair follicle development. Sci Signal 6, ra8, doi: 10.1126/scisignal.2003638 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Faubert B, Solmonson A & DeBerardinis RJ Metabolic reprogramming and cancer progression. Science 368, doi: 10.1126/science.aaw5473 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sciacovelli M, Schmidt C, Maher ER & Frezza C Metabolic Drivers in Hereditary Cancer Syndromes. Annual Review of Cancer Biology 4, 77–97, doi: 10.1146/annurev-cancerbio-030419-033612 (2020). [DOI] [Google Scholar]
- 100.Kaelin William G. & McKnight Steven L. Influence of Metabolism on Epigenetics and Disease. Cell 153, 56–69, doi: 10.1016/j.cell.2013.03.004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Clark GR et al. Germline FH Mutations Presenting With Pheochromocytoma. The Journal of Clinical Endocrinology & Metabolism 99, E2046–E2050, doi: 10.1210/jc.2014-1659 (2014). [DOI] [PubMed] [Google Scholar]
- 102.Baysal BE et al. Mutations in SDHD, a Mitochondrial Complex II Gene, in Hereditary Paraganglioma. Science 287, 848–851, doi: 10.1126/science.287.5454.848 (2000). [DOI] [PubMed] [Google Scholar]
- 103.Vanharanta S et al. Early-Onset Renal Cell Carcinoma as a Novel Extraparaganglial Component of SDHB-Associated Heritable Paraganglioma. The American Journal of Human Genetics 74, 153–159, doi: 10.1086/381054 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Janeway KA et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proceedings of the National Academy of Sciences 108, 314–318, doi: 10.1073/pnas.1009199108 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tomlinson IP et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30, 406–410, doi: 10.1038/ng849 (2002). [DOI] [PubMed] [Google Scholar]
- 106.Gimenez-Roqueplo A-P et al. The R22X Mutation of the SDHD Gene in Hereditary Paraganglioma Abolishes the Enzymatic Activity of Complex II in the Mitochondrial Respiratory Chain and Activates the Hypoxia Pathway. The American Journal of Human Genetics 69, 1186–1197, doi: 10.1086/324413 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Armstrong N et al. SDHB knockout and succinate accumulation are insufficient for tumorigenesis but dual SDHB/NF1 loss yields SDHx-like pheochromocytomas. Cell Reports 38, doi: 10.1016/j.celrep.2022.110453 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Selak MA et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 7, 77–85, doi: 10.1016/j.ccr.2004.11.022 (2005). [DOI] [PubMed] [Google Scholar]
- 109.Isaacs JS et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 8, 143–153, doi: 10.1016/j.ccr.2005.06.017 (2005). [DOI] [PubMed] [Google Scholar]
- 110.Sciacovelli M et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537, 544–547, doi: 10.1038/nature19353 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sulkowski PL et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat Genet 50, 1086–1092, doi: 10.1038/s41588-018-0170-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sulkowski PL et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med 9, doi: 10.1126/scitranslmed.aal2463 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Guzy RD, Sharma B, Bell E, Chandel NS & Schumacker PT Loss of the SdhB, but Not the SdhA, Subunit of Complex II Triggers Reactive Oxygen Species-Dependent Hypoxia-Inducible Factor Activation and Tumorigenesis. Molecular and Cellular Biology 28, 718–731, doi: 10.1128/mcb.01338-07 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Alderson NL et al. S-(2-Succinyl)cysteine: A novel chemical modification of tissue proteins by a Krebs cycle intermediate. Archives of Biochemistry and Biophysics 450, 1–8, doi: 10.1016/j.abb.2006.03.005 (2006). [DOI] [PubMed] [Google Scholar]
- 115.Ooi A et al. An Antioxidant Response Phenotype Shared between Hereditary and Sporadic Type 2 Papillary Renal Cell Carcinoma. Cancer Cell 20, 511–523, doi: 10.1016/j.ccr.2011.08.024 (2011). [DOI] [PubMed] [Google Scholar]
- 116.Adam J et al. Renal Cyst Formation in Fh1-Deficient Mice Is Independent of the Hif/Phd Pathway: Roles for Fumarate in KEAP1 Succination and Nrf2 Signaling. Cancer Cell 20, 524–537, doi: 10.1016/j.ccr.2011.09.006 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sciacovelli M & Frezza C Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radical Biology and Medicine 100, 175–181, doi: 10.1016/j.freeradbiomed.2016.04.025 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Platero-Luengo A et al. An O2-Sensitive Glomus Cell-Stem Cell Synapse Induces Carotid Body Growth in Chronic Hypoxia. Cell 156, 291–303, doi: 10.1016/j.cell.2013.12.013 (2014). [DOI] [PubMed] [Google Scholar]
- 119.Bardella C, Pollard PJ & Tomlinson I SDH mutations in cancer. Biochimica et Biophysica Acta (BBA) - Bioenergetics 1807, 1432–1443, doi: 10.1016/j.bbabio.2011.07.003 (2011). [DOI] [PubMed] [Google Scholar]
- 120.Medeiros BC et al. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 31, 272–281, doi: 10.1038/leu.2016.275 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yan H et al. IDH1andIDH2Mutations in Gliomas. New England Journal of Medicine 360, 765–773, doi: 10.1056/NEJMoa0808710 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ward PS et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting α-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell 17, 225–234, doi: 10.1016/j.ccr.2010.01.020 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Watanabe T, Nobusawa S, Kleihues P & Ohgaki H IDH1 Mutations Are Early Events in the Development of Astrocytomas and Oligodendrogliomas. The American Journal of Pathology 174, 1149–1153, doi: 10.2353/ajpath.2009.080958 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lu C et al. Induction of sarcomas by mutant IDH2. Genes & Development 27, 1986–1998, doi: 10.1101/gad.226753.113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Amatangelo MD et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 130, 732–741, doi: 10.1182/blood-2017-04-779447 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fukumoto K, Nguyen TB, Chiba S & Sakata-Yanagimoto M Review of the biologic and clinical significance of genetic mutations in angioimmunoblastic T-cell lymphoma. Cancer Sci 109, 490–496, doi: 10.1111/cas.13393 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Butzmann A et al. A comprehensive analysis of RHOA mutation positive and negative angioimmunoblastic T-cell lymphomas by targeted deep sequencing, expression profiling and single cell digital image analysis. Int J Mol Med 46, 1466–1476, doi: 10.3892/ijmm.2020.4686 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xie M et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nature Medicine 20, 1472–1478, doi: 10.1038/nm.3733 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Locasale JW et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nature Genetics 43, 869–874, doi: 10.1038/ng.890 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lin A-P et al. D2HGDH regulates alpha-ketoglutarate levels and dioxygenase function by modulating IDH2. Nature Communications 6, doi: 10.1038/ncomms8768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gorelick AN et al. Respiratory complex and tissue lineage drive recurrent mutations in tumour mtDNA. Nat Metab 3, 558–570, doi: 10.1038/s42255-021-00378-8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.DeBerardinis RJ & Chandel NS Fundamentals of cancer metabolism. Science Advances 2, doi: 10.1126/sciadv.1600200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Labuschagne CF, Zani F & Vousden KH Control of metabolism by p53 – Cancer and beyond. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 1870, 32–42, doi: 10.1016/j.bbcan.2018.06.001 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hingorani SR et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450, doi: 10.1016/s1535-6108(03)00309-x (2003). [DOI] [PubMed] [Google Scholar]
- 135.Morris JP, Wang SC & Hebrok M KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nature Reviews Cancer 10, 683–695, doi: 10.1038/nrc2899 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Carrer A et al. Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis. Cancer Discov 9, 416–435, doi: 10.1158/2159-8290.CD-18-0567 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Agathocleous M et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476–481, doi: 10.1038/nature23876 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bonnay F et al. Oxidative Metabolism Drives Immortalization of Neural Stem Cells during Tumorigenesis. Cell 182, 1490–1507.e1419, doi: 10.1016/j.cell.2020.07.039 (2020). [DOI] [PubMed] [Google Scholar]
- 139.Schell John C. et al. A Role for the Mitochondrial Pyruvate Carrier as a Repressor of the Warburg Effect and Colon Cancer Cell Growth. Molecular Cell 56, 400–413, doi: 10.1016/j.molcel.2014.09.026 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bensard CL et al. Regulation of Tumor Initiation by the Mitochondrial Pyruvate Carrier. Cell Metab 31, 284–300 e287, doi: 10.1016/j.cmet.2019.11.002 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Perez-Ramirez CA & Christofk HR Challenges in Studying Stem Cell Metabolism. Cell Stem Cell 28, 409–423, doi: 10.1016/j.stem.2021.02.016 (2021). [DOI] [PubMed] [Google Scholar]
- 142.Viale A et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514, 628–632, doi: 10.1038/nature13611 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Janiszewska M et al. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes & Development 26, 1926–1944, doi: 10.1101/gad.188292.112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kuntz EM et al. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nature Medicine 23, 1234–1240, doi: 10.1038/nm.4399 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Vazquez F et al. PGC1α Expression Defines a Subset of Human Melanoma Tumors with Increased Mitochondrial Capacity and Resistance to Oxidative Stress. Cancer Cell 23, 287–301, doi: 10.1016/j.ccr.2012.11.020 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rao S et al. AIF-regulated oxidative phosphorylation supports lung cancer development. Cell Research 29, 579–591, doi: 10.1038/s41422-019-0181-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Raffel S et al. BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 551, 384–388, doi: 10.1038/nature24294 (2017). [DOI] [PubMed] [Google Scholar]
- 148.Schaaf MB, Garg AD & Agostinis P Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death & Disease 9, doi: 10.1038/s41419-017-0061-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schito L & Semenza GL Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends in Cancer 2, 758–770, doi: 10.1016/j.trecan.2016.10.016 (2016). [DOI] [PubMed] [Google Scholar]
- 150.Lee P, Chandel NS & Simon MC Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nature Reviews Molecular Cell Biology 21, 268–283, doi: 10.1038/s41580-020-0227-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ito K & Suda T Metabolic requirements for the maintenance of self-renewing stem cells. Nature Reviews Molecular Cell Biology 15, 243–256, doi: 10.1038/nrm3772 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chakraborty AA et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 363, 1217–1222, doi: 10.1126/science.aaw1026 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Intlekofer Andrew M. et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metabolism 22, 304–311, doi: 10.1016/j.cmet.2015.06.023 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Oldham William M., Clish Clary B., Yang Y & Loscalzo J Hypoxia-Mediated Increases in l -2-hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell Metabolism 22, 291–303, doi: 10.1016/j.cmet.2015.06.021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Shim E-H et al. l-2-Hydroxyglutarate: An Epigenetic Modifier and Putative Oncometabolite in Renal Cancer. Cancer Discovery 4, 1290–1298, doi: 10.1158/2159-8290.Cd-13-0696 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wahl DR & Venneti S 2-Hydoxyglutarate: D/Riving Pathology in gLiomaS. Brain Pathol 25, 760–768, doi: 10.1111/bpa.12309 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Vladoiu MC et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 572, 67–73, doi: 10.1038/s41586-019-1158-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Michealraj KA et al. Metabolic Regulation of the Epigenome Drives Lethal Infantile Ependymoma. Cell 181, 1329–1345.e1324, doi: 10.1016/j.cell.2020.04.047 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mack SC et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506, 445–450, doi: 10.1038/nature13108 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Panwalkar P et al. Targeting integrated epigenetic and metabolic pathways in lethal childhood PFA ependymomas. Sci Transl Med 13, eabc0497, doi: 10.1126/scitranslmed.abc0497 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sullivan MR et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8, doi: 10.7554/eLife.44235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Pan M et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol 18, 1090–1101, doi: 10.1038/ncb3410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ishak Gabra MB et al. Dietary glutamine supplementation suppresses epigenetically-activated oncogenic pathways to inhibit melanoma tumour growth. Nature Communications 11, doi: 10.1038/s41467-020-17181-w (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Baksh SC et al. Extracellular serine controls epidermal stem cell fate and tumour initiation. Nature Cell Biology, doi: 10.1038/s41556-020-0525-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Loenarz C & Schofield CJ Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends in Biochemical Sciences 36, 7–18, doi: 10.1016/j.tibs.2010.07.002 (2011). [DOI] [PubMed] [Google Scholar]
- 166.Cimmino L et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 170, 1079–1095.e1020, doi: 10.1016/j.cell.2017.07.032 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rappez L et al. SpaceM reveals metabolic states of single cells. Nature Methods 18, 799–805, doi: 10.1038/s41592-021-01198-0 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jang C et al. Metabolite Exchange between Mammalian Organs Quantified in Pigs. Cell Metabolism 30, 594–606.e593, doi: 10.1016/j.cmet.2019.06.002 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A & Wargo JA The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 33, 570–580, doi: 10.1016/j.ccell.2018.03.015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Janney A, Powrie F & Mann EH Host-microbiota maladaptation in colorectal cancer. Nature 585, 509–517, doi: 10.1038/s41586-020-2729-3 (2020). [DOI] [PubMed] [Google Scholar]
- 171.Louis P, Hold GL & Flint HJ The gut microbiota, bacterial metabolites and colorectal cancer. Nature Reviews Microbiology 12, 661–672, doi: 10.1038/nrmicro3344 (2014). [DOI] [PubMed] [Google Scholar]
- 172.Smith PM et al. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science 341, 569–573, doi: 10.1126/science.1241165 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chang PV, Hao L, Offermanns S & Medzhitov R The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proceedings of the National Academy of Sciences 111, 2247–2252, doi: 10.1073/pnas.1322269111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bell HN et al. Reuterin in the healthy gut microbiome suppresses colorectal cancer growth through altering redox balance. Cancer Cell 40, 185–200 e186, doi: 10.1016/j.ccell.2021.12.001 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kadosh E et al. The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 586, 133–138, doi: 10.1038/s41586-020-2541-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Furman D et al. Chronic inflammation in the etiology of disease across the life span. Nature Medicine 25, 1822–1832, doi: 10.1038/s41591-019-0675-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Greten FR & Grivennikov SI Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 51, 27–41, doi: 10.1016/j.immuni.2019.06.025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lindemans CA et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 528, 560–564, doi: 10.1038/nature16460 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gaber T, Strehl C & Buttgereit F Metabolic regulation of inflammation. Nature Reviews Rheumatology 13, 267–279, doi: 10.1038/nrrheum.2017.37 (2017). [DOI] [PubMed] [Google Scholar]
- 180.Guerra C et al. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell 11, 291–302, doi: 10.1016/j.ccr.2007.01.012 (2007). [DOI] [PubMed] [Google Scholar]
- 181.Alonso-Curbelo D et al. A gene–environment-induced epigenetic program initiates tumorigenesis. Nature 590, 642–648, doi: 10.1038/s41586-020-03147-x (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Morris J. P. t., Cano DA, Sekine S, Wang SC & Hebrok M Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. 120, 508–520, doi: 10.1172/JCI40045 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Schwitalla S et al. Intestinal Tumorigenesis Initiated by Dedifferentiation and Acquisition of Stem-Cell-like Properties. Cell 152, 25–38, doi: 10.1016/j.cell.2012.12.012 (2013). [DOI] [PubMed] [Google Scholar]
- 184.Li F et al. FBP1 loss disrupts liver metabolism and promotes tumorigenesis through a hepatic stellate cell senescence secretome. Nature Cell Biology 22, 728–739, doi: 10.1038/s41556-020-0511-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Arnold M et al. Obesity and cancer: An update of the global impact. Cancer Epidemiology 41, 8–15, doi: 10.1016/j.canep.2016.01.003 (2016). [DOI] [PubMed] [Google Scholar]
- 186.Calle EE, Rodriguez C, Walker-Thurmond K & Thun MJ Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of U.S. Adults. New England Journal of Medicine 348, 1625–1638, doi: 10.1056/NEJMoa021423 (2003). [DOI] [PubMed] [Google Scholar]
- 187.Tajan M & Vousden KH Dietary Approaches to Cancer Therapy. Cancer Cell 37, 767–785, doi: 10.1016/j.ccell.2020.04.005 (2020). [DOI] [PubMed] [Google Scholar]
- 188.Schulz MD et al. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 514, 508–512, doi: 10.1038/nature13398 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Beyaz S et al. Dietary suppression of MHC class II expression in intestinal epithelial cells enhances intestinal tumorigenesis. Cell Stem Cell 28, 1922–1935.e1925, doi: 10.1016/j.stem.2021.08.007 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Park EJ et al. Dietary and Genetic Obesity Promote Liver Inflammation and Tumorigenesis by Enhancing IL-6 and TNF Expression. Cell 140, 197–208, doi: 10.1016/j.cell.2009.12.052 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Chung KM et al. Endocrine-Exocrine Signaling Drives Obesity-Associated Pancreatic Ductal Adenocarcinoma. Cell 181, 832–847.e818, doi: 10.1016/j.cell.2020.03.062 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Beyaz S et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 531, 53–58, doi: 10.1038/nature17173 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ringel AE et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 183, 1848–1866.e1826, doi: 10.1016/j.cell.2020.11.009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Mana MD et al. High-fat diet-activated fatty acid oxidation mediates intestinal stemness and tumorigenicity. Cell Reports 35, doi: 10.1016/j.celrep.2021.109212 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Cheng C-W et al. Ketone Body Signaling Mediates Intestinal Stem Cell Homeostasis and Adaptation to Diet. Cell 178, 1115–1131.e1115, doi: 10.1016/j.cell.2019.07.048 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Dmitrieva-Posocco O et al. β-Hydroxybutyrate suppresses colorectal cancer. Nature 605, 160–165, doi: 10.1038/s41586-022-04649-6 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Geidl-Flueck B et al. Fructose- and sucrose- but not glucose-sweetened beverages promote hepatic de novo lipogenesis: A randomized controlled trial. Journal of Hepatology 75, 46–54, doi: 10.1016/j.jhep.2021.02.027 (2021). [DOI] [PubMed] [Google Scholar]
- 198.Febbraio MA & Karin M “Sweet death”: Fructose as a metabolic toxin that targets the gut-liver axis. Cell Metabolism 33, 2316–2328, doi: 10.1016/j.cmet.2021.09.004 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Taylor SR et al. Dietary fructose improves intestinal cell survival and nutrient absorption. Nature 597, 263–267, doi: 10.1038/s41586-021-03827-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Goncalves MD et al. High-fructose corn syrup enhances intestinal tumor growth in mice. Science 363, 1345–1349, doi: 10.1126/science.aat8515 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Luengo A, Gui DY & Vander Heiden MG Targeting Metabolism for Cancer Therapy. Cell Chemical Biology 24, 1161–1180, doi: 10.1016/j.chembiol.2017.08.028 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Possemato R et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350, doi: 10.1038/nature10350 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Maddocks ODK et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493, 542–546, doi: 10.1038/nature11743 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Gao X et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 572, 397–401, doi: 10.1038/s41586-019-1437-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Bian Y et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 585, 277–282, doi: 10.1038/s41586-020-2682-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lim AR, Rathmell WK & Rathmell JC The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. eLife 9, doi: 10.7554/eLife.55185 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Courtney KD et al. Isotope Tracing of Human Clear Cell Renal Cell Carcinomas Demonstrates Suppressed Glucose Oxidation In Vivo. Cell Metabolism 28, 793–800.e792, doi: 10.1016/j.cmet.2018.07.020 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sack LM et al. Profound Tissue Specificity in Proliferation Control Underlies Cancer Drivers and Aneuploidy Patterns. Cell 173, 499–514.e423, doi: 10.1016/j.cell.2018.02.037 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hoadley KA et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 173, 291–304.e296, doi: 10.1016/j.cell.2018.03.022 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Carmona-Fontaine C et al. Metabolic origins of spatial organization in the tumor microenvironment. Proceedings of the National Academy of Sciences 114, 2934–2939, doi: 10.1073/pnas.1700600114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tasdogan A et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 577, 115–120, doi: 10.1038/s41586-019-1847-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]