

Abstract
Centrosomes together with the mitotic spindle ensure the faithful distribution of chromosomes between daughter cells, and spindle orientation is a major determinant of cell fate during tissue regeneration. Spindle defects are not only an impetus of chromosome instability but are also a cause of developmental disorders involving defective asymmetric cell division. In this work, we demonstrate BCCIP, especially BCCIPα, as a previously unidentified component of the mitotic spindle pole and the centrosome. We demonstrate that BCCIP localizes proximal to the mother centriole and participates in microtubule organization and then redistributes to the spindle pole to ensure faithful spindle architecture. We find that BCCIP depletion leads to morphological defects, disoriented mitotic spindles, chromosome congression defects, and delayed mitotic progression. Our study identifies BCCIP as a novel factor critical for microtubule regulation and explicates a mechanism utilized by BCCIP in tumor suppression.
Keywords: Centrosomes, Mother Centriole, Spindle Pole, Mitotic Spindle, Spindle Orientation, Aneuploidy, Microtubules, Acetyl Tubulin, Dynactin, Chromosome Congression
INTRODUCTION
During mitosis, both the faultless segregation of newly duplicated chromosomes and the proper positioning of daughter cells requires an elegant mitotic apparatus, a complex microtubule-based protein machine organized in a bipolar fashion (1). The assembly of the mitotic apparatus occurs de novo once, and only once per cell cycle and requires a high level of cooperation between microtubules, centrosomes, microtubule associated proteins (MAPs), and molecular motors (1, 2). Factors that compromise the reliability of the mitotic apparatus cause aneuploidy, a hallmark of cancer and the rate-limiting step in tumorigenic transformation (2–6). Faithful spindle assembly is critical not only for chromosome distribution, but also for the three dimensional orientation of the spindle (3, 7). Mitotic spindle orientation is regulated by the interplay between centrosomes, microtubules, and molecular motors and is critical for stem cell polarity and tissue regeneration (8, 9). This pathway also plays a pivotal role in cell division directed differentiation (8, 9). In addition, a link between the fidelity of spindle orientation and tumor formation has been recognized in the context of cancer stem cell renewal (10). Thus, the characterization of factors which destabilize the mitotic apparatus is not only of importance to understand the nature of aneuploid diseases, such as cancer, but also for stem cell renewal, tissue development and regeneration.
The principal microtubule organizing center of the cell is known as the centrosome. It consists of an orthogonal pair of centrioles enveloped by a mesh of an electron-dense material known as the pericentriolar matrix (11–13).
One centriole, known as the mother centriole, is one full cell cycle older than its counterpart and contains unique protein complexes responsible for the organizing the cell’s microtubule network into a single point-like focus (11). This function, known as microtubule anchoring, is strictly associated with the mother centriole and is paramount for directing cell polarity, shape, and motility as well as orienting the cell axis during division (8–11, 14). During mitosis, centrosomes play an integral role in chromosome capture by nucleating soluble tubulin subunits into the polymeric microtubules that comprise the spindle. Following microtubule nucleation, centrosomes are focused by a series of motor proteins into two distinct spindle poles containing a meshwork of microtubule regulators. The focusing of each centrosome into a distinct spindle pole matrix is thought to increase spindle tension and chromosome segregation fidelity by transducing negative end motor force (1, 15, 16). Among these constituents, the minus-end directed motor dynein is vital for pole establishment (1, 15). Dynein activities are regulated through its processivity factor, dynactin, a component also found in the mother centriole that regulates the centrosome’s microtubule anchoring and stabilizing capabilities (17–20). Dynein/dynactin also cooperate with minus-end MAPs, such as NuMa, which sequester, stabilize, and bundle microtubules at the poles (8, 16, 21). Thus, the interplay between centrosomes, molecular motors, and MAPs is intimately linked to ensure faithfulness of mitosis.
BCCIP was initially identified as a BRCA2 and p21 interacting protein and is essential for cell viability in mice and budding yeast (22–27). Despite a high degree of evolutionary conservation across all eukaryotes, the structure and function of the BCCIP gene is not fully understood. Canonically, BCCIP is thought to regulate DNA damage response, suppress spontaneous DNA damage, and modulate the G1/S transition through the cell cycle (23–26, 28). Concurrently, this view of BCCIP has also been expanded to include roles in cytoskeletal rearrangement, ribosome biogenesis, and nuclear export (22–27, 29–31). In Homo sapiens two major BCCIP isoforms that result from alternative splicing exist; designated BCCIPα (322 amino acids) and BCCIPβ (314aa) (32). These two isoforms share an identical conserved domain spanning ~258aa but are diversified by unique C-termini (32). Thus, it is likely that in humans BCCIPα and BCCIPβ have evolved to further specify the function of the sole BCCIP gene present in other eukaryotes. BCCIP loss has been implicated in numerical chromosome instability, polyploidization, and partial and non-permanent loss of BCCIP can spur tumorigenesis in mice (26, 29).
In this work we demonstrate that BCCIP, especially BCCIPα, associates with centrosomes, spindle poles, and the mitotic cell cortex. We identify BCCIP as a new component of the centrosome and the mitotic spindle pole, and functions in regulating microtubule anchoring, microtubule stability, spindle architecture, and spindle orientation. These newly identified functions appear to be independent of BCCIP’s role in DNA damage response. Our work not only identifies a critical component the mother centriole that plays a role in interphase microtubule organization, but also dynein/dynactin mediated spindle assembly. These data suggest an additional mechanism by which BCCIP contributes to not only genomic stability but also to organismal development.
RESULTS
Preferential localization of BCCIP to the mother centriole
BRCA2 and BRCA1 are bona-fide components of the microtubule-organizing center and BCCIP has been demonstrated to interact with BRCA2 (30, 33, 34). In HT1080 cells, we observed a clear localization of BCCIP in both the interphase centrosome and the mitotic spindle poles as judged by BCCIP co-localization with γ-tubulin or α-tubulin (Figure 1a). Interestingly, during late prophase, immunofluorescent staining revealed that the concentration of centrosomal BCCIP was enhanced relative to interphase cells and that BCCIP appeared to expand its presence to the crescent shaped spindle pole matrix (Figure 1a). The specificity of the BCCIP antibody was fully validated by Western blots, antigen-absorption followed by immunofluorescent staining (see Supplement Figure S1a, S1b), and the localization of BCCIP to centrosomes was confirmed with several independent antibodies in different human and mouse cells (Figure S1c, S1d, S1e).
Figure 1. BCCIP is associated with the centrosome and the mitotic apparatus.

A: The localization of BCCIP to centrosomes and spindle poles. HT1080 cells were with probed with anti-BCCIP and anti-γ-Tubulin (interphase centrosomes; top two rows) or with anti-BCCIP anti-α-tubulin (mitotic spindles; bottom 4 rows). Shown are representative images of cells at indicated cell cycle phase.
B: Purification of centrosome isolates. HeLa cells were synchronized in mitosis, collected, and lysed. Centrosomes were separated by sucrose gradient centrifugation (see Materials and Methods). Following density gradient centrifugation, centrosome fractions were collected, boiled in SDS sample buffer, and subjected to SDS-PAGE and western blot. The assayed fractions were probed with the indicated antibodies. The concentration of the sucrose fraction increases from left to right (40%–70%). WCE: whole cell extract.
C: BCCIP is associated with purified centrosomes. Centrosome isolates were pooled, sedimented onto a coverslip, and immuno-probed with the indicated antibodies. The boxed image is zoomed to demonstrate the decoration of BCCIP around the orthogonal centriole pair.
D: Preferential localization of BCCIP to the mother centriole. GFP-EB1 expressing HT1080 cells were immuno-stained for γ-Tubulin (blue, a marker equally present on both centrioles) and BCCIP (red). Images from a representative pair of centrioles as viewed from three different angles is depicted, as well as a cartoon illustrating the arrangements of γ-Tubulin, EB1, and BCCIP. MC: mother centriole, DC: daughter centriole.
E: The relative distribution of proteins between paired centrioles. The fluorescent intensities of γ-Tubulin, BCCIP, and EB1 among 46 centriole pairs was measured. Shown are the fluorescent intensity ratios and the clustered plot of ratios among all the centriole pairs.
A 3D view of the BCCIP localization to interphase centrioles and mitotic spindles can be found in Supplement S2, movies M1 and M2.
To confirm the observations, we first purified centrosomes through use of sucrose gradient centrifugation. Following ultracentrifugation, gradient fractions were collected and the centrosome-enriched fraction was verified by immunoblotting for the centrosome markers γ-tubulin, Aurora-A, and CDC2. As shown in Figure 1b, the centrosome fractions were devoid of the cytosolic and nuclear markers (GAPDH and PCNA), but enriched with several centrosome components (γ-tubulin, Aurora-A, and CDC2), as well as BCCIP. Furthermore, when the centrosome fraction was centrifuged through a glycerol cushion onto a coverslip and stained for the centrosome markers Plk1 and γ-Tubulin, we observed that BCCIP, but not CoxIV (a non-centrosome protein), was retained within the centrosome complex (Figure 1c).
In interphase cells, we noticed that BCCIP tended to be co-enriched with EB1 within one centriole, and was relatively reduced within its cohort (Figure 1d). In order to quantitate this phenomenon we acquired 0.2 micron centrosome Z-stacks and measured the fluorescent intensities of γ-tubulin, BCCIP, or EB1 within each centriole. We then calculated the protein fluorescent intensity ratio between the paired EB1-high (mother) and EB1-low (daughter) centrioles. As shown in Figure 1e, the distribution of γ-tubulin between the paired centrioles was relatively identical, with an average ratio and standard error values of 1.05±0.02 (n=46). However, the EB1 and BCCIP ratios between the same centriole pairs demonstrated values of 1.56±0.08 and 1.78±0.17 respectively; both significantly higher than that of γ-tubulin (p=6.8E-08, 8.5E-05 respectively, Student-t test). These results suggest that BCCIP exhibits a localization bias within EB1-enriched mother centrioles. Three-dimensional reconstruction of the centrosome complex revealed that BCCIP sheathed, but did not overlap the appendage marker EB1 (Figure 1d, Supplement S2 and Movie M1), which suggests that BCCIP is not a subdistal appendage component, but is more likely a physical tether between the microtubule minus end and the subdistal appendages. Altogether, these data firmly establish that BCCIP is a component of the centrosome and mitotic spindle pole, and that most of BCCIP is confined proximal to, but not within the subdistal appendages of the mother centriole in interphase.
The human specific BCCIPα is the dominant centrosome and spindle pole associated isoform, but this association is mediated through a shared domain between BCCIPα and BCCIPβ
Human cells express two isoforms of BCCIP created by alternative splicing, designated BCCIPα and BCCIPβ (26). These two isoforms are largely identical with the exception of a variable C-terminus (26). This discrepancy led us to ask the question if human cells exhibited isoform specific localization patterns. To address this question we expressed YFP-BCCIPα or YFP-BCCIPβ in cells, and were surprised to notice that BCCIPα clearly associated with spindle poles and fibers (Figure 2a), while BCCIPβ only weakly appeared on spindle poles (Figure 2a). Next, we examined the distribution and retention of BCCIP isoforms in the centrosome complex after treatments with buffers of increasing ionic strength (see Materials and Method). As shown in Figure 2b, BCCIPα was more abundant than BCCIPβ in the centrosome preparation. Although both isoforms could be removed from the centrosome complex with weak detergents, BCCIPα appeared to be more resistant to extraction than BCCIPβ. In contrast, core centrosome components, such as γ-tubulin and HSP90 could only be removed with harsher chaotropic reagents, as previously demonstrated (35). Taken together, these results demonstrate that in human cells both isoforms of BCCIP are capable of associating with the centrosome and the spindle pole, but BCCIPα is the more dominant isoform. Intriguingly, the sole BCCIP isoform in mice, which resembles human BCCIPβ, can fully localize to spindle poles endogenously in mice (Figure S1d), and when transiently expressed in human cells (Figure S1e). These findings suggest that humans have likely evolved a preferred spindle pole associated BCCIP isoform but in the absence of BCCIPα, BCCIPβ (or BCCIPβ-like homologs) demonstrates a comparable localization to centrosomes and mitotic spindle poles.
Figure 2. BCCIPα is the predominant centrosome associated isoform.

A: The localization of exogenous BCCIP isoforms to spindle poles. HT1080 cells stably expressing YFP-BCCIPα, YFP-BCCIPβ, or YFP (negative control) were stained for γ-tubulin (red, centrosomes) and counterstained with DAPI. Illustrated are the representative images at different stages of mitosis.
B: BCCIP has a labile association with centrosomes. Centrosome fractions were pelleted and incubated with the indicated buffers, re-pelleted by centrifugation, and the supernatant was collected. The remaining pellet fraction (P) was resuspended in boiling loading buffer and both supernatant and pellet fractions were subjected to SDS-PAGE and western blot. The crude extract (CE) was run concurrently as reference. P: the pelleted centrosomes after buffer treatment; S: the stripped-off proteins in the solution. 1D, 2D, 3D, and Urea indicate the buffers with increasing ionic strength (see Materials and Methods).
C: Amino acids 111-257 of BCCIP mediate its localization to spindle poles. The illustrated panel of YFP-tagged full-length and truncated BCCIP proteins were transiently expressed in BCCIP knockdown 293T cells. Cells were co-probed with α-tubulin to assess spindle pole localization.
D: Expression verification of exogenous YFP-BCCIP fragments. The expression levels of the same panel of BCCIP fragments as in panel-2C verified by Western blot.
Next, we sought to determine the domain that mediated the localization of BCCIP to the spindle pole. To answer this question, we transiently expressed a panel of shRNA-resistant YFP-BCCIP fragments in BCCIP knockdown cells (Figure 2c, 2d). We observed that the smallest fragment capable of binding to the spindle pole spanned amino acids 111-257, which contains a putative and conserved coiled coil domain (Figure 2c). Deletion of either the coiled coil (BCCIP-SR3 in Figure 2c) or the region upstream of the coiled coil (BCCIP-Δ2 in Figure 2c) markedly reduced the association of BCCIP with the spindle pole. Interestingly, although BCCIPα and BCCIPβ have distinct C-termini (26), loss of the C-terminus of BCCIPα had no effect on spindle pole localization. These data imply that the unique C-terminal of BCCIPα does not promote its association with the spindle pole, but rather it is likely that C-terminus elements found in BCCIPβ restrict its binding, consistent with the finding that the sole mouse BCCIP isoform is able to localize to the spindle pole (Figure S1d, S1e).
BCCIP is recruited to the spindle pole matrix and centrosome by microtubules and dynein/dynactin activity
The spindle pole matrix consists of a meshwork of microtubules, centrosomes, microtubule associated proteins, and molecular motors and depends on an intact microtubule network together with retrograde motor transport for assembly (1, 15). Conversely, centrosome core components are associated with the centrosome constitutively and do not depend on microtubule flux (35, 36). The observation that BCCIP localized to both interphase centrosomes and mitotic spindle poles (Figure 1a) prompted us to determine if the localization of BCCIP was dependent on microtubules. To answer this question, we first challenged cells with nocodoazole, a microtubule depolymerizing agent, or taxol, a drug that inhibits the disassembly of microtubules (Figure 3a). We observed that nocodazole completely disassembled the spindle, reducing centrosomes to their respective centrioles, and eliminated most but not all of the endogenous BCCIP signal at the centrosome as visualized by colocalization with either α-tubulin or γ-tubulin staining (Figure 3a, middle row). Alternatively, taxol induced the formation of multiple psuedo-asters and abolished the tight association of the centrosomes with the minus end of the spindle. We observed that in this condition BCCIP was strongly recruited to the taxol stabilized microtubule bundles, but largely lost from the centrosomes (lower panel Figure 3a). Identical results were reproduced by YFP tagged BCCIPα (Figure S3a). Next, we recapitulated these results biochemically by precipitating microtubules from mitotic lysates with taxol and observed that both BCCIPα and BCCIPβ were enriched in the taxol-treated microtubule pellet fraction, but not in the nocodazole-treated sample (Figure 3b). In addition, transiently expressed YFP-BCCIPα and YFP-BCCIP (aa111-257), but not YFP alone, had equal affinity to microtubules (Figure S3b), confirming a requirement for this domain to associate with the spindle pole.
Figure 3. BCCIP binds to microtubules and is recruited to spindle poles by dynein/dynactin activity.

A: The localization of BCCIP to spindle poles requires microtubules. U2OS cells were treated with nocodazole or taxol and were co-stained with BCCIP and γ-tubulin (left group) or with BCCIP and α-tubulin (right group). When the spindle is depolymerized by nocodazole, α-tubulin stains the nocodazole resistant centriole microtubules. The arrows indicate the small fraction of BCCIP that remains stably associated with centrosomes following treatments.
B: BCCIP co-precipitates with polymerized microtubules. Mitotic cell lysates were pre-cleared by ultracentrifugation. The soluble lysates were then treated with taxol (Tax) to repolymerize microtubules or nocodazole (Noc) to prevent repolymerization. Following ultracentrifugation, microtubules are in the pellet (P), and soluble tubulin is retained in the supernatant (S). Equal portion of supernatant and pellet fractions were subjected to western blot and with the indicated antibodies. Sup: supernatant; WCE: whole cell extract; N: Nocodazole Treated; T: Taxol Treated.
C: BCCIP complexes with microtubules, centrosomes, and dynactin. HeLa mitotic extract was incubated with GST-BCCIPα, GST-BCCIPβ, or GST and subjected to glutathione-bead pulldown. 1% of the input and 5% of the pulldown was subjected to western blot and probed with the indicated antibodies.
D: BCCIP co-localizes with dynactin at the mother centriole, spindle pole, and cell cortex. Shown are representative confocal images demonstrating the co-localization of BCCIP with dynactin at the mother centriole during interphase and the spindle pole and cell cortex during mitosis.
E & F: Inhibition of dynein by Ciliobrevin D disrupts the distribution of BCCIP at the spindle poles. U2OS cells were treated with the dynein inhibitor Ciliobrevin D and stained with the indicated antibodies. Shown are the representative images (3E) and the quantified BCCIP positive areas from spindle poles (3F).
G & H: Over-expression of p50/dynamitin diminishes the levels of spindle pole associated BCCIP. HeLa cells were transfected with GFP-p50/dynamitin or GFP and the BCCIP intensity at the poles was quantified. Shown are representative images (3G) and quantification of the BCCIP intensity at the poles.
We then reasoned that because the deposition of BCCIP to the poles required an intact microtubule network, BCCIP might be localized through the activities of the major minus end motor protein dynein/dynactin, a multi-protein complex compromised of at least seven subunits, including p150 glued and Arp-1 (18, 37). In order to test this hypothesis, we performed a GST-pull down of mitotic cell lysates with BCCIPα and BCCIPβ, and GST (Figure 3c). Intriguingly, BCCIPα, but not BCCIPβ or GST itself was sufficient to pull down the dynactin components, p150 glued and Arp-1, and dynactin and BCCIP were associated with a complex that contained centrosomal γ-tubulin and α/β-tubulin dimers (Figure 3c). In addition, BCCIP and p150 glued colocalized proximal to the mother centriole during interphase, as well as the spindle pole and cortex during mitosis (Figure 3d).
Last, to determine if dynein activity was required for proper BCCIP targeting during mitosis, we incubated cells with the dynein specific inhibitor Ciliobrevin-D (CD). In this condition we observed that the normally compact focus of BCCIP at the spindle poles was broadened and increased compared to the control, suggesting that dynein motility plays a critical role in deposition of BCCIP in the cell (Figure 3e, 3f). To recapitulate this finding genetically we over-expressed the p50/dynamitin subunit of dynactin, which results in a well characterized dominant negative dispersion of the dynactin complex and subsequent inhibition of the retrograde transport of dynactin cargo (15, 38). We found that transient overexpression of the dynactin subunit p50/dynamitin markedly disrupted the spindle pole associated BCCIP fraction (Figure 3g, 3h), suggesting a model where BCCIP associates with dynein/dynactin in a microtubule dependent manner.
BCCIP is required for the microtubule organization at the spindle pole and centrosome
The formation of the microtubule array is comprised of three independent but interconnected steps: first the nucleation of nascent tubulin by γ-tubulin complexes, second the elongation, stabilization, and minus end capping of the microtubule polymer by minus end associated MAPs, and third the anchoring of the growing microtubule to the subdistal appendages of the mother centriole (39, 40). We then hypothesized that constitutively associated centrosome fraction of BCCIP might be involved in the regulation of minus end microtubule dynamics, given that BCCIP also associates with microtubules. In order to test this hypothesis, a microtubule regrowth experiment was performed. We first treated mitotic cells with nocodazole over ice to depolymerize microtubules followed by washout with pre-warmed media to promote microtubule regrowth. We observed no remarkable difference in the microtubule nucleation stage of regrowth (5 minutes recovery) in BCCIP deficient cells. Consistent with this finding, levels of the microtubule nucleating factors γ-tubulin and pericentrin were identical at the poles in control and BCCIP knockdown cells (data not shown). Despite this result, as shown in Figures 4a and 4b, after 30 minutes of recovery, while most of the control cells were able to form a well-focused bipolar spindle, BCCIP deficient spindles remained unorganized and contained a diminished amount of spindle microtubules. To further confirm this finding, we then filmed GFP-tubulin expressing control and BCCIP knockdown cells after overnight treatment with nocodazole and a 1-hour cold shock. We observed that following recovery, a bipolar spindle was re-established at roughly 40 minutes in control cells while this number was increased to 70 minutes (p=0.033) in BCCIP deficient cells (Figure 4c). These data strongly demonstrate that BCCIP-deficiency compromises spindle assembly independent of microtubule nucleation.
Figure 4. BCCIP loss negatively impacts microtubule retention.

A–C: Recovery of mitotic spindle formation after nocodazole treatment. Control and BCCIP knockdown cells on different slides were treated with nocodozole, washed and allow reformation of spindles. Panels 4A and 4B are the representative image sets and the quantified α-Tubulin intensity at 5 and 30 mins after recovery from nocodazole treatment. Panel 4C is the time used to re-establish bipolar spindle after nocodazole washout are quantified using time-lapse analysis from individual mitotic control or BCCIP knockdown cells.
D–H: Recovery of interphase centrosome microtubule after nocodazole treatment. Control and GFP-labeled BCCIP knockdown cells are mixed and seeded on the same coverslip, immune-fluorescent staining was performed at indicated times after washing off the nocodazole treatment, and the intensity of α-tublin in GFP positive (BCCIP knockdown) and GFP-negative (BCCIP normal) cells were measured and compared. 4D shows the verification of distinguishable BCCIP levels between GFP-negative (control) and GFP-positive (BCCIP knockdown) cells by anti-BCCIP staining in interphase cell. The blue arrowhead indicates a representative GFP-negative control cell expressing normal levels of BCCIP and the white arrow indicates a GFP-positive cell with BCCIP knockdown. 4E are representative images of α-Tubulin intensity in control (GFP negative) and knockdown (GFP positive) cells at indicated times after nocodazole washout and recovery. Blue arrowheads indicate centrosomal α-Tubulin intensity in control cells while white arrows indicate centrosomal α-Tubulin intensity in GFP-positive BCCIP knockdown cells. 4F is the quantified intensity of α-tubulin following recovery from nocodazole. The α-Tubulin signal was quantified in pre-determined area around each centrosome in control and BCCIP knockdown cells and plotted at the indicated time points. 4G are representative images of a control cell (blue arrowhead) that exhibits a well-formed radial microtubule organization, adjacent to a BCCIP knockdown cell (white arrow) that lacks a normal radial microtubule focus. 4H shows the percentages of interphase cells lacking a normal radial microtubule organization (unfocused microtubules) as represented in panel 4G. Depicted is data obtained from 232 control and 259 knockdown cells.
Next, because BCCIP is localized to proximal to the mother centriole and binds dynactin, a microtubule anchoring factor (18, 19), we hypothesized that the interphase centrosomal fraction of BCCIP might be involved in microtubule organization. In order to test this hypothesis, we then seeded a mixture GFP-tagged BCCIP knockdown and wild type (GFP negative) cells onto the same coverslip, and verified that the BCCIP-normal and BCCIP knockdown cells could be readily distinguished based on the GFP signals on the same slide (Figure 4d). Cells were then treated identically to Figure 4a/b. This approach was adapted to eliminate potential sampling bias due to the fast nature of microtubule regrowth in interphase cells. As shown in Figures 4e and 4f, asters of roughly equal intensity formed in both control and BCCIP deficient cells at two minutes, demonstrating that BCCIP depletion does not impact centrosome nucleation, mirroring our findings during mitosis. However at five minutes after recovery, the microtubule intensity around each centrosome was significantly reduced in BCCIP deficient cells. This delayed reformation of microtubule was recovered by 20 minutes, but in this state we observed that a significant portion of BCCIP deficient cells exhibited abnormal morphology (Figure 4f, 20 minutes), and lacked a sharply focused radial array of centrosome microtubules (Figure 4g and 4h). These data (Figure 4d–4h) suggest that interphasic BCCIP deficient cells are defective in organizing microtubules, and suggest that BCCIP fulfills a role in the regulation of microtubule anchorage/organization.
BCCIP deficiency reduces tubulin acetylation
Microtubule growth from the centrosome is a multistep process that involves the coordination of nucleating complexes, minus-end associated MAPs, and centrosomal anchoring proteins (11). Because the organization centrosomal microtubules was affected by BCCIP depletion, we theorized that BCCIP might also have a microtubule stabilizing role (14). Such a case has previous been observed in the p150 subunit of dynactin, an anchoring factor which also regulates microtubule stability (20). K-40 acetyl tubulin has been regarded as a surrogate marker for stable microtubules (20, 41–43), and thus we chose to examine the K-40 acetylation status in control and BCCIP deficient cells. As shown in Figure 5a, BCCIP loss resulted in a loss of K-40 acetyl tubulin in both BCCIP knockdown HeLa cells and in MEF cells where BCCIP had been deleted. To verify the reduction of acetyl-tubulin in BCCIP knockdown cells, control and BCCIP knockdown HeLa cells were mixed at 1:1 ratio and co-stained with acetyl-tubulin and BCCIP on the same slide. As shown in the Figure 5b, the BCCIP deficient cells (with weak green signals) had significantly lower level of acetyl-tubulin (red signal) than the BCCIP proficient cells (with strong green signals and arrowed) in the same field.
Figure 5. BCCIP deficiency decreases levels of stable, acetylated microtubules.

A: BCCIP knockdown reduces microtubule acetylation. Control and BCCIP knockdown HeLa cells or MEF control and MEF BCCIP knockout cells were lysed and blotted for the stable microtubule marker, acetyl-tubulin.
B: Levels of acetylated-tubulin in control and BCCIP knockdown cells. HeLa cells expressing BCCIP-shRNA (weak green, un-arrowed) were mixed with control cells (strong green, arrowed), and stained for stained for BCCIP and acetyl tubulin.
C: The reduced acetyl-tubulin in BCCIP deficient cell can be rescued by RNAi-resistant BCCIPα. RNAi-resistant YFP-BCCIPα, YFP-BCCIPβ, and YFP were re-expressed in BCCIP knockdown cells. These YFP-positive cells were mixed with non-transfected knockdown or control cells (YFP-negative cells). Total fluorescent intensity of acetyl tubulin in BCCIP knockdown (YFP-negative) and BCCIP re-expressed (YFP positive) cells on the same staining slide was quantified. Shown are relative intensities of acetyl-tubulin in wild type cells (first column from left), BCCIP knockdown cells (2nd column from left), and the knockdown cells expressing different YFP-tagged proteins. Representative images of rescue can be viewed in Supplement Figure S4.
To further address which specific isoform of BCCIP resulted in a loss of acetyl-tubulin, we then expressed shRNA resistant YFP-BCCIPα, YFP-BCCIPβ, or YFP in BCCIP knockdown cells (see Figure S4 for representative images), and compared the relative intensity of acetyl-tubulin with the knockdown or control cells. As shown in Figure 5c, the BCCIP knockdown cells demonstrated reduced levels of acetyl-tubulin compared to wild type cells. YFP-BCCIPα, but not YFP-BCCIPβ or YFP was capable of recovering this marker (Figure 5c). These data suggest that BCCIP, particularly BCCIPα, confers microtubule stability.
Spindle defects in BCCIP-deficient cells
Dynein, dynactin, and spindle pole auxiliary proteins, such as NuMa play an essential role in mitosis by regulating spindle length, architecture, and positioning (8, 9, 18). The association between BCCIP and dynactin components, together with the spindle pole localization of BCCIP (Figure 3c) led us to ask the question if BCCIP loss resulted in mitotic spindle defects. First we investigated the architecture of the mitotic spindle in BCCIP-deficient cells. As shown in Figures 6a, the spindles in BCCIP deficient HeLa cells appeared bipolar, but collapsed, leading to a reduced pole-to-pole distance (Figure 6b). Close examination of the minus end of BCCIP-deficient spindles revealed a broadening at the poles, and the normally sharp focus at minus end of the spindle was stretched in BCCIP deficient cells (Figure 6a). Additionally the spindle arc angle (see orange Arc in Figure 6a cartoon), or the angle between the distal points of the spindle microtubules was significantly widen at the spindle poles (Figure 6a and 6c) and spindle pole components were stretched and splayed in BCCIP deficient cells (see green line in Figure 6a cartoon, and Figure 6d). These defects were also observed in BCCIP knockout MEF cells, which displayed a similar spindle pole broadening phenotype and exhibited extreme spindle pole fragmentation (bottom row of Figure 6a). These abnormalities canonically match defects in microtubule focusing and the phenotype that results from loss of dynein/dynactin/NuMa associated activities (8, 9, 18, 38).
Figure 6. Abnormal spindle architecture in BCCIP deficient cells.
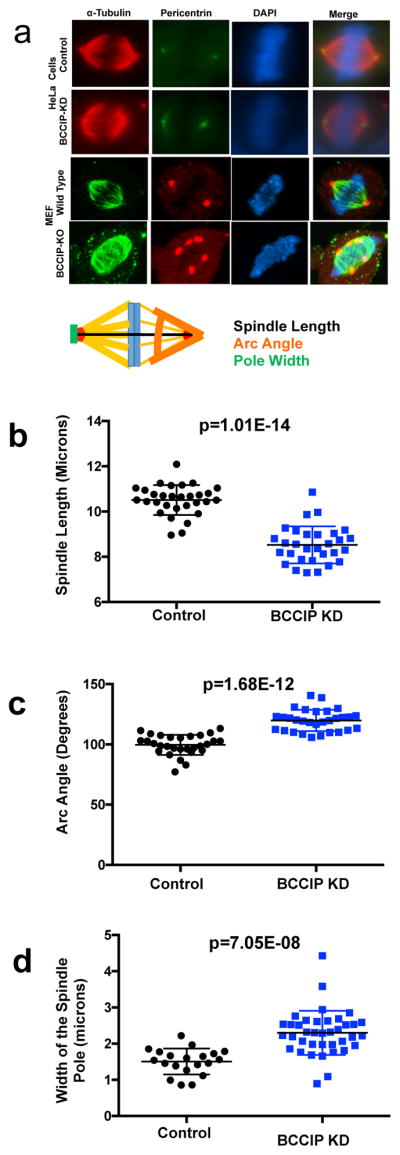
BCCIP knockdown HeLa or BCCIP knockout MEF cells were immunostained with α-tubulin and pericentrin and analyzed at metaphase. The distance between the paired centrosomes, the angles of spindle arcs and the width of the spindle poles were assessed. Representative image sets and a cartoon illustration of the assessed geometric measurements are shown in Panel-A. The distributions and averages of the spindle length, arc angles, and the width of the spindle poles are shown in panels B, C, and D respectively.
Disorientation of spindles in BCCIP deficient cells
Dynein/dynactin cooperates with spindle pole components such as NuMa to direct motor force from the spindle pole to the cell cortex, which in turn properly orients the spindle (8). This process plays a critical role in the fate of nascent cells during tissue regeneration and maintains stemness during asymmetric cell division (9). We hypothesized that, because the dynein network plays an essential role in spindle orientation (8), BCCIP silencing might confer similar defects. In order to test this hypothesis we immunostained cells for pericentrin and analyzed 0.2 micron Z-stacks of the spindle poles with respect to the petri dish plane. Next, we calculated the three dimensional tilt of mitotic spindles using the geometrics illustrated in Figure 7a. We observed that control metaphase cells typically exhibited a small spindle angle, and cells were oriented so that division occurred completely parallel to the culture dish surface (Figure 7b). As demonstrated in the representative images (Figure 7b), the two spindle poles (pericentrin foci) of control cells reached maximum intensity within the same confocal plane, indicating both poles exhibited a limited angle between them. In stark contrast to the control cells, the BCCIP deficient cells exhibited pericentrin foci that resided in two distant confocal planes. Representative 3D images of spindle orientation can be found in Supplement S5 Movie-M3, and M4. As quantified in Figure 7c, the spindle angles of BCCIP deficient spindles were significantly increased in comparison to the control, suggesting that the BCCIP deficiency imparts spindle orientation defects. In order to determine if this phenotype was specifically induced by loss of BCCIPα, we then performed a rescue experiment by re-expressing shBCCIP resistant flag-BCCIPα, BCCIPβ, or empty vector in BCCIP knockdown cells (Figure 7b, bottom 3 rows). We observed that the defect in spindle orientation was largely rescued by BCCIPα expression but not by BCCIPβ (Figure 7c, Figure S5, Movie M5–M7), confirming that it is the BCCIPα isoform that is required for maintaining proper spindle orientation. These spindle orientation defects were further verified by time-lapse imaging which revealed a significant portion of BCCIP deficient cells completed division in a manner in which one daughter cell tended to reside outside of the focal plane of its cohort (see Figure S6a).
Figure 7. BCCIP loss induces spindle orientation defects.

A: Measurement of spindle angles. Image stacks of mitotic HeLa cells grown parallel to the culture surface were obtained by confocal imaging. The Z-planes containing maximal spindle pole intensities were recorded as Z1 and Z2 and the Z-distance between Z1 and Z2 was used to measure the depth (or the vertical distance) between the spindle poles (ΔZ shown in illustration). Maximal projections were utilized to determine the horizontal distance between the spindle poles (x-value as illustrated). Consequently, the spindle angles relative to the culture surface can be determined as α=Sin−1(ΔZ/x).
B: Representative images of spindle pole planes (Z1 & Z2). Spindle poles (pericentrin foci) reside in the same Z-planes in control cells but often reside two different confocal planes (Z1 or Z2) for BCCIP deficient cells, reflecting an increase in the spindle angle.
C: The distribution of spindle angles is increased by BCCIP depletion. The spindle angles from control (i), BCCIP knockdown (ii), and knockdown cells expressing RNAi resistant BCCIPα (iii) or BCCIPβ (iv) or control vector (v) were assessed. Plotted are the distributions of the spindle angles among indicated cells. The p-values of t-test between the two indicated pairs are shown in the table.
D: reduced levels of cortical dynactin in BCCIP deficient cells. Control and BCCIP deficient HeLa cells were co-seeded to coverslips, arrested in metaphase with MG-132, and co-stained with p150 glued (green) and BCCIP (red). Shown are the representative images of BCCIP normal (up right corner) and BCCIP knockdown (lower left corner) cells on the same staining field, and the zoomed images demonstrating the p150 staining at the spindle pole and the cortex. Yellow arrows indicate the presence (control) or lack (BCCIP-knockdown) of cortex p150 staining in the control and BCCIP deficient cells.
A fraction of mitotic dynein/dynactin is deposited to the cell cortex and this is deposition essential to produce the motor force that buttresses and orients the spindle (8, 16, 21). The abnormal organization of the mitotic spindle in BCCIP deficient cells together with the cortical localization of BCCIP (Figure 3d) lead us to ask the question if cortical dynactin was mislocalized in BCCIP deficient mitotic cells. We first observed that BCCIP deficient spindles were often severely displaced from their normal central distribution within the mitotic cytoplasm (Figure S6b, S6c), which is indicative of astral microtubule defects (44). Next, we investigated if this phenotype was concomitant with misdistribution of cortical dynactin. As shown in Fig 7d, BCCIP deficient cells demonstrated a decreased level of dynactin at the cell cortex. Thus, it is likely that the disorganization of the microtubule network in BCCIP deficient cells also results in aberrant trafficking of dynein/dynactin and displaces it from the cortex, which in turn leads to spindle orientation defects.
Lagging chromosomes and reduced kinetochore tension in BCCIP deficient cells
Mitotic chromosomes are aligned at the metaphase plate before the onset of anaphase. This alignment is critical for ensuring the equivalent distribution of sister chromatids into two identical daughter cells. Spindle pole focusing defects have been demonstrated to impair chromosome congression as a result of displacing pole directed motor force, and silencing of the dynein epistasis group leads to decreased spindle tension, chromosome congression defects, and delayed mitotic completion (15, 16, 37, 45, 46). Inspection of the mitotic chromosomes in BCCIP deficient metaphase cells demonstrated that 24% (10 out of 41) knockdown cells contained lagging chromosomes; chromatin bodies completely disassociated from the metaphase plate. These lagging chromosomes were undetected among control cells (0 out of 52). Next, in order to quantify chromosome alignment, we measured the chromosome congression index; a representation of the cell’s ability to capture and move chromosomes (44). We found that BCCIP knockdown significantly increased the congression index, indicating poor metaphase chromosome alignment. These chromosome congression defects were rescued by re-expression of RNAi-resistant BCCIP proteins (Figure 8a, 8b). We then reasoned that these defects could be resultant from two distinct possibilities: defective poleward pulling forces or defective kinetochore microtubule attachments. We first measured spindle tension, which represents the ability of the cell to generate the robust poleward pulling forces necessary to move chromosomes. Spindle-kinetochore tension can be represented by the distance between sister kinetochore pairs; the greater distance between sister kinetochore foci the greater amount of tension transduced by the spindle (47–50). Therefore, we utilized the distance between sister kinetochores as a surrogate for spindle tension, using the well-established method of Waters et al. (49, 50). We observed a significant decrease in the distance between sister chromatid CREST foci in BCCIP knockdown HeLa (Figure 8c, 8d) and U2OS cells (Figure S7a, 7b), demonstrating that BCCIP deficiency compromises robust pulling forces, which is consistent with the notion that BCCIP functions in coordination with dynein/dynactin/NuMa (Figure 3) (9, 38). We then challenged BCCIP deficient and control cells with a ten-minute cold shock using the reported procedure (44). This procedure is sufficient to destabilize all microtubules except kinetochore-associated microtubules (stable k-fibers), unless these attachments were rendered unstable (44). This assay revealed no change to k-fiber stability in BCCIP deficient cells (data not shown), indicating that microtubule-kinetochore attachments occur normally during BCCIP silencing. Taken as a whole, these results indicate that the chromosome movement is defective in BCCIP deficient cells.
Figure 8. The forces required for chromosome movement are defective in BCCIP deficient cells.

A & B: Abnormal congression of metaphase chromosomes in BCCIP deficient cells. Metaphase chromosome width (parallel to the spindle poles) and length (perpendicular to the spindle poles) was obtained as illustrated. The ratio between the width and length was utilized to calculate the chromosome congression index (W/L) and plotted in (8B) for control (i), BCCIP knockdown (ii), and knockdown cells that express RNAi resistant BCCIPα (iii) or BCCIPβ (iv) or control vector (v). The p-values of t-test between selected cells are shown in the table bellow the graphic.
C & D: Reduced intra-kinetochore distance in BCCIP deficient cells. Cells were stained with CREST (red) and α-tubulin (Green). Unambiguous kinetochore CREST pairs were identified, and the distance between CREST foci (representing the distance between sister-chromatid kinetochores) was measured. (8C) shows the representative slice views of control and BCCIP knockdown HeLa cells, a pair of identified sister bivalents (boxed and zoomed), and the maximum projections depicting accumulation of α-tubulin and CREST stacks of the confocal slice views. (8D) illustrates the distance distribution based on measurement of more than 200 pairs. Results using U2OS cells can be found in Supplement S7.
Delayed completion of mitosis in BCCIP deficient cells
We then predicted that the defects in spindle and chromosome geometry in BCCIP deficient cells might lead to impaired mitotic progression. To verify this, we utilized the reversible CDK-1 inhibitor RO-3306 to synchronize BCCIP deficient and BCCIP competent cells at the G2/M boundary overnight. Following incubation, cells were released from the block, and fixed stepwise at different time points following the release. Mitotic cells were judged by staining with the pre-anaphase marker pH3-T11, which is rapidly dephosphorylated upon anaphase onset. We observed that BCCIP deficient and control cells entered mitosis at roughly the same rate (Figure 9a), suggesting that mitotic entry is not impeded by BCCIP loss, which is consistent with a previous report (26). However, at the 90 and 120 minute time points following release from the block, control cells roughly returned to their baseline levels of mitosis while BCCIP knockdown cells remained in M-phase (Figure 9a). These data demonstrate that BCCIP deficient cells have a delay in mitosis, and that this delay occurs during metaphase. To verify this finding, we performed a similar experiment, but substituted RO-3306 by nocodazole to block cells in mitosis, collected lysates from control and knockdown cells after drug washout, and subjected the lysates to western blot for cyclin-B, another mitotic marker that is rapidly degraded upon anaphase onset (51, 52). As shown in Figure 9b, levels of cyclin-B level peaked at 0–30 minutes and thereafter sharply decreased in control cells, while in knockdown cells the peak cyclin B fractions shifted to between 30–60 minutes and reduced at slower rate. We then used live cell imaging to determine the precise defects observed in BCCIP depleted cells. We generated stably expressing GFP-Tubulin cells lines and observed the duration of mitosis from nuclear envelope breakdown to the formation of a visible mid-body. Cells were then transduced with empty control lentiviral particles or particles expressing shRNAs specific to BCCIP. We observed that the mitotic time was significantly extended in knockdown cells, and re-expression of BCCIPα, but not BCCIPβ or empty vector, could largely rescue the extended mitotic time in the BCCIP knockdown cells (Figure 9c). Representative time-lapse images for mitosis are shown in Supplement S8.
Figure 9. Delayed mitotic completion in BCCIP deficient cells.

A & B: BCCIP loss delays anaphase onset. HCT116 cells were blocked at the G2/M boundary by RO-3306 treatment, released, and immuno-stained with pH3-T11. (9A) shows the percentages of mitotic cells at different times after the release from the G2/M block. (9B) Depicts a separate set of experiments where cells were blocked in M phase with nocodazole, released, and subjected to western blot at the indicated time points. NT: untreated asynchronized cells used as a loading reference.
C & D: Delayed completion of mitosis in BCCIP deficient cells. U2OS cells were filmed overnight in an incubated live cell microscope. Frames were acquired every 5 minutes, and the length of mitosis was quantified from the onset of nuclear envelope break down to the formation of a visible midbody. Shown in (9C) is mitotic time in control (i), BCCIP knockdown (ii), and knockdown cells rescued with RNAi-resisitant BCCIPa (iii) or BCCIPb (iv) or control vector (v). The p-values of t-test between the indicated cells are shown. Verification of the re-expression of exogenous BCCIP in the BCCIP knockdown cells is shown in (9D).
E: BCCIP deficient synergizes with spindle poison to delay mitotic completion. Control and BCCIP knockdown HT1080 cells treated with nocodazole over ice to completely depolymerize the mitotic spindle. The cells were then washed rapidly three times with warm media and filmed until mitosis was completed.
To further confirm that the mitotic delay experienced by BCCIP deficient cells was specifically due to improper spindle assembly, we disassembled spindle microtubules with nocodazole treatment and cold shock, washed the drug away with warm media, and immediately began filming cells at 37°C. Again, we found that BCCIP deficient cells took longer time than the control to complete mitosis after recovery from disassembly of the spindle (Figure 9e). Therefore, the mitotic delay experienced by BCCIP deficient cells is dependent on proper spindle assembly.
DISCUSSION
In this study we identify BCCIP, especially BCCIPα, as novel microtubule associated protein localizes to the interphase centrosome and the mitotic spindle poles. We conclude that BCCIP is critical for microtubule organizing and anchoring activities during interphase and this function is later co-opted to organize and stabilize the spindle pole during mitosis. In the absence of BCCIP, the interphasic microtubule network fails to maintain its normal association with the centrosome, leading to a general disorganization and destabilization of microtubule arrays, concomitant with an increase of morphologically abnormal cells and decreased acetyl tubulin. This observation is consistent with BCCIP’s association with p150 glued/dynactin and localization to the mother centriole. During mitosis, the microtubule binding abilities of BCCIP are directed by the minus end motor dynein to coordinate the microtubule’s minus end with the centrosome in order to generate spindle tension. These observations are consistent with the collapsed, defocused spindles observed during BCCIP silencing which display decreased bivalent distance and are sequestered in mitosis. It is important to note that these defects are a phenocopy silencing of the dynein/dynactin/NuMa epistasis group. The totality of this evidence coupled with the physical association of BCCIP with dynactin suggests that these proteins lie within the same pathway.
The coordinated activity of molecular motors with minus end MAPs is required to focus microtubule minus ends (1). Silencing of dynein, dynactin, as well as spindle pole MAPs, such as NuMA, results in spindle splaying, lagging chromosomes, and delayed mitotic progression in cultured cells (16, 37). Genetic deletion of these factors in vertebrates results in embryonic lethality and/or aneuploidy, suggesting this network is essential for both the rapid cell divisions that characterize embryonic development and for the maintenance of genome integrity (13, 15, 38). We suggest that the microtubule organizing function of BCCIP acts as a safeguard against tumorigenesis and its loss is another root cause of the excessive level of aneuploidy observed in BCCIP deficient cells by previous works (25, 26).
During development, cell division is critical for regulating not only cell number but also cell diversity (53). While symmetric cell division facilitates rapid clonal expansion, asymmetric division is responsible for cell lineage diversification (53). For example, vertebrate neurogenesis is heralded by a sequence of symmetric and asymmetric cell divisions exquisitely orchestrated to generate the remarkable cellular diversity and complex tissue architecture of the brain (53, 54). Consequently, defects in genes that regulate the orientation of the mitotic spindle and the fidelity of the centrosome are endemic to human brain diseases (9, 53–55). Among the most prominent is primary microcephaly (MCPH), a condition that results in an abnormally small brain and other neurological disorders. Remarkably, all identified MCPH genes are centrosome and mitotic spindle regulators, and their knockdown in both cultured cells and in mice induces abnormal organization of interphase microtubule arrays and mitotic defects (53, 56, 57). We have previously observed that BCCIP knockdown leads to microcephaly and altered cell differentiation in the neural cortex in mice (58) and therefore propose that the microcephaly experienced in BCCIP deficient mice may be related in part to defective of asymmetric division.
It is interesting to note that of two BCCIP isoforms, BCCIPα is the predominant centrosome and microtubule associated isoform. Despite this observation, the recruitment of BCCIPα to spindle poles is dependent on amino acids 111-257; a region shared by both BCCIPα and BCCIPβ. It is of note that this domain predicts a highly conserved coiled coil; a major structural motif responsible for pericentriolar matrix anchoring (59). We demonstrate that BCCIPα is the predominant isoform that associates with the spindle and centrosome in vivo, yet the C-terminal domains that diversify BCCIPα from BCCIPβ have little role in spindle targeting. This is consistent with the observation that in mouse cells where BCCIPα is not available, the BCCIPβ-like isoform of BCCIP localizes to the centrosome and spindle poles in an identical manner to human BCCIPα. Despite this observation, in nearly all of our biochemical experiments, we were able to detect an association of human BCCIPβ with spindle components following cell lysis, which supports the notion that the inhibition of targeting of BCCIPβ to the centrosome may only be relevant in vivo, and is possibly influenced by transacting elements that are lost after the cell lysis during biochemical manipulation.
In summary, our study has established BCCIP as a previously unidentified regulator of spindle assembly that cooperates with the dynein epistatic group to ensure the fidelity of mitosis. Our data not only describes a new functional aspect of the BCCIP gene but also expands the list of factors critical for mitotic progression, spindle orientation, and microtubule organizing.
MATERIALS AND METHODS
Microscopes
Confocal microscopy image capture and analysis was performed on a Nikon A1 and the Nikon elements software suite. Otherwise, for standard epifluorescence, a Nikon eclipse TS100 microscope was used in conjunction with ImageJ. Live microscopy was performed on a Zeiss Axiovert 200M and analysis was achieved using Axiovision software.
Cell culture, expression of transgenes, and drug treatment
All cell culture reagents were purchased from Sigma-Aldrich (St. Louis, MO), except stated specifically. HT1080, Cos-7, 293T, U2OS, and HeLa cells were cultured in α-Dulbecco’s Modified Eagle’s Medium (αMEM), with 10% fetal bovine serum, 20 mM glutamine, and 1% penicillin-streptomycin. Mouse embryonic fibroblasts were isolated from as described previously (25) and routinely maintained in the same media as above. For transgene expression, cells were seeded overnight and were transfected at 80% confluence (100mm dish) with 10μg plasmid DNA using the RU-50 transfection reagent (Syd-Labs, MB088-450-20, MA) according to the manufacturer’s instructions. Twenty-four hours after transfection, the media was aspirated, and cells were processed for specific assays such as immunofluorescence staining or western blotting. To establish cells with stable transgene expression including YFP/Flag-BCCIP or GFP-tubulin, the transfected cells were subjected to antibiotic selection including puromycin (Sigma 2μg/ML) or G418 (400 μg/ML) depending on the vector, starting at 48 hours after transfection. Positive single clones were obtained and the population was expanded to provide a stable cell lines.
Plasmid vectors and production of retrovirus and lentivirus
The pLXSN vector (CloneTech, 631509) and its derivative pLXSP (30) was utilized for retroviral packaging. A panel of YFP-BCCIP fragments from a BCCIP cDNA as EcoRI/BamHI fragments were cloned into the pLXSN-YFP retroviral backbone. In order to mutate wild type BCCIPα in the pLXSN-BCCIP vector, BCCIP deletion fragments were created using a PCR splicing strategy (60). The YFP deletion constructs were also inserted as an EcoRI/NotI fragment into the pCMV-Myc vector (Clontech, 631604) for transient expression. The procedure to package retrovirus has been previously described procedures (30). The backbone of the H1P-HygroEGFP lentivirus vector (61) was modified by replacing the HygroEFGP sequence with that of GFP or Puromycin marker, resulting in H1P-shRNA-GFP or H1P-shRNA-Pur vectors. The following pairs of oligonucleotides were synthesized: 5′GGCCTTCTCCTAAGTGAAATTCAAGAGATTTCACTTAGGAGAAGGCCTTTTTTG3′ and its reverse complement of 5′CAAAAAAGGCCTTCTCCTAAGTGAAATCTCTTGAATTTCACTTAGGAGAAGGCC 3′. A total of 5mM of each oligo was mixed, heated at 95°C, and allowed to cool to anneal complementary oligos. The resulting double stranded nucleotide was cloned into the vectors through the XbaI and EcoRI sites, resulting H1P-shBCCIP552-GFP and H1P-shBCCIP552-Pur. In this construct, the BCCIP shRNA expression is under the control of the hH1P promoter, and the expressed shRNA targets the common region of 5′GGCCUUCUCCUAAGUGAAAGA3′ starting at location 552nt in the BCCIPα and BCCIPβ RNAs. To generate lentivirus particles, the 293T cells were seeded at 70% confluence. The next day, cells were cotransfected with 6μg H1P-shBCCIP552-GFP or H1P-shBCCIP552-Pur, 3μg psPAX2 (Addgene #12260), and 3μg pMD2G (Addgene #12259). At 48-hour post transfection, virus-containing supernatant was collected, filtered through a 0.45μM nylon mesh, and adjusted to 8 μg/ML polybrene (Sigma 107689). Target cells (HeLa, HT1080, and U2OS) were incubated with viral supernatant overnight. 18 hours later, the supernatant was aspirated and the cells were allowed to recover overnight. Infection efficiency was evaluated by observing GFP expression 72 hours after the initial infection. Alternatively, stable shBCCIP expressing cells were selected in puromyicin (Sigma 2μg/ML) for 48 hours when the H1P-shBCCIP552-Pur vector was used. The H1P-shRNA-GFP or H1P-shRNA-Pur vector backbone was used as negative control. Knockdown cells were discarded following 3 passages.
Antibodies and Western blots
The rabbit anti-BCCIP BR5 and S1472-2 antibodies were custom made using recombinant BCCIP protein as the antigens. The commercial antibodies purchased for fluorescent immunostaining and western blot include: α-Tubulin (Sigma DM1A, 1:500), γ-Tubulin (Sigma GTU-88, 1:1000), K-40 Acetyl α-Tubulin (Sigma 6-11B-1, 1:500), Phospho H3-T11 (Cell Signaling #9764 1:500), Plk-1 (Santa Cruz Monoclonal F-8 1:100), GAPDH (Cell Signaling #14C10, 1:100, or Santa Cruz 6C5 1:2000), Pericentrin (Covance PRB-432C, 1:300), CREST (Immunovision HCT0100, 1:1000), CENP-E (Santa Cruz H-300, 1:500), GST (Santa Cruz B-14 1:500), CoxIV (Cell Signaling 3E11 1:100), Lamin A/C (Cell Signaling 4C11 1:1000), PCNA (Invitrogen PC-10 1:1000), GFP (Santa Cruz sc-8334 1:1000), Aurora-A (Cell Signaling 1G4 1:500), HSP90 (Cell Signaling C45G5 1:1000), CDC2 (Invitrogen A17 1:1000), Flag (Cell Signaling 1:500 #2368) Cyclin B (Santa Cruz GNS1 1:200) p150 (BD Labs 1:100 #610473) Arp1 (1:300 Sigma A5601).
To perform western blots, cells were lysed in RIPA buffer (50 mM Tris HCl, pH 7.4, with 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate 1mM Leupeptin, 1mM Aprotinin, 20mM PMSF). Lysates were subjected to electrophoresis and transferred to nitrocellulose. The membranes were blocked in 1% milk for 1 hour, and incubated overnight with the specified antibodies. Following incubation, membranes were washed four times in 0.1% Tween-20-TBST, and incubated for one hour with HRP anti-mouse or anti-rabbit IgG secondary antibodies (Sigma 1:2500). Membranes were then washed as above and proteins were detected using ECL (Promega).
Immunofluorescence staining
To perform immunofluorescent (IF) staining, cells were seeded onto poly-L-lysine treated coverslips in a 6-well plate at 60% confluence. 48 hours after seeding, the media was aspirated and cells were extracted in 37°C D-BRB80 buffer (50μg/ML Digitonin, 80mM PIPES [PH 6.9], 30% glycerol, 1mM EGTA, 1mM MgCl2) or 37°C T-BRB80 (0.3% Triton-X100, 80mM PIPES [PH 6.9], 30% glycerol, 1mM EGTA, 1mM MgCl2) for one minute. The extraction buffer was aspirated and cells were then fixed at 37°C in 4% PFA (paraformaldehyde) for 20 minutes. Following fixation slides were blocked in 0.3% Triton, 5% bovine serum albumin (BSA) for 1 hour (IF block buffer). Cells were immunostained overnight in blocking buffer with the indicated antibodies. The slides were then washed thrice in PBS + 0.1% Triton X100 for 5 minutes. Slides were incubated with 1:1000 dilution of FITC or TRITC anti-mouse, anti-rabbit, or anti-human conjugated secondary antibodies (1:1000, Sigma) for one hour in blocking buffer. The slides were washed as above, and mounted onto coverslips with Vectashield mounting media containing 4′,6-diamidino-2-phenylindole (DAPI).
Exogenous expression of RNAi-resistant BCCIP
Because the shRNA expressed from the lentivirus was designed to target the BCCIP mRNA at the following site: GGGCCUUCUCCUAAGUGAAAGA, transgenic RNAi-resistant constructs were created by mutating four nucleotides in the shRNA targeted region of BCCIP cDNA to 5′-GGGCTTCTGCTCAGCGAAAGA-3′. This produces silent point mutations in the BCCIP cDNAs (designated BCCIPαM4, and BCCIPβM4), which codes for exogenous BCCIP that is resistant to shRNA targeted at the same site of the endogenous BCCIP. In this study, we express these shBCCIP552-resistant BCCIPαM4 and BCCIPβM4 variants in BCCIP knockdown cells in order to test which BCCIP isoform can rescue the defects caused by BCCIP knockdown and to rule out off-target effect.
Measurement of Centriole Bias
In order to determine marker co-localization, GFP-EB1 expressing cells were co-stained with BCCIP and γ tubulin. Centriole bias was calculated by obtaining 0.2-micron centrosome stacks in T-BRB80 PFA fixed cells that exhibited two clearly separate γ tubulin puncta. Image-J was then utilized to measure the fluorescent intensity of each marker in individual γ tubulin foci, and the fluorescent intensity of each marker was divided by one another to obtain a ratio. γ-tubulin was utilized as a reference (no bias; ratio of 1), while EB1 was utilized as a mother centriole marker as previously reported (17, 39, 40).
Quantitative comparison of immunofluorescent intensity between two cell types
In order to reliably and quantitatively compare the intensity of fluorescent signals between cells of two different genotypes, we co-seeded cells of two different genotypes at a 1:1 ratio, and proceeded with immunofluorescent staining. For example, in order quantitatively compare acetyl-tubulin intensity between BCCIP knockdown and knockdown cells complemented with exogenous YFP-BCCIPα, YFP-BCCIPα expressing cells (green cells) were mixed with BCCIP knockdown cells (no color). The mixed cells were grown to 80% confluency, extracted with T-BRB80, and fixed in fresh PFA. Microscope fields containing both YFP expressing (complemented) and YFP negative cells were imaged for acetyl-tubulin intensity, using the 60X objective of a Nikon eclipse TS100 microscope. Only images containing both cell types (as confirmed by BCCIP staining or GFP markers) were scored. This procedure uses stringent internal control within the same individual slides and allows us to eliminate any potential discrepancy that may be associated with variation of staining and image acquisition procedures. On each image, the polygon tool in Image-j was utilized to measure the fluorescent intensity in the cells containing high levels of BCCIP to cells where BCCIP staining was not visible. Fluorescent intensity was calculated using the following formula (Fluorescent intensity = Integrated Density - (Area of selected x Mean fluorescence of background readings). Intensities of >100 cells were quantified for each cell types, the fluorescent intensity of all slides was pooled, and subjected to Student’s t-test to determine the statistical significance. In some cases, Nikon-elements AR software suite was utilized to calculate the fluorescent density from the regions of interest of an image.
Protein interaction assays: GST-fusion protein pull down
Because the locations of endogenous α, β, and γ tubulins overlaps with that of the 50kD IgG used in a routine immunoprecipitation experiments and this precludes the accurate detection of the amount of tubulin co-precipitated by BCCIP, we used GST-BCCIP fusion proteins to co-precipitate endogenous tubulin. GST-BCCIP proteins were incubated with 1mg spindle fraction prepared from mitotic cell lysates as described elsewhere (62). We added 50μl glutathione resin (Novagen) to this mixture and incubated overnight. The beads were spun down and washed 5 times in PBS 0.1% Triton, and eluted 3 times in 10mM reduced glutathione. To test the direct binding between BCCIP and tubulin, 10μg GST tagged BCCIPα, BCCIPβ, or GST were incubated with 5μg of purified bovine tubulin (cytoskeleton) in T-BRB80 without glycerol. The eluted GST-BCCIP bound proteins along with the input were then subjected to SDS PAGE and western blotting.
Isolation of mitotic centrosomes
The mitotic centrosomes were isolated using a procedure described previously, with some modifications (33, 63). Briefly, HeLa cells were grown in ten 150 mm dishes until 80% confluence and were arrested in 5mM thymidine overnight. The thymidine containing media was aspirated, cells were washed 3 times in PBS, and cells were incubated overnight in fresh media containing 100ng/ML nocodazole (Sigma). The next day the media was adjusted to 5μg/ML nocodazole, 1μg/ML Cytochalasin-D (Sigma) and cells were incubated an additional hour. Cells were typsinzed and resuspended in ice-cold media, pelleted, and washed sequentially in ice cold PBS, ice-cold PBS diluted tenfold in water, and ice-cold water in 8% sucrose. Following the last wash cells were lysed in centrosome lysis buffer (0.5% Triton-X100, 1mM Tris PH 7.0). The remaining procedure was performed identically to procedures outlined elsewhere (33, 63). For immunofluorescence, the peak centrosome fraction was collected, diluted in 5ML 10mM PIPES, and centrifuged at 10,000xG (SW-41 Rotor) through a 30% glycerol onto a coverslip. The coverslip was carefully removed, fixed in 100% methanol, and processed with the immunofluorescence procedure described above.
Differential extraction of centrosome proteins
To evaluate the association strength of centrosome proteins, the same assay as reported was used (35). Briefly, centrosome fractions were pooled, diluted in 5ml 10mM PIPES, and divided into four microcentrifuge tubes. Centrosomes were pelleted at 20,000xg and the supernatant was aspirated. Pellets were incubated with 1D Buffer (0.5% Triton X-100), 2D Buffer (0.5% Triton-X100 and 0.5% deoxycholate), 3D buffer (0.5% Triton-X100, 0.1% SDS and 0.5% deoxycholate) or 8 M urea. Pellet and supernatant fractions were separated by centrifugation at 20,000xg, the supernatant was collected, and the pellet was solubilized in boiling Laemmli buffer (30 mM Tris-HCl, pH 6.8, 1% SDS, 10% (w/v) glycerol, 0.01% bromophenol blue, 10% β-mercaptoethanol). Equal portions of pellet and supernatant were utilized for western blotting.
Microtubule spin down assay
To measure the interaction of BCCIP with mitotic microtubules, we modified the procedure described by Young et al (36). Briefly, HeLa or Cos-7 cells expressing BCCIP or BCCIP fragments were grown to 80% confluency in 150mm dishes, blocked with 5mM thymidine overnight, washed, and released into fresh media containing 100 ng/ml nocodazole. Eighteen hours later, cells were harvested by trypsinization, the nocodazole was removed and cells were lysed and sonicated in tubulin lysis buffer (50 mM Tris HCl, pH 7.4, with 75 mM NaCl, 80mM PIPES, 1 mM MgCl2 and 0.3% Triton X-100, 1mM Leupeptin, 1mM Aprotinin, 20mM PMSF). The lysate was then cleared by centrifugation for 20 minutes at 16000xg. The supernatant was then collected, adjusted with 20 units of benzonase (sigma), and the lysate was precleared again by centrifugation at 100,000xg in an SW41 rotor for 1 hour. The clarified, high speed lysate (containing the non-polymerized tubulin) was adjusted to 100mM DTT, 1mM EGTA, 100 μg/ml bovine tubulin (cytoskeleton), 1mM GTP, 50 μM taxol (Sigma) or the same buffer containing 10μg/ml nocodazole without taxol and centrifuged for 30 minutes at 50,000xg in an SW41 rotor over a 30% sucrose cushion containing taxol or nocodazole. The supernatant was collected, the pellet was washed once in tubulin lysis buffer containing taxol or nocodazole, and resuspended in boiling Laemmli buffer. The pellet represents the mitotic microtubule fraction and associated proteins. Equal portion of supernatant and pellet fractions were analyzed by western blotting.
Microtubule regrowth assay and quantification of focused MTs
A previously reported assay was performed was utilized to determine the kinetics of minus end microtubule assembly (39, 64). GFP-tagged BCCIP knockdown cells were mixed 50/50 with cells transduced with an empty shRNA cassette and seeded onto coverslips. Cells were treated with 5 μg/ml nocodazole, ice-chilled for 1 hour, and fixed at either 2, 5, 10, and 20 minutes following nocodazole washout with pre-warmed media. Centrosome aster intensity was measured by using the α-Tubulin signal around a predefined area from each centrosome with the Image-j circle tool. Only images containing knockdown cells adjacent to control cells (as assessed by GFP) were scored.
Analysis of spindle poles and mitotic spindles
In order to quantify the spindle defects in an unbiased manner, we selected only metaphase cells in which a clear chromosome congression was detected at the metaphase plate. This was accomplished by a 3 hour MG-132 block. The pole-to-pole distance was assessed by utilizing Image-J to measure the pole-to-pole distance between pericentrin foci. To measure spindle arc, the angle tool was utilized in Image-J. The second point of the angle tool was consistently placed on the central pericentrin focus and the first and second points were placed 3 microns in length parallel to the α-Tubulin stained mitotic spindle. For the analysis of pole splaying, we measured the length of pericentrin foci at the poles in the control and BCCIP knockdown condition. For quantification of spindle pole components, the polygon tool in ImageJ was utilized to trace and measure the intensity of indicated markers at the poles.
Measurement of spindle tilt and off-center spindles
We utilized the method described by Hori et al., including synchronization of metaphase cells (14). Briefly, cells were plated on collagen treated coverslips and fixed in 100% methanol, stained with pericentrin, and a series of 0.2-micron stacks were obtained. The signal maxima and the central maxima of pericentrin foci was utilized to measure the horizontal (X) and vertical (Z) distance between the poles, respectively, and spindle tilt was calculated as a function of θ=sin−1(Z/X). In order to measure the deviation of the mitotic spindle from the centroid of the cell, we measured the distance of the spindle pole to the closest cortex and acquired a ratio between these values. The higher ratio between cortex distances was consistently used to plot position.
Measurement of distance between kinetochore sister bivalents and chromosome congression
In order to measure metaphase chromosome congression cells were treated by MG-132 for 3 hours. Image processing and analysis followed that of Green and Kaplan (44).
The assay developed by Waters et al. (49, 50) was used to measure the distance between unambiguous sister kinetochore pairs identified CREST staining. Images were collected in a single focal plane for BCCIP control and BCCIP knockdown cells and the distance between kinetochore bivalents was measured suing Image-J.
Measurement of mitotic time, bipolarity, spindle orientation, and nocodazole recovery using time-lapse imaging of live cells
HT1080 or U2OS cells stably expressing GFP-tubulin were transduced with H1P-shBCCIP522-Pur lentiviral particles or control lentivirus. Cells were transferred to an incubated microscope (Zeiss Axiovert 200M) and filmed every 5 minutes using the 20X objective overnight. The duration of mitosis was quantified by the disappearance of the nuclear envelope to the presence of a visible mid body. In some experiments, cells were challenged by 1mg/ml nocodazole overnight, chilled on ice for one hour the next day, and mitotic cells were filmed following 3 washes in prewarmed media. Spindle bipolarity onset was quantified by measuring the amount of time cells took to assemble a well-defined metaphase spindle.
Statistics analysis
Data value of individual cells, the average and standard deviations are plotted. Two-tailed and unpaired Student’s t-test was used to determine the statistical significance between two different cell populations.
Supplementary Material
Acknowledgments
This research was supported by NIH R01CA156706 and R01CA195612-01 to ZS, by New Jersey Commission on Cancer Research Pre-Doctoral fellowship (DFHS15PPC042) to SH, and by the Histopathology & Imaging shared resources of The Cancer Institute of New Jersey (P30CA072720).
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Merdes A, Cleveland DW. Pathways of spindle pole formation: different mechanisms; conserved components. J Cell Biol. 1997;138(5):953–6. doi: 10.1083/jcb.138.5.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hernandez P, Tirnauer JS. Tumor suppressor interactions with microtubules: keeping cell polarity and cell division on track. Dis Model Mech. 2010;3(5–6):304–15. doi: 10.1242/dmm.004507. [DOI] [PubMed] [Google Scholar]
- 3.Pellman D. Cell biology: aneuploidy and cancer. Nature. 2007;446(7131):38–9. doi: 10.1038/446038a. [DOI] [PubMed] [Google Scholar]
- 4.Chandhok NS, Pellman D. A little CIN may cost a lot: revisiting aneuploidy and cancer. Curr Opin Genet Dev. 2009;19(1):74–81. doi: 10.1016/j.gde.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 5.Namba M, Mihara K, Fushimi K. Immortalization of human cells and its mechanisms. Crit Rev Oncog. 1996;7(1–2):19–31. doi: 10.1615/critrevoncog.v7.i1-2.20. [DOI] [PubMed] [Google Scholar]
- 6.Ried T, Hu Y, Difilippantonio MJ, Ghadimi BM, Grade M, Camps J. The consequences of chromosomal aneuploidy on the transcriptome of cancer cells. Biochim Biophys Acta. 2012;1819(7):784–93. doi: 10.1016/j.bbagrm.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cimini D, Degrassi F. Aneuploidy: a matter of bad connections. Trends Cell Biol. 2005;15(8):442–51. doi: 10.1016/j.tcb.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 8.Yang Y, Liu M, Li D, Ran J, Gao J, Suo S, et al. CYLD regulates spindle orientation by stabilizing astral microtubules and promoting dishevelled-NuMA-dynein/dynactin complex formation. Proc Natl Acad Sci U S A. 2014;111(6):2158–63. doi: 10.1073/pnas.1319341111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Godin JD, Colombo K, Molina-Calavita M, Keryer G, Zala D, Charrin BC, et al. Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron. 2010;67(3):392–406. doi: 10.1016/j.neuron.2010.06.027. [DOI] [PubMed] [Google Scholar]
- 10.Thoma CR, Toso A, Gutbrodt KL, Reggi SP, Frew IJ, Schraml P, et al. VHL loss causes spindle misorientation and chromosome instability. Nature cell biology. 2009;11(8):994–1001. doi: 10.1038/ncb1912. [DOI] [PubMed] [Google Scholar]
- 11.Bornens M. Centrosome composition and microtubule anchoring mechanisms. Curr Opin Cell Biol. 2002;14(1):25–34. doi: 10.1016/s0955-0674(01)00290-3. [DOI] [PubMed] [Google Scholar]
- 12.Nigg EA, Stearns T. The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nature cell biology. 2011;13(10):1154–60. doi: 10.1038/ncb2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukasawa K. Oncogenes and tumour suppressors take on centrosomes. Nature reviews Cancer. 2007;7(12):911–24. doi: 10.1038/nrc2249. [DOI] [PubMed] [Google Scholar]
- 14.Hori A, Ikebe C, Tada M, Toda T. Msd1/SSX2IP-dependent microtubule anchorage ensures spindle orientation and primary cilia formation. EMBO Rep. 2014;15(2):175–84. doi: 10.1002/embr.201337929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khodjakov A, Cole RW, Oakley BR, Rieder CL. Centrosome-independent mitotic spindle formation in vertebrates. Curr Biol. 2000;10(2):59–67. doi: 10.1016/s0960-9822(99)00276-6. [DOI] [PubMed] [Google Scholar]
- 16.Haren L, Gnadt N, Wright M, Merdes A. NuMA is required for proper spindle assembly and chromosome alignment in prometaphase. BMC Res Notes. 2009;2:64. doi: 10.1186/1756-0500-2-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Louie RK, Bahmanyar S, Siemers KA, Votin V, Chang P, Stearns T, et al. Adenomatous polyposis coli and EB1 localize in close proximity of the mother centriole and EB1 is a functional component of centrosomes. J Cell Sci. 2004;117(Pt 7):1117–28. doi: 10.1242/jcs.00939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quintyne NJ, Schroer TA. Distinct cell cycle-dependent roles for dynactin and dynein at centrosomes. J Cell Biol. 2002;159(2):245–54. doi: 10.1083/jcb.200203089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo J, Yang Z, Song W, Chen Q, Wang F, Zhang Q, et al. Nudel contributes to microtubule anchoring at the mother centriole and is involved in both dynein-dependent and -independent centrosomal protein assembly. Mol Biol Cell. 2006;17(2):680–9. doi: 10.1091/mbc.E05-04-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim DJ, Martinez-Lemus LA, Davis GE. EB1, p150Glued, and Clasp1 control endothelial tubulogenesis through microtubule assembly, acetylation, and apical polarization. Blood. 2013;121(17):3521–30. doi: 10.1182/blood-2012-11-470179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kline-Smith SL, Walczak CE. Mitotic spindle assembly and chromosome segregation: refocusing on microtubule dynamics. Mol Cell. 2004;15(3):317–27. doi: 10.1016/j.molcel.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 22.Audhya A, Emr SD. Regulation of PI4,5P2 synthesis by nuclear-cytoplasmic shuttling of the Mss4 lipid kinase. EMBO J. 2003;22(16):4223–36. doi: 10.1093/emboj/cdg397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan J, Wray J, Meng X, Shen Z. BCCIP is required for the nuclear localization of the p21 protein. Cell Cycle. 2009;8(18):3019–24. [PMC free article] [PubMed] [Google Scholar]
- 24.Lu H, Guo X, Meng X, Liu J, Allen C, Wray J, et al. The BRCA2-interacting protein BCCIP functions in RAD51 and BRCA2 focus formation and homologous recombinational repair. Mol Cell Biol. 2005;25(5):1949–57. doi: 10.1128/MCB.25.5.1949-1957.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu H, Huang YY, Mehrotra S, Droz-Rosario R, Liu J, Bhaumik M, et al. Essential roles of BCCIP in mouse embryonic development and structural stability of chromosomes. PLoS Genet. 2011;7(9):e1002291. doi: 10.1371/journal.pgen.1002291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu H, Yue J, Meng X, Nickoloff JA, Shen Z. BCCIP regulates homologous recombination by distinct domains and suppresses spontaneous DNA damage. Nucleic acids research. 2007;35(21):7160–70. doi: 10.1093/nar/gkm732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mao N, Zhou Q, Kojic M, Perez-Martin J, Holloman WK. Ortholog of BRCA2-interacting protein BCCIP controls morphogenetic responses during DNA replication stress in Ustilago maydis. DNA repair. 2007;6(11):1651–60. doi: 10.1016/j.dnarep.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meng X, Yue J, Liu Z, Shen Z. Abrogation of the transactivation activity of p53 by BCCIP down-regulation. J Biol Chem. 2007;282(3):1570–6. doi: 10.1074/jbc.M607520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang YY, Dai L, Gaines D, Droz-Rosario R, Lu H, Liu J, et al. BCCIP Suppresses Tumor Initiation but Is Required for Tumor Progression. Cancer Res. 2013;73(23):7122–33. doi: 10.1158/0008-5472.CAN-13-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Yuan Y, Huan J, Shen Z. Inhibition of breast and brain cancer cell growth by BCCIPalpha, an evolutionarily conserved nuclear protein that interacts with BRCA2. Oncogene. 2001;20(3):336–45. doi: 10.1038/sj.onc.1204098. [DOI] [PubMed] [Google Scholar]
- 31.Wyler E, Wandrey F, Badertscher L, Montellese C, Alper D, Kutay U. The beta-isoform of the BRCA2 and CDKN1A(p21)-interacting protein (BCCIP) stabilizes nuclear RPL23/uL14. FEBS Lett. 2014;588(20):3685–91. doi: 10.1016/j.febslet.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 32.Meng X, Liu J, Shen Z. Genomic structure of the human BCCIP gene and its expression in cancer. Gene. 2003;302(1–2):139–46. doi: 10.1016/s0378-1119(02)01098-3. [DOI] [PubMed] [Google Scholar]
- 33.Hsu LC, White RL. BRCA1 is associated with the centrosome during mitosis. Proc Natl Acad Sci U S A. 1998;95(22):12983–8. doi: 10.1073/pnas.95.22.12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang HF, Takenaka K, Nakanishi A, Miki Y. BRCA2 and nucleophosmin coregulate centrosome amplification and form a complex with the Rho effector kinase ROCK2. Cancer Res. 2011;71(1):68–77. doi: 10.1158/0008-5472.CAN-10-0030. [DOI] [PubMed] [Google Scholar]
- 35.Keryer G, Di Fiore B, Celati C, Lechtreck KF, Mogensen M, Delouvee A, et al. Part of Ran is associated with AKAP450 at the centrosome: involvement in microtubule-organizing activity. Mol Biol Cell. 2003;14(10):4260–71. doi: 10.1091/mbc.E02-11-0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Young A, Dictenberg JB, Purohit A, Tuft R, Doxsey SJ. Cytoplasmic dynein-mediated assembly of pericentrin and gamma tubulin onto centrosomes. Mol Biol Cell. 2000;11(6):2047–56. doi: 10.1091/mbc.11.6.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raaijmakers JA, Tanenbaum ME, Medema RH. Systematic dissection of dynein regulators in mitosis. J Cell Biol. 2013;201(2):201–15. doi: 10.1083/jcb.201208098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Howell B, McEwen B, Canman J, Hoffman D, Farrar E, Rieder C, et al. Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J Cell Biol. 2001;155(7):1159–72. doi: 10.1083/jcb.200105093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delgehyr N, Sillibourne J, Bornens M. Microtubule nucleation and anchoring at the centrosome are independent processes linked by ninein function. J Cell Sci. 2005;118(Pt 8):1565–75. doi: 10.1242/jcs.02302. [DOI] [PubMed] [Google Scholar]
- 40.Mogensen MM, Malik A, Piel M, Bouckson-Castaing V, Bornens M. Microtubule minus-end anchorage at centrosomal and non-centrosomal sites: the role of ninein. J Cell Sci. 2000;113(Pt 17):3013–23. doi: 10.1242/jcs.113.17.3013. [DOI] [PubMed] [Google Scholar]
- 41.Song Y, Brady ST. Post-translational modifications of tubulin: pathways to functional diversity of microtubules. Trends Cell Biol. 2015;25:125–136. doi: 10.1016/j.tcb.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shida T, Cueva JG, Xu Z, Goodman MB, Nachury MV. The major alpha-tubulin K40 acetyltransferase alphaTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc Natl Acad Sci U S A. 2010;107(50):21517–22. doi: 10.1073/pnas.1013728107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vos MD, Martinez A, Elam C, Dallol A, Taylor BJ, Latif F, et al. A role for the RASSF1A tumor suppressor in the regulation of tubulin polymerization and genomic stability. Cancer Res. 2004;64(12):4244–50. doi: 10.1158/0008-5472.CAN-04-0339. [DOI] [PubMed] [Google Scholar]
- 44.Green RA, Kaplan KB. Chromosome instability in colorectal tumor cells is associated with defects in microtubule plus-end attachments caused by a dominant mutation in APC. J Cell Biol. 2003;163(5):949–61. doi: 10.1083/jcb.200307070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heald R, Tournebize R, Habermann A, Karsenti E, Hyman A. Spindle assembly in Xenopus egg extracts: respective roles of centrosomes and microtubule self-organization. J Cell Biol. 1997;138(3):615–28. doi: 10.1083/jcb.138.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heald R, Tournebize R, Blank T, Sandaltzopoulos R, Becker P, Hyman A, et al. Self-organization of microtubules into bipolar spindles around artificial chromosomes in Xenopus egg extracts. Nature. 1996;382(6590):420–5. doi: 10.1038/382420a0. [DOI] [PubMed] [Google Scholar]
- 47.King JM, Nicklas RB. Tension on chromosomes increases the number of kinetochore microtubules but only within limits. J Cell Sci. 2000;113(Pt 21):3815–23. doi: 10.1242/jcs.113.21.3815. [DOI] [PubMed] [Google Scholar]
- 48.DeLuca JG, Dong Y, Hergert P, Strauss J, Hickey JM, Salmon ED, et al. Hec1 and nuf2 are core components of the kinetochore outer plate essential for organizing microtubule attachment sites. Mol Biol Cell. 2005;16(2):519–31. doi: 10.1091/mbc.E04-09-0852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Waters JC, Chen RH, Murray AW, Salmon ED. J Cell Biol. 1998;141(5):1181–91. doi: 10.1083/jcb.141.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waters JC, Skibbens RV, Salmon ED. J Cell Sci. 1996;109(Pt 12):2823–31. doi: 10.1242/jcs.109.12.2823. [DOI] [PubMed] [Google Scholar]
- 51.Jackman M, Lindon C, Nigg EA, Pines J. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nature cell biology. 2003;5(2):143–8. doi: 10.1038/ncb918. [DOI] [PubMed] [Google Scholar]
- 52.Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Curr Biol. 2012;22(22):R966–80. doi: 10.1016/j.cub.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 53.Thornton GK, Woods CG. Primary microcephaly: do all roads lead to Rome? Trends Genet. 2009;25(11):501–10. doi: 10.1016/j.tig.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Basto R, Lau J, Vinogradova T, Gardiol A, Woods CG, Khodjakov A, et al. Flies without centrioles. Cell. 2006;125(7):1375–86. doi: 10.1016/j.cell.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 55.Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nature reviews Cancer. 2005;5(10):773–85. doi: 10.1038/nrc1714. [DOI] [PubMed] [Google Scholar]
- 56.Gruber R, Zhou Z, Sukchev M, Joerss T, Frappart PO, Wang ZQ. MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1-Cdc25 pathway. Nature cell biology. 2011;13(11):1325–34. doi: 10.1038/ncb2342. [DOI] [PubMed] [Google Scholar]
- 57.Zhou ZW, Tapias A, Bruhn C, Gruber R, Sukchev M, Wang ZQ. DNA damage response in microcephaly development of MCPH1 mouse model. DNA repair. 2013;12(8):645–55. doi: 10.1016/j.dnarep.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 58.Huang YY, Lu H, Liu S, Droz-Rosario R, Shen Z. Requirement of mouse BCCIP for neural development and progenitor proliferation. PLoS One. 2012;7(1):e30638. doi: 10.1371/journal.pone.0030638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salisbury JL. Centrosomes: coiled-coils organize the cell center. Curr Biol. 2003;13(3):R88–90. doi: 10.1016/s0960-9822(03)00033-2. [DOI] [PubMed] [Google Scholar]
- 60.Heckman KL, Pease LR. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc. 2007;2(4):924–32. doi: 10.1038/nprot.2007.132. [DOI] [PubMed] [Google Scholar]
- 61.Ivanova N, Dobrin R, Lu R, Kotenko I, Levorse J, DeCoste C, et al. Dissecting self-renewal in stem cells with RNA interference. Nature. 2006;442(7102):533–8. doi: 10.1038/nature04915. [DOI] [PubMed] [Google Scholar]
- 62.Skop AR, Liu H, Yates J, 3rd, Meyer BJ, Heald R. Dissection of the mammalian midbody proteome reveals conserved cytokinesis mechanisms. Science. 2004;305(5680):61–6. doi: 10.1126/science.1097931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mitchison TJ, Kirschner MW. Isolation of mammalian centrosomes. Methods Enzymol. 1986;134:261–8. doi: 10.1016/0076-6879(86)34094-1. [DOI] [PubMed] [Google Scholar]
- 64.Sehrawat S, Ernandez T, Cullere X, Takahashi M, Ono Y, Komarova Y, et al. AKAP9 regulation of microtubule dynamics promotes Epac1-induced endothelial barrier properties. Blood. 2011;117(2):708–18. doi: 10.1182/blood-2010-02-268870. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.