

Abstract
Background:
In 2014, germline signal transducer and activator of transcription (STAT) 3 gain-of-function (GOF) mutations were first described to cause a novel multisystem disease of early-onset lymphoproliferation and autoimmunity.
Objective:
This pivotal cohort study defines the scope, natural history, treatment, and overall survival of a large global cohort of patients with pathogenic STAT3 GOF variants.
Methods:
We identified 191 patients from 33 countries with 72 unique mutations. Inclusion criteria included symptoms of immune dysregulation and a biochemically confirmed germline heterozygous GOF variant in STAT3.
Results:
Overall survival was 88%, median age at onset of symptoms was 2.3 years, and median age at diagnosis was 12 years. Immune dysregulatory features were present in all patients: lymphoproliferation was the most common manifestation (73%); increased frequencies of double-negative (CD4−CD8−) T cells were found in 83% of patients tested. Autoimmune cytopenias were the second most common clinical manifestation (67%), followed by growth delay, enteropathy, skin disease, pulmonary disease, endocrinopathy, arthritis, autoimmune hepatitis, neurologic disease, vasculopathy, renal disease, and malignancy. Infections were reported in 72% of the cohort. A cellular and humoral immunodeficiency was observed in 37% and 51% of patients, respectively. Clinical symptoms dramatically improved in patients treated with JAK inhibitors, while a variety of other immunomodulatory treatment modalities were less efficacious. Thus far, 23 patients have undergone bone marrow transplantation, with a 62% survival rate.
Conclusion:
STAT3 GOF patients present with a wide array of immune-mediated disease including lymphoproliferation, autoimmune cytopenias, and multisystem autoimmunity. Patient care tends to be siloed, without a clear treatment strategy. Thus, early identification and prompt treatment implementation are lifesaving for STAT3 GOF syndrome
Keywords: STAT3, gain of function, lymphoproliferation, cytopenia, autoimmunity, immune dysregulation, immunodeficiency, precision medicine
Investigations of early-onset lymphoproliferation and multisystem autoimmunity have identified monogenic variants leading to disruption of the immune response, several of which feature significant immune dysregulation. The involvement of several and variable organ systems leads to presentation to different subspecialty clinics, causing a clinical quandary in establishing a unifying diagnosis. A streamlined approach to diagnosis and molecular characterization of these patients has the potential to lead to precision therapies. One example is signal transducer and activator of transcription (STAT) 3 gain-of-function (GOF) syndrome, which was recently recognized as an autosomal dominant primary immune regulatory disorder.1,2 Importantly, understanding mechanisms of disease pathogenesis has led to the use of gene/pathway-guided therapies that specifically target these mechanistic causes, resulting in improved outcomes.1,3–7
STAT3 is 1 of 7 members of the STAT protein family and is a critical regulator of cellular survival, proliferation, differentiation, and effector function. STAT3 is activated downstream of many cytokines including the IL-6 and IL-10 families of cytokines, as well as IL-21, IL-23, and IL-27. Binding of cytokines to their respective specific surface receptors leads to activation of Janus activating kinases (JAK), followed by JAK-mediated STAT3 phosphorylation and translocation of phosphorylated STAT3 into the nucleus, and phosphorylated STAT3 binding to specific DNA sequences resulting in transcriptional changes. Before the discovery of STAT3 GOF syndrome, heterozygous dominant negative (DN) variants in STAT3 were an established cause of autosomal dominant hyper-IgE syndrome (AD-HIES) or Job syndrome (OMIM 147060). Somatic STAT3 GOF variants are closely associated with large granular lymphocytic (LGL) leukemia,8 lymphoma,9 and solid tumors, including hepatocellular adenoma.10 Patients with germline STAT3 GOF variants experience heterogenous multisystem disease that can include early-onset autoimmunity, lymphoproliferation, susceptibility to infection, and growth failure.11–13 Over the last 7 years, case reports have described the clinical features of at least 50 patients with STAT3 GOF.3,4,11–34 The immune phenotype of STAT3 GOF patients is variable, yet hypogammaglobulinemia and lymphopenia are common features along with elevated double-negative (CD4−CD8−) TCRab+ T-cell (DNT) populations and reduced Treg cells. Treatment of clinical symptoms of STAT3 GOF is challenging, often requiring multiple immunosuppressive medications. Recently, success with targeted therapy using JAK inhibitors and/or anti–IL-6R blockade has been reported.3,27
The clinical phenotype of STAT3 GOF is diverse, with varied presentation to different specialists. Therefore, is critical to define the scope, natural history, treatment, and overall survival (OS) of a large cohort of STAT3 GOF patients. We describe an international cohort of individuals with STAT3 GOF syndrome to aid with improved recognition of common and rare presentations, report novel variants, and enhance clinical management.
METHODS
Patients
Patient data were solicited through academic organizations and when specific centers reached out to the principal investigators for functional validation of novel STAT3 variants identified in individual patients. Inclusion criteria included symptoms of immune dysregulation and a germline heterozygous variant in STAT3. All variants were confirmed to be GOF at baseline using a previously reported standardized luciferase assay.13 Deidentified data were collected on each patient using a detailed questionnaire completed by investigators at each institution (see Table E1 in this article’s Online Repository at www.jacionline.org). All studies involving human subjects were performed in accordance with site-specific institutional review board–approved protocols, as well as guidelines in the 1964 Declaration of Helsinki and its later amendments, with written informed consent obtained from the patients.
Statistical analysis
Data were collected and organized by Microsoft Excel (Microsoft, Redmond, Wash), and statistical analysis was performed by SPSS v26 (IBM, Armonk, NY). Association between categorical variables was investigated by chi-square or Fisher exact test with continuity correction. The OS from first clinical manifestation was plotted using the Kaplan-Meier method, and log rank statistics were used to investigate differences among median OS based on explanatory variables. Analyses with P <.05 were considered statistically significant. Exploratory data analysis was conducted without multiple comparisons adjustment to the P value.
RESULTS
Patients
We identified 191 patients with STAT3 GOF variants from 33 different countries (Fig 1). The cohort consisted of 115 novel and 76 previously published patients (Fig 2). There was a male predominance, with a male-to-female ratio of 1.25. Median age at onset of disease symptoms was 2.3 years. The median age at diagnosis was 12 years. The first reports of STAT3 GOF were published in 2014–15;11–13 therefore, this cohort contains both originally described patients and those diagnosed after the initial discovery of STAT3 GOF syndrome. To keep the data consistent, we did not include unaffected healthy carriers of STAT3 GOF variants in the analysis, although some have been reported,13,19 because many patients were diagnosed using proband-only gene panels, and family variant segregation was not always performed.
FIG 1.
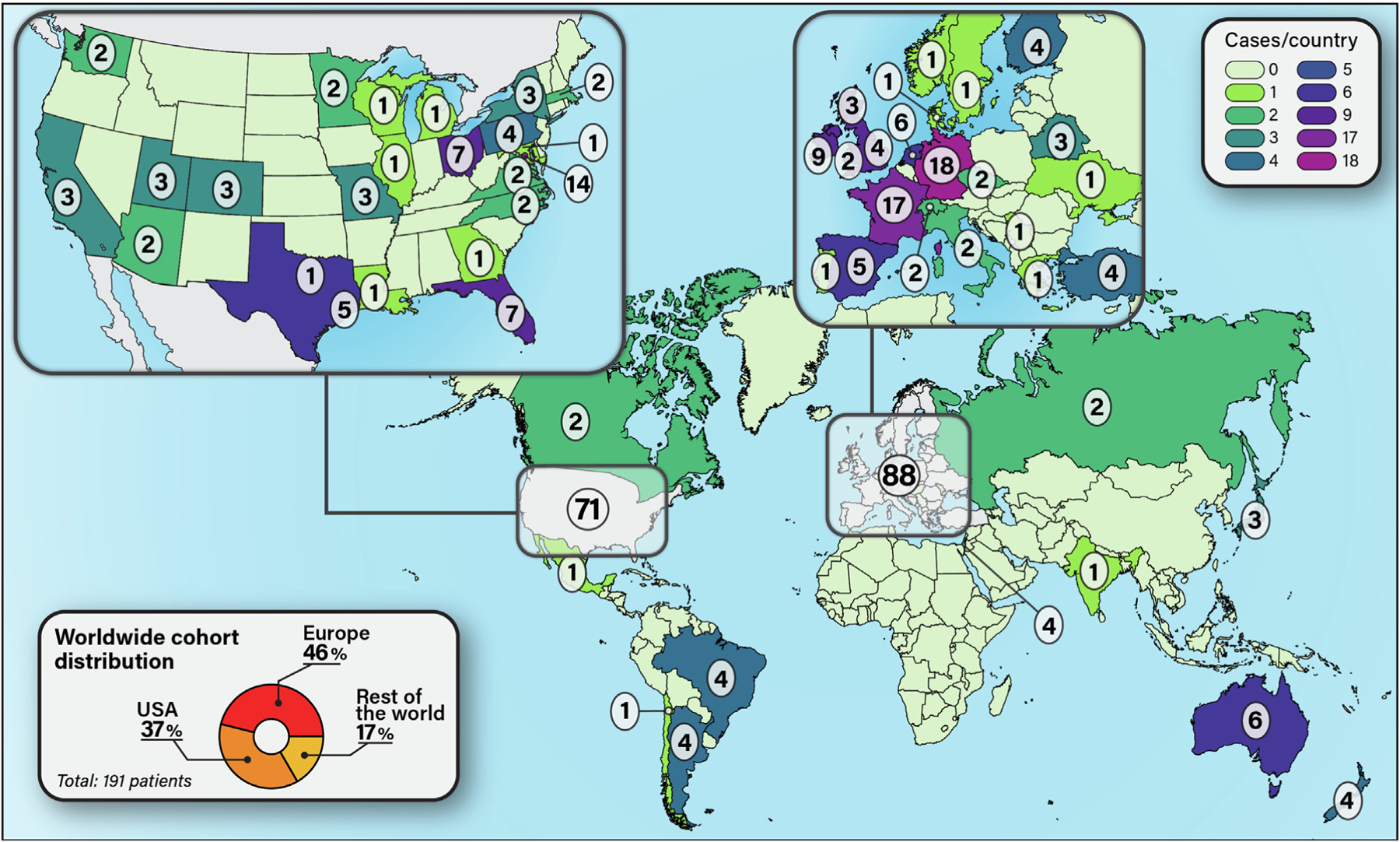
Country of origin for each STAT3 GOF patient. Thirty-seven percent of patients were from the United States, 46% from Europe, and 17% from the rest of the world.
FIG 2.
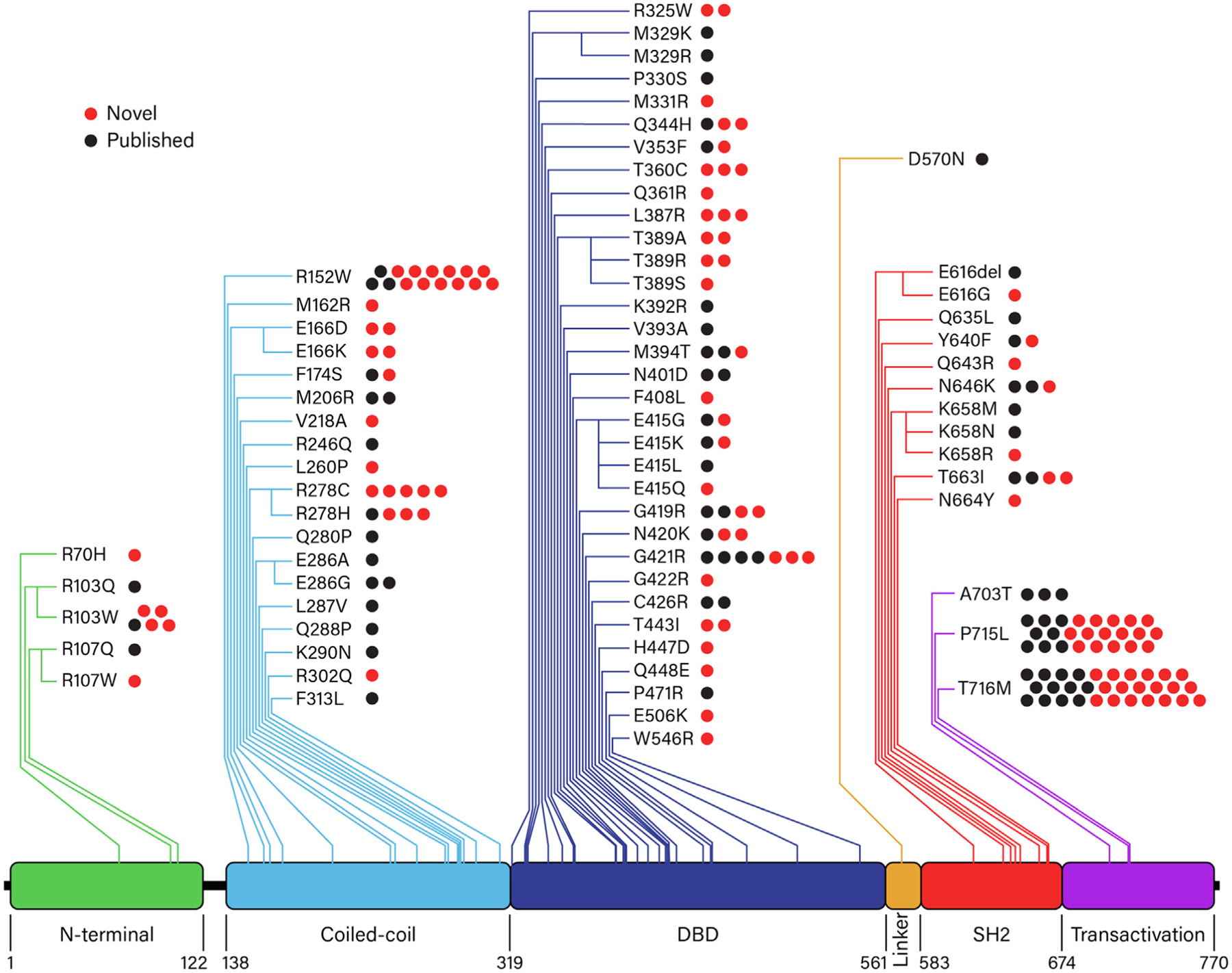
STAT3 GOF mutations. Seventy-two unique STAT3 variants spanned all domains of the STAT3 molecule. Red dots indicate novel patients; black dots, previously published patients. A, Alanine (Ala); C, cysteine (Cys); D, aspartic acid (Asp); E, glutamic acid (Glu); F, phenylalanine (Phe); G, glycine (Gly); H, histidine (His); I, isoleucine (Ile); K, lysine (Lys); L, leucine (Leu); M, methionine (Met); N, asparagine (Asn); P, prolie (Pro); Q, glutamine (Gln); R, arginine (Arg); S, serine (Ser); T, threonine (Thr); V, valine (Val); W, tryptophan (Trp); Y, tyrosine (Tyr).
STAT3 variants
We identified 72 unique STAT3 variants, spanning all domains of the STAT3 protein, in the 191 patients in this cohort (Fig 2; see Table E2 in the Online Repository at www.jacionline.org). All variants, except a single amino acid deletion (Glu616del), are missense mutations. Sixty-eight of the 72 unique variants are absent from population databases such as gnomAD (see Table E3 in the Online Repository).35 Germline STAT3 GOF variants are predominately located in the DNA binding (n = 33, 46%) and coiled-coil (n = 19, 26%) domains. Only 3 STAT3 GOF variants were found in the transactivation domain, but interestingly, these variants accounted for nearly a third of patients (30%, 58 patients), with 2 variants being the most commonly identified: Pro715Leu (24 patients, 13%) and Thr716Met (31 patients, 16%). The third most common variant was Arg152Trp in the coiled-coil domain (15 patients, 8%). Variants in the amino-terminal domain were the least common, with 5 different variants being identified in only 9 patients (5%). GOF variants in the SH2 domain were rare, with 11 unique SH2 domain variants in 17 patients (9%).36 Somatic GOF variants primarily localized in the SH2 domain of STAT3 have previously been reported to cause LGL leukemia. Interestingly, 6 of the 9 germline SH2 variants described here correspond to somatic variants identified in patients with LGL leukemia (in addition to the Phe174Ser variant in the C-C domain).8,37,38
Clinical manifestations
We organized clinical manifestations in the following disease categories: lymphoproliferative disease, autoimmune cytopenias, growth delay, enteropathy, skin disease, pulmonary disease, endocrinopathy, arthritis, hepatitis, neurologic disease, vasculopathy/vasculitis, renal disease, and malignancy (Fig 3; see, in the Online Repository at www.jacionline.org, Figs E1–E3 and Table E4). Only 3 patients had 1 disease category involved: respectively, 2 lineage autoimmune cytopenias, autoimmune hemolytic anemia, and growth failure. Fourteen percent of patients had 2 disease categories that most commonly included lymphoproliferation and autoimmune cytopenias. Most patients had 5 or more disease categories affected. Discrete facial features have not been previously described in patients with STAT3 GOF variants as they have been with STAT3 DN variants. However, patients with STAT3 GOF syndrome have characteristic round facies with cupped ears, a prominent forehead, a thin upper lip, and a smooth philtrum. These features were present irrespective of age, sex, or race (Fig 4, A).
FIG 3.
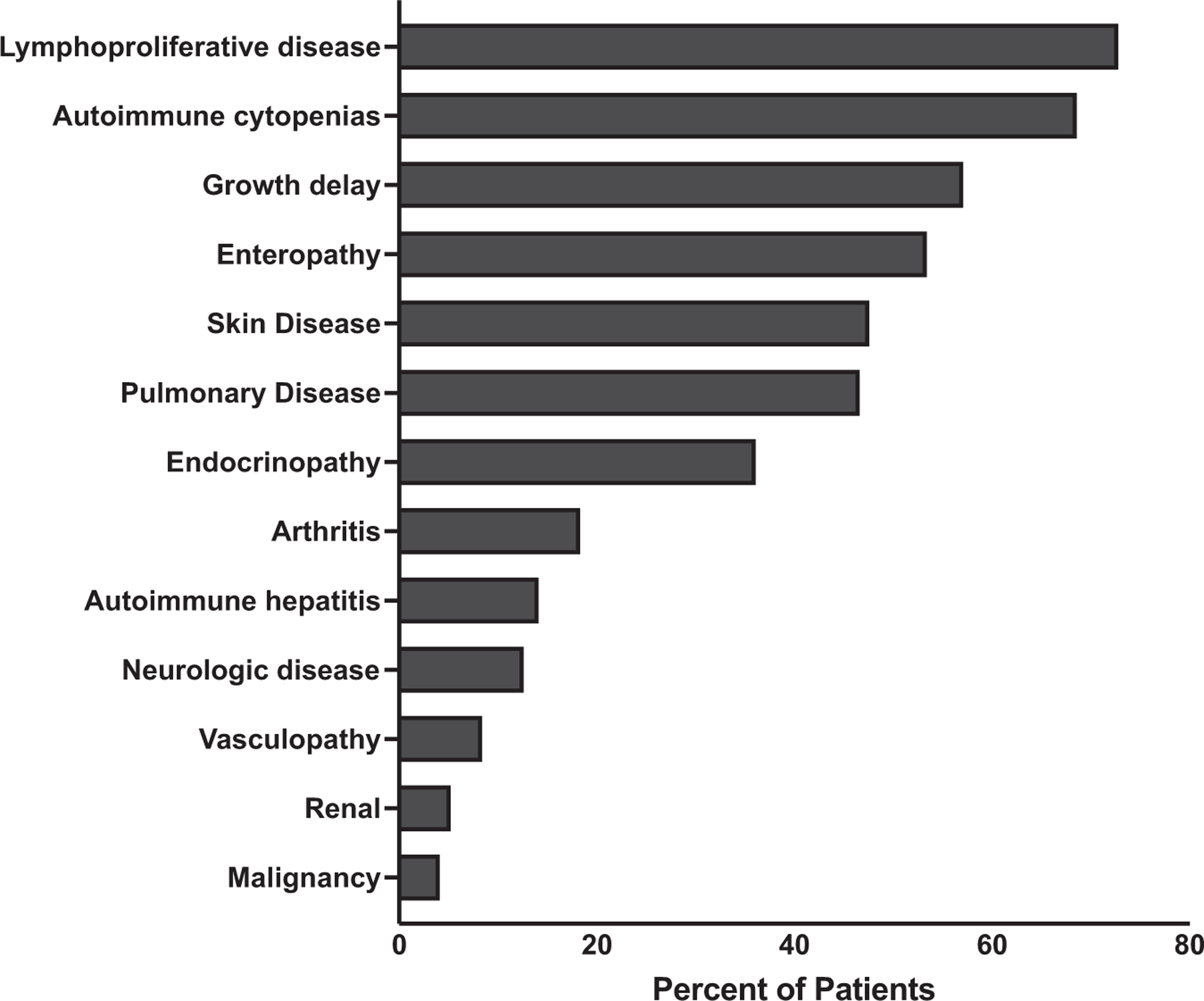
Clinical manifestations of STAT3 GOF patients. Percentage is shown of each clinical manifestation within the cohort of 191 patients.
FIG 4.

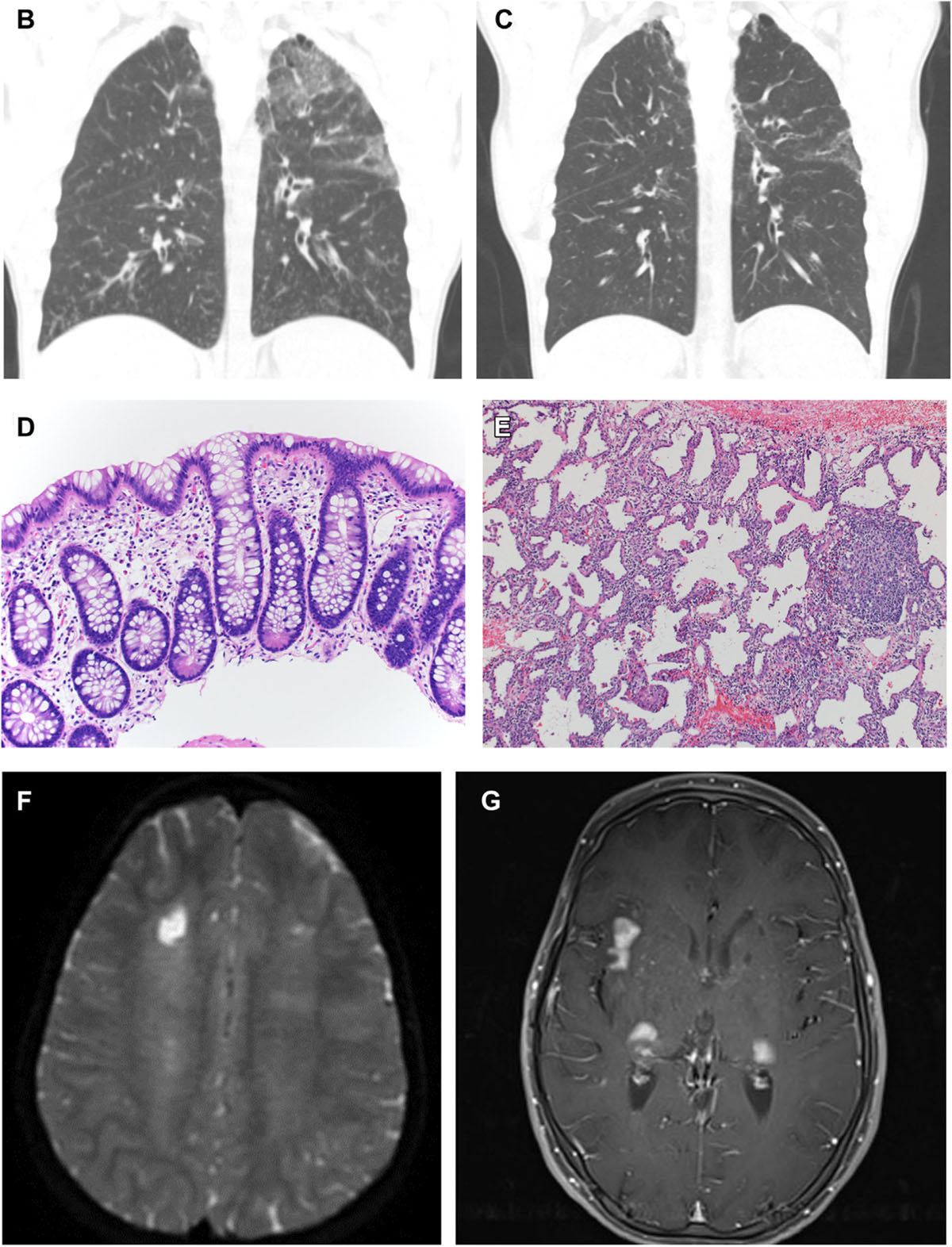
Clinical images of STAT3 GOF patients. (A) Characteristic facial appearance includes round face, prominent forehead, cupped ears, and smooth philtrum. (B) Interstitial infiltrate demonstrates nodules in right upper lung, bronchiectasis, and airspace disease (patient 57). (C) Same patient as (B) after treatment with tofacitinib for 5 years showing resolution of ground-glass opacities and pulmonary nodules. (D) Rectosigmoid biopsy sample reveals mild eosinophilia in the lamina propria and chronic changes in the form of Paneth cell metaplasia (patient 61). (E) Lung biopsy of right lower lobe reveals nodular and linear septal inflammation, predominantly by lymphocytes and histiocytes, without interstitial fibrosis (patient 61) (hematoxylin and eosin, original magnification 100 ×). (F) Chronic ischemic lesion is in the white matter of the middle frontal gyrus and right semioval center, and extensive leukoencephalopathy is evident of probable microvascular origin (patient 152). (G) Subcortical and cortical enhancing lesions are in the presylvian regions, anterior temporal lobes, and pons (patient 110).
Seventy-three percent of patients had evidence of lymphoproliferation most commonly manifesting as diffuse lymphadenopathy (75.5%) and/or splenomegaly (72%). Biopsies of lymph nodes often revealed follicular hyperplasia. Malignancy occurred in 12 patients;4,12,13 all are alive (Fig E1, B).
Autoimmune cytopenias were the second most common clinical manifestation. Most patients had 2 or 3 cell lineages affected. Coomb-positive anemia, anti-platelet antibodies, or anti-granulocyte antibodies were present in 41% of those tested.
Pulmonary disease typically diagnosed as interstitial lung disease (ILD) occurred in 43% of patients (Fig 4, B and C). Of patients with ILD, 26% had bronchiectasis. Patients with B-cell lymphopenia (30%, P = .003) and hypogammaglobulinemia (22.6%, P = .061), excluding those who received B-cell–depleting agents, were more likely to have bronchiectasis. In 13 patients, pulmonary disease led to fibrosis and restrictive lung disease. Two patients underwent lung transplantation for ILD; 1 patient is alive.
Growth failure remained a frequent feature of STAT3 GOF syndrome, affecting 57% of patients. Further complicating growth failure, more than half of the patients with growth failure had concurrent enteropathy (54.5%, P < .001). The majority of gastrointestinal manifestations began in the first 2 years of life. Fifty-three percent of patients had enteropathy most commonly diagnosed as celiac disease. Symptoms included chronic diarrhea, failure to thrive, achalasia, pseudo-obstruction, and features of colitis with 8 patients requiring total parenteral nutrition. When performed, the most common constellation of findings in the upper gastrointestinal tract was villous atrophy with small bowel thickening, and lymphocytic infiltration (Fig 4, D and E).
Autoimmune hepatitis occurred in 14% of patients. Regarding other liver-related disease, 3 patients developed portal hypertension and 1 developed hepatopulmonary syndrome. Two patients received liver transplants. Within several months, 1 patient had return of hepatitis and liver synthetic dysfunction and died of multiorgan failure resulting from sepsis.3
In line with significant risk of autoimmunity, patients also developed autoimmune endocrinopathies most commonly hypothyroidism, growth hormone deficiency, and early-onset diabetes (Fig E1, D). Other autoimmune manifestations of STAT3 GOF syndrome included arthritis, skin disease, vasculitis, and renal disease. Eighteen percent of patients developed arthritis that was characterized as polyarticular.
Dermatologic complications are a common feature of disease. Interestingly, this is also the case for AD-HIES, in which a distinctive eczematoid dermatitis emerges, typically presenting at birth.39 Forty-eight percent of patients had skin lesions, with eczema (60%), psoriasis (12%), and alopecia (10%) being the most common findings.
In this cohort, the first cases of systemic vasculopathy are reported (7%). These vascular lesions included central nervous system vasculitis and strokes, pulmonary vasculitis, vascular malformations, Moyamoya malformation in 2 patients, and systemic vasculitis. Renal manifestations were immune mediated, and 10 patients developed chronic renal disease. Several other systemic autoimmune conditions were reported (Fig E1, H).
Neurologic and specifically brain abnormalities or central nervous system insults have not previously been highlighted in STAT3 GOF syndrome but now include subcortical and cortical enhancing lesions in the perisylvian regions, anterior temporal lobes, and pons (Fig 4, F and G). One patient developed bilateral facial muscle weakness resulting from myositis and muscle atrophy. Approximately 13% had neurologic complaints, and clinical symptoms were variable (Fig E1, I).
Immunologic abnormalities
Blood lymphocyte subsets were measured in 171 patients (see Fig E4 and Tables E5 and E6 in the Online Repository at www.jacionline.org). This analysis includes new and previously reported data.3,11–13,15,16,18–20,23,26–28,31,34,40 CD4+ T, B, and NK cell lymphopenia were common. Lymphopenia and reduced functional responses to mitogens were more likely to occur in patients concurrently or previously treated with chronic or pulse steroids. Approximately half of patients had humoral deficiency.41
As a marker of systemic autoimmunity,42,43 Treg cells (CD4+CD25+FOXP3+) and IL-17–producing (CD4+IL17+) T cells were quantified in 69 and 41 patients, respectively. Treg cell percentages were low in 39% and IL-17–producing T cells were elevated in 27% of those tested.44
The severe lymphoproliferation in STAT3 GOF syndrome has been described as a mimic of autoimmune lymphoproliferative disease involving defective Fas-induced apoptosis.13 Therefore, we collected data from patients who had increased proportions of DNTs (CD4−CD8−TCRab+), a cellular characteristic of autoimmune lymphoproliferative disease, or sensitivity to Fas-induced apoptosis, or both. Eighty-nine patients with lymphoproliferation had DNTs assessed, and of those, 82% had increased DNTs45 (P = .001). Of the patients with lymphoproliferation and elevated DNTs, Fas-induced apoptosis was abnormal in 4 patients (although Fas apoptosis was only performed in 27 patients). This assessment supports the notion that the presence of increased numbers of DNTs46,47 is more suggestive of lymphoproliferation driven by STAT3 GOF rather than expansion due to impaired Fas-mediated immune regulation.
Infections
Mild to severe infections occurred in 72% of patients (see Tables E7 and E8 in the Online Repository at www.jacionline.org). Bacterial infections were most common (80%), followed by viral (61%), fungal (25%), opportunistic (7%), and mycobacterial (6%) infections. Patients with hypogammaglobulinemia were most likely to develop bacterial infections (73.6%, P = .006). Those with CD8+ (60.5%, P = .033) and/or CD4+ T-cell lymphopenia (56.5%, P =.018) were more likely to develop viral infections, and those with CD3+ (21.9%, P = .007) and/or CD4+ T-cell lymphopenia (21.7%, P = .002) were more likely to develop fungal infections.
Treatment
Given the severity of immune-mediated disease, various forms of treatment were used to control the clinical manifestations of patients with STAT3 GOF variants (Fig 5; see Table E4 and Table E9 in the Online Repository at www.jacionline.org). Therapies included immunomodulation, hormone replacement (insulin, growth, and thyroid hormone), and antimicrobial treatments. Overall, 77% of patients received at least 1 systemic immune-suppressing medication and 67% received immunoglobulin supplementation. Indications for immunoglobulin therapy included hypogammaglobulinemia, antimicrobial prophylaxis, and treatment for autoimmune cytopenias. Twenty-two percent of patients received hormone replacement and 43% received antimicrobial prophylaxis.
FIG 5.
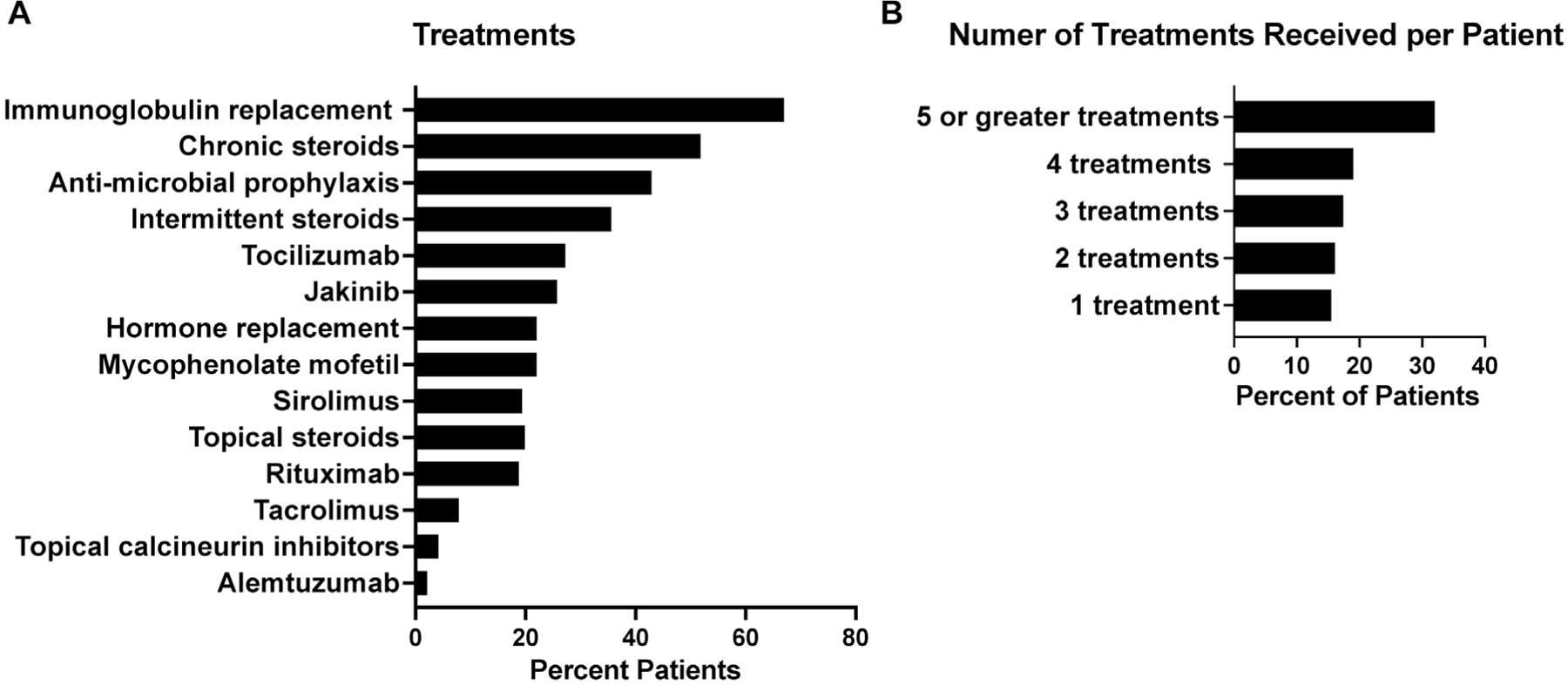
Treatment provided to STAT3 GOF patients. Data were available from 164 patients. (A) Percentage of each treatment provided to patients. (B) Percentage of patients receiving 1, 2, 3, 4, or 5 or more treatments.
Most patients required 5 or more treatment modalities to treat disease manifestations (Fig 5, B). In >30% of cases, corticosteroids were provided as initial treatment that escalated to corticosteroid-sparing agents and finally precision therapy with tocilizumab and/or JAK inhibitor therapy (jakinibs) (21%). The most commonly used immune modulators were chronic systemic corticosteroids and topical corticosteroids. The disease of most patients was refractory to corticosteroid treatment, requiring second- and third-line agents to control immune dysregulation. These additional therapeutic agents included rituximab, mycophenolate mofetil, sirolimus, tacrolimus, and alemtuzumab. The disease of more than half of patients with autoimmune cytopenias failed to respond to treatment with intravenous immunoglobulin and chronic corticosteroids. Rituximab was provided to 26% of patients with autoimmune cytopenias, most in combination with mycophenolate mofetil or sirolimus. Of these patients, 79% experienced a partial response, but half required additional therapies. Splenectomy was performed in 6 patients, leading to resolution of cytopenias and/or lymphoproliferation.
Thirty-six patients received jakinibs as salvage therapy (age 3.5–48 years; median 13 years), which successfully controlled cytopenias. Tocilizumab and/or jakinibs were used to treat refractory ILD in 8 patients3,27 and led to sufficient improvement in symptoms to permit withdrawal of supplemental oxygen in 7. Arthritis was the primary indication for tocilizumab and/or jakinib therapy in 6 patients; 66% had improvement of arthritis. Tocilizumab led to disease improvement in 1 patient with optic neuritis. Ruxolitinib was initiated and led to clinical improvement for 1 patient with a cerebral vascular lesion (Fig 4, F) in whom therapy with corticosteroids had failed, whereas another only minimally improved with pulse corticosteroids and high-dose intravenous immunoglobulin (Fig 4, G). Disease manifestations including diabetes mellitus,48 pulmonary fibrosis, and pulmonary hypertension did not reverse substantially with receipt of precision therapy with tocilizumab and/or jakinibs.
Receipt of intravenous chronic steroids (57%, P < .001), pulse steroids (54%, P = .035), and tacrolimus (73%, P = .018) was associated with development of viral infections. Additionally, all 4 patients who received alemtuzumab developed viral infections (P = .023). Of the steroid-sparing agents, patients receiving mycophenolate mofetil were more likely to develop fungal infections (31%, P = .006). Unwanted adverse effects of tocilizumab and/or jakinibs were rare. Two patients developed significantly elevated liver transaminases after tocilizumab, and 1 patient with trilineage cytopenias had worsened bleeding episodes with jakinib therapy, leading to its discontinuation. There was no association of infection, including viral infections, in patients receiving tocilizumab and/or jakinibs.
Twenty-three patients underwent hematopoietic cell transplantation (HCT) to treat treatment-refractory life-threatening disease manifestations (unpublished data).3,11–13,18,19,26,28,31 Indications for HCTwere severe enteropathy, cytopenias, lymphoproliferative disease, and hemophagocytic lymphohistiocytosis. OS was 62%.
Domain–phenotype correlations
We sought to determine if there were any specific correlations between the domain in which the protein change occurred in each patient and corresponding clinical phenotype and survival. Although no major gene domain–phenotype correlation could be gleaned from these data, we observed the following: 56% of patients with SH2 domain variants (P =.008) had endocrinopathy as opposed to 44% in the coiled-coil (CCD), 43% in the DNA binding domain (DBD), 24% in the transactivation domain (TAD), and none in the N-terminal domain. Cytopenias and lymphoproliferative disease were the most common features in variants across all domains. Cytopenias occurred in 89% of patients with variants in the N-terminal domain (P = .025), 79% in the TAD, 76% in the CCD, 63% in the SH2 domain, and 55% in the DBD. Lymphoproliferative disease occurred in 91% of patients with variants in the coiled-coil domain and 100% with variants in the N-terminal domain (P = .001).
Survival
With a median age at presentation of 2.3 years, overall cumulative survival of the cohort was 88% (Fig 6, A). However, there were specific parameters that significantly affected survival. Twenty-three patients died, 10 of whom received HCT. Mean age at death for non-HCT patients was 23 years (4 months to 52 years). Causes of death of the 13 non-HCT patients included renal failure, progressive respiratory failure, severe enteropathy, and multiorgan failure. One patient died of progressive myositis leading to respiratory failure, and one died of complications after lung transplantation. Survival was affected by time from first clinical presentation to genetic diagnosis, but not age at presentation and disease burden (see Fig E5, A–D, in the Online Repository at www.jacionline.org), when assessed by the number of treatments received. Since the original reports of STAT3 GOF were published in 2014–15, a delay in definitive diagnosis was present for many subjects in the cohort whose symptoms manifested before the disease’s discovery. Therefore, we sought to understand how such a delay may affect survival. When the time from presentation to diagnosis was 3 to 12 years, OS was negatively correlated (15.68 years, 95% confidence interval [CI] 14.45 to 16.91, P < .001). When diagnosis was delayed beyond 12 years from onset, survival was less affected, likely because patients had a milder phenotype. Providing 5 or more treatments was negatively associated with survival (P <.001) and was a better indicator of disease burden than number of disease manifestations involved.
FIG 6.
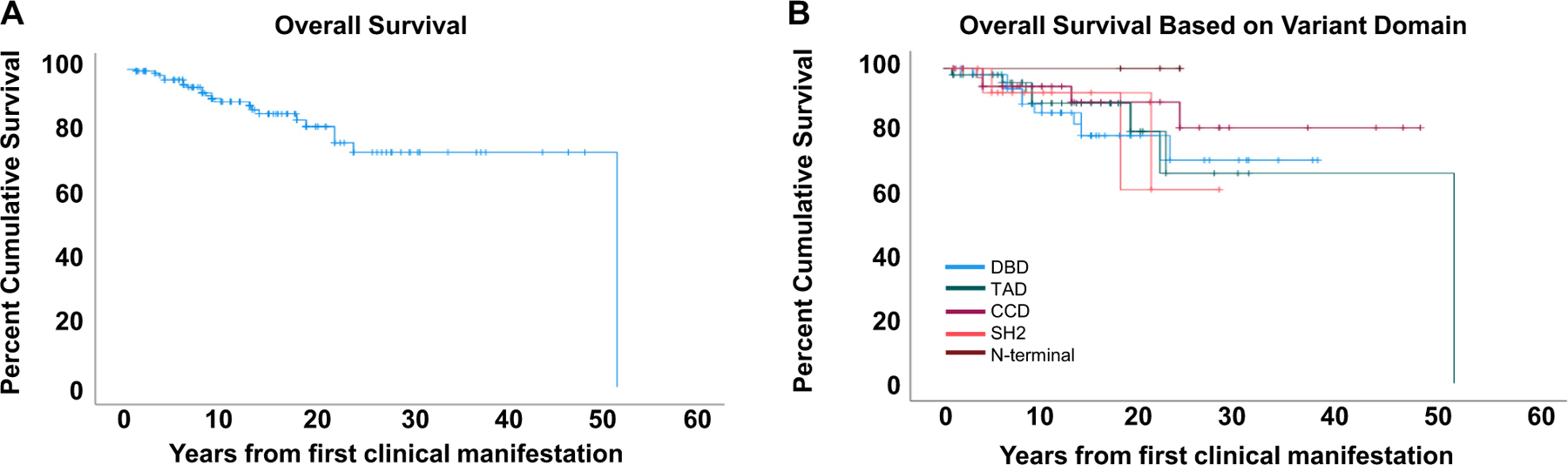
Survival curves of STAT3 GOF patients. (A) OS of STAT3 GOF patients. (B) OS of STAT3 GOF patients based on variant domain. Purple, CCD; blue, DBD; brown, N-terminal domain; red, SH2; green, TAD; P = .0677.
We further analyzed whether the presence of specific disease manifestations affected survival (Fig E6, A–N). Although pulmonary disease was not the most common manifestation of STAT3 GOF syndrome, progressive respiratory disease leading to oxygen dependence was highly associated with death (13.25 years, 95% CI 7.85 to 18.65, P = .002) (Fig 7, A). Enteropathy (median OS not reached, P =.034) (Fig 7, B; see also Fig E6, D, in the Online Repository at www.jacionline.org), autoimmune hepatitis (24.0 years, 95% CI 12.62 to 35.38, P = .004) (Fig 7, C, Fig E6, F), growth failure (median OS not reached, P = .026) (Fig E6, C), and dependence on total parenteral nutrition (median OS not reached, P = .024) (Fig 7, D, and Fig E6, E) were associated with early death. Although the association of ILD with survival was not statistically different (median OS not reached, P = .799) (Fig E6, H), oxygen dependency highly correlated with worse survival (13.25 years, 95% CI 7.85 to 18.65, P = .002) (Fig 7, A; see also Fig E6, I), suggesting that severity of lung disease is a poor prognostic indicator. Finally, the location of the STAT3 variant by protein domain did not correlate with OS (Fig 6, B).
FIG 7.
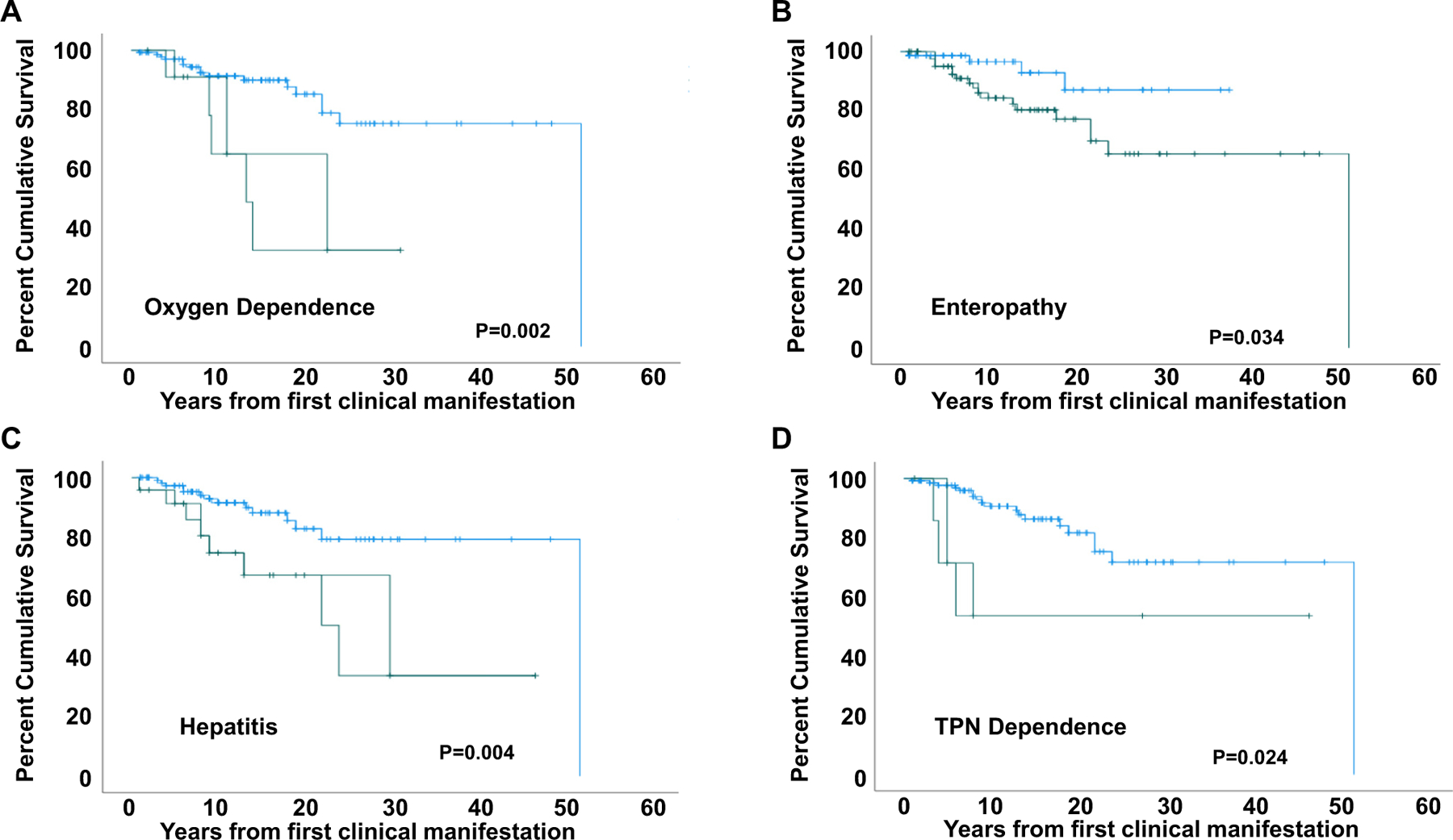
Survival according to clinical manifestations. Survival is significantly affected by oxygen dependance (A), enteropathy (B), autoimmune hepatitis (C), and total parenteral nutrition (TPN) dependance (D). Green, present; blue, not present.
DISCUSSION
To our knowledge, this international cohort of 191 patients is the largest series of patients with germline STAT3 GOF variants reported to date. With this large data set we, were able to establish the spectrum of systemic autoimmunity that is often diagnosed as classic autoimmune conditions, determine the frequency of major disease manifestations, uncover unrecognized clinical symptoms, identify biomarkers for disease such as immunologic parameters, and develop recommendations regarding best treatments under a unifying diagnosis and mechanism of disease.
STAT3 GOF syndrome is a severe progressive disease with early onset affecting several organ systems. Interestingly, genotype did not predict survival; nor was it revealing as a prognostic indicator of disease manifestations. Establishing a diagnosis of STAT3 GOF is complicated by the heterogeneity of presenting symptoms, resulting in patients presenting across myriad medical specialties, which in turn results in siloed care. As reported in the initial descriptions of STAT3 GOF11–13 and herein, nonmalignant lymphoproliferation, early-onset autoimmune cytopenias, enteropathy, and growth delay are the most common features of disease. The presence of more than one of these symptoms in a patient, especially if severe, chronic, or difficult to control, should prompt evaluation for STAT3 GOF. Diagnosis can be achieved by genetic sequencing of STAT3. In addition, it is important to note that the constellation of features most common in STAT3 GOF syndrome (lymphoproliferation, autoimmune cytopenias, enteropathy, ILD, growth failure) are not unique to STAT3 GOF but rather are shared by many primary immune regulatory disorders caused by other gene defects (eg, FOXP3, CTLA4, LRBA, PIK3CD). These features should prompt an evaluation for primary immune regulatory disorders including STAT3 GOF, and this is best achieved by an unbiased genetic approach—either a targeted panel or by whole exome sequencing or whole genome sequencing to assess for structural variants.
Currently, functional assessment for GOF of a novel variant is only available on a research basis. The immune phenotype of patients with this disease is variable, as were perturbations in TH17 and Treg cell quantities historically shown to influence autoimmunity,49–51 and thus did not aid in diagnosis. There were not sufficient numbers of subjects with Treg and TH17 cells quantified to show a consistency in the expected phenotype in Treg and TH17 subsets—that is, a reduction in Treg cells and an elevation in TH17 cells.13 However, many patients who had αβ DN T cells measured had elevated levels, suggesting that patients without an apoptotic defect and elevated αβ DN T cells should be assessed for STAT3 GOF syndrome.
In AD-HIES (STAT3 DN) and somatic STAT3 GOF variants, there is an increased risk of lymphoma,52 possibly related to up-regulation of programmed death ligand 1 (PD-L1),53 and LGL leukemia,8 respectively. Because severe lymphoproliferation, as seen in this cohort, typically raises concern for malignant transformation, the low incidence of malignancy in this cohort compared to what has been observed in AD-HIES is interesting. Reduced STAT1 expression has been previously described to cause poor upregulation of programmed cell death 1 (PD-1)/PD-L1. Because STAT1 expression is low in STAT3 GOF,13,53 poor PD-1/PD-L1 expression in STAT3 GOF could explain both the prevalence of autoimmunity and protection from malignancy.
Growth failure was observed in more than half of patients, raising concern for defects in growth hormone signaling, which requires STAT5b.54 STAT3 regulates STAT5b, and this has been postulated as one of the important mechanisms by which these patients experience growth delay.26
Analysis of this large cohort revealed several new features of STAT3 GOF syndrome, including discrete facies, neurologic insults, renal disease, and vasculopathy. STAT3 plays an important role in endothelial and vascular smooth muscle cell differentiation, migration, and proliferation.55 Enhanced STAT3 activity has been implicated in cardiovascular disease as well as vasculitic diseases including microscopic polyangiitis, other vasculitis, and glomerulonephritis.56–59 Additionally, STAT3 hyperactivation leads to reduced pulmonary artery endothelial nitric oxide synthase, low levels of which highly correlate with development of pulmonary hypertension.60 Indeed, treatment with jakinibs has led to clinical improvement in patients with giant cell arteritis and is a therapeutic target being explored for the treatment of cardiovascular diseases.55,58
The heterogeneity of symptoms in STAT3 GOF syndrome patients led to a variety of treatments. Standard immunosuppressants were often ineffective in controlling autoimmune symptoms, and most patients required more than 5 agents. Although systemic corticosteroids remain the standard first line of treatment, both corticosteroids and corticosteroid-sparing immuno-suppression led to increased bacterial, viral, and fungal infections. Treatments targeting IL-6 and the JAK-STAT pathway have shown promising clinical efficacy in a small number of previously published patients with STAT3 GOF3,27 and in this cohort. Although it is not clear whether JAK inhibition alone is sufficient, using jakinibs as the backbone of therapy is the currently preferred approach, with addition of an IL-6 blocking agent to achieve synergistic control not achieved with JAK inhibition alone. Most patients continued targeted therapy even with remission of clinical symptoms. In addition, jakinibs may be used as a bridge to HCT in severe disease. HCT was the treatment of choice for patients with severe, progressive disease, but it remains unclear whether early intervention with this definitive therapy is the optimal choice. For now, HCT remains a treatment option for patients at risk of severe disease progression.
Given the wide spectrum of autoimmune disease manifestations, high morbidity, premature death, and availability of precision therapy, recognition and diagnosis of STAT3 GOF syndrome is essential and relevant to the broader medical community. In an effort to halt disease progression, a diagnosis should prompt consideration for precision therapy (JAK-STAT inhibition, IL-6 blockade). Jakinibs and/or tocilizumab are approved by the US Food and Drug Administration for treating autoimmune diseases61–63 and have been effective at reversing disease-related sequelae in STAT GOF disorders.3,64,65 The long-term efficacy of these agents in treating and preventing disease-related sequelae requires further investigation. Early identification and treatment implementation are lifesaving, so it is essential that this disorder is considered in patients with lymphoproliferation and severe and/or multisystem autoimmune disease.
Supplementary Material
Key messages.
Identification of STAT3 GOF syndrome may improve outcomes in patients with multisystem immune-mediated disease by aiding in the selection of appropriate therapy.
Over 70% of individuals with STAT3 GOF had lymphoproliferation and autoimmune cytopenias; initiation of targeted therapy led to improved patient outcomes.
Acknowledgments
Supported in part by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health. S.B. was supported by the Scientific and Technological Research Council of Turkey (318S202). S.E. is supported by the German Research Foundation (DFG) under Germany’s excellence strategy (CIBBS-EXC-2189-Project ID 390939984) and the BMBF GAIN consortium (01GM1910A). L.R.F.S. is supported by the Jeffrey Modell Foundation and Baylor College of Medicine Chao Physician Scientist Award. G.S. was supported by the Germany’s Excellence Strategy–EXC 2155 “RESIST” (39087428) and the BMBF German Auto-Immunity Network (GAIN) (01GM1910E). T.P.V. was supported by the Arthritis National Research Foundation. B.G. is supported by the Deutsche Forschungsgemeinschaft (GR1617/14-1/iPAD; SFB1160/2_B5; RESIST-EXC 2155-Project ID 390874280; and CIBSS-EXC-2189-Project ID 390939984), and the BMBF (GAIN) 01GM1910A. S.G.T. was supported by a Principal Research Fellowship (1042925) and Leadership 3 Investigator grant (1176665) from the National Health and Medical Research Council of Australia and CIRCA Investigators I.C., M.O’S., S.B., J.S., K.P., D.S., and S.G.T. are funded by grants awarded by the Jeffrey Modell Foundation and the Jon Brown Fook Foundation.
We thank Helen Matthews for regulatory support.
Abbreviations used
- AD-HIES
Autosomal dominant hyper-IgE syndrome
- CI
Confidence interval
- DN
Dominant negative
- DNT
Double-negative T cell
- GOF
Gain of function
- HCT
Hematopoietic cell transplantation
- ILD
Interstitial lung disease
- JAK
Janus kinase
- Jakinib
JAK inhibitor
- LGL
Large granular cell
- OS
Overall survival
- PD-1
Programmed cell death 1
- PD-L1
Programmed death ligand 1
- STAT
Signal transducer and activator of transcription
The STAT3 Working Group members are as follows:
Svetlana Aleshkevich, MD (Belarusian Research Center for Pediatric Oncology, Hematology and Immunology, Minsk, Belarus); Luis M. Allende, PhD (Cincinnati Children’s Hospital Medical Center, University of Cincinnati College of Medicine, Cincinnati, Ohio); T. Prescott Atkinson, MD, PhD (Division of Pediatric Allergy and Immunology, University of Alabama, Birmingham, Ala); Faranaz Atschekzei, MD, PhD (Department of Clinical Immunology and Rheumatology, Hanover Medical School, Hanover, Germany); Sezin Aydemir, MD (Department of Pediatrics, Division of Allergy and Immunology, Istanbul University–Cerrahpasa, Cerrahpasa Medical School, Istanbul, Turkey); Utku Aygunes, MD (Department of Pediatric Hematology Oncology, Cumhuriyet University, Sivas, Turkey); Vincent Barlogis, MD, PhD (Department of Pediatric Hematology, La Timone Hospital, Marseille University Hospital, Marseille, France); Ulrich Baumann, PhD (Department of Pediatric Pneumology, Allergy and Neonatology, Hanover Medical School, Hanover, Germany); John Belko, MD (Department of Pediatric Hematology/Oncology, Kaiser Permanente, Sacramento, Calif); Liliana Bezrodnik, MD (University Hospital of Wales, Cardiff, Wales, United Kingdom); Ariane Biebl, MD (Department of Pediatric and Adolescent Medicine, Kepler University Hospital Linz, Linz, Austria); Lori Broderick, MD, PhD (Department of Pediatrics, University of California San Diego, La Jolla, Calif, and Rady Children’s Hospital San Diego, Division of Pediatric Allergy, Immunology, and Rheumatology, San Diego, Calif); Nancy J. Bunin, MD (Perelman School of Medicine at University of Pennsylvania, Division of Bone Marrow Transplantation, Children’s Hospital of Philadelphia, Philadelphia, Pa); Maria Soledad Caldirola, PhD (University Hospital of Wales, Cardiff, Wales, United Kingdom); Martin Castelle, MD (Pediatric Hematology-Immunology and Rheumatology Department, Necker-Enfants-Malades University Hospital, Assistance Publique Hôpitaux de Paris, Paris, France); Fatih Celmeli, MD (Division of Pediatric Allergy and Immunology, Antalya Education and Research Hospital, University of Medical Science, Antalya, Turkey); Louis-Marie Charbonnier, PhD (Division of Immunology, Boston Children’s Hospital, Department of Pediatrics, Harvard Medical School, Boston, Mass); Talal A. Chatila, MD MSc (Division of Immunology, Boston Children’s Hospital, Department of Pediatrics, Harvard Medical School, Boston, Mass); Deepak Chellapandian, MD (Department of Pediatrics, Division of Allergy and Immunology, University of South Florida, Tampa, Fla); Haluk Cokugras, MD (Department of Pediatrics, Division of Allergy and Immunology, Istanbul University–Cerrahpasa, Cerrahpasa Medical School, Istanbul, Turkey); Niall Conlon, PhD (Department of Immunology, St James’s Hospital and School of Medicine, Trinity College, Dublin, Ireland); Fionnuala Cox, PhD (Children’s Health Ireland at Crumlin, Dublin, Ireland); Etienne Crickx, MD, PhD (Assistance Publique-Hôpitaux de Paris, Department of Internal Medicine, Hôpital Henri-Mondor, Université Paris-Est, Créteil, France); Buket Dalgic, MD (Gazi University, School of Medicine, Department of Pediatrics, Division of Gastroenterology, Hepatology and Nutrition, Ankara, Turkey); Virgil ASH Dalm, MD, PhD (Department of Internal Medicine, Division of Clinical Immunology and Department of Immunology, Erasmus University Medical Center, Rotterdam, Netherlands); Silvia Danielian, PhD (Servicio de Immunología y Reumatología, Hospital Nacional de Pediatría Prof Dr Juan P. Garrahan, Buenos Aires, Argentina); Nerea Dominguez-Pinilla, MD (Hospital Virgen de la Salud, Toledo, Instituto de Investigación Sanitaria [imas12], Madrid, Spain); Tal Dujovny, MD (Division of Rheumatology and Immunology, Department of Pediatrics, Washington University School of Medicine, St Louis, Mo); Mikael Ebbo, MD, PhD (Department of Internal Medicine, La Timone University Hospital, Assistance Publique-Hôpitaux de Marseille, Aix Marseille Université, Marseille, France); Ahmet Eken, MD (Erciyes University Medical Faculty, Department of Medical Biology & Genome and Stem Cell Center [Genkok], Kayseri, Turkey); Brittany Esty, MD (Division of Immunology, Boston Children’s Hospital, Department of Pediatrics, Harvard Medical School, Boston, Mass); Alexandre Fabre, MD, PhD (Pediatric Multidisciplinary Department APHM, Timone Enfant, Marseille, France); Alain Fischer, MD, PhD (Laboratory of Immunogenetics of Pediatric Autoimmune Disease, Institut Imagine, INSERM UMR1163, Paris, France); Mark Hannibal, MD, PhD (Department of Pediatrics, C. S. Mott Children’s Hospital, Michigan Medicine, Ann Arbor, Mich); Laura Huppert, MD (Department of Medicine, University of California, San Francisco, Calif); Marc D. Ikeda, MD (Department of Allergy, The Permanente Medical Group; and Division of Rheumatology/Allergy and Clinical Immunology, University of California, Davis, School of Medicine, Sacramento, Calif); Stephen Jolles, MD (University Hospital of Wales, Cardiff, United Kingdom); Kent W. Jolly, MD (Department of Pediatric Hematology-Oncology and BMT, Cleveland Clinic, Cleveland, Ohio); Neil Jones, MD (Department of Pediatrics, Paracelsus Medical University, Salzburg, Austria); Musa Kakakukcu, MD (Pediatric Hematology Oncology & Erciyes Pediatric HSCT Unit, Erciyes University, Kayseri, Turkey); Maria Kanariou, MD, PhD (Department of Immunology-Histocompatibility, Specialized Center & Referral Center for Primary Immunodeficiencies - Paediatric Immunology, “Aghia Sophia” Children’s Hospital, Athens, Greece); Elif Karakoc-Aydiner, MD (Marmara University School of Medicine, Pediatric Allergy and Immunology, Istanbul, Turkey); Theoni Karamantziani, MD (Department of Pediatric Hematology-Oncology [TAO] and First Department of Pediatrics, Aghia Sophia Children’s Hospital, Athens, Greece); Charikleia Kelaidi, MD (Department of Pediatric Hematology-Oncology [TAO], Aghia Sophia Children’s Hospital, Athens, Greece); Mary Keogan, MD (Beaumont Hospital, Dublin, Ireland); Ayşenur Pac Kisaarslan, MD (Erciyes University, Pediatric Rheumatology, Kayseri, Turkey); Ayca Kiykim, MD (Department of Pediatrics, Division of Allergy and Immunology, Istanbul University–Cerrahpasa, Cerrahpasa Medical School, Istanbul, Turkey); Kosmas Kotsonis, MD (Department of Pediatric Hematology-Oncology [TAO] and First Department of Pediatrics, Aghia Sophia Children’s Hospital, Athens, Greece); Natalia Kuzmenko, MD (Dmitry Rogachev National Medical and Research Center for Pediatric Hematology, Oncology and Immunology, Moscow, Russia); Sylvie Leroy, MD (Pulmonology and Allergology Department, CHU de Nice, Nice, France); Harry Lesmana, MD (Kaiser Permanente Pediatric Rheumatology, Sacramento, Calif); Dimitra Lianou, MD (Department of Pediatric Hematology-Oncology [TAO] and First Department of Pediatrics, Aghia Sophia Children’s Hospital, Athens, Greece); Hilary Longhurst, PhD (Department of Immunology, Auckland District Health Board, and Department of Medicine, University of Auckland, Auckland City Hospital, Grafton, Auckland, New Zealand); Myriam Ricarda Lorenz, PhD (Institute for Transfusion Medicine, University of Ulm, Ulm, Germany); Patrick Maffucci, MD, PhD (Department of Anesthesiology, Mount Sinai School of Medicine, New York, NY); Ania Manson, PhD (BartsHealth NHS Trust, London, England, United Kingdom); Sarah Marchal, MD (Institute for Transfusion Medicine, University of Ulm, Ulm, Germany); Marion Malphettes, MD (Department of Clinical Immunology, Hôpital Saint-Louis, Assistance Publique Hôpitaux de Paris, Université Paris Diderot, Paris, France); Lia Furlaneto Marega, PhD (Pediatric Allergy and Immunology/Center of Investigation in Pediatrics, Faculty of Medical Sciences, State University of Campinas–Unicamp, São Paulo, Brazil); Andrea A. Mauracher, MD, PhD (Division of Immunology, University Children’s Hospital Zurich, Children’s Research Center [CRC], Zurich, Switzerland); Martin Van Hagen MD, PhD (Department of Internal Medicine, Division of Clinical Immunology and Department of Immunology, Erasmus University Medical Center, Rotterdam, Netherlands); Holly Miller DO (Center for Cancer and Blood Disorders, Phoenix Children’s Hospital, Phoenix, Ariz); Joy Mombourquette, MD (Kaiser Permanente Pediatric Rheumatology, Sacramento, Calif); Noel G. Morgan, PhD (Institute of Biomedical and Clinical Science, University of Exeter Medical School, Exeter, United Kingdom); Anna Mukhina, MD (Dmitry Rogachev National Medical and Research Center for Pediatric Hematology, Oncology and Immunology, Moscow, Russia); Aladjidi Nathalie, MD (Centre de Référence National des Cytopénies Autoimmunes de l’Enfant (CEREVANCE), Bordeaux, France; Pediatric Oncology Hematology Unit, University Hospital, Plurithématique CIC (CICP), Centre d’Investigation Clinique (CIC) 1401, INSERM Bordeaux, France); Brigitte Nelken, MD (Department of Pediatric Hematology, Jeanne de Flandre Hospital, CHU Lille, Lille, France); David Nolan, MD (Department of Immunology, Royal Perth Hospital, Perth, Australia); Anna-Carin Norlin, MD, PhD (Department of Infectious Diseases, Karolinska University Hospital, Stockholm, Sweden); Matias Oleastro, MD (Servicio de Immunología y Reumatología, Hospital Nacional de Pediatría Prof. Dr. Juan P. Garrahan, Buenos Aires, Argentina); Alper Ozcan, MD (Erciyes University, Pediatric Hematology Oncology & Erciyes Pediatric HSCT Unit, Kayseri, Turkey); Marlene Pasquet, MD, PhD (Pediatric Oncology Immunology Hematology Unit, Children’s University Hospital, Toulouse, France); José Roberto Pegler, MD (Instituto Da Crianca do Hospital, Universidade de São Paulo, São Paulo, Brazil); Capucine Picard, MD, PhD (Université de Paris, Imagine Institut, Study Center for Primary Immunodeficiencies, Necker-Enfants Malades University Hospital, Paris, France); Sophia Polychronopoulou, MD, PhD (Department of Pediatric Hematology-Oncology [TAO], Aghia Sophia Children’s Hospital, Athens, Greece); Pierre Quartier, MD (Pediatric Hematology-Immunology and Rheumatology Department, Necker-Enfants-Malades University Hospital, Assistance Publique Hôpitaux de Paris, Paris, France); Juan Francisco Quesada, MD (Genetics Department at University Hospital 12 Octubre, Madrid, Spain); Jan Ramakers, MD (Department of Pediatrics, Hospital Universitari Son Espases; and Multidisciplinary Group for Research in Paediatrics, Balearic Islands Health Research Institute [IdISBa], Palma, Spain); Katrina L. Randall MBBS, PhD (Australian National University Medical School, Australian National University, Canberra, Australia); V. Koneti Rao, MD (Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Md); Allison Remiker, MD (Division of Pediatric Hematology and Oncology, Medical College of Wisconsin, Milwaukee, Wisc); Geraldine Resin, MD (Pediatric Unit Cayenne General Hospital, Cayenne, French Guiana, France); Peter Richmond, MD (Division of Immunology, University Children’s Hospital Zurich, CRC, Zurich, Switzerland; Division of Paediatrics, School of Medicine, University of Western Australia, Perth, Australia); Frederic Rieux-Laucat, PhD (Laboratory of Immunogenetics of Pediatric Autoimmune Diseases, Imagine Institute, UMR 1163 INSERM, and Paris University, Paris, France); Yulia Rodina, MD (Dmitry Rogachev National Medical and Research Center for Pediatric Hematology, Oncology and Immunology, Moscow, Russia); Pierre Rohrlich, MD, PhD (Pediatric Hematology Department, CHU de Nice, Nice, France); Johnathan Sachs, MD (Division of Allergy and Immunology, Phoenix Children’s Hospital, Phoenix, Ariz); Inga Sakovich, PhD (Belarusian Research Center for Pediatric Oncology, Hematology and Immunology, Minsk, Belarus); Christopher Santarlas, BS (Department of Pediatrics, Division of Allergy and Immunology, University of South Florida, Tampa, Fla); Sinan Sari, MD (Department of Pediatrics, Division of Gastroenterology, Hepatology and Nutrition, School of Medicine, Gazi University, Ankara, Turkey); Gregory Sawicki, MD (Division of Immunology, Boston Children’s Hospital, Department of Pediatrics, Harvard Medical School, Boston, Mass); Uwe Schauer, MD (University Children’s Hospital, Ruhr University Bochum, Bochum, Germany); Selma C. Scheffler Mendoza, MD, MS (Clinical Immunology Service, National Institute of Pediatrics, Mexico City, Mexico); Oksana Schvetz, MD (Dmitry Rogachev National Medical and Research Center for Pediatric Hematology, Oncology and Immunology, Moscow, Russia); Reinhold Ernst Schmidt, MD (St. Vincent’s Clinical School, Faculty of Medicine, UNSW Sydney, Sydney, Australia); Klaus Schwarz, MD (Institute for Transfusion Medicine, University of Ulm; and Institute for Clinical Transfusion Medicine and Immunogenetics Ulm, German Red Cross Blood Service, Baden-Württemberg-Hessen, Ulm, Germany); Anna Sediva, MD, PhD (Department of Immunology, Charles University in Prague, Second Faculty of Medicine and University Hospital Motol, Prague, Czech Republic); Kyle Sinclair, MD (Health Fairview Clinic Department of Rheumatology, Fridley, Minn); Mary Slatter, MD (Newcastle, England, United Kingdom); John Sleasman, MD (Department of Pediatric Allergy and Immunology, Duke University, Durham, NC); Katerina Stergiou, MD (Department of Pediatric Hematology-Oncology [TAO] and First Department of Pediatrics, Aghia Sophia Children’s Hospital, Athens, Greece); Narissara Suratannon, MD (Center of Excellence for Allergy and Clinical Immunology, Division of Allergy, Immunology and Rheumatology, Department of Pediatrics, Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand, King Chulalongkorn Memorial Hospital, the Thai Red Cross Society, Bangkok, Thailand); Kay Tanita, MD, PhD (Department of Pediatrics and Developmental Biology, Tokyo Medical and Dental University, Tokyo, Japan); Grace Thompson, MD (Immunology Department, Sir Charles Gairdner Hospital, Nedlands, Australia); Stephen Travis, MD (Department of Internal Medicine, University of Oklahoma, Oklahoma City, Okla); Timothy Trojan, MD (Allergy Partners of Oklahoma, Stillwater, Okla); Maria Tsinti, MD, PhD (Department of Pediatric Hematology-Oncology [TAO] and First Department of Pediatrics, Aghia Sophia Children’s Hospital, Athens, Greece); Ekrem Unal, MD (Erciyes University, Pediatric Hematology Oncology & Erciyes Pediatric HSCT Unit, Kayseri, Turkey); Luciano Urdinez, MD (Servicio de Inmunologıa y Reumatologıa, Hospital Nacional de Pediatrıa Prof Dr Juan P. Garrahan, Buenos Aires, Argentina); Felisa Vazquez-Gomez, MD (Pediatric Oncology, Hematology and Stem Cell Transplant Department, Hospital Infantil Universitario Niño Jesus, Madrid, Spain); Mariana Villa, MD (Hospital de Pediatría Juan P. Garrahan, Argentina); Michael Weinreich, MD (Division of Intramural Research, National Institute of Allergy and Infectious Diseases [NIAID], National Institutes of Health [NIH], Bethesda, Md); Mitchell J. Weiss, MD, PhD (Department of Hematology, St Jude Children’s Research Hospital, Memphis, Tenn); Benjamin L. Wright, MD (Division of Allergy and Immunology, Phoenix Children’s Hospital, Phoenix, Ariz); Ebru Yilmaz, MD (Erciyes University, Pediatric Hematology Oncology & Erciyes Pediatric HSCT Unit, Kayseri, Turkey); Radana Zachova, MD (Department of Immunology, Charles University in Prague, Second Faculty of Medicine and University Hospital Motol, Prague, Czech Republic); and Yu Zhang, PhD (Division of Intramural Research, NIAID, NIH, Bethesda, Md).
Footnotes
Disclosure of potential conflict of interest: A. Kumar reports speakers’ bureau for SOBI; L. Forbes Satter reports consultancy for Enzyvant, Grifols, CSL Behring, Takeda, and ADMA; J. W. Leiding is a full-time employee and shareholder of Bluebirdbio and speaker and consultant for Sobi and Horizon Therapeutics; and T. Vogel has consulted for SOBI, Novartis, Pfizer, and Moderna. The rest of the authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J Clin Immunol 2020;40:66–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan AY, Torgerson TR. Primary immune regulatory disorders: a growing universe of immune dysregulation. Curr Opin Allergy Clin Immunol 2020;20:582–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forbes LR, Vogel TP, Cooper MA, Castro-Wagner J, Schussler E, Weinacht KG, et al. Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J Allergy Clin Immunol 2018;142:1665–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hadjadj J, Aladjidi N, Fernandes H, Leverger G, Magérus-Chatinet A, Mazerolles F, et al. Pediatric Evans syndrome is associated with a high frequency of potentially damaging variants in immune genes. Blood 2019;134:9–21. [DOI] [PubMed] [Google Scholar]
- 5.Heimall JR, Hagin D, Hajjar J, Henrickson SE, Hernandez-Trujillo HS, Tan Y, et al. Use of genetic testing for primary immunodeficiency patients. J Clin Immunol 2018;38:320–9. [DOI] [PubMed] [Google Scholar]
- 6.Singh JA, Solomon DH, Dougados M, Felson D, Hawker G, Katz P, et al. Development of classification and response criteria for rheumatic diseases. Arthritis Rheum 2006;55:348–52. [DOI] [PubMed] [Google Scholar]
- 7.Stray-Pedersen A, Sorte HS, Samarakoon P, Gambin T, Chinn IK, Coban Akdemir ZH, et al. Primary immunodeficiency diseases: genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol 2017;139:232–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koskela HL, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmäki H, Andersson EI, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med 2012;366:1905–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kucuk C, Jiang B, Hu X, Zhang W, Chan JK, Xiao W, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-T or NK cells. Nat Commun 2015;6:6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bioulac-Sage P, Sempoux C, Balabaud C. Hepatocellular adenomas: morphology and genomics. Gastroenterol Clin North Am 2017;46:253–72. [DOI] [PubMed] [Google Scholar]
- 11.Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, De Franco E, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet 2014;46:812–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haapaniemi EM, Kaustio M, Rajala HL, van Adrichem AJ, Kainulainen L, Glumoff V, et al. Autoimmunity, hypogammaglobulinemia, lymphoproliferation, and mycobacterial disease in patients with activating mutations in STAT3. Blood 2015;125:639–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood 2015;125:591–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Besnard C, Levy E, Aladjidi N, Stolzenberg MC, Magerus-Chatinet A, Alibeu O, et al. Pediatric-onset Evans syndrome: heterogeneous presentation and high frequency of monogenic disorders including LRBA and CTLA4 mutations. Clin Immunol 2018;188:52–7. [DOI] [PubMed] [Google Scholar]
- 15.Fabre A, Marchal S, Barlogis V, Mari B, Barbry P, Rohrlich PS, et al. Clinical aspects of STAT3 gain-of-function germline mutations: a systematic review. J Allergy Clin Immunol Pract 2019;7:1958–69.e9. [DOI] [PubMed] [Google Scholar]
- 16.Giovannini-Chami L, Vogel TP, Forbes LR, Fabre A, Trojani MC, Leroy S, et al. STAT3 gain of function: a new aetiology of severe rheumatic disease. Rheumatology (Oxford) 2019;58:365–7. [DOI] [PubMed] [Google Scholar]
- 17.Gonzalez-Mancera MS, Johnson B, Mirsaeidi M. STAT3 gain-of-function mutation in a patient with pulmonary Mycobacterium abscessus infection. Respir Med Case Rep 2020;30:101125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutiérrez M, Scaglia P, Keselman A, Martucci L, Karabatas L, Domené S, et al. Partial growth hormone insensitivity and dysregulatory immune disease associated with de novo germline activating STAT3 mutations. Mol Cell Endocrinol 2018;473: 166–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jägle S, Heeg M, Grün S, Rensing-Ehl A, Maccari ME, Klemann C, et al. Distinct molecular response patterns of activating STAT3 mutations associate with penetrance of lymphoproliferation and autoimmunity. Clin Immunol 2020;210:108316. [DOI] [PubMed] [Google Scholar]
- 20.Khoury T, Molho-Pessach V, Ramot Y, Ayman AR, Elpeleg O, Berkman N, et al. Tocilizumab promotes regulatory T-cell alleviation in STAT3 gain-of-function–associated multi-organ autoimmune syndrome. Clin Ther 2017;39:444–9. [DOI] [PubMed] [Google Scholar]
- 21.Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova JL, et al. Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol 2016;7:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mauracher AA, Eekels JJM, Woytschak J, van Drogen A, Bosch A, Prader S, et al. Erythropoiesis defect observed in STAT3 GOF patients with severe anemia. J Allergy Clin Immunol 2020;145:1297–301. [DOI] [PubMed] [Google Scholar]
- 23.Nabhani S, Schipp C, Miskin H, Levin C, Postovsky S, Dujovny T, et al. STAT3 gain-of-function mutations associated with autoimmune lymphoproliferative syndrome like disease deregulate lymphocyte apoptosis and can be targeted by BH3 mimetic compounds. Clin Immunol 2017;181:32–42. [DOI] [PubMed] [Google Scholar]
- 24.Parlato M, Charbit-Henrion F, Abi Nader E, Begue B, Guegan N, Bruneau J, et al. Efficacy of ruxolitinib therapy in a patient with severe enterocolitis associated with a STAT3 gain-of-function mutation. Gastroenterology 2019;156: 1206–10.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Russell MA, Pigors M, Houssen ME, Manson A, Kelsell D, Longhurst H, et al. A novel de novo activating mutation in STAT3 identified in a patient with common variable immunodeficiency (CVID). Clin Immunol 2018;187:132–6. [DOI] [PubMed] [Google Scholar]
- 26.Sediva H, Dusatkova P, Kanderova V, Obermannova B, Kayserova J, Sramkova L, et al. Short stature in a boy with multiple early-onset autoimmune conditions due to a STAT3 activating mutation: could intracellular growth hormone signalling be compromised? Horm Res Paediatr 2017;88:160–6. [DOI] [PubMed] [Google Scholar]
- 27.Silva-Carmona M, Vogel TP, Marchal S, Guesmi M, Dubus JC, Leroy S, et al. Successful treatment of interstitial lung disease in STAT3 gain-of-function using JAK inhibitors. Am J Respir Crit Care Med 2020;202:893–7. [DOI] [PubMed] [Google Scholar]
- 28.Suh YW, Horton JC. Papilledema from gain-of-function mutations in the STAT3 gene. Ophthalmic Genet 2019;40:165–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takagi M, Hoshino A, Yoshida K, Ueno H, Imai K, Piao J, et al. Genetic heterogeneity of uncharacterized childhood autoimmune diseases with lymphoproliferation. Pediatr Blood Cancer 2018;65(2):. [DOI] [PubMed] [Google Scholar]
- 30.Tanita K, Sakura F, Nambu R, Tsumura M, Imanaka Y, Ohnishi H, et al. Clinical and immunological heterogeneity in Japanese patients with gain-of-function variants in STAT3. J Clin Immunol 2021;41:780–90. [DOI] [PubMed] [Google Scholar]
- 31.Todaro F, Tamassia N, Pinelli M, Moratto D, Dotta L, Grassi A, et al. Multisystem autoimmune disease caused by increased STAT3 phosphorylation and dysregulated gene expression. Haematologica 2019;104:e322–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Velayos T, Martínez R, Alonso M, Garcia-Etxebarria K, Aguayo A, Camarero C, et al. An activating mutation in STAT3 results in neonatal diabetes through reduced insulin synthesis. Diabetes 2017;66:1022–9. [DOI] [PubMed] [Google Scholar]
- 33.Weinreich MA, Vogel TP, Rao VK, Milner JD. Up, down, and all around: diagnosis and treatment of novel STAT3 variant. Front Pediatr 2017;5:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wienke J, Janssen W, Scholman R, Spits H, van Gijn M, Boes M, et al. A novel human STAT3 mutation presents with autoimmunity involving Th17 hyperactivation. Oncotarget 2015;6:20037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaimowitz NS, Branch J, Reyes A, Vargas-Hernández A, Orange JS, Forbes LR, et al. A novel STAT3 mutation in a Qatari patient with hyper-IgE syndrome. Front Pediatr 2019;7:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andersson E, Kuusanmäki H, Bortoluzzi S, Lagström S, Parsons A, Rajala H, et al. Activating somatic mutations outside the SH2-domain of STAT3 in LGL leukemia. Leukemia 2016;30:1204–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vogel TP, Milner JD, Cooper MA. The ying and yang of STAT3 in human disease. J Clin Immunol 2015;35:615–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sowerwine KJ, Holland SM, Freeman AF. Hyper-IgE syndrome update. Ann N Y Acad Sci 2012;1250:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seddiki N, Santner-Nanan B, Tangye SG, Alexander SI, Solomon M, Lee S, et al. Persistence of naive CD45RA+ regulatory T cells in adult life. Blood 2006;107: 2830–8. [DOI] [PubMed] [Google Scholar]
- 41.Shearer WT, Rosenblatt HM, Gelman RS, Oyomopito R, Plaeger S, Stiehm ER, et al. Lymphocyte subsets in healthy children from birth through 18 years of age: the Pediatric AIDS Clinical Trials Group P1009 study. J Allergy Clin Immunol 2003;112:973–80. [DOI] [PubMed] [Google Scholar]
- 42.Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G. Past, present, and future of regulatory T cell therapy in transplantation and autoimmunity. Front Immunol 2019;10:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tabarkiewicz J, Pogoda K, Karczmarczyk A, Pozarowski P, Giannopoulos K. The role of IL-17 and Th17 lymphocytes in autoimmune diseases. Arch Immunol Ther Exp (Warsz) 2015;63:435–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garcia-Prat M, Alvarez-Sierra D, Aguilo-Cucurull A, Salgado-Perandres S, Briongos-Sebastian S, Franco-Jarava C, et al. Extended immunophenotyping reference values in a healthy pediatric population. Cytometry B Clin Cytom 2019; 96:223–33. [DOI] [PubMed] [Google Scholar]
- 45.Oliveira JB, Bleesing JJ, Dianzani U, Fleisher TA, Jaffe ES, Lenardo MJ, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood 2010;116:e35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rensing-Ehl A, Völkl S, Speckmann C, Lorenz MR, Ritter J, Janda A, et al. Abnormally differentiated CD4+ or CD8+ T cells with phenotypic and genetic features of double negative T cells in human Fas deficiency. Blood 2014;124:851–60. [DOI] [PubMed] [Google Scholar]
- 47.Maccari ME, Fuchs S, Kury P, Andrieux G, Volkl S, Bengsch B, et al. A distinct CD381CD45RA1 population of CD4+, CD8+, and double-negative T cells is controlled by FAS. J Exp Med 2021;218:e20192191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wegehaupt O, Muckenhaupt T, Johnson MB, Schwab KO, Speckmann C. Ruxolitinib controls lymphoproliferation and diabetes in a STAT3-GOF patient. J Clin Immunol 2020;40:1207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired TH17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 2008;452:773–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuwabara T, Ishikawa F, Kondo M, Kakiuchi T. The role of IL-17 and related cytokines in inflammatory autoimmune diseases. Mediators Inflamm 2017;2017: 3908061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dejaco C, Duftner C, Grubeck-Loebenstein B, Schirmer M. Imbalance of regulatory T cells in human autoimmune diseases. Immunology 2006;117:289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumánovics A, Perkins SL, Gilbert H, Cessna MH, Augustine NH, Hill HR. Diffuse large B cell lymphoma in hyper-IgE syndrome due to STAT3 mutation. J Clin Immunol 2010;30:886–93. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y, Ma CA, Lawrence MG, Break TJ, O’Connell MP, Lyons JJ, et al. PD-L1 up-regulation restrains Th17 cell differentiation in STAT3 loss- and STAT1 gain-of-function patients. J Exp Med 2017;214:2523–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kofoed EM, Hwa V, Little B, Woods KA, Buckway CK, Tsubaki J, et al. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med 2003;349: 1139–47. [DOI] [PubMed] [Google Scholar]
- 55.Dutzmann J, Daniel JM, Bauersachs J, Hilfiker-Kleiner D, Sedding DG. Emerging translational approaches to target STAT3 signalling and its impact on vascular disease. Cardiovasc Res 2015;106:365–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arakawa T, Masaki T, Hirai T, Doi S, Kuratsune M, Arihiro K, et al. Activation of signal transducer and activator of transcription 3 correlates with cell proliferation and renal injury in human glomerulonephritis. Nephrol Dial Transplant 2008;23: 3418–26. [DOI] [PubMed] [Google Scholar]
- 57.Wilde B, Hoerning A, Kribben A, Witzke O, Dolff S. Signal tranducers and activators of transcription: expression and function in anti-neutrophil cytoplasmic antibody-associated vasculitis. Mol Med Rep 2014;9:2316–20. [DOI] [PubMed] [Google Scholar]
- 58.Zhang H, Watanabe R, Berry GJ, Tian L, Goronzy JJ, Weyand CM. Inhibition of JAK-STAT signaling suppresses pathogenic immune responses in medium and large vessel vasculitis. Circulation 2018;137:1934–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weimbs T, Talbot JJ. STAT3 signaling in polycystic kidney disease. Drug Discov Today Dis Mech 2013;10:e113–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med 1995;333:214–21. [DOI] [PubMed] [Google Scholar]
- 61.Loh ML, Tasian SK, Rabin KR, Brown P, Magoon D, Reid JM, et al. A phase 1 dosing study of ruxolitinib in children with relapsed or refractory solid tumors, leukemias, or myeloproliferative neoplasms: a Children’s Oncology Group phase 1 consortium study (ADVL1011). Pediatr Blood Cancer 2015;62: 1717–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov 2017;16:843–62. [DOI] [PubMed] [Google Scholar]
- 63.Verstovsek S, Passamonti F, Rambaldi A, Barosi G, Rosen PJ, Rumi E, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer 2014;120:513–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Al Shehri T, Gilmour K, Gothe F, Loughlin S, Bibi S, Rowan AD, et al. Novel gain-of-function mutation in STAT1 sumoylation site leads to CMC/CID phenotype responsive to ruxolitinib. J Clin Immunol 2019;39:776–85. [DOI] [PubMed] [Google Scholar]
- 65.Chaimowitz NS, Ebenezer SJ, Hanson IC, Anderson M, Forbes LR. STAT1 gain of function, type 1 diabetes, and reversal with JAK inhibition. N Engl J Med 2020; 383:1494–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.