

Abstract
TP53 does not fully comply with the Knudson model [Knudson, A. G., Jr. (1971) Proc. Natl. Acad. Sci. USA 68, 820–823] in that a reduction of constitutional expression of p53 may be sufficient for tumor predisposition . This finding suggests a gene-dosage effect for p53 function. To determine whether TP53 gene dosage affects the transcriptional regulation of target genes, we performed oligonucleotide-array gene expression analysis by using human cells with wild-type p53 (p53 +/+), or with one (p53 +/−), or both (p53 −/−) TP53 alleles disrupted by homologous recombination. We identified 35 genes whose expression is significantly correlated to the dosage of TP53. These genes are involved in a variety of cellular processes including signal transduction, cell adhesion, and transcription regulation. Several of them are involved in neurogenesis and neural crest migration, developmental processes in which p53 is known to play a role. Motif search analysis revealed that of the genes highly expressed in p53 +/+ and +/− cells, several contain a putative p53 consensus binding site (bs), suggesting that they could be directly regulated by p53. Among those genes, we chose CSPG2 (which encodes versican) for further study because it contains a bona fide p53 bs in its first intron and its expression highly correlates with TP53 dosage. By using in vitro and in vivo assays, we showed CSPG2 to be directly transactivated by p53. In conclusion, we developed a strategy to demonstrate that many genes are affected by TP53 gene dosage for their expression. We report several candidate genes as potential downstream targets of p53 in nonstressed cells. Among them, CSPG2 is validated as being directly transactivated by p53. Our method provides a useful tool to elucidate additional mechanisms by which p53 exerts its functions.
The tumor suppressor protein p53 is a transcription factor involved in important cellular processes such as cell cycle checkpoint regulation, DNA damage response and repair, apoptosis and senescence (reviewed in ref. 1). The best characterized biochemical function of p53 is its sequence-specific transactivation activity. A consensus binding site (bs) has been defined and contains two copies of the 10 base pair motif RRRC(A/T)(T/A)GYYY (R = A or G; Y = C or T) separated by 0–13 base pairs (2) and is found in the regulatory regions of target genes. Recent studies using global approaches to identify p53-regulated genes have identified numerous putative target genes that may be directly or indirectly regulated by p53 (3, 4). These studies have revealed heterogeneity of the gene expression changes induced by p53 overexpression or by p53 activation after genotoxic insults, in that the genes transcriptionally regulated by p53 vary depending on the cell type, inducing agent, and amount of p53 in the cells.
p53 −/− mice develop tumors at a very early age and succumb by 10 months of age. Heterozygous mice (p53 +/−) have a 50% chance of developing tumors by 18 months of age, and almost all are dead by 2 years of age (5). About half of the tumors that develop in p53 +/− mice do not show loss of the wild-type (wt) allele (6, 7). The wt allele is retained and encodes a functional protein able to bind DNA and activate transcription of target genes (7). In humans, germ-line TP53 mutations account for 70–85% of classical Li–Fraumeni cancer predisposition syndrome (LFS) cases (8). The LFS tumor spectrum resembles that of p53 +/− mice and it has been shown that in LFS patients only ≈50% of tumors show loss of heterozygosity (LOH) at the TP53 locus (9). A decrease in constitutional expression of p53 should not be sufficient for tumor formation, according to the “two-hit” model for tumorigenesis proposed by Knudson (10). However, the finding that tumors arising in p53 +/− individuals often retain the wt allele does not support this model. Instead, a reduction in p53 level is sufficient to alter the response to genotoxic stress of specific cell types in mice (11) and might be involved in the predisposition to tumor formation in both mice and humans.
We hypothesize that the dosage of TP53 affects its transcriptional regulatory activity in that the amount of p53 in the cell may determine which downstream target genes are activated, or repressed, and the extent of their regulation in the absence of DNA damage. To verify our hypothesis, we analyzed human cancer cells with wt p53 (p53 +/+), or with one (p53 +/−) or both (p53 −/−) TP53 alleles somatically knocked-out by using oligonucleotide-based DNA microarrays. We report here that cells with different TP53 genotypes can indeed be discriminated on the basis of global expression levels. By using statistical analysis and computational motif searches, we identified a subset of genes significantly correlated with TP53 status. Among the genes highly expressed in p53 +/+ cells, but expressed lower in p53 +/− cells, is CSPG2, encoding the extracellular matrix protein versican. We demonstrate that CSPG2 is a direct target of p53. This study demonstrates the applicability of microarray analysis to the identification of subtle changes in gene expression, such as those associated with heterozygous changes in tumor-suppressor-like genes.
Materials and Methods
Cell Lines and Treatments.
The human colorectal carcinoma-derived cell lines with a somatic knock-out of p53 (p53 +/+, p53 +/−, and p53 −/−) (12) were kindly provided by B. Vogelstein (The Johns Hopkins University, Baltimore), and cultivated in McCoy's 5A medium supplemented with 10% FBS (Invitrogen). The Calu6 lung carcinoma, Saos-2 osteosarcoma, and MCF7 breast cancer cell lines were obtained from the American Type Culture Collection (ATCC) and maintained in DMEM/high glucose (4.5 g/liter) with 10% FBS. MCF7-E6 cells (13) were a gift of A. Fornace (National Institutes of Health, Bethesda), and were also grown in DMEM. Growth arrest was achieved by a combination of high cell density and serum starvation (0.25% FBS for 48 h; 0-h time point). Cells were released from growth arrest by replating them in the presence of serum and then harvested 12 h later for mRNA and protein extractions (12-h time point). An aliquot of cells was fixed in ethanol for flow cytometric analysis of DNA content. Cells were infected with recombinant, replication-deficient adenovirus, either containing wt p53 (Ad-wtp53) or empty (Ad), kindly provided by G. Leone, (Ohio State University). Adenoviral vectors were diluted in cell culture medium and infections were performed in the absence of FBS or antibiotics for 24 h at 37°C. For γ-irradiation, cells were treated with 6 Gy of γ-rays by using a cesium-137 source. For UV irradiation, cells were treated with 10 J/m2. Adriamycin (500 ng/ml) and etoposide (500 μM) were added to the media for the indicated time.
Expression Profiling.
Total mRNA was extracted by using Trizol reagent (Invitrogen) and an RNeasy kit (Qiagen, Valencia, CA). cRNA target was prepared as reported (14) and hybridized to the GeneChip HuGeneFL oligonucleotide arrays (Affymetrix, Santa Clara, CA), which contain probes for 6,606 unique genes. The cRNA samples were each hybridized to two GeneChips. Scanned output files were visually inspected for hybridization artifacts and scaled to an average intensity of 1,500 per gene. The expression estimates for each gene were obtained according to the Li–Wong “full” model (15) and were further normalized by using simple quadratic scaling on all genes, with the median expression for each gene as a baseline (16). The raw data are available through the link www.ncbi.nlm.nih.gov/geo/, accession no. GSE90.
TaqMan Real-Time PCR Assay.
Relative expression levels and differences among p53 +/+, p53 +/− and p53 −/− cells were validated by TaqMan real-time PCR (17). The approach is detailed in Supporting Materials and Methods, which is published as supporting information on the PNAS web site, www.pnas.org. Briefly, the comparative CT method was used to determine the ratio of target and 18S rRNA endogenous control.
Statistical Analyses.
All statistical comparisons were performed by using SPLUS 6.0 for the 12 h condition alone. Each gene was examined for concordance with the patterns in Fig. 1b. For Patterns 1 and 4, genes exhibiting Pearson correlation coefficients >0.95 were retained. For the remaining patterns, pairwise t tests were performed comparing the expression of p53 −/− to p53+/−, and p53 +/− to p53+/+. Genes were retained if the t test was nominally significant at P < 0.05 for the two conditions expected to differ (according to the pattern) and P > 0.05 for the two conditions expected not to differ. As single samples of RNA were produced in each condition, the interpretation of the P values is over repeated arrays from the RNA sample. Although our criteria were somewhat arbitrary and numerous multiple comparisons were performed, the intent was to produce a manageable list of candidate genes for further study. The choice of detection threshold relied on published results on the relationship between average expression and standard error (16), as well as the observation that TP53 estimated expression was near the 25th percentile in the p53 −/− condition. The significance of the changes in CSPG2 expression levels identified by real-time PCR was determined by using a Student's t test (one-tailed). Hierarchical cluster analysis was performed as outlined in the supporting information. Briefly, genes passing a variation filter were selected and two-way hierarchical clustering was performed by using the programs cluster and treeview (18).
Fig 1.

Gene expression analysis of p53 +/+, p53 +/−, and p53 −/− cells. (a) Dendrogram showing the similarities in the expression patterns of the samples (also see Fig. 4). (b) Expression patterns within genotypes at the 12-h time point. Schematic representation of the patterns of gene expression defined by significance testing (see Materials and Methods). A detection threshold for gene expression estimates was defined (dotted line).
Algorithm for Motif Searching.
A computer algorithm was developed to identify putative p53 bs at the genome level. A p53 consensus bs consists of two 10-bp half-sites having the consensus sequence 5′-RRRC(A/T)(T/A)GYYY-3′, with spacing in between the two decamers. We used a weight matrix (19) to capture the degeneracy in the consensus sequence; each element of the matrix representing the relative base frequency at a given position in the known bs. The weight matrix has been derived by using a set of 56 experimentally known p53 response elements (http://genemap.med.ohio-state.edu/p53). By using the weight matrix, the algorithm calculates a likelihood score for each half-site. The score ranges from 0 to 1, with 1 representing the optimal p53-binding site. We allowed a spacing of 0–17 bp between the two half-sites. We ran the algorithm on the genomic regions around the first exon of each gene of interest, by using the first exon annotations (http://genemap.med.ohio-state.edu). Additional filters are used to capture only the highly likely candidate p53-responsive genes. It is known that the 4 nucleotides CWWG in each palindrome most closely interact with the p53 protein (20). Hence, we allowed at most 1 mismatch of these 8 nucleotides, and a total of 4 mismatches of the 20 nucleotides.
Plasmid Vector Construction and Luciferase Assays.
The 1.26-kb genomic fragment containing the CSPG2 promoter from nucleotide position −466 bp, exon 1, and the first 511 bp of intron 1 was obtained by PCR, using the following primers: forward, GCT AGC CTC CCG AGA AGA AGT GAT CG; reverse, GCT AGC GAA CAC CAG GCA CTG ACC AC, subcloned into the pGL2-basic vector (Promega) and designated as CSPG2-prom. The fragment not containing the bs was obtained by digesting CSPG2-prom with XhoI to excise the region containing the bs and then religating the vector (CSPG2-promΔbs). Double-stranded oligonucleotides corresponding to the CSPG2 p53-binding site (CSPG2-bs) or a mutated version of it (CSPG2-mut) (Fig. 2a) were subcloned into the pGL2-promoter vector (Promega). The nucleotide sequence of all constructs was confirmed by automated sequencing. Cells were plated in six-well plates (1 × 105 per well) and transfected 24 h later with 0.5 μg of reporter construct, together with 250 ng of pCMV-wtp53 (W) or pCMV-p53–248W (M) expression vector by using the FuGENE 6 reagent (Roche Applied Science, Indianapolis). Normalization of transfection efficiency was obtained by cotransfection with the pCMV-β-gal construct constitutively expressing the β-galactosidase gene. Cells were harvested in reporter lysis buffer (Promega) 48 h after transfection, and luciferase activity was quantified by using a luminometer. Representative results of triplicate measurements of duplicate experiments with mean and standard deviation are shown in the figures.
Fig 2.

CSPG2 contains a functional p53 bs. (a) Schematic representation of the reporter gene constructs used. The two decamers that constitute the identified p53 bs in CSPG2 intron 1 are underlined. SV, minimal promoter. (b) Activation of the CSPG2-prom construct by wt, but not mutant, p53. pGL2-P, CSPG2-prom, or CSPG2-promΔbs were cotransfected, along with a construct encoding wt p53 (W), mutant p53–248W (M), or pCMV empty expression vector (C) in Saos-2 cells. (c) Saos-2 cells were cotransfected with CSPG2-bs, CSPG2-mut, or pGL2-P empty vector, and W, M, or C. wt, but not mutant, p53 activates transcription of a heterologous promoter in the presence of the CSPG2 p53 bs. Mutations in the CSPG2 p53 bs abolish this activation. (d and e) MCF7 and MCF7-E6 cells were transfected with the constructs in a. When CSPG2-prom was transfected into MCF7-E6 cells (p53 null), no increase in the luciferase activity was observed. In contrast, transfection of this construct into MCF7 cells showed a marked increase in luciferase activity. Similar results were observed when transfecting CSPG2-bs. For b–e, luciferase activity is shown as relative light units (RLU), and for Saos2 cells it is shown as fold induction relative to the activity of the reporter vector in the absence of p53. (f) EMSA was done by using radiolabeled CSPG2-bs, CSPG2-mut, and p53R2 oligonucleotides with nuclear extracts from p53 +/+ cells treated with 500 μM etoposide.
Electrophoretic Mobility-Shift Assay (EMSA).
The double-stranded oligonucleotides CSPG2-bs, CSPG2-mut, and the p53R2 p53 bs (p53R2) (21) were end-labeled with polynucleotide kinase by using [γ-32P]ATP. p53 +/+ cells were treated with 500 μM etoposide for 48 h. Nuclear extracts were prepared by the high-salt extraction of nuclei (22). Binding reactions with labeled oligonucleotides and nuclear extracts were performed essentially as described (23). One microgram of monoclonal antibody against p53, Pab421, or Pab1801 (Santa Cruz Biotechnology), was included in the binding reaction where indicated. The reaction mix was incubated at room temperature for 15 min. Labeled oligomers (0.5 ng) and competitors (100×) were added and incubated at 30°C for 15 min. DNA-protein complexes were resolved on 4% polyacrylamide gels in 0.5X TBE buffer. The gels were dried and autoradiographed.
Western Blotting.
Cell lysates were prepared as reported (24) and 40 μg of total protein was resolved by SDS/PAGE (4–12% gradient gels, Bio-Rad). Primary antibodies (DO-1 for p53, F-5 for p21WAF1, and D-10 for β-tubulin) were from Santa Cruz Biotechnology. Equal loading of proteins was monitored by hybridizing the same membrane with β-tubulin antibody.
Results and Discussion
Global Expression Profiling of Isogenic Cell Lines with Different TP53 Status.
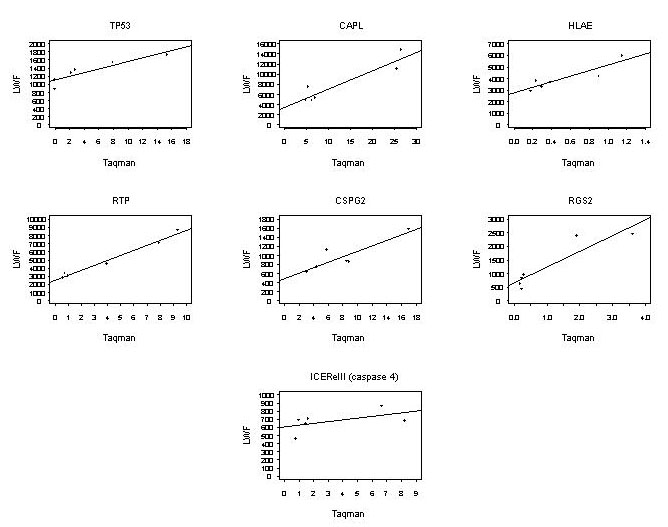
The HCT116 colorectal cancer cell line containing wt p53 was previously used for targeted homologous recombination of the TP53 alleles (12). p53 +/+, p53 +/−, and p53 −/− cells were growth-arrested (0-h), then released (12-h), and their mRNA was studied by means of global gene expression. Duplicate hybridizations of a single cRNA sample from each condition resulted in the mean correlation of gene expression of r = 0.983 across all genes, compared with r = 0.856 for nonduplicate arrays. Validation of microarray gene expression results was carried out by TaqMan real-time PCR for seven genes (see Fig. 3, which is published as supporting information on the PNAS web site). When we performed hierarchical clustering using >5,000 genes that passed our filtering criteria, the three cell lines clustered in separate branches in the dendrogram that summarizes the similarities in gene expression patterns among samples (Fig. 1a and Fig. 4, which is published as supporting information on the PNAS web site). This result supports the hypothesis that cells differing in the number of functional TP53 alleles can be distinguished, based on gene expression.
Effect of TP53 Gene Dosage on Gene Expression.
To select the genes of greatest interest, we defined six expression patterns for genes that are differentially expressed among the cell lines (Fig. 1b). The patterns correspond to what might be described as codominant, dominant, and recessive effects of wt TP53 alleles on gene expression. It is notable that TP53 itself showed the highest correlation coefficient with TP53 status of any gene on the array (pattern 1 in Fig. 1b). We further validated TP53 expression profiles by using real-time PCR (Fig. 3), and the correlation between TaqMan and GeneChip expression estimates for TP53 was 0.9. For each pattern, lists of genes were produced in significance testing as described in the Materials and Methods. To increase our chances of discovering new p53 targets, we further restricted our study to genes that had essentially undetectable expression in one or more of the p53 conditions as presumed by the genetic model (illustrated in Fig. 1b). We used a detection threshold of the 25th percentile for all expression estimates, based on previous results (16). The combination of significance testing and detection threshold for the 12-h time point resulted in a list of 35 genes of interest (Table 1), representing only 0.5% of the genes on the array. With such an abbreviated list it is feasible to perform careful computational identification of p53 bs, as described below. It is evident from Table 1, that in nonstressed cells, TP53 gene dosage affects the transcription of a plethora of genes involved in many cellular functions including signal transduction, cell adhesion, and transcription regulation. The majority of the published transcriptional targets of p53, except p21WAF1 and MDM2 (see Table 1), did not show a high correlation with TP53 status and were not significantly differentially expressed among the three cell lines. These genes were identified as p53-responsive genes on ectopic overexpression of p53 or on treatment with DNA damaging agents. Our experimental system compares the constitutional levels of gene expression in cells that differ for the number of wt TP53 alleles without external perturbation, and therefore the results we obtained are not entirely surprising.
Table 1.
List of 35 genes selected using the criteria illustrated in Fig. 1b
| Probe set ID | Gene symbol | Gene ontology bio process, cell component, molecular function | Correlation | P value |
|---|---|---|---|---|
| Pattern 1 (6 genes) | ||||
| M22898_at | TP53 | Apoptosis, cell cycle checkpoint, DNA repair, transcription factor, tumor suppressor | 0.9858 | 0.0003 |
| K02766_at | C9 | Complement component, hemolysin | 0.9315 | 0.0069 |
| U44429_at | TPD52L1 | Oncogenesis | 0.9113 | 0.0114 |
| M16987_at | F5 | Blood coagulation | 0.8894 | 0.0177 |
| M64358_at | RHOM3A | Unknown function | 0.888 | 0.0181 |
| M26061_at | PDE6A | Vision, cGMP-specific phosphodiesterase | 0.8653 | 0.026 |
| Pattern 2 (1 gene) | ||||
| X82539_at | MAGEB1 | Tumor antigen | 0.0416 | |
| Pattern 3 (21 genes) | ||||
| U16306_at | CSPG2 | Cell adhesion, developmental processes, cell recognition | 0.0002 | |
| M96843_at | ID2B | Dominant-negative helix–loop–helix transcription factor, DNA binding, protein binding, developmental processes | 0.0041 | |
| L25798_at | HMGCS1 | Lipid metabolism | 0.0085 | |
| X57025_at | IGF1 | Signal transduction, positive control of cell proliferation, cell motility | 0.0135 | |
| D82345_at | TMSNB | Unknown function | 0.0164 | |
| U12535_at | EPS8 | Signal transduction, cell proliferation, EGF receptor signaling pathway, SH3/SH2 adaptor protein | 0.0169 | |
| Z25521_s_at | CD47 | Signal transduction | 0.0199 | |
| L22075_at | GNA13 | Signal transduction, cell motility, heterotrimeric G-protein GTPase, α subunit | 0.0237 | |
| U18242_at | CAMLG | Signal transduction, defense response | 0.0243 | |
| L19871_at | ATF3 | Cell-matrix adhesion, integrin-mediated signaling pathway, oncogenesis, transcription corepressor | 0.0305 | |
| S41458_at | PDE6B | Phototransduction, visible light, vision, cGMP-specific phosphodiesterase | 0.0316 | |
| M86852_at | PXMP3 | Protein-peroxisome, targeting peroxisome, organization, and biogenesis | 0.034 | |
| Y11897_at | CHIC1 | Unknown function | 0.0359 | |
| U44772_at | PPT1 | Neurogenesis | 0.036 | |
| U63295_at | SIAH1 | Axon guidance, embryogenesis and morphogenesis, apoptosis, neurogenesis | 0.0378 | |
| X05997_at | LIPF | Hydrolase, lipid degradation, glycoprotein | 0.0387 | |
| X66087_at | MYBL1 | Transcription regulation from Pol II promoter, transcription-activating factor | 0.04 | |
| Y08991_at | PIK3R4 | Nonselective vesicle transport enzyme activator | 0.0409 | |
| Z46788_at | CYLC2 | Cell shape, cell size control, cytoskeletal structural protein | 0.0434 | |
| Z15108_at | PIKCZ | Antiapoptosis, signal transduction, protein phosphorylation, protein kinase | 0.0436 | |
| D87127_at | TLOC1 | Cotranslational membrane targeting | 0.0483 | |
| Pattern 4 (3 genes) | ||||
| AB000220_at | SEMA3C | Axon guidance, cell growth and maintenance, immune response, drug resistance | −0.9711 | 0.0012 |
| M60047_at | HBP17 | Signal transduction, negative control of cell proliferation, cell–cell signaling, heparin binding | −0.8764 | 0.022 |
| X02874_at | OAS1 | Nucleobase, nucleoside, nucleotide, and nucleic acid metabolism | −0.8326 | 0.0397 |
| Pattern 5 (2 genes) | ||||
| D38491_at | KIAA0117 | RNA-binding protein | 0.0019 | |
| U11313_at | SCP2 | Acyl-CoA metabolism, peroxisome organization and biogenesis, sterol carrier protein X-related thiolase | 0.0085 | |
| Pattern 6 (2 genes) | ||||
| U67988_at | DLGAP1 | Synaptic transmission protein binding | 0.0089 | |
| U79289_at | LOC148357 | Unknown function | 0.0094 | |
| Known targets | ||||
| U33202_s_at | MDM2 | Negative control of cell proliferation oncogenesis | 0.8824 | 0.0199 |
| U33203_s_at | MDM2 | Negative control of cell proliferation oncogenesis | 0.8821 | 0.02 |
| U09579_at | CDKN1A | Induction of apoptosis, cell cycle arrest, negative control of cell proliferation, regulation of CDK activity | 0.8785 | 0.0213 |
Gene annotation based on the GeneOntology Consortium (www.geneontology.org) and Affymetrix (www.affymetrix.com/analysis/index.affx).
Pearson's correlation coefficient for correlation with p53 status.
Genes with P values <0.05 were considered. Known p53 target genes that show a high correlation with p53 status but did not meet the threshold criteria are reported at the bottom.
Functional Analysis of the Genes Affected by the Dosage of TP53.
As shown in Table 1, most of the genes we identified as being differentially expressed among the three cell lines belong to expression pattern 3 (21 genes). This finding suggests that whether there are one or two functional TP53 alleles makes a difference in the ability to regulate gene expression, perhaps because p53 needs to reach a threshold to properly regulate some of its transcriptional targets. Note that 11 of 21 genes in pattern 3 belong to the cluster of genes highly expressed in p53 +/+ cells when hierarchical clustering is performed (Fig. 4a). Of the genes in pattern 3, it is interesting to highlight SIAH1. This gene is the human homologue of the Drosophila seven in absentia (Sina) gene and is a putative tumor suppressor gene (25). SIAH1 is up-regulated during the first few hours of apoptosis induced by wt p53 (26), thereby supporting our finding that SIAH1 transcriptional regulation may depend on the amount of p53. Several of the genes listed in Table 1 play a role in neurogenesis or neural crest migration, or are highly expressed in the nervous system, such as PPT1, CSPG2, Id2, SEMA3C, IGF1, and CHIC1.
In p53-deficient embryos, a subset presents with a spectrum of developmental defects, including failure in neural tube closure, which leads to exencephaly and embryonic death, and craniofacial abnormalities (27, 28). This result suggests that p53 may play a role in neurogenesis, but there is functional redundancy and other genes can compensate for its absence. Transgenic mouse models having a reporter gene under the control of a p53-regulated promoter, such as the mdm2 promoter, have identified high transcriptional activity of p53 in the brain and haploinsufficiency in the level of p53-mediated response to γ-irradiation of heterozygous embryos (29). These and our data support the hypothesis that the dose of TP53 affects the transcriptional regulation of downstream genes. Some of these genes require two intact TP53 alleles, or two mutant alleles, for their expression in nonstressed cells.
Identification of Genes Directly Regulated by p53.
Our experimental approach allowed us to identify genes whose transcriptional regulation is affected by TP53 gene dosage. Of those genes, some might be directly regulated by p53 through binding to a p53 bs. We selected the genes whose expression level was increased in the presence of p53 (patterns 1–3) and analyzed 10 kb of the genomic sequence around the first exon of each of these genes (3 kb upstream and about 7 kb downstream to exon 1) for the presence of a consensus p53 bs by using a similarity matrix. For each putative p53 bs we obtained a score as a measure of its similarity to the published consensus bs: the higher the score for a particular sequence, the closer to the published consensus (optimal consensus score = 1). No more than four mismatches to the canonical binding site were allowed. The efficiency of the computational analysis was evaluated by using previously identified functional p53 bs. The p53 bs of MDM2, p21WAF1, IGFBP3, and GADD45 were recognized by the algorithm (data not shown). Of the 27 genes analyzed (excluding TP53), we found 10 that contained promising putative p53-binding sites (Table 3, which is published as supporting information on the PNAS web site). The genes with the highest summed score (and fewer mismatches) were CSPG2 and GNA13. The CSPG2 putative p53 response element is located in the 5′ portion of the first intron, 424 bp from the end of the untranslated exon 1. MDM2 and IGFBP3, two known p53 target genes, also possess a functional p53 bs in intron 1 (30, 31) The putative p53 bs of GNA13 is about 2.2 kb upstream of the transcriptional start site, similar to the position of the p53 response element in the p21WAF1 gene promoter (32). CSPG2 was the most highly significant gene in pattern 3, and showed the second-highest similarity score in Table 3. This gene thus emerged as the clearest candidate gene to validate experimentally. We performed TaqMan real-time PCR using the same mRNA that had been used for the microarray analysis to confirm the TP53 dose-dependent regulation of CSPG2 expression. The results agreed well with the microarray data (Fig. 3). The use of the heterozygous cells, in addition to p53 +/+ and p53 −/− cells, allowed us to identify the TP53 dose-dependent regulation of CSPG2 expression.
p53 Activates the Transcription of the CSPG2 Gene in Reporter Gene Assays.
To verify that the identified CSPG2 p53 bs is functional, we performed reporter gene assays. The CSPG2 promoter has been characterized and the region from −466 to +244 relative to the transcription start site in exon 1 shows maximal promoter activity in reporter gene assays (33). We cloned a 1.26-kb fragment, comprising the genomic region from −466 to the putative CSPG2 p53 bs in intron 1, upstream to a promoterless luciferase reporter gene (pGL2-basic vector) to obtain the construct designated CSPG2-prom (Fig. 2a). A similar construct without the p53 bs was also made (CSPG2-promΔbs) (not shown). Luciferase assays showed that the CSPG2-prom construct was activated more than 200-fold by cotransfection with wt p53 (Wp53) in Saos-2 p53-null cells compared with the control plasmid (pGL2-P) (Fig. 2b). Mutant p53 did not activate CSPG2-prom. The CSPG2-promΔbs construct was not activated by wt p53, indicating that the identified CSPG2 p53 bs is required for the p53-dependent activation of the reporter gene. To confirm that the identified putative p53 bs mediates the p53-induced transactivation of CSPG2, a copy of the 21-bp CSPG2 response element (CSPG2-bs) or a mutated oligonucleotide (CSPG2-mut) was cloned upstream to a luciferase gene, preceded by the minimal promoter element of the pGL2-promoter vector (Fig. 2a). Cotransfection of the CSPG2-bs construct and the wt p53 expression vector in Saos2 cells increased promoter activity by more than 15-fold (Fig. 2c). Mutation of the putative CSPG2 p53-binding site (CSPG2-mut) abolished transactivation by p53. No activation was observed on cotransfection of the mutant p53 construct.
We also cotransfected the constructs in Fig. 2a into MCF7 cells, harboring wt p53, and into MCF7-E6 cells constitutively expressing the E6 protein from human papilloma virus (HPV) type 16, and therefore p53-null. Transfection of CSPG2-prom in MCF7 cells gives high luciferase activity compared with the pGL2-P empty vector, because of the endogenous wt p53 (Fig. 2d). MCF7-E6 cells fail to activate the CSPG2-prom construct. Transfection of CSPG2-promΔbs gives a level of luciferase activity comparable to pGL2-P in both cell lines. Luciferase activity is dramatically increased on the transfection of CSPG2-bs into MCF7, but not in MCF7-E6 cells compared with vector control (Fig. 2e). In conclusion, the CSPG2 p53 bs is a functional p53 response element.
p53 Protein Binds to the CSPG2 Intron 1 Response Element in Vitro.
To directly show the interaction of p53 protein and the CSPG2 p53 bs, an EMSA was performed. The 21-bp p53-binding site in CSPG2-bs was used as a probe (Fig. 2a) and nuclear extracts from p53 +/+ cells treated with etoposide (which increases the level of p53 protein) were used in the binding reaction. We used CSPG2-mut as a negative control probe and an oligonucleotide corresponding to the p53 bs in the p53R2 gene (p53R2) (21) as a positive control. The mobility of the labeled CSPG2-bs probe was shifted in the presence of nuclear extracts containing activated p53 (Fig. 2f, lane 6) and formed a binding complex with p53 itself. The binding complex was diminished by increasing amounts of self-competitor (lane 12) as well as by consensus p53 bs (lane 11), but not by the nonspecific competitor CSPG2-mut (lane 13). No shifted bands were observed when the CSPG2-mut oligonucleotide probe was used. Supershift of the band observed in the presence of the CSPG2-bs oligonucleotide when anti-p53 monoclonal antibodies Pab421 and Pab1801 were used demonstrates that p53 specifically binds the CSPG2-bs sequence.
Induction of Endogenous CSPG2 Gene Expression by p53.
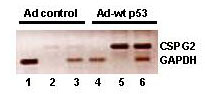
To determine whether the endogenous level of CSPG2 is affected by p53, we infected p53-null Calu6 cells with adenoviral constructs. While on infection of the empty vector (Ad) we observed no activation of CSPG2 expression, on infection of a vector overexpressing wt p53 (Ad-p53) we observed a substantial increase of endogenous CSPG2 expression (Fig. 5, which is published as supporting information on the PNAS web site). These experiments demonstrate a p53-dependent induction of CSPG2 in Calu6 cells. Infected cells express p53 at levels likely to be higher than those encountered in vivo. To assess CSPG2 transcriptional activation at more physiologic levels of p53, we treated cells with DNA-damaging agents known to stabilize p53. We treated p53 +/+, p53 +/−, and p53 −/− cells with radiation (UV and γ) and with the drugs Adriamycin and etoposide. The results obtained by TaqMan real-time PCR show that CSPG2 expression is induced in a p53-dependent fashion on treatment with γ-rays or Adriamycin (Table 2). No induction of CSPG2 expression was observed after UV or etoposide treatment (data not shown). To verify that in our experimental conditions p53 was stabilized and also active in its transactivation ability, we assessed the induction of p21WAF1 in the same samples by Western blotting. In all of the conditions used, p53 was able to transactivate p21WAF1 (data not shown). Therefore, we conclude that CSPG2 expression is induced in a p53-dependent manner after specific genotoxic stress.
Table 2.
Relative expression level of CSPG2
| Sample | Relative expression level | P |
|---|---|---|
| p53 +/+ untreated | 1.00 ± 0.043 | |
| p53 +/+ adriamycin 24 h | 4.315 ± 0.501 | 0.0002 |
| p53 +/− untreated | 1.766 ± 0.262 | |
| p53 +/− adriamycin 24 h | 2.326 ± 0.418 | 0.0602 |
| p53 −/− untreated | 1.181 ± 0.138 | |
| p53 −/− adriamycin 24 h | 1.129 ± 0.154 | 0.3452 |
| p53 +/+ untreated | 1.05 ± 0.367 | |
| p53 +/+ 6 Gy 6 h | 1.059 ± 0.258 | 0.4873 |
| p53 +/+ 6 Gy 12 h | 2.313 ± 0.142 | 0.0026 |
| p53 +/+ 6 Gy 24 h | 5.044 ± 0.586 | 0.0003 |
Cells were treated with 500 ng/ml adriamycin or γ-irradiation (6 Gy) and mRNA was isolated at the indicated times after treatment.
Values represent the means of two independent experiments in triplicate ± standard deviation, which were standardized by using the 18S rRNA internal control.
P values were calculated by Student's t test comparing treated and untreated samples.
Conclusions
Gene expression array analysis allowed us to compile a list of genes regulated by TP53 gene dosage in the absence of cellular stress. Among them, we found genes involved in a variety of cellular processes. In particular, we noted the transcriptional regulation of genes encoding proteins highly expressed in the nervous system, involved in neural crest cell migration and neural tube closure. p53 plays a role in neurogenesis, as indicated by the fact that a subset of p53-deficient mice show defects in neural tube closure and other abnormalities (27, 28). p53 is not required to promote the normal developmental program, but may be involved in fine tuning the transcriptional regulation of genes required for neural crest cell migration, developmentally regulated apoptosis, or other cellular mechanisms. It is therefore possible that some of the genes listed in Table 1 mediate important developmentally related functions of p53.
Recently, it has been shown that a decrease in the constitutional expression level of the APC gene predisposes to familial adenomatous polyposis (34), demonstrating that small quantitative changes in gene expression are involved in complex human diseases. A reduced level of p53 predisposes to tumor formation in mice (6, 7) and possibly in humans. The fact that the transcriptional regulation of a subset of genes, including CSPG2, SIAH1, and others, seems to require a double dosage of wt TP53 suggests that these downstream targets could be involved in tumor predisposition.
By using a motif identification approach, we identified potential direct targets of p53. We demonstrated that CSPG2, a gene highly expressed in p53 +/+ but expressed lower in p53 +/− cells, is a direct target of p53. CSPG2/versican is an anti-cell adhesive molecule that negatively regulates neural crest cell migration and axon outgrowth (35). High expression of CSPG2 has been found in the peritumoral stromal tissue of early stage prostate cancers, and of breast cancers, and it is associated with an aggressive tumor behavior (36, 37). Also, CSPG2 is silenced in colorectal cancer because of promoter hypermethylation (38, 39), suggesting that, depending on the tissue, this protein may either inhibit or stimulate cell motility and local invasion. In colorectal cancer, CSPG2 methylation is an early event possibly involved in the predisposition to tumorigenesis or its progression (39). Our finding that the dosage of TP53 affects CSPG2 expression supports the hypothesis of such a role for this gene.
Our analysis of nonstressed cells highlights additional regulatory pathways that may be of importance to the understanding of p53 function in the normal cycle of cells. Furthermore, the knock-out design offers an opportunity to assess the requirement for one or two (haploinsufficiency) functional alleles of TP53 to mediate the transcriptional regulation of specific genes. The fascinating possibility that some of the genes we identified might be involved in the predisposition to tumor formation warrants further studies.
Supplementary Material
Acknowledgments
We are grateful to Dr. B. Vogelstein, Dr. A. Fornace, Dr. A. Wani, and Dr. G. Leone for providing cell lines and reagents, Dr. R. Opavski for help with the β-galactosidase assays, and a reviewer who provided important suggestions. This work was supported in part by Grant P30 CA16058 from the National Cancer Institute, Bethesda.
Abbreviations
bs, binding site
wt, wild type
EMSA, electrophoretic mobility-shift assay
References
- 1.Bargonetti J. & Manfredi, J. J. (2002) Curr. Opin. Oncol. 14 86-91. [DOI] [PubMed] [Google Scholar]
- 2.el-Deiry W. S., Kern, S. E., Pietenpol, J. A., Kinzler, K. W. & Vogelstein, B. (1992) Nat. Genet. 1 45-49. [DOI] [PubMed] [Google Scholar]
- 3.Zhao R., Gish, K., Murphy, M., Yin, Y., Notterman, D., Hoffman, W. H., Tom, E., Mack, D. H. & Levine, A. J. (2000) Genes Dev. 14 981-993. [PMC free article] [PubMed] [Google Scholar]
- 4.Yu J., Zhang, L., Hwang, P. M., Rago, C., Kinzler, K. W. & Vogelstein, B. (1999) Proc. Natl. Acad. Sci. USA 96 14517-14522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donehower L. A., Harvey, M., Slagle, B. L., McArthur, M. J., Montgomery, C. A., Jr., Butel, J. S. & Bradley, A. (1992) Nature 356 215-221. [DOI] [PubMed] [Google Scholar]
- 6.Harvey M., McArthur, M. J., Montgomery, C. A., Jr., Bradley, A. & Donehower, L. A. (1993) FASEB J. 7 938-943. [DOI] [PubMed] [Google Scholar]
- 7.Venkatachalam S., Shi, Y. P., Jones, S. N., Vogel, H., Bradley, A., Pinkel, D. & Donehower, L. A. (1998) EMBO J. 17 4657-4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malkin D., Li, F. P., Strong, L. C., Fraumeni, J. F., Jr., Nelson, C. E., Kim, D. H., Kassel, J., Gryka, M. A., Bischoff, F. Z., Tainsky, M. A., et al. (1990) Science 250 1233-1238. [DOI] [PubMed] [Google Scholar]
- 9.Varley J. M., Thorncroft, M., McGown, G., Appleby, J., Kelsey, A. M., Tricker, K. J., Evans, D. G. & Birch, J. M. (1997) Oncogene 14 865-871. [DOI] [PubMed] [Google Scholar]
- 10.Knudson A. G. (1971) Proc. Natl. Acad. Sci. USA 68 820-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clarke A. R., Purdie, C. A., Harrison, D. J., Morris, R. G., Bird, C. C., Hooper, M. L. & Wyllie, A. H. (1993) Nature 362 849-852. [DOI] [PubMed] [Google Scholar]
- 12.Bunz F., Dutriaux, A., Lengauer, C., Waldman, T., Zhou, S., Brown, J. P., Sedivy, J. M., Kinzler, K. W. & Vogelstein, B. (1998) Science 282 1497-1501. [DOI] [PubMed] [Google Scholar]
- 13.Fan S., Smith, M. L., Rivet, D. J., Jr., Duba, D., Zhan, Q., Kohn, K. W., Fornace, A. J., II & O'Connor, P. M. (1995) Cancer Res. 55 1649-1654. [PubMed] [Google Scholar]
- 14.Virtaneva K., Wright, F. A., Tanner, S. M., Yuan, B., Lemon, W. J., Caligiuri, M. A., Bloomfield, C. D., de la Chapelle, A. & Krahe, R. (2001) Proc. Natl. Acad. Sci. USA 98 1124-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li C. & Wong, W. H. (2001) Proc. Natl. Acad. Sci. USA 98 31-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemon, W. J., Palatini, J. J. T., Krahe, R. & Wright, F. A. (2002) Bioinformatics 18, in press. [DOI] [PubMed]
- 17.Heid C. A., Stevens, J., Livak, K. J. & Williams, P. M. (1996) Genome Res. 6 986-994. [DOI] [PubMed] [Google Scholar]
- 18.Eisen M. B., Spellman, P. T., Brown, P. O. & Botstein, D. (1998) Proc. Natl. Acad. Sci. USA 95 14863-14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stormo G. D. & Fields, D. S. (1998) Trends Biochem. Sci. 23 109-113. [DOI] [PubMed] [Google Scholar]
- 20.Cho Y., Gorina, S., Jeffrey, P. D. & Pavletich, N. P. (1994) Science 265 346-355. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka H., Arakawa, H., Yamaguchi, T., Shiraishi, K., Fukuda, S., Matsui, K., Takei, Y. & Nakamura, Y. (2000) Nature 404 42-49. [DOI] [PubMed] [Google Scholar]
- 22.Dignam J. D., Lebovitz, R. M. & Roeder, R. G. (1983) Nucleic Acids Res. 11 1475-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nevins J., DeGregori, J., Jakoi, L. & Leone, G. (1997) Methods Enzymol. 283 205-219. [DOI] [PubMed] [Google Scholar]
- 24.Pellegata N., Cajot, J. & Stanbridge, E. (1995) Oncogene 11 337-349. [PubMed] [Google Scholar]
- 25.Medhioub M., Vaury, C., Hamelin, R. & Thomas, G. (2000) Int. J. Cancer 87 794-797. [PubMed] [Google Scholar]
- 26.Amson R. B., Nemani, M., Roperch, J. P., Israeli, D., Bougueleret, L., Le Gall, I., Medhioub, M., Linares-Cruz, G., Lethrosne, F., Pasturaud, P., et al. (1996) Proc. Natl. Acad. Sci. USA 93 3953-3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sah V. P., Attardi, L. D., Mulligan, G. J., Williams, B. O., Bronson, R. T. & Jacks, T. (1995) Nat. Genet. 10 175-180. [DOI] [PubMed] [Google Scholar]
- 28.Armstrong J. F., Kaufman, M. H., Harrison, D. J. & Clarke, A. R. (1995) Curr. Biol. 5 931-936. [DOI] [PubMed] [Google Scholar]
- 29.Gottlieb E., Haffner, R., King, A., Asher, G., Gruss, P., Lonai, P. & Oren, M. (1997) EMBO J. 16 1381-1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Juven T., Barak, Y., Zauberman, A., George, D. L. & Oren, M. (1993) Oncogene 8 3411-3416. [PubMed] [Google Scholar]
- 31.Bourdon J. C., Deguin-Chambon, V., Lelong, J. C., Dessen, P., May, P., Debuire, B. & May, E. (1997) Oncogene 14 85-94. [DOI] [PubMed] [Google Scholar]
- 32.el-Deiry W. S., Tokino, T., Velculescu, V. E., Levy, D. B., Parsons, R., Trent, J. M., Lin, D., Mercer, W. E., Kinzler, K. W. & Vogelstein, B. (1993) Cell 75 817-825. [DOI] [PubMed] [Google Scholar]
- 33.Naso M. F., Zimmermann, D. R. & Iozzo, R. V. (1994) J. Biol. Chem. 269 32999-33008. [PubMed] [Google Scholar]
- 34.Yan H., Dobbie, Z., Gruber, S. B., Markowitz, S., Romans, K., Giardiello, F. M., Kinzler, K. W. & Vogelstein, B. (2002) Nat. Genet. 30 25-26. [DOI] [PubMed] [Google Scholar]
- 35.Landolt R. M., Vaughan, L., Winterhalter, K. H. & Zimmermann, D. R. (1995) Development (Cambridge, U.K.) 121 2303-2312. [DOI] [PubMed] [Google Scholar]
- 36.Ricciardelli C., Mayne, K., Sykes, P. J., Raymond, W. A., McCaul, K., Marshall, V. R., Tilley, W. D., Skinner, J. M. & Horsfall, D. J. (1997) Clin. Cancer Res. 3 983-992. [PubMed] [Google Scholar]
- 37.Ricciardelli C., Brooks, J. H., Suwiwat, S., Sakko, A. J., Mayne, K., Raymond, W. A., Seshadri, R., LeBaron, R. G. & Horsfall, D. J. (2002) Clin. Cancer Res. 8 1054-1060. [PubMed] [Google Scholar]
- 38.Toyota M., Ho, C., Ahuja, N., Jair, K. W., Li, Q., Ohe-Toyota, M., Baylin, S. B. & Issa, J. P. (1999) Cancer Res. 59 2307-2312. [PubMed] [Google Scholar]
- 39.Issa J. P., Ahuja, N., Toyota, M., Bronner, M. P. & Brentnall, T. A. (2001) Cancer Res. 61 3573-3577. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}