

Abstract
Transgenic (Tg) mouse lines that express chimeric mouse–human prion protein (PrP), designated MHu2M, are susceptible to prions from patients with sporadic Creutzfeldt–Jakob disease (sCJD). With the aim of decreasing the incubation time to fewer than 200 days, we constructed transgenes in which one or more of the nine human residues in MHu2M were changed to mouse. The construct with murine residues at positions 165 and 167 was expressed in Tg(MHu2M,M165V,E167Q) mice and resulted in shortening the incubation time to ≈110 days for prions from sCJD patients. The construct with a murine residue at position 96 resulted in lengthening the incubation time to more than 280 days for sCJD prions. When murine residues 96, 165, and 167 were expressed, the abbreviated incubation times for sCJD prions were abolished. Variant CJD prions showed prolonged incubation times between 300 and 700 days in Tg(MHu2M) mice on first passage and incubation times of ≈350 days in Tg(MHu2M,M165V,E167Q) mice. On second and third passages of variant CJD prions in Tg(MHu2M) mice, multiple strains of prions were detected based on incubation times and the sizes of the protease-resistant, deglycosylated PrPSc fragments. Our discovery of a previously undescribed chimeric transgene with abbreviated incubation times for sCJD prions should facilitate studies on the prion species barrier and human prion diversity.
Human prion diseases include Creutzfeldt–Jakob disease (CJD), kuru, and fatal insomnia (FI; ref. 1). Sporadic (s) CJD accounts for ≈85% of all cases of human prion disease, familial (f) CJD for 10–15%, and infection from exogenous prions for <1% (2). Prions consist solely of a pathogenic prion protein, denoted PrPSc, that is derived from the cellular isoform PrPC (1, 3). Replication of prions occurs when PrPSc stimulates conversion of PrPC into nacent PrPSc. It seems likely that a cofactor participates in the formation of PrPSc (4, 5); one such cofactor, provisionally designated protein X, has been invoked to explain dominant-negative inhibition of prion replication. Using scrapie-infected neuroblastoma (ScN2a) cells, the putative binding site of protein X on PrP was mapped (6).
Transmission of human prions to wild-type (wt) rodents was difficult because of the “species barrier” (7) that appears to be modulated by (i) the degree of homology between PrP sequences (8–10), (ii) cofactors such as protein X, and (iii) the prion strain. Although incubation times of ≈200 days in transgenic (Tg) mice greatly accelerated studies of human prion disease, the prolonged duration has hampered many studies (4, 11, 12).
Within a single species, strains of prions stably transmit a particular disease phenotype, characterized by the length of the incubation time, clinical signs as well as patterns of PrPSc deposition and neuropathological lesions (13, 14). A growing body of evidence argues that strain-specific properties of prions are enciphered in the conformation of PrPSc (15–19). Strains of sCJD prions have been classified according to the amino acid at position 129 [methionine (M) or valine (V)] and with respect to the electrophoretic gel mobility (21 or 19 kDa, as type 1 or type 2, respectively) of the deglycosylated, protease-resistant (r) PrPSc fragment (20). Employing this analysis, 70% of sCJD cases are either MM1 or MV1, and 24% are either VV2 or MV2. In addition to the sCJD strains, a new strain of CJD prions, termed variant CJD (vCJD), has been found (21). This strain is thought to have originated in cattle with bovine spongiform encephalopathy (BSE) and was transmitted to humans through ingestion of BSE-tainted beef products (18, 22, 23).
To define the molecular basis of the species barrier and to produce mice with shorter incubation times, we mutagenized the chimeric mouse–human (MHu2M) transgene. Substitution of mouse residues at positions 165 and 167 in the Hu2 insert reduced the incubation time by nearly 50% for sCJD and fCJD prions. Whether these substitutions shorten the incubation time by optimizing the interaction of the transgene product with protein X is unknown. Investigations of vCJD prions serially passaged in Tg(MHu2M) mice demonstrated the existence of multiple strains.
Methods
Production of Tg Mice.
In MHu2M, the N and C termini are from mouse PrP and the central “Hu2” insert corresponding to residues 96–167, is from human PrP (11). Tg mice were produced as described (8, 24), except that purified cos.SHa.Tet fragments containing the respective constructs were microinjected into the pronuclei of fertilized oocytes from FVB/Prnp0/0 mice (25). Constructs were cloned by using mismatch primer mutagenesis (8). Genomic DNA isolated from weanling animals was screened for the incorporation of the transgene by using probes hybridizing to the 3′ UTR of the cos.SHa.Tet vector (1). Tg(HuPrP,M129)440/Prnp0/0 mice express HuPrPC with M at 129 and at 2× the level found in human brain. Tg(HuPrP,V129)152/Prnp0/0 mice express HuPrPC with V at 129 and at 4–8× the level found in the human brain. In Tg(MHu2M)5378/Prnp0/0 mice, PrPC is expressed at 2× the level found in Syrian hamster brain.
Human Brain Inocula.
Patient diagnoses were confirmed by histopathology, immunohistochemistry, and detection of human PrPSc by Western blotting (20, 26). Patients were genotyped (4) by DNA sequencing of the entire ORF after extracting genomic DNA either from leukocytes collected during life or from frozen brain after death.
Preparation of Brain Homogenates and Bioassays.
Ten percent homogenates of sCJD human brains or Tg mouse brains were prepared as described (4). Bioassays were performed as described (27). After onset of disease symptoms, mice were killed and their brains were immediately frozen or fixed in formalin. For neuropathology, brains were immersion-fixed in a 10% buffered formalin solution and embedded in paraffin. Immunohistochemistry for PrP on formalin-fixed, paraffin-embedded tissue sections was performed using the hydrolytic autoclaving technique (28). Histoblot analysis was performed as described (29).
Results
Tg Mice Expressing MHu2M PrP.
sCJD transmits to Tg(MHu2M)5378/Prnp0/0 mice with an incubation time of ≈200 days (11) (Fig. 4a, which is published as supporting information on the PNAS web site, www.pnas.org); these mice are subsequently referred to as Tg5378 mice. Because an inverse relationship was found previously between the level of PrP transgene expression and the length of the incubation time (30), six Tg lines expressing higher levels of MHu2M were constructed and inoculated with sCJD(MM1) prions. The lines with 8× expression levels had incubation periods of 160 ± 3 days (n/n0 = 10/10), 177 ± 4 days (n/n0 = 10/10), and 209 ± 4 days (n/n0 = 10/10). The lines with 32× expression levels showed longer incubation periods of 233 ± 4 days (n/n0 = 10/10), 236 ± 5 days (n/n0 = 11/11), and 239 ± 4 days (n/n0 = 11/11). Thus, raising the level of MHu2M expression did not reduce the incubation time.
Mutagenesis of the MHu2M Transgene.
Because of the limitations posed by the production of Tg mouse lines, we decided to change some of nine human residues in the Hu2 insert of MHu2M to those encoded by MoPrP (Fig. 5, which is published as supporting information on the PNAS web site). Five single substitutions were made: three at the N-terminal positions S96N, M108L, and M111V, and two at the C-terminal positions M165V and E167Q. We also combined some of the five mutated residues to create double and triple substitutions.
Abbreviated Incubation Times in Tg(MHu2M,M165V,E167Q) Mice.
Because none of the single substitutions except possibly residue M165V seemed promising, we constructed Tg mice expressing the doubly substituted MHu2M(M165V,E167Q) transgene. The first line, Tg(MHu2M,M165V,E167Q)22372/Prnp0/0, subsequently referred to as Tg22372 mice, express chimeric PrP at a level of 1–2×. Tg22372 mice were inoculated with brain homogenates from 10 different sCJD cases (Table 1, see a representative case depicted in Fig. 4d), one case with fCJD(E200K) (Fig. 4e), one case with familial FI [FFI(MV2,D178N)] (Fig. 4f), and one case of sporadic FI (sFI; ref. 31). Strikingly, the mean incubation times after inoculation with sCJD(MM1) prions were reduced by nearly 50% compared with those in Tg5378 mice; they ranged from 106 to 114 days (Table 1). No sCJD(VV2) cases have transmitted disease at >450 days after inoculation. fCJD(E200K) prions transmitted disease in 109 days on first passage and in 100 days on second passage, similar to sCJD(MM1) prions. FFI and sFI prions transmitted inefficiently to Tg22372 mice; the animals exhibited incubation times of 263 ± 15 days (n/n0 = 5/6) and 303 ± 20 days, respectively. On second passage of FFI prions in Tg22372 mice, the incubation time was reduced from 263 days to 97 days (Fig. 4f). Uninoculated Tg22372 mice remained healthy for more than 620 days.
Table 1.
Transmission of human prions to transgenic mice expressing MHu2M(M165V,E167Q), MHu2M, HuPrP(M129), or HuPrP(V129)
| Inoculum | Incubation period in days ± SEM (n/n0*)
|
|||
|---|---|---|---|---|
| MHu2M(M165V,E167Q) Tg22372 | MHu2M Tg5378 | HuPrP(M129) Tg440 | HuPrP(V129) Tg152 | |
| MM1† | ||||
| RG | 106 ± 2 (13/13)‡ | 191 ± 3 (10/10) | 165 ± 4 (7/7) | 263 ± 2 (6/6) |
| EC | 114 ± 2 (7/7)§ | 157 ± 3 (7/7) | 254 ± 6 (9/9) | |
| HS | 111 ± 2 (7/7)§ | 196 ± 4 (8/8) | 163 ± 2 (9/9) | |
| Ho | 205 ± 7 (6/6) | 155 ± 3 (8/8) | ||
| DG | 106 ± 2 (7/7)§ | |||
| AM | 112 ± 2 (8/8)¶ | |||
| MM2 | ||||
| A88-418 | >680 (0/10) | 232 ± 5 (3/3) | 368 ± 19 (9/9) | |
| 094-3 | >650 (0/7) | >580 (0/8) | 556 ± 63 (5/5) | |
| sFI-St‖ | 303 ± 20 (4/6) | 221 ± 6 (4/4) | 699 ± 30 (2/5) | |
| vCJD-RU96/45** | 335 ± 23 (7/7) | 647 ± 35 (2/7) | ||
| vCJD-RU96/02** | 380 ± 10 (6/6) | 563 ± 201 (4/7) | ||
| MV1 | ||||
| WP | 124 ± 3 (7/7)§ | 214 ± 3 (8/8) | ||
| Ghi | 215 ± 4 (5/5) | |||
| Ro | 176 ± 2 (9/9) | |||
| MV2 | ||||
| 093-25 | >640 (0/10) | 350 ± 38 (3/6) | 209 ± 3 (7/7) | |
| A94-311 | >640 (0/10) | 419 ± 13 (9/9) | 206 ± 3 (6/6) | |
| 096-48 | >640 (0/10) | 307 ± 27 (7/9) | 231 ± 4 (5/5) | |
| AMB | >450 (0/10) | |||
| VV2 | ||||
| RP | >450 (1/10) | 531 ± 46 (3/14) | 248 ± 12 (3/7) | 223 ± 7 (7/7) |
| A90-332 | >500 (0/8) | 448 ± 34 (3/7) | 195 ± 3 (8/8) | |
| 094-87 | 433 (1/10) | 378 ± 7 (3/7) | 198 ± 5 (9/9) | |
| GF | >450 (2/10) | |||
n, number of diseased animals; n0, number of inoculated animals.
Strain typing as described in ref. 20.
§¶ Number of animals that died by a cause other than prion disease: 5 (‡), 3 (§), 2 (¶).
Inoculum from patient with sporadic FI.
Inocula from patients with vCJD.
These strain-specific patterns of incubation times from different inocula are similar to those found with Tg5378 mice. When brain homogenates from patients with fCJD(E200K) were inoculated into Tg5378 mice, the incubation time was 170 ± 2 days (n/n0 = 10/10) and did not change on second passage (Fig. 1b). Inoculation of Tg5378 mice with brain homogenates from three patients with FFI(MV2,D178N) produced incubation times of 247 ± 6 days (n/n0 = 7/7), 206 ± 7 days (7/7), and 193 ± 5 days (9/9) on first passage, which shortened on second passage to 123 ± 3 days (7/7), 132 ± 5 days (9/9), and 136 ± 1 days (6/6), respectively (see a representative case depicted in Fig. 4c). Interestingly, the shortening of incubation times on second passage demonstrates a similar transmission barrier for FFI prions in both Tg5378 and Tg22372 mice (compare Fig. 4 c and f).
Figure 1.

Western blots of brain homogenates from inocula and inoculated Tg5378 and Tg22372 mice. (a) Immunoblots of rPrPSc (Upper) and deglycosylated rPrPSc (Lower) from brain homogenates, inoculated first and second passages, respectively, of sCJD(MM1) (lanes 1–3), fCJD(E200K) (lanes 4–6), FFI (lanes 7–9), and vCJD (lanes 10–12) into Tg5378 mice. In the first lane of each triplet (lanes 1, 4, 7, and 10), PrPSc in homogenates of the diseased human brains is shown; in the second lane of each triplet (lanes 2, 5, 8, and 11), PrPSc in brain homogenates of Tg5378 mice inoculated with diseased human brains is shown; and in the third lane of each triplet (lanes 3, 6, 9, and 12), PrPSc in brain homogenates from Tg5378 mice inoculated with diseased Tg5378 brain homogenates (from first passage of human prions) is shown. (b) Undigested brain homogenates from sCJD (RG) (lane 1); sCJD (EC) (lane 2); sCJD (WP) (lane 3), and Tg22372 mice inoculated with sCJD (RG) (lane 4); sCJD (EC) (lane 5); and sCJD (WP) (lane 6). Lane 7 shows an uninoculated Tg22372 mouse. Lanes 8–14 show the same brain homogenates as lanes 1–7, respectively, but brain homogenates were digested with proteinase K. Lanes 15–17 are the same as lanes 8–10, respectively, except the film was exposed longer to detect the weak band of PrPSc in lane 16. The blot clearly shows the overexpression of PrP in the brains of transgenic mice relative to human brains (compare lane 7 with lanes 1–3). (c) Brain homogenates from sCJD(MM1) (RG) (lanes 1, 5, and 9) and from Tg22372 mice inoculated with sCJD (RG) on first passage (lanes 2, 6, and 10) and second passage (lanes 3, 7, and 11). Brain homogenate from the second passage of sCJD (RG) into Tg5378 mice are shown in lanes 4, 8, and 12. Lanes 1–4 show undigested brain homogenates, lanes 5–8 show brain homogenates after 100 μg/ml PK digestion, and lanes 9–12 show brain homogenates after both PK digestion and PNGase F digestion. The glycoform pattern of PrPSc changes from the original sCJD (RG) pattern (lane 5) to the first passage (lane 6), remains the same on second passage (lane 7) but changes when passaged into Tg5378 mice (lane 8). The gel mobility of deglycosylated rPrPSc does not change (compare lane 9 to lanes 10–12).
Additional Tg(MHu2M,M165V,E167Q)Prnp0/0 mice were derived from two other founders. The chimeric MHu2M(M165V,E167Q) was expressed at 4–8× as measured by mRNA expression. Many of these Tg(MHu2M,M165V,E167Q)Prnp0/0 mice developed ataxia between 120 and 150 days of age and thus, neither line was inoculated. Mice killed at 200 days of age exhibited widespread astrocytic gliosis in the caudate and putamen, but no neuronal loss was observed. Faint, minimal punctate deposits of mutant PrPSc detected by immunostaining with the chimeric human–mouse recombinant Fab D18 and the 3F4 mAb were found only in the caudate and putamen (data not shown). The hippocampus and cerebellum showed no pathologic changes.
Biochemical Characteristics of Chimeric Mouse–Human Prions.
For sCJD and fCJD(E200K) prions, the molecular size of the deglycosylated rPrPSc fragment was 21 kDa (type 1) (Fig. 1a, lanes 1 and 4). The same-sized PrPSc fragments were found in the brains of Tg5378 mice after first and second passages of either sCJD or fCJD(E200K) prions (Fig. 1a, lanes 2, 3, 5, and 6). For FFI, the molecular size of deglycosylated rPrPSc was 19 kDa (type 2) (Fig. 1a, lane 7). Although the incubation times for FFI prions decreased nearly 50% on second passage in Tg5378 mice (Fig. 4c), the molecular size of the deglycosylated, rPrPSc fragment remained 19 kDa (Fig. 1a, lanes 8 and 9). These findings clearly dissociate the size of the deglycosylated, rPrPSc fragment from the incubation time.
Western blots of brain homogenates from sCJD patients and from corresponding inoculated Tg22372 mice demonstrated rPrPSc (Fig. 1b). Three different cases of sCJD (Fig. 1b, lanes 1–3, 8–10, and 15–17) were passaged into Tg22372 mice and produced MHu2M(M165V,E167Q) prions (Fig. 1b, lanes 4–6 and 11–13). Uninoculated mice did not produce MHu2M(M165V,E167Q) prions (Fig. 1b, lanes 7 and 14). Brain homogenates from sCJD(MM1) (RG) inoculated into Tg22372 mice (Fig. 1c, lanes 2, 6, and 10) were repassaged into the same line (Fig. 1c, lanes 3, 7, and 11), and into Tg5378 mice (Fig. 1c, lanes 4, 8, and 12). On passage into Tg22372 mice, slight changes of the glycosylation pattern were seen (compare Fig. 1c, lane 5 with lanes 6 and 7), but there were no changes in deglycosylated rPrPSc (Fig. 1c, lanes 9–12).
MHu2M(M165V,E167Q) prions from sCJD(MM1) (RG) inocula produced incubation times of 95 ± 2 days when passaged into Tg22372 mice (Fig. 4d), and of 166 ± 3 days when passaged into Tg5378 mice. MHu2M(M165V,E167Q) prions could not be passaged into Tg(MoPrP)4053/FVB mice overexpressing wt mouse PrP, indicating that chimeric MHu2M(M165V,E167Q) prions are unlikely to cross this transmission barrier.
PrPSc Deposition in Tg22372 Mice.
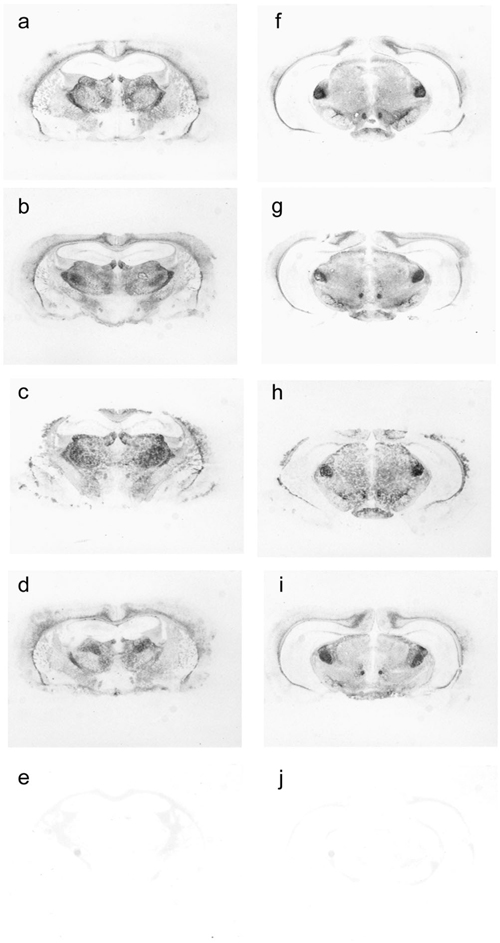
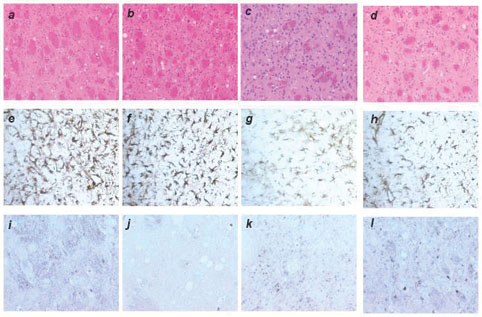
Tg22372 mice were inoculated with sCJD(MM1), fCJD, FFI, sFI, or vCJD prions. Histoblotting of brain sections showed that each of these inocula produced a different pattern of PrPSc deposition. Brains from Tg22372 mice infected with prions from six cases of sCJD(MM1) showed similar patterns of PrPSc deposition (Fig. 2 a and b; and Fig. 6, which is published as supporting information on the PNAS web site). PrPSc was deposited in the habenula, thalamus, colliculi nucleus, and midbrain tegmentum as well as the inner half of the cerebral cortex (Fig. 2 a and b). Some, but not all, white matter tracts were also immunopositive, including the corpus callosum (Fig. 2a) and the habenulopeduncular tract of the midbrain. The internal capsule and cerebral peduncles were negative. In contrast to sCJD prions, fCJD(E200K) prions produced widespread accumulation of PrPSc, in the cerebral cortex and hippocampus as well as much of the thalamus and hypothalamus, and most nuclei of the brainstem (Fig. 2 c and d). FFI prions produced the most intense immunostaining of the thalamus, lateral hypothalamus and brainstem (Fig. 2 e and f). sFI prions also produced intense PrPSc immunostaining of the thalamus (Fig. 2g), but little staining was found in the brainstem (Fig. 2h). In Tg22372 mice inoculated with vCJD prions, intense immunostaining in the tegmentum of the brainstem was found with less intensity in the thalamus and hypothalamus, but no PrPSc was seen in the cerebral cortex (Fig. 2 i and j). Punctate PrPSc deposits that correspond to PrP amyloid were distributed over the surface of the hippocampus (Fig. 2i). PrP immunopositive amyloid plaques (Fig. 6 l and p), particularly in the subcallosal region overlying the hippocampus, were associated with intense reactive astrocytic gliosis (Fig. 6n).
Figure 2.

PrPSc deposition shown by histoblotting in Tg22372 mice inoculated with different forms of CJD and FI. Coronal sections through the thalamic-hippocampal area (a, c, e, g, and i) or midbrain-pons area (b, d, f, h, and j) are depicted. Histoblots showing PrPSc deposition patterns after inoculation with sCJD(MM1) (RG) (a and b), fCJD(E200K) (c and d), FFI (e and f), sFI (g and h), and vCJD (i and j).
Like the similar patterns of PrPSc deposition, the intensity and distribution of vacuolation as well as reactive astrocytic gliosis in Tg22372 mice were indistinguishable for the six different cases of sCJD(MM1) (Figs. 7 and 8, which are published as supporting information on the PNAS web site). Moreover, the brains of Tg22372 and Tg5378 mice inoculated with the same CJD strains were virtually indistinguishable on pathologic examination. For each sCJD inoculum, the vacuolation scores (percentage of an area occupied by vacuoles) for the inner half of the cerebral cortex were 2× greater than those for the outer half. The degree of vacuolation in the thalamus, caudate nucleus and brainstem was similar (Fig. 7). No vacuolation was found in the cerebellar cortex with any of the sCJD inocula. Immunohistochemistry for PrPSc by hydrolytic autoclaving showed deposits ranging from finely granular (synaptic-like) to coarse and primitive (plaque-like); however, differences among inocula did not seem to be significant (Fig. 7). No differences between first and second passage of sCJD (RG) in Tg22372 mice were found on neuropathological examination (Fig. 7).
The combined findings for six sCJD(MM1) cases suggest that the prions in each case might be similar or indistinguishable strains. However, additional studies are required to test this conjecture, including end-point titrations, which yield dose-response curves. The slopes of these curves have been used to distinguish different prion strains (32).
Single and Triple Substitutions in MHu2M Transgenes.
Tg mouse lines were generated expressing MHu2M(M165V) and MHu2M(E167Q). Tg(MHu2M,E167Q)18990/Prnp0/0 mice overexpressing MHu2M(E167Q) at a level of ≈8× developed spontaneous neurodegenerative disease with frequent symptoms of dysmetria and proprioceptive deficits and less frequently, hindlimb paresis ending in death at ≈300 days of age. Neuropathological examination showed no vacuolation or obvious nerve cell loss; however, generalized reactive astrocytic gliosis accentuated in the caudate nucleus, brainstem, and cortex was found (Fig. 9, which is published as supporting information on the PNAS web site). PrP staining showed unusual punctate, perinuclear deposits in the pyramidal cell layer and in the hippocampus. No rPrPSc was detected in brain homogenates (data not shown) and no infectivity was found by inoculating Tg5378 mice, which have remained well for >524 days.
Tg(MHu2M,M165V)17030/Prnp0/0 mice inoculated with sCJD(MM1) (RG) prions had shortened incubation times of 105 ± 2 days (n/n0 = 10/10) on first passage and 92 ± 2 days (n/n0 = 10/10) on second passage. However, a second line, Tg(MHu2M,M165V)17000/Prnp0/0, had an incubation time of 246 ± 3 days (n/n0 = 8/8) when inoculated with sCJD(MM1) (RG) prions. Both Tg lines express MHu2M(M165V) at 2–4×.
Tg mouse lines expressing either MHu2M(S96N) or MHu2M(S96N,M165V,E167Q) were not fully established. Instead, individual founders and F1 littermates were inoculated with sCJD(MM1) (RG) prions. Six inoculated Tg(MHu2M,S96N) 28386/Prnp0/0 mice developed disease at 285 ± 8 days. Two Tg(MHu2M,S96N,M165V,E167Q)34098/Prnp0/0 mice inoculated with sCJD(MM1) (RG) prions developed disease after 205 and 364 days.
Four Tg mouse founders and their F1 littermates expressing MHu2M(S96N,M108L,M111V) had incubation times of 160 ± 3 days (n/n0 = 4/4), 170 ± 5 days (3/3), 229 ± 9 days (3/3), 195 ± 20 days (2/2) when inoculated with sCJD(MM1) (RG) prions. The differences in incubation times between groups may be due to different levels of PrP expression, but in all four experiments, the Tg(MHu2M,S96N,M108L,M111V)34098/Prnp0/0 mice showed shorter incubation times than the 285 days noted above for Tg(MHu2M,S96N)28386/Prnp0/0 mice.
Transmission of sCJD Prions to Tg(HuPrP) Mice.
Tg5378 and Tg(HuPrP,M129)440/Prnp0/0 mice with PrP transgenes encoding M at position 129 had shorter incubation times for sCJD(MM1) prions compared with Tg(HuPrP,V129)152/Prnp0/0 mice (Table 1). Conversely, only Tg(HuPrP,V129)152/Prnp0/0 mice with a transgene encoding V at position 129 could be reproducibly infected with sCJD(VV2) prions (Table 1). Whereas Tg(HuPrP,V129)152/Prnp0/0 mice were generally susceptible to human prions with M at position 129, Tg(HuPrP,M129)440/Prnp0/0 and Tg5378 mice were marginally susceptible to human prions with V at position 129 (Table 1, compare MM1, MV2, and VV2). Presumably, these results reflect the same mechanism that is responsible for the increased frequency of sCJD in patients that are homozygous for M or V at position 129 (9). For sCJD(MV2) prions, the incubation times were shortest in Tg(HuPrP,V129)152/Prnp0/0 mice. Whether this reflects the fact that PrPSc in these inocula originated from PrPC containing V129 (Table 1) is uncertain. sCJD(MV1) prions transmitted to Tg mice with incubation times similar to those found with sCJD(MM1) prions (Table 1).
Transmission of vCJD Prions to Tg Mice.
Tg22372 mice inoculated with brain homogenates of two patients with vCJD exhibited incubation times of 335 ± 23 days and 380 ± 10 days, with the first mouse showing signs of neurologic dysfunction at 254 days after inoculation (Table 1). In contrast, vCJD prions inoculated into Tg5378 mice resulted in prolonged incubation times between 300 and 725 days, with only 25% of the mice becoming ill by 500 days (Fig. 3a).
Figure 3.

Serial transmission of vCJD prions into Tg5378 mice. (a) Survival curve of Tg5378 mice inoculated with vCJD. The black line with the filled circles represents the first passage into Tg5378 mice, the black line with the open squares depicts the second passage, the blue line shows the third passage of the long-incubating substrain, and the red line illustrates the third passage of the short-incubating substrain. (b) Immunoblots of deglycosylated rPrPSc in selected brains from the third passage of vCJD into Tg5378 mice. The three sets of numbers above the immunoblot represent, from top to bottom, incubation times for first, second, and third passage. For the third passage, deglycosylated rPrPSc from brains of two different mice are shown. Deglycosylated rPrPSc from human patients with sCJD(MM1) (lane 1) and vCJD (lane 2) are depicted. Tg mice that were classified as short incubation time on second passage (lanes 3–6, red line in a) and as long incubation time on second passage (lanes 7–10, blue line in a). Deglycosylated rPrPSc has the electrophoretic mobility of either 21 kDa (lanes 3–5 and 9) or 19 kDa (lanes 6–8 and 10). The original vCJD prions used to inoculate the mice were either from the frontal lobe (FL) (lanes 3 and 4) or from the thalamus (TH) (lanes 5–10) of the human patient.
Inocula prepared from different mouse brains, all of which received the same human vCJD inoculum on first passage, produced incubation times ranging between 156 and 308 days on second passage. On second passage of MHu2M(vCJD) prions, the incubation times in Tg5378 mice shortened substantially and showed a biphasic pattern, with mean incubation times of ≈160 days and ≈290 days (Fig. 3a), indicating a transmission barrier for at least two different strains of vCJD prions. On third passage, the incubation time of the shorter strain (≈160 days) remained constant, whereas the longer strain (≈290 days) showed a modest shortening of ≈60 days. Of note, all inocula derived from the frontal lobe of one vCJD patient (RU 96/02) resulted in short (≈160 days) incubation times on third passage, whereas inocula from the thalamus of another vCJD patient (RU 96/07) resulted in both long (250–320 days) and short incubation times on third passage. Because only a total of eight second-passage mouse brains inoculated with prions from two vCJD patients were passaged a third time, we are reluctant to assign strain-specific characteristics to particular brain regions. In our initial study of vCJD passaged in Tg5378 mice, we found that on first and second passage, a deglycosylated rPrPSc fragment of 19 kDa was produced (Fig. 1a, lanes 10–12), which has been found in all vCJD cases examined to date. In third-passage mouse brain homogenates, we were surprised to find deglycosylated rPrPSc fragments of 19 kDa in some brains and of 21 kDa in others (Fig. 3b). There was no correlation between the incubation time and the fragment size. In fact, different Tg5378 mice inoculated with the same second-passage mouse brain homogenate had similar incubation times on third passage but different sizes of deglycosylated rPrPSc fragments (Fig. 3b, compare lane 5 with 6, and lane 9 with 10).
Discussion
Mutants of Chimeric PrP with Abbreviated Incubation Times.
Unable to shorten the incubation time by increasing the expression of MHu2M, we asked whether changing some residues encoded by the transgene might lead to a shortening of the incubation time. Faced with a very large number of possible mutations that could only be assessed by construction of Tg mice, we focused on the nine residues that distinguish MHu2MPrP from MoPrP. Because MHu2MPrP is converted to PrPSc in the presence of MoPrP but not HuPrP (4), we decided to revert some of the Hu residues at the N- and C-terminal ends of Hu2 insert (Fig. 5). Changing the two human residues at positions 165 and 167 at the C terminus of the Hu2 insert yielded a transgene product that is responsive to sCJD(MM1) prions, with incubation times between 106 and 124 days. sCJD(MM1) is the most prevalent subclass of sCJD, accounting for ≈68% of cases (20). sCJD(VV2) prions did not transmit as rapidly as sCJD(MM1) prions, presumably reflecting the fact that residue 129 in MHu2M(M165V,E167Q) is M (Table 1). The importance of homology at codon 129 between the recipient and prions in the inoculum also seems to be reflected by the differences in incubation times between Tg(HuPrP,M129)440/Prnp0/0 and Tg(HuPrP,V129)152/Prnp0/0 mice that received the same inoculum (Table 1).
Mutants of Chimeric PrP with Prolonged Incubation Times.
The S96N mutation in MHu2MPrP resulted in a prolongation of the incubation time, which was partially counteracted by introduction of the M108L and M111V mutations (Fig. 5). When the M165V and E167Q mutations were combined with the S96N mutation, the abbreviated incubation times were abolished. This finding suggests that both the N- and C-terminal domains of the Hu2 insert modulate prion replication. Because the M108L and M111V mutations counteracted the prolonged incubation time produced by the S96N mutation, it is reasonable to ask whether either one or both of these mutations might further shorten the abbreviated incubation times observed with M165V and E167Q. It is noteworthy that M108L and M111V mutations have been shown to modulate transmission of prions to Syrian hamsters (33, 34).
Neurodegeneration in Tg Mice Expressing Chimeric PrP.
Two Tg lines expressing MHu2MPrP(M165V,E167Q) at ≈4× were constructed. The majority of the offspring from each of these lines developed ataxia between 120 and 150 days of age. Interestingly, the E167Q mutation alone in Tg(MHu2M,E167Q)18990/Prnp0/0 mice expressing the transgene product at ≈8× resulted in spontaneous neurodegeneration at ≈300 days of age.
The neurodegeneration observed in Tg mice expressing high levels of either MHu2M(M165V,E167Q) or MHu2M(E167Q) is not unlike some other mutant PrPs expressed at high levels on the Prnp0/0 background (35–39). Of note, the neurodegeneration caused by either truncated MoPrP(Δ32–121) or MoPrP(Δ32–134) could be prevented by coexpression of wt MoPrP (40). This is reminiscent of the rescue of mice overexpressing the PrP paralog doppel (Dpl) in the cerebellum; coexpression of either Mo or SHa PrP prevented neurodegeneration (41–43). Whether the coexpression of MoPrP in Tg mice expressing high levels of MHu2M(M165V,E167Q) will prevent neurodegeneration and still enable abbreviated incubation times is unknown. In earlier studies with Tg(MHu2M) mice, the incubation times were similar regardless of whether MoPrP was coexpressed (4, 11).
Species Barriers and Prion Strains.
Our results show that homology of the PrP sequence between the host PrPC and the infecting prion is an imperfect predictor of prion transmission. For example, some sCJD(MM2) prions transmitted inefficiently to Tg(HuPrP,M129)440/Prnp0/0 (Table 1) and vCJD prions also transmitted quite inefficiently to Tg5378 mice (Fig. 3). In contrast, vCJD prions readily transmit to Tg(BoPrP)Prnp0/0 mice, whereas sCJD prions do not (18). Our findings emphasize the need to consider the tertiary structures of PrPC and PrPSc with respect to prion transmission. Although in many cases, homologous PrPC is able to adopt the conformation of PrPSc, some strains, such as vCJD, are clear exceptions. The efficient transmission of vCJD prions in Tg(BoPrP)Prnp0/0 mice and the rather inefficient transmission to Tg5378 mice (Fig. 3a) argue that the conformation of the BSE prion strain was maintained on passage from cattle to humans.
In earlier studies, it was recognized that passage of prions across a species barrier often produced new prion strains (44). Later, the species barrier was shown to be caused by species-specific differences in the PrP sequence of the recipient host and the donor inoculum (10, 30). In the studies reported here, we used the transmission barrier for vCJD prions in Tg5378 mice (Fig. 3a) to isolate multiple strains of prions. On second passage in Tg5378 mice, at least two distinct strains of vCJD prions were evident based on the biphasic distribution of incubation times. On third passage, the two vCJD strains were analyzed by determining the molecular size of the rPrPSc fragment after deglycosylation (Fig. 3b). Our findings show no correlation between the size of the PrPSc fragment and the incubation time, but clearly indicate that several vCJD prion strains can be generated under these circumstances.
Concluding Remarks.
Our discovery that Tg22372 mice have incubation times of ≈100 days for sCJD(MM1) prions represents a substantial advance in understanding the molecular basis of the species barrier. In addition, this discovery has practical importance in that end-point titrations of human brain homogenates can be accomplished in <250 days. Such titrations are critical for characterizing human prion strains as well as for calibrating sensitive immunoassays (45).
Supplementary Material
Acknowledgments
We thank Cynthia Cowdrey for technical assistance as well as Pierre Lessard and Mike Scott for discussions. This work was supported by grants from the National Institutes of Health as well as by a gift from the G. Harold and Leila Y. Mathers Charitable Foundation. C.K. was supported by a postdoctoral fellowship from the Swiss National Foundation.
Abbreviations
- PrP
prion protein
- PrPC
normal cellular isoform
- PrPSc
disease-associated isoform
- Tg
transgenic
- CJD
Creutzfeldt–Jakob disease
- vCJD
variant CJD
- sCJD
sporadic CJD
References
- 1.Prusiner S B. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prusiner S B. Annu Rev Microbiol. 1989;43:345–374. doi: 10.1146/annurev.mi.43.100189.002021. [DOI] [PubMed] [Google Scholar]
- 3.Kellings K, Prusiner S B, Riesner D. Philos Trans R Soc London B. 1994;343:425–430. doi: 10.1098/rstb.1994.0039. [DOI] [PubMed] [Google Scholar]
- 4.Telling G C, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen F E, DeArmond S J, Prusiner S B. Cell. 1995;83:79–90. doi: 10.1016/0092-8674(95)90236-8. [DOI] [PubMed] [Google Scholar]
- 5.Perrier V, Kaneko K, Safar J, Vergara J, Tremblay P, DeArmond S J, Cohen F E, Prusiner S B, Wallace A C. Proc Natl Acad Sci USA. 2002;99:13079–13084. doi: 10.1073/pnas.182425299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaneko K, Zulianello L, Scott M, Cooper C M, Wallace A C, James T L, Cohen F E, Prusiner S B. Proc Natl Acad Sci USA. 1997;94:10069–10074. doi: 10.1073/pnas.94.19.10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pattison I H. In: Slow, Latent and Temperate Virus Infections, National Institute of Neurological Diseases and Blindness Monograph. Gajdusek D C, Gibbs C J Jr, Alpers M P, editors. Washington, DC: U. S. Government Printing; 1965. , No. 2, pp. 249–257. [Google Scholar]
- 8.Scott M, Foster D, Mirenda C, Serban D, Coufal F, Wälchli M, Torchia M, Groth D, Carlson G, DeArmond S J, Westaway D, Prusiner S B. Cell. 1989;59:847–857. doi: 10.1016/0092-8674(89)90608-9. [DOI] [PubMed] [Google Scholar]
- 9.Palmer M S, Dryden A J, Hughes J T, Collinge J. Nature. 1991;352:340–342. doi: 10.1038/352340a0. [DOI] [PubMed] [Google Scholar]
- 10.Scott M, Ridley R M, Baker H F, DeArmond S J, Prusiner S B. In: Prion Biology and Diseases. Prusiner S B, editor. Plainview, NY: Cold Spring Harbor Lab. Press; 1999. pp. 307–347. [Google Scholar]
- 11.Telling G C, Scott M, Hsiao K K, Foster D, Yang S-L, Torchia M, Sidle K C L, Collinge J, DeArmond S J, Prusiner S B. Proc Natl Acad Sci USA. 1994;91:9936–9940. doi: 10.1073/pnas.91.21.9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asante E A, Linehan J M, Desbruslais M, Joiner S, Gowland I, Wood A L, Welch J, Hill A F, Lloyd S E, Wadsworth J D, Collinge J. EMBO J. 2002;21:6358–6366. doi: 10.1093/emboj/cdf653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruce M E, McBride P A, Farquhar C F. Neurosci Lett. 1989;102:1–6. doi: 10.1016/0304-3940(89)90298-x. [DOI] [PubMed] [Google Scholar]
- 14.Hecker R, Taraboulos A, Scott M, Pan K-M, Torchia M, Jendroska K, DeArmond S J, Prusiner S B. Genes Dev. 1992;6:1213–1228. doi: 10.1101/gad.6.7.1213. [DOI] [PubMed] [Google Scholar]
- 15.Bessen R A, Marsh R F. J Virol. 1994;68:7859–7868. doi: 10.1128/jvi.68.12.7859-7868.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Telling G C, Parchi P, DeArmond S J, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner S B. Science. 1996;274:2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 17.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen F E, Prusiner S B. Nat Med. 1998;4:1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 18.Scott M R, Will R, Ironside J, Nguyen H-O B, Tremblay P, DeArmond S J, Prusiner S B. Proc Natl Acad Sci USA. 1999;96:15137–15142. doi: 10.1073/pnas.96.26.15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peretz D, Williamson R A, Legname G, Matsunaga Y, Vergara J, Burton D, DeArmond S J, Prusiner S B, Scott M R. Neuron. 2002;34:921–932. doi: 10.1016/s0896-6273(02)00726-2. [DOI] [PubMed] [Google Scholar]
- 20.Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, et al. Ann Neurol. 1999;46:224–233. [PubMed] [Google Scholar]
- 21.Will R G, Ironside J W, Zeidler M, Cousens S N, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith P G. Lancet. 1996;347:921–925. doi: 10.1016/s0140-6736(96)91412-9. [DOI] [PubMed] [Google Scholar]
- 22.Collinge J, Sidle K C L, Meads J, Ironside J, Hill A F. Nature. 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 23.Bruce M E, Will R G, Ironside J W, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, et al. Nature. 1997;389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 24.Scott M R, Köhler R, Foster D, Prusiner S B. Protein Sci. 1992;1:986–997. doi: 10.1002/pro.5560010804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Büeler H, Fisher M, Lang Y, Bluethmann H, Lipp H-P, DeArmond S J, Prusiner S B, Aguet M, Weissmann C. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- 26.DeArmond S J, Kretzschmar H A, Prusiner S B. In: Greenfield's Neuropathology. 7th Ed. Graham D I, Lantos P L, editors. London: Hodder Arnold; 2002. pp. 273–323. [Google Scholar]
- 27.Carlson G A, Ebeling C, Yang S-L, Telling G, Torchia M, Groth D, Westaway D, DeArmond S J, Prusiner S B. Proc Natl Acad Sci USA. 1994;91:5690–5694. doi: 10.1073/pnas.91.12.5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muramoto T, Kitamoto T, Tateishi J, Goto I. Brain Res. 1992;599:309–316. doi: 10.1016/0006-8993(92)90406-y. [DOI] [PubMed] [Google Scholar]
- 29.Taraboulos A, Jendroska K, Serban D, Yang S-L, DeArmond S J, Prusiner S B. Proc Natl Acad Sci USA. 1992;89:7620–7624. doi: 10.1073/pnas.89.16.7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prusiner S B, Scott M, Foster D, Pan K-M, Groth D, Mirenda C, Torchia M, Yang S-L, Serban D, Carlson G A, et al. Cell. 1990;63:673–686. doi: 10.1016/0092-8674(90)90134-z. [DOI] [PubMed] [Google Scholar]
- 31.Mastrianni J A, Nixon R, Layzer R, Telling G C, Han D, DeArmond S J, Prusiner S B. N Engl J Med. 1999;340:1630–1638. doi: 10.1056/NEJM199905273402104. [DOI] [PubMed] [Google Scholar]
- 32.Dickinson A G, Meikle V M. Genet Res. 1969;13:213–225. doi: 10.1017/s0016672300002895. [DOI] [PubMed] [Google Scholar]
- 33.Priola S A, Caughey B, Race R E, Chesebro B. J Virol. 1994;68:4873–4878. doi: 10.1128/jvi.68.8.4873-4878.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Supattapone S, Muramoto T, Legname G, Mehlhorn I, Cohen F E, DeArmond S J, Prusiner S B, Scott M R. J Virol. 2001;75:1408–1413. doi: 10.1128/JVI.75.3.1408-1413.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Telling G C, Haga T, Torchia M, Tremblay P, DeArmond S J, Prusiner S B. Genes Dev. 1996;10:1736–1750. doi: 10.1101/gad.10.14.1736. [DOI] [PubMed] [Google Scholar]
- 36.Muramoto T, DeArmond S J, Scott M, Telling G C, Cohen F E, Prusiner S B. Nat Med. 1997;3:750–755. doi: 10.1038/nm0797-750. [DOI] [PubMed] [Google Scholar]
- 37.DeArmond S J, Sánchez H, Yehiely F, Qiu Y, Ninchak-Casey A, Daggett V, Camerino A P, Cayetano J, Rogers M, Groth D, et al. Neuron. 1997;19:1337–1348. doi: 10.1016/s0896-6273(00)80424-9. [DOI] [PubMed] [Google Scholar]
- 38.Hegde R S, Tremblay P, Groth D, Prusiner S B, Lingappa V R. Nature. 1999;402:822–826. doi: 10.1038/45574. [DOI] [PubMed] [Google Scholar]
- 39.Supattapone S, Bouzamondo E, Ball H L, Wille H, Nguyen H-O B, Cohen F E, DeArmond S J, Prusiner S B, Scott M. Mol Cell Biol. 2001;21:2608–2616. doi: 10.1128/MCB.21.7.2608-2616.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shmerling D, Hegyi I, Fischer M, Blattler T, Brandner S, Gotz J, Rulicke T, Flechsig E, Cozzio A, von Mering C, et al. Cell. 1998;93:203–214. doi: 10.1016/s0092-8674(00)81572-x. [DOI] [PubMed] [Google Scholar]
- 41.Nishida N, Tremblay P, Sugimoto T, Shigematsu K, Shirabe S, Petromilli C, Erpel S P, Nakaoke R, Atarashi R, Houtani T, et al. Lab Invest. 1999;79:689–697. [PubMed] [Google Scholar]
- 42.Moore R C, Mastrangelo P, Bouzamondo E, Heinrich C, Legname G, Prusiner S B, Hood L, Westaway D, DeArmond S J, Tremblay P. Proc Natl Acad Sci USA. 2001;98:15288–15293. doi: 10.1073/pnas.251550798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rossi D, Cozzio A, Flechsig E, Klein M A, Rulicke T, Aguzzi A, Weissmann C. EMBO J. 2001;20:694–702. doi: 10.1093/emboj/20.4.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kimberlin R H, Cole S, Walker C A. J Gen Virol. 1987;68:1875–1881. doi: 10.1099/0022-1317-68-7-1875. [DOI] [PubMed] [Google Scholar]
- 45.Safar J G, Scott M, Monaghan J, Deering C, Didorenko S, Vergara J, Ball H, Legname G, Leclerc E, Solforosi L, et al. Nat Biotechnol. 2002;20:1147–1150. doi: 10.1038/nbt748. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}