

Abstract
In vitro evolution was used to develop a DNA enzyme that catalyzes the site-specific depurination of DNA with a catalytic rate enhancement of about 106-fold. The reaction involves hydrolysis of the N-glycosidic bond of a particular deoxyguanosine residue, leading to DNA strand scission at the apurinic site. The DNA enzyme contains 93 nucleotides and is structurally complex. It has an absolute requirement for a divalent metal cation and exhibits optimal activity at about pH 5. The mechanism of the reaction was confirmed by analysis of the cleavage products by using HPLC and mass spectrometry. The isolation and characterization of an N-glycosylase DNA enzyme demonstrates that single-stranded DNA, like RNA and proteins, can form a complex tertiary structure and catalyze a difficult biochemical transformation. This DNA enzyme provides a new approach for the site-specific cleavage of DNA molecules.
Keywords: depurination, DNA cleavage, DNA repair, in vitro evolution, nucleic acid catalysis
Although the sugar-phosphate backbone of DNA is highly resistant to hydrolysis, DNA is not impervious to degradation by other means. Damage can occur to either the sugar or nucleobase components of DNA, compromising its strand continuity and information content. Various environmental factors, such as high temperature, oxidative conditions, ionizing radiation, and reactive chemical agents, all can lead to alteration of DNA (1). One of the simplest and best studied forms of DNA damage is spontaneous depurination. This reaction involves hydrolytic cleavage of the N-glycosidic bond of a purine nucleoside (Fig. 1), giving rise to an apurinic (AP) site (compound 1). The reaction is catalyzed by acid and facilitated by heating. It proceeds by a SN1-like loss of the purine to give an oxocarbenium ion, which subsequently is trapped with water.
Figure 1.

DNA strand scission resulting from depurination and subsequent β-elimination at the AP site. Hydrolysis at the C1′ position of deoxyguanosine results in release of guanine and formation of an AP site (1). The α- and β-hemiacetals are in equilibrium with the open chain aldehyde, which is susceptible to β-elimination that results in cleavage of the adjacent 3′ phosphoester (2). This product in turn undergoes cleavage of the 5′ phosphoester under alkaline conditions.
Production of AP sites in DNA can result in the loss of genetic information, both because AP sites themselves are mutagenic (2, 3) and because these sites can lead to DNA strand scission (4). As illustrated in Fig. 1, hydrolysis of the N-glycosidic bond unmasks the latent aldehyde functionality at the C1′ position, rendering the 3′-phosphate group susceptible to β-elimination (compound 2). It has been estimated that about 10,000 AP sites are produced in a typical mammalian cell each day (5, 6). Not surprisingly, organisms have evolved elaborate biochemical pathways to repair and/or minimize the effects of these lesions (7–9).
Although the primary role of DNA is information storage, it also has structural and functional properties. In recent years, the laboratory evolution of catalytic DNA molecules has demonstrated that single-stranded DNA is capable of forming tertiary structural motifs that catalyze a variety of chemical transformations with rate enhancements comparable to those of naturally occurring enzymes (10, 11). It is reasonable to suppose that more complex DNA enzymes could be developed, including ones that function analogously to DNA repair enzymes.
Starting with a population of 1014 random-sequence DNA molecules, in vitro evolution was used to obtain a DNA enzyme that catalyzes the hydrolysis of the N-glycosidic bond of a specific purine nucleoside within DNA, leading to strand scission. This DNA enzyme was made to operate on a separate DNA substrate with multiple turnover. It exhibited a catalytic rate enhancement of 8.8 × 105 compared with the uncatalyzed rate of depurination under the same reaction conditions. The DNA enzyme contains 93 nucleotides and is the most structurally complex catalytic DNA discovered to date. Analysis of its biochemical properties and the reaction products provided strong evidence that the enzyme cleaves DNA by a sequential mechanism involving depurination followed by cleavage at the AP site.
Materials and Methods
Nucleotides and Enzymes.
Nucleoside phosphoramidites were purchased from Glen Research (Sterling, VA). The lactose phosphoramidite was synthesized as described elsewhere (12). Standard oligodeoxynucleotides were purchased from Operon Technologies (Alameda, CA). Modified oligonucleotides were synthesized on a Pharmacia Gene Assembler Special automated DNA synthesizer. The starting pool of random-sequence oligonucleotides was prepared by Integrated DNA Technologies (Coralville, IA). All oligonucleotides were purified by denaturing PAGE and desalted on a Sephadex G-25 column (Amersham Pharmacia). dNTPs were from Amersham Pharmacia, dideoxynucleoside 5′-triphosphates were from United States Biochemical, and [γ-32P]ATP (7 μCi/pmol) and [α-32P]dATP (3 μCi/pmol) were from ICN. Taq DNA polymerase was from Stratagene, Sequenase T7 DNA polymerase was from United States Biochemical, and T4 polynucleotide kinase was from New England Biolabs.
In Vitro Selection.
A starting pool of DNA was prepared to select for cleavage of the β-1,4-O-glycosidic bond joining the galactose and glucose subunits of a lactose moiety embedded within DNA. The pool was constructed by cycled primer extension of 250 pmol of 5′-biotin-d(GAGCTACGTTT-Lac-TTTGGAAGAGATGGCGAC)-3′ (primer 1) on 22.5 pmol of 5′-d(GTGCCAAGCTTACCG-N85-GTCGCCATCTCTTCC)-3′ (N = A, C, T, or G) in a 500-μl reaction mixture containing 0.1 unit/μl Taq DNA polymerase/1.5 mM MgCl2/50 mM KCl/10 mM Tris⋅HCl (pH 8.3)/0.01% gelatin/0.2 mM each of the four dNTPs. Reactions were initiated by adding Taq polymerase at 92°C, followed by five temperature cycles of 92°C for 2 min, 45°C for 2 min, and 72°C for 4 min. Then 250 pmol of 5′-d(GTGCCAAGCTTACCG)-3′ (primer 2) and 13 pmol of [α-32P]dATP (3 μCi/pmol) were added and five cycles of PCR were carried out (92°C for 2 min, 48°C for 2 min, and 72°C for 4 min). The products were ethanol precipitated and single-stranded lactose-containing DNA was isolated by PAGE, eluted from the gel, and purified by Sephadex chromatography. A total reaction volume of 29 ml (58 reaction mixtures) yielded ≈10 copies each of 1014 different purified DNA molecules.
The starting pool was incubated in a 4-ml reaction mixture containing 50 mM MgCl2/150 mM NaCl/0.01% SDS/50 mM 1,4-piperazine-diethanesulfonic acid (pH 6.5) at 37°C for 36 h. The products were ethanol precipitated and the cleaved molecules were isolated by PAGE, with an authentic product in an adjacent lane of the gel serving as a marker. The cleaved molecules were eluted from the gel, purified by Sephadex chromatography, and amplified by PCR (30 cycles of 92°C for 1 min, 48°C for 1 min, and 72°C for 1 min) by using primers 1 and 2, thereby generating the pool for the next round of in vitro selection. Nine additional rounds were carried out under identical conditions as above, except that the scale was reduced to ≈250 pmol of input DNA. DTT (0.1 mM) was included in the reaction mixture during rounds 3, 5, 7, and 9 to prevent selection of molecules that cleaved DNA through an oxidative mechanism.
Optimization of N-Glycosylase Activity.
The molecules obtained after the 10th round of in vitro selection were subjected to mutagenic PCR, introducing random mutations at a frequency of ≈0.7% per nucleotide position (13). The PCR primers were 5′-biotin-d(GAGCTACGTTTTTTTGGAAGAGATGGCGAC)-3′ (primer 3) and 5′-d(AACAACAACXXXXXGTGCCAAGCTTACCG-3′ (primer 4; X = dSpacer nucleotide analogue; Glen Research). The products were ethanol precipitated and the shorter 5′-biotinylated strand was isolated by PAGE. The DNA was electroeluted at 4°C (Elutrap, Schleicher & Schuell) and purified by Sephadex chromatography. Approximately 300 pmol of this material was included in a mixture containing 150 mM NaCl/1 mM Na2EDTA/0.01% SDS/50 mM Hepes (pH 7.5) and immobilized on a 300-μl bed volume of Ultralink Streptavidin Plus (Pierce). The matrix was washed with five 150-μl volumes of loading buffer, two 150-μl volumes of ice cold 0.1 M NaOH/150 mM NaCl, and 12 150-μl volumes of loading buffer at 37°C.
The DNA-catalyzed reaction was carried out in two successive 150-μl volumes of a mixture containing 10 mM MgCl2/150 mM NaCl/1 mM spermine/0.01% SDS/50 mM Hepes (pH 7.5), each incubated at 37°C for 18 h. The eluate was collected and ethanol precipitated in the presence of 100 pmol each of primers 3 and 4. The cleaved molecules were isolated by PAGE, eluted from the gel, purified by Sephadex chromatography, and amplified by PCR. Rounds 2–10 were carried out similarly, except that the reaction time was reduced progressively from 36 h to 15 min. Mutagenic PCR was performed after rounds 3 and 6. DTT (0.1 mM) was included in the reaction mixture during rounds 3–5, 7, 9, and 10.
Analysis of Individual Clones.
After 10 rounds of in vitro selection, the population was amplified by PCR by using primer 2 and a nonbiotinylated form of primer 3. The PCR product was cloned by using the TOPO TA Cloning Kit and TOP10F′ competent cells (Invitrogen). Individual colonies were isolated on agar plates and used to inoculate 3-ml cultures, from which plasmid DNA was isolated (14) and sequenced by using the dideoxy chain termination method (15). Internally 32P-labeled single-stranded DNA was prepared from individual clones by PCR by using primers 3 and 4 in the presence of 40 μM dATP (6 μCi/nmol) and 200 μM concentrations of each of the other three dNTPs. The 28th clone obtained after the 10th round of selection for N-glycosylase activity (designated 10-28) was mutagenized at a frequency of ≈10% per nucleotide position by using hypermutagenic PCR (16), employing primers 3 and 4. Three additional rounds of in vitro selection were carried out and the resulting molecules again were cloned and sequenced.
Kinetic Analysis.
Unless stated otherwise, all DNA-catalyzed reactions were carried out in the presence of ≈3 nM internally 32P-labeled DNA catalyst/2 mM CaCl2/10 mM NaCl/10 mM Mes (pH 5.2) at 25°C. Reactions were initiated by mixing the DNA and a solution containing all of the other reaction components. Aliquots were taken from the mixture at various times and quenched by the addition of excess Na2EDTA or Na2EGTA. Strand scission at the AP site was driven to completion by subsequent treatment with 0.1 M piperidine at 95°C for 30 min. Radiolabeled substrate and products were separated by PAGE and quantitated by using a Molecular Dynamics PhosphorImager. Each value for kobs was determined based on six data points.
When measuring pH dependence, the reactions were performed in the presence of either 10 mM NaOAc (pH 3.6–5.2) or 10 mM Mes (pH 5.2–6.6), with the total ionic strength held constant. When measuring calcium dependence, the concentration of CaCl2 ranged from 0.001–40 mM. Values for kcat and Km were determined under multiple-turnover conditions employing 5′-32P-labeled substrate. Values for kobs were obtained for a range of concentrations of S that spanned Km, always with the concentration of S in ≥10-fold excess over the concentration of E and with the concentration of E at least 5-fold lower than Km. Each value for kobs was based on six data points obtained over the first 10–15% of the reaction. kcat and Km values were calculated from a standard Michaelis–Menten saturation plot that contained 42 data points.
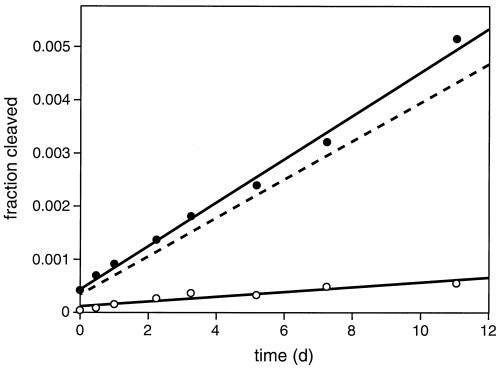
The uncatalyzed rate of depurination of the target deoxyguanosine was determined by incubating 1 μM 5′-32P-labeled substrate under standard reaction conditions. Aliquots were taken over 11 days and were either untreated or cleaved with piperidine, and then analyzed by PAGE. The value for kuncat was obtained from the slope of a best fit line of the fraction cleaved as a function of time, subtracting the amount of cleavage in the untreated sample.
Analysis of Reaction Products.
A large-scale reaction was performed by using 0.5 nmol of DNA enzyme and 2 nmol of substrate, which were incubated under standard reaction conditions for 20 h, and then treated with piperidine as described above. The two oligomeric products were purified by PAGE and subsequent chromatography on Nensorb 20 (NEN). The identity of these products was confirmed by matrix-assisted laser desorption ionization–time of flight mass spectrometry. The sample was irradiated with a nitrogen laser (Laser Science, Cambridge, MA) at 337 nm and the released ions were accelerated at 30,000 V and differentiated by using a PerSeptive Biosystems (Framingham, MA) Voyager-STR mass spectrometer with delayed extraction.
The reaction intermediate compound 1 was identified by covalent modification of the C1′ aldehyde by using 5-(((2-carbohydrazino)methyl)thio)acetylaminofluorescein (17). The DNA-catalyzed reaction was quenched by adding 2 mM Na2EDTA/70 mM 4-(2-hydroxyethyl)-piperazine-1-propanesulfonic acid (pH 7.0), and then the hydrazinofluorescein compound was added at a 2 mM concentration and the mixture was incubated at 37°C for 2 h. The products were separated by PAGE and the fluorescinated material was visualized by UV light. The released guanine was isolated by HPLC by using a reversed-phase column (Vydac, Hesperia, CA) and an isocratic gradient of 100 mM HCOONH4 (pH 6.9) at 1.0 ml/min, monitoring UV absorbance at 280 nm. Coinjection of the reaction product with authentic guanine (Sigma) demonstrated comigration of the two compounds.
Results
Emergence of N-Glycosylase Activity.
The initial goal of the project was to develop a DNA enzyme that cleaves the β-1,4-O-glycosidic bond between the galactose and glucose subunits of lactose. An oligonucleotide primer was prepared that contained an internal lactose residue (12), surrounded by deoxynucleotides of fixed sequence. The primer was extended on a synthetic DNA template to add 85 random-sequence deoxynucleotides and a fixed primer binding site to the 3′ end. A pool of ≈10 copies each of ≈1014 different extended molecules was incubated for 36 h in the presence of 10 mM MgCl2 at pH 6.5 and 37°C. Molecules that underwent cleavage in the vicinity of the lactose residue were selected on the basis of their mobility in a denaturing polyacrylamide gel. The selected molecules were eluted from the gel, purified by size-exclusion chromatography, and amplified by PCR. The lactose moiety was reintroduced during PCR amplification to complete one round of in vitro selection.
After 10 rounds of in vitro selection, the population of molecules appeared to be capable of performing a self-cleavage reaction to give the desired products. Further analysis revealed, however, that cleavage was occurring not at the glycosidic bond within lactose, but at a particular deoxyguanosine residue located several nucleotides downstream. The site of cleavage was localized to residue G17 by dideoxy sequencing to the 5′ end of the 3′ cleavage product. The reaction was not inhibited by 5 mM DTT, indicating that it did not involve oxidative cleavage of DNA, as had been observed in a previous in vitro selection experiment (18). Most significantly, the rate of the reaction was unchanged when the lactose moiety was replaced by thymidine.
A pool of DNA molecules that contained thymidine in place of lactose were 5′-32P-labeled and allowed to undergo cleavage. The products were analyzed in a high-resolution polyacrylamide gel by comparing their mobility to a ladder generated by partial digestion of the DNA with P1 nuclease. The labeled 5′ product migrated slightly faster than 5′-pGAGCTACGTTTTTTTG-3′, consistent with an oligodeoxynucleotide of the same length that has a 3′-terminal phosphate. This is the product one would expect from a reaction involving depurination of residue G17, followed by strand scission at the AP site. In support of this hypothesis, the extent of cleavage was markedly enhanced by incubating the reaction products with either spermine or piperidine. Furthermore, as will be discussed below, the identity of the cleavage products was consistent with a reaction mechanism involving depurination and subsequent β-elimination. Rather than generating DNA catalysts with O-glycosidase activity, the in vitro selection procedure produced DNAs with site-specific N-glycosylase activity. In light of this result, the selection strategy was changed to select intentionally for N-glycosylase activity.
Direct Selection for N-Glycosylase Activity.
The population obtained after the 10th round of in vitro selection was randomized at a frequency of ≈0.7% per nucleotide position by using mutagenic PCR (13). The lactose-containing primer was replaced by a 5′-biotinylated all-DNA primer that contained thymidine in place of lactose. A population of ≈1014 different molecules was prepared, immobilized on a streptavidin support, and allowed to react for 36 h in the presence of 10 mM MgCl2 and 1 mM spermine at pH 7.5 and 37°C. Any molecules that underwent cleavage at the target site were released from the support and collected in the eluate. The eluted molecules were reselected on the basis of their mobility in a denaturing polyacrylamide gel. Cleaved DNAs of the proper length were eluted from the gel, purified by size-exclusion chromatography, and amplified by PCR. This in vitro selection procedure was repeated for 10 rounds, progressively decreasing the reaction time from 36 h to 15 min. Mutagenic PCR was performed after rounds 3 and 6 to maintain sequence diversity in the population of evolving molecules.
After the 10th round of selection for N-glycosylase activity, individuals were cloned from the population and sequenced. Comparative sequence analysis revealed the presence of four major classes of molecules. Single-stranded DNA was prepared from representative members of each class and assayed for catalytic activity. The efficiency of self-cleavage was strongly dependent on the presence of spermine, suggesting that the evolved molecules continued to catalyze the depurination step, but had become dependent on spermine for the subsequent β-elimination step. Molecules from class I exhibited well-behaved first-order kinetics, with an observed rate of ≈0.1/min. A representative member of this class, the 28th clone obtained after the 10th round of selection (designated 10-28), was chosen for further study.
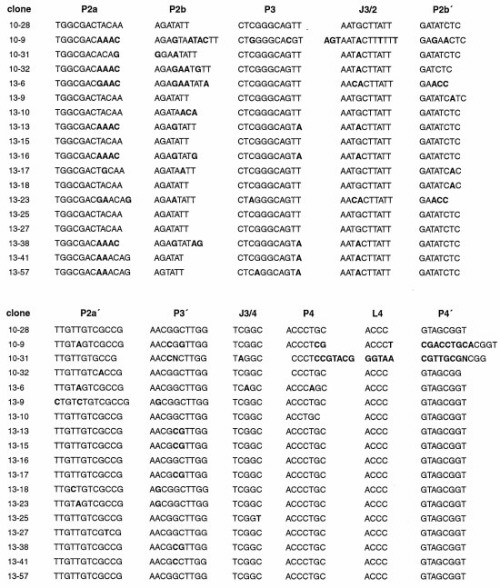
The 10-28 N-glycosylase was randomly mutagenized at a frequency of ≈10% per nucleotide position (16). Starting with this pool of molecules, three additional rounds of in vitro selection were carried out to refine the catalytic motif. A secondary structural model of the 10-28 catalyst was developed based on examination of the sequences of individuals that were isolated from the selected population (Fig. 2). According to this model, both the 5′ and 3′ ends of the molecule are engaged in short hairpin structures (the P1 and P4 stems, respectively). The deoxyguanosine residue that is the site of depurination (G17) is located within the hairpin loop at the 5′ end. The two hairpins are joined by stems of 18 and 9 bp (P2 and P3, respectively), which are connected together at both ends. The secondary structural model is supported by sequence covariation among the selected clones (see the supplemental Fig. 5 on the PNAS web site, www.pnas.org). However, this covariation is not sufficient to confirm the model.
Figure 2.

Secondary structural model of the 10-28 N-glycosylase. The site of depurination (G17) is highlighted. For multiple-turnover experiments, the DNA was divided within the P2 stem after residue A35. Other nucleotide positions that are discussed in the text are numbered. See supplemental Fig. 5 for the sequences of selected variants of the 10-28 clone.
Catalytic Behavior of the N-Glycosylase.
The DNA-catalyzed DNA-cleavage reaction involves depurination of a particular deoxyguanosine residue followed by β-elimination at the AP site. The depurination reaction was studied in isolation by using piperidine to drive the second step to completion. Before piperidine treatment, <2% of the molecules were cleaved, and no further DNA-catalyzed depurination occurred during the piperidine treatment. The rate of depurination of the 10-28 catalytic DNA was ≈0.2/min, measured in the presence of 2 mM Ca2+ and 10 mM NaCl at pH 5.2 and 25°C. The reaction reached a maximum extent of ≈90% (Fig. 3A). The uncatalyzed rate of depurination of the target deoxyguanosine, measured under the same reaction conditions, was 2.5 × 10−7/min (see Materials and Methods and supplemental Fig. 6). This corresponds to a catalytic rate enhancement, kcat/kuncat, of 8.8 × 105 for DNA-catalyzed cleavage of the N-glycosidic bond of residue G17.
Figure 3.

Catalytic activity of the 10-28 N-glycosylase. (A) Autoradiogram depicting the time course of the self-cleavage reaction, employing internally 32P-labeled DNA under standard reaction conditions (see Materials and Methods). Reaction products were treated with piperidine before electrophoresis in a 5% denaturing polyacrylamide gel. Unclv, the 128-nucleotide starting DNA; and clv, the 111-nucleotide 3′ product. The 5′ product is unlabeled because it was contained within the DNA primer. (B) pH dependence of the reaction carried out in the presence of 2 mM Ca2+ and either 10 mM NaOAc (●) or 10 mM Mes (○).
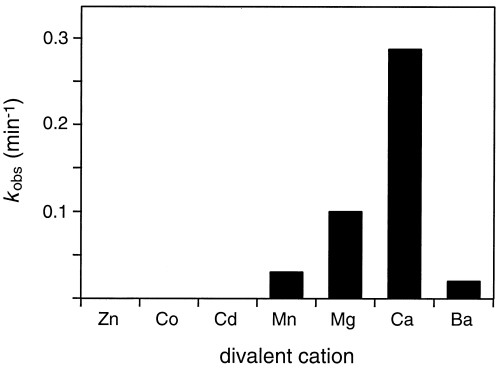
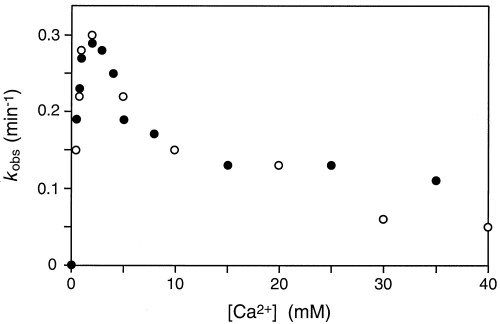
Analysis of the pH dependence of the 10-28 catalyst revealed that it had appreciable activity only in the pH range 4–6, with maximum activity at about pH 5 (Fig. 3B). Activity was dependent on the presence of a divalent metal cation, which could be either Ca2+, Mg2+, Mn2+, or Ba2+ (in order of reactivity; see supplemental Fig. 7). No activity was observed in the presence of Zn2+, Co2+, or Cd2+. A plot of kobs versus the concentration of Ca2+ demonstrated that the reaction rate was maximal in the presence of ≈2 mM Ca2+, dropping off significantly at higher Ca2+ concentrations (see supplemental Fig. 8). Activity was greater at 25°C than at 37°C, even though the in vitro selection procedure was carried out at 37°C. The lower temperature may favor proper folding of the catalytic DNA under the conditions of low ionic strength.
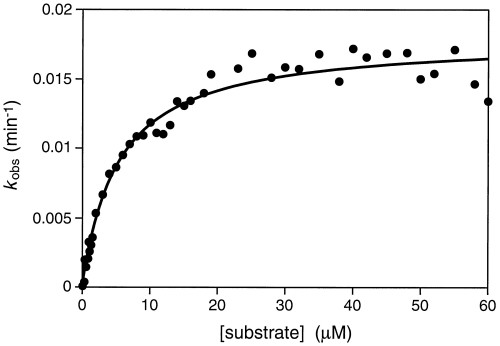
The 10-28 catalyst was engineered to operate with multiple turnover on a separate DNA substrate by dividing it within the P2 stem, after residue A35 (Fig. 2). The resulting 93mer DNA enzyme catalyzed depurination of residue G17 within the 35mer DNA substrate. The reaction proceeded with a kcat of 0.018/min and Km of 5.4 μM (kcat/Km = 3,300 M−1/min), measured in the presence of 2 mM Ca2+ at pH 5.2 and 25°C (see supplemental Fig. 9). The enzyme underwent 44 turnovers over the course of a 3-day incubation. Catalytic turnover likely was limited by the rate of release of the depurinated product rather than the rate of the chemical step. In support of this view, when the molecule was divided after residue T42 rather than A35, thereby extending the interaction between enzyme and substrate by 7 bp, the turnover rate decreased by about 3-fold.
There was no detectable activity when the 10-28 catalyst was divided after residue A23. This would have required the DNA enzyme to recognize its substrate entirely through tertiary interactions. On the other hand, removing the first eight nucleotides at the 5′ end of the substrate reduced activity by about 30-fold, suggesting that one or more of these residues are involved in tertiary interactions with the enzyme.
Several variant substrates were tested in which either the stem or loop portion of the P1 hairpin was modified (Fig. 2). Replacing the T10⋅G22 and T12⋅G20 pairs by C⋅G pairs greatly reduced catalytic activity, whereas replacing them by T⋅A pairs eliminated activity altogether. Inverting the T11⋅A21 pair to an A⋅T pair had no effect, demonstrating that a run of consecutive pyrimidine⋅purine pairs is not essential for catalysis. Replacing the hairpin loop, 5′-TGG-3′, by an internal bulge loop having the sequence 5′-TAAGCCCCTTGG-3′, 5′-TTAAGCCCCTTGG-3′, or 5′-AAAAGCCCCTTGG-3′ (G17 underlined), reduced activity by only about 10-fold. When the bulge loop was replaced by Watson–Crick pairs, activity was lost. Replacing the target deoxyguanosine by deoxyadenosine, 7-deazadeoxyguanosine, or 8-oxodeoxyguanosine eliminated activity, demonstrating that the enzyme is specific for excision of guanine at the target position.
Several enzyme variants were tested to evaluate the proposed secondary structure model (Fig. 2). Deleting the single unpaired residue within the P2 stem (C71) had no effect on catalytic activity. Removing the 3′ hairpin, after residue G93, eliminated activity. Each of the three conserved deoxycytidines within the 3′ hairpin loop was mutated independently to deoxyadenosine. The A109 mutant exhibited a 7-fold reduced catalytic rate, whereas the A107 and A108 mutants had no detectable activity.
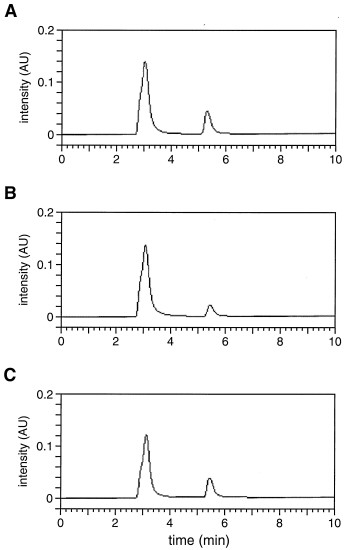
To provide direct evidence that the DNA enzyme cleaves DNA by a depurination/β-elimination mechanism, it was essential to characterize the depurinated intermediate compound 1, the released guanine, the 5′ oligodeoxynucleotide product bearing a 3′ phosphate, and the 3′ oligodeoxynucleotide product bearing a 5′ phosphate. The depurinated intermediate was identified by chemoselective modification of the C1′ aldehyde by using 5-(((2-carbohydrazino)methyl)thio)acetylaminofluorescein, then separating the materials by PAGE and visualizing them under UV light. The DNA-catalyzed reaction was carried out in the absence of spermine and without subsequent piperidine treatment so that the depurinated intermediate remained uncleaved. The reacted, but not the unreacted, substrate was labeled by the hydrazinofluorescein compound. The released guanine was analyzed by reversed-phase HPLC. Its identity was confirmed by UV spectroscopic analysis and HPLC comigration with authentic material (see supplemental Fig. 10). No other nucleobase products were observed on the HPLC chromatogram. The two oligodeoxynucleotide products were purified by PAGE and their identity was confirmed both by comparing their gel mobility to authentic material and by analysis with matrix-assisted laser desorption ionization–time of flight mass spectrometry (Fig. 4).
Figure 4.

Matrix-assisted laser desorption ionization mass spectra of the two oligodeoxynucleotide products resulting from cleavage of the 35mer DNA substrate. (A) The 5′ product, containing 16 nucleotides and a 3′-terminal phosphate, has an expected m/z for the principle product ion of 4,973. (B) The 3′ product, containing 18 nucleotides and a 5′-terminal phosphate, has an expected m/z of 5,646.
Discussion
The 10-28 N-glycosylase DNA enzyme is the most structurally complex catalytic DNA discovered to date. This 93-nucleotide DNA molecule catalyzes excision of a specific guanine within a DNA substrate to generate an AP site. In the presence of an amine, such as spermine or piperidine, the depurinated DNA undergoes strand cleavage at the AP site through a β-elimination mechanism. In the intramolecular reaction format, the enzyme catalyzes deoxyguanosine depurination with a kobs of 0.2/min, measured in the presence of 2 mM Ca2+ at pH 5.2 and 25°C. This corresponds to a catalytic rate enhancement of about 106-fold compared with the uncatalyzed rate of reaction. The rate of catalytic turnover in the intermolecular reaction was about 20-fold slower than the rate of cleavage in the intramolecular reaction, which likely was caused by the slow rate of release of the depurinated product.
A secondary structural model of the 10-28 N-glycosylase was formulated based on sequence analysis of selected variants of the 10-28 clone (Fig. 2). This model has not been confirmed by biophysical studies and must be regarded as tentative. Nonetheless, the extensive regions of Watson–Crick complementarity and the demonstration of catalytic turnover when the enzyme and substrate were separated on the basis of the P2 pairing are consistent with the model. The site of depurination (G17) is located within the loop portion of the 5′ hairpin structure. This loop must dock into the catalytic core of the enzyme, although there are no obvious Watson–Crick pairing interactions to bring this about. It is important to keep in mind that at pH 5, which provides optimal enzymatic activity, some of the deoxyadenosine and deoxycytidine residues will be protonated at N1 and N3, respectively. This raises the possibility that nonstandard pairing interactions, such as A+⋅C pairs and C+⋅G⋅C triples, play an important role in defining the tertiary structure of the enzyme.
The original goal of the project was to select for an O-glycosidase DNA enzyme that could cleave a target disaccharide. Instead, N-glycosylase activity emerged from the in vitro evolution process. In retrospect, this outcome was made more likely by two factors: the lower stability of an N-glycosidic bond within DNA compared with an O-glycosidic bond within a disaccharide, and the particular way in which the selection scheme was implemented. The spontaneous rate of depurination of DNA, extrapolating to physiological salt concentrations at pH 7.4 and 37°C, is about 1–2 × 10−9/min (6). Recent measurements of the spontaneous rate of hydrolysis of various O-glycosides under similar conditions provided a value for the β-linked glucopyranosides of 2.8 × 10−13/min (19), which is much slower than previous estimates (20). Thus, the uncatalyzed rate of cleavage of the N-glycosidic bond of deoxyguanosine is about 104-fold faster than that of the O-glycosidic bond of lactose.
It also appears that the long reaction times and mildly acidic conditions that were used during the first 10 rounds of in vitro selection were favorable for the emergence of N-glycosylases. During those rounds, the reaction was carried out at pH 6.5 for 36 h in an attempt to facilitate what was expected to be the slow cleavage of the O-glycosidic bond. Reacted molecules were selected on the basis of their ability to promote cleavage at or near the target lactose moiety. The reaction conditions provided an opportunity for depurination as well, which is favored under acidic conditions. To be selected, the depurinated molecules would have to have undergone subsequent β-elimination to give a product with approximately the same gel mobility as the product of the O-glycosidase reaction. During the initial rounds of selection, DNA-cleavage activity was inefficient because AP sites that were generated by depurination were cleaved slowly under the mildly acidic conditions. During the later rounds, N-glycosylase activity was selected intentionally by adding spermine to the reaction mixture to ensure that the β-elimination step would not be rate limiting.
The discovery by in vitro evolution of a DNA enzyme that efficiently cleaves the N-glycosidic bond of a deoxynucleoside expands the known catalytic repertoire of nucleic acids. RNA enzymes have been described that catalyze the reverse reaction, the synthesis of a nucleoside from an activated sugar and a base (21). In contemporary biochemistry, however, the reactions of nucleoside biosynthesis and degradation are catalyzed by protein enzymes. N-glycosylase enzymes occur widely in biology. In protozoa, the nucleoside hydrolases operate in the salvage pathway of nucleotide biosynthesis by catalyzing the hydrolysis of the N-glycosidic bond of spent nucleotides (22) In humans, the enzyme purine nucleoside phosphorylase is critical for the catabolism of purine nucleosides. It catalyzes displacement of purine bases from ribo- and deoxyribonucleotides by attack of a phosphate group at the C1′ position (23) Various cytotoxic ribosome-inactivation proteins, such as the ricin A-chain, act by cleaving the N-glycosidic bond of a particular nucleotide within ribosomal RNA (24, 25). There also are numerous N-glycosylase enzymes involved in base excision repair, each with the ability to excise particular damaged DNA bases (8, 9).
Despite the diversity of targets and biological contexts, all of the N-glycosylase protein enzymes have a similar mechanism of action (26). Each displays specificity for a certain nucleobase or structurally-related class of base analogues. Each facilitates loss of the base from the C1′ position of a nucleoside, typically by protonation of N7 of a purine or protonation of a keto oxygen of a pyrimidine. Each facilitates delivery of a nucleophilic group, such as water or a phosphate anion, to the C1′ position to generate the sugar product. Conversely, the biosynthesis of a nucleoside N-glycosidic bond involves displacement of a suitable leaving group by the incoming nitrogenous base.
In view of the mechanistic similarities of N-glycosylase proteins and the likely mechanism of the nucleoside synthase ribozyme (21) it is reasonable to propose that the 10-28 N-glycosylase DNA enzyme catalyzes depurination by specific recognition of the target deoxyguanosine, protonation of the N7 position, and delivery of a water molecule to the C1′ position. The DNA enzyme is specific for deoxyguanosine, although the way in which it recognizes this residue is not known. Many protein enzymes that are involved in DNA modification and repair bind to DNA with the target residue “flipped” out of the duplex (9, 27). The DNA enzyme may employ a similar means of substrate recognition that takes advantage of the special position of the target deoxyguanosine within either a hairpin loop or internal bulge loop.
DNA-catalyzed cleavage of the N-glycosidic bond occurs under acidic conditions, suggesting that it behaves similarly to nonenzymatic acid-catalyzed depurination (28), involving protonation of N7 of the target guanosine. This position normally has a pKa of 2.4 (29). The fact that the 10-28 N-glycosylase has a pH optimum of about five suggests that it creates a local environment in which the pKa of N7 is shifted upward. There is a precedent for such pKa shifts in structured RNAs (30, 31). Alternatively, there may be no pKa shift and the enzyme simply becomes less active below pH 5 because of structural changes or some other perturbation. If the DNA enzyme facilitates protonation of N7 of the target deoxyguanosine, the lack of observed activity when this residue is replaced by either deoxyadenosine or 7-deazadeoxyguanosine is not surprising. The pKa of N7 of deoxyadenosine is <1 and substitution of a carbon atom at the N7 position of 7-deazadeoxyguanosine would prevent protonation altogether. The hydrolysis reaction is completed by delivery of a water molecule to the C1′ position, either as the guanine base departs or after the sugar oxocarbenium ion has formed. There is insufficient information to distinguish between these two possibilities, but the requirement for a divalent metal cation suggests that a metal may help to position a water molecule for nucleophilic attack on the C1′ carbon.
The 10-28 DNA N-glycosylase is a complex DNA enzyme that is capable of catalyzing a difficult biochemical transformation that is relevant to DNA repair. This activity opens new avenues for research into DNA-catalyzed DNA modification. Considering that the reaction is specific for a particular nucleobase, it may be possible to use the 10-28 N-glycosylase enzyme as a starting point to evolve other N-glycosylases that are specific for modified bases, such as 8-oxoguanine or 1, N6-ethenoadenine. It also may be possible, through in vitro evolution, to alter the preferred reaction conditions of the 10-28 enzyme so that it can operate at more physiological pH and temperature. This might allow DNA-catalyzed depurination/β-elimination of specific lesions within genomic DNA. In vitro evolution strategies are likely to yield other nucleic acid enzymes that catalyze reactions relevant to DNA modification and repair. Like the 10-28 N-glycosylase, these enzymes will allow one to compare structural and mechanistic features of repair enzymes that are composed of either protein or nucleic acid.
Supplementary Material
Acknowledgments
We thank Gary Siuzdak and Klas Broo for assistance with mass spectrometry analysis and John Tainer for helpful discussions. This work was supported by National Institutes of Health Grant AI30882 and National Aeronautics and Space Administration Grant NAG5–3647. T.L.S. was supported by a postdoctoral fellowship from the National Aeronautics and Space Administration Specialized Center for Research and Training in Exobiology.
Abbreviation
- AP
apurinic
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Lindahl T. Nature (London) 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 2.Schaaper R M, Kunkel T A, Loeb L A. Proc Natl Acad Sci USA. 1983;80:487–491. doi: 10.1073/pnas.80.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shibutani S, Takeshita M, Grollman A P. J Biol Chem. 1997;272:13916–13922. doi: 10.1074/jbc.272.21.13916. [DOI] [PubMed] [Google Scholar]
- 4.Lindahl T, Andersson A. Biochemistry. 1972;11:3618–3623. doi: 10.1021/bi00769a019. [DOI] [PubMed] [Google Scholar]
- 5.Lindahl T, Nyberg B. Biochemistry. 1972;11:3610–3618. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 6.Nakamura J, Walker V E, Upton P B, Chiang S-Y, Kow Y W, Swenberg J A. Cancer Res. 1998;58:222–225. [PubMed] [Google Scholar]
- 7.Friedberg E C, Walker G C, Siede W. DNA Repair and Mutagenesis. Washington, DC: Am. Soc. Microbiol.; 1995. pp. 135–190. [Google Scholar]
- 8.McCullough A K, Dodson M L, Dodson R S. Annu Rev Biochem. 1999;68:255–285. doi: 10.1146/annurev.biochem.68.1.255. [DOI] [PubMed] [Google Scholar]
- 9.Mol C D, Parikh S S, Putnam C D, Lo T P, Tainer J A. Annu Rev Biophys Biomol Struct. 1999;28:101–128. doi: 10.1146/annurev.biophys.28.1.101. [DOI] [PubMed] [Google Scholar]
- 10.Sen D, Geyer C R. Curr Opin Chem Biol. 1998;2:680–687. doi: 10.1016/s1367-5931(98)80103-8. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Breaker R R. Curr Opin Struct Biol. 1999;9:315–323. doi: 10.1016/S0959-440X(99)80042-6. [DOI] [PubMed] [Google Scholar]
- 12.Sheppard, T. L., Wong, C.-H. & Joyce, G. F. (2000) Angew. Chemie Intl. Ed. Engl., in press. [PubMed]
- 13.Cadwell R C, Joyce G F. PCR Methods Applic. 1992;2:28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- 14.Tsang J, Joyce G F. Biochemistry. 1994;33:5966–5973. doi: 10.1021/bi00185a038. [DOI] [PubMed] [Google Scholar]
- 15.Sanger F, Nicklen S, Coulson A R. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vartanian J-P, Henry M, Wain-Hobson S. Nucleic Acids Res. 1996;24:2627–2631. doi: 10.1093/nar/24.14.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boturyn D, Constant J-F, Defrancq E, Lhomme J, Barbin A, Wild C P. Chem Res Toxicol. 1999;12:476–482. doi: 10.1021/tx980275g. [DOI] [PubMed] [Google Scholar]
- 18.Carmi N, Balkhi S R, Breaker R R. Proc Natl Acad Sci USA. 1998;95:2233–2237. doi: 10.1073/pnas.95.5.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolfenden R, Lu X, Young G. J Am Chem Soc. 1998;120:6814–6815. [Google Scholar]
- 20.Legler G. Adv Carbohydr Chem Biochem. 1990;48:319–384. doi: 10.1016/s0065-2318(08)60034-7. [DOI] [PubMed] [Google Scholar]
- 21.Unrau P J, Bartel D P. Nature (London) 1998;395:260–263. doi: 10.1038/26193. [DOI] [PubMed] [Google Scholar]
- 22.Degano M, Almo S C, Sacchettini J C, Schramm V L. Biochemistry. 1998;37:6277–6285. doi: 10.1021/bi973012e. [DOI] [PubMed] [Google Scholar]
- 23.Erion M D, Stoeckler J D, Guida W C, Walter R L, Ealick S E. Biochemistry. 1997;36:11735–11748. doi: 10.1021/bi961970v. [DOI] [PubMed] [Google Scholar]
- 24.Barbieri L, Battelli M G, Stirpe F. Biochim Biophys Acta. 1993;1154:237–282. doi: 10.1016/0304-4157(93)90002-6. [DOI] [PubMed] [Google Scholar]
- 25.Chen X-Y, Link T M, Schramm V L. Biochemistry. 1998;37:11605–11613. doi: 10.1021/bi980990p. [DOI] [PubMed] [Google Scholar]
- 26.David S S, Williams S D. Chem Rev (Washington, DC) 1998;98:1221–1261. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- 27.Roberts R J, Cheng X. Annu Rev Biochem. 1998;67:181–198. doi: 10.1146/annurev.biochem.67.1.181. [DOI] [PubMed] [Google Scholar]
- 28.Zoltewicz J A, Clark D F, Sharpless T W, Grahe G. J Am Chem Soc. 1970;92:1741–1750. doi: 10.1021/ja00709a055. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki T, Ohsumi S, Makino K. Nucleic Acids Res. 1994;22:4997–5003. doi: 10.1093/nar/22.23.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Connell G J, Yarus M. Science. 1994;264:1137–1141. doi: 10.1126/science.7513905. [DOI] [PubMed] [Google Scholar]
- 31.Legault P, Pardi A. J Am Chem Soc. 1997;119:6621–6628. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}