

Abstract
Internalization and proteolytic degradation of epidermal growth factor (EGF) receptor (R) following ligand binding is an important mechanism for regulating EGF-stimulated signals. Using pharmacological and RNA interference inhibition of p38 mitogen-activated protein kinase, we show that p38 is required for efficient EGF-induced EGFR destruction but not internalization. In the absence of p38 activity, EGF fails to stimulate the ubiquitin ligase Cbl or ubiquitinylation of EGFR, and internalized EGFR accumulates in intracellular vesicles containing caveolin-1. These effects are accompanied by loss of EGFR phosphorylation on Y1045, a phosphorylation site required for Cbl activation. Furthermore, similar to cells treated with p38 inhibitors, intestinal epithelial cells expressing Y1045F EGFR mutants show increased proliferation but not migration in response to EGF, thus uncoupling these biological responses. Together these data position p38 as a modulator of ligand-stimulated EGFR processing and demonstrate that this processing has a profound impact on the cellular outcome of EGFR signaling.
Keywords: EGF receptor, intestinal epithelium, p38 MAPK, ubiquitinylation, wound healing
Introduction
Maintenance of the mammalian intestinal epithelium, a self-renewing single-cell barrier that protects the organism from potentially toxic gut contents and pathogenic intestinal microflora, depends on minutely coordinated cell migration/restitution and proliferation. Signaling through epidermal growth factor (EGF) receptor (R) is a key regulator of these processes in epithelial cells (Carpenter and Cohen, 1981; Polk, 1998). EGFR is a transmembrane tyrosine kinase glycoprotein with a ligand-binding ectodomain, a single hydrophobic membrane-spanning region, and a cytoplasmic tail containing the kinase domain as well as tyrosine residues which are themselves targets of EGFR- and other kinase-mediated phosphorylation. The receptor is expressed in a wide variety of tissues, where it is activated by a suite of related soluble growth factors including EGF and transforming growth factor-α (Harris et al, 2003). Ligand binding promotes formation of receptor dimers, increased tyrosine kinase activity, and phosphorylation of up to 11 cytoplasmic tyrosine residues on EGFR (Olayioye et al, 2000). When phosphorylated, these tyrosines provide docking sites for downstream effectors and regulators, such as Shc, Grb2, and c-Cbl. In intestinal epithelial cells, EGF exposure promotes cell migration and wound healing (Blay and Brown, 1985; Polk, 1998), suggesting that EGFR stimulation may be beneficial in pathologies such as ulcerative colitis, which involve wounded epithelia. However, EGF is involved in neoplastic transformation (Potten et al, 1995; Janmaat and Giaccone, 2003) as well as restitution. Thus, a thorough dissection of the molecular pathways determining the cellular outcome of EGFR activation is necessary for the development of therapies based on these signaling cascades.
Central to appropriate regulation of EGFR activity is the process by which EGFR triggers its own internalization and destruction (Carpenter and Cohen, 1976; Levkowitz et al, 1998), providing for signal termination. Following ligand binding to EGFR, the Cbl ubiquitin ligase is tyrosine phosphorylated by the receptor. This requires prior EGFR phosphorylation on tyrosine 1045 and/or serines 1046/1047 (Oksvold et al, 2003; Grovdal et al, 2004). Tyrosine phosphorylation of Cbl increases its ubiquitin ligase activity by >1000-fold, and Cbl ubiquitinylates EGFR on multiple sites (Mosesson et al, 2003). Recruitment of additional adapter proteins such as Cin85 and endophilins results in translocation of EGFR-containing complexes to the endosomal compartment (Dikic and Giordano, 2003). Receptors are subsequently sorted by incompletely understood mechanisms through distinct vesicles for either recycling to the plasma membrane or destruction (Bucci et al, 2000; de Renzis et al, 2002). Although a requisite role for Cbl ubiquitin ligase activity in EGFR internalization is controversial (Oksvold et al, 2003), EGFR ubiquitinylation does appear to be required for efficient receptor destruction (Longva et al, 2002; Grovdal et al, 2004). The physiological importance of this mechanism is underlined by the fact that mutations in Cbl, which impair its ability to desensitize EGFR signaling, may be oncogenic (Levkowitz et al, 1999; Waterman et al, 1999).
An unanswered question about EGFR regulation is the extent to which downstream signaling cascades feed back to affect receptor desensitization. One candidate feedback regulator of EGFR is the p38 kinase. A member of the mitogen-activated protein kinase (MAPK) signaling family, p38 is associated with cellular stress responses and apoptosis (Xia et al, 1995; Daly et al, 1999), but is also involved in wound healing in epithelial cells (Dieckgraefe et al, 1997; Sharma et al, 2003). Recent work from our laboratory identified p38 as a switch determining whether EGF treatment of intestinal epithelial cells results in migration or proliferation (Frey et al, 2004). Furthermore, p38 activity modulates activation of EGFR targets in other signaling cascades (Sharma et al, 2003; Frey et al, 2004); for example, young adult mouse colon (YAMC) cells treated with EGF in the presence of a p38 inhibitor show dramatically sustained extracellular signal-regulated kinase (ERK)1/2 activation as compared with cells given EGF alone.
This study tests the hypothesis that p38 MAPK regulates EGFR activity. We show that p38 is required for EGF-stimulated Cbl activation, EGFR ubiquitinylation, and destruction of receptor. In contrast, loss of p38 activity does not affect ligand-driven EGFR internalization, and EGFR molecules protected by p38 inhibition accumulate in intracellular recycling vesicles. This recapitulates the effect of an EGFR mutation, Y1045F, that prevents Cbl activation. In this regard, the effects of Y1045F EGFR on EGF-stimulated proliferation and restitution are similar to those of p38 inhibition. Taken together, the data support regulation of EGFR desensitization as the means by which p38 exerts its influence on EGF-stimulated cell migration and proliferation, and implicate EGFR Y1045 as a molecular switch coordinating these processes.
Results
p38 MAPK activity is required for EGF-stimulated EGFR downregulation
To determine whether p38 is involved in feedback regulation of EGFR, YAMC cells were exposed to 10 ng/ml EGF with or without pretreatment with the p38 inhibitor SB202190. As shown in Figure 1A, p38 blockade did not attenuate rapid (5 min) EGF-stimulated EGFR activation, as determined by overall receptor tyrosine phosphorylation, phosphorylation on the major in vivo autophosphorylation site Y1173, and phosphorylation of the downstream signaling targets, ERK1/2. In contrast, over longer EGF treatments, SB202190 efficiently blocked ligand-driven EGFR downregulation. Continuous EGF exposure for 4 or 24 h (identical results obtained; only 4 h shown) resulted in a >85% loss of EGFR in cultures pretreated with vehicle alone (Figure 1B), but SB202190 substantially attenuated this downregulation. In addition, an appreciable portion of the preserved EGFR was still tyrosine-phosphorylated (Figure 1B, third panel). These effects result from altered degradation rather than a change in EGFR synthesis, as CHX blockade of protein translation did not interfere with SB202190 preservation of EGFR (Supplementary Figure 1). Similar results were obtained with another p38 inhibitor, SB220025 (data not shown). Protection of EGFR from ligand-induced destruction was also observed in the A431, MDCK, and Cos-7 cell lines (Figure 1B, lower panels), indicating that this effect is not restricted to a particular cell line or tissue.
Figure 1.
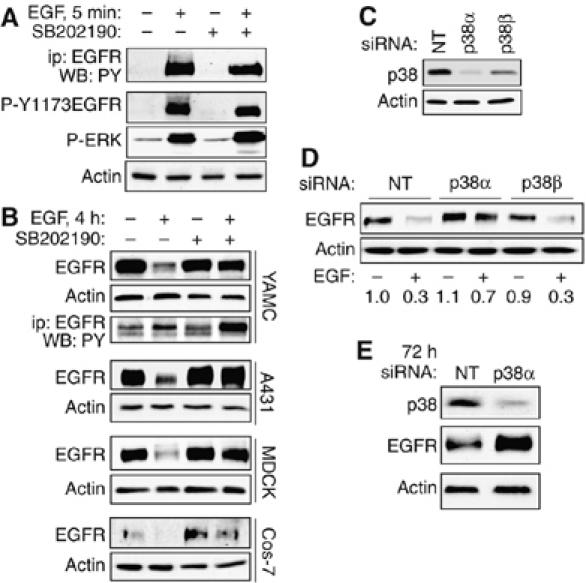
p38 is required for EGFR downregulation. (A) YAMC cell cultures were stimulated with 10 ng/ml EGF for 5 min with or without 30 min p38 inhibitor (SB202190, 10 μM) pretreatment. Top panel: EGFR immunoprecipitated from cell lysates was subjected to SDS–PAGE and Western blot analysis using anti-PY. Bottom three panels: whole-cell lysates were subjected to SDS–PAGE and Western blotting using antibodies specific for phospho (P)-Y1173 EGFR, P-ERK1/2, and actin (loading control). (B) Indicated cell lines were exposed to EGF (all 10 ng/ml except A431 100 ng/ml) with or without SB202190 for 4 h. EGFR and actin levels were determined by Western blot analysis of whole-cell lysates. Third panel: PY in EGFR immunoprecipitates from YAMC cells. (C) YAMC cells were transfected for 48 h with 100 nM siRNA pools against p38α or p38β, with a nontargeting pool (NT) as control. Whole-cell lysates were subjected to Western blot analysis for p38 expression to determine the relative contribution of different isoforms to the immunoreactive band. (D) Transfected YAMC cells were exposed to 10 ng/ml EGF for 4 h. Whole-cell lysates were subjected to Western blot analysis for EGFR. Numbers are averaged densitometry (relative to control) from three experiments. (E) Cells were transfected with NT or p38α siRNA for 72 h. Levels of p38 and EGFR were determined by Western blotting.
Loss of p38α from colonic epithelial cells results in accumulation of EGFR
To identify which p38 isoform is involved in EGFR downregulation, YAMC cells were transfected with siRNA pools selectively targeting either p38α or p38β. Western blot analysis of these cells (Figure 1C) indicated that p38α is the primary species detected in YAMC cells by an antibody which recognizes both p38α and p38β, although both isoform-specific siRNA pools decreased the total p38 signal to some extent. Incubation with p38α, but not p38β, siRNA for 48 h attenuated ligand-stimulated receptor loss (Figure 1D), confirming the results obtained by chemical inhibition and implicating p38α as the primary isoform regulating EGFR levels. A small increase in basal EGFR in p38α siRNA-transfected cells was also observed, and long-term incubation with p38α siRNA resulted in a substantial (2.1±0.2-fold from three experiments) increase in EGFR expression (Figure 1E). This may result from blockade of basal receptor turnover stimulated by constitutive release of low levels of EGFR ligands.
p38 regulates EGFR phosphorylation on Y1045
Our initial data indicated that p38 blockade does not inhibit overall tyrosine phosphorylation of EGFR (Figure 1A), but the possibility remained that changes in specific tyrosines could be masked by the overall phosphotyrosine (PY) content of the receptor. Therefore, we treated YAMC cells with EGF from 3 to 20 min with or without SB202190 and performed Western blot analysis using antibodies directed against individual EGFR phosphosites. Specificity of these antibodies was confirmed in EGFR−/− mouse colon epithelial (MCE) cells re-expressing respective Y>F mutant EGFR constructs (RS Dise, T Yamaoka, and DB Polk, unpublished results). Phosphorylation of most tyrosines examined (Y845, Y992, Y1068, Y1086, Y1148, and Y1173) was not blocked by SB202190 (Figure 2A). In contrast, phosphorylation of Y1045 in response to EGF was completely abolished by p38 blockade.
Figure 2.
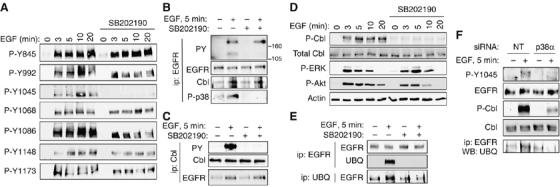
p38 blockade selectively interrupts ligand-driven phosphorylation of EGFR Y1045, c-Cbl activation, and EGFR ubiquitinylation. (A) YAMC cell cultures were exposed to EGF and/or SB202190 (10 μM, 30 min pretreatment) for the indicated times; lysates were subjected to Western blot analysis for EGFR phosphorylation using site-specific antibodies as indicated. (B) Tyrosine-phosphorylated proteins, the EGFR-directed ubiquitin ligase Cbl, and active p38 were detected in EGFR immunoprecipitates using antibodies specific for PY, total Cbl protein, or phospho (P)-p38, respectively. (C) Cbl tyrosine phosphorylation, Cbl expression, and EGFR association with Cbl were analyzed by Western blot of Cbl immunoprecipitates. (D) Lysates from YAMC cells exposed to EGF and/or SB202190 were subjected to Western blot analysis for P-Cbl, total Cbl, P-ERK, and P-Akt. (E) EGFR and ubiquitinylated proteins were immunoprecipitated from YAMC cells treated with EGF for 5 min with or without p38 inhibitor. Immunocomplexes were separated by SDS–PAGE and subjected to Western blot analysis for ubiquitin and EGFR. (F) YAMC cells were transfected for 48 h with 100 nM nontargeting (NT) or p38α siRNA. P-Y1045 EGFR, EGFR, P-Cbl, Cbl, and ubiquitinylated EGFR levels after 5 min EGF exposure were determined as above.
EGF-induced c-Cbl phosphorylation and EGFR ubiquitinylation require p38 activity
Y1045 is thought to be important for EGF-induced c-Cbl activation and subsequent EGFR ubiquitinylation (Grovdal et al, 2004). Thus, we examined the phosphorylation state of the Cbl ubiquitin ligase following EGF exposure in the presence and absence of p38 inhibitor. In EGFR immunoprecipitates blotted for PY (Figure 2B), an EGF-stimulated band of approximately the same mobility as Cbl (∼120 kDa) disappeared in the presence of SB202190. In addition, Cbl immunoprecipitated from EGF-treated YAMC cells showed robust tyrosine phosphorylation, which was completely ablated by p38 inhibitor (Figure 2C). Interestingly, a band recognized by phospho-p38 antibody co-immunoprecipitated with EGFR and Cbl in EGF-treated cells (Figure 2B), suggesting that active p38 complexes with EGFR/Cbl. Western blot analysis using an antibody against phospho-Cbl confirmed that EGF's ability to stimulate Cbl phosphorylation was completely blocked by SB202190 despite robust activation of other EGFR-signaling targets such as ERK1/2 (Figure 2D) or Akt, and phosphorylation of EGFR itself (Figure 2A). However, total Cbl association with EGFR was not affected by SB202190 (Figure 2B and C) or p38 siRNA (Supplementary Figure 2), in agreement with reports showing that EGFR mutants incapable of phosphorylating Cbl can still bind the ubiquitin ligase and undergo ligand-induced internalization but not degradation.
As EGFR-stimulated Cbl tyrosine phosphorylation in YAMC cells requires p38 activity, we asked whether the same is true of EGFR ubiquitinylation by immunoprecipitating with anti-EGFR or -ubiquitin from YAMC cells exposed to EGF with or without SB202190. EGFR immunocomplexes from EGF-treated cells contain ubiquitin and vice versa, and ubiquitinylation of the receptor is abolished by pretreatment with SB202190 (Figure 2E). A requirement for p38 in the Cbl-mediated EGFR ubiquitinylation pathway was also observed in A431, MDCK, and Cos-7 cells (Supplementary Figure 3), indicating that this effect is not restricted to colon epithelial cells.
To confirm the specificity of the results obtained with SB202190, YAMC cells were transfected with nontargeting or p38α-directed siRNA. The p38α siRNA blocked EGF-induced EGFR Y1045 and Cbl phosphorylation as well as EGFR ubiquitinylation (Figure 2F), validating the chemical inhibitor data.
p38 inhibitor blockade of EGFR ubiquitinylation is independent of receptor internalization
To rule out a defect in intracellular transport as the basis of our results, we performed experiments at 4°C. When EGF is incubated with YAMC cells at 4°C, immunofluorescence localization shows that internalization of EGFR is blocked (Figure 3A), but acute signaling events such as phosphorylation of p38 (Figure 3B), EGFR, and Cbl (Figure 3C) are preserved. Under these conditions, SB202190-mediated blockade of Cbl phosphorylation and EGFR ubiquitinylation was still observed (Figure 3C), demonstrating that these effects are prior to internalization and not a result of altered endocytic transport. To provide further evidence that SB202190-induced blockade of EGFR downregulation is not a result of altered internalization rates, quantitative internalization assays using biotin-labeled EGF were performed with and without SB202190 pretreatment. The p38 inhibitor did not block endocytosis of EGF-Biotin/EGFR complexes (Figure 3D).
Figure 3.
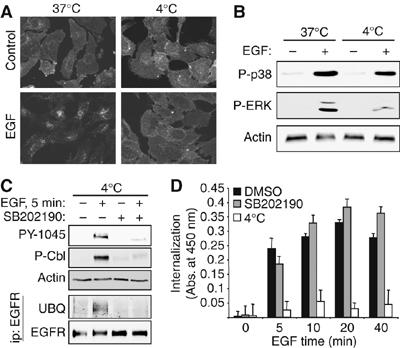
p38 regulation of EGFR ubiquitinylation is independent of receptor internalization. (A) YAMC cells were treated with EGF for 20 min at 37 or 4°C and immunostained for EGFR localization. (B) YAMC cells treated with EGF at 37 or 4°C were subjected to Western blot analysis for p38 and ERK activation. (C) Lysates from EGF-treated YAMC cells (with or without 30 min SB202190 pretreatment) were subjected to immunoblot analysis for EGFR Y1045 and Cbl phosphorylation. EGFR was immunoprecipitated from lysates and immunocomplexes were blotted for ubiquitin. (D) YAMC cells were pretreated with SB202190 for 30 min and exposed to biotin-labeled EGF for the indicated times, then EGFR internalization was determined as described in Materials and methods. Specificity of signal was confirmed by competition with 10-fold excess unlabeled EGF (not shown); positive control for inhibition of uptake was incubation at 4°C. No significant decreases (all P>0.05) with SB202190 were observed.
p38 is required for maintenance of ‘degradation-competent' EGFR after internalization
In our previous studies we showed that EGF activates p38 in a biphasic manner, with both the acute and sustained phases required for EGF-stimulated wound healing in YAMC cells (Frey et al, 2004). To determine whether sustained p38 activity is similarly required for ligand-induced EGFR downregulation, YAMC cells were exposed to EGF for 4 h with p38 inhibitor added either 30 min before or at various times after EGF. SB202190 added 15 min, 1, or 2 h after the beginning of EGF treatment still attenuated EGFR loss (Figure 4A). Furthermore, addition of SB202190 for the last 5 min of 15 or 60 min EGF exposure reversed stimulated phosphorylation of EGFR Y1045 and Cbl, as well as EGFR ubiquitinylation (Figure 4B and C). Thus, inactivation of p38 even after receptor internalization blocks these processes.
Figure 4.
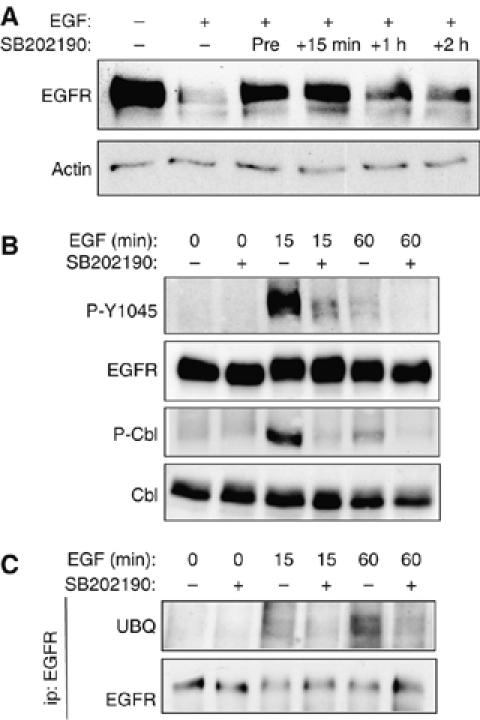
Sustained p38 activity is required for ligand-induced EGFR degradation. (A) YAMC cell cultures were treated with 10 ng/ml EGF continuously for 4 h, with either 10 μM SB202190 pretreatment (30 min before and throughout assay) or SB202190 added ‘late' (15 min, 1, or 2 h after beginning of EGF treatment). EGFR downregulation was assessed by Western blot analysis. (B, C) YAMC cells were exposed to EGF for the indicated times, with or without SB202190 addition for last 5 min of treatment only. Whole-cell lysates (B) or EGFR immunoprecipitates (C) were analyzed by Western blot for P-Y1045 EGFR, P-Cbl, EGFR, Cbl, and ubiquitin.
p38 activity is required for EGF-induced EGFR degradation but not internalization
Several recent reports show that phenylalanine mutation of EGFR on tyrosine 1045 (Y1045F) produces a receptor which, like EGFR in p38 inhibitor-treated cells, is deficient in its ability to phosphorylate Cbl. Furthermore, like EGFR in cells given SB202190, Y1045F EGFR is not efficiently ubiquitinylated nor rapidly downregulated following ligand binding (Grovdal et al, 2004; Ravid et al, 2004). Interestingly, this EGFR mutant internalizes normally but then accumulates in early endosomes and never transits to the lysosome. As p38 activity is required for EGFR tyrosine phosphorylation on Y1045, we asked whether p38 inhibition also arrests intracellular trafficking of internalized EGFR. YAMC cells were exposed to EGF in the presence or absence of SB202190, fixed, and subjected to immunofluorescence localization analysis for EGFR. Some cultures were loaded with lysotracker dye, which accumulates in acidic vesicles and thus identifies lysosomal compartments. In cells treated with EGF alone, EGFR is internalized, transits through an intracellular vesicular compartment, and is lost from the cells within 4 h (Figure 5A). In contrast, cells pretreated with p38 inhibitor before EGF exposure internalize EGFR normally, but the internalized receptor accumulates in intracellular vesicles. EGFR in vehicle-treated cells exposed to EGF transits to a lysotracker-positive compartment, indicating recruitment to the lysosome for degradation; this is not seen in the presence of SB202190 (Figure 5B).
Figure 5.
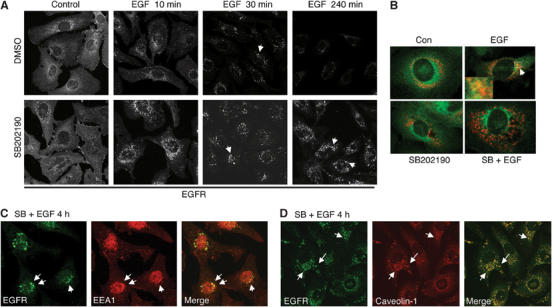
p38 inhibition causes accumulation of internalized receptor in the endosomal compartment. To visualize the internalization process of EGFR after ligand treatment, YAMC cell cultures were treated with EGF in the presence or absence of SB202190. (A) Cells were fixed at the indicated times, immunostained for EGFR, and visualized by confocal laser scanning microscopy. Arrows, EGFR-containing vesicles. (B) Cells were exposed to EGF for 1 h in the presence of 50 nM Lysotracker Red dye and immunostained for EGFR. Arrows indicate EGFR/Lysotracker colocalization. (C) YAMC cells treated with EGF and SB202190 for 4 h were immunostained for EGFR (green) and the early endosomal marker EEA1 (Red). Arrows indicate colocalized EGF and EEA1. (D) YAMC cells treated with EGF and SB202190 for 4 h were immunostained for EGFR (green) and caveolin-1 (Red). Arrows indicate colocalized EGFR and caveolin.
To investigate the identity of the EGFR-containing vesicles, we immunostained cells treated with EGF and SB202190 for trafficking markers. A subset of the EGFR-containing vesicles at 4 h contain the early endosomal marker EEA1 (Figure 5C), paralleling the results reported in Y1045F mutant EGFR cells (Grovdal et al, 2004; Ravid et al, 2004). Additionally, a majority of EGFR-containing vesicles were associated with caveolin-1 (Figure 5D), a mediator of cell surface receptor trafficking and recycling pathways (Gagescu et al, 2000; Parton, 2004).
Hyperactivation of p38 results in loss of EGFR
The data described above show that p38 MAPK is required for EGFR downregulation in response to ligand exposure. To determine whether p38 stimulation itself is sufficient to trigger EGFR downregulation, we treated YAMC cells with the antibiotic anisomycin (which activates p38 and other stress kinases), or transfected cells with a constitutively active form of the upstream kinase of p38, MKK6. Both of these methods stimulated p38, as shown by phosphorylation of its target ATF-2, and decreased EGFR protein levels (Figure 6A). Furthermore, SB202190 significantly attenuated anisomycin-driven EGFR loss (Figure 6B). Anisomycin treatment also stimulated phospho-Y1045 EGFR and phospho-Cbl (Figure 6C). However, EGFR ubiquitinylation was promoted only weakly, in accord with a recent report describing ubiquitinylation-independent EGFR degradation by anisomycin (Vergarajauregui et al, 2006). p38 appears to participate in multiple EGFR regulatory mechanisms, the ligand-dependent mechanism described in this paper and the tyrosine kinase/Cbl-independent mechanism described by others.
Figure 6.
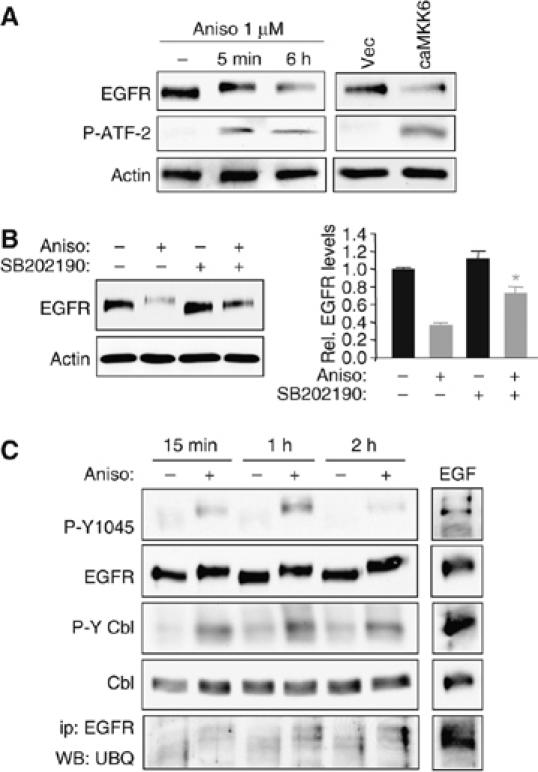
Forced p38 activation promotes EGFR degradation in YAMC cells. (A) YAMC cells were exposed to the p38-activating antibiotic anisomycin (Aniso; 1 μM) for the indicated times, or transfected with a constitutively active MKK6 construct (caMKK6) or vector control (Vec). Western blot analysis was performed for EGFR levels, and for phospho-ATF-2 as a p38 activity readout. (B) YAMC cells were given anisomycin for 6 h with or without 30 min SB202190 pretreatment; EGFR expression was determined by Western blotting. Densitometry shows averaged results from three experiments. *P<0.001 versus anisomycin alone. (C) Cells were treated with anisomycin or vehicle for the indicated times. Whole-cell lysates were subjected to Western blot analysis for P-Y1045 EGFR, P-Cbl, EGFR, and Cbl; EGFR immunoprecipitates were probed for ubiquitin. EGF lane (5 min exposure) is included as positive control.
EGFR Y1045 phosphorylation serves as a molecular switch regulating migration
As detailed above, the effects of p38 inhibition on EGFR internalization and degradation are remarkably similar to published reports regarding Y1045F EGFR (Grovdal et al, 2004; Ravid et al, 2004). Thus, we asked whether the previously reported effects of p38 blockade in abrogating EGF-stimulated restitution, but potentiating EGF-driven proliferation (Frey et al, 2004), might be accounted for by the observed loss of Y1045 phosphorylation and Cbl activation (Figure 2). We subjected EGFR−/− MCE cells stably transfected with wild-type (WT) or Y1045F-mutated EGFR to in vitro wound healing and proliferation assays. Y1045F add-back cells activate p38 in response to EGF (Figure 7A; for additional analysis of p38 activation by EGFR Y>F mutants, see Supplementary Figure 4), suggesting that p38 is upstream of EGFR Y1045 phosphorylation (Figure 2A), but as predicted they fail to phosphorylate Cbl or show increased migration in response to EGF as compared to WT add-back cells (Figure 7B). In contrast, the Y1045F mutation does not interfere with EGFR/Cbl association (Supplementary Figure 2) or EGF-induced cell proliferation (Figure 7C). Thus, this mutant phenocopies the effects of p38 inhibition in YAMC cells (Frey et al, 2004). The influence of p38 blockade on EGF-stimulated cell growth and migration may be accounted for by deregulation of EGFR localization and destruction following internalization. Furthermore, these findings demonstrate uncoupling of EGFR-stimulated cellular migration from proliferation and support Y1045 as a molecular switch controlling EGF-induced wound healing.
Figure 7.
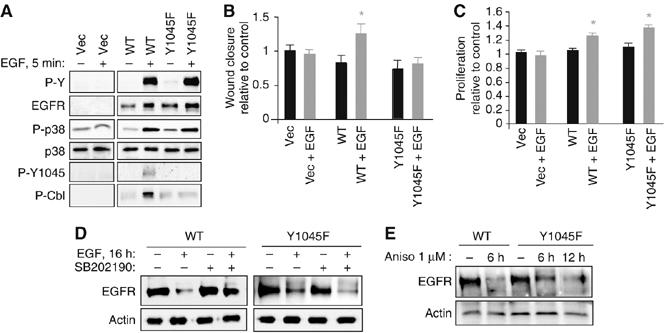
EGFR Y1045F mutation uncouples EGF-stimulated cell migration from proliferation in mouse colon epithelial cells, similar to p38 inhibition. (A) EGFR−/− MCE cells transfected with WT EGFR, Y1045F EGFR, or vector (Vec) were treated with EGF for 5 min; EGFR expression and phosphorylation, p38 activation, and Cbl phosphorylation were determined by Western blotting with antibodies specific to EGFR, PY, phospho (P)-p38, P-Y1045 EGFR, and P-Cbl. (B, C) EGFR−/− MCE cells transfected with vector, WT EGFR, or Y1045F mutant EGFR were exposed to EGF and subjected to (B) in vitro wound healing or (C) proliferation assays. *P<0.005 versus relevant control. (D) WT or Y1045F EGFR-expressing cells were exposed to EGF for 4 h with or without 30 min SB202190 pretreatment and Western blot analysis was performed for EGFR levels. (E) Cells were exposed to 1 μM anisomycin for the indicated times and EGFR levels in whole-cell lysates were determined.
Others have reported that Y1045F EGFR does eventually downregulate in response to ligand, but more slowly and presumably via a different mechanism than WT EGFR (Ravid et al, 2004). As shown in Figure 7D, Y1045F EGFR was indeed downregulated by long-term (16 versus >4 h in Figure 1) EGF exposure. However, this depletion was not sensitive to SB202190, in contrast to the WT receptor. Thus, Y1045F EGFR is apparently cleared by a separate mechanism not dependent on p38. Y1045F EGFR was also sensitive to anisomycin on a longer time course than WT EGFR (Figure 7E), likely due to the degradation pathway described by Vergarajauregui et al.
Discussion
This report constitutes the first demonstration of a novel regulatory pathway in which EGFR-stimulated p38 MAPK activity is required for ligand-driven ubiquitinylation and destruction of activated receptor. Inhibition of p38α in the context of EGF exposure blocked EGFR Y1045 phosphorylation, activation of Cbl, and EGFR ubiquitinylation. As a result, tyrosine-phosphorylated EGFR failed to transit to acidic lysosomes and was preserved in the cell for a sustained period. These data phenocopy the EGFR Y1045F mutant, which is internalized but not ubiquitinylated or efficiently degraded in response to ligand exposure (Grovdal et al, 2004). Conversely, MCE cells expressing Y1045F EGFR responded to EGF in the same way as cells treated with p38 inhibitor: migration was impaired but stimulated proliferation was potentiated (Figure 7 and Frey et al, 2004). Thus, interrupting EGFR phosphorylation of Cbl recapitulates the effects of p38 inhibition on intestinal epithelial cells, suggesting that EGFR Y1045 phosphorylation and Cbl activation are key targets of the p38-mediated migration/proliferation switch.
A role for phosphorylation of EGFR Y1045 and Cbl in determining either a migratory or proliferative phenotype following EGFR activation may be important in multiple physiological contexts. The oncogenic potential of some Cbl truncations correlates with failure to ubiquitinylate EGFR (Levkowitz et al, 1999), although it must be noted that RING finger mutations abolishing EGFR desensitization are not always sufficient for transformation (Thien et al, 2001). With regard to cell motility, loss of Cbl during fly oogenesis deregulates tyrosine kinase-mediated directional sensing, interfering with coordinated cell migration (Jekely et al, 2005), and studies of bone morphogenesis show Cbl dependence for migration of osteoclasts (Chiusaroli et al, 2003).
Several lines of initial evidence correlated Cbl activity and EGFR ubiquitinylation to both receptor internalization and degradation. For example, mutation of EGFR S1046/47 inhibits both ubiquitinylation and internalization (Oksvold et al, 2003), and interruption of EGFR/Grb2/Cbl complexes with Sprouty overexpression blocks both ubiquitinylation and clathrin-coated pit endocytosis of EGFR (Stang et al, 2004). However, recent results from several labs indicate that while receptor ubiquitinylation is required for transit to the lysosome and degradation, it is dispensable for internalization. Both Y1045F EGFR (Ravid et al, 2004) and EGFR mutated on kinase domain lysine residues targeted for polyubiquitinylation (Huang et al, 2006) are defective in transit to the lysosome without any change in endocytosis rates. Our results are in agreement with these data in that p38 inhibition selectively targets Cbl activity and EGFR ubiquitinylation/degradation without affecting internalization. It should be noted, however, that both mono- and polyubiquitinylation of EGFR have been reported (Mosesson et al, 2003; Huang et al, 2006), and the possibility remains that, for example, monoubiquitinylation promotes internalization while polyubiquitinylation is required for degradation.
Our results imply that simple EGFR activation at the cell membrane is insufficient to stimulate wound closure. Rather, receptor activation followed by modification and appropriate subcellular localization or termination of the signal seems to be required for efficient restitution. Internalized EGFR has access to a restricted or altered panel of effector molecules; for example, a study using fluorescent probes for phosphatidylinositol-4,5-diphosphate in fibroblasts showed that EGFR in the endosome does not effectively stimulate phospholipase C activity despite robust activation of other targets (Haugh and Meyer, 2002). Other work on MDCK cells demonstrates that endosomal EGFR signals select for proliferation and survival (Pennock and Wang, 2003). Transit time through specific compartments may greatly affect the outcome of a signal. Thus, p38's influence on EGFR ubiquitinylation, which in turn affects intracellular EGFR sorting, could have a significant impact on cell physiology. Furthermore, the need for sustained p38 activity to maintain a ‘downregulation-competent' receptor (Figure 4) emphasizes the importance of the duration as well as strength of MAPK signals.
p38 may play additional roles in EGFR processing through modulation of the vesicular compartment containing the receptor. Studies on opioid receptor endocytosis showed that p38α can phosphorylate and alter the activity of the early endosomal marker EEA1 (Mace et al, 2005), which colocalizes with some preserved EGFR-containing vesicles in our study. In addition, EGFR phosphorylation is altered in vitro by direct interaction with trafficking proteins such as caveolin-1 (Couet et al, 1997).
An interesting context for the results in this paper is recent observations that p38 can also promote a separate mechanism of ligand-independent EGFR internalization in response to cellular stress. In HeLa cells, anisomycin treatment (Vergarajauregui et al, 2006), inflammatory cytokines, or UV irradiation (Zwang and Yarden, 2006) initiate p38-dependent EGFR internalization that does not require ubiquitinylation or degradation in the lysosome. Similarly, Y1045F EGFR, which lacks the ability to activate the Cbl-mediated pathway, is degraded by anisomycin albeit more slowly than wild-type receptor (Figure 7E). It is interesting that both ligand-stimulated degradation and the stress–response internalization pathway appear to utilize the same MAPK, especially considering that our data demonstrate a requirement for p38 in EGFR ubiquitinylation and destruction, but not internalization, in ligand-stimulated desensitization. As yet it is unclear what, if any, interaction or crosstalk occurs between the two mechanisms. While anisomycin promotes some Y1045 EGFR phosphorylation, Cbl activation, and EGFR ubiquitinylation in YAMC cells (Figure 6C), the receptor loss observed appears greater than can be explained by this modest response and is likely at least in part the result of the Cbl-independent mechanism described by Vergarajauregui et al (2006).
A central question raised by these studies is the mechanism by which p38 promotes EGF-induced EGFR Y1045 phosphorylation and Cbl activation. The simplest possibility is a model in which p38 directly associates with EGFR/Cbl complexes (Figure 2B) and phosphorylates EGFR on Ser/Thr residues in a fashion permissive for Y1045 phosphorylation. T669 on EGFR can be a substrate for p38α in vitro (Morrison et al, 1993; Chen et al, 2000), and p38-dependent phosphorylation of EGFR on other residues has recently been demonstrated (Zwang and Yarden, 2006). Phosphorylation of EGFR in the juxtamembrane region by protein kinase C exerts a regulatory effect on the receptor (Hunter et al, 1984), and similarly p38-mediated phosphorylation might drive a conformational change increasing the availability of Y1045. Alternatively, p38 may inhibit phosphatases acting on EGFR or Cbl. In this regard, p38 is required for nitric oxide-stimulated EGFR activation in A431 cells in an orthovanadate-sensitive manner (Ruano et al, 2003). Also interesting, given that EGF-stimulated p38 activation in YAMC cells is Src-dependent (Frey et al, 2004), is work showing that EGFR activation by oxidative injury in renal epithelial cells requires Src-stimulated p38 activation (Zhuang et al, 2005). Further investigation is needed to determine the relative roles of direct EGFR phosphorylation by p38 versus regulation of other kinases and phosphatases in both ligand-induced downregulation and response to stimuli such as oxidative injury or UV irradiation.
Establishing a role for p38 MAPK in EGFR downregulation adds an additional dimension to the already-described link between this MAPK and the pathogenesis of ulcerative colitis and other inflammatory bowel diseases. Hyperactivation of p38 has been reported clinically in these disorders; furthermore, p38 promotes production of inflammatory cytokines such as TNFα (Waetzig et al, 2002). The potential importance of EGFR in colitis has been demonstrated by clinical trials showing a therapeutic effect of topical EGF in ulcerative colitis patients (Sinha et al, 2003). Furthermore, mice harboring defects or deletions in EGFR or EGFR ligands exhibit a worsened phenotype in experimental colitis models (Procaccino et al, 1994). Thus, p38-mediated regulation of EGFR may impact growth factor-based therapy for inflammatory conditions. It should be noted, however, that studies of p38 inhibitors in experimental colitis models have yielded ambiguous results (Arulampalam and Pettersson, 2002) with one group reporting improvement (Hollenbach et al, 2004), and another improvement in some parameters but worsening of others (ten Hove et al, 2002). It is possible that alternative targets uncovered by investigation of p38-stimulated pathways in intestinal epithelial cells may provide avenues of therapeutic intervention.
In summary, we propose a novel mechanism of feedback regulation through which the p38 MAPK signaling cascade is required for termination of upstream EGFR signaling. In this model, EGF-driven phosphorylation of EGFR on Y1045, as well as subsequent Cbl activation, EGFR ubiquitinylation, and destruction of activated receptor, are dependent on p38α activity. These data provide an explanation for p38-mediated regulation of EGF-stimulated epithelial wound healing and proliferation, and suggest potential future avenues of investigation into EGFR trafficking and intracellular signaling. Furthermore, they provide evidence that phosphorylation of Y1045 on EGFR serves as a molecular switch regulating ligand-stimulated migration that is uncoupled from EGF-driven proliferation.
Materials and methods
Antibodies, probes, growth factors and inhibitors
Lysotracker Red dye was obtained from Invitrogen Molecular Probes (Carlsbad, CA). Anisomycin and cycloheximide were purchased from Sigma. Mouse EGF was a gift from Stanley Cohen (Vanderbilt University). SB202190 and SB220025 were purchased from Calbiochem (San Diego, CA). In all experiments using SB202190, unless otherwise noted, cells were pretreated for 30 min and control cells were given an equivalent concentration of vehicle. Optimal concentration of SB202190 for inhibition of EGF-stimulated p38 activity was determined by a concentration curve for blockade of ATF-2 phosphorylation (Supplementary Figure 5). For antibody sources, please see Supplementary data.
Cell culture
Conditionally immortalized YAMC and EGFR−/−MCE cells were derived by Dr Robert Whitehead of the Vanderbilt DDRC novel cell line core (Whitehead et al, 1993). These cells express heat-labile simian virus 40 large T antigen under the control of an interferon (IFN)-γ-inducible promoter. Cells were maintained on rat tail collagen (Mediatech, Herndon, VA)-coated plates in RPMI 1640 with 5% fetal bovine serum (FBS), 5 U/ml mouse IFN-γ (Intergen, Norcross, GA), 100 U/ml penicillin and streptomycin, 5 μg/ml insulin, 5 μg/ml transferrin, and 5 ng/ml selenous acid (BD Biosciences, San Jose, CA) at 33°C (permissive conditions). In preparation for experiments, cells were shifted to RPMI 1640 containing only 0.5% FBS, streptomycin, and penicillin at 37°C (nonpermissive conditions) for 16 h.
A431, MDCK, and Cos-7 cells were maintained in Dulbecco's modified Eagle's medium with 4 mM L-glutamine and 10% FBS at 37°C. In preparation for experiments, cells were transferred to medium with 4 mM L-glutamine and 0.5% FBS for 16 h.
EGFR internalization assays
EGFR endocytosis rates were determined using the method of de Wit et al (2000). Briefly, YAMC cells were plated at 20 000 cells/well in 96-well dishes, grown for 24 h, and shifted to nonpermissive conditions for 16 h. Cultures were washed and treated with 20 ng/ml biotin-labeled EGF (Invitrogen Molecular Probes) in PBS with 0.9 mM CaCl2, 0.5 mM MgCl2, and 5 mM glucose, followed by acid wash in 125 mM NaCl, 25 mM acetic acid, pH 3 to strip off surface-bound EGF. Cells were fixed in PBS containing 3% paraformaldehyde, 0.25% glutaraldehyde, and 0.25% Triton X-100, treated with 50 mM glycine, blocked, and incubated with streptavidin–HRP. Internalized HRP was detected by o-Phenylenediamine dihydrochloride (Sigma) absorbance at 450 nm.
Cell lysates, SDS–PAGE, and Western blotting
Lysates were prepared by scraping in modified RIPA buffer (50 mM Tris, pH 7.4, 150 mm NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.2% sodium deoxycholate, 0.1% SDS, and 0.1% protease and phosphatase inhibitor cocktails (Sigma)). Aliquots of lysates (30 μg) boiled in Laemmli sample buffer were separated on SDS–polyacrylamide gels (6, 7.5, or 10% gel as appropriate) and blotted on nitrocellulose membranes as previously described (Frey et al, 2004). Loading was routinely monitored by fast green staining and Western blot analysis for actin and at least one additional unaffected protein.
Immunoprecipitation
To recover EGFR, ubiquitinylated proteins, or Cbl, YAMC cells were scraped on ice in RIPA buffer without SDS or deoxycholate. In total, 1 mg of each precleared lysate was nutated for 1 h at 4°C with 2 μg of appropriate antibody and then for 1 h with Protein A/G PLUS-Agarose beads (Santa Cruz). Beads were collected, washed 3 × with lysis buffer, and eluted by boiling in Laemmli sample buffer.
Migration assays and cell wounding
Cells were plated at confluent density on 35 mm dishes coated with fibronectin. Eight circular wounds of equal size (∼1.5 mm2) were made in each monolayer as previously described (Frey et al, 2004). Wounds were photographed over time and measured using ImageJ software (NIH, Bethesda, MD). Percent closure for each wound was determined at 8 h; values are averages of all eight wounds.
Proliferation assays
Cells were plated in 96-well dishes (5 × 103 per well) under nonpermissive conditions. Cultures were exposed to EGF or vehicle for 24 h. Cells were counted at 0 and 24 h using an MTS-based colorimetric proliferation assay kit (Promega Corp.). Reported values reflect averages of at least 12 replicate wells.
Constructs, transfections, and mutant cell lines
Constitutively active MKK6 was the gift of Graham Carpenter (Vanderbilt University). Pooled siRNAs targeting mouse p38α and p38β were purchased from Dharmacon (Lafayette, CO). All transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Mutant Y1045F-EGFR was made from pcDNA3.1/Zeo(−)/EGFR (human) using QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) (primer sequence 5′-AGCTTCTTGCAGCGATTCAGCTCAGACCCCACA-3′). EGFR−/− MCE cells were stably transfected with pcDNA3.1/Zeo(−) empty vector (Invitrogen), pcDNA3.1/Zeo(−)/wild-type EGFR, or pcDNA3.1/Zeo(−)/Y1045F-EGFR using Zeocin selection. Selected populations were collected and stained with anti-EGFR 528-PE-conjugated antibody, then sorted at the Nashville VA Medical Center Flow Cytometry Special Resource Center using a Becton-Dickinson FACSAria. Multiple sorting rounds were performed until a population of cells 100% positive for PE was obtained. Other mutant-expressing lines were generated using similar methods.
Immunofluorescence localization analysis and microscopy
For analysis of EGFR, EEA1, and caveolin expression/localization, cells grown on collagen-coated Lab-Tek chamber slides (Nalge Nunc International, Rochester, NY) were fixed in 4% paraformaldehyde, washed in PBS and equilibrated in PBSt (PBS plus 0.2% Triton X-100). Primary antibodies were added for 2 h in a humidified chamber, followed by two 5 min PBS washes, 40 min exposure to secondary antibodies, and two more 5 min PBS washes. Slides were mounted using Vectashield (Vector Labs, Burlingame, CA) and viewed on either a Zeiss (Thornwood, NY) LSM 5-510 confocal microscope or an Axiovert 200 microscope equipped with ApoTome optical sectioning and appropriate filters.
Replicates and statistical analysis
All data are representative of at least three independent experiments. Statistical significance of differences between mean values was assessed with paired Student's t-test analysis. Minimum level of significance was set at 0.05.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary data
Acknowledgments
This work was supported by NIH Award DK54993 (DBP), by a Crohn's and Colitis Foundation Research Fellowship Award (MRF) and by the Vanderbilt University Digestive Disease Research Center (NIH Grant DK58404). Confocal LSM imaging was performed through the use of the VUMC Cell Imaging Shared Resource (supported by NIH Grants CA68485, DK20593, DK58404, HD15052, DK59637 and EY08126). We thank the late Holger Kulessa, Ambra Pozzi, Graham Carpenter, and Jim Goldenring for thoughtful discussions and members of the Polk laboratory, especially Wei Tong, for technical advice and support.
References
- Arulampalam V, Pettersson S (2002) Uncoupling the p38 MAPK kinase in IBD: a double edged sword? Gut 50: 446–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blay J, Brown KD (1985) Epidermal growth factor promotes the chemotactic migration of cultured rat intestinal epithelial cells. J Cell Physiol 124: 107–112 [DOI] [PubMed] [Google Scholar]
- Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B (2000) Rab7: a key to lysosome biogenesis. Mol Biol Cell 11: 467–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter G, Cohen S (1976) 125I-labeled human epidermal growth factor. Binding, internalization, and degradation in human fibroblasts. J Cell Biol 71: 159–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter G, Cohen S (eds). (1981) EGF: Receptor Interactions and the Stimulation of Cell Growth. London: Chapman and Hall [Google Scholar]
- Chen G, Porter MD, Bristol JR, Fitzgibbon MJ, Pazhanisamy S (2000) Kinetic mechanism of the p38-alpha MAP kinase: phosphoryl transfer to synthetic peptides. Biochemistry 39: 2079–2087 [DOI] [PubMed] [Google Scholar]
- Chiusaroli R, Sanjay A, Henriksen K, Engsig MT, Horne WC, Gu H, Baron R (2003) Deletion of the gene encoding c-Cbl alters the ability of osteoclasts to migrate, delaying resorption and ossification of cartilage during the development of long bones. Dev Biol 261: 537–547 [DOI] [PubMed] [Google Scholar]
- Couet J, Sargiacomo M, Lisanti MP (1997) Interaction of a receptor tyrosine kinase, EGF-R, with caveolins. Caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. J Biol Chem 272: 30429–30438 [DOI] [PubMed] [Google Scholar]
- Daly JM, Olayioye MA, Wong AM, Neve R, Lane HA, Maurer FG, Hynes NE (1999) NDF/heregulin-induced cell cycle changes and apoptosis in breast tumour cells: role of PI3 kinase and p38 MAP kinase pathways. Oncogene 18: 3440–3451 [DOI] [PubMed] [Google Scholar]
- de Renzis S, Sonnichsen B, Zerial M (2002) Divalent Rab effectors regulate the sub-compartmental organization and sorting of early endosomes. Nat Cell Biol 4: 124–133 [DOI] [PubMed] [Google Scholar]
- de Wit R, Hendrix CM, Boonstra J, Verkleu AJ, Post JA (2000) Large-scale screening assay to measure epidermal growth factor internalization. J Biomol Screen 5: 133–140 [DOI] [PubMed] [Google Scholar]
- Dieckgraefe BK, Weems DM, Santoro SA, Alpers DH (1997) ERK and p38 MAP kinase pathways are mediators of intestinal epithelial wound-induced signal transduction. Biochem Biophys Res Commun 233: 389–394 [DOI] [PubMed] [Google Scholar]
- Dikic I, Giordano S (2003) Negative receptor signalling. Curr Opin Cell Biol 15: 128–135 [DOI] [PubMed] [Google Scholar]
- Frey MR, Golovin A, Polk DB (2004) Epidermal growth factor-stimulated intestinal epithelial cell migration requires Src family kinase-dependent p38 MAPK signaling. J Biol Chem 279: 44513–44521 [DOI] [PubMed] [Google Scholar]
- Gagescu R, Demaurex N, Parton RG, Hunziker W, Huber LA, Gruenberg J (2000) The recycling endosome of Madin–Darby canine kidney cells is a mildly acidic compartment rich in raft components. Mol Biol Cell 11: 2775–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grovdal LM, Stang E, Sorkin A, Madshus IH (2004) Direct interaction of Cbl with pTyr 1045 of the EGF receptor (EGFR) is required to sort the EGFR to lysosomes for degradation. Exp Cell Res 300: 388–395 [DOI] [PubMed] [Google Scholar]
- Harris RC, Chung E, Coffey RJ (2003) EGF receptor ligands. Exp Cell Res 284: 2–13 [DOI] [PubMed] [Google Scholar]
- Haugh JM, Meyer T (2002) Active EGF receptors have limited access to PtdIns(4,5)P(2) in endosomes: implications for phospholipase C and PI 3-kinase signaling. J Cell Sci 115: 303–310 [DOI] [PubMed] [Google Scholar]
- Hollenbach E, Neumann M, Vieth M, Roessner A, Malfertheiner P, Naumann M (2004) Inhibition of p38 MAP kinase- and RICK/NF-kB-signaling suppresses inflammatory bowel disease. FASEB J 18: 1550–1552 [DOI] [PubMed] [Google Scholar]
- Huang F, Kirkpatrick D, Jiang X, Gygi S, Sorkin A (2006) Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol Cell 21: 737–748 [DOI] [PubMed] [Google Scholar]
- Hunter T, Ling N, Cooper JA (1984) Protein kinase C phosphorylation of the EGF receptor at a threonine residue close to the cytoplasmic face of the plasma membrane. Nature 311: 480–483 [DOI] [PubMed] [Google Scholar]
- Janmaat ML, Giaccone G (2003) The epidermal growth factor receptor pathway and its inhibition as anticancer therapy. Drugs Today (Barc) 39 (Suppl C): 61–80 [PubMed] [Google Scholar]
- Jekely G, Sung HH, Luque CM, Rorth P (2005) Regulators of endocytosis maintain localized receptor tyrosine kinase signaling in guided migration. Dev Cell 9: 197–207 [DOI] [PubMed] [Google Scholar]
- Levkowitz G, Waterman H, Ettenberg SA, Katz M, Tsygankov AY, Alroy I, Lavi S, Iwai K, Reiss Y, Ciechanover A, Lipkowitz S, Yarden Y (1999) Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol Cell 4: 1029–1040 [DOI] [PubMed] [Google Scholar]
- Levkowitz G, Waterman H, Zamir E, Kam Z, Oved S, Langdon WY, Beguinot L, Geiger B, Yarden Y (1998) c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev 12: 3663–3674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longva KE, Blystad FD, Stang E, Larsen AM, Johannessen LE, Madshus IH (2002) Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J Cell Biol 156: 843–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace G, Miaczynska M, Zerial M, Nebreda AR (2005) Phosphorylation of EEA1 by p38 MAP kinase regulates mu opioid receptor endocytosis. EMBO J 24: 3235–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison P, Takishima K, Rosner MR (1993) Role of threonine residues in regulation of the epidermal growth factor receptor by protein kinase C and mitogen-activated protein kinase. J Biol Chem 268: 15536–15543 [PubMed] [Google Scholar]
- Mosesson Y, Shtiegman K, Katz M, Zwang Y, Vereb G, Szollosi J, Yarden Y (2003) Endocytosis of receptor tyrosine kinases is driven by monoubiquitylation, not polyubiquitylation. J Biol Chem 278: 21323–21326 [DOI] [PubMed] [Google Scholar]
- Oksvold MP, Thien CB, Widerberg J, Chantry A, Huitfeldt HS, Langdon WY (2003) Serine mutations that abrogate ligand-induced ubiquitination and internalization of the EGF receptor do not affect c-Cbl association with the receptor. Oncogene 22: 8509–8518 [DOI] [PubMed] [Google Scholar]
- Olayioye MA, Neve RM, Lane HA, Hynes NE (2000) The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J 19: 3159–3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton RG (2004) Caveolae meet endosomes: a stable relationship? Dev Cell 7: 458–460 [DOI] [PubMed] [Google Scholar]
- Pennock S, Wang Z (2003) Stimulation of cell proliferation by endosomal epidermal growth factor receptor as revealed through two distinct phases of signaling. Mol Cell Biol 23: 5803–5815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polk DB (1998) Epidermal growth factor receptor-stimulated intestinal epithelial cell migration requires phospholipase C activity. Gastroenterology 117: 493–502 [DOI] [PubMed] [Google Scholar]
- Potten CS, Owen G, Hewitt D, Chadwick CA, Hendry H, Lord BI, Woolford LB (1995) Stimulation and inhibition of proliferation in the small intestinal crypts of the mouse after in vivo administration of growth factors. Gut 36: 864–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procaccino F, Reinshagen M, Hoffmann P, Zeeh JM, Lakshmanan J, McRoberts JA, Patel A, French S, Eysselein VE (1994) Protective effect of epidermal growth factor in an experimental model of colitis in rats. Gastroenterology 107: 12–17 [DOI] [PubMed] [Google Scholar]
- Ravid T, Heidinger JM, Gee P, Khan EM, Goldkorn T (2004) c-Cbl-mediated ubiquitinylation is required for epidermal growth factor receptor exit from the early endosomes. J Biol Chem 279: 37153–37162 [DOI] [PubMed] [Google Scholar]
- Ruano MJ, Hernandez-Hernando S, Jimenez A, Estrada C, Villalobo A (2003) Nitric oxide-induced epidermal growth factor-dependent phosphorylations in A431 tumour cells. Eur J Biochem 270: 1828–1837 [DOI] [PubMed] [Google Scholar]
- Sharma GD, He J, Bazan HE (2003) p38 and ERK1/2 coordinate cellular migration and proliferation in epithelial wound healing: evidence of cross-talk activation between MAP kinase cascades. J Biol Chem 278: 21989–21997 [DOI] [PubMed] [Google Scholar]
- Sinha A, Nightingale J, West KP, Berlanga-Acosta J, Playford RJ (2003) Epidermal growth factor enemas with oral mesalamine for mild-to-moderate left-sided ulcerative colitis or proctitis. N Engl J Med 349: 350–357 [DOI] [PubMed] [Google Scholar]
- Stang E, Blystad FD, Kazazic M, Bertelsen V, Brodahl T, Raiborg C, Stenmark H, Madshus IH (2004) Cbl-dependent ubiquitination is required for progression of EGF receptors into clathrin-coated pits. Mol Biol Cell 15: 3591–3604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Hove T, van den Blink B, Pronk I, Drillenburg P, Peppelenbosch MP, van Deventer SJH (2002) Dichotomal role of inhibition of p38 MAPK with SB 203580 in experimental colitis. Gut 50: 507–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thien CB, Walker F, Langdon WY (2001) RING finger mutations that abolish c-Cbl-directed polyubiquitination and downregulation of the EGF receptor are insufficient for cell transformation. Mol Cell 7: 355–365 [DOI] [PubMed] [Google Scholar]
- Vergarajauregui S, San Miguel A, Puertollano R (2006) Activation of p38 mitogen-activated protein kinase promotes epidermal growth factor receptor internalization. Traffic 7: 686–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waetzig GH, Seegert D, Rosenstiel P, Nikolaus S, Schreiber S (2002) p38 mitogen-activated protein kinase is activated and linked to TNF-α signaling in inflammatory bowel disease. J Immunol 168: 5342–5351 [DOI] [PubMed] [Google Scholar]
- Waterman H, Levkowitz G, Alroy I, Yarden Y (1999) The RING finger of c-Cbl mediates desensitization of the epidermal growth factor receptor. J Biol Chem 274: 22151–22154 [DOI] [PubMed] [Google Scholar]
- Whitehead RH, VanEeden PE, Noble MD, Ataliotis P, Jat PS (1993) Establishment of conditionally immortalized epithelial cell lines from both colon and small intestine of adult H-2Kb-tsA58 transgenic mice. Proc Natl Acad Sci USA 90: 587–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270: 1326–1331 [DOI] [PubMed] [Google Scholar]
- Zhuang S, Yan Y, Han J, Schnellmann RG (2005) p38 kinase-mediated transactivation of the epidermal growth factor receptor is required for dedifferentiation of renal epithelial cells after oxidant injury. J Biol Chem 280: 21036–21042 [DOI] [PubMed] [Google Scholar]
- Zwang Y, Yarden Y (2006) p38 MAP kinase mediates stress-induced internalization of EGFR: implications for cancer chemotherapy. EMBO J 25: 4195–4206 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary data