

Abstract
We use a free energy functional theory to elucidate general properties of heterogeneously ordering, fast folding proteins, and we test our conclusions with lattice simulations. We find that both structural and energetic heterogeneity can lower the free energy barrier to folding. Correlating stronger contact energies with entropically likely contacts of a given native structure lowers the barrier, and anticorrelating the energies has the reverse effect. Designing in relatively mild energetic heterogeneity can eliminate the barrier completely at the transition temperature. Sequences with native energies tuned to fold uniformly, as well as sequences tuned to fold reliably by a single or a few routes, are rare. Sequences with weak native energetic heterogeneity are more common; their folding kinetics is more strongly determined by properties of the native structure. Sequences with different distributions of stability throughout the protein may still be good folders to the same structure. A measure of folding route narrowness is introduced that correlates with rate and that can give information about the intrinsic biases in ordering arising from native topology. This theoretical framework allows us to investigate systematically the coupled effects of energy and topology in protein folding and to interpret recent experiments that investigate these effects.
Keywords: protein folding, energy landscapes, φ values, free energy functional
The energy landscape has been a central paradigm in understanding the physical principles behind the self organization of biological molecules (1–4). A central feature of landscapes of biomolecules that has emerged is that the process of evolution, in selecting for sequences that fold reliably to a stable conformation within a biologically relevant time, induces a new energy scale into the landscape (5–7). In addition to the ruggedness energy scale already present in heteropolymers, it now has the overall topography of a funnel (2, 8–10). A sequence with a funneled landscape has a low-energy native state occupied with large Boltzmann weight at temperatures high enough that folding kinetics is not dominated by slow escape from individual traps.
As an undesigned heteropolymer with a random unevolved sequence is cooled, it becomes trapped into one of many structurally different low-energy states, similar to the phase transitions seen in spin glasses, glasses, and rubber. The low-temperature states typically look like a snapshot of the high-temperature collapsed states but have dramatically slower dynamics. On the other hand, when a designed heteropolymer or protein is cooled, it reliably and quickly finds the dominant low-energy structure(s) corresponding to the native state, in a manner similar to the phase transition from the gas or liquid to the crystal state. As in crystals, the low-temperature states typically have a lower symmetry group than the many high-temperature states (11). Connections have been made between native structural symmetry and robustness to mutations of proteins (11–13). Funnel topographies are maximized in atomic clusters when highly symmetric arrangements of the atoms are possible, as in van der Waals clusters with “magic numbers” (14, 15), and similar arguments have been applied to proteins (11), where funneled landscapes are directly connected to mutational robustness (16).
It is appealing to make the connection between symmetry and designability of native structures to the actual kinetics of the folding process, arguing that symmetry or uniformity in ordering the protein maximizes the number of folding routes and thus the ease of finding a candidate folding nucleus, thus maximizing the folding rate. Explicit signatures of multiple folding routes as predicted by the funnel theory (17, 18) have been seen in simulations of well-designed proteins (8, 19–23) as well as experiments on several small globular proteins (24–26). However, these folding routes are not necessarily equivalent. There is an accumulating body of experimental (27–31) and simulation (22, 32–42) evidence that shows varying degrees of heterogeneity in the ordering process. These data refine the funnel picture by focusing on which parts of the protein most effectively contribute to ordering and on the effects of native topology and native energy distribution on rates and stability. The ensemble of foldable sequences with a given ratio of TF/TG > 1 has a wide distribution of mean first passage times (17, 33, 43), indicating that several other properties of the sequence and structure contribute to folding thermodynamics and kinetics. These include topological properties of the native structure (11, 44–51) (e.g., mean loop length 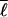 , dispersion in loop length δℓ, and kinetic accessibility of the native structure), the distribution over contacts of total native energy in the protein, and the coupling of contact energetics with native topology.
, dispersion in loop length δℓ, and kinetic accessibility of the native structure), the distribution over contacts of total native energy in the protein, and the coupling of contact energetics with native topology.
In this paper, we integrate the above sundry observations into a theory that explicitly accounts for native heterogeneity, structural and energetic, in the funnel picture. We introduce a simple field theory with a nonuniform order parameter to study fluctuations away from uniform ordering, through free energy functional methods introduced earlier by Wolynes and collaborators (36, 48)a. The theory is in agreement with simulations also performed in this paper. We organize the paper as follows. First we outline the calculation and results. Next we derive and use an approximate free energy functional that captures the essence of the problem. Then we conclude and suggest future research, leaving technical aspects of the derivation for the methods section.
Outline
The free energy functional description allows, in principle, for a fairly complete understanding of the folding process for a particular sequence; this includes effects caused by the three-dimensional topological native structure, possible misfolded traps, and heterogeneity among the energies of native contacts. We model a well-designed minimally frustrated protein with an approximate functional, but many of the results we obtain are quite general. We find that for a well-designed protein, gains in loop entropy and/or core energy always dominate over losses in route entropy, so the thermodynamic folding barrier is always reduced by any preferential ordering in the protein.b However, as long as ordering heterogeneity is not too large, there are still many folding routes to the native structure, and the funnel picture is valid. When there are very few routes to the native state because of large preferential ordering, folding is slow and multiexponential at temperatures where the native structure is stable. In this scenario, the rate is governed by the kinetic traps along the path induced, rather than the putative thermodynamic barrier that is absent. Several physically motivated arguments giving the above results are described in the supplementary material (www.pnas.org.).
To analyze the effects of native energetic as well as structural heterogeneity on folding, we coarsely describe the native structure through its distributions of native contact energies {ɛi} and native loop lengths {ℓi}. Here, ɛi is the solvent-averaged effective energy of contact i, and ℓi is the sequence length pinched off by contact i. The labeling index i runs from 1 to M, where M = zN is the total number of contacts, N is the length of the polymer, and z, the number of contacts per residue. In the spirit of the density functional theory of fluids (52), we introduce a coarse-grained free energy functional F({Qi(Q)}|{ɛi}, {ℓi}) approximating the physics of secondary (as, e.g., along a helix) and tertiary (nonlocal) contacts in ordering. Q is defined as the overall fraction of native contacts made, used here to stratify the configurations with given similarity to the native state. The fraction of time contact i is made in the subensemble of states at Q is Qi (Q). From a knowledge of this functional, all relevant thermodynamic functions can be calculated in general, such as transition-state entropies and energies, barrier heights, and surface tensions. Moreover, derivatives of the functional give the equilibrium distribution and correlation functions describing the microscopic structure of the inhomogeneous system, as we see below.
Given all the contact energies {ɛi} and loop lengths {ℓi} for a protein, the thermal distribution of contact probabilities {Qi(Q)} is found by minimizing the free energy functional F({Qi(Q)} | {ɛi}, {ℓi}) subject to the constraint that the average probability is Q, i.e., ΣiQi = MQ (Q parameterizes the values of the Q′is).c Because in the model the probability of a contact to be formed is a function of its energy and loop length, we can next consider the minimized free energy as a function of the contact energies for a given native topology: F({ɛi} | {ℓi}). Then we can seek the special distribution of contact energies {ɛi★(ℓi)} that minimizes or maximizes the thermodynamic folding barrier to a particular structure by finding the extremum of F†({ɛi} | {ℓi}) with respect to the contact energies ɛi, subject to the constraint of fixed native energy, Σiɛi = Mɛ̄ = EN. This distribution, when substituted into the free energy, gives in principle the extremum free energy barrier as a function of native structure F†({ℓi}), which might then be optimized for the fastest/slowest folding structure and its corresponding barrier. We found that in fact the only distribution of energies for which the free energy was an extremum is the distribution that maximizes the barrier by tuning all the contact probabilities to the same value.
Methods
We derive an approximate free energy functional, which takes account for ordering heterogeneity, starting from a contact Hamiltonian 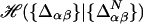 of the form
of the form
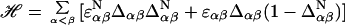 |
1 |
Here the double sum is over residue indices, 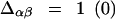 if residues α and β (do not) contact each other and
if residues α and β (do not) contact each other and 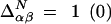 if these residues (do not) contact each other in the native configuration. Weighting the contacts by native energies
if these residues (do not) contact each other in the native configuration. Weighting the contacts by native energies 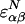 and nonnative energies ɛαβ gives the energy for a particular configuration.d To obtain the thermodynamics, we proceed by obtaining the distribution of state energies in the microcanonical ensemble by averaging nonnative interactions over a Gaussian distribution of variance
and nonnative energies ɛαβ gives the energy for a particular configuration.d To obtain the thermodynamics, we proceed by obtaining the distribution of state energies in the microcanonical ensemble by averaging nonnative interactions over a Gaussian distribution of variance 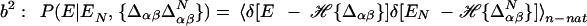 .e The averaging results in a Gaussian distribution having mean Σiɛi𝒬i and variance Mb2(1 − Q), where
.e The averaging results in a Gaussian distribution having mean Σiɛi𝒬i and variance Mb2(1 − Q), where 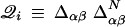 , counts native contacts present in the configuration state inside the stratum Q. From this distribution, the log density of states is obtained in terms of the configurational entropy of stratum Q, 𝒮({𝒬i}|Q), and the free energy functional F({𝒬i}|Q) obtained by performing the usual Legendre transform to the canonical ensemble (cf. Eq. 4).f
, counts native contacts present in the configuration state inside the stratum Q. From this distribution, the log density of states is obtained in terms of the configurational entropy of stratum Q, 𝒮({𝒬i}|Q), and the free energy functional F({𝒬i}|Q) obtained by performing the usual Legendre transform to the canonical ensemble (cf. Eq. 4).f
We express the free energy in terms of an arbitrary distribution of contact probabilities—the distribution of {Qi} that minimizes F({Qi}|Q) is the (most probable) thermal distribution.g For the ensemble of configurations at Q, we define the entropy that corresponds to the multiplicity of contact patterns as 𝒮ROUTE({Qi}|Q) (>0) and the configurational entropy lost from the coil state to induce a contact pattern {Qi} as 𝒮BOND({Qi}|{ℓi}, Q) (<0). We make no capillarity or spinodal assumption and treat the route entropy as the entropy of a binary fluid mixture (10, 53), modified by a prefactor λ(Q) ≡ 1 − Qα, which measures the number of combinatoric states reduced by chain topology: residues connected by a chain have less mixing entropy than if they were freeh:
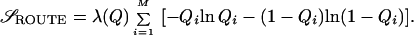 |
2 |
We introduce a measure of “routing” ℛ(Q) by expanding the entropy to lowest orderi: 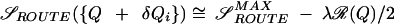 , where we have defined ℛ(Q) by ℛ(Q) = 〈δQ2〉/〈δQ2〉MAX, which is the variance of contact probabilities normalized by the maximal variance,j in the limit ℛ(Q) = 0, the uniformly ordering system has the maximal route entropy. When Qi = 0 or 1 only, ℛ(Q) = 1, 𝒮ROUTE = 0, and only one route to the native state is allowed.k
, where we have defined ℛ(Q) by ℛ(Q) = 〈δQ2〉/〈δQ2〉MAX, which is the variance of contact probabilities normalized by the maximal variance,j in the limit ℛ(Q) = 0, the uniformly ordering system has the maximal route entropy. When Qi = 0 or 1 only, ℛ(Q) = 1, 𝒮ROUTE = 0, and only one route to the native state is allowed.k
In the supplementary material (www.pnas.org), we derive a form for the configurational entropy loss to fold to a given topological structure by accounting for the distribution of entropy losses to form bonds or contacts because of the distribution of sequence lengths in that structure. We let the effective sequence (loop) length between residues i and j, ℓEFF(|i − j|, Q) be a function of Q (this is a mean-field approximation), and we take the entropy loss to close this loop to be of the Flory form ∼(3/2)ln(a/ℓEFF). The requirement that the entropy be a state function restricts the possible functional form of the effective loop length. The result of the derivation for the contact entropy loss to form state {Qi} is:
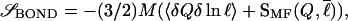 |
3 |
where 〈δQδ ln ℓ〉 = (1/M)Σi(Qi − Q)(ln ℓi − 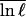 ) is the correlation between the fluctuations in contact probability and log loop length, and SMF(Q,
) is the correlation between the fluctuations in contact probability and log loop length, and SMF(Q,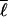 ) is the mean-field bond entropy loss (described in supplemental data) and is a function only of Q and the mean loop length
) is the mean-field bond entropy loss (described in supplemental data) and is a function only of Q and the mean loop length 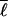 . By Eq. 3, the entropy is raised above that of a symmetrically ordering system when shorter ranged contacts have higher probability to be formed; this effect lowers the barrier. Eqs. 2–4 together give expression (6) for the free energy F({Qi(Q)}|{ɛi}, {ℓi}) of a well-designed protein that orders heterogeneously.
. By Eq. 3, the entropy is raised above that of a symmetrically ordering system when shorter ranged contacts have higher probability to be formed; this effect lowers the barrier. Eqs. 2–4 together give expression (6) for the free energy F({Qi(Q)}|{ɛi}, {ℓi}) of a well-designed protein that orders heterogeneously.
The lattice protein used in Fig. 1 to check the theory is a chain of 27 monomers constrained to the vertices of a three-dimensional cubic lattice. Details of the model and its behavior can be found in refs. 8, 19, 21, 32, 42, and 43. Monomers have nonbonded contact interactions with a Gō potential (native interactions only).l Coupling energies were chosen for row 1 of Fig. 1 by first running a simulated annealing algorithm to find the set {ɛi★} that makes all the Qi({ɛi★}) = Q‡ at the barrier peak. Energies are always constrained to sum to a fixed total native energy: Σiɛi = Mɛ̄. Then energies were relaxed by letting ɛi = ɛi★ + α(ɛ̄i − ɛi★). The values α = 1, 1.35, and 2.05 were used in rows 2, 3, and 4, respectively.
Figure 1.

The effects of heterogeneity in contact probability (increased, Top to Bottom) on barrier height F‡, folding temperature TF, and ordering heterogeneity are summarized here; plots are for simulations of a 27-mer lattice Gō model (yellow) to the same native structure (given in ref. 21), and for the analytic theory in the text (red). The simulation results make no assumptions as to the nature of the configurational entropy; the theoretical results use the approximate state function of Eq. 3, along with a cutoff used for the shorter loops so the bond entropy loss for each loop is always ≤0 (the same loop length distribution is used as in the lattice structure. In the top row, energies are tuned for both simulation and theory to fully symmetrize the funnel: Qi(ɛi★) = Q; second row: energies are then relaxed for the simulation results so they are all equal: ɛi = ɛ̄; energies in the theory are relaxed the same way until a comparable TF is achieved; third row: energies are then further tuned to a distribution ɛi ≅ ɛio that kills the barrier (there are many such distributions; all that is necessary is sufficient contact heterogeneity). The top three rows are funneled folding mechanisms with many routes to the native structure. Bottom row: energies are tuned to induce a single or a few specific routes for folding. All the while, the energies are constrained to sum to EN: Σiɛi = EN. The free energy profile F(Q) (in units of ɛ̄) is plotted in the left column at the folding transition temperature TF, which is given. The next column shows the distribution of thermodynamic contact probabilities Qi(Q‡) ≡ φ′ at the barrier peak [we use the notation φ′, because this is a thermodynamic rather than kinetic measurement; however, for well-designed proteins, the two are strongly correlated with coefficient ≈0.85 (42)]. Only simulation results are shown to keep the figure easy to read; the theory gives φ′ distributions within ∼10%, as may be inferred from their similar route measures. The next column shows the route measure ℛ(Q) of Eq. 5 and gives the dispersion in native energies required to induce the scenario of that row [ℛ(0, 1) = 0/0 is undefined and so is omitted from the simulation plots; it is defined in the theory through the limits Q → 0,1]. The right column shows schematically the different folding routes as heterogeneity is increased; from a maximum number of routes through Q† to essentially just one route. Top row: In the uniformly ordering funnel, we can see first that P(φ′) is a δ function, and ℛ(Q‡) = 0 (cf. Eq. 5), so ordering at the transition state (or barrier peak Q†) is essentially homogeneous. The number of routes through the bottleneck (cf. Eq. 2) is maximized, as schematically drawn (Right). Branches are drawn in the routes to illustrate the minimum of ℛ(Q) at Q‡. The free energy barrier is maximized (Eq. 10), thus the stability of the native state at fixed temperature and native energy is maximized, and so the folding temperature TF at fixed native energy is maximized. TF in the simulation is defined as the temperature where the native state (Q = 1) is occupied 50% of the time. In the theory, at TF the probability for Q ≥ 0.8 is 0.5. A very large dispersion in energies is required to induce this scenario. Some contact energies are nearly zero; others are several times stronger than the average. Second row: In the uniform native energy funnel, the barrier height is roughly halved while hardly changing TF for the following reason. In a Gō model, as the contact energies are relaxed from {ɛi★} to a uniform value ɛi = ɛ̄, the energy of the transition state is essentially constant: initially the energy is ΣiQi★(Q†)ɛi★ = Q Σi ɛi★ = QEN and as the contact energies are relaxed to a uniform value ΣiQiɛ̄ = ɛ̄ΣiQi = QEN once again. However, the transition-state entropy increases and obtains its maximal value when ɛi = ɛ̄, because then all microstates at Q† are equally probable, because the probability of occupying a microstate is pi ∼ exp(−Ei(Q†)/T) = exp(−QEN/T)/Z = 1/Ω(Q†). The thermal entropy −Σipilogpi then equals the configurational entropy log Ω(Q†) (its largest possible value). Thus, as contact energies are relaxed from ɛi★ where they are anticorrelated to their loop lengths (more negative energies tend to be required for longer loops to have equal free energies) to ɛ̄ where they are uncorrelated to their loop lengths, the barrier initially decreases because the total entropy of the bottleneck increases (drawn schematically on the right); i.e., increases in polymer halo entropy are more important than decreases in route entropy. The system is still sufficiently two state that TF is hardly changed. P(φ′) is broad, indicating inhomogeneity in the transition state, solely in this scenario because of the topology of the native structure: all contacts are equivalent energetically. Routing is more pronounced when ɛi = ɛ̄, ℛ(Q) is a measure of the intrinsic fluctuations in order because of the natural inhomogeneity present in the native structure. Different structures will have different profiles, and it will be interesting to see how this measure of structure couples with thermodynamics and kinetics of folding. Loops and dead ends in the schematic drawings are used to illustrate local decreases and increases in ℛ(Q); these fluctuations are captured by the theory only when the routing becomes pronounced (bottom row). The solid curves presented for the theory are shown for a reduction in TF comparable to the simulations. There is still some energetic heterogeneity present, as indicated. When ɛi = ɛ̄ in the theory (dashed curves), the fluctuations in Qi are somewhat larger than the simulation values, and the entropic heterogeneity is sufficient to kill the barrier—the free energy is downhill at TF ≅ 0.5ɛ̄. The free energy barrier results from a cancellation of large terms and is significantly more sensitive than intensive parameters such as route measure ℛ(Q). Third row: In approaching the zero-barrier funnel scenario for the simulation, the energies are further perturbed and now begin to anticorrelate with contact probability (and tend to correlate with loop length); i.e., more probable contacts (which tend to have shorter loops) have stronger energies. For the theory, not as much heterogeneity is required. Contact energies are still correlated with formation probability, as indicated by the signs in parentheses. The free energy barrier continues to decrease until some set of energies {ɛio}, where the barrier at TF vanishes entirely. All the while, the transition temperature TF decreases only ∼10%, so that slowing of dynamics (as TF approaches TG) would not be a major factor. At this point, the φ′ distribution at the barrier position Q‡(ɛ̄) is essentially bimodal, but the distribution at Q‡({ɛio}) (Inset) is less so because of transition state drift towards lower Q values (the Hammond effect). A relatively small amount of energetic heterogeneity is needed to kill the barrier at TF. There are still many routes to the native state, because ℛ(Q‡) ≈ 0.3 − 0.4, but some contacts are fully formed in the transition state (some φ′ ≅ 1). Bottom row: As the energies continue to be perturbed to values that cause folding to occur by a single dominant route rather than a funnel mechanism, folding becomes strongly downhill at the transition temperature, which drops more sharply towards TG: to induce a single pathway here, TF must be decreased to about 1/4 the putative estimate of TG (about TF({ɛ̄})/1.6; see ref. 9). In this scenario, the actual shape of the free energy profile depends strongly on which route the system is tuned to; nonnative interactions not included here become important. Contact participation at the barrier is essentially one or zero, and the route measure at the barrier is essentially one. The entropy at the bottleneck is relatively small (the halo entropy of a single native core). The energetic heterogeneity necessary to achieve this scenario is again very large, comparable to what is needed to achieve a uniform funnel.
Free Energy Functional
By averaging a contact Hamiltonian over nonnative interactions, we can derive an approximate free energy functional for a well-designed protein (see Methods). We analyze here heterogeneity in minimally frustrated sequences, where the roughness energy scale b is smaller than the stability gap ɛ̄. The general form of the free energy functional is:
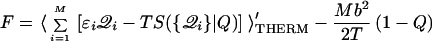 |
4 |
where 𝒬i = (0, 1) counts native contacts in a configurational state (so the sum on ɛi𝒬i gives the states energy), summing S({𝒬i}|Q) gives the states configurational entropy, and then this is thermally averaged over all states restricted to have MQ contacts. The second term accounts for low-energy nonnative traps.
The study of the configurational entropy is a fascinating but complicated problem detailed in Methods. In summary, this entropy functional generalizes the Flory mean-field result (53, 54) to account for the topological heterogeneity inherent in the native structure and a finite average return length for that structure [contact order (47)], as well as to account for the number of folding routes to the native structure. The amount of route diversity or narrowness in folding can be quantified in terms of the relative fluctuations of contact formation δQ = Qi(Q) − Q:
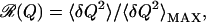 |
5 |
which is useful for our analysis below. Our resulting analytic expression for the free energy of a protein that folds heterogeneously ism:
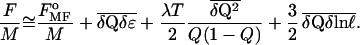 |
6 |
Here FMFo(Q) is the uniform-field free energy function (similar to that obtained previously in ref. 10). The free energy functional is approximate in that it results from an integration over a local free energy density whose only information about the surrounding medium is through the average field present (Q), F = Σifi(Qi, Q). Cooperative entropic effects caused by local correlations (48, 55) between contacts would be an important extension of the model and have been treated elsewhere in similar models (48). Inspection of Eq. 6 shows that as heterogeneity increases, the effect on the barrier is a competition between energetic and polymer entropy gains (second and fourth terms) and route entropy losses (third term), as described above.
Minimizing the free energy (6) at fixed Q, δ(F + μΣjQj) = 0 gives a Fermi distribution for the most probable bond occupation probabilities {Qi★} for a given {ɛi} and {ℓi}:
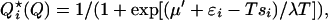 |
7 |
where the Lagrange multiplier μ′ ∼ −(1/M)∂F/∂Q is related to the effective force on the potential F(Q). Positive second variation of F indicates the extremum is in fact a minimum.
Optimizing Rates, Stability, and Entropy
We now consider the effects on the free energy when the native interactions between residues are changed in a controlled manner. The theory predicts a barrier at the transition temperature of a few kBT, in general agreement with experiments on small single-domain proteins. The barrier height is fairly small compared to the total thermal energy of the system, reflecting the exchange of entropy for energy as the protein folds. However, the barrier height can vary significantly depending on which parts of the protein are more stable in their local native structure. At uniform stability, we find that the largest barrier (for a given total native energy) is about twice as large as the barrier when stability is governed purely by the three-dimensional native structure, i.e., when all interaction energies are equal. Increasing heterogeneity, by energetically favoring regions of the protein that are already entropically likely to order, systematically decreases the barrier and in fact can eliminate the barrier entirely if the heterogeneity is large enough. See Fig. 1.
We seek to relax the values of {ɛj} at fixed native energy EN = Σjɛj to the distribution {ɛi★({ℓj})} that extremizes the free energy barrier, by finding the solution of Σi[δF‡/δɛi − p]δɛi = 0 for arbitrary and independent variations δɛi in the energies. It can be shown that δF/δɛi = ∂F/∂ɛi + μ(δ/δɛi) ΣjQj; however, the second term is zero, because δQ/δɛi = 0, so by Eq. 4 δF/δɛi = Qi: the contact probability plays the role of the local density, and the perturbation δɛi the role of an external field, as in liquid state theory. At the extremum, all contact probabilities are equal: Qi = p = Q†, which in our model means that longer loops have lower (stronger) energies: δɛi = Tδsi = −(3/2)Tδlnℓi; there is full symmetry in the ordering of the protein at the extremum. Evaluating the second derivative mechanical-stability matrix shows Qi = Q‡ to be an unstable maximum:
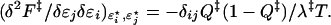 |
8 |
This is clearly negative, meaning that tuning the energies so that Qi = Q‡ maximizes the free energy at the barrier peak. Because the change in the unfolded state (at Q ≈ 0) is much weaker than at the transition state, the barrier height itself is essentially maximized. Substituting Eq. 8 into a Taylor expansion of the free energy at the extremum (and using λ‡ = λ(Q‡) ≈ 1 − Q‡) gives for the rate:
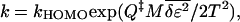 |
9 |
which is to be compared with Eq. 1 in the supplementary material (www.pnas.org) (obtained by an argument using the random energy model). In terms of the route narrowness measure ℛ(Q), the change in free energy barrier on perturbation is
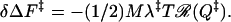 |
10 |
A variance in contact participations 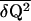 = 0.05, which is about 20% of the maximal dispersion (≈1/4, taking Q‡ ≈ 1/2), lowers the barrier by about 0.1 NkBT or about 5kBT for a chain of length N ≈ 50 [believed to model a protein with ∼100 aa (9)].
= 0.05, which is about 20% of the maximal dispersion (≈1/4, taking Q‡ ≈ 1/2), lowers the barrier by about 0.1 NkBT or about 5kBT for a chain of length N ≈ 50 [believed to model a protein with ∼100 aa (9)].
We can extend the analysis by perturbing about a structure with mean loop length 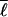 and including effects on the barrier caused by dispersion in loop length and correlations between energies and loop lengths. A perturbation expansion of the free energy gives to lowest order:
and including effects on the barrier caused by dispersion in loop length and correlations between energies and loop lengths. A perturbation expansion of the free energy gives to lowest order:
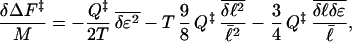 |
11 |
indicating that the free energy barrier is lowered additionally by structural variance in loop lengths and also when shorter range contacts become stronger energetically (δℓi < 0 and δɛi < 0) or longer range contacts become weaker energetically (δℓi > 0 and δɛi > 0), i.e., in the model the free energy is lowered additionally when fluctuations are correlated so as to increase further the variance in contact participations. This effect has been seen in experiments by the Serrano group (46, 56).
To test the validity of the theory, we compare the analytical results obtained from our theory with the results from simulation of a 27-mer lattice protein model. The comparison is shown on Fig. 1, where a full analysis is performed. All energies are in units of the mean native interaction strength ɛ̄.
The rate dependence on heterogeneity should be experimentally testable by measuring the dependencies of folding rate at the transition temperature of a well-designed protein on the dispersion of φ values. It is important that before and after the mutation(s), the protein remain fast folding to the native structure without “off-pathway” intermediates and that its native state enthalpy remain approximately the same, perhaps by tuning environmental variables.
Conclusions and Future Work
In this paper, we have introduced refinement and insight into the funnel picture by considering heterogeneity in the folding of well-designed proteins. We have explored in minimally frustrated sequences how folding is effected by heterogeneity in native contact energies as well as the entropic heterogeneity inherent in folding to a specific three-dimensional native structure. Specifically, we examined the effects on the folding free energy barrier, distribution of participations in the transition-state ensemble TSE′,n as well as the diversity or narrowness of folding routes. For the ensemble of sequences having a given TF/TG, homogeneously ordering sequences have the largest folding free energy barrier. For most structures, where topological factors play an important role, this regime is achieved by introducing a large dispersion in the distribution of native contact energies, which in practice would be almost impossible to achieve. As we reduce the dispersion in the contact energy distribution to a uniform value ɛ̄, the dispersion of contact participations increases, and thus the number of folding routes decreases, the free energy barrier decreases, and the total configurational entropy at the TSE′ initially increases because of polymer halo effects. The folding temperature is only mildly affected; the prefactor appearing in the rate is probably only mildly affected also, because it is largely a function of TF/TG and polymer properties (21). Tuning the interaction energies further results in more probable contacts having stronger energy. Route diversity decreases to moderate values—there are still many routes to the native state, and TF/TG is still sufficiently greater than one. The barrier eventually decreases to zero, at relatively mild dispersion in native contact energy. The funnel picture, with different structural details, is valid for the above wide range of native contact energy distributions. However, tuning the energies further so that probable contacts have even lower energy eventually induces the system to take a single or very few folding routes at the transition temperature. A large dispersion of energies is required to achieve this, and in this regime the folding temperature drops well below the glass temperature range, where folding rates are extremely slow.
Because fine tuning interactions on the funnel may affect the rate, sequences may be designed to fold both faster or slower to the same structure of a wild-type sequence, depending on how the interaction strengths correlate with the entropic likelihood of contact formation. Folding rates in mutant proteins that exceed those of the wild type have been receiving much interest in recent experiments (46, 56–59). Enhancement (or suppression) of folding rate to a given structure arising from changes in sequence is modeled in our theory through changes in native interactions; our results are supported fully by the experiments cited above. The fact that a minimally frustrated protein is robust to perturbations in the interactions means that at least the folding scenarios depicted in the center two rows of Fig. 1 are feasible within the ensemble of sequences that fold to the given structure. However, the number of sequences should be maximal when all the native interactions are near their average, and the actual width of the native interactions depends on the true potential energy function. Fluctuations in rate because of the weakening or strengthening of nonnative traps by sequence perturbations is an interesting topic of future research. The enhancements or reductions in rate we have explored are mild compared to the enhancement by minimal frustration (funneling the landscape): the fine tuning of rates may be a phenomenon manifested by in vitro or in machina evolution rather than in vivo evolution. Nevertheless, rate tuning and folding heterogeneity may become an important factor for larger proteins, where, e.g., stabilizing partially native intermediates may increase the overall rate or prevent aggregation. Given that a sequence is minimally frustrated, heterogeneity or broken-ordering symmetry in fact aids folding, similar to the enhancement of nucleation rates seen in other disordered media (60). Similar effects have been observed in Monte Carlo simulations of sequence evolution, when the selection criteria involves a fast-folding rate (33). Here we see how such phenomena can arise from general considerations of the energy landscape theory. The notion that rates increase with heterogeneity at little expense to native stability contrasts with the view that nonuniform ordering exists merely as a residual signature of incomplete evolution to a uniformly folding state. Adjusting the backbone rigidity or the nonadditivity of interactions (10, 61) can also modify the barrier height, possibly as much as the effects we are considering here. There may also be functional reasons for nonuniform folding—malleability or rigidity requirements of the active site may inhibit or enhance its tendency to order. The amount of route narrowness in folding was introduced as a thermodynamic measure through the mean square fluctuations in a local order parameter. The route measure may be useful in quantifying the natural kinetic accessibility of various structures. Although structural heterogeneity is essentially always present, the flexibility inherent in the number of letters of the sequence code limits the amount of native energetic heterogeneity possible. However, some sequence flexibility is in fact required for funnel topographies (62) and so is probably present at least to a limited degree. We have seen here how a very general theoretical framework can be introduced to explain and understand the effects of local heterogeneity in native stability and structural topology on such quantities as folding rates, transition temperatures, and the degree of routing in the funnel-folding mechanism. Such a theory should be a useful guide in interpreting and predicting experimental results on many fast-folding proteins.
Supplementary Material
Acknowledgments
We thank Peter Wolynes, Hugh Nymeyer, Cecilia Clementi, and Chinlin Guo for their generous and insightful discussions. This work was initiated while S.S.P. was a graduate student with Peter Wolynes. This work was supported by National Science Foundation (NSF) Grant MCB9603839 and NSF Bio-Informatics Fellowship DBI9974199.
Abbreviations
- TSE′
ensemble of configurations at the free energy barrier peak
Footnotes
We treat only native couplings in detail, accounting for nonnative interactions as a uniform background field. Additionally, the correlation between contacts (i, j) is a function only of the overall order Q in our theory. This is analogous to the Hartree approximation in the one-electron theory of solids, where electrons mutually interact only through an averaged field; extensions of our theory to include correlation mediated by native structure may be examined within the density-functional framework and are a topic of future research. On the other hand, tests of the theory by simulation (Fig. 1) produce qualitatively the same results, so the conclusions are not affected by including correlations to any order.
Folding heterogeneity affects the free energy in three ways: (i) The number of folding routes to the native state decreases; this effect increases the folding barrier; (ii) the conformational entropy of polymer loops increases, because native cores with larger halo entropies are more strongly weighted. This decreases the folding barrier. (iii) making likely contacts stronger in energy lowers the thermal energy of partially native structures; this decreases the folding barrier.
This procedure is analogous to finding the most probable distribution of occupation numbers, and thus the thermodynamics, by maximizing the microcanonical entropy for a system of particles obeying a given occupation statistics; here, the effective particles (the contacts) obey Fermi–Dirac statistics; Eq. 7.
A similar derivation of the free energy for a uniform order parameter Q was calculated in ref. 10.
This approach assumes minimal frustration, in that native heterogeneity is retained explicitly, and nonnative heterogeneity is averaged over; phenomena specific to a particular set of nonnative energies, e.g., “off-pathway” intermediates, are smoothed over in this procedure.
Note that in Eq. 4, we explicitly include the trace over configurations at overall order Q. The Qi that minimize F are the thermal values.
In the contact representation, the averaged bond occupation probabilities Qi = 〈𝒬i〉TH are analogous to the averaged number density operator in an inhomogeneous fluid: 〈n(x)〉TH = 〈Σiδ(xi − x)〉TH.
The value α = 1.37 gives the best fit to the lattice 27-mer data for the route entropy, whereas α ≅ 1.0 best fits the 27-mer free energy function. We generally use α ≅ 1.0, because the 27-mer is small; for larger systems, α is smaller: more polymer is buried, and thus it is more strongly constrained by surrounding contacts.
We avoid the word “pathway,” because several definitions exist in the literature; here a single route is unambiguously defined through the limit 𝒮ROUTE → 0.
That is, if MQ contacts were made with probability 1, and M − MQ contacts were made with probability 0, then 〈(Qi − Q)2〉MAX = (1/M)(MQ(1 − Q)2 + (M − MQ)Q2) = Q(1 − Q). Thus ℛ(Q) is between 0 and 1.
That is, because all Qi are only zero or one at any degree of nativeness, each successive bond added must always be the same one, so folding is then a random walk on the potential defined by that single route (chain entropy is still present). ℛ(Q) is in the spirit of a Debye–Waller factor applied to folding routes.
Corner, crankshaft, and end moves are allowed. Free energies and contact probabilities are obtained by equilibrium Monte Carlo sampling by using the histogram method (43). Sampling error is <5%.
We have expanded the route entropy Eq. 2 to second order in this expression for clarity; in deriving the results of the theory, the full expression is used.
We use a prime, because we actually look at the barrier peak along the Q coordinate.
References
- 1.Onuchic J N, Luthey-Schulten Z, Wolynes P G. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 2.Dill K A, Chan H S. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 3.Veitshans T, Klimov D, Thirumalai D. Folding Des. 1997;2:1–22. doi: 10.1016/S1359-0278(97)00002-3. [DOI] [PubMed] [Google Scholar]
- 4.Gruebele M. Annu Rev Phys Chem. 1999;50:485–516. doi: 10.1146/annurev.physchem.50.1.485. [DOI] [PubMed] [Google Scholar]
- 5.Bryngelson J D, Wolynes P G. Proc Natl Acad Sci USA. 1987;84:7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldstein R A, Luthey-Schulten Z A, Wolynes P G. Proc Natl Acad Sci USA. 1992;89:4918–4922. doi: 10.1073/pnas.89.11.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shakhnovich E I, Gutin A M. Proc Natl Acad Sci USA. 1993;90:7195–7199. doi: 10.1073/pnas.90.15.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leopold P E, Montal M, Onuchic J N. Proc Natl Acad Sci USA. 1992;89:8721–8725. doi: 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Onuchic J N, Wolynes P G, Luthey-Schulten Z, Socci N D. Proc Natl Acad Sci USA. 1995;92:3626–3630. doi: 10.1073/pnas.92.8.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plotkin S S, Wang J, Wolynes P G. J Chem Phys. 1997;106:2932–2948. [Google Scholar]
- 11.Wolynes P G. Proc Natl Acad Sci USA. 1996;93:14249–14255. doi: 10.1073/pnas.93.25.14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Helling R, Tang C, Wingreen N. Science. 1996;273:666–669. doi: 10.1126/science.273.5275.666. [DOI] [PubMed] [Google Scholar]
- 13.Nelson E D, Teneyck L F, Onuchic J N. Phys Rev Lett. 1997;79:3534–3537. [Google Scholar]
- 14.Wales D J, Scheraga H A. Science. 1999;285:1368–1372. doi: 10.1126/science.285.5432.1368. [DOI] [PubMed] [Google Scholar]
- 15.Ball K D, Berry R S, Kunz R E, Li F Y, Proykova A A, Wales D J. Science. 1996;271:963–966. [Google Scholar]
- 16.Pande V S, Grosberg A Y, Tanaka T. J Chem Phys. 1995;103:9482–9491. [Google Scholar]
- 17.Bryngelson J D, Wolynes P G. J Phys Chem. 1989;93:6902–6915. [Google Scholar]
- 18.Bryngelson J D, Onuchic J N, Socci N D, Wolynes P G. Proteins. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 19.S̆ali A, Shakhnovich E, Karplus M. Nature (London) 1994;369:248–251. doi: 10.1038/369248a0. [DOI] [PubMed] [Google Scholar]
- 20.Boczko E M, Brooks C L. Science. 1995;269:393–396. doi: 10.1126/science.7618103. [DOI] [PubMed] [Google Scholar]
- 21.Socci N D, Onuchic J N, Wolynes P G. J Chem Phys. 1996;104:5860–5868. [Google Scholar]
- 22.Lazaridis T, Karplus M. Science. 1997;278:1928–1931. doi: 10.1126/science.278.5345.1928. [DOI] [PubMed] [Google Scholar]
- 23.Pande V S, Rokhsar D S. Proc Natl Acad Sci USA. 1999;96:1273–1278. doi: 10.1073/pnas.96.4.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burton R E, Huang G S, Daugherty M A, Calderone T, Oas T G. Nat Struct Biol. 1997;4:305–310. doi: 10.1038/nsb0497-305. [DOI] [PubMed] [Google Scholar]
- 25.Oliveberg M, Tan Y, Silow M, Fersht A. J Mol Biol. 1998;277:933–943. doi: 10.1006/jmbi.1997.1612. [DOI] [PubMed] [Google Scholar]
- 26.Goldbeck R A, Thomas Y G, Chen E, Exquerra R M, Kligar D S. Proc Natl Acad Sci USA. 1999;96:2782–2787. doi: 10.1073/pnas.96.6.2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fersht A R, Matouschek A, Serrano L. J Mol Biol. 1992;224:771–782. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- 28.Radford S A, Dobson M, Evans P A. Nature (London) 1992;358:302–307. doi: 10.1038/358302a0. [DOI] [PubMed] [Google Scholar]
- 29.Bai Y, Sosnick T R, Mayne L, Englander S W. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinez J C, Pisabarro M T, Serrano L. Nat Struct Biol. 1998;5:721–729. doi: 10.1038/1418. [DOI] [PubMed] [Google Scholar]
- 31.Grantcharova V P, Santiago J V, Baker D, Riddle D S. Nat Struct Biol. 1998;5:714–720. doi: 10.1038/1412. [DOI] [PubMed] [Google Scholar]
- 32.Abkevich V I, Gutin A M, Shakhnovich E I. Biochemistry. 1994;33:10026–10036. doi: 10.1021/bi00199a029. [DOI] [PubMed] [Google Scholar]
- 33.Gutin A M, Abkevich V I, Shakhnovich E I. Proc Natl Acad Sci USA. 1995;92:1282–1286. doi: 10.1073/pnas.92.5.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panchenko A R, Luthey-Schulten Z, Wolynes P G. Proc Natl Acad Sci USA. 1996;93:2008–2013. doi: 10.1073/pnas.93.5.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Onuchic J N, Socci N D, Luthey-Schulten Z, Wolynes P G. Folding Des. 1996;1:441–450. doi: 10.1016/S1359-0278(96)00060-0. [DOI] [PubMed] [Google Scholar]
- 36.Shoemaker B A, Wang J, Wolynes P G. Proc Natl Acad Sci USA. 1997;94:777–782. doi: 10.1073/pnas.94.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Portman J J, Takada S, Wolynes P G. Phys Rev Lett. 1998;81:5237–5240. [Google Scholar]
- 38.Klimov D, Thirumalai D. J Mol Biol. 1998;282:471–492. doi: 10.1006/jmbi.1998.1997. [DOI] [PubMed] [Google Scholar]
- 39.Sheinerman F B, Brooks C L. Proc Natl Acad Sci USA. 1998;95:1562–1567. doi: 10.1073/pnas.95.4.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Micheletti C, Banavar J R, Maritan A, Seno F. Phys Rev Lett. 1999;82:3372–3375. [Google Scholar]
- 41.Shea J E, Onuchic J N, Brooks C L. Proc Natl Acad Sci USA. 1999;96:12512–12517. doi: 10.1073/pnas.96.22.12512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nymeyer H, Socci N D, Onuchic J N. Proc Natl Acad Sci USA. 2000;97:634–639. doi: 10.1073/pnas.97.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Socci N D, Onuchic J N. J Chem Phys. 1995;103:4732–4744. [Google Scholar]
- 44.Abkevich V I, Gutin A M, Shakhnovich E I. J Mol Biol. 1995;252:460–471. doi: 10.1006/jmbi.1995.0511. [DOI] [PubMed] [Google Scholar]
- 45.Betancourt M R, Onuchic J N. J Chem Phys. 1995;103:773–787. [Google Scholar]
- 46.Viguera A R, Villegas V, Aviles F X, Serrano L. Folding Des. 1996;2:23–33. doi: 10.1016/S1359-0278(97)00003-5. [DOI] [PubMed] [Google Scholar]
- 47.Plaxco K W, Simons K T, Baker D. J Mol Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 48.Shoemaker B A, Wang J, Wolynes P G. J Mol Biol. 1999;287:675–694. doi: 10.1006/jmbi.1999.2613. [DOI] [PubMed] [Google Scholar]
- 49.Alm E, Baker D. Proc Natl Acad Sci USA. 1999;96:11305–11310. doi: 10.1073/pnas.96.20.11305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muñoz V, Eaton W A. Proc Natl Acad Sci USA. 1999;96:11311–11316. doi: 10.1073/pnas.96.20.11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clementi C, Jennings P A, Onuchic J N. Proc Natl Acad Sci USA. 2000;97:5871–5876. doi: 10.1073/pnas.100547897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Percus J K. In: The Liquid State of Matter: Fluids, Simple and Complex. Montroll E, Lebowitz J, editors. Amsterdam: North–Holland; 1982. [Google Scholar]
- 53.Plotkin S S, Wang J, Wolynes P G. Phys Rev E. 1996;53:6271–6296. doi: 10.1103/physreve.53.6271. [DOI] [PubMed] [Google Scholar]
- 54.Flory P J. J Am Chem Soc. 1956;78:5222–5235. [Google Scholar]
- 55.Dill K A, Fiebig K M, Chan H S. Proc Natl Acad Sci USA. 1993;90:1942–1946. doi: 10.1073/pnas.90.5.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muñoz V, Serrano L. Folding Des. 1996;1:R71–R77. doi: 10.1016/S1359-0278(96)00036-3. [DOI] [PubMed] [Google Scholar]
- 57.Hagen S J, Hofrichter J A, Szabo A, Eaton W A. Proc Natl Acad Sci USA. 1996;93:11615–11617. doi: 10.1073/pnas.93.21.11615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim D E, Gu H, Baker D. Proc Natl Acad Sci USA. 1998;95:4982–4986. doi: 10.1073/pnas.95.9.4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brown B M, Sauer R T. Proc Natl Acad Sci USA. 1999;96:1983–1988. doi: 10.1073/pnas.96.5.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karpov V G, Oxtoby D W. Phys Rev B. 1996;54:9734–9745. doi: 10.1103/physrevb.54.9734. [DOI] [PubMed] [Google Scholar]
- 61.Kolinski A, Godzik A, Skolnick J. J Chem Phys. 1993;98:7420–7433. [Google Scholar]
- 62.Wolynes P G. Nat Struct Biol. 1997;4:871–874. doi: 10.1038/nsb1197-871. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.