

Abstract
To better define the molecular basis of multiple myeloma (MM), we performed unsupervised hierarchic clustering of mRNA expression profiles in CD138-enriched plasma cells from 414 newly diagnosed patients who went on to receive high-dose therapy and tandem stem cell transplants. Seven disease subtypes were validated that were strongly influenced by known genetic lesions, such as c-MAF– and MAFB-, CCND1- and CCND3-, and MMSET-activating translocations and hyperdiploidy. Indicative of the deregulation of common pathways by gene orthologs, common gene signatures were observed in cases with c-MAF and MAFB activation and CCND1 and CCND3 activation, the latter consisting of 2 subgroups, one characterized by expression of the early B-cell markers CD20 and PAX5. A low incidence of focal bone disease distinguished one and increased expression of proliferation-associated genes of another novel subgroup. Comprising varying fractions of each of the other 6 subgroups, the proliferation subgroup dominated at relapse, suggesting that this signature is linked to disease progression. Proliferation and MMSET-spike groups were characterized by significant overexpression of genes mapping to chromosome 1q, and both exhibited a poor prognosis relative to the other groups. A subset of cases with a predominating myeloid gene expression signature, excluded from the profiling analyses, had more favorable baseline characteristics and superior prognosis to those lacking this signature.
Introduction
Multiple myeloma (MM) is a malignancy of antibody-secreting, terminally differentiated B cells that home to and expand in the bone marrow, with symptoms related to anemia, immunosuppression, bone destruction, and renal failure.1,2 Bone lesions developing adjacent to plasma cell foci result from the activation of osteoclasts and inactivation of osteoblasts.3-5 Many of the pathogenetic mechanisms of this clinically heterogeneous malignancy have been unraveled by application of molecular genetics.6-8 While sharing most of the genetic lesions seen in MM,9-11 monoclonal gammopathy of undetermined significance (MGUS) rarely progresses to overt MM.12 The universal activation of 1 of the 3 cyclin D genes is consistent with this being an initiating event in MM.13 Nonhyperdiploid MM, present in 40%, is characterized by transcriptional activation of CCND1, CCND3, MAF, MAFB, or FGFR3/MMSET genes (resulting from translocations involving the immunoglobulin heavy chain locus).8 While hyperdiploidy and CCND1 activation confer a favorable prognosis, MAF, MAFB, or FGFR3/MMSET activation and deletion of chromosomes 13 and 17 are associated with poor prognosis.14-25
Although high-dose therapy has markedly improved MM prognosis,26-28 individual patients' survival remains variable29,30 and cannot be accurately predicted with current prognostic models.31,32 In lymphoma and leukemia, microarray profiling has helped establish clinically relevant disease subclassifications.33-41 In MM, such an approach has identified genes involved in pathogenesis and “drugable” pathways.5,21,42-50 Toward the development of a prognostically relevant molecular classification of MM, gene expression profiling was performed on CD138-enriched plasma cells from 414 newly diagnosed patients treated with high-dose melphalan-based tandem transplants.
Patients, materials, and methods
Patients: training and test sets
Gene expression profiling of highly purified bone marrow plasma cells was performed in 414 newly diagnosed patients with MM. The training set consisted of 256 cases enrolled on total therapy 2 (TT2),28 representing a subset of all 668 patients. There were similar baseline and outcome features in those with and without array profiling performed (data not shown). No difference was observed in event-free or overall survival among the 256 patients relative to the total population of 668, whether or not they were randomized to thalidomide (data not shown). With a median follow-up of 36 months, 110 disease-related events and 72 disease-related deaths have occurred. The test set comprised 158 patients enrolled in total therapy 3 (TT3) and served to validate the gene expression model developed with total therapy 2 samples; the short median follow-up of 12 months precluded an analysis of survival differences based on gene expression classes in the test set. Baseline clinical characteristics of training and test sets are presented in Table S1, which is available on the Blood website (see the Supplemental Tables link at the top of the online article). The institutional review board of the University of Arkansas for Medical Sciences had approved the research studies, and all subjects had provided written informed consent to both treatment protocols and sample procurement, in accordance with the Declaration of Helsinki.
Plasma cell selection and filtering of samples for analysis
Plasma cells were enriched by anti-CD138 immunomagnetic bead selection of mononuclear cell fractions of bone marrow aspirates in a central laboratory. In all, 351 and 214 consecutive patients were evaluated in the training and test sets, respectively. All samples applied to microarray contained more than 85% plasma cells as determined by 2-color flow cytometry (CD38+ and CD45–/dim) performed after selection. We have previously reported that CD138 selection can result in cell populations with varying degrees of contamination with cells of the myeloid lineage and/or normal plasma cells.13 To maintain consistency and ensure faithful assessment of the MM transcriptome, we eliminated samples with high degree of contamination of either of these 2 cell types as assessed by gene expression signatures. In all, 95 of the 351 training set samples and 56 of the 214 test set samples were excluded from the unsupervised clustering and subsequent gene expression–based classifications.
All primary microarray data presented in this paper have been deposited in the NIH Gene Expression Omnibus (GEO; National Center for Biotechnology Information [NCBI], http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE2658.
Gene expression profiling and data analyses
Gene expression profiling was performed with the Affymetrix U133Plus2.0 microarray platform (Santa Clara, CA) using methods previously described.42 All data used in these analyses were derived with the Affymetrix Microarray Suite GCOS1.1 software. Affymetrix signals were transformed by the log-base 2 for each sample. The genes used in the analysis were chosen as follows. Genes having a present detection call in fewer than 3% of the samples and duplicate genes with a smaller standard deviation were removed from analysis up-front. A total of 1559 unique genes exhibiting highly variable expression (standard deviation > 1.34) across the training set was retained. Hierarchic clustering of average linkage with the centered correlation metric51 was used to identify disease subgroups.
Genes that were uniquely overexpressed or underexpressed in specific subgroups defined by the unsupervised hierarchic clustering analysis were selected using significance analysis of microarray (SAM)52 and chi-square analysis, with a 1000-permutation adjustment. The method of the nearest shrunken centroid identified a subgroup of 50 overexpressed (Table S2) and 50 underexpressed (Table S3) genes unique to each subgroup, and these genes were then used to develop a class predictor using prediction analysis for microarrays (PAMs) in R version 2.1.1 (PAM, Stanford, CA).53 The prediction error was calculated by means of 10-fold cross-validation within the training set followed by use in the test set. Supervised cluster analysis of known classes was performed using GeneCluster2 (Broad Institute, Cambridge, MA).54
An expression-based proliferation index (PI) was calculated using the normalized value of 11 genes associated with proliferation (TOP2A, BIRC5, CCNB2, NEK2, ANAPC7, STK6, BUB1, CDC2, C10orf3, ASPM, and CDCA1) scaled to the maximum value among 22 plasma cell samples from 22 healthy donors (defined PI = 1), 414 newly diagnosed myelomas, and 45 myeloma cell lines.13,55 The normalized subgroup PI was shown the relative alteration to NPC by adjusting NPC PI as 1. The one-way ANOVA test for difference of PI across the groups was used.
Survival distributions were presented with the use of the Kaplan-Meier method and compared with the log-rank test. Statistical tests were performed with the software package SPSS 12.0 (SPSS, Chicago, IL).
Results
Identification and validation of 7 subgroups in newly diagnosed myeloma based on common gene expression signatures
Unsupervised hierarchic cluster analysis produced 2 major dendrogram branches with 7 subbranches, which in turn were strongly influenced by the coordinated overexpression of specific genes, many with anchoring genes such as c-MAF and MAFB, CCND1, CCND3, ASS, IL6R, MMSET, FGFR3, CCNB2, FRZB, and DKK1 (Figure 1).
Figure 1.
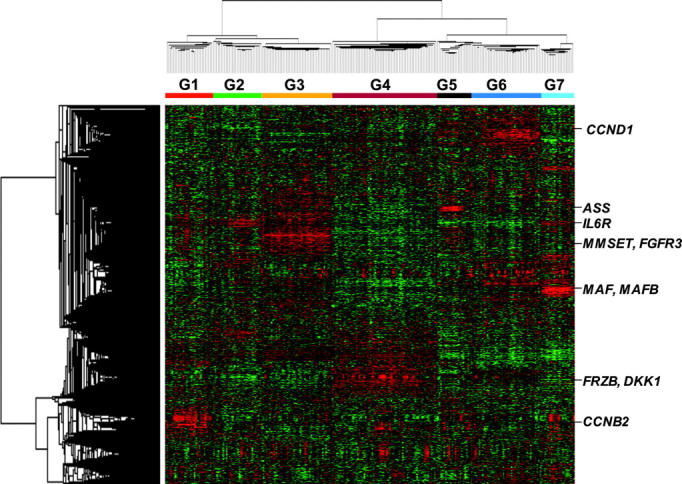
Gene expression patterns in malignant plasma cells reveals that myeloma consists of 7 subgroups. Two-dimensional unsupervised hierarchic cluster analysis of 1559 highly variable genes (rows) in CD138-enriched plasma cells from 256 newly diagnosed multiple myeloma cases (columns). A mean-centered gene expression is depicted by a normalized-signal pseudocolor scale. Red and green indicate overexpressed and underexpressed genes, respectively. The sample dendrogram at the top and gene dendrogram to the side reflect the relatedness of the samples. Note that the dendrogram branches are strongly influenced by noticeable clusters of overexpressed genes. Subgroup designations 1 through 7, from left to right, are indicated under the dendrogram. Subgroup-specific gene clusters are indicated by colored bars to the right of the dendrogram.
Application of the PAM model to the training set classified 98% of the samples correctly based on the original unsupervised hierarchic clustering subgroup designations (Table 1). A colorgram of the expression levels of the 700 PAM genes across the training cohort provides a visual reference of the unique gene expression patterns distinguishing the 7 subgroups (Figure 2A). Application of the PAM model to an independent test set produced a resultant colorgram of the 7 classes and the expression levels of the 700-classifier genes (Figure 2B). Although training and test sets contained a different total number of cases, their proportions in each of the 7 subgroups were comparable in training and test sets (Table S4).
Table 1.
Confusion matrix of subgroup designations by unsupervised hierarchic clustering and the PAM model in the training set
|
Unsupervised hierarchic clustering group designation
|
||||||||
|---|---|---|---|---|---|---|---|---|
| PAM designation | G1 | G2 | G3 | G4 | G5 | G6 | G7 | Class error rate |
| PR | 29 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| LB | 1 | 28 | 0 | 2 | 0 | 0 | 0 | 0.097 |
| MS | 0 | 0 | 44 | 0 | 0 | 0 | 0 | 0 |
| HY | 0 | 0 | 0 | 66 | 0 | 0 | 0 | 0 |
| CD-1 | 0 | 0 | 0 | 1 | 20 | 1 | 0 | 0.091 |
| CD-2 | 0 | 0 | 0 | 0 | 0 | 43 | 0 | 0 |
| MF | 1 | 0 | 0 | 0 | 0 | 0 | 20 | 0.048 |
Figure 2.
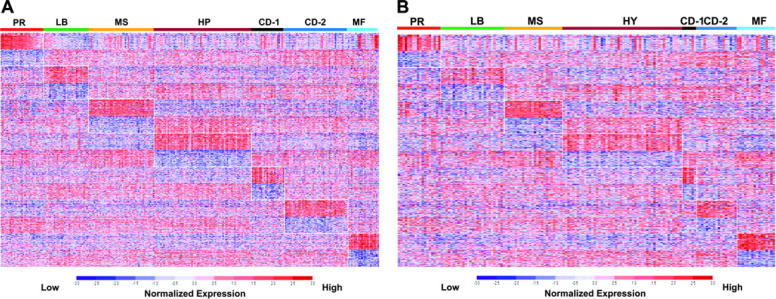
Supervised clustering with SAM/PAM subgroup–defined genes in training and test sets. A supervised clustergram of the expression of 700 genes (50 SAM-defined overexpressed and underexpressed genes from each of the 7 subgroups) across the training set of 256 cases (A) and the test set of 158 cases (B). Genes are indicated along the vertical axis and samples on the horizontal axis. The normalized expression value for each gene is indicated by a color, with red representing high expression and blue representing low expression.
Genetic signatures of expression-defined subgroups
Translocations between the immunoglobulin heavy chain locus and CCND1, CCND3, c-MAF, MAFB, FGFR3, and MMSET represent recurrent genetic lesions in approximately 40% of MM.8 As a result of the juxtaposition of powerful immunoglobulin enhancer elements, hyperexpression of these genes is readily detectable in microarray studies.43 Such spiked expression was a characteristic feature of 4 of the 7 subgroups in both the training (Figure 3A) and test (Figure 3B) sets.
Figure 3.
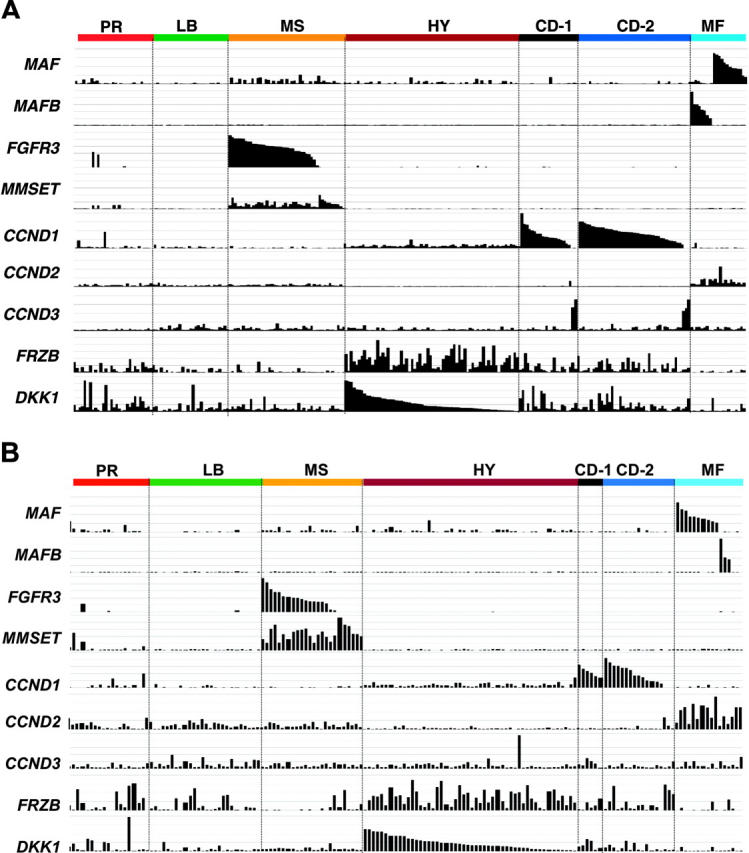
Subgroups are characterized by unique expression patterns. The Affymetrix signal (expression level: vertical axis) of MAF, MAFB, FGFR3, MMSET, CCND1, CCND2, CCND3, FRZB, and DKK1 from the 256 and 158 cases based on the clustergram sample distribution from Figure 2A and B, respectively. The expression levels for each gene are proportional to the height of each bar (representing a single patient sample). Note that spiked expression of CCND1, MAF and MAFB, and FGFR3 and MMSET is strongly correlated with specific subgroup designations. Also note that cases retaining the MMSET spike but lacking FGFR3 spikes maintain similar cluster designation, and MAF and MAFB spikes cluster in the same subgroups. Several MMSET spike–positive cases cluster in the proliferation subgroup. CCND2 expression was mutually exclusive of CCND1 expression. While highly correlated with the hyperdiploid subgroup, FRZB and DKK1 were both significantly underexpressed in groups LB and MF.
The t(14;16)(q32;q23) and t(14;20)(q32;q11) translocations result in activation of c-MAF and MAFB proto-oncogenes, respectively, and are together seen in approximately 6% of cases. Although mutually exclusive, MAF and MAFB spikes clustered together in group 7 (Figure 3A-B), suggesting that ectopic expression of the MAF family of transcription factors results in dysregulation of common downstream targets, justifying an MF (MAF/MAFB) subgroup designation. It is noteworthy that 3 cases within the MF subgroup lacked c-MAF or MAFB spikes, suggesting that other MAF family genes may be activated in these cases. Hurt et al50 have reported that CCND2, CX3CR1, and ITGB7 are targets of the c-MAF transcription factor. Indeed, SAM analysis revealed that CX3CR1 and ITGB7 were among the top 50 overexpressed genes unique to the MF group. CCND2, not on the SAM list, was also expressed in other subgroups, although its expression was highest in the MF subgroup (Figure 3A-B). Additional genes with high SAM scores uniquely overexpressed in the MF group and representing known and putative targets of these transcription factors included the recently identified large MAF family target NUAK1/ARK556 as well as NTRK2, ARID5A, SMARCA1, TLR4, SPP1, and G6MB6. The Wnt signaling antagonist SFRP2 was also uniquely overexpressed in this group. Of the SAM-defined underexpressed genes, TNF-induced gene, TNFAIP8, was the most significant. Another notable underexpressed gene in the MF group was DKK1, the overexpression of which has been implicated in MM-related bone disease3; indeed, the MF group as a whole exhibited a relatively low incidence of bone lesions (Figure 3).
The reciprocal t(4;14)(p16;q32) translocation results in the hyperactivation of both the FGFR3 and MMSET genes. The majority of cases with spiked FGFR3 or MMSET expression clustered together in one subgroup in both training (Figure 3A) and test (Figure 3B) sets. Of importance, 25% of these cases exhibited only a MMSET spike.57 Conversely, loss of MMSET expression in FGFR3-positive tumors was not observed. Thus, consistent with a central role of MMSET in driving downstream transcriptional events, cases with MMSET spikes but lacking FGFR3 spikes clustered together with samples exhibiting activation of both genes (Figure 3A-B). Since the MMSET spike represents a dominant feature of group 3, this group was designated as the MS (MMSET) group. While FGFR3 and WHSC1/MMSET represented the top-ranked overexpressed genes in the MS group, other notable genes included the cadherin family member, desmoglein 2 (DSG2), Wnt receptors FZD2 and FZD8, and the B-cell oncogene PBX1. Significant underexpressed genes of potential relevance included the adhesion molecules ICAM4, N-cadherin (CDH2), cadherin 7 (CDH7), and the B-cell differentiation transcription factor PAX5.
Two cyclin D family members are activated by translocations in MM: cyclin D1 by the t(11;14)(q13;q32) in 17% and CCND3 by t(6;14)(p21;q32) in 2%. As with MAF and MAFB spikes, cases with CCND1 and CCND3 spikes clustered together in the training (Figure 3A) and test (Figure 3B) sets, suggesting that activation of 2 cyclin D orthologs results in dysregulation of common downstream transcriptional programs (Figure 3). Unlike MAF and MAFB and FGFR3 and MMSET spikes, which together comprised a single cluster group, CCND1 and CCND3 each were contained in 2 distinct groups in both training (Figure 3A) and test (Figure 3B) sets, and were termed CD-1 (group 5) and CD-2 (group 6) (CCND1/CCND3). In the original unsupervised cluster analysis, CD-1 and CD-2 groups were difficult to distinguish from each other in the sample dendrogram. However, a subset of cases in this branch contained a group of patients with a distinct expression signature anchored by the gene argininosuccinate synthetase (ASS) (Figure 1). SAM analysis identified 158 genes that were common to these 2 groups (Table S5) but also 123 genes that were significantly differentially expressed between the 2 groups (Table S6). Taken together, these data provide strong evidence for the existence of 2 different forms of CCND1/CCND3 spike–positive MM. Relative to other groups, including CD-2, the human homologue of the Drosophila KELCH gene, Kelch-like 4 (KLHL4), was the most significantly overexpressed gene in CD-1; other genes included INHBE, the FYN proto-oncogene, CEBPB (NF-IL6), and EVER1 and EVER2, 2 cytoplasmic proteins that colocalize with calnexin, an integral membrane protein located in the endoplasmic reticulum. The most significantly overexpressed gene in CD-2 was MS4A1/CD20; CD-2 cases also overexpressed the early B-cell marker VPREB and the B-cell transcription factor PAX5. The CD-1 group lacked expression of CD59, a potent inhibitor of the complement membrane attack complex, a novel Notch protein of unknown function, NOTCH2NL, and the Notch target gene HES1.
Hyperdiploidy, most often associated with trisomies of chromosomes 3, 5, 7, 9, 11, 15, 19, and 21, represents 1 of 2 central genetic pathways in the development of MM, and this type of disease has been previously shown to have a distinct gene expression signature.13 Present in nearly 60%, a hyperdiploid signature was characteristic of group 4 in both training and test sets and was associated with hyperdiploid karyotypes in more than 90% of the cases (Table 2). Genes overexpressed in the hyperdiploid (HY) group included guanine nucleotide binding protein, gamma 11 (GNG11), TRAIL (TNFSF10), the Wnt signaling antagonists FRZB (sFRP3) and DKK1, and the MIP1-alpha chemokine receptor CCR5. Overexpression of several interferon-induced genes including OAS2, IFI27, and IFI35 was also characteristic of this group. Significantly underexpressed genes in the HY group relative to the other groups included CD52 and genes mapping to chromosome 1q TAGLN2, CKS1B, and OPN3 whose overexpression has been linked to a poor survival (F.Z. and J.D.S., unpublished data, July 2004).
Table 2.
Percentages of hyperdiploidy and nonhyperdiploid karyotypes in training (TR) and test (TE) sets
| PR | LB | MS | HY | CD-1 | CD-2 | MF | |
|---|---|---|---|---|---|---|---|
| TT2 | |||||||
| Hyperdiploid | 55 | 67 | 43 | 93 | 20 | 20 | 25 |
| Nonhyperdiploid | 45 | 33 | 57 | 7 | 80 | 80 | 75 |
| TT3 | |||||||
| Hyperdiploid | 53 | 25 | 33 | 86 | 0 | 50 | 17 |
| Nonhyperdiploid | 47 | 75 | 67 | 14 | 100 | 50 | 83 |
Values indicate the percentage of the total number of cytogenetics abnormality cases within each subgroup having the variable indicated.
P < .001 for both TT2 and TT3 groups.
Group 2 was characterized by the elevated expression of endothelin 1(EDN1), which has been implicated in inducing the osteoblastic phenotype of prostate cancer metastases and negatively regulating the expression of DKK1,58,59 the chemokine receptor CCR2, the BCL2-interacting killer (apoptosis-inducing) gene BIK, HES5, HIF1A, and SMAD1. In contrast to the HY group, interferon-induced genes IFI27, IFI35, IFIT5, STAT1, and STAT2 were underexpressed in this group. As in MS and MF groups, group 2 expressed relatively high levels of the IL6LR and low levels of the WNT signaling antagonists FRZB (P < .001) and DKK1 (P < .001) relative to the other groups. Overexpression of these latter genes has been linked to the presence of focal bone disease and osteolytic lesions.3 Consistent with the low expression of DKK1, group 2 had a significantly lower number of magnetic resonance imaging (MRI)–defined focal lesions than seen in the other groups in both the training and test cohorts (Table 2). In lieu of any clear genetic signatures distinguishing this group, we termed it the low bone disease (LB) group.
Group 1 of the unsupervised clustering dendrogram was characterized by the overexpression of numerous cell cycle– and proliferation-related genes (eg, CCNB2, CCNB1, MCM2, CDCA2, BUB1, CDC2, TYMS) and cancer–testis antigen genes (eg, MAGEA6, MAGEA3, GAGE1, GAGE4). This group also had a significantly higher gene expression–defined proliferation index (PI) than the other groups in both training and test sets, justifying its designation as proliferation (PR) subgroup. All the MM subgroups defined here had a higher PI than plasma cells from healthy donors. In addition, the PR group had a PI similar to that of human MM cell lines (P < .001) (data not shown). Metaphase cytogenetic abnormalities were present in an extraordinarily high 69% of cases in the training group and 83% in the test group (the mean for the remaining newly diagnosed cases was approximately 20%). Both hyperdiploid and nonhyperdiploid cases were equally common, with and without concomitant spikes (Table 2). The training and test sets contained 6 MMSET, 3 CCND1, and 2 MAF spikes. Consistent with the emergence of a PR signature overtime, a number of diagnostic samples in virtually all subgroups, especially in the MF group, exhibited overexpressed subsets of genes defining the PR subgroup (Figure 3A-B).
Cyclin D expression in subgroups
Dysregulated expression of 1 of the 3 cyclin D genes (CCND1, CCND2, or CCND3) is a feature of virtually every case of newly diagnosed MM.13 Hyperactivated expression of CCND1 and CCND2 was seen in more than 95% of the cases studied here, but relative levels and distribution varied across the subgroups in both training and test sets. Expression of 1 of the 3 cyclin D genes is mutually exclusive, and a trend was noted for CCND2 to be expressed in the PR, LB, and MS groups (an occasional CCND1 spike in the PR group may reflect MM progression) and was expressed at highest levels in the MF group. As expected, ectopic low-level expression of CCND1 was observed in the HY group.13 CCND1 and CCND3 genes exhibited mutually exclusive spiked expression in the CD-1 and CD-2 groups (except for one CCND3 case in the training set clustering in the HY group).
Laboratory characteristics in the 7 subgroups in the training and test cohorts
Significant differences in standard laboratory features were noted across subgroups (Table 3): the PR group had a higher incidence of abnormal cytogenetics (P < .001), higher serum levels of B2M (P < .001) and LDH (P < .001), and lower levels of albumin (P < .033). Low albumin and high B2M levels were also observed in MS and MF groups, respectively. A striking difference across subgroups was the lower incidence of MRI-defined bone lesions in LB (30% in the training set and 21% in test set) compared with the remaining groups (P < .01). Thus, gene expression patterns and clinical parameters are highly correlated; the higher incidence of high-risk variables in the PR and MS groups is consistent with their poor prognosis (Figure 4, Table 3).
Table 3.
Correlation of clinical features across GEP-defined subgroups in training (TR) and test (TE) cohorts
|
PR
|
LB
|
MS
|
HY
|
CD-1*
|
CD-2
|
MF
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TR | TE | P | TR | TE | P | TR | TE | TR | TE | TR | TE | TR | TE | P | TR | TE | P | |
| B2M level of 339 nM or higher | 66 | 61 | NS | 35 | 41 | NS | 30 | 29 | 27 | 36 | 36 | 33 | 42 | 41 | NS | 52 | 44 | < .01 |
| LDH level of 190 U/L or higher | 48 | 56 | NS | 29 | 22 | NS | 26 | 38 | 26 | 18 | 36 | 17 | 40 | 29 | NS | 45 | 38 | < .01 |
| Albumin level of less than 35 g/L | 55 | 39 | .023 | 29 | 41 | NS | 55 | 67 | 38 | 44 | 32 | 17 | 30 | 35 | NS | 29 | 25 | .033 |
| Cytogenetic abnormalities | 69 | 83 | .021 | 19 | 44 | < .01 | 48 | 50 | 41 | 44 | 45 | 17 | 12 | 12 | NS | 38 | 38 | < .01 |
| MR1 focal lesions, 3 or more | 83 | 71 | .044 | 30 | 21 | NS | 52 | 61 | 57 | 62 | 71 | 67 | 69 | 46 | .001† | 40 | 46 | < .01 |
For the MS, HY, and MF groups, the P values were not significant; for the CD-1 group, the P values were not applicable. The total P represents the test for differences in the variable across the subgroups. This was performed on the mean of training and test set percentages. The total P was less than .01 for all groups. Values indicate the percentage of the total number of cases within each subgroup having the variable indicated. Statistical tests were performed for differences in the percentage of cases between the training and test sets.
NS indicates not significant.
Due to small number of CD-1 cases (n = 6) in the TT3 cohort, a statistical analysis was not performed. Differences were established by chi-square test.
Figure 4.
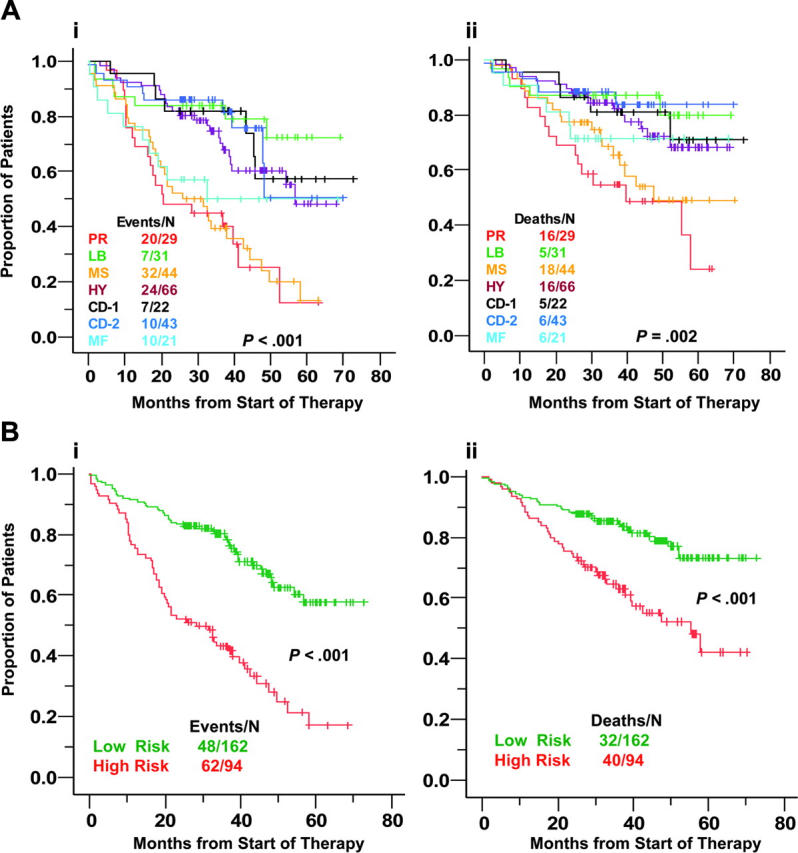
Molecular subgroups show differences in event-free and overall survival. (A) Kaplan-Meier estimates of event-free (i) and overall (ii) survival in the 7 subgroups showed that the 3-year actuarial probabilities of event-free survival were favorable at 84% in low bone disease (LB); 72% in hyperdiploid (HY); 82% in CD-1; and 86% in CD-2. High-risk was associated with proliferation (PR), MMSET (MS), and MAF/MAFB (MF), with 3-year estimates of event-free survival of 44% in PR and 39% in MS and 50% in MF. With respect to overall survival, the 3-year actuarial probabilities were 55% for PR, 69% for MS, 71% for MF, 81% for CD1, 84% for HY, 87% for LB, and 88% in CD2. (B) Event-free (i) and overall (ii) survival analysis of low-risk (HR, CD1, CD2, LB) and high-risk (PR, MF, MS) groups.
Event-free survival and overall survival differ in the 7 subgroups, and molecular class designation is an independent predictor on multivariate analysis
With a 36-month median follow-up on the training cohort, HY, CD-1, CD-2, and LB subgroups were associated with superior event-free (Figure 4A) and overall (Figure 4B) survival relative to the PR, MS, and MF groups. Kaplan-Meier plots suggested a natural cut between high-risk and low-risk diseases, with CD-1, CD-2, LB, and HY groups representing low risk and MF, MS, and PR cases, high risk (Figure 4B), with 48-month estimates of event-free survival of 68% versus 31% (P < .001) and of overall survival of 79% versus 51% (P < .001), respectively. On multivariate analysis, these genetic groups, along with abnormal cytogenetics and elevation of serum levels of B2M and LDH, were significant independent predictors of survival (Table 4).
Table 4.
Multivariate proportional hazards analysis
|
Event-free survival
|
Survival | ||||
|---|---|---|---|---|---|
| %* | HR | P | HR | P | |
| PR, MS, and MF versus other | 37 | 2.41 | < .001 | 1.98 | .004 |
| Any cytogenetic abnormalities | 38 | 1.60 | .014 | 1.98 | .005 |
| β2-microglobulin level of 339 nM or higher | 44 | 1.47 | .061 | 1.55 | .092 |
| LDH level of 190 μ/L or higher | 34 | 169 | .011 | 1.94 | .009 |
n = 255 (of 256 patients); 1 patient is missing cytogenetics. There were 115 progression events and 74 deaths. The following variables were not significant: (1) with the sample of 255, age of 65 years or older was not significant in either analysis (P = .07 for EFS); (2) C-reactive protein level of 4 or higher; and (3) albumin level of 3.5 g/dL or less.
This column refers to the percentage of 255 cases having the incidence of each clinic feature specified in the first column of each row.
Chromosome distribution of SAM-defined overexpressed genes
The chromosome map positions of all SAM-defined overexpressed genes for each of the 7 subgroups were determined. In the case of hyperdiploid MM, a significantly higher number of overexpressed genes mapped to chromosomes 3, 5, 7, 9, 11, 15, and 19 (data not shown). The number of overexpressed genes mapping to chromosome 1q was significantly higher in the poor-risk PR and MS groups (Table 5); the number of overexpressed genes mapping to chromosome 1p was significantly higher in the CD-2 group (Table 5).
Table 5.
Number of SAM-defined overexpressed genes mapping to chromosome 1q and 1p in the 7 subgroups
|
No. genes
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Chromosome | Total | PR | LB | MS | HY | CD-1 | CD-2 | MF | P |
| 1q | 2003 | 178 | 52 | 162 | 22 | 21 | 37 | 38 | < .001 |
| 1p | 2453 | 39 | 20 | 69 | 67 | 38 | 132 | 46 | < .001 |
Myeloid gene signature in CD138-selected cells is associated with a good prognosis
As recognized here, and in previous microarray analyses of CD138-selected plasma cells,13 a myeloid gene expression signature was detectable in a substantial proportion of cases in both training and test sets. This signature was sufficiently strong to confound unsupervised hierarchic cluster analyses, and these cases were removed prior to unsupervised hierarchic cluster analysis. A comparison revealed that the 95 cases in the training set with a myeloid signature displayed more favorable baseline features and better survival than the 256 cases lacking this signature. In this excluded group, abnormal cytogenetics, IgA subtype, and higher levels of bone marrow plasmacytosis as well as serum creatinine and B2M were all significantly less common (P < .05). Moreover, event-free survival (P = .017) and overall survival (P = .046) were superior in the excluded groups. Higher levels of bone marrow plasmacytosis and B2M were also significantly less frequent in the myeloid signature-positive subgroup of the test set (P < .05). However, there was no difference between the retained and excluded cases with respect to albumin, LDH, and number of MRI-defined focal lesions in either the training or test sets.
Discussion
Although presenting with the same histologic diagnosis, MM displays an enormous genomic complexity as well as marked variation in clinical characteristics and patient survival.1 For treatment advances to occur, clinical outcome data have to be interpreted within the framework of genetic entities, as has been proven useful in leukemia and lymphoma. Here, we have provided a comprehensive and integrated view of the myeloma transcriptome in highly enriched tumor cells from a large cohort of newly diagnosed patients. Based on concordant gene expression signatures, predominantly driven by recurrent translocations and hyperdiploidy, MM could be characterized as belonging to 7 distinct molecular entities. HY, CD-1, CD-2, and LB subgroups were associated with superior event-free and overall survival following high-dose therapy and stem cell transplantation.
Although the molecular classification of newly diagnosed disease presented here was validated, the associations between classes and survival are likely to be dependent on the type of therapy used. It is our belief that the relationship between subgroups and survival should form the basis for modification and continual evolution of therapies toward subgroup-specific trials. While many of the subgroups defined in this paper are doing extraordinarily well on TT2, the MS and PR groups do not appear to benefit from this therapeutic strategy. However, future therapies that might exploit molecular insights presented here should lead to an improvement in outcome for patients with these types of disease. Indeed, while there is no significant difference in the EFS and OS of 6 of the 7 groups treated on TT3 relative to TT2, the MS group has a significantly longer EFS (P = .04) and trend for better OS (P = .06) on TT3 relative to TT2 (J.D.S., unpublished data, 2006). This benefit will have to be confirmed with longer follow-up of the test set.
When viewed in the context of our previous gene expression classification studies (4 subgroups among 74 cases applying a first generation microarray with only 8000 gene features,42 the validated classification system presented here, with 7 rather than 4 groups, can be explained by an increase both in gene and sample number, enabling the distinction of rare entities (eg, MF, CD-1, CD-2, and PR groups) with very distinct GEP signatures. In a supervised microarray analysis, we previously reported that activation of one of the cyclin D genes is a universal event in MM,13 leading to a so-called TC classification system (based on cyclin D gene expression and recurrent translocation spikes). As a result of dysregulation of similar transcriptional programs, CCND1 and CCND3 translocations were noted here to exhibit a joint signature. The current unsupervised hierarchic cluster approach yielded important novel information without imposing any predetermined structure on the data; thus, PR and LB groups and the differentiation of the CD-1 and CD-2 groups were identified.
A significant proportion (27%) of newly diagnosed myeloma could not be analyzed for a myeloma signature due to an overwhelming myeloid/normal plasma cell gene expression signature in the post–CD138-selected cells. Postselection flow cytometry clearly showed that while the cases lacking a myeloid gene expression signature were predominantly CD38+/CD45–, the cases with a myeloid gene signature contained both CD38+/CD45– and CD38+/CD45dim cells (data not shown). The presence of this myeloid cell gene expression signature in CD138-selected cells from healthy donors13 suggests that this reflects a copurification of myeloid cells rather than an aberrant expression of myeloid genes in malignant plasma cells. As myeloid cells do not express CD138, one possible explanation for this finding is that the anti-CD138 antibody binds to immunoglobulin Fc receptors that are highly expressed on cells of the myeloid lineage, which are then collected in the separation procedure. The patients with a myeloid expression signature in the CD138-selected fractions often presented with macrofocal bone marrow involvement with low or even absent diffuse infiltration, as in subjects with MGUS (data not shown). The plasma cell yield in randomly obtained samples depends on whether focal lesions were targeted. CT-guided fine-needle biopsies of focal lesions, recognized on magnetic resonance imaging examination, will help distinguish whether enriched plasma cells express signatures of the 7 subgroups described here or whether this type of myeloma constitutes a novel subclass. Nevertheless, the presence of a myeloid gene signature appears to hold important clinical information. In addition to lower levels of bone marrow plasmacytosis, this group was associated less frequently with cytogenetic abnormalities and elevations of B2M and creatinine, and most importantly enjoyed superior EFS and OS than the groups lacking this feature. In view of its macrofocal disease presentation, it is noteworthy that the total number of MRI-defined focal lesions did not exceed those cases lacking the myeloid signature. While not appropriate for molecular profiling of the MM transcriptome, the presence of a myeloid gene signature in CD138-selected cells from newly diagnosed MM would denote a favorable disease course.
Several interesting features of the molecular subgroups defined here are worth noting. Relative to the other 5 groups, genes uniquely underexpressed in both CD-1 and CD-2 subgroups included IL6R, HOXB7, BMPR1A, the mitotic cyclin, CCNE, and the cyclin-dependent kinase, CDK6, which has recently been shown to uniquely interact with cyclin D2 in MM plasma cells.60 In a comparison of differentially expressed genes in CD-1 and CD-2 groups, CD-2 was characterized by overexpression of TNFRSF7 (CD27), the SDF-1 receptor CXCR4, CD20, BTG2, and CD38, whereas ASS, INHBE, the proto-oncogene FYN, NID2, and SET7, a gene with homology to MMSET, were overexpressed in CD-1. The biologic and clinical relevance of CD-1 and CD-2 groups is currently unclear, as there was no difference in clinical parameters or survival between the 2 groups. However, CD-2, but not CD-1 was associated with elevated expression of CD20, which has previously been shown to be associated with the t(11;14)(q13; q32)61 and highly correlated with CD20 protein expression in MM plasma cells.42,62 These tumors also expressed other markers of more immature B cells including PAX5 and the surrogate immunoglobulin light chain VPREB. Another striking genetic difference between CD-1 and CD-2 was the significant difference in the elevated expression of genes from the p arm, but not the q arm, of chromosome 1 in CD-2.
Hyperdiploidy is a distinct genetic entity with a good prognosis and largely devoid of common recurrent immunoglobulin-mediated translocations.17-19 The HY MM subgroup was mainly characterized by overexpressed genes derived from the odd number chromosomes 3, 5, 7, 9, 11, 15, 19, and 21; however, this signature was also observed in cases not showing hyperdiploidy by flow cytometry (data not shown); such diploid and hypodiploid cases may be derived through a similar genetic mechanism (trisomies of odd chromosomes), although clonal evolution may result in loss of DNA on other chromosomes so that the DNA complement is essentially diploid. The assignment of both hyperdiploid and nonhyperdiploid cases to the PR group suggests that a simple recognition of hyperdiploidy is insufficient for proper risk assessment: those with a proliferation signature and hyperdiploidy would be at higher risk than those with an HY signature alone.
An important question concerns the influence of various types of genetic insults in the etiology of MM and their subsequent effects on the transcriptome. Using high-resolution array comparative genomic hybridization (aCGH), mRNA microarray, interphase fluorescence in situ hybridization (FISH), and novel bioinformatics approaches, we recently identified 4 different MM subtypes based on recurrent DNA copy number changes.63 Using gene expression profiling as a surrogate to validate the aCGH-defined groups, we were able to confirm the existence of 2 forms of hyperdiploid disease, one containing gains of 1q, deletion of chromosome 13, and absence of trisomy of chromosome 11.63 In this study, we could distinguish only hyperdiploid MM as a single group without the ability to separate out the 2 unique subtypes identified by aCGH. These subgroups and additional groups are likely to emerge as more sophisticated data mining tools are applied to this large dataset.
A striking feature common to the 2 high-risk groups, MS and PR, was the significant number of overexpressed genes mapping to 1q. While elevated expression of 1q genes is apparent de novo in the MS group, the elevated expression of 1q genes is not apparent in the 5 remaining groups and thus appears to be coincident with the acquisition of a proliferation signature. Major questions emerging from these observations are (1) whether there is a common mechanism by which the 1q genes are activated de novo in the MS group and acquired during disease progression in the other groups, and (2) if there are genes mapping to 1q that contribute to the proliferation signature. Indeed, a central role for chromosome 1q abnormalities in myelomagenesis has been suggested. Tandem duplications and jumping translocations of 1q21 occur frequently in this malignancy,64-66 and gain of 1q is one of the most common abnormalities in MM.67-70 As mentioned above, a form of hyperdiploidy characterized by gains of 1q was found to have a poorer clinical outcome than hyperdiploid disease lacking this feature.63 High-resolution aCGH studies also identified a nonhyperdiploid entity characterized by an amplicon at 1q21.63 Using correlations of gene expression extremes with survival in the training cohort, we recently found that high-risk disease was linked to overexpression of 1q genes and reduced expression of 1p genes (J.D.S., manuscript in preparation). Recent studies using aCGH71 and interphase FISH72 have revealed that gains/amplification of 1q21 accompany the progression of smoldering to overt MM. We have recently shown that gains/amplifications of 1q21 were linked to inferior survival in patients treated on TT2, and the incidence and magnitude of 1q21 gains increased from diagnosis to relapse in patients on this trial.72 Thus, it appears that gain/amplification of 1q21 may be a key genetic event in MM pathogenesis and progression. Whether abnormalities of 1q are a marker of or contribute to disease progression is not currently clear.
An important unanswered question is whether a majority of, if not all, MM cases will eventually acquire a PR class designation. Support for this concept comes from data derived from applying the PAM model to relapsed cases. While present in approximately 18% of newly diagnosed MM, a PR signature was found in 45% of 83 relapses (J.D.S., unpublished data, November 2005). Further evidence for acquisition of a PR signature during disease progression comes from data showing that while 30 of 35 cases with paired baseline and relapse samples maintain the same class designation at relapse, 1 MS and 4 HY cases at diagnosis shifted to a PR classification at relapse (J.D.S., unpublished data). Overexpression of proliferation-associated genes in cases within each of the 6 other subgroups (Figure 2A-B), presence of spikes in the PR group, and class shift to PR with disease progression all suggest that the acquisition of the PR designation is a feature that will emerge in most if not all relapsing MM cases.
The lack of progress in prolonging survival in patients with high-risk MM, now best identified by molecular tests, and the possibility that all MM will evolve to acquire an aggressive PR-like disease should encourage development of therapeutics that target the molecular pathways unique to high-risk disease elucidated through genomic profiling.
Supplementary Material
Acknowledgments
We would like to recognize the efforts of other members of the Donna D. and Donald M. Lambert Laboratory of Myeloma Genetics: Erming Tian, Christopher Adams, Adam Hicks, Bob Kordsmeier, Christopher Randolph, Owen Stephens, David R. Williams, Yan Xaio, and Hongwei Xu. We would also like to thank Clyde Bailey for database management and the nurses and administrative staff of the Myeloma Institute in their supportive role.
Prepublished online as Blood First Edition Paper, May 25, 2006; DOI 10.1182/blood-2005-11-013458.
Supported by National Institutes of Health grants CA55819 (J.D.S., J.C., F.Z., G.T., R.W., and B.B.) and CA97513 (J.D.S.) and by the Fund to Cure Myeloma and Peninsula Community Foundation.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Barlogie B, Shaughnessy J, Munshi N, Epstein J. Plasma cell myeloma. In: Beutler E, Lichtman M, Coller B, Kipps T, Seligsohn U, eds. Williams Hematology. 6th ed. New York, NY: McGraw-Hill; 2001: 1279-1304.
- 2.Anderson K, Shaughnessy J Jr, Barlogie B, Harousseau J, Roodman G. Multiple myeloma. Hematology (Am Soc Hematol Educ Program). 2002; 214-240. [DOI] [PubMed]
- 3.Roodman GD. Biology of osteoclast activation in cancer. J Clin Oncol. 2001;19: 3562-3571. [DOI] [PubMed] [Google Scholar]
- 4.Heider U, Hofbauer LC, Zavrski I, Kaiser M, Jakob C, Sezer O. Novel aspects of osteoclast activation and osteoblast inhibition in myeloma bone disease. Biochem Biophys Res Commun. 2005;338: 687-693. [DOI] [PubMed] [Google Scholar]
- 5.Stewart JP, Shaughnessy JD Jr. Role of osteoblast suppression in multiple myeloma. 2006;98: 1-13. [DOI] [PubMed] [Google Scholar]
- 6.Fonseca R, Barlogie B, Bataille R, et al. Genetics and cytogenetics of MM: a workshop report. Cancer Res. 2004;64: 1546-1558. [DOI] [PubMed] [Google Scholar]
- 7.Shaughnessy J, Barlogie B. Interpreting the molecular biology and clinical behavior of multiple myeloma through global gene expression profiling. Immunol Rev. 2003;94: 140-163. [DOI] [PubMed] [Google Scholar]
- 8.Kuehl WM, Bergsagel PL. Multiple myeloma: evolving genetic events and host interactions. Nature Rev Cancer. 2002;2: 175-187. [DOI] [PubMed] [Google Scholar]
- 9.Fonseca R, Bailey RJ, Ahmann GJ, et al. Genomic abnormalities in monoclonal gammopathy of undetermined significance. Blood. 2002;100: 1417-1424. [PubMed] [Google Scholar]
- 10.Kaufmann H, Ackermann J, Baldia C, et al. Both IGH translocations and chromosome 13q deletions are early events in monoclonal gammopathy of undetermined significance and do not evolve during transition to multiple myeloma. Leukemia. 2004;18: 1879-1882. [DOI] [PubMed] [Google Scholar]
- 11.Chng WJ, Van Wier SA, Ahmann GJ, et al. A validated FISH trisomy index demonstrates the hyperdiploid and non-hyperdiploid dichotomy in MGUS. Blood. 2005;106: 2156-2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346: 564-569. [DOI] [PubMed] [Google Scholar]
- 13.Bergsagel PL, Keuhl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J. Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood. 2005;106: 296-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smadja NV, Fruchart C, Isnard F, et al. Chromosomal analysis in multiple myeloma: cytogenetic evidence of two different diseases. Leukemia. 1998;12: 960-969. [DOI] [PubMed] [Google Scholar]
- 15.Wuilleme S, Robillard N, Lode L, et al. Ploidy, as detected by fluorescence in situ hybridization, defines different subgroups in multiple myeloma. Leukemia. 2005;19: 275-278. [DOI] [PubMed] [Google Scholar]
- 16.Cremer FW, Bila J, Buck I, et al. Delineation of distinct subgroups of multiple myeloma and a model for clonal evolution based on interphase cytogenetics. Genes Chromosomes Cancer. 2005;44: 194-203. [DOI] [PubMed] [Google Scholar]
- 17.Smadja NV, Bastard C, Brigaudeau C, Leroux D, Fruchart C. Hypodiploidy is a major prognostic factor in multiple myeloma. Blood. 2001;98: 2229-2238. [DOI] [PubMed] [Google Scholar]
- 18.Smadja NV, Leroux D, Soulier J, et al. Further cytogenetic characterization of multiple myeloma confirms that 14q32 translocations are a very rare event in hyperdiploid cases. Genes Chromosomes Cancer. 2003;38: 234-239. [DOI] [PubMed] [Google Scholar]
- 19.Fonseca R, Blood E, Rue M, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003;101: 4569-4575. [DOI] [PubMed] [Google Scholar]
- 20.Soverini S, Cavo M, Cellini C, et al. Cyclin D1 overexpression is a favorable prognostic variable for newly diagnosed multiple myeloma patients treated with high-dose chemotherapy and single or double autologous transplantation. Blood. 2003;102: 1588-1594. [DOI] [PubMed] [Google Scholar]
- 21.Keats JJ, Reiman T, Maxwell CA, et al. In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood. 2003;101: 1520-1529. [DOI] [PubMed] [Google Scholar]
- 22.Gertz MA, Lacy MQ, Dispenzieri A, et al. Clinical implications of t(11;14)(q13;q32), t(4;14)(p16.3; q32), and -17p13 in myeloma patients treated with high-dose therapy. Blood. 2005;106: 2837-2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zojer N, Konigsberg R, Ackermann J, et al. Deletion of 13q14 remains an independent adverse prognostic variable in multiple myeloma despite its frequent detection by interphase fluorescence in situ hybridization. Blood. 2000;95: 1925-1930. [PubMed] [Google Scholar]
- 24.Fonseca R, Harrington D, Oken MM, et al. Biological and prognostic significance of interphase fluorescence in situ hybridization detection of chromosome 13 abnormalities (delta13) in multiple myeloma: an eastern cooperative oncology group study. Cancer Res. 2002;62: 715-720. [PubMed] [Google Scholar]
- 25.Shaughnessy J, Jacobson J, Sawyer J, et al. Continuous absence of metaphase abnormalities especially of chromosome 13 and hypodiploidy assures long term survival in MM treated with total therapy I: interpreted in the context of gene expression profiling. Blood. 2003;101: 3849-3856. [DOI] [PubMed] [Google Scholar]
- 26.Barlogie B, Shaughnessy J, Tricot G, et al. Treatment of multiple myeloma. Blood. 2004;103: 20-32. [DOI] [PubMed] [Google Scholar]
- 27.Attal M, Harousseau JL, Facon T, et al. Single versus double autologous stem-cell transplantation for multiple myeloma. N Engl J Med. 2003;349: 2495-2502. [DOI] [PubMed] [Google Scholar]
- 28.Barlogie B, Tricot G, Aniassie E, et al. Thalidomide and hematopoietic-cell transplantation for multiple myeloma. N Engl J Med. 2006;354: 1021-1030. [DOI] [PubMed] [Google Scholar]
- 29.Kyle RA. Long-term survival in multiple myeloma. N Engl J Med. 1983;308: 314-316. [DOI] [PubMed] [Google Scholar]
- 30.Tricot G, Spencer T, Sawyer J, et al. Predicting long-term (> or = 5 years) event-free survival in multiple myeloma patients following planned tandem autotransplants. Br J Haematol. 2002;116: 211-217. [DOI] [PubMed] [Google Scholar]
- 31.Jacobson J, Hussein M, Barlogie B, Durie B, Crowley J. A new staging system for multiple myeloma patients based on the Southwest Oncology Group (SWOG) experience. Br J Haematol. 2003;122: 441-450. [DOI] [PubMed] [Google Scholar]
- 32.Barlogie B. Thalidomide and CC-5013 in multiple myeloma: the University of Arkansas experience. Semin Hematol. 2003;40(suppl 4): 33-38. [DOI] [PubMed] [Google Scholar]
- 33.Alizadeh A, Eisen M, Davis R, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403: 503-511. [DOI] [PubMed] [Google Scholar]
- 34.Shipp M, Ross K, Tamayo P, et al. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med. 2002;8: 68-74. [DOI] [PubMed] [Google Scholar]
- 35.Yeoh E, Ross M, Shurtleff S, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1: 133-143. [DOI] [PubMed] [Google Scholar]
- 36.Ross M, Zhou X, Song G, et al. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood. 2003;102: 2951-2959. [DOI] [PubMed] [Google Scholar]
- 37.Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 2003;198: 851-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bullinger L, Dohner K, Bair E, et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350: 1605-1616. [DOI] [PubMed] [Google Scholar]
- 39.Valk P, Verhaak R, Beijen M, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350: 1617-1628. [DOI] [PubMed] [Google Scholar]
- 40.Lossos I, Czerwinski D, Alizadeh A, et al. Prediction of survival in diffuse large-B-cell lymphoma based on the expression of six genes. N Engl J Med. 2004;350: 1828-1837. [DOI] [PubMed] [Google Scholar]
- 41.Dave S, Wright G, Tan B, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med. 2004;351: 2159-2169. [DOI] [PubMed] [Google Scholar]
- 42.Zhan F, Hardin J, Kordsmeier B, et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood. 2002;99: 1745-1757. [DOI] [PubMed] [Google Scholar]
- 43.De Vos J, Thykjaer T, Tarte K, et al. Comparison of gene expression profiling between malignant and normal plasma cells with oligonucleotide arrays. Oncogene. 2002;21: 6848-6857. [DOI] [PubMed] [Google Scholar]
- 44.Chauhan D, Auclair D, Robinson E, et al. Identification of genes regulated by dexamethasone in multiple myeloma cells using oligonucleotide arrays. Oncogene. 2002;21: 1346-1358. [DOI] [PubMed] [Google Scholar]
- 45.Mitsiades C, Mitsiades N, McMullan C, et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc Natl Acad Sci U S A. 2004;101: 540-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhan F, Tian E, Bumm K, Smith R, Barlogie B, Shaughnessy J. Gene expression profiling of human plasma cell differentiation and classification of multiple myeloma based on similarities to distinct stages of late-stage B-cell development. Blood. 2003;101: 1128-1140. [DOI] [PubMed] [Google Scholar]
- 47.Tarte K, Zhan F, De Vos J, Klein B, Shaughnessy J. Gene expression profiling of plasma cells and plasmablasts: toward a better understanding of the late stages of B-cell differentiation. Blood. 2003;102: 592-600. [DOI] [PubMed] [Google Scholar]
- 48.Magrangeas F, Nasser V, Avet-Loiseau H, et al. Gene expression profiling of multiple myeloma reveals molecular portraits in relation to the pathogenesis of the disease. Blood. 2003;101: 4998-5006. [DOI] [PubMed] [Google Scholar]
- 49.Davies F, Dring A, Li C, et al. Insights into the multistep transformation of MGUS to myeloma using microarray expression analysis. Blood. 2003;102: 4504-4511. [DOI] [PubMed] [Google Scholar]
- 50.Hurt E, Wiestner A, Rosenwald A, et al. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell. 2004;5: 191-199. [DOI] [PubMed] [Google Scholar]
- 51.Eisen M, Spellman P, Brown P, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95: 14863-14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tusher V, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98: 5116-5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci U S A. 2002;99: 6567-6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286: 531-537. [DOI] [PubMed] [Google Scholar]
- 55.Perou C, Jeffrey S, van de Rijn M, et al. Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci U S A. 1999;96: 9212-9217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki A, Iida S, Kato-Uranishi M, et al. ARK5 is transcriptionally regulated by the large-MAF family and mediates IGF-1-induced cell invasion in multiple myeloma: ARK5 as a new molecular determinant of malignant multiple myeloma. Oncogene. 2005;24: 6936-6944. [DOI] [PubMed] [Google Scholar]
- 57.Santra M, Zhan F, Tian E, Barlogie B, Shaughnessy J. A subset of multiple myeloma harboring the t(4;14)(p16;q32) translocation lack FGFR3 expression but maintain an IGH/MMSET fusion transcript. Blood. 2003;101: 2374-2376. [DOI] [PubMed] [Google Scholar]
- 58.Yin JJ, Mohammad KS, Kakonen SM, et al. A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proc Natl Acad Sci U S A. 2003;100: 10954-10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clines GA, Mohaddad KS, Wessner LL, Chirgwin JM, Guise TA. Endothelin-1 stimulates bone formation by regulating osteoblast secretion of the paracrine regulators IL-6, Cyr61, CTGF and Dkk1. J Bone Min Res. 2005;20(suppl 1): S249. [Google Scholar]
- 60.Ely S, Di Liberto M, Niesvizky R, et al. Mutually exclusive cyclin-dependent kinase 4/cyclin D1 and cyclin-dependent kinase 6/cyclin D2 pairing inactivates retinoblastoma protein and promotes cell cycle dysregulation in multiple myeloma. Cancer Res. 2005;65: 11345-11353. [DOI] [PubMed] [Google Scholar]
- 61.Robillard N, Avet-Loiseau H, Garand R, et al. CD20 is associated with a small mature plasma cell morphology and t(11;14) in multiple myeloma. Blood. 2003;102: 1070-1071. [DOI] [PubMed] [Google Scholar]
- 62.Lin P, Mahdavy M, Zhan F, Zhang HZ, Katz RL, Shaughnessy JD. Expression of PAX5 in CD20-positive multiple myeloma assessed by immunohistochemistry and oligonucleotide microarray. Mod Pathol. 2004;17: 1217-1222. [DOI] [PubMed] [Google Scholar]
- 63.Carrasco R, Tonon G, Huang Y, et al. High-resolution genomic profiles defines distinct clinicopathogenetic subgroups of multiple myeloma patients. Cancer Cell. 2006;4: 313-325. [DOI] [PubMed] [Google Scholar]
- 64.Sawyer JR, Tricot G, Mattox S, Jagannath S, Barlogie B. Jumping translocations of chromosome 1q in multiple myeloma: evidence for a mechanism involving decondensation of pericentromeric heterochromatin. Blood. 1998;91: 1732-1741. [PubMed] [Google Scholar]
- 65.Le Baccon P, Leroux D, Dascalescu C, et al. Novel evidence of a role for chromosome 1 pericentric heterochromatin in the pathogenesis of B-cell lymphoma and multiple myeloma. Genes Chromosomes Cancer. 2001;32: 250-264. [DOI] [PubMed] [Google Scholar]
- 66.Sawyer JR, Tricot G, Lukacs JL, et al. Genomic instability in multiple myeloma: evidence for jumping segmental duplications of chromosome arm 1q. Genes Chromosomes Cancer. 2005;42: 95-106. [DOI] [PubMed] [Google Scholar]
- 67.Liebisch P, Wendl C, Wellmann A, et al. High incidence of trisomies 1q, 9q, and 11q in multiple myeloma: results from a comprehensive molecular cytogenetic analysis. Leukemia. 2003;17: 2535-2537. [DOI] [PubMed] [Google Scholar]
- 68.Gutierrez NC, Garcia JL, Hernandez JM, et al. Prognostic and biologic significance of chromosomal imbalances assessed by comparative genomic hybridization in multiple myeloma. Blood. 2004;104: 2661-2666. [DOI] [PubMed] [Google Scholar]
- 69.Avet-Loiseau H, Andree-Ashley LE, Moore D II, et al. Molecular cytogenetic abnormalities in multiple myeloma and plasma cell leukemia measured using comparative genomic hybridization. Genes Chromosomes Cancer. 1997;19: 124-133. [DOI] [PubMed] [Google Scholar]
- 70.Cigudosa JC, Rao PH, Calasanz MJ, et al. Characterization of nonrandom chromosomal gains and losses in multiple myeloma by comparative genomic hybridization. Blood. 1998;91: 3007-3010. [PubMed] [Google Scholar]
- 71.Rosinol L, Carrio A, Blade J, et al. Comparative genomic hybridisation identifies two variants of smoldering multiple myeloma. Br J Haematol. 2005;130: 729-732. [DOI] [PubMed] [Google Scholar]
- 72.Hanamura I, Stewart JP, Huang Y. Frequent gain of chromosome band 1q21 in plasma cell dyscrasias detected by fluorescence in situ hybridization: incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem cell transplantation. Blood. Prepublished on May 16, 2006, as DOI 10.1182/blood-2006-03-009910. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.