

Abstract
We present a general algorithm to detect genes differentially expressed between two nonhomogeneous time-series data sets. As increasing amounts of high-throughput biological data become available, a major challenge in genomic and computational biology is to develop methods for comparing data from different experimental sources. Time-series whole-genome expression data are a particularly valuable source of information because they can describe an unfolding biological process such as the cell cycle or immune response. However, comparisons of time-series expression data sets are hindered by biological and experimental inconsistencies such as differences in sampling rate, variations in the timing of biological processes, and the lack of repeats. Our algorithm overcomes these difficulties by using a continuous representation for time-series data and combining a noise model for individual samples with a global difference measure. We introduce a corresponding statistical method for computing the significance of this differential expression measure. We used our algorithm to compare cell-cycle-dependent gene expression in wild-type and knockout yeast strains. Our algorithm identified a set of 56 differentially expressed genes, and these results were validated by using independent protein-DNA-binding data. Unlike previous methods, our algorithm was also able to identify 22 non-cell-cycle-regulated genes as differentially expressed. This set of genes is significantly correlated in a set of independent expression experiments, suggesting additional roles for the transcription factors Fkh1 and Fkh2 in controlling cellular activity in yeast.
Keywords: DNA microarray, splines, cell cycle, yeast
Gene expression data can be divided into two classes: static and time-series data. In static expression experiments, a snapshot of gene expression levels is taken [for example, expression levels of tumor cells from different cancer types (1)]. In time-series expression experiments, a temporal process is measured [for example, infection (2), response to environmental conditions (3), or the cell cycle (4-6)]. One of the key issues in time-series gene expression analysis is the identification of genes with altered expression between samples. For instance, one would like to identify genes that have changed significantly after an experimental treatment or that differ between normal and diseased cells. In clinical research, such differentially expressed genes can serve as disease-specific markers or as predictors of the clinical outcome of a treatment (2, 7-9). In knockout experiments, differentially expressed genes represent first- or second-order downstream effects of the knocked-out gene (5, 6), and their identification allows the discovery of genetic interaction networks.
Recently a number of algorithms for analyzing various features of time-series expression data have been introduced, but none of these algorithms are directly applicable to detecting genes that are differentially expressed. Ramoni et al. (10) investigated the clustering of time-series expression data based on dynamics. Qian et al. (11) used local alignment algorithms to study time-shifted and inverted gene expression profiles. Holter et al. (12) used a time translational matrix to model the temporal relationships between different modes of the singular value decomposition. Although these algorithms are useful for identifying patterns in a single expression experiment, they cannot be used directly to compare two time-series experiments. Other researchers have focused on interpolating time-series expression data. Aach and Church (13) used linear interpolation to align cell-cycle expression experiments. D'haeseleer et al. (14) used spline interpolation on individual genes, and Zhao et al. (15) used a custom-tailored model to interpolate cell-cycle experiments. Although interpolation and alignment are important preprocessing steps, it is not clear how they can be used to identify differentially expressed genes. In this article we use an interpolation method that we developed previously [Bar-Joseph et al. (16)], which combines spline interpolation with clustering (see Methods) as a preprocessing step.
Many algorithms have already been introduced for identifying genes differentially expressed between two experiments in the static expression case (1). However, due to differences in sampling rates and variations in the timing of biological processes (see Table 1), such methods cannot be applied directly to time-series expression data. Previously reported algorithms for identifying differentially expressed genes in time series data essentially applied static analysis methods, used ad hoc methods that are not generally applicable, or were highly tailored for a specific data set. Previously reported methods include cluster analysis (5), generalized singular value decomposition (17), pointwise comparison (2, 9), and custom-tailored models (8). Although these methods have achieved some success, they suffer from many problems. As we demonstrate in Results and Discussion, cluster analysis fails to detect differentially expressed genes that belong to clusters for which most genes do not change. Generalized singular value decomposition [presented by Alter et al. (17)] can be used to detect differences between sets of genes but is not appropriate for comparing individual genes. Further, this method requires that the data sets being compared contain the same number of experiments (or time points), which is clearly not the case in general (see Table 1). Direct pointwise comparison of samples, essentially a static analysis method, is problematic because it does not take into account the dynamic nature of the experiments and is unable to distinguish between systematic changes and random noise. Further, due to the inconsistencies in time-series data obtained from the different sources mentioned above, in many cases direct pointwise comparison is not possible. Finally, custom-tailored models clearly do not present a general solution, because they require significant assumptions about the shape of the expression profiles being compared (e.g., linear or quadratic models). In most cases, a priori knowledge that would justify using highly specific models is unavailable. Even in cases in which some genes are known to change in a certain way over time (e.g., a sinusoidal model for the cell cycle), using a highly specific model for the shape of expression profiles will result in missing changes in many genes that are not behaving according to the assumed model.
Table 1. Time-series expression experiments.
| Cells (ref.) | Method of arrest | Duration, min | Cell-cycle length, min | Sampling rate |
|---|---|---|---|---|
| WT alpha (4) | Alpha mating factor | 0-119 | 64 | Every 7 min |
| WT cdc15 (4) | Temperature-sensitive cdc15 mutant | 10-290 | 112 | Every 20 min for 1 hr, every 10 min for 3 hr, every 20 min for the final hr |
| WT cdc28 (23) | Temperature-sensitive cdc28 mutant | 0-160 | 85 | Every 10 min |
| fkh1/fkh2 knockout (5) | Alpha mating factor | 0-210 | 105 | Every 15 min until 165 min, then after 45 min |
| yox1/yhp1 knockout (6) | Alpha mating factor | 0-120 | 60 | Every 10 min |
A summary of five different time-series expression experiments that were performed to study the cell cycle in yeast is shown. Note that the sampling rates are not always uniform and vary among the different experiments. Even under identical arrest methods, the sampling rates differed significantly (ranging from 7 to 10 to 15 min in the three alpha experiments). In addition, the cell-cycle duration differs depending on the experimental conditions.
Here we present a general algorithm that fully exploits information in time-series gene expression data to detect differentially expressed genes. Our algorithm represents the two expression profiles to be compared with aligned continuous curves and computes a global difference measure between these two curves. This collapses the information about differential expression into a single number, which allows for a statistically principled comparison that requires a minimum of unwarranted assumptions about the underlying form of the data. To determine the significance of this global difference, we combine a noise model for individual samples (which is easy to compute) with a global error measurement that captures the temporal difference between two expression profiles. Thus, it can assign significance to temporal expression differences while requiring only a minimum number of expression measurement repeats. This latter point is important, because repeating time-series experiments can be prohibitively expensive, and in most publicly available data sets there are no or very few repeats for all the time points measured (2, 4, 5).
Methods
Data Preprocessing: Continuous Representation and Alignment. In previous work we described a method for representing expression profiles with aligned continuous curves (16). In this article we use our continuous representation and alignment algorithms as a preprocessing step so that we can make time-series experiments comparable when these experiments have different sampling rates and variations in the timing of the underlying biological process. Here we briefly outline this preprocessing step. To obtain a continuous time formulation, we use cubic splines to represent gene expression curves. Cubic splines are a set of piecewise cubic polynomials and are frequently used for fitting time series and other noisy data. In this work we use B-splines, a type of spline that is mathematically convenient for data approximation (18). B-splines are described as a linear combination of a set of basis polynomials. By knowing the value of these splines at a set of control points, one can generate the entire set of polynomials from these basis functions. We assume that a gene can be represented by a spline curve and additional noise using the following equation
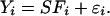 |
1 |
Here Yi is the expression profile for gene i, Fi is a vector of spline control points for gene i, and S is a matrix of spline basis functions evaluated at the sampling points of the experiment. εi is a vector of the noise terms, which is assumed to be normally distributed with mean 0. Due to noise and missing values, determining the parameters of Eq. 1 (Fi and εi) for each gene separately may lead to overfitting. Instead, when estimating these splines from expression data, we constrain the control point values of genes in the same class (coexpressed genes) to co-vary, and thus we use other coexpressed genes to overcome noise and missing values in a single gene. In previous work (16) we showed that this method provides a superior fit for time-series expression data when compared to all other previous methods.
Because the rate at which similar underlying biological processes unfold differs across genetic variants and environmental conditions (13), prior to comparing two time series-experiments we align them temporally. Our alignment algorithm warps the time scale of a reference realization of a biological process to align it with that of a second data set measuring the same process under different conditions. Using splines, we can use a linear warping function to obtain an optimal alignment by adjusting shift and stretch parameters to minimize a global error function. In previous work (16) we showed that this method obtains both significant and biologically meaningful results.
Hypothesis Testing for Differentially Expressed Genes. Following spline assignment and alignment, each gene is represented by two continuous curves (one for each experiment). Denote the first (reference) curve as C1 and the second (test) curve as C2 (for example, C1 could be the WT expression profile, and C2 is a knockout profile). Given C1 and C2, we would like to answer the following question: Is the difference between the two expression profiles for a certain gene significant? This problem can be formulated as a hypothesis-testing problem, with two hypotheses:
H0: C2 is a noisy realization of C1.
H1: The two curves are independent.
Under the null hypothesis, we assume that C2 can be represented by the same spline curve as C1 and that any difference between the two profiles is a result of noise in the measurement of the test experiment. Under the alternative hypothesis we assume that both C1 and C2 can be represented by a spline curve, although we do not assume anything about the relationship between the two curves.
The hypothesis test can be performed by looking at the ability of each hypothesis to explain the difference between the two curves. By using log a likelihood ratio test this could be written as
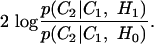 |
2 |
Straightforward comparison of the two curves will not work well in our case. Unlike regular curves, the expression profile curves were derived from very few sample points, and this fact should be taken into account when computing the significance of the difference between the curves. In addition, most comparison methods require additional information about the curves (such as a noise model for the entire curve), which is not available in our case because of the small number of full repeats. Thus we present a method that allows us to compute these conditional probabilities even when only a few repeats exist.
Noise Model for Individual Samples. Here we assume that noise in individual measurements is normally distributed with mean 0 and variance σ2 (we relax this assumption later). Because noise in individual expression measurements is assumed to be independently varying, σ2 can be computed even if few repeats exist. Denote by Y1 and Y2 the actual expression values measured in the reference and test experiments, respectively. Let Yt1 be the expression value at time t in the reference experiment. Then p(x|Yt1, σ2) is the probability of obtaining expression measurement x at time t. Comparing Y1 and Y2 directly is not possible because of their different sampling rates and temporal expression variations. However, we can sample C2 at the same time points as the actual reference experiment to obtain a set of values that are comparable to Y1. Let t1... tm be the set of time points that were measured in the reference experiment. For a curve C, denote by C(t) the value of C at time t. Set Y ′2 = {C2(t1)... C2(tm)}, i.e., Y′2 is the vector of points sampled from C2 at the reference experiment points. To compute the conditional probability under the alternative hypothesis (p(C2|C1, H1)), we use Y′2, and recall that under H1, C1 and C2 are independent. Thus, we can set
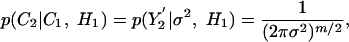 |
where we set the means at the sampled points.
Although a sample-based method works well for the alternative hypothesis, under the null hypothesis this method suffers from a number of drawbacks. First, it ignores systematic differences between the curves (for example, if one were always higher or lower). Second, in many cases sampling rates for time-series data are nonuniform. Using a sampling-based method, we assign an equal weighting to each sampled point, which does not reflect the actual time each point represents.
Combining a Sample Noise Model with Global Difference. Instead of directly using samples from C2 to compute p(C2|C1, H0), we use the global difference between the two expression curves C1 and C2, which is defined as
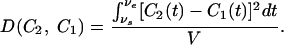 |
Here, vs and ve are the start and end of the interval in which the two curves can be compared (the alignment interval), and V = ve - vs. Note that D(C2, C1) is proportional to the averaged squared distance between the two curves. This is a suitable difference measure for the following reasons. First, D(C2, C1) depends on the actual duration over which the curves can be compared and thus is less sensitive to sampling rates. In addition, D(C2, C1) can distinguish between consistent differences and random oscillations around the reference samples and thus is sensitive to actual systematic differences.
We now discover a new curve, C, that best explains the difference between C1 and C2. Let e2 = D(C1, C2). Setting p(C2|C1, H0) = p(e2|Y1, σ2, H0) leads to a framework that combines the individual error model (σ2) with a global difference measurement (e2) that, as discussed above, correctly captures the differences between the two curves. For a curve C, set YC = {C(t1)... C(tm)}. To find the maximum-likelihood assignment of p(e2|Y1, σ2, H0), we need to find a curve C with the same global distance (e2) from C1 that maximizes the probability that C is a noisy realization of C1. Formally, this could be stated as the following maximization problem:
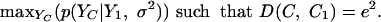 |
3 |
That is, we are looking for a curve C that satisfies the global error constraint (D(C, C1) = D(C1, C2)) such that, when sampling from C at time points that were used in the reference experiment (YC), we get values that are as close as possible to the values measured in the reference experiment. Using the maximum-likelihood assignment instead of simply the original C2 guarantees that the computations of the significance of differential expression will err on the conservative side. That is, only a global error value e2 that cannot be adequately explained by the “best” (maximum-likelihood) curve C will be considered significant. The above maximization problem can be solved by working on the spline representation for each curve and rewriting Eq. 3 in terms of the spline control points (see Appendix I, which is published as supporting information on the PNAS web site, www.pnas.org).
The Complete Algorithm. Fig. 1 presents the complete algorithm we use for identifying differentially expressed genes in time-series data. The input to the algorithm is the set of genes G, the two expression datasets E1 and E2, and a significance threshold ε. Following spline assignment and alignment, we solve Eq. 3 for each gene and use the solution (denoted Yδ) to compute the log-likelihood ratio for that gene (see Appendix I). Finally, to perform a significance test, we use the χ2 distribution with q degrees of freedom (where q is the number of spline control points used by the curves).
Fig. 1.

Complete algorithm for identifying differentially expressed genes in time-series data.
The computational complexity of this algorithm is linear in the number of genes we are testing. Our algorithm is asymmetric and relies on the use of a reference curve. It is also possible to present a symmetric version of this algorithm as we show in Appendix II, which is published as supporting information on the PNAS web site. In addition, the algorithm discussed above can be modified such that it can use variances that depend on expression value magnitudes. Such a method reduces the effect of experimental artifacts and associated high variance, allowing us to accurately detect significant changes and ignore changes that are a result of noise (see Appendix III, which is published as supporting information on the PNAS web site).
Results and Discussion
We have tested our algorithm using synthetic data and cell-cycle expression data. As we show below, our algorithm generated biologically meaningful results that improved on prior methods for comparing time-series expression data sets.
Synthetic Data. To test our algorithm and determine significance thresholds under a variety of expression profiles and noise models, we first tested our algorithm on synthetic data. We generated a reference curve and two other curves. Next we sampled the reference curve and added random noise to these samples. We then used our algorithm to compare a curve generated by using splines from the sampled points with the reference curve and the other generated curves. We repeated this process with a number of different noise models. In all cases, using a 0.005 P-value cutoff our algorithm correctly identified the tested curve as similar to the reference curve and significantly different from the two other curves (see Fig. 4 and Appendix IV, which are published as supporting information on the PNAS web site).
Yeast Cell-Cycle and Knockout Data. To test our algorithm on biological data sets and compare our algorithm with algorithms that have been used in the past, we used a data set from Zhu et al. (5). These authors performed an experiment in which two yeast transcriptional factors (Fkh1 and Fkh2) involved in regulating the cell cycle were knocked-out and a time series of gene expression levels was measured in synchronized cells. Focusing on 800 previously identified cell-cycle-regulated genes [from Spellman et al. (4)], the authors used hierarchical clustering to compare their results with a time-series WT data set from ref. 4. Using this method Zhu et al. identified two clusters (Clb2 and Sic1) that contain genes affected by the knockout. They were able to demonstrate direct binding of Fkh2 only to promoters of genes from the Clb2 cluster, and therefore they suggest that the forkhead proteins affect the Sic1 cluster genes indirectly through Swi5 and Ace2. Note that because the two experiments used different sampling rates, direct pointwise comparison of the samples is not possible. Independently, Simon et al. (19) used DNA-binding experiments to identify genes that are regulated by nine cell-cycle transcription factors including Fkh1, Fkh2, Swi5, and Ace2. We applied our algorithm to the two time-series data sets (WT and mutant) and have used the binding data to verify our results.
Identifying Differentially Expressed Genes Controlled by Fkh1 and Fkh2. We used our algorithm to identify genes that are differentially expressed in the Fkh1/Fkh2 knockout experiment when compared to the WT experiment. Of the 800 cell-cycle-regulated genes, our algorithm identified 56 genes as differentially expressed with P < 0.005 (see www.psrg.lcs.mit.edu/DiffExp/DiffExp.html for a discussion about the value-specific variance used). In Table 2 we present the top 30 genes (in decreasing significance) identified by our algorithm. As can be seen, many of the genes identified by our algorithm are confirmed by independent binding experiments. Although many of the genes come from the two primary phases that are either directly controlled by Fkh1/Fkh2 (G2/M) or indirectly controlled (M/G1), there are a number of genes from other cell-cycle phases, suggesting a role for the forkhead transcription factors in G1 and S phases. These results are supported by the genome-wide binding data that describes association of Fkh1 and Fkh2 with genes expressed in G1 and S (19). To verify our results on a global scale, we computed the percentage of genes in our list that are bound by each of the nine cell-cycle factors (using a binding P-value cutoff of 0.005) and compared this result to the percentage obtained using the entire set of 800 cell-cycle-regulated genes. We then computed a P value for the enrichment of each factor for differentially expressed genes using the hypergeometric distribution. We expected to find enrichment for three types of factors:
Table 2. Differentially expressed cycling genes.
| Gene name | P value | Phase | Previously identified? | Comments |
|---|---|---|---|---|
| Cwp1 | 1 × 10−16 | S/G2 | No | Bound by Fkh2 and Ace2 |
| Cts1 | 1.4 × 10−15 | G1 | Yes | Bound by Fkh1 and Fkh2 |
| Ole1 | 3.5 × 10−14 | M/G1 | No | Bound by Swi5 |
| Egt2 | 1.7 × 10−10 | M/G1 | Yes | Bound by Ace2 and Swi5 |
| Scw11 | 1.8 × 10−10 | G1 | Yes | Bound by Fkh1, Fkh2, Ace2, and Swi5 |
| YER124C | 8 × 10 × −9 | G1 | Yes | Bound by Fkh1, Fkh2, and Ace2 |
| Ald6 | 1.2 × 10−8 | S/G2 | No | |
| YHR143W | 2.7 × 10−8 | G1 | Yes | Bound by Fkh1, Fkh2, and Ace2 |
| Pho5 | 3.4 × 10−8 | G2/M | No | |
| YLR194C | 3.5 × 10−8 | M/G1 | No | Bound by Swi5 |
| YBR158W | 5.5 × 10−8 | M/G1 | Yes | Bound by Fkh1, Fkh2, Ndd1, Ace2, and Swi5 |
| YNL058C | 9.7 × 10−8 | G2/M | Yes | Bound by Ndd1 and Mcm1 |
| Clb2 | 4.4 × 10−7 | G2/M | Yes | Bound by Fkh1, Fkh2, Ndd1, and Mcm1 |
| Dip5 | 5.1 × 10−7 | G2/M | No | |
| YPL158W | 5.3 × 10−7 | M/G1 | No | Bound by Swi5 |
| YNL078W | 8.5 × 10−7 | M/G1 | No | Bound by Ace2 and Swi5 |
| Pry3 | 9.1 × 10−7 | G1 | Yes | Bound by Fkh1, Fkh2, Ace2, and Swi5 |
| Utr2 | 1.34 × 10−6 | M/G1 | No | Bound by Fkh1, Fkh2, and Mcm1 |
| YDR055W | 1.36 × 10−6 | M/G1 | No | Bound by Swi5 |
| YGL184C | 1.5 × 10−6 | S | No | |
| Clb1 | 1.8 × 10−6 | G2/M | Yes | |
| Pbi2 | 2.6 × 10−6 | S | No | |
| Sic1 | 7.5 × 10−6 | M/G1 | No | Bound by Swi5 |
| Pho11 | 1.53 × 10−5 | G2/M | No | Bound by Fkh1 |
| Pho12 | 1.77 × 10−5 | G2/M | No | |
| Mnn1 | 2.1 × 10−5 | G1 | No | |
| Bud9 | 2.4 × 10−5 | G1 | Yes | Bound by Fkh1, Fkh2, Mcm1, Ace2, and Swi5 |
| Pry1 | 4.6 × 10−5 | G2/M | No | Bound by Fkh2 Ndd1, Mcm1, and Ace2 |
| Pir1 | 4.7 × 10−5 | M/G1 | Yes | Bound by Mcm1 and Swi5 |
| Ash1 | 5.3 × 10−5 | M/G1 | Yes | Bound by Swi5 |
Top 30 differentially expressed cell-cycle genes identified, ordered by significance P value. Phase is based on assignment by Spellman et al. (4). The previously identified column is based on a list of genes extracted from Zhu et al. (5). Binding information is taken from Simon et al. (19) by using a P-value cutoff of 0.005. Note that many of the genes in this list that are bound by one of the effected factors were not identified by using the cluster-based method.
Fkh1 and Fkh2, which should bind directly to regulated genes;
Swi5 and Ace2, which should bind the promoters of the indirectly regulated genes; and
Mcm1 and Ndd1, which are cofactors of Fkh2 in the regulation of G2/M genes (20) and therefore should bind at least a subset of the Fkh2 target genes.
As can be seen in Fig. 2, the set of genes identified by our algorithm agrees very well with the binding data. Factors 1-3 were significantly enriched for genes in the identified set; on the other hand, there was no significant enrichment for binding of Swi4, Mbp1, and Swi6. Overall, the expression changes of 37 of the 56 genes (66%) can be explained by the binding of the six factors listed above (P = 4 × 10-11). A number of other genes can be explained by using prior knowledge.
Fig. 2.

Differentially expressed cell cycle genes. (A) Percentage of genes bound by the nine factors in the entire set of 800 cell-cycle-regulated genes and the set identified by our algorithm. As can be seen, all the relevant factors are significantly enriched for genes in the selected set (P values are in parentheses after the factor name). (B) Results of clustering the 56 selected genes. Cluster 1 is composed of genes that are affected directly and clusters 2 and 3 contain genes with second-order effects. See Results and Discussion.
To examine the different ways in which these 56 genes were affected, we clustered their expression profiles in the knockout experiment (using the K-means algorithm) into three different groups. Most of the genes (45 of 56) belonged to cluster 1. As can be seen in Fig. 2, this cluster had a flat profile, indicating that genes in this cluster lost their cycling ability in the knockout experiment. The second and third clusters contained fewer genes (eight and three) and represent either loss of cycling early on (cluster 2) or seeming participation in only one cycle instead of the two that are covered by the experiment duration. Interestingly, 8 of the 11 genes in the second and third clusters (>70%) were bound by Swi5 or Ace2 (compared with only 10% in the entire set of 800 cycling genes), suggesting that these clusters are composed of genes with expression changes caused by second-order downstream effects of the knockout of Fkh1/2. In contrast, genes in the first cluster were more likely to be bound by Fkh1/2 or one of their two cofactors (44% versus 20% in the entire set of 800 cycling genes).
Comparison with Clustering-Based Methods. As mentioned above, due to the differences in sampling rates and the different cell-cycle durations (see Table 1) in the two time-series experiments, all previously reported methods for identifying differentially expressed genes other than clustering-based analysis cannot be applied to these data sets. Thus we compared our results with the list of 42 genes identified in the original paper by Zhu et al. (5) using cluster analysis. The two lists overlapped in 21 genes. As can be seen in Table 2, many of the genes identified by our algorithm and not detected by hierarchical clustering seem to be controlled by one of the factors affected by the knockout, indicating that they were identified correctly by our method. In addition, many of them seem to be losing their cycling ability (see Fig. 3 Upper). It is likely that these genes are missed when using cluster analysis, because most of the other genes in their clusters did not change significantly. In contrast, following alignment, many of the 21 genes identified by Zhu et al. that were not detected by our algorithm do not seem to be changing in expression between the two experiments.
Fig. 3.

Genes identified by our algorithm that were missed by the clustering method used in ref. 5. (Upper) Five of the cell-cycle-regulated genes. WT expression is represented by the solid line, and knockout by the dashed line. A P value appears to the right of the gene name. As can be seen, all these genes displayed significant reduction in their cycling ability. In addition, all the above genes are bound by Fkh1/2, Ace2, or Swi5, indicating that our algorithm identified a relevant set of genes. (Lower) Five of the noncycling genes. Note that some of these genes seem to be cycling in the knockout experiment, whereas they are not cycling in the WT experiment (see Results and Discussion).
Analysis of Non-Cell-Cycle-Regulated Genes. Although the main function of the Fkh1/2 transcription factors is in regulating the cell cycle, they are also involved in other functions including mating type switching and cell morphology. We used our algorithm to identify differentially expressed genes in the set of 5,000 genes that are not cell-cycle-regulated. Due to the size of this set, we used a more stringent P value of 0.001 (thus, in random data we would expect only five genes to be identified as significantly differentially expressed). Our algorithm identified 22 genes as significantly changing in the knockout data. We note that Zhu et al. (5) did not identify any noncycling genes as differentially expressed, perhaps because of the limitation of the cluster analysis method.
In sharp contrast with the cycling genes, all except one of the promoters of the affected non-cycling genes are not bound by any of the cell-cycle transcription factors (see Table 3), suggesting that these genes are not controlled directly by the forkhead proteins or by their direct targets (Swi5 and Ace2). Fkh1/2 double-null mutation has global affects on cell growth; the cells show pseudohyphal and invasive growth phenotypes, unusual cell morphology, and slow growth rates (5, 21). Thus, some of the changes in gene expression in the mutant cells may be due to the overall changes in the cell rather than the direct effects of Fkh1/2.
Table 3. Differentially expressed noncycling genes.
| Gene name | P value |
|---|---|
| YDR098C | 1.9 × 10−10 |
| YER037W | 4.7 × 10−8 |
| Ura1 | 6.4 × 10−8 |
| YCR007C | 2 × 10−7 |
| Gdh2 | 3.6 × 10−7 |
| Cit1 | 1.8 × 10−6 |
| YOL164W | 1.4 × 10−5 |
| YFR026C | 1.4 × 10−5 |
| Ade5,7 | 4 × 10−5 |
| Mdh2 | 7.5 × 10−5 |
| Ura3 | 1.5 × 10−4 |
| Ctr1 | 1.8 × 10−4 |
| Cit2 | 1.9 × 10−4 |
| YNL279W | 2.2 × 10−4 |
| YLR162W | 4.3 × 10−4 |
| Fig1 | 5.1 × 10−4 |
| Hxt6 | 5.2 × 10−4 |
| Fre7 | 5.4 × 10−4 |
| Pot1 | 5.5 × 10−4 |
| Acb1 | 6.2 × 10−4 |
| YMR040W | 9.1 × 10−4 |
| Ade1 | 9.9 × 10−4 |
Shown are the 22 noncycling genes identified by our algorithm as differentially expressed, ordered according to their significance P value. Of these 22 genes, only Fig1 is bound by one of the cell-cycle activators (Swi5) that were profiled by Simon et al. (19). As discussed in Results and Discussion, most of these genes are significantly correlated with yeast response to stress in a set a stress-related expression experiments.
To investigate the set of genes identified by our algorithm further, we looked at a large collection of gene expression experiments (see www.psrg.lcs.mit.edu/DiffExp/DiffExp.html). Following Hughes et al. (22), we looked for experiments in which these genes were significantly correlated in the following way. For each expression experiment we performed a hypergeometric test to compute the significance for the up- (or down-) regulation of the genes in the set detected by our algorithm in that experiment. We include an experiment in the selected set if
The P value for either up- or down-regulation is <0.0001, and
At least 25% of the genes in our set are up- or down-regulated in that experiment.
Based on these criteria, 20 expression experiments were selected (see Table 4, which is published as supporting information on the PNAS web site). These experiments come from six different data sets, indicating that our results are not an artifact of a specific hybridization method. We also carried out a randomization analysis to test the significance of these results and concluded that, if random sets of genes of equal size are used, no experiments are selected. This indicates that the set of genes identified by our algorithm is significantly associated with the experiments selected by this method.
The experiments that were selected fall mainly into the categories red/ox stress, response to α factor, response to zinc depletion, and starvation. These finding suggests that
our algorithm finds relevant sets of differentially expressed genes,
Fkh1/2 may be involved in cellular pathways that are associated with the conditions under which these experiments were carried out,
because some of the noncycling genes are cycling in the knockout experiment, whereas they are flat under WT conditions (see Fig. 3), the effects of the identified conditions may vary along the cell cycle.
Our ability to raise such hypotheses indicates the importance of algorithms that are specifically designed to analyze time-series gene expression data.
Extensions to Other Data Sets. We used transcription factor knockout data to test our algorithm and to show that it correctly detects differentially expressed genes that were not detected by using prior methods. In addition, we have shown that by focusing on the set of genes detected by our algorithm, we can correctly detect first- and second-order effects of the experimental condition. Our algorithm can be used to analyze many biological systems, including infectious and other diseases, and cell behavior under different treatments that have been studied by using time-series expression data. For such systems, there is usually no independent high-throughput data source that can be used to validate sets of differentially expressed genes. Thus, when analyzing such systems, it is important to use computational methods that have been shown to produce correct results such as the algorithm described here.
Supplementary Material
Acknowledgments
We thank Richard Young, Tony Lee, and Chen-Hsiang Yeang for comments on earlier drafts of this manuscript. Z.B.-J. is supported by the Program in Mathematics and Molecular Biology at Florida State University through the Burroughs Wellcome Fund Interfaces Program. G.G. is supported by a National Defense Engineering and Science graduate fellowship.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Golub, T. R., Slonim, D. K., Tamayo, P., Huard, C., Gassenbeek, M., Mesirov, J., Coller, H., Loh, M., Downing, J. R. & Caliqiuri, M. A., et al. (1999) Science 286, 531-537. [DOI] [PubMed] [Google Scholar]
- 2.Nau, G. J., Richmond, J. F. L., Schlesinger, A., Jennings, E. G., Lander, E. S. & Young, R. A. (2002) Proc. Natl. Acad. Sci. USA 99, 1503-1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gasch, A. P., Spellman, P. T., Kao, C. M., Carmel-Harel, O., Eisen, M. B., Storz, G., Botstein, D. & Brown, P. O. (2000) Mol. Biol. Cell 11, 4241-4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spellman, P. T., Sherlock, G., Zhang, M. Q., Iyer, V. R., Anders, K., Eisen, M. B., Brown, P. O., Botstein, D. & Futcher, B. (1998) Mol. Biol. Cell 9, 3273-3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu, G., Spellman, P. T., Volpe, T., Brown, P. O., Botstein, D., Davis, T. N. & Futcher, B. (2000) Nature 406, 90-94. [DOI] [PubMed] [Google Scholar]
- 6.Pramilla, T., Miles, S., GuhaThakurta, D., Jemiolo, D. & Breeden, L. L. (2002) Genes Dev. 16, 3034-3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Troyanskaya, O. G., Garber, M. E., Brown, P. O., Botstein, D. & Altman, R. B. (2002) Bioinformatics 18, 1454-1461. [DOI] [PubMed] [Google Scholar]
- 8.Xu, X. L., Olson, J. M. & Zhao, L. P. (2002) Hum. Mol. Genet. 11, 1977-1985. [DOI] [PubMed] [Google Scholar]
- 9.Huang, Q., Liu, D., Majewski, P., Schulte, L. C., Korn, J. M., Young, R. A., Lander, E. S. & Hacohen, N. (2001) Science 294, 870-875. [DOI] [PubMed] [Google Scholar]
- 10.Ramoni, M. F., Sebastiani, P. & Kohane, I. S. (2002) Proc. Natl. Acad. Sci. USA 99, 9121-9126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian, J., Dolled-Filhart, M., Lin, J., Yu, H. & Gerstein, M. (2001) J. Mol. Biol. 314, 1053-1066. [DOI] [PubMed] [Google Scholar]
- 12.Holter, N. S., Maritan, A., Cieplak, M., Fedoroff, N. V. & Banavar, J. R. (2001) Proc. Natl. Acad. Sci. USA 98, 1693-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aach, J. & Church, G. M. (2001) Bioinformatics 17, 495-508. [DOI] [PubMed] [Google Scholar]
- 14.D'haeseleer, P., Wen, X., Fuhrman, S. & Somogyi, R. (1999) Pac. Symp. Biocomput. 41-52. [DOI] [PubMed]
- 15.Zhao, L. P., Prentice, R. & Breeden, L. (2001) Proc. Natl. Acad. Sci. USA 98, 5631-5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bar-Joseph, Z., Gerber, G., Jaakkola, T. S., Gifford, D. K. & Simon, I. (2003) J. Comput. Biol. 3-4, 341-356. [DOI] [PubMed] [Google Scholar]
- 17.Alter, O., Brown, P. O. & Botstein, D. (2003) Proc. Natl. Acad. Sci. USA 100, 3351-3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogers, D. & Adams, J. (1990) Mathematical Elements for Computer Graphics (McGraw-Hill, New York), pp. 247-375.
- 19.Simon, I., Barnett, J., Hannett, N., Harbison, C. T., Rinaldi, N. J., Volkert, T. L., Wyrick, J. J., Zeitlinger, J., Gifford, D. K., Jaakkola, T. S. & Young, R. A. (2001) Cell 106, 697-708. [DOI] [PubMed] [Google Scholar]
- 20.Koranda, M., Schleiffer, A., Endler, L. & Ammerer, G. A. (2000) Nature 406, 94-98. [DOI] [PubMed] [Google Scholar]
- 21.Hollenhorst, P. C., Bose, M. E., Mielke, M. R., Muller, U. & Fox, C. A. (2000) Genetics 154, 1533-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hughes, T. R., Marton, M. J., Jones, A. R., Roberts, C. J., Stoughton, R., Armour, C. D., Bennett, H. A., Coffey, E., Dai, H., He, Y. D., et al. (2000) Cell 102, 109-126. [DOI] [PubMed] [Google Scholar]
- 23.Cho, R. J., Campbell, M. J., Winzeler, E. A., Steinmetz, L., Conway, A., Wodicka, L., Wolfsberg, T. G., Gabrielian, A. E., Landsman, D., Lockhart, D. J. & Davis, R. W. (1998) Mol. Cell 2, 65-73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}