

Abstract
The G534E polymorphism (Marburg I [MI]) of factor VII–activating protease (FSAP) is associated with carotid stenosis and cardiovascular disease. We have previously demonstrated that FSAP is present in atherosclerotic plaques and it is a potent inhibitor of vascular smooth muscle proliferation and migration in vitro. The effect of wild-type (WT)- and MI-FSAP on neointima formation in the mouse femoral artery after wire-induced injury was investigated. Local application of WT-FSAP led to a 70% reduction in the neointima formation, and this effect was dependent on the protease activity of FSAP. MI-FSAP did not inhibit neointima formation in vivo. This is due to a reduced proteolytic activity of MI-FSAP, compared to WT-FSAP, toward platelet-derived growth factor BB, a key mediator of neointima development. The inability of MI-FSAP to inhibit vascular smooth muscle accumulation explains the observed linkage between the MI-polymorphism and increased cardiovascular risk. Hence, FSAP has a protective function in the vasculature, and analysis of MI polymorphism is likely to be clinically relevant in restenosis.
The chances of restenosis are ∼40% with balloon angioplasty and ∼25% after stenting. With the advent of “drug-eluting” stents the extent of restenosis can be reduced to 0–5% (1). Identification of patients at risk for developing restenosis will lead to better patient treatment based on individual needs, and this has stimulated a search for markers in addition to classical risk factors such as hypertension and diabetes (2).
A newly identified plasma protein called factor VII–activating protease (FSAP) is known to activate prourokinase (pro-uPA) and is thus a new member of the fibrinolysis pathway (3). A polymorphism in FSAP gene, G534E, also called the Marburg I (MI) polymorphism, is found in ∼5% of the population, and it is associated with atherosclerosis leading to carotid stenosis (4) cardiovascular disease (5) and possibly thromboembolic disorders (6). MI-FSAP has a weaker pro-uPA activation potential than WT-FSAP but seems to be equipotent with WT-FSAP with respect to factor VII activation (7). FSAP is present in atherosclerotic plaques (8), and it is a potent inhibitor of platelet-derived growth factor BB (PDGF-BB)–mediated vascular smooth muscle cell (VSMC) proliferation and migration in vitro (8). Here we demonstrate that FSAP is a potent inhibitor of neointima formation in vivo. Moreover, the MI isoform of FSAP is not active in this respect. Together with a mechanistic insight into the inhibition of neointima formation, these results provide a clear rationale for using the MI-FSAP as a diagnostic tool to predict the development of postangioplasty restenosis. Application of FSAP may represent a novel therapeutic approach to prevent restenosis.
RESULTS AND DISCUSSION
Isolation and characterization of MI-FSAP and its comparison with WT-FSAP
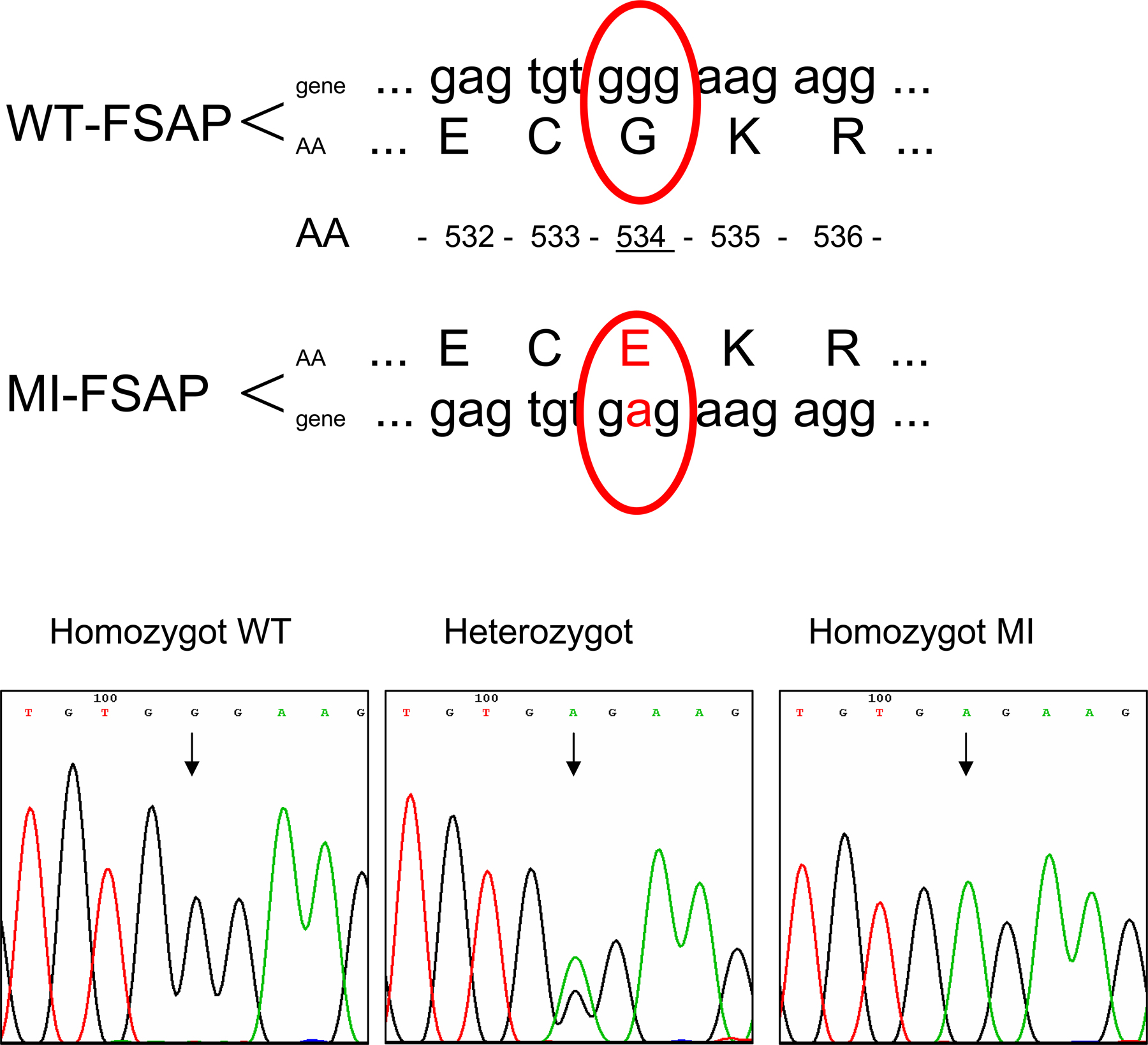
The reduced ability of MI-FSAP to activate pro-uPA (7) was used to screen 1,000 subjects for the homozygous MI genotype. Genomic DNA was sequenced to confirm the MI homozygous genotype in a singular subject (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20052546/DC1), and MI-FSAP was isolated and compared with WT-FSAP prepared under identical conditions. The size and immunoreactivity of both isoforms were identical as was the autocatalytic conversion of the single-chain form into the two-chain form (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20052546/DC1). Chymotrypsin digestion followed by matrix-assisted laser desorption time of flight spectroscopy (MALDI-TOF) analysis showed that there was an alteration in the molecular weight of a peptide caused by the amino acid difference G534E (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20052546/DC1). With purified proteins we could confirm that MI-FSAP had reduced proteolytic activity toward its direct chromogenic substrate (Fig. 1 A). WT- and MI-FSAP had a Vmax of 10,577 ± 2,103 and 3,917 ± 848 μmole/min/mg enzyme and a Km of 40 ± 27 and 27 ± 4 μM, respectively. Pro-uPA activation was also weaker with MI-FSAP compared with WT-FSAP (Fig. 1 B). Heparin and PDGF-BB binding characteristics were identical for WT- and MI-FSAP (Fig. 1, C and D). FSAP cleaved PDGF-BB, and this was observed only under reducing conditions but not under nonreducing conditions (Fig. 1 E). 125I–PDGF-BB was also cleaved by WT-FSAP to a limited extent, and under reducing conditions, smaller molecular weight bands were observed (Fig. 1 F). The rate of cleavage by WT-FSAP was much faster than by MI-FSAP (Fig. 1 F). Native PDGF-BB cleavage was observed after 15 min at a ratio of protease to PDGF-BB of 3:1 (Fig. S3). In our previous report, we only used nonreducing conditions and hence this cleavage was not observed (8). In conclusion, the alteration of an amino acid in the serine protease domain of MI-FSAP resulted in a loss of proteolytic activity, whereas the binding characteristics were unchanged. PDGF-BB is specifically cleaved and inactivated by WT-FSAP to a greater extent than by MI-FSAP.
Figure 1.

Enzymatic and binding properties of WT- and MI-FSAP. (A) WT- and MI-FSAP (0.33 μg/ml each) were incubated with increasing concentrations of the chromogenic substrate (H-D-Ile-Pro-Arg-pNA) in the presence of heparin (10 μg/ml), and the maximal velocity (MaxV) was measured in a kinetic plate reader (mean ± SD of triplicate wells). (B) Pro-uPA activation (MaxV) was measured, as described previously (3), in the presence of increasing concentrations of WT- and MI-FSAP in the presence of heparin. (C) Indicated concentrations of FSAP were immobilized, and biotinylated heparin-BSA (0.5 μg/ml) was used as a ligand and its binding was detected with streptavidin-coupled peroxidase (mean ± SD of triplicate wells). (D) PDGF-BB (1 μg/ml) was immobilized, and FSAP (0.5 μg/ml) was used as a ligand in the absence or presence of heparin (10 μg/ml) and its binding was detected with anti-FSAP antibody (mean ± SD of triplicate wells). (E) Mixtures of FSAP (or PPACK-FSAP) (10 μg/ml), buffer control, heparin (10 μg/ml), PDGF-BB (1 μg/ml), and aprotinin (15 μg/ml) were incubated for 1 h at 37°C, and Western blot was performed with an anti–PDGF-BB antibody under reducing or nonreducing conditions. (F) 125I–PDGF-BB was incubated with WT- or MI-FSAP in the presence of heparin, and after SDS-PAGE under reducing conditions autoradiography was performed. The effect of different concentrations of FSAP was examined at the 60-min time point (top), and the time course (bottom) was analyzed at FSAP concentration of 1 μg/ml.
Endogenous FSAP in the injured vessels
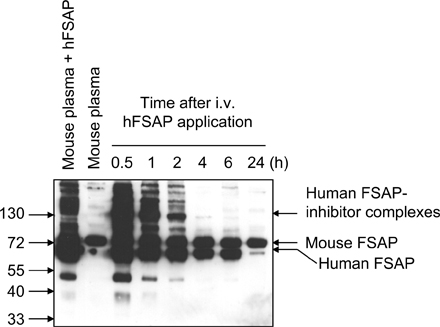
In Western blots, an anti–mouse FSAP antibody could detect FSAP in mouse plasma in its single-chain form, FSAP inhibitor complexes, and degradation products after autoactivation with polyanions (Fig. 2 A, left). These results indicate that there is a substantial amount of FSAP in mouse plasma. Mouse FSAP was also detected in 293 cells transfected with the active site mutant H399F-FSAP by Western blotting and by immunocytochemistry (Fig. 2, A and B). Only a faint scattered staining was observed with an anti–mouse FSAP antibody in the media of normal arteries (Fig. 2 C, top left). In the developing neointima of mechanically injured vessels, there was generally more staining (Fig. 2 C, bottom right). Under identical conditions, intense staining was observed in the liver, which is the primary source of FSAP expression in the mouse (Fig. 2 C, top right). No FSAP mRNA transcripts could be detected in femoral or carotid arteries, monocytes, or VSMCs, but high levels were found in mouse liver (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20052546/DC1). Hence, FSAP in the vasculature, when present, seems not to be produced locally but is derived from the circulation. In normal human arteries, there was no FSAP immunostaining; however, in human atherosclerotic plaques there were foci of FSAP immunostaining in regions of macrophage- and VSMC-derived foam cells and less FSAP was present in medial VSMCs (8). Hence, there are some parallels but also differences in the staining pattern of FSAP in mouse and human arteries.
Figure 2.

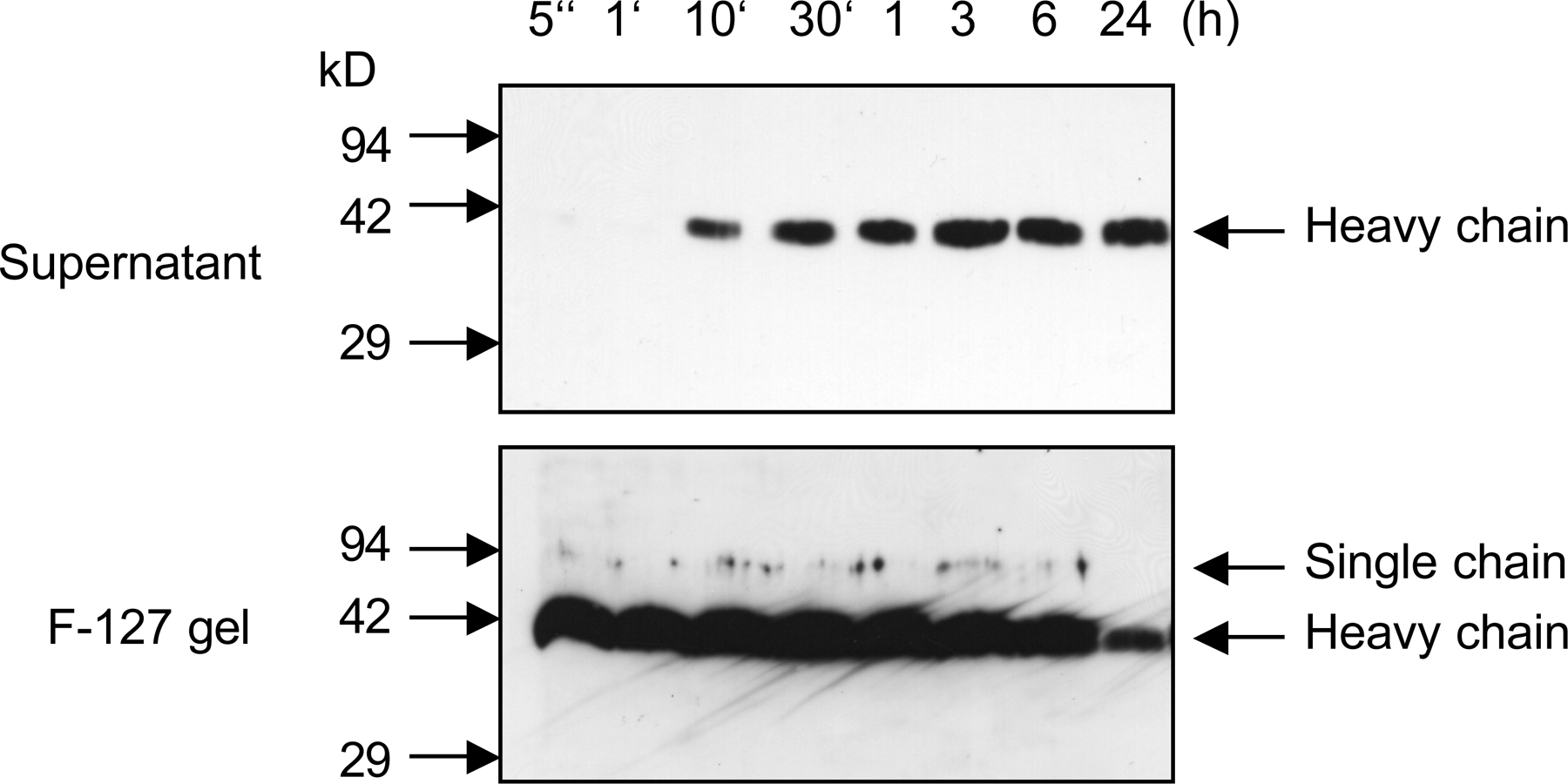
Release, activation, and localization of exogenously applied human FSAP and endogenous mouse FSAP. (A) Mouse plasma with and without polyinosinic-polycytidylic acid (11) (50 μg/ml) (polyanions) was subjected to precipitation with acetic acid (left). HEK-293 cells were transfected with vector (pIRESpuro3) alone or vector encoding mouse FSAP (H399F), and the conditioned medium was collected (right). These samples were analyzed by Western blotting with an anti–mouse FSAP antibody. (B) For immunofluorescence analysis HEK-293 cells were transfected with an empty vector or that encoding mouse FSAP and were stained with an anti–mouse FSAP antibody. (C) Mice were killed after 7 or 21 d after wire-induced injury of the femoral artery. Contralateral vessel and liver were also excised for analysis. Immunofluorescence analysis for mouse FSAP was performed with an anti–mouse FSAP antibody, and nuclei were stained with DAPI. There was no immunostaining reaction with the negative control antibody. (D) FSAP was added to pluronic F-127 gel in PBS at 4°C, and then the mixture was shifted to 37°C in order for it to gel, and it was covered with a buffer. The supernatants and the pluronic gels were removed at the indicated times and analyzed for human FSAP using a combination of mouse monoclonal antibodies against the light (mAb 677) and heavy chain (mAb 1189) of FSAP. Pluronic F-127 gel influences the migration properties of proteins in SDS-PAGE and gives rise to artifacts as indicated in the figure. (E) FSAP (1 μg) was added to 100 μl pluronic F-127 gel in PBS at 4°C, and then the mixture was applied to a mouse artery in situ in vivo. At the indicated times, the pluronic gel was recovered and analyzed for the presence of human FSAP by Western blotting using biotinylated mAb 1189. (F) After injury, 100 μl of pluronic F-127 was applied, either with FSAP (1 μg) or buffer control. The mice were killed after 12 h, 48 h, or 21 d. Control sections were from the respective contralateral artery. Immunofluorescence analysis for human FSAP was performed with directly labeled anti–human FSAP mAb 677.
Application of exogenous human FSAP to the injured vessels
To investigate the effect of FSAP on neointima formation, it was applied to injured arteries in a gel to attain high, sustainable, local concentrations. Before application of FSAP in the thermosensitive pluronic F-127 gel to mechanically injured arteries, the stability and diffusibility of FSAP was analyzed in vitro and in vivo. There was a slow sustained release of intact FSAP from the pluronic F-127 gel over a period of 24 h in vitro (Fig. 2 D, supernatant). FSAP released from the gel was in the active two-chain form (Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20052546/DC1). Maximal re-lease was at ∼24 h, a time frame where initiation of early events takes place that trigger the process of neointima formation. When applied abluminally to mouse arteries in vivo, most of the FSAP was released from the gel within 1 h. No FSAP inhibitor complexes were observed in the gel in vivo (Fig. 2 E). There was a complete resorption of the gel after 24–48 h. In contrast, application of intravenous FSAP lead to the formation of complexes with inhibitors present in the plasma (Fig. S6, available at http://www.jem.org/cgi/content/full/jem.20052546/DC1). Immunofluorescence analysis of injured arteries indicated that the exogenously applied human FSAP was released from the gel and associated with the vasculature because there was increased FSAP immunoreactivity in vessels after 12 and 48 h. Nontreated vessels showed virtually no immunoreactivity (Fig. 2 F). The monoclonal antibody (mAb 677) used for these experiments did not cross-react with mouse FSAP. Hence, active two-chain FSAP was constantly released from the gel, and it was present at a crucial phase of the initiation of neointima formation (9). The abluminally applied exogenous FSAP may be more effective compared with its endogenous circulating counterpart, because it is relatively protected against endogenous protease inhibitors present in the plasma and may diffuse more easily into the vessel wall.
Comparison of MI-FSAP and WT-FSAP in cell proliferation, p42/44 mitogen-activated protein kinase phosphorylation, and neointima formation
The influence of WT-FSAP on neointimal thickening, after direct application to the denuded artery in a thermosensitive gel, was determined 3 wk after injury. FSAP application led to a dose-dependent decrease in the intima to media ratio (Fig. 3 A), and maximal inhibition (∼70%) was achieved at 0.5–1 μg FSAP per mouse. FSAP application did not influence the medial area, but only the intimal area was reduced (Fig. S7, available at http://www.jem.org/cgi/content/full/jem.20052546/DC1). Because the in vitro inhibitory effect of FSAP on VSMCs was neutralized after protease inactivation (8), we used the active site–inhibited Phe-Pro-Arg- chloromethylketone (PPACK)-FSAP (Fig. S8, available at http://www.jem.org/cgi/content/full/jem.20052546/DC1) for comparison. PPACK-FSAP did not inhibit neointima formation (Fig. 3 A). Because FSAP was applied locally to the injured artery, we hypothesize that FSAP diffuses into the artery and thereby mediates its inhibitory effect rather than having a more systemic mode of action. This was investigated by applying FSAP to the contralateral uninjured artery and not directly to the denuded artery. Distant application of FSAP on the contralateral artery was ineffective in reducing the intima to media ratio (Fig. S7), indicating a local mode of action of FSAP.
Figure 3.

Comparison of WT- and MI-FSAP on DNA synthesis, p42/p44 MAPK phosphorylation, and neointima formation. (A) After injury, 100 μl of pluronic F-127 was applied to each artery either containing human single-chain FSAP (0.2, 0.5, 1 μg), buffer, or PPACK–two-chain FSAP (1 μg). The intima to media ratio is indicated (mean ± SD, n = 6 mice). *, P < 0.05; n.s., lack of significant difference compared with control. (B) In the hematoxylin and eosin (H &E)-stained sections, an open arrow indicates the internal elastic lamina and a filled arrow indicates the external elastic lamina. The neointima is indicated with the character “N”. α-Smooth muscle actin was detected with a directly Cy3-conjugated mAb (red) and vWF was stained with a FITC-conjugated secondary antibody (green), both with DAPI counterstaining. PCNA was stained brown with a biotin-streptavidin-peroxidase system. (C) After injury, pluronic gel containing either buffer, WT-FSAP, or MI-FSAP (1 μg) was applied, and neointima formation was determined (mean ± SD, n = 6 mice). *, P < 0.05; n.s., lack of significant difference compared with control. (D) DNA synthesis (depicted as absorbance [mean ± SD of triplicate wells]) in VSMCs was analyzed in the absence (white bars) or presence (grey bars) of 20 ng/ml PDGF-BB and either control buffer, WT-FSAP, or MI- FSAP (10 μg/ml) in the presence of heparin (10 μg/ml). (E) Mixtures in the absence (−) or presence (+) of PDGF-BB (20 ng/ml) and either no further addition, buffer control, WT-FSAP, or MI-FSAP (10 μg/ml) in the presence of heparin (10 μg/ml) were preincubated for 1 h at 37°C in serum-free medium and then added to the cells for 15 min. Western blotting was performed to detect phosphorylated MAPK p42/p44 (top) and total MAPK (middle). Optical density was determined to estimate the extent of p42/p44 MAPK phosphorylation expressed as a ratio of phosphorylated to nonphosphorylated MAPK (bottom).
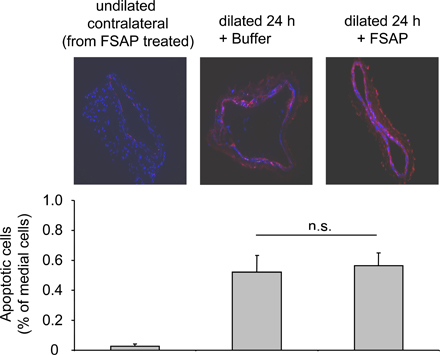
The immunofluorescence staining of α-smooth muscle actin (Fig. 3 B, red) was significantly reduced in FSAP-treated vessels compared with controls. The extent of staining correlated with the degree of neointimal thickening, indicating that FSAP inhibits the accumulation of VSMCs in the neointima. Reendothelialization of vessels after injury was not influenced by FSAP (Fig. 3 B, green; Fig. S7 D). FSAP significantly reduced the number of proliferating cell nuclear antigen (PCNA; or Ki76)-positive cells in the neointima and media by 70% (Fig. 3 B, brown; Fig. S7 D). Injury-induced apoptosis was not modulated by FSAP (Fig. S9, available at http://www.jem.org/cgi/content/full/jem.20052546/DC1). Hence, the proteolytic activity of FSAP is required for inhibition of cell proliferation in vivo and the associated neointima formation.
In the vascular injury model, there was no substantial inhibition of neointima formation with MI-FSAP (Fig. 3 C). In vitro, PDGF-stimulated DNA synthesis was inhibited strongly by WT-FSAP, but the inhibition by MI-FSAP was much weaker (Fig. 3 D). Similarly, PDGF-BB–stimulated phosphorylation of p42/p44 mitogen-activated protein kinase (MAPK) (extracellular signal-regulated kinase) was inhibited by WT-FSAP but not by MI-FSAP (Fig. 3 E). The residual proteolytic activity in MI-FSAP accounts for the partial inhibition of PDGF-BB in very sensitive assays such as DNA synthesis and MAPK phosphorylation (Fig. 3 D, and E), but the consequences of this proteolysis are not apparent in the relatively insensitive neointima formation model (Fig. 3 C). Hence, only WT-FSAP but not MI-FSAP seems to inhibit VSMC activation and neointima formation.
FSAP did not inhibit DNA synthesis or phosphorylation of p42/p44 MAPK induced by insulin-like growth factor (IGF)-1, thrombin, sphingosine-1-phosphate (S1P), TGF-β, basic fibroblast growth factor (bFGF), or hepatocyte growth factor (HGF) (Fig. 4). If at all, the effect of some of these growth factors was stronger in the presence of FSAP in accordance with previously published results on fibroblasts (10). Thus, PDGF-BB is the only growth regulator that is specifically cleaved and inhibited by WT-FSAP but not MI-FSAP. Hence, this difference might be central to their distinct effects on VSMC proliferation and neointima formation.
Figure 4.

Effect of FSAP on growth factor–induced DNA synthesis and phosphorylation of MAPK in VSMCs. (A) PDGF-BB (20 ng/ml), IGF-1 (100 ng/ml), thrombin (1 U/ml), S1P (200 nM), TGF-β (20 ng/ml), bFGF (50 ng/ml), and FCS (10% vol/vol) were preincubated with single-chain FSAP (10 μg/ml) (hatched bars) or vehicle (dotted bars) in the presence of heparin (25 μg/ml) for 60 min at 37°C and then added to serum-starved VSMCs. The extent of DNA synthesis was determined (mean ± SD of triplicate wells). (B) PDGF-BB, bFGF, IGF-1, TGF-β, bFGF, S1P, and HGF (20 ng/ml) were preincubated with FSAP (10 μg/ml) in the presence of heparin (25 μg/ml) for 60 min at 37°C and then added to serum-starved VSMCs for 15 min. The extent of phosphorylation of p42/p44 MAPK was determined by Western blotting. The top panel represents phosphorylated MAPK, and the bottom panel represents total MAPK.
Conclusions
FSAP is not produced locally in the vasculature but is taken up from the circulation. The inactive zymogen is activated by polyanions such as heparin, hyaluronans, and nucleic acids (11) that are released as a consequence of vascular injury or during the process of remodeling (12). Once activated, its actions are further regulated by the local protease inhibitors. Active FSAP could inhibit VSMC proliferation and migration and vascular lesion formation by inhibiting the PDGF-BB. Several investigations have indicated that PDGF-BB inhibition alone may be sufficient to inhibit neointimal growth (13). Therapeutic application of FSAP may represent a novel approach to prevent vascular proliferative disease such as restenosis. The information that the MI isoform of FSAP exhibits altered proteolysis of PDGF-BB provides a direct mechanistic link to vascular lesion formation in patients that harbor this polymorphism. Because MI polymorphism is present in 5% of the European population, our results provide a rationale for the development of MI as a risk predictor in restenosis.
MATERIAL AND METHODS
Materials.
The isolation of FSAP from human plasma, polyclonal antibody, and monoclonal antibodies to human FSAP, preparation of active site– inhibited PPACK-FSAP, and the conversion of single-chain latent FSAP into the two-chain active form and their characterization has been described previously (8). Rabbit polyclonal antibody to mouse FSAP was raised using the NH2-terminal peptide (SLMSFIAPPDC) as an antigen. The species specificity of anti–human FSAP and anti–mouse FSAP was determined by Western blotting and immunofluorescence analysis of human-FSAP– and mouse-FSAP–transfected 293 cells. Pluronic F-127 was from Sigma-Aldrich.
Mouse femoral artery injury model of neointimal hyperplasia.
Experiments were performed on C57/BL6 mice (six mice per treatment group) according to local ethical guidelines based on procedures described by Sata et al. (9). Immediately after dilatation, the artery was covered in 100 μl of a thermosensitive polymer (pluronic F-127 gel; Sigma-Aldrich) containing the test substances. The mice were killed by intraperitoneal administration of an overdose of Nembutal at the time points indicated (after 12 h, 48 h, or 3 wk). At death, the mice were perfused via the left ventricle with 0.9% (wt/vol) NaCl solution followed by perfusion fixation with 2% (wt/vol) paraformaldehyde in PBS (pH 7.4). The femoral artery was excised, postfixed in 2% (wt/vol) paraformaldehyde for 30 min, and embedded in Tissue Tek OCT embedding medium (Miles Laboratories), snap frozen, and stored at −80°C.
Immunohistochemistry and morphometry.
The whole artery was cut in 6-μm serial sections, and six sections per artery from regular intervals were fixed with acetone and stained with hematoxylin and eosin. For morphometric analysis, KS300 software (Carl Zeiss MicroImaging, Inc.) was used to measure external elastic lamina, internal elastic lamina, lumen circumference, and medial and neointimal area. For immunohistochemistry, slides were fixed in acetone (except for mouse FSAP analysis where paraformaldehyde was used), preincubated with 10% normal goat serum, and then incubated with antibodies against von Willebrand factor (vWF; Dako) or mouse FSAP. Ensuing incubations were performed with Cy5- or Cy3- coupled secondary antibodies. Monoclonal antibodies to human FSAP (mAb 677) and smooth muscle α-actin (Dako) were labeled directly with Alexa 488 or Cy3, respectively (Molecular Probes). Immunostaining for PCNA was performed by using a PCNA staining kit (Zymed Laboratories) with nuclear DAPI (Linaris) counterstain and analyzed under fluorescence/light microscopy. Negative controls were conducted by substituting the primary antibody through an appropriate species and isotype-matched control antibody. Reendothelialization (luminal circumference staining positive for vWF) was quantified using a scale from 0 (no staining) to 6 (complete staining of luminal circumference). Six sections per artery from regular intervals were evaluated from six mice per group.
Identification of a subject with MI homozygote genotype, isolation, and characterization of FSAP.
Because MI-FSAP has reduced ability to activate pro-uPA, we used an ELISA to screen for reduced FSAP activity in plasma. Activity was determined with the direct chromogenic substrate H-D-Ile-Pro-Arg-pNA (Chromogenix) and pro-uPA activation with the chromogenic substrate pyro-Glu-Gly-Arg-pNA (Chromogenix) and FSAP antigen levels described previously (7). From the pool of 1,000 subjects, a 45-yr-old female with a history of bleeding tendency was found to have normal FSAP antigen levels but no pro-uPA–activating potential. The bleeding tendency was likely to be caused by a concomitant vWF defect. Sequencing of the exon XIII of FSAP from the genomic DNA indicated that the subject was homozygous for the MI genotype. With respect to the second single nucleotide polymorphism, Q379E, the subject was heterozygous (7). Plasma from this subject was purified over an anti-FSAP antibody column, and the purified protein was analyzed by electrophoresis, Western blotting, pro-uPA activation tests, enzymatic activity assays, peptide finger printing, and MALDI-TOF.
Western blotting and immunofluorescence analysis of mouse and human FSAP.
Single-chain FSAP (10 μg/ml) and 25% (wt/vol) pluronic 127 gel were allowed to solidify for 30 min at 37°C. Thereafter, an equal volume of serum-free DMEM was added to the solidified gel. At the indicated times (5 s to 24 h), the buffer and the solidified gel were separated and mixed with SDS sample buffer and boiled. For the analysis of FSAP the samples were either nonreduced or reduced with β-mercaptoethanol (10%, vol/vol). For Western blot analysis of human FSAP a mixture of two monoclonal antibodies, raised against FSAP (mAb677 against the light chain and mAb1189 against the heavy chain), was used as described previously (8). In vivo analysis of FSAP release from gel was investigated by placing the FSAP gel mix directly on the arteries in situ for the indicated times.
Mouse plasma with and without activation by polyanions and supernatants from HEK-293 cells transfected with an empty vector (pIRESpuro3) or that encoding mouse FSAP (H399F) was analyzed by Western blotting using the mouse-FSAP–specific antibody after euglobin precipitation with acetic acid (10 mM). For immunofluorescence analysis HEK-293 cells were transfected and after 48 h cells were fixed with paraformaldehyde, permeabilized, and stained with a mouse FSAP-specific antibody.
DNA synthesis assay.
DNA synthesis assays were based on the uptake of BrdU during S-phase and quantitative binding of a monoclonal anti-BrdU antibody (Roche Diagnostics). Mouse VSMCs were cultivated for 2 d in a 96-well microtiterplate (Nunc). Subsequently, the cells were cultivated in serum-free medium for 1 d. Thereafter, the medium was exchanged against serum-free DMEM containing 0.1% FCS (vol/vol), and test substances were added as indicated in the Fig. 3 D and Fig. 4 A legends and the incorporation of BrdU was determined.
Phosphorylation of MAPK p42/p44.
VSMCs were prepared as for DNA synthesis assays and then stimulated for 15 min with the appropriate agonist mixture. The experiments were stopped by adding SDS sample buffer containing 1 mM orthovanadate, and the samples were processed for Western blotting. Detection of the phosphorylated forms of MAPK p42 and p44 (MAPK-p42/p44) was performed with an antibody against MAPK-p42/p44 (New England Biolabs, Inc.). Total MAPK was also determined to show no quantitative changes in the amount of total cellular MAPK (New England Biolabs, Inc.).
Cleavage of PDGF-BB by FSAP and binding of FSAP to heparin or PDGF-BB.
PDGF-BB (1 μg/ml) was incubated with FSAP (10 μg/ml) or PPACK-FSAP or control buffer in Tris, pH 7.4, 100 mM NaCl, 2 mM CaCl2 for 1 h at 37°C in the absence or presence of heparin (10 μg/ml) or aprotinin (10 μg/ml) as indicated in Fig. 1 E, and the reaction was stopped with SDS sample buffer. Western blots were performed under reducing (β-mercaptoethanol, 10%, vol/vol) or nonreducing conditions, and PDGF-BB was detected. Alternatively, experiments were performed with 125I–PDGF-BB (GE Healthcare), and cleavage was followed by SDS-PAGE and autoradiography. PDGF-BB was immobilized in a Maxisorp microtiter 96-well plate (Nunc) at a concentration of 1 μg/ml (50 μl solution) overnight at 4°C in 50 mM NaHCO3 buffer, pH 9.6. The plate was blocked with 3% (wt/vol) BSA in Tris, pH 7.4, 100 mM NaCl. FSAP (0.5 μg/ml) in the presence or absence of heparin (10 μg/ml) was added to the wells with 0.3% (wt/vol) BSA for 2 h at 22°C. After extensive washing, bound FSAP was detected with a mAb followed by peroxidase-linked secondary antibody. Binding of biotinylated heparin-albumin conjugate to immobilized FSAP was performed in a similar manner. The binding of ligands to BSA-coated wells was used as a blank in all the experiments and was subtracted to obtain specific binding.
Statistical analysis.
For the studies with experimental animals the data between the study groups were analyzed by ANOVA followed by pair-wise comparison with Fisher's least significant difference test. All calculations were made with the Statgraphics plus statistical package (Manugistics).
Online supplemental material.
Fig. S1 shows the determination of the genotype of different subjects with respect to the single nucleotide polymorphism G534E. Fig. 2 shows a biophysical characterization of purified WT- and MI-FSAP. Fig. 3 shows a characterization of FSAP-mediated cleavage of PDGF-BB. Fig. 4 displays an analysis of FSAP mRNA by RT-PCR in mouse vascular cells and tissue. Fig. 5 displays the release and activation of FSAP from pluronic gel. Fig. 6 shows a characterization of human FSAP in mouse plasma. Fig. 7 shows the influence of FSAP on neointima formation in the mouse femoral artery after wire-induced injury. Fig. 8 shows a characterization of PPACK-FSAP. Fig. 9 displays the influence of FSAP on apoptosis in the mouse femoral artery after wire-induced injury. Figs. S1–S9 are available at http://www.jem.org/cgi/content/full/jem.20052546/DC1.
Supplemental Material
Acknowledgments
We thank Dr. Monica Linder, Susanne Tannert-Otto, Thomas Schmidt Wöll, and Stefanie Wolfram for their excellent assistance.
Grant support was from the Deutsche Forschungsgemeinschaft (Bonn, Germany) to S.M. Kanse (Ka 1468/4-1 and SFB547:C14).
The authors have no conflicting financial interests.
References
- 1.Woods, T.C., and A.R. Marks. 2004. Drug-eluting stents. Annu. Rev. Med. 55:169–178. [DOI] [PubMed] [Google Scholar]
- 2.Monraats, P.S., N.M. Pires, W.R. Agema, A.H. Zwinderman, A. Schepers, M.P. de Maat, P.A. Doevendans, R.J. de Winter, R.A. Tio, J. Waltenberger, et al. 2005. Genetic inflammatory factors predict restenosis after percutaneous coronary interventions. Circulation. 112:2417–2425. [DOI] [PubMed] [Google Scholar]
- 3.Roemisch, J., A. Feussner, and H.A. Stohr. 2001. Quantitation of the factor VII- and single-chain plasminogen activator-activating protease in plasmas of healthy subjects. Blood Coagul. Fibrinolysis. 12:375–383. [DOI] [PubMed] [Google Scholar]
- 4.Willeit, J., S. Kiechl, T. Weimer, A. Mair, P. Santer, C.J. Wiedermann, and J. Roemisch. 2003. Marburg I polymorphism of factor VII–activating protease: a prominent risk predictor of carotid stenosis. Circulation. 107:667–670. [DOI] [PubMed] [Google Scholar]
- 5.Ireland, H., G.J. Miller, K.E. Webb, J.A. Cooper, and S.E. Humphries. 2004. The factor VII activating protease G511E (Marburg) variant and cardiovascular risk. Thromb. Haemost. 92:986–992. [DOI] [PubMed] [Google Scholar]
- 6.Hoppe, B., F. Tolou, H. Radtke, H. Kiesewetter, T. Dorner, and A. Salama. 2005. Marburg I polymorphism of factor VII-activating protease is associated with idiopathic venous thromboembolism. Blood. 105:1549–1551. [DOI] [PubMed] [Google Scholar]
- 7.Roemisch, J., A. Feussner, C. Nerlich, H.A. Stoehr, and T. Weimer. 2002. The frequent Marburg I polymorphism impairs the pro-urokinase activating potency of the factor VII activating protease (FSAP). Blood Coagul. Fibrinolysis. 13:433–441. [DOI] [PubMed] [Google Scholar]
- 8.Kannemeier, C., N. Al-Fakhri, K.T. Preissner, and S.M. Kanse. 2004. Factor VII activating protease (FSAP) inhibits growth factor-mediated cell proliferation and migration of vascular smooth muscle cells. FASEB J. 18:728–730. [DOI] [PubMed] [Google Scholar]
- 9.Sata, M., Y. Maejima, F. Adachi, K. Fukino, A. Saiura, S. Sugiura, T. Aoyagi, Y. Imai, H. Kurihara, K. Kimura, et al. 2000. A mouse model of vascular injury that induces rapid onset of medial cell apoptosis followed by reproducible neointimal hyperplasia. J. Mol. Cell. Cardiol. 32:2097–2104. [DOI] [PubMed] [Google Scholar]
- 10.Etscheid, M., N. Beer, and J. Dodt. 2005. The hyaluronan-binding protease upregulates ERK1/2 and PI3K/Akt signalling pathways in fibroblasts and stimulates cell proliferation and migration. Cell. Signal. 17:1486–1494. [DOI] [PubMed] [Google Scholar]
- 11.Nakazawa, F., C. Kannemeier, A. Shibamiya, Y. Song, E. Tzima, U. Schubert, T. Koyama, M. Niepmann, H. Trusheim, B. Engelmann, and K.T. Preissner. 2005. Extracellular RNA is a natural cofactor for the (auto-)activation of Factor VII-activating protease (FSAP). Biochem. J. 385:831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riessen, R., T.N. Wight, C. Pastore, C. Henley, and J.M. Isner. 1996. Distribution of hyaluronan during extracellular matrix remodeling in human restenotic arteries and balloon-injured rat carotid arteries. Circulation. 93:1141–1147. [DOI] [PubMed] [Google Scholar]
- 13.Englesbe, M.J., S.M. Hawkins, P.C. Hsieh, G. Daum, R.D. Kenagy, and A.W. Clowes. 2004. Concomitant blockade of platelet-derived growth factor receptors alpha and beta induces intimal atrophy in baboon PTFE grafts. J. Vasc. Surg. 39:440–446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}