

Abstract
Protein post-translational modifications are an important biological regulatory mechanism, and the rate of their discovery using high throughput techniques is rapidly increasingly. To make use of this wealth of sequence data, we introduce a new general strategy designed to predict a variety of post-translational modifications in several organisms. We used the motif-x program to determine phosphorylation motifs in yeast, fly, mouse, and man and lysine acetylation motifs in man. These motifs were then scanned against proteomic sequence data using a newly developed tool called scan-x to globally predict other potential modification sites within these organisms. 10-fold cross-validation was used to determine the sensitivity and minimum specificity for each set of predictions, all of which showed improvement over other available tools for phosphoprediction. New motif discovery is a byproduct of this approach, and the phosphorylation motif analyses provide strong evidence of evolutionary conservation of both known and novel kinase motifs.
Few if any proteins are unaffected by protein post-translational modifications (PTMs).1 These modifications serve not only to diversify the chemical and physical repertoire of the individual amino acids but also act as key agents of protein regulation that have been implicated in nearly every facet of modern cellular biology (1). Although in the past the identification of such modifications and their precise location along the protein backbone was a difficult and time-consuming task, the advent of high throughput techniques, most notably tandem mass spectrometry, has led to the identification of well over 40,000 precisely localized sites of modification in the past 5 years alone (2–4). The most significant increase in data has come in the field of protein phosphorylation where whole proteome scale studies are routinely reaching several thousand unique and novel sites across a wide range of species (5–8), and recently a large new data set has become available containing thousands of human lysine acetylation sites (2).
Although impressive in magnitude and often exciting because of the implication of aberrant phosphorylation in a variety of human diseases, the number of novel PTMs identified in such large scale studies also demonstrates the fact that our knowledge of all PTMs is not yet near the point of saturation. Also there are a number of other modification types for which large enzyme families are known to exist (e.g. ubiquitin ligases and acetyltransferases) but for which little substrate PTM data exist in any organism. To inform directed biological experimentation for proteins of interest, we would ideally like to know all of the modification types, the sites of the modifications, and the enzyme responsible for each modification.
Until such a time when all modifications can be easily measured, computational methods of prediction can be crucial to inform hypothesis-driven biology. The current state of the art in mass spectrometry provides uneven sequence coverage of proteins because of systematic biases that are not completely understood, and sequence coverage typically varies widely between 20 and 40% (9, 10). Increasing protein coverage by mass spectrometry is an active area of research, and reasons for this reduced coverage may include sample preparation biases, mass spectrometer instrumentation limitations (including limited sensitivity or limited mass range), and failures involving spectral analysis. Thus, even as we begin to amass modification data, computational tools will still fill the need to predict PTMs in sequences refractory to direct measurement.
Historically the most studied PTM has been phosphorylation, which can be used as an example of approaches to the general prediction of PTMs. To date, tools for the prediction of phosphorylation sites have largely fallen into two general approaches. In one approach, the kinase (or enzyme-specific) approach, tools have been based on the principle that each kinase has its own unique sequence specificity. This principle is strongly supported by biological and crystallographic studies examining kinase substrate recognition (11, 12). By using kinase-substrate data available from literature searches, databases (such as Phospho.ELM (13)), or combinatorial peptide library screens, these tools have been able to get kinase-specific signatures that can be used to predict other substrates of a particular kinase (14–17). Although such tools have utilized the information contained within kinase-specific motifs, they are limited by the amount of available data for each kinase. For example, in the case of protein kinase A (PKA), the kinase with the greatest number of known substrate phosphorylation sites, fewer than 400 sites are currently known (13). Furthermore these sites come from a wide variety of organisms, forcing such tools to operate under the assumption that kinase specificities are universal, thereby making organism-specific prediction virtually impossible. Combinatorial peptide library screening approaches to phosphorylation prediction (16) do not suffer from a data deficiency; however, their high cost per experiment and in vitro basis have limited their predictive abilities to only a fraction of all known kinases.
In the other approach, a kinase-independent (or enzyme-independent) approach, several new phosphorylation prediction tools have been developed that do not rely on kinase-specific data. These tools are aimed at using mass spectrometry data, which contains only phosphorylation sites without regard to the responsible enzyme. Some of these tools use neural networks (18) or support vector machines (19), which, generally speaking, do not need to model the properties of substrate recognition inherent in an enzyme-specific approach. Although not an intrinsic limitation, even though abundant data exists, these tools have used only a small percentage of publicly accessible data from a narrow set of organisms, limiting the current scope of their predictions.
The novel approach presented here combines the best of both the enzyme-specific and enzyme-independent prediction methodologies. We first used a previously developed tool known as motif-x (20) that has been successfully used to find motifs in a number of large scale phosphorylation studies (21–24). Briefly the motif-x algorithm discovers overrepresented motifs in user-provided data sets, and the output of the motif discovery process is a set of all motifs containing highly significant residues in fixed positions (for example RRXS) and an accompanying position weight matrix (PWM) containing statistical information for motif positions that did not meet significance thresholds. Many motifs found using motif-x have been shown to directly correspond to particular known kinase or kinase family recognition sites. motif-x is used here in a meta-analysis to automatically partition all available data of a particular modification type and organism into individual motifs. To maximize the yield of motif discovery, we used nearly all publicly available modification data from large data sets that, when merged, contain over 50,000 unique sites of modification.
Although the motif-x algorithm was designed to discover overrepresented short linear motifs within a given data set, a previously lacking feature of motif-x was the ability to predict additional instances of a motif within a user-provided proteomic sequence. However, version 2.0 of motif-x along with a companion program called scan-x that can locate and score motifs in any protein sequence is about to be released.2 In this study we utilized features of this new version of motif-x and demonstrate a simple and effective way to use the accompanying scan-x program to predict several protein post-translational modifications in the proteomes of a number of organisms.
In several cases, this study marks the first attempt to predict a particular modification in a given organism (e.g. serine and threonine phosphorylation in Drosophila and tyrosine phosphorylation in human). In the case of lysine acetylation, this study marks the first computationally based determination of acetylation motifs and the first attempt to predict these modifications in any species.
Because this strategy first involves the discovery of motifs and because motifs have also been shown to have a direct biological relationship to specific enzymes or enzyme families, the benefits of enzyme-specific prediction are retained despite having started with PTM data sets where no specific modifying enzymes were known. In this approach, prediction ability can go beyond the limitations of previous enzyme-specific approaches because even completely novel motifs for unknown enzymes (e.g. phosphorylation motifs with no known kinase) can be used for prediction. In addition, by discovering motifs within large organism-specific data sets, most discovered motifs similarly contain large numbers of supporting examples, and in contrast to other enzyme-specific approaches, this abundance of data allows predictions to be made that are completely organism-specific. The approach described here is also an improvement over previous enzyme-independent approaches because it provides users with the actual extracted motifs used to make each prediction, and this often allows for inference of the responsible modifying enzyme even in the absence of actual enzyme-specific data.
EXPERIMENTAL PROCEDURES
Data Harvesting—
Data were obtained from several sources including Swiss-Prot/UniProt (3) (version 54.3, October 2, 2007) downloaded from the ExPASy FTP site, PhosphoSite (2) (provided by Cell Signaling Technology, Inc. as of September 18, 2007), PhosphoPep (4) (as of December 16, 2007), Phospho.ELM (13) (version 7), and data accompanying Smith et al. (23), Molina et al. (25), Yang et al. (26), and Wang et al. (27). Custom Perl scripts were written to generate lists of peptide sequences with known modifications from each database (UniProt and PhosphoSite) or to automatically download peptides from the Web sites (Phospho.ELM and PhosphoPep). These peptides were ultimately reformatted as 15-mers centered upon the modified residue and then used directly to derive training and cross-validation data sets as described next.
Derivation of the Training and Cross-validation Data Sets—
Training and cross-validation data sets were derived from the various databases of known PTMs (as described above), and the background proteomic sequences were derived from NCBI (National Center for Biotechnology Information), IPI, SGD, and FlyBase Drosophila databases. After grouping by organism and type of modification, each of these groupings was processed as follows. (a) The background database was converted to 15-mers centered on every residue relevant to the modification type (i.e. all tyrosines for a tyrosine phosphorylation analysis, all lysines for an acetylated lysine analysis, etc.). (b) The background data set was reduced by removing all foreground peptide sequences to yield mutually exclusive foreground and background data sets. (c) The foreground 15-mer data set was reduced by homology filtering as described below. (d) The foreground sequences were randomized and divided into 10 equally sized groups. (e) The background sequences were randomized and divided into 10 equally sized groups, and each was arbitrarily paired with a foreground group resulting in 10 pairs of foreground/background sequences. (f) For a given pair j from 1 to 10, (i) all pairs except for pair j was repooled with the other nine foreground and nine background data sets into training foreground and training background data sets, respectively, that excluded all peptides in pair j; (ii) these repooled training foreground and training background data sets were then used as input to motif-x to find motifs with the significance parameters described below; (iii) the foreground of pair j was then labeled as positive, and the background of pair j was labeled as negative,3 and scan-x was run on all of these labeled positive and negative data sets; (iv) scan-x determined a score for each peptide (as described below for any query sequence); (v) these scores were subject to a series of fixed integer threshold values from −100 to +994 to bifurcate the data for each threshold level into those labeled positives and negatives that scored above each threshold (i.e. the TPs and FPs for that threshold level) and those labeled positives and negatives that scored below each threshold (i.e. the FNs and TNs for that threshold level); and (vi) finally the size of each of the four classes for each threshold level (TP, TN, FP, and FN) was used to compute sensitivities and specificities (see Equations 2 and 3) for each fixed threshold for the test set j and its mutually exclusive training set. (g) Step f was repeated 10 times, once for each test pair j with its mutually exclusive pooled training sets. (h) Finally for each of the 10 runs, the sensitivity and specificity were averaged across each threshold value from −100 to +99 to determine the expected performance of the method on unknown data sets for each organism and modification type and to derive a receiver operating characteristic (ROC) curve (supplemental Table 8 and Fig. 3).
Homology Filtering—
Existing PTM substrate databases contain many homologous sequences. Such homologous sequences in cross-validation data, although perhaps reflective of realistic sequences, would not give the most conservative estimates of sensitivity and specificity in a cross-validation study because they can behave as self-consistency data points and thereby increase apparent algorithmic performance. Therefore, as a first step in preparation of our training sets (prior to randomization and division into 10-fold cross-validation sets) we removed presumptively homologous sequences that were more than 60% identical to another sequence in the foreground data set. Note that it is not necessary to remove all peptides that are similar but just enough of them to assure that the remaining data set does not contain homologs after processing. For instance, if two peptides are nearly identical but share no identity with any other peptides in the data set, elimination of just one (not both) will satisfy this requirement. The algorithm proceeds in a stepwise manner by first eliminating sequences that were different from another in exactly one position. By definition, each potential homolog is found as two or more peptides, and the algorithm preferentially eliminates the sequence that is most similar to all other sequences in the set and leaves the one(s) that is most different from all others in the set. Elimination proceeds one peptide at a time, re-evaluating after each peptide is removed. Once no further homologs of distance 1 remain, homologs of distance 2 are eliminated and so forth until all peptides are different from one another in at least six positions. Because peptides in this study were 15-mers, this roughly corresponded to the remaining peptides having an identity level of <60% (no more than nine of 15 residues being identical). We feel that this identity level selects for peptides that are evolutionarily distant and not prone to contamination of cross-validation data sets.
Running motif-x on the Data—
To carry out the prediction procedure as described in the text, each of the training data sets needed to be deconvoluted into constitutive motifs using the motif-x algorithm. This was accomplished using a prerelease version of the motif-x version 2.0 Web site2 using the following parameters: foreground format, prealigned; central character, S, T, Y, or K (dependent on analysis); width, 15; significance, 1e−10; and background database, IPI human proteome, IPI mouse proteome, SGD yeast proteome, or FlyBase Drosophila proteome (dependent on analysis). The fixed residues of the motifs are those that have attained a user-defined level of significance at a given position. A very stringent significance threshold was used to ensure the validity of the extracted motifs (typically a p value <1e−6 or in this study 1e−10). Following Bonferroni correction, this significance threshold corresponded to a p value <3e−8.
Sequence Logos—
Values in the non-fixed positions of the motif are also a function of probability of occurrence of each residue, and they contribute to the weight of values of the PWM. This entire motif and PWM can be visually represented in a novel sequence logo, which is exemplified in Figs. 1–3 and supplemental Tables 1–7, that will be available in motif-x version 2.0. These sequence logos have an intuitive interpretation in which residues above the horizontal axis are overrepresented with respect to the background, and those residues below the axis are underrepresented with respect to the background. More significant residues are taller than residues that are less significant, and the most significant residues are placed closest to the horizontal axis. Locked positions are shown as full height and are the only residues shown for that position.
Fig. 1.

Overall schematic of the prediction methodology. Starting with a sequence-based input data set of modification sites from any given organism, the motif-x algorithm is used to deconvolute the data into constitutive motifs. These motifs can then be used as input to the scan-x program, which scans the motifs against any proteomic FASTA-formatted data set and scores each hit. Those sequences exceeding a selected stringency threshold are output as potential modification sites. The modification sites are denoted with an asterisk, and the “fixed” positions in the motifs used for scan-x are highlighted here in blue.
Fig. 2.

Sample scan-x scoring procedure for the TUS1 protein by the RXXSXXS phosphorylation motif. Starting with a motif obtained as output from the motif-x program (e.g. RXXSXXS) and a sequence to be scanned (e.g. TUS1), the scan-x program first identifies syntactic matches (i.e. protein regions matching the fixed positions in the motif). The PWM formulation of the sequence logo is then used to score the matching segment with the total scan-x score being equal to the sum of the scores for the residues at each position. In this example, 15 positions are scored because the initial RXXSXXS motif had a total width of 15 (including the three fixed positions that are given scores of 0). Peptide scores may be converted to expected false positive rates by querying the appropriate cross-validation ROC curve data. Here serine 241 yielded a scan-x score of 8.546, whereas serine 270 yielded a scan-x score of 1.261. The expected false positive rates of these two serines are 1.8 and 8.9%, respectively. Note that RXXSXXS is a novel phosphorylation motif discovered by the motif-x algorithm and that serine 241 was recently confirmed to be a true phosphorylation site in the literature (42).
Fig. 3.

ROC curves for yeast, fly, mouse, and human modification data sets. These curves show the trade-off between prediction sensitivity and specificity for each of the data sets analyzed in this study. Each curve was computed using 10 unique training set/test set combinations according to the cross-validation procedure outlined in the text. Each curve thus represents the average sensitivity and specificity for threshold values across 10 cross validation runs. The data points shown for each ROC curve correspond to sensitivity and specificity values calculated at scan-x stringency thresholds varying between −100 and +99. Raw data for these curves are provided in supplemental Table 8.
Residual Motifs—
Another feature of motif-x version 2.0 is the introduction of the “residual motif.” The residual motif is not a real motif, but it has a PWM and accompanying logo representing the remainder of peptides in the foreground data set that could not be deconvoluted into a discrete motif where at least one non-central residue/position attained the specified significance level. Thus the PWM for the residual motif has only the central residue fixed and contains statistical scores representing the residual peptides in all of the non-central residues/positions in an exact analogy to the statistical representation of peptides in the PWM of a real motif.
PWM Weights and Scoring a Sequence by scan-x—
Each position/residue score in the PWM is derived during the motif discovery process using a prerelease of the next version of the motif-x program and is approximately a log transformation of the binomial probability of that residue occurring at that position k of N times given a background probability of p. Here k is the number of occurrences of that residue at that position in all of the peptides of the foreground data set that syntactically matched the fixed positions of the motif, N is the total number of peptides in the foreground data set matching that syntactic motif, and p is the empirical probability of that residue at that position across all of the peptides in the background data set that also exactly syntactically match the motif.
More precisely, to handle under- and overrepresentation of each residue at each position, actually two binomial probabilities are computed: the probability Pover, the binomial probability of k or more of N occurrences of the residue at that position, and the probability Punder, the binomial probability of k or less of N occurrences of the residue at that position, both given the empirical background probability p. These probabilities are combined into a log odds ratio to arrive at the final log10-transformed score for each possible residue at each non-fixed position in the PWM.
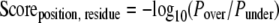 |
(Eq. 1) |
This scoring method was chosen for the following reasons. (i) It is mathematically well behaved across the full set of values of k (0 through N), (ii) it ranges from negative to positive values when a residue is under- or overrepresented, respectively, (iii) it is 0 when k/N = p (that is when a residue is neither under- nor overrepresented at a given position), and (iv) it quickly approaches −log10(Pover) or +log10(Punder) when k/N is even slightly higher or lower than p, respectively, so it grossly approximates these values and gives the logo representation of the PWM an intuitive interpretation as described above.
To create a score for a potential hit in a particular query sequence for a motif being scanned, first a syntactic match with the fixed positions of the motif must be made. That is, each locked position of the motif must exactly match corresponding residues in the query sequence. Then each actual position/residue of the query sequence is used to look up the corresponding Scoreposition, residue in the PWM of the motif for each of the non-locked residue positions of the motif. These individual scores (15 in this case) are then summed across the entire motif width to arrive at the final score for the motif at that query position. Fixed positions are assigned a value of 0.
Comparison with Other Algorithms—
The scan-x prediction tool was compared with three other Web-based phosphorylation prediction tools: NetPhosYeast, Phosida, and Scansite. In the case of NetPhosYeast, 2,000 random serine and threonine phosphorylation sites from yeast, each of width 15, were uploaded to the server. Additionally 2,000 random serine and threonine sites from the SGD yeast proteome not known to be phosphorylated were uploaded separately to serve as an approximation of the actual negatives in the same way they were used to validate our method. Sensitivity was computed as the total number of correctly identified phosphorylation sites from the positive data set divided by the total positive data set size (i.e. 2,000). Specificity was calculated as the total number of negative sites that were not predicted to be phosphorylated divided by the total negative data set size. To assess the Phosida tool, 1,345 random human and 793 random mouse serine and threonine phosphorylation sites were uploaded as a positive test set. Again an approximation to actual negative data sets was provided by extracting random serine and threonine sites not known to be phosphorylated from the human and mouse IPI proteomes. All positive and negative peptides had a width of 15. To run Phosida at the highest stringency a precision level of 100% was selected prior to running the software. Sensitivity and specificity were calculated as described for NetPhosYeast. Scansite was run using all available kinase position-specific scoring matrices on the same human positive and negative data sets used for the Phosida validation. In the case of serine and threonine phosphorylation, Scansite was run at “high stringency.” Again sensitivity and specificity were calculated based on the total number of correctly identified positive and negative hits divided by the total data set size (a correctly identified site needed to match one or more serine or threonine kinase profiles). To compare our tyrosine phosphorylation results to that of Scansite, 866 random human tyrosine phosphorylation sites and 866 random human tyrosine sites not currently known to be phosphorylated (all of width 15) were used as actual positive and an approximation of actual negative data sets, respectively. The uploaded data were then run at “medium stringency.” To be considered a positive match, a site needed to match one of the tyrosine kinase profiles. Sensitivity and specificity were calculated using the same procedure as for the serine and threonine data sets.
Analysis of Newly Published Data Sets—
The analysis of newly published phosphorylation data from Albuquerque et al. (5) in yeast and Zhai et al. (8) in fly was accomplished by taking only sites reported in those studies that had a high degree of localization confidence (i.e. PLscore > 7 in Albuquerque et al. (5) and Ascore > 13 in Zhai et al. (8)). Each phosphorylation site was extended from the appropriate proteome to contain at least seven residues upstream and downstream of the phosphorylation site. Phosphorylation sites that were already included in our total phosphorylation training data set for yeast and fly were removed. Presumed negative data included all those serine and threonine sites in the respective proteomes that were not observed to be phosphorylated (either in our training data or in the newly published studies). The scan-x program was then run on each of the data sets with both positive and negative data using all of the motifs shown in supplemental Tables 1 and 2. The scan-x threshold used for the yeast data was 5.2 for all motifs with the exception of the residual motifs, which had a threshold of 35.2. For the fly data, the threshold used was 7.1 for all motifs with the exception of the residual motifs, which had a threshold of 37.1. Sensitivity and specificity were calculated according to Equations 2 and 3.
Web Site Availability—
The motif-x Web site version 2.0, which includes some of the features used in this study, will be available for non-commercial and academic use. Details on other features of motif-x version 2.0 and the use of the new scan-x program are described elsewhere in more detail.2 The ability to scan proteins of interest using motifs discovered using the methods in this study will be made available from the motif-x Web site and may be periodically updated as new training data sets become available.
Programming Details—
The motif-x Web site, the core engine, and supporting programs were all written in the Perl programming language. Certain computationally intensive or specialized portions of the system such as the computation of binomial probabilities, the creation of graphical motif logos, and homology filtering were partially written in the C programming language. The motif-x Web site is hosted on a large multinode Linux and PC-based computer cluster hosted by Harvard Medical School's Research Information Technology Group.
RESULTS
Overall Strategy—
The first step in any protein post-translational modification prediction strategy involves the establishment of training data sets that serve as the foundation upon which all future predictions will be made. In an effort to be as comprehensive as possible, training data were harvested from a wide variety of sources including Swiss-Prot (3), Phospho.ELM (13), PhosphoPep (4), and PhosphoSite (2), which yielded a total of 51,808 unique sites of modification split up among four commonly researched species (yeast, fly, mouse, and human) and two major modification types (phosphorylation and acetylation). In addition, several large scale studies whose data sets were not yet incorporated into these other data sets were used as sources (23, 25–27). Specific statistics on the training sets are provided in Table I.
Table I.
Post-translational modification training data statistics
| Organism and data origina | Modification type | Total number of unique modification sitesb |
|---|---|---|
| Saccharomyces cerevisiae | ||
| Swiss-Prot (99.7%, 99.2%) | Serine phosphorylation | 3,882 (3,808) |
| Phospho.ELM (0.8%, 0.3%) | ||
| Swiss-Prot (99.8%, 99.3%) | Threonine phosphorylation | 833 (814) |
| Phospho.ELM (0.7%, 0.2%) | ||
| Drosophila melanogaster | ||
| PhosphoPep (100%, 100%) | Serine phosphorylation | 8,849 (8,607) |
| PhosphoPep (100%, 100%) | Threonine phosphorylation | 2,485 (2,459) |
| Mus musculus | ||
| PhosphoSite (84.5%, 45.2%) | Serine phosphorylation | 6,887 (6,410) |
| Swiss-Prot (48.6%, 12.2%) | ||
| Smith et al. (4.7%, 2.9%) | ||
| Phospho.ELM (3.4%, 0.3%) | ||
| PhosphoSite (85.2%, 47.2%) | Threonine phosphorylation | 1,617 (1,523) |
| Swiss-Prot (44.7%, 11.9%) | ||
| Smith et al. (2.9%, 2.4%) | ||
| Phospho.ELM (6.2%, 0.6%) | ||
| Homo sapiens | ||
| PhosphoSite (78.7%, 25.0%) | Serine phosphorylation | 11,741 (10,640) |
| Swiss-Prot (64.0%, 17.7%) | ||
| Phospho.ELM (11.2%, 2.2%) | ||
| Wang et al. (1.6%, 0.8%) | ||
| Molina et al. (8.8%, 0.1%) | ||
| Yang et al. (3.3%, 0.03%) | ||
| PhosphoSite (86.0%, 38.7%) | Threonine phosphorylation | 3,028 (2,815) |
| Swiss-Prot (46.4%, 9.9%) | ||
| Phospho.ELM (14.1%, 2.5%) | ||
| Wang et al. (1.3%, 0.7%) | ||
| Molina et al. (8.3%, 0.1%) | ||
| Yang et al. (4.7%, 0.1%) | ||
| PhosphoSite (98.6%, 87.0%) | Tyrosine phosphorylation | 9,524 (8,662) |
| Swiss-Prot (9.6%, 0.8%) | ||
| Phospho.ELM (4.8%, 0.5%) | ||
| Wang et al. (0.05%, 0.03%) | ||
| Molina et al. (0.7%, 0.01%) | ||
| PhosphoSite (100%, 100%) | Lysine acetylation | 2,962 (2,737) |
Values in parentheses indicate the percentage of total sites contained within the given data set followed by the percentage of total sites unique to a given data set. Percentages of total sites from different databases can add up to >100%, and percentages of unique sites will total ≤100% because the same peptides can be found from multiple data sources. Citations are as follows: Smith et al. (23), Molina et al. (25), Yang et al. (26), and Wang et al. (27).
Values in parentheses indicate number of unique modification sites following the homology filtering procedure.
The overall prediction methodology is summarized in Fig. 1. Training data sets are first separated based on organism and modification type as shown in Table I. The motif-x algorithm is then run on each of these data sets with a proteomic background and at a stringent significance threshold to extract only high confidence motifs (p value <1e−10, which is a Bonferroni-corrected equivalent of the already highly significant p value <3e−8; see also supplemental Tables 1–5). A whole proteome sequence data set on which to make predictions is then selected for searching by scan-x using these discovered motifs. All subsequences within the proteomic data sets that match one or more of the motifs are scored by scan-x. Residue heights in the motif logos are a function of their binomial probabilities, and scoring a sequence simply involves taking the sum of residue heights within the sequence based on the appropriate residues and positions in the corresponding motif (see Fig. 2 and “Experimental Procedures”). Scores are indicative of the degree of the match of a sequence with the training set used to derive the motif, and sequences are deemed “predicted substrates” for the modification in question if they meet or exceed a particular score threshold.
Cross-validation—
When carrying out a procedure to predict additional modification sites in a proteome of interest, it is necessary to fairly assess the degree of confidence one may have in the results. Typically this can be accomplished through a cross-validation methodology in which a certain percentage of the training data is set aside as a test data set and used to compute the sensitivity and specificity of the prediction procedure. Here we used a 10-fold cross-validation strategy in which the total positive and negative training set was subdivided into 10 training and test sets such that each test set was mutually exclusive from its associated training set. To ensure that sequences in the cross-validation data sets were not homologous to sequences in the training sets (resulting in unfairly advantageous prediction results) the training data sets were first filtered to remove all sequences with greater than 60% identity (see “Experimental Procedures”).
Negative data were obtained by selecting all of those residue-specific sites in the appropriate proteome not found in our positive training data (e.g. negative data for serine and threonine phosphorylation in the yeast proteome comprised all serine and threonine residues in the proteome not included in the positive training data). The assumption that all residues except those currently known to be modified are actually negative is highly conservative, and were it in fact true, it would obviate the need for a prediction methodology altogether. As such, our calculated false positive rate is guaranteed to represent an overestimate of the false positive rate of the methodology (or equivalently an underestimate of the specificity of the methodology).
motif-x was run on each of the 10 training sets (with the actual positive data as the foreground and the presumed negative data as the background), and extracted motifs were scanned against the mutually exclusive test sets to make predictions. For each cross-validation run, and for each of 200 integer threshold values (ranging from −100 to +99), the sensitivity and specificity of the method was calculated according to the following formulas.
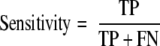 |
(Eq. 2) |
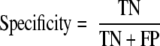 |
(Eq. 3) |
Average values of sensitivity and specificity at each threshold were then calculated based on the data obtained from the 10 cross-validation sets. Plotting these data on coordinate axes resulted in the standard ROC curves shown in Fig. 3 for each of the modification data sets. Converting the sensitivity and specificity values provided in the ROC curve into scoring thresholds thus allows the prediction procedure to be optimized for a user-defined stringency (see supplemental Table 8 for raw ROC curve data and a threshold conversion chart).
Although other studies have reported the accuracy (Equation 4) of their prediction strategies, we have opted to refrain from reporting this value because it represents a weighted average of the sensitivity and specificity and therefore can change with the sizes of the data sets used. In our case, the proteome scale size of our negative data set gives us values for accuracy that mimic specificity values almost exactly (i.e. when specificity is 95%, accuracy is also ∼95%). Thus, accuracy values can be arbitrarily large or small because negative data set size can vary from one study to another. As such, we believe that accuracy is not an appropriate metric for comparison.
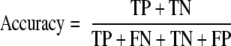 |
(Eq. 4) |
ROC curve analyses indicated that for serine and threonine phosphorylation at 95% specificity sensitivities vary from 32 to 38%. In the case of tyrosine phosphorylation and lysine acetylation only 16 and 18% sensitivity is achieved at 95% specificity. Although these sensitivities may appear low, they nevertheless translate into tens of thousands of as yet unidentified high confidence post-translational modification prediction sites in each of the analyzed organisms (Table II). Even when specificity is raised to the 99% level, over 80,000 novel total prediction sites can still be made. At best, standard tryptic digestion followed by tandem mass spectrometry results in protein coverages between 80 and 90% on a non-complex sample with more standard protein coverages in the 20–40% range for a complex sample (9, 10, 28). If one also takes into account the fact that mass spectrometers are concentration-sensitive instruments and that many interesting biological post-translational modification events occur only transiently or on low abundance proteins, it is clear that a significant portion of these sites are unlikely to be detected using standard high throughput techniques. In silico predictions may be the only practical way to find many of these modification sites.
Table II.
Number of novel proteomic predictions made at the 95 and 99% specificity levels
| Organism and modification type | Number of unique high confidence predictions (95/99%) |
|---|---|
| S. cerevisiae | |
| Serine and threonine phosphorylation | 22,919/4,697 |
| D. melanogaster | |
| Serine and threonine phosphorylation | 48,293/8,936 |
| M. musculus | |
| Serine and threonine phosphorylation | 97,488/18,941 |
| H. sapiens | |
| Serine and threonine phosphorylation | 104,349/24,514 |
| Tyrosine phosphorylation | 23,900/5,521 |
| Lysine acetylation | 54,436/18,392 |
Comparison with Other Prediction Algorithms—
Comparison of the prediction methodology presented here with two recently published tools aimed at phosphorylation prediction based on large scale data indicates a substantial improvement in specificity and sensitivity. NetPhosYeast is an artificial neural network-based serine and threonine phosphorylation predictor designed specifically for yeast (18). When applied to positive and negative yeast data used for our own cross-validation, NetPhosYeast achieved a sensitivity of 93.7% with a specificity of 39.2%. The low specificity of NetPhosYeast coupled with its single stringency level makes it a difficult tool to use for the experimentalist seeking to identify phosphorylation sites with a low false positive rate.
Phosida is another recently published phosphorylation prediction tool aimed at human and mouse serine and threonine phosphorylation prediction using a support vector machine strategy (19). When tested against a sampling of positive and negative sites from our human serine and threonine cross-validation data set it achieved sensitivity and specificity values of 12.2 and 97.3%, respectively. These values represent the Phosida algorithm being run at its maximal stringency. By comparison, our scan-x approach achieved a sensitivity of 23.3% at an equivalent specificity of 97.3%. In the mouse data set, Phosida fared somewhat better, achieving a sensitivity of 22.1% at a specificity of 97.1% compared with the scan-x sensitivity of 27.2% at the equivalent specificity value.
Note that some of our test peptides were likely found in the NetPhosYeast and Phosida training sets, and as such, our cross-validation data sets may not be as truly challenging as they ideally could be. The sensitivity bias toward these methods suggests that we are also somewhat underestimating the relative improvement of scan-x over these methodologies.
In addition to the aforementioned tools that aim to predict phosphorylation sites based on large scale mass spectrometry data we also chose to compare our prediction results with the Scansite program, which uses position-specific scoring matrices (PSSMs) derived experimentally for individual kinases to make phosphorylation predictions (16). To date, Scansite has PSSMs for 26 kinases, making it one of the most widely used and versatile tools for phosphorylation prediction. Although Scansite is not organism-specific and is meant to be used in a kinase-specific manner, running Scansite with all 26 PSSMs gives users a global view of protein phosphorylation. When applied to our human serine and threonine phosphorylation test sets, Scansite achieved a sensitivity of 13.2% and a specificity of 97.6% compared with the scan-x sensitivity of 21.4% at the equivalent specificity.
We were also able to compare our human tyrosine phosphorylation predictions against the Scansite tyrosine kinase prediction tool. At medium stringency, Scansite achieved a sensitivity of 5.1% and a specificity of 97.5%. In comparison, scan-x yielded a 9.1% sensitivity at the same specificity level. High stringency could not be used in Scansite because the resulting sample size was too small to accurately measure specificity. Because several of the motifs discovered in these proteome wide data sets do not correspond to those of any known kinase, this lower sensitivity of Scansite may be due in part to a lack of complete kinase-specific data and not to the Scansite algorithm itself.
Performance under Varying Biological Conditions—
During preparation of this manuscript, two additional phosphorylation studies were published that nearly doubled the number of known phosphorylation sites in yeast (Albuquerque et al. (5)) and fly (Zhai et al. (8)). These yeast and fly studies resulted in 3,579 and 6,671 novel phosphorylation sites, respectively, that were not included in our total training sets. The novel phosphorylation sites contained within these studies represented challenging data sets on which to test the predictive capacity of our approach because the sites were extracted from cells grown under specific conditions or stimuli that were not reflective of the conditions under which our complete yeast and fly training sets were obtained.
The Albuquerque et al. (5) study investigated phosphorylation sites carried out under DNA damage conditions by treating cells with methyl methanesulfonate, an agent known to activate a number of damage-specific kinase pathways. In fact, differences in kinase activation between the Albuquerque et al. (5) study and our training data set can be confirmed by inspecting those motifs that were extracted from each of those data sets (compare supplemental Tables 1 and 6). Of 25 total motifs that were extracted from both data sets, only 11 motifs were shared between both data sets. Interestingly the Albuquerque et al. (5) data set contained several unique and novel motifs including SXXS, SXXN, SXP, and SF (phosphorylated residues are underlined) that are likely to be involved in the DNA damage pathway (the SF motif was in fact recently hypothesized to be a substrate specificity for the kinase Rad53, which is known to become activated in response to methyl methanesulfonate treatment in yeast (29)).
The Zhai et al. (8) study, which was carried out on Drosophila embryos, also investigated phosphorylation events that differed from our training data in which phosphorylation was observed in Drosophila Kc167 cells (4). As in the yeast data set, differential kinase activity is noticeable with the majority of the motifs extracted found solely in either the Zhai et al. (8) data or in our fly training set (compare supplemental Tables 2 and 7). Some interesting novel motifs that were extracted exclusively from the Zhai et al. (8) study include RSP, KSP, RTP, KTP, NXS, RXXSXXS, SXXSL, RRS, RXXTP, SXSP, SXXS, SXXS, and SXXXSP.
Despite the unique conditions under which the Albuquerque et al. (5) and Zhai et al. (8) studies were run, the scan-x algorithm was still able to predict ∼27% of the phosphorylation sites from each of the studies at a 95% specificity rate. More specifically, using the thresholds derived for 95% specificity (see supplemental Table 8) scan-x predicted 27.7% of the phosphorylation sites in the Albuquerque et al. (5) study with an expected specificity of 94.2%. Similarly scan-x predicted 27.1% of the phosphorylation sites in the Zhai et al. (8) study with an expected specificity of 95.3%. Although these sensitivity values represent a modest decrease from the expected sensitivity values of 37.7% for yeast and 31.8% for fly, given the unique nature of these new data sets, they in fact serve to highlight the robustness of the prediction procedure. In time, as new protein modification studies are added to our training sets with a wide variety of experimental conditions, we expect that the discrepancy between our predicted sensitivity and actual sensitivity to approach 0.
scan-x Predictions—
Application of the aforementioned prediction strategy on the full training data set resulted in a total of 81,001 predicted modification sites at the 99% specificity level (summarized in Table II and available upon request). Literature-based corroboration of our predictions were difficult to obtain because the overwhelming majority of known phosphorylation sites have come from large scale mass spectrometry studies and were thus already included in our training set. Following are a few instances, however, where it was possible to independently confirm these predictions in the literature.
For example, in yeast, serine 191 of the Sic1 protein, serine 77 of the Fip1 protein, and both serines 133 and 134 of the Grx4 protein have all been experimentally verified as either in vivo or in vitro kinase substrates (30–32) and were all predicted phosphorylation sites by scan-x. In fly, scan-x predicted two phosphorylation sites on the slowpoke channel-binding protein, Slob. The first of these sites on serine 54 was detected using the RXXS motif that has been verified to be a true phosphorylation site involved in regulating the kinase activity of the Slob protein (33).
In mouse, a number of phosphorylation predictions could be supported by sites shown to be phosphorylated on homologous human proteins, including serine residues 28, 98, and 637 in RalBP1, a protein shown to be involved in cancer cell proliferation. (scan-x also predicts a patch of serine phosphorylation sites on RalBP1 between residues 541 and 545; however, these residues lie in a region of the protein that upon tryptic digestion would create a peptide too large to be sequenced using standard tandem mass spectrometry methods.) Additionally serine residues 602 in mouse protein BRD4 (bromodomain-containing 4) and 906 in mouse protein Delangin have both been shown to be phosphorylated on homologous residues in their respective human counterparts (25, 34). Increased confidence in these predictions is gained from the fact that no human data were used in the training set for these predictions, yet the sequences of these predicted targets are nearly identical to their homologous human sequences in the vicinity of the putative PTM.
In the human data set, our phosphorylation prediction of serine 419 on CCDC6 (coiled coil domain-containing protein 6) was very recently verified by a large scale tandem mass spectrometry study whose data were not included in our training set (35). We were also able to validate several tyrosine phosphorylation predictions in the human data set, including tyrosine 348 on PSD-93 (postsynaptic density protein 93) in which the homologous site has been identified as the primary site of Fyn phosphorylation in mouse (36). Similarly of a total of 24 tyrosine residues in the CENTD3 protein (centaurin δ3), three were predicted to be phosphorylation sites by scan-x. Two of these sites have been verified in the literature (Tyr-1403 by homology with mouse and Tyr-1408 by homology with mouse and more recently by mass spectrometry in human) (2, 37). Finally scan-x also correctly predicted the phosphorylation of tyrosine 277 in TFII-I, a multifunctional transcription factor involved in the regulation of cell proliferation and whose defect contributes to Williams-Beuren syndrome (38, 39).
We were unfortunately unable to find independent literature-based evidence for our acetylation predictions because prior to the PhosphoSite data set only several dozen human non-histone acetylation sites were known. To illustrate this point, a PubMed search at the time of preparation of this manuscript for the phrase “lysine acetylation” came up with only 90 hits in the literature, whereas a similar search for the phrase “tyrosine phosphorylation” came up with 16,125 hits.
Analysis of Phosphorylation and Acetylation Motifs—
In addition to prediction of modification sites, this study also provided a unique opportunity to compare independently derived phosphorylation motifs across a wide range of organisms because extraction of these motifs was already carried out as a first step in the prediction procedure (Fig. 1 and supplemental Tables 1–5). As can be expected, a large proportion of these motifs correspond to the motif signatures of well known protein kinases. These data provide strong evidence for the conservation of kinase specificity throughout evolutionary history. Notable examples that were found in at least three of the four species examined in this study include the motifs RRX(S/T), (S/T)DXE, PX(S/T)P, and (S/T)PX(K/R) corresponding to the consensus sequences for PKA, casein kinase II (CK II), MAPK, and cyclin-dependent kinase (CDK), respectively. Perhaps more interesting, however, are those motifs that were independently extracted in the data sets for different organisms yet have no corresponding kinase known to specifically phosphorylate such a sequence. These include RXXSXXS (observed in yeast, mouse, and human), RXXSP (observed in fly, mouse, and human), RSXS (observed in yeast, mouse, and human), TPP (observed in fly, mouse, and human), and SPXXXX(K/R) (observed in fly and human). Inspection of the sequence logos for each of these motifs reveals strong evolutionary conservation of residue preferences in the non-fixed motif positions of the PWMs. For example, the TPP motif shows preference for basic residues at the −3 position, whereas the RXXSXXS motif shows preference for hydrophobic residues at the +1 position. The similarity of the sequence logos across species also validates the use of these PWMs for post-translational modification prediction.
Table III outlines all of the motifs that were extracted from two or more of the organismal data sets. The table only includes those motifs that were exact matches across the sets despite the fact that several additional groupings could likely be made through examination of the sequence logos (compare, for example, motif 1.01 in both supplemental Tables 3 and 4).
Table III.
Identical motifs found in at least two of the four serine and threonine phosphorylation data sets using the motif-x algorithm
PKG, protein kinase G; G-CK, Golgi casein kinase; CaMK II, calcium/calmodulin-dependent protein kinase II.
| Motifa | Potential kinase(s) | Found in yeast | Found in fly | Found in mouse | Found in human |
|---|---|---|---|---|---|
| SP | Pro-directed | + | + | + | + |
| TP | Pro-directed | + | + | + | + |
| RRXS | PKA | + | + | + | + |
| RRXT | PKA | + | + | − | − |
| RXS | PKA | + | + | + | + |
| KXXS | PKA | + | + | − | + |
| KRXS | PKA | − | + | − | + |
| RXXS | PKA/CaMK II | + | + | + | + |
| RXXT | PKA/CaMK II | + | + | + | + |
| RKXS | PKA/PKG | + | + | − | − |
| RXXXS | PKG | + | + | − | − |
| PXSP | MAPK | − | + | + | + |
| PXTP | MAPK | − | + | + | + |
| SPXR | CDK | − | − | + | + |
| SPXK | CDK | − | + | − | + |
| SXE | G-CK | − | − | + | + |
| SDXE | CK II | + | + | + | + |
| TDXE | CK II | − | − | + | + |
| SDXD | CK II | + | + | + | + |
| SEXE | CK II | + | − | + | − |
| SXXE | CK II | + | − | − | + |
| SDDE | CK II | − | + | − | + |
| SDXEXE | CK II | − | − | + | + |
| SXDE | Novel/CK II | − | + | + | + |
| DS | Novel/CK II | − | + | + | − |
| DSEXE | Novel/CK II | − | − | + | + |
| SXXS | Novel/CK I | − | − | + | + |
| TPP | Novel | − | + | + | + |
| RXXSP | Novel | − | + | + | + |
| RSXS | Novel | + | − | + | + |
| RXXSXXS | Novel | + | − | + | + |
| SPXXXXK | Novel | − | + | − | + |
| GS | Novel | − | + | + | − |
Phosphorylated residues are underlined.
The large magnitude of the PhosphoSite database allowed us an unprecedented opportunity to deconvolute the human tyrosine phosphorylation data set into 16 motifs (supplemental Table 5). A majority of these motifs exhibited known canonical features of many tyrosine kinases including acidic character surrounding the phosphorylation site (especially at position −3) and proline and/or hydrophobic residues at the +3 position. However, several motifs such as KXXY (a novel motif), which did not fit this standard profile, and NPXY (a known ligand for a number of phosphotyrosine-binding domains) were also extracted.
Although previously fewer than 150 lysine acetylation sites were known in the human proteome, the PhosphoSite database has increased this number ∼20-fold using proprietary methods, enabling us to computationally extract acetylation motifs for the first time (Fig. 4). Extracted motifs include KK, KR, KF, KY, KXF, GK, KXXXK, and KXXK. Inspection of several of the motifs in Fig. 4 suggests a preference for glycine and lysine in the residues immediately surrounding the acetylation site as well as aromatic residues at the +1 position. These motifs may represent differences in acetyltransferase enzyme specificities. For example, a gene ontology analysis of proteins bearing the KY acetylation motif indicated a significant overenrichment of mitochondrial proteins, suggesting that a unique acetyltransferase with a preference for tyrosine at the +1 position is active in the mitochondrial compartment (data not shown). The motif results presented here are consistent with the general residue preferences adjacent to acetylation sites found in a recent survey of acetylation in the mouse proteome (40).
Fig. 4.

Human acetylation motifs and their corresponding sequence logos extracted from the PhosphoSite database. The database contained 2,962 unique actual acetylation sites. Running motif-x on these sites results in a deconvolution of the data into eight motif groups plus a residual motif (group 9). Sequence logos in which the heights of the residues are approximately proportional to their binomial probabilities are shown for each corresponding motif. The residual motif group contains all of those sites that were not able to fall into one of the other motif classes and from which another significant motif could not be found (see “Experimental Procedures”).
Certain similarities exist between the tyrosine phosphorylation and lysine acetylation data sets. First, despite the large size of both of these data sets (n = 9,524 and n = 2,962 unique sites for the tyrosine phosphorylation and lysine acetylation data sets, respectively), the number of motifs extracted was relatively small compared with comparably sized serine and threonine phosphorylation data sets. Second, the extracted motifs from these data sets also revealed a lack of specificity (i.e. contained fewer fixed positions) compared with the serine and threonine phosphorylation results. Finally the proportion of sequences that could not be deconvoluted into a motif class (which is referred to as the residual) was substantially higher in the tyrosine phosphorylation and lysine acetylation data sets compared with the serine and threonine phosphorylation data sets. Taken together, these findings suggest either that tyrosine kinase and lysine acetyltransferases are fairly nonspecific or that information beyond local primary structure may be necessary for their specificities. This finding is consistent with the decreased ROC curve performance on both of these subsets (Fig. 3).
DISCUSSION
As our understanding of the role played by post-translational modifications in all aspects of cellular biology continues to grow at a fast pace, tools for the prediction of such sites will become increasingly valuable especially in hypothesis-driven studies. For example, in a typical 500-amino acid human protein, ∼60 serine and threonine residues can be expected. Experimentally testing each of these sites for phosphorylation is currently time-consuming and expensive. Thus, a predictive strategy that can reduce the number of testable sites to any degree would be beneficial to the researcher. Although mass spectrometry has been the enabling technology for our predictive tool, its current inability to achieve full protein coverage and its likelihood of missing labile or low abundance modification sites necessitates alternate strategies to uncover the complete protein “modify-ome.”
The data presented here represent a unique approach to the prediction of protein post-translational modifications from large scale data sets that builds upon the motif-x algorithm that extracts motifs from data sets of known PTMs. This research represents a first step toward the goal of high specificity and sensitivity post-translational modification prediction and uses a simple yet effective approach. The scan-x algorithm, using only sequence information immediately adjacent to the modification site (particularly for serine and threonine phosphorylation), was able to make a large number of predictions at very high specificity, reinforcing the importance of local residues in the modification process. Nevertheless it is also our opinion that to achieve sensitivities and specificities much higher than the 60–80% range it will be necessary to take into account a wide variety of additional factors including protein interaction data, structural data, localization data, homology data, the existence of other PTMs proximal to one another, and enzymatic processivity. Additionally allowing for variable motif widths, conservative amino acid substitutions within motifs, and motif-dependent scoring thresholds are three improvements to the methodology that are likely to result in enhanced prediction performance.
Although improvements can be made, the increase in specificity and sensitivity achieved by scan-x over competing approaches likely benefits from the unique motif-based strategy used. The deconvolution of correlated residues surrounding protein modification sites by the motif-x program closely mimics the biological situation in which specific sequence-based recognition determinants are used by enzymes to modify a wide variety of protein substrates. Retaining these residue correlations in the prediction methodology not only allows users of scan-x to infer responsible modifying enzymes but also allows for improved discrimination of protein targets by filtering those results that do not syntactically match a given motif completely. In addition, the use of high stringency motifs in the prediction procedure acts as a filter through which incorrectly assigned modification sites are unlikely to pass, thus ensuring the limited propagation of any PTM errors from large scale mass spectrometry experiments (which may be included in our training set) into the prediction results.
This study also exemplifies how large scale, enzyme-independent, sequence data sets could be used to understand post-translational modification motifs. Here we have carried out the first computational prediction of the motif specificities of the acetyltransferase family of proteins. This has led to the discovery of eight potential acetylation motifs that are completely novel.
In the case of protein phosphorylation, despite the fact that our approach does not use kinase-specific data, many of the motifs extracted using motif-x are nearly identical to known kinase motif signatures (20). This was most recently confirmed in a recent study aiming to create an atlas of linear kinase motifs derived from kinase-specific data (41). This motif atlas, and its corresponding bioinformatics tool NetPhorest, may potentially be used in conjunction with scan-x to assign kinases to predicted phosphorylation sites. Furthermore we have provided strong evidence supporting the view that kinase specificities have remained well conserved through evolutionary history. Such a finding is not surprising when one considers the fact that the kinase enzyme-to-substrate interaction is usually a one-to-many relationship. Thus, allowable variations in kinase residues affecting substrate selectivity may be evolutionarily constrained because changes have the potential to have a deleterious effect on a large range of protein substrates.
We have also shown that there exist at least seven well conserved motifs for which a kinase has yet to be identified. Determining the identities of such kinases is in of itself an interesting scientific challenge, and without in silico discovery of motifs from phosphorylation data sets such as those provided by motif-x, predictions for sites bearing these motif signatures would likely remain difficult to detect.
To make the scan-x tool as versatile as possible, in addition to browsing the scan-x analyses carried out for this study, users will be able to run scan-x analyses on their own input data sets. Users that have unique modification data sets (e.g. from organisms or modifications not covered here or in response to particular stimuli) can predict additional modification sites in their proteins or proteome of interest. For example, a researcher studying the differential phosphorylation effects of a particular kinase-influencing drug is able to upload into motif-x those phosphorylation sites that are up-regulated upon exposure to the drug and use scan-x to scan the resulting motifs against the appropriate proteome to find additional potential downstream targets of the drug. In such an analysis, the cutoff for high stringency may not be readily apparent without calibrating sensitivities and specificities by performing a cross-validation analysis similar to what was done in this study; however, those sequences bearing scores that meet or exceed the scores for the upper quartile of known targets are likely to be high confidence hits.
We believe that using the added predictive functionality of scan-x with the already widely used motif-x tool will provide the necessary bridge between those who work on the proteomic scale and those who work on the protein scale. Although protein phosphorylation has been the most widely characterized protein post-translational modification over the past decade, there is little evidence to suggest that its place in the realm of protein modifications is unique in either prevalence or biological importance. Thus, future work will be aimed at the improvement of the predictive performance of the methodology as well as the addition of updated data from a wide variety of modification types as data sets for them become more abundant. It is the concerted interaction of numerous protein modifications that likely contribute to a significant amount of phenotypic variability (both beneficial and detrimental), and it is therefore our hope that protein modification prediction can also become a useful tool for interpreting diversity in human populations and in those of other species.
Supplementary Material
Acknowledgments
We thank John Aach and Desmond Lun for valuable discussions regarding statistical analysis of the data. In addition, we thank John Rush and Peter Hornbeck of Cell Signaling Technology, Inc. for assistance in providing phosphorylation and acetylation sites contained within the PhosphoSite database and Harvard Medical School Research Information Technology Group for hosting the motif-x Web site and maintaining the Orchestra computer cluster on which it runs.
Footnotes
Published, MCP Papers in Press, October 28, 2008, DOI 10.1074/mcp.M800332-MCP200
The abbreviations used are: PTM, post-translational modification; PKA, protein kinase A; PWM, position weight matrix; CK II, casein kinase II; CK I, casein kinase I; TP, true positive; FP, false positive; TN, true negative; FN, false negative; ROC, receiver operating characteristic; PSSM, position-specific scoring matrix; RalBP1, RalA-binding protein 1; MAPK, mitogen-activated protein kinase; CDK, cyclin-dependent kinase; SGD, Saccharomyces genome database; IPI, International Protein Index.
M. F. Chou, D. Schwartz, and G. M. Church, manuscript in preparation.
It is impossible at this time to truly determine actual negative data sets, and we have tried a number of approaches to this problem, but ultimately any method is an overestimate of the number of actual negatives. This is readily apparent based upon the fact that each new mass spectrometry study reveals a significant number of new modifications in each organism under study. Therefore, all specificity numbers are underestimates of the actual specificity and should not be taken to be absolutely quantitative. Nevertheless they allow relative comparisons between algorithms and parameter choices for a given algorithm.
Empirical studies showed that increasing the stringency of the so-called residual motif (the catchall motif for all peptides that cannot otherwise be deconvoluted into a motif class) by adding a constant offset of +30 to its threshold cutoff value yielded more specific predictions than when using a threshold identical to that of the other motifs. Therefore, the range of thresholds for residual motifs actually ranged from −70 to +129, and the threshold for all other motifs ranged from −100 to +99. Thus when interpreting data in supplemental Table 8, implicitly a row in the table for threshold value t should really be considered as the threshold value t for all motifs except for the residual motif for which the threshold value was instead set to t + 30.
This work was supported, in whole or in part, by National Institutes of Health Grant GM068763 and Grant EY07110-17 from the NEI. This work was also supported by the United States Department of Energy Genomes to Life program. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
REFERENCES
- 1.Hunter, T. ( 2007) The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol. Cell 28, 730–738 [DOI] [PubMed] [Google Scholar]
- 2.Hornbeck, P. V., Chabra, I., Kornhauser, J. M., Skrzypek, E., and Zhang, B. ( 2004) PhosphoSite: a bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics 4, 1551–1561 [DOI] [PubMed] [Google Scholar]
- 3.UniProt Consortium ( 2008) The universal protein resource (UniProt). Nucleic Acids Res. 36, D190–D195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bodenmiller, B., Malmstrom, J., Gerrits, B., Campbell, D., Lam, H., Schmidt, A., Rinner, O., Mueller, L. N., Shannon, P. T., Pedrioli, P. G., Panse, C., Lee, H. K., Schlapbach, R., and Aebersold, R. ( 2007) PhosphoPep—a phosphoproteome resource for systems biology research in Drosophila Kc167 cells. Mol. Syst. Biol. 3, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Albuquerque, C. P., Smolka, M. B., Payne, S. H., Bafna, V., Eng, J., and Zhou, H. ( 2008) A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol. Cell. Proteomics 7, 1389–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olsen, J. V., Blagoev, B., Gnad, F., Macek, B., Kumar, C., Mortensen, P., and Mann, M. ( 2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 7.Sugiyama, N., Nakagami, H., Mochida, K., Daudi, A., Tomita, M., Shirasu, K., and Ishihama, Y. ( 2008) Large-scale phosphorylation mapping reveals the extent of tyrosine phosphorylation in Arabidopsis. Mol. Syst. Biol. 4, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhai, B., Villen, J., Beausoleil, S. A., Mintseris, J., and Gygi, S. P. ( 2008) Phosphoproteome analysis of Drosophila melanogaster embryos. J. Proteome Res. 7, 1675–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biringer, R. G., Amato, H., Harrington, M. G., Fonteh, A. N., Riggins, J. N., and Huhmer, A. F. ( 2006) Enhanced sequence coverage of proteins in human cerebrospinal fluid using multiple enzymatic digestion and linear ion trap LC-MS/MS. Brief. Funct. Genomics Proteomics 5, 144–153 [DOI] [PubMed] [Google Scholar]
- 10.Creese, A. J., and Cooper, H. J. ( 2007) Liquid chromatography electron capture dissociation tandem mass spectrometry (LC-ECD-MS/MS) versus liquid chromatography collision-induced dissociation tandem mass spectrometry (LC-CID-MS/MS) for the identification of proteins. J. Am. Soc. Mass Spectrom. 18, 891–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brinkworth, R. I., Breinl, R. A., and Kobe, B. ( 2003) Structural basis and prediction of substrate specificity in protein serine/threonine kinases. Proc. Natl. Acad. Sci. U. S. A. 100, 74–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manning, B. D., and Cantley, L. C. ( 2002) Hitting the target: emerging technologies in the search for kinase substrates. Sci. STKE 2002, PE49. [DOI] [PubMed] [Google Scholar]
- 13.Diella, F., Gould, C. M., Chica, C., Via, A., and Gibson, T. J. ( 2008) Phospho.ELM: a database of phosphorylation sites—update 2008. Nucleic Acids Res. 36, D240–D244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blom, N., Sicheritz-Ponten, T., Gupta, R., Gammeltoft, S., and Brunak, S. ( 2004) Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4, 1633–1649 [DOI] [PubMed] [Google Scholar]
- 15.Neuberger, G., Schneider, G., and Eisenhaber, F. ( 2007) pkaPS: prediction of protein kinase A phosphorylation sites with the simplified kinase-substrate binding model. Biol. Direct 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Obenauer, J. C., Cantley, L. C., and Yaffe, M. B. ( 2003) Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31, 3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xue, Y., Li, A., Wang, L., Feng, H., and Yao, X. ( 2006) PPSP: prediction of PK-specific phosphorylation site with Bayesian decision theory. BMC Bioinformatics 7, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ingrell, C. R., Miller, M. L., Jensen, O. N., and Blom, N. ( 2007) NetPhosYeast: prediction of protein phosphorylation sites in yeast. Bioinformatics 23, 895–897 [DOI] [PubMed] [Google Scholar]
- 19.Gnad, F., Ren, S., Cox, J., Olsen, J. V., Macek, B., Oroshi, M., and Mann, M. ( 2007) PHOSIDA (phosphorylation site database): management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 8, R250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartz, D., and Gygi, S. P. ( 2005) An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 23, 1391–1398 [DOI] [PubMed] [Google Scholar]
- 21.Matsuoka, S., Ballif, B. A., Smogorzewska, A., McDonald, E. R., III, Hurov, K. E., Luo, J., Bakalarski, C. E., Zhao, Z., Solimini, N., Lerenthal, Y., Shiloh, Y., Gygi, S. P., and Elledge, S. J. ( 2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 [DOI] [PubMed] [Google Scholar]
- 22.Mukherji, M., Brill, L. M., Ficarro, S. B., Hampton, G. M., and Schultz, P. G. ( 2006) A phosphoproteomic analysis of the ErbB2 receptor tyrosine kinase signaling pathways. Biochemistry 45, 15529–15540 [DOI] [PubMed] [Google Scholar]
- 23.Smith, J. C., Duchesne, M. A., Tozzi, P., Ethier, M., and Figeys, D. ( 2007) A differential phosphoproteomic analysis of retinoic acid-treated P19 cells. J. Proteome Res. 6, 3174–3186 [DOI] [PubMed] [Google Scholar]
- 24.Wilson-Grady, J. T., Villen, J., and Gygi, S. P. ( 2008) Phosphoproteome analysis of fission yeast. J. Proteome Res. 7, 1088–1097 [DOI] [PubMed] [Google Scholar]
- 25.Molina, H., Horn, D. M., Tang, N., Mathivanan, S., and Pandey, A. ( 2007) Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 104, 2199–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang, F., Stenoien, D. L., Strittmatter, E. F., Wang, J., Ding, L., Lipton, M. S., Monroe, M. E., Nicora, C. D., Gristenko, M. A., Tang, K., Fang, R., Adkins, J. N., Camp, D. G., II, Chen, D. J., and Smith, R. D. ( 2006) Phosphoproteome profiling of human skin fibroblast cells in response to low- and high-dose irradiation. J. Proteome Res. 5, 1252–1260 [DOI] [PubMed] [Google Scholar]
- 27.Wang, Y., Ding, S. J., Wang, W., Jacobs, J. M., Qian, W. J., Moore, R. J., Yang, F., Camp, D. G., II, Smith, R. D., and Klemke, R. L. ( 2007) Profiling signaling polarity in chemotactic cells. Proc. Natl. Acad. Sci. U. S. A. 104, 8328–8333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peterman, S. M., Dufresne, C. P., and Horning, S. ( 2005) The use of a hybrid linear trap/FT-ICR mass spectrometer for on-line high resolution/high mass accuracy bottom-up sequencing. J. Biomol. Tech. 16, 112–124 [PMC free article] [PubMed] [Google Scholar]
- 29.Smolka, M. B., Albuquerque, C. P., Chen, S. H., and Zhou, H. ( 2007) Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc. Natl. Acad. Sci. U. S. A. 104, 10364–10369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kubinski, K., Domanska, K., Sajnaga, E., Mazur, E., Zielinski, R., and Szyszka, R. ( 2007) Yeast holoenzyme of protein kinase CK2 requires both beta and beta’ regulatory subunits for its activity. Mol. Cell. Biochem. 295, 229–236 [DOI] [PubMed] [Google Scholar]
- 31.Lopreiato, R., Facchin, S., Sartori, G., Arrigoni, G., Casonato, S., Ruzzene, M., Pinna, L. A., and Carignani, G. ( 2004) Analysis of the interaction between piD261/Bud32, an evolutionarily conserved protein kinase of Saccharomyces cerevisiae, and the Grx4 glutaredoxin. Biochem. J. 377, 395–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verma, R., Annan, R. S., Huddleston, M. J., Carr, S. A., Reynard, G., and Deshaies, R. J. ( 1997) Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science 278, 455–460 [DOI] [PubMed] [Google Scholar]
- 33.Zeng, H., Fei, H., and Levitan, I. B. ( 2004) The slowpoke channel binding protein Slob from Drosophila melanogaster exhibits regulatable protein kinase activity. Neurosci. Lett. 365, 33–38 [DOI] [PubMed] [Google Scholar]
- 34.Yu, L. R., Zhu, Z., Chan, K. C., Issaq, H. J., Dimitrov, D. S., and Veenstra, T. D. ( 2007) Improved titanium dioxide enrichment of phosphopeptides from HeLa cells and high confident phosphopeptide identification by cross-validation of MS/MS and MS/MS/MS spectra. J. Proteome Res. 6, 4150–4162 [DOI] [PubMed] [Google Scholar]
- 35.Zahedi, R. P., Lewandrowski, U., Wiesner, J., Wortelkamp, S., Moebius, J., Schutz, C., Walter, U., Gambaryan, S., and Sickmann, A. ( 2008) Phosphoproteome of resting human platelets. J. Proteome Res. 7, 526–534 [DOI] [PubMed] [Google Scholar]
- 36.Nada, S., Shima, T., Yanai, H., Husi, H., Grant, S. G., Okada, M., and Akiyama, T. ( 2003) Identification of PSD-93 as a substrate for the Src family tyrosine kinase Fyn. J. Biol. Chem. 278, 47610–47621 [DOI] [PubMed] [Google Scholar]
- 37.Stacey, T. T. I., Nie, Z., Stewart, A., Najdovska, M., Hall, N. E., He, H., Randazzo, P. A., and Lock, P. ( 2004) ARAP3 is transiently tyrosine phosphorylated in cells attaching to fibronectin and inhibits cell spreading in a RhoGAP-dependent manner. J. Cell Sci. 117, 6071–6084 [DOI] [PubMed] [Google Scholar]
- 38.Perez Jurado, L. A., Wang, Y. K., Peoples, R., Coloma, A., Cruces, J., and Francke, U. ( 1998) A duplicated gene in the breakpoint regions of the 7q11.23 Williams-Beuren syndrome deletion encodes the initiator binding protein TFII-I and BAP-135, a phosphorylation target of BTK. Hum. Mol. Genet. 7, 325–334 [DOI] [PubMed] [Google Scholar]
- 39.Roy, A. L. ( 2007) Signal-induced functions of the transcription factor TFII-I. Biochim. Biophys. Acta 1769, 613–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim, S. C., Sprung, R., Chen, Y., Xu, Y., Ball, H., Pei, J., Cheng, T., Kho, Y., Xiao, H., Xiao, L., Grishin, N. V., White, M., Yang, X. J., and Zhao, Y. ( 2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618 [DOI] [PubMed] [Google Scholar]
- 41.Miller, M. L., Jensen, L. J., Diella, F., Jorgensen, C., Tinti, M., Li, L., Hsiung, M., Parker, S. A., Bordeaux, J., Sicheritz-Ponten, T., Olhovsky, M., Pasculescu, A., Alexander, J., Knapp, S., Blom, N., Bork, P., Li, S., Cesareni, G., Pawson, T., Turk, B. E., Yaffe, M. B., Brunak, S., and Linding, R. ( 2008) Linear motif atlas for phosphorylation-dependent signaling. Sci. Signal. 1, ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshida, S., Kono, K., Lowery, D. M., Bartolini, S., Yaffe, M. B., Ohya, Y., and Pellman, D. ( 2006) Polo-like kinase Cdc5 controls the local activation of Rho1 to promote cytokinesis. Science 313, 108–111 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.