

Abstract
13C-selective NMR, combined with inhibitor perturbation experiments, shows that the Cɛ1—H proton of the catalytic histidine in resting α-lytic protease and subtilisin BPN′ resonates, when protonated, at 9.22 ppm and 9.18 ppm, respectively, which is outside the normal range for such protons and ≈0.6 to 0.8 ppm further downfield than previously reported. They also show that the previous α-lytic protease assignments [Markley, J. L., Neves, D. E., Westler, W. M., Ibanez, I. B., Porubcan, M. A. & Baillargeon, M. W. (1980) Front. Protein Chem. 10, 31–61] were to signals from inactive or denatured protein. Simulations of linewidth vs. pH demonstrate that the true signal is more difficult to detect than corresponding signals from inactive derivatives, owing to higher imidazole pKa values and larger chemical shift differences between protonated and neutral forms. A compilation and analysis of available NMR data indicates that the true Cɛ1—H signals from other serine proteases are similarly displaced downfield, with past assignments to more upfield signals probably in error. The downfield displacement of these proton resonances is shown to be consistent with an H-bond involving the histidine Cɛ1—H as donor, confirming the original hypothesis of Derewenda et al. [Derewenda, Z. S., Derewenda, U. & Kobos, P. M. (1994) J. Mol. Biol. 241, 83–93], which was based on an analysis of literature x-ray crystal structures of serine hydrolases. The invariability of this H-bond among enzymes containing Asp-His-Ser triads indicates functional importance. Here, we propose that it enables a reaction-driven imidazole ring flip mechanism, overcoming a major dilemma inherent in all previous mechanisms, namely how these enzymes catalyze both the formation and productive breakdown of tetrahedral intermediates.
The H-bonds between Asp-His‡ and His-Ser of the catalytic triad have long occupied center stage in discussions of serine hydrolase structure and mechanism. Derewenda et al. (1) have recently proposed the existence of an additional H-bond involving the triad, one connecting the Cɛ1—H proton of the catalytic His and a backbone carbonyl oxygen atom. In a comparative analysis of the available structures of serine hydrolases containing the Asp-His-Ser triad, these authors invariably found a backbone carbonyl oxygen atom sufficiently close and favorably aligned for H-bonding to the Cɛ1—H proton of His57.
The idea that carbon-bound protons can act as H-bond donors was first proposed more than 60 years ago (2, 3). Pauling (4) offered C—H⋅⋅⋅O bonding as explanation for the 51°C elevation in boiling point of acetyl chloride over that of trifluoroacetyl chloride. Sutor proposed the existence of C—H donated H-bonds in biological molecules in the early 1960s when she noticed “short” C⋅⋅⋅O (<3.3 Å) and O⋅⋅⋅H (<2.6 Å) distances in crystal structures of nucleic acid bases (5). Donahue, believing that O⋅⋅⋅H approach of as little as 2.2 Å was possible without attractive interactions, derided the idea, pushing it into the background for a decade (6). However, theoretical studies by Kollman (7) supported Sutor's claim, and neutron diffraction studies provided evidence for C—H⋅⋅⋅O bonds in low molecular weight compounds, including sugars, amino acids, and dipeptides (8). Many short C—O distances with orientations favorable for H-bonding of C—H⋅⋅⋅O have since been discovered in x-ray structures of nucleic acids (9) and proteins (10). Thus, however slowly, the notion that carbon-bound protons can act as H-bond donors has become accepted (11).
NMR chemical shifts are determined by electronic currents in a molecule. Because H-bonds affect electronic structure, chemical shifts have great potential for identifying and evaluating H-bonds, especially in macromolecules where other spectroscopic methods are impractical. A substantial body of data, for example, shows that, for N—H groups in proteins, H-bonding deshields both the proton and nitrogen atoms (12). For backbone N—H groups, downfield displacements are typically 0.5 to 3 ppm for the 1H signal, and about twice as much for the corresponding 15N signals. The magnitude of the displacement is related to the strength of the H-bond (12). N—H groups of histidine can show somewhat larger H-bonding effects, as demonstrated by 1H (13–16) and 15N (17, 18) NMR studies of α-lytic protease, I. 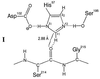
C—H protons, in principle, should experience similar deshielding on H-bonding. Experimental demonstrations of such H-bonded shifts, however, are lacking. Derewenda's proposal (1) suggests that the Cɛ1—H protons of catalytic His in serine proteases should exhibit such displacement. We report that this is indeed the case, perhaps not detected earlier owing to a misassignment problem discussed herein§ (20–26). The fact that α-lytic protease and subtilisin, proteases of independent ancestry (27), both exhibit the H-bond deshielding indicates that the finding is likely to hold for other serine proteases not yet examined. Thus, the results presented here confirm Derewenda's hypothesis and document the H-bond-induced displacement of a C—H proton in a protein. We also discuss the Cɛ1—H proton H-bond as a functional element of catalytic triad-containing proteases, proposing a mechanism involving a reaction-driven “ring flip,” which may resolve a long-standing dilemma posed by Wang (28), Polgar (29), and Jencks (30) of how hydrolase-catalyzed reactions proceed in the forward direction at all.¶
Materials and Methods
α-Lytic protease (EC 3.4.21.12) containing histidine uniformly 13C and 15N labeled (Cambridge Isotopes, Andover, MA) was purified from a histidine auxotroph of Lysobacter enzymogenes (31) and desalted as previously described (17, 32). Enzyme activity was measured at 410 nm by using Ac-Ala-Pro-Ala-p-nitroanilide (Ac-Ala-Pro-Ala-pNa) (Bachem) (31). Expression of His-labeled subtilisin bacterial proteinase novo (BPN)′97 was accomplished via fermentation of a His auxotrophic strain of Bacillus subtilis DB104 in a minimal medium containing uniformly 13C- and 15N-labeled His (33). Purification of labeled subtilisin BPN′ 97 was done by dialysis into a 10-mM Tris solution (pH 6.0) and subsequent passage over a CM52 cellulose ion exchange column, with elution of the protein in 0.01 M Mes and 0.1 M KCl. Enzyme activity was measured via a colorimetric assay employing the substrate succinyl-l-Ala-l-Ala-l-Pro-l-Phe-paranitroanilide (Suc-Ala-Ala-Pro-Phe-pNa) (33). Diisopropylfluorophosphate was purchased from Sigma. Gels (15% SDS/polyacrylamide) were run in standard fashion by using a Bio-Rad Minigel Aparatus. Methoxysuccinyl (MeOSuc)-Ala-Ala-Pro-boroVal‖ and MeOSuc-Ala-Ala-Pro-boroPhe were synthesized as previously described (34).
Unless otherwise specified, enzyme NMR samples were ≈2 mM in 90% H2O/10% D2O, and any inhibited sample contained 2- to 3-fold excess of inhibitor. α-Lytic protease samples contained 0.1 M KCl. NMR experiments were carried out on a Bruker (Billerica, MA) AMX-500 spectrometer equipped with a 5-mm triple resonance inverse probe. 1H 1D 13C-selective heteronuclear multiple-quantum correlation spectroscopy (HMQC) spectra used delay d2 = 0.5/(1JCH) = 2.5 ms. 1H chemical shifts were referenced to 2,2-dimethyl-2-silapentane-5-sulfonate (DSS).
Results and Discussion
Cɛ1—H Proton Chemical Shifts of the Catalytic Histidine in α-Lytic Protease.
α-Lytic protease contains only one histidine. 1D 13C-selective HMQC spectra of {13C, 15N} histidine-labeled α-lytic protease should therefore show only two signals. Fig. 1 shows that this is indeed the case for a freshly prepared, fully active sample of resting enzyme. The more downfield signal belongs to the histidine Cɛ1—H proton, the one more upfield to a Cδ2—H proton. The Cɛ1—H signal moves from 9.22 ppm at pH 4.5 (Fig. 1A) (Table 1) to 8.15 ppm at pH 9.0 (Fig. 1B), whereas the Cδ2—H signal moves from 6.92 ppm at pH 4.5 (Fig. 1A) to 6.38 ppm at pH 9.0 (Fig. 1B). Both 1H signals delineate titration curves with pKa values of ≈7.0. Therefore, the positions in the spectra shown in Fig. 1 A and B correspond closely to the chemical shifts of the titrating histidine in the fully protonated and fully neutral forms, respectively. The low pH** chemical shift of 9.22 ppm is unusual for a Cɛ1—H proton; to our knowledge, there have been no other reports of a chemical shift above 9.0 ppm for any resting serine protease (35) or histidine model compound (Table 1).
Figure 1.

1D 1H/13C HMQC NMR spectra of 2 mM {13C, 15N} His-labeled α-lytic protease in 0.1 M KCl and 90% H2O/10% D2O at 30°C. (A) Resting α-lytic protease at pH 4.5 (imidazole protonated). (B) Resting α-lytic protease at pH 9.0 (imidazole unprotonated). (C) α-Lytic protease inhibited with ≈2-fold excess MeOSuc-Ala-Ala-Pro-boroVal (AAPbV) at pH 4.5 [imidazole-protonated and spectrum pH-independent over accessible range (3.5–10.5)].
Table 1.
Chemical shifts of histidine Cɛ1—H protons
| Sample | Imidazolium ion, ppm | Imidazole form, ppm | Δδ, ppm |
|---|---|---|---|
| α-Lytic protease* | 9.22 | 8.15 | 1.07 |
| α-Lytic protease + AAPbV† | 9.35 | — | NA‡ |
| α-Lytic protease + DIFP | 8.35§ | 7.37¶ | 0.98 |
| Subtilisin BPN′97 (D99K)‖ | 9.18 | — | — |
| Subtilisin BPN′97 (N155A) + AAPbF** | 9.45 | — | NA‡ |
| Histidine‡‡ | 8.60 | 7.65 | 0.95 |
| N-acetyl-histidine‡‡ | 8.40 | 7.45 | 0.95 |
13C/15N His-labeled, 30°C (35).
13C/15N His (AAPbV, Ki = 6.7 × 10−9 M, (36), 30°C.
Not applicable. Chemical shift constant over pH ≈ 3 to 10.5.
(15Nδ1, 15Nɛ2) His, 35°C.
13C/15N His, 35°C.
13Cɛ1 His, 20 mM CaCl2, 0.1 M NaCl, 25°C (37).
An amount equal to 0.2 ppm subtracted to compensate for different reference (38).
Previous reports have assigned the Cɛ1—H proton of His57 in α-lytic protease to signals with low pH chemical shifts ranging from 8.0 to 8.71 ppm (Table 2). Such shifts are more typical of Cɛ1—H protons than the 9.22 ppm reported here. The addition of MeOSuc-Ala-Ala-Pro-boroVal (AAPbV) (18, 36), a potent, transition-state analog inhibitor of α-lytic protease, causes the two pH-dependent signals of Fig. 1 A and B to be replaced by two pH-independent signals with chemical shifts of 9.35 ppm and 6.76 ppm for Cɛ1—H and Cδ2—H, respectively (Fig. 1C). This result is consistent with previous NMR studies of α-lytic protease (31) and establishes the assignment of the 9.22 and 9.35 ppm signals to the Cɛ1—H protons of His57 in the resting and inhibited enzymes, respectively.
Table 2.
Literature 1H chemical shifts assigned to low pH form of serine protease active site imidazolium Cɛ1—H protons and pKa values
| Sample | δ, ppm | pKa | Ref. |
|---|---|---|---|
| Inactive or degraded forms | |||
| α-Lytic protease | 8.0 | 5.6 | Westler & Markley§ |
| α-Chymotrypsin | 8.54 | 6.18 | Markley & Ibanez (20) |
| α-Lytic protease | 8.70 | 6.5 | Markley (21) |
| α-Lytic protease, “fresh” | 8.65 | 6.5 | Markley et al. (22) |
| α-Lytic protease, “aged” | 8.71 | 5.9 | Markley et al. (22) |
| α-Lytic protease | 8.65 | 6.5 | Westler et al. (23) |
| Subtilisin Carlsberg | 8.6 | 7.22 | Jordan et al. (24) |
| Subtilisin BPN′ | 8.61 | 7.23 | Bycroft & Fersht (25) |
| Questionable | |||
| Porcine trypsin | 8.32 | 4.5 (slow) 5.0 (fast) | Markley & Porubcan (26) |
| Not in question | |||
| Chymotrypsinogen | 9.21 | 7.33 | Markley & Ibanez (20) |
| Porcine trypsinogen | 9.12 | 7.67 | Porubcan et al. (39) |
| Bovine trypsinogen | 9.12 | 7.72 | Porubcan et al. (39) |
| α-Lytic protease + DIFP | 8.38 | 8.16 | Extracted from Porubcan et al. (40) |
| Subtilisin E + AAPbF | 9.20 | NA | Bao et al. (41) |
| α-Chymotrypsin + AAPbF | 9.24 | NA | Bao et al. (41) |
| α-Chymotrypsin + TFMK | 8.97–9.18 | 10.7–12.1 | Lin et al. (42) |
| α-Chymotrypsin | 9.20 | — | Bao et al. (43) |
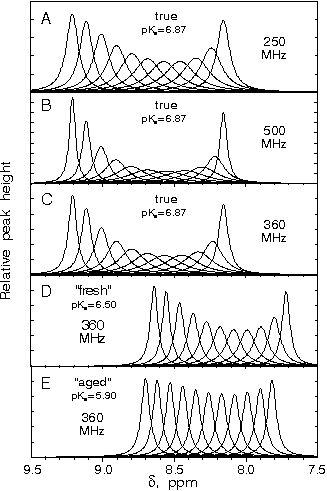
After having been frozen for several years, the enzyme sample that produced Fig. 1 A and B displays two additional sets of histidine Cɛ1—H and Cδ2—H proton signals (Fig. 2A), with low pH chemical shifts ≈8.7 ppm. The properties of the new Cɛ1—H signals match those of signals previously described by Markley et al. (22) as “fresh” and “aged” forms of α-lytic protease. Raising the pH (Fig. 2 B and C) shows a general broadening of all peaks, followed by line narrowing at high pH (9.50). Over the time course of the titration depicted in Fig. 2 A–E (≈1 wk), the amount of both “fresh” and “aged” forms increases, demonstrating the decay of true α-lytic protease to these forms, although enzyme assays showed little decrease in activity until ≈30% decrease occurring between Fig. 2 D and E. Addition of AAPbV to the sample of Fig. 2E abolishes the remaining enzyme activity. However, only the true signal becomes pH independent (Fig. 2 F and G). The “fresh” and “aged” signals continue to exhibit the same pH dependence as in the noninhibited sample, indicating that they represent degraded or inactive forms of α-lytic protease. The “fresh” and “aged” forms do not seem to be low molecular fragments on the basis of linewidth and SDS/PAGE gels.
Figure 2.

1D 1H/13C HMQC NMR spectra vs. pH of partially denatured ≈1 mM {13C, 15N} His-labeled α-lytic protease at 25°C. (F and G) Sample after inhibition with ≈2-fold excess of MeOSuc-Ala-Ala-Pro-boroVal (AAPbV). The asterisks denote extraneous peaks because of denatured or inactive enzyme. This sample, which had been frozen for 3 yr, remained at each pH value for 24 h or longer.
Cɛ1—H Proton Chemical Shifts of the Catalytic Histidine in Subtilisin BPN′ 97 (D99K).
BPN′ 97 D99K is an autolysis-resistant variant of subtilisin mutated at a site remote (>12 Å) from the active site. Subtilisin contains six histidines, so that the 1D 13C-selective HMQC spectra of {13C, 15N} histidine-labeled subtilisin BPN′ are more complicated than those of α-lytic protease. However, only one of the six His Cɛ1—H proton signals resonates downfield of 9.0 ppm at low pH (pH 5.6). This signal, at 9.18 ppm, is from the catalytic His (37). As was the case for α-lytic protease, the Cɛ1—H proton resonances in subtilisin Carlsberg (24) and BPN′ (25) were also previously misassigned to signals with more conventional low pH chemical shifts (Table 2). Consonni et al. (44) have demonstrated that these assignments were to autolyzed fragments of enzyme. Bao et al. (41) have recently reported a chemical shift of 9.20 ppm for the MeOSuc-Ala-Ala-Pro-boroPhe (AAPbF) complex of subtilisin E. To the best of our knowledge, our finding of a Cɛ1—H proton chemical shift (9.18 ppm) for a resting, active subtilisin has not been elsewhere reported. Here we also report a Cɛ1—H proton pH-independent chemical shift for the subtilisin BPN′/AAPbF complex of 9.45 ppm, to our knowledge the most downfield yet observed (Table 1).
Historical Misassignment of Histidine Cɛ1—H Protons in Serine Proteases.
Table 2 summarizes most of the Cɛ1—H proton chemical shifts reported over the past 25 years for the protonated His forms of serine proteases. Any value less than ≈9.1 ppm for a resting active enzyme or zymogen is probably erroneous. The inherent instability of proteases, combined with drastic “preexchange” treatments [up to 80°, pH ≤ 3 for hours (26), sometimes 6 M guanidinium chloride (20)] explains why many previous NMR studies reported signals from inactive species. However, it does not explain why previous researchers failed to detect signals from the true form, particularly because the samples were reported to have substantial enzyme activity. As demonstrated in the supplementary material (in the form of Figs. 4–6, Tables 3–6, additional text, and a movie, which are published on the PNAS web site, www.pnas.org), the true signals are inherently broader than those from the inactive species, particularly through the mid-range of pH titrations, owing to fast-exchange broadening (45) caused by the diffusion-controlled imidazole protonation rate (46). Most previous studies also used resolution enhancement techniques in acquisition (25) and/or processing (20) that discriminate against the broader, true signals, selecting instead for fragments and inactive forms.
Origin of the Unusual Cɛ1—H Proton Chemical Shifts for Catalytic Histidines in Serine Proteases.
Because the misassignment problem was confined to active enzymes, the Cɛ1—H proton chemical shift data appeared to indicate that the magnetic environment of the Cɛ1—H proton was substantially different (Δδ ≈ 0.6 ppm) between zymogens and active enzymes (39), prompting speculation about the nature of the apparent structural difference. Differences in anisotropic shielding by the neighboring Ser214 carbonyl group was proffered as the source of the chemical shift difference. Anisotropic shieldings of −0.7 ppm and “essentially zero” ppm were calculated for zymogen and active enzyme, respectively. In light of present results, these calculations (39) are obviously in error.
Anisotropic shielding from the serine carbonyl is, nevertheless, a possible contributor to the unusual shift of the Cɛ1—H protons in resting enzymes, zymogens, and boronic acid complexes, representing displacements of 0.8 to 1.0 ppm, respectively, from the ≈8.4 ppm of protonated but not H-bonded model systems, such as diisopropylfluorophosphate-inhibited α-lytic protease (47) and Ac-His (Table 2). Three contemporary models that have been applied to calculation of both Cα—H and amide N–H protons shielding in proteins are those of Ösapay and Case (48), Herranz et al. (49), and Williamson and Asakura (50) [also Asakura et al. (51)]. A detailed analysis is given in the supplementary material. These calculations demonstrate that anisotropic shielding by the adjacent peptide group accounts for but a fraction of the experimentally observed displacement (≥0.8 ppm). The bulk of the displacement we attribute to the H-bond proposed by Derewenda. This is a significant H-bond shift and, in our view, represents an energy sufficient to bind His57 to Ser214 for vital mechanistic purposes.
Mechanistic Implications of Histidine Cɛ1—H Proton-Donated H-bonds.
Derewenda et al. (1) suggested three possible roles for the His Cɛ1—H⋅⋅⋅O⩵C H-bond: (i) to aid in prealigning and maintaining the catalytic His in ideal position to activate Ser195; (ii) to increase the electronegativity of Nɛ2; and (iii) to facilitate deprotonation at Nɛ2 by creating a more even distribution of positive charge on the imidazolium ion. We believe these factors would have too small an effect to account for the invariance of the Cɛ1 H-bond. We propose instead that the donor and acceptor groups of the Cɛ1 H-bond supply needed components for a “reaction-driven,” catalytically functional rotation of ≈180° about the His Cβ—Cγ bond, i.e., “flipping” the imidazole ring (Fig. 3).
Figure 3.

Proposed model of reaction-driven imidazole ring flip mechanism in serine hydrolases containing the catalytic triad. Shaded species are unproductive. Relative abundance of imidazole rotamers represented only qualitatively by magnitudes of double arrows, so arrow lengths should not be interpreted literally. The mechanism requires only an increasing trend in the flipped/normal ratio in moving from resting enzyme to TI-1 to acyl enzyme. See supplementary material for an animated video loop with sound effects.
X-ray crystallography (52) and NMR spectroscopy (14, 17) have shown that the catalytic histidine in resting serine proteases is ideally poised to activate the Ser hydroxyl group for nucleophilic attack on the substrate. However, if the His is optimally aligned to accept the serine proton to form tetrahedral intermediate 1 (TI-1, Fig. 3), then it must also be optimally aligned to donate the proton back to serine, expelling the alkoxide ion and regenerating the starting materials, rather than expelling the amine anion (R′—NH−) and proceeding to acyl enzyme (28–30). Here a significant problem arises because, in the absence of other factors, expulsion of the alkoxide ion would be greatly favored over expulsion of the amine anion, as the alkoxide ion (pKb ≈ 1) is inherently a much better leaving group than the amine anion (pKb ≈ 16) (53). With a catalytic advantage added to the already favored back reaction, it is difficult to understand how the reaction can proceed in the forward direction.
Recognizing this problem, Wang (28) proposed that the catalytic His is aligned midway between the Ser and the leaving nitrogen, and that proton transfers occur along bent H-bonds. Kraut (54) proposed that the catalytic histidine may be rigidly held in position to H-bond with the leaving nitrogen atom. The Kraut proposal recognizes the importance of catalyzing the breakdown of TI-1 in the productive direction but gives up entirely on general-base assistance in its formation. Satterthwait and Jencks (30) proposed that the imidazole ring oscillates, alternately aligning the His Nɛ2 between Ser and the leaving amide. On the basis of NMR evidence, Bachovchin proposed that formation of TI-1 triggers movement of the catalytic histidine (47). All these proposals make the same basic tradeoff, sacrificing catalytic assistance in formation of TI-1 for assistance in its breakdown in the productive direction.
Our ring flip mechanism (Fig. 3), solves the above dilemma, and assigns a role to the His Cɛ1—H⋅⋅⋅O⩵C H-bond commensurate with its invariance. It involves two positions of the imidazole ring differing by ≈180° rotation about the Cβ—Cγ bond, which we shall refer to as the normal (e.g., 1) and flipped (e.g., 2) rotamers, respectively. Note that there are four main positions (labeled clockwise a through d) in which H-bonds between the imidazole ring and the active site can occur. Both 1 and 2 make favorable contacts at positions a, b, and c. A favorable interaction is present at position d in 1, the His Cɛ1—H-donated H-bond. However, an unfavorable contact exists at d in 2, i.e., the unshared electron pair of Nɛ2 oriented toward an unshared electron pair on the carbonyl oxygen of Ser214. The interaction at d could easily determine relative rotameric stability. For example, if the H-bond energy at 1d were as little as −2 to −3 kcal/mol, and the repulsion of electron pairs in 2d were equally unfavorable, i.e., +2 to +3 kcal/mol, the difference in this interaction alone would cause 1 to be favored over 2 by 1,000- to 20,000-fold. This energy difference would be sufficient to prevent observation of 2 in resting enzymes by NMR or x-ray diffraction.
However, once Nɛ2 accepts the proton from Ser, and TI-1 is formed, the situation changes (3 and 4, Fig. 3). The unfavorable interaction at d in the flipped rotamer 2 becomes favorable in the flipped rotamer 4, because Nɛ2 now carries a proton. Consequently, 4 will not be as unfavored relative to 3 as 2 is to 1. Such a “reaction-driven” ring flip would have important catalytic consequences, because the flipped rotamer 4 is as ideally suited to catalyzing the breakdown of TI-1 in the forward direction as the normal rotamer 1 is in catalyzing its formation. In 4, not only is Nδ1—H ideally aligned to expel the amine anion, the nonlabile Cɛ1—H proton is also ideally positioned to H-bond with the Ser195 oxygen, where it can serve only to inhibit expulsion of the alkoxide ion by not giving up its proton and by blocking access to any labile proton donors.
By estimates analogous to those made above for resting enzyme, we might expect formation of the TI-1 to stabilize the flipped relative to the normal rotamer by ≈1,000- to 20,000-fold, which would make the ratio of 4:3 ≈1,000- to 20,000-fold greater than that of 2:1. It is conceivable that this effect could make 4 favored over 3, but 4 need not be favored over 3 for the effect to be catalytically functional. For example, if the effect were such as to make the ratio of 4:3 = 0.01, we would expect a maximum of 1 in every 100 TI-1s to go forward to product. Nevertheless, conversion of 1 per 100 represents a substantial advantage over 1 per 105 to 106, which might be the case without the ring flip.
After formation and productive breakdown of TI-1, the enzyme must hydrolyze the acyl enzyme. Here the ring flip mechanism offers further advantages: with an electron pair on Nδ1 oriented toward solvent, 6 is ideally suited to catalyzing the next step—activation of water for attack on the acyl enzyme. Return of 6 to the more abundant but less catalytically useful rotamer, 5, at this stage would be hindered by the unfavorable interaction between the unshared pair on Nδ1 and the catalytic Asp. After activation of water, 6 becomes 8, and 8 is then free to return to the normal rotamer 7, which brings Nɛ2—H back into alignment with Ser195, where it can expel the alkoxide ion, thus completing the reaction and returning the enzyme to the initial state.
The energetics of the H-bonding interactions at positions a through d during catalysis, as outlined above, appear sufficient but may not be the sole driving force for the histidine flip mechanism. Because the positive charge on the imidazolium ring does not lie along the rotational axis, the charge–charge electrostatic interactions that develop on formation of TI-1 between imidazolium and oxyanion will favor the flipped rotamer, the direction needed to contribute to the ring flip mechanism.
Experimental evidence that the flipped rotamer can exist in serine protease-active sites has recently been supplied by an x-ray crystal structure of subtilisin BPN′ in 50% dimethylformamide (DMF) at low pH by Kidd et al. (55). In this structure, the imidazole ring is rotated 164°, rather than 180°, but nevertheless the contacts at points a–d are present. Kidd et al. sketch in both Cɛ1—H- and Cδ2—H-donated H-bonds to the active site Asp and Ser groups in the flipped structure without comment. Nor do Kidd et al. make any mention of H-bonding to the backbone Ser carbonyl group, but it is well oriented for H-bonding to the Nɛ2—H proton, and at an N–O distance we measured at 3.14 Å–0.09 Å shorter than the C–O distance of the normal rotamer.
The “low-barrier” H-bond (LBHB) hypothesis (56–58) of an unusually strong Asp-His H-bond (at position a in Fig. 3), promoted by some researchers in the face of unanswered experimental (16,59) and theoretical (19, 60, 61) evidence to the contrary, offers no solution to the dilemma discussed above. In fact, if the Asp-His H-bond were unusually strong at low pH, it is difficult to understand why the ring would flip in the less polar solvent of 50% DMF, as observed in the x-ray crystal structure of subtilisin BPN′ (55), because the LBHB hypothesis predicts an increase in bond energy with lower dielectric constant (58).
Supplementary Material
Acknowledgments
We thank Charles A. Kettner for providing boronic acid inhibitors, and Bruce A. Malcolm for helpful discussions.
Abbreviations
- MeOSuc
methoxysuccinyl
- HMQC
heteronuclear multiple-quantum correlation spectroscopy
- TI
tetrahedral intermediate
- BPN
bacterial proteinase novo
Footnotes
Both single and triple letter codes are used for amino acids.
Presented in part as a poster entitled “Experimental Evidence for a His-57 Cɛ1—H⋅⋅⋅O⩵C Hydrogen Bond in the Active Site of α-Lytic Protease” at the XVII International Conference on Magnetic Resonance in Biological Sciences, 1996, Keystone, CO.
The prefix b or boro indicates replacement of the amino acid carboxylate group by B(OH)2.
“Low pH” and “high pH” are shorthand for “protonated” and “unprotonated” His.
Westler, W. M. & Markley, J. L. (1978) Fed. Proc. 37, 1795 (abstr.).
References
- 1.Derewenda Z S, Derewenda U, Kobos P M. J Mol Biol. 1994;241:83–93. doi: 10.1006/jmbi.1994.1475. [DOI] [PubMed] [Google Scholar]
- 2.Glasstone S. Trans Faraday Soc. 1937;33:200–214. [Google Scholar]
- 3.Dulmage W J, Lipscomb W N. Acta Crystallogr. 1951;4:330–334. [Google Scholar]
- 4.Pauling L. The Nature of the Chemical Bond. 3rd Ed. Ithaca, NY: Cornell Univ. Press; 1960. [Google Scholar]
- 5.Sutor D J. Nature (London) 1962;195:68–69. [Google Scholar]
- 6.Donohue J. In: Structural Chemistry and Molecular Biology. Rich A, Davidson N, editors. San Francisco: Freeman; 1968. pp. 443–465. [Google Scholar]
- 7.Kollman P, McKelvey J, Johansson A, Rothenberg S. J Am Chem Soc. 1975;97:955–965. [Google Scholar]
- 8.Taylor R, Kennard O. J Am Chem Soc. 1982;104:5070–5076. [Google Scholar]
- 9.Desiraju G R. Acc Chem Res. 1991;24:290–296. [Google Scholar]
- 10.Derewenda Z S, Lee L, Derewenda U. J Mol Biol. 1995;252:248–262. doi: 10.1006/jmbi.1995.0492. [DOI] [PubMed] [Google Scholar]
- 11.Scheiner S. Hydrogen Bonding: A Theoretical Perspective. New York: Oxford Univ. Press; 1997. [Google Scholar]
- 12.Wishart D S, Sykes B D, Richards F M. J Mol Biol. 1991;222:311–333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- 13.Robillard G, Shulman R G. J Mol Biol. 1974;86:519–540. doi: 10.1016/0022-2836(74)90178-8. [DOI] [PubMed] [Google Scholar]
- 14.Bachovchin W W. Proc Natl Acad Sci USA. 1985;82:7948–7951. doi: 10.1073/pnas.82.23.7948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsilikounas E, Kettner C A, Bachovchin W W. Biochemistry. 1992;31:12839–12846. doi: 10.1021/bi00166a019. [DOI] [PubMed] [Google Scholar]
- 16.Ash E L, Sudmeier J L, De Fabo E C, Bachovchin W W. Science. 1997;278:1128–1132. doi: 10.1126/science.278.5340.1128. [DOI] [PubMed] [Google Scholar]
- 17.Bachovchin W W, Roberts J D. J Am Chem Soc. 1978;100:8041–8047. [Google Scholar]
- 18.Bachovchin W W. Stable Isotope Applications in Biomolecular Structure and Mechanisms. Los Alamos, NM: Los Alamos National Laboratory; 1994. [Google Scholar]
- 19.Warshel A. J Biol Chem. 1998;273:27035–27038. doi: 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- 20.Markley J L, Ibanez I B. Biochemistry. 1978;17:4627–4640. doi: 10.1021/bi00615a008. [DOI] [PubMed] [Google Scholar]
- 21.Markley J L. Biological Applications of Magnetic Resonance. New York: Academic; 1979. pp. 397–461. [Google Scholar]
- 22.Markley J L, Neves D E, Westler W M, Ibanez I B, Porubcan M A, Baillargeon M W. Front Protein Chem. 1980;10:31–61. [Google Scholar]
- 23.Westler W M, Markley J L, Bachovchin W W. FEBS Lett. 1982;138:233–235. doi: 10.1016/0014-5793(82)80449-3. [DOI] [PubMed] [Google Scholar]
- 24.Jordan F, Polgar L, Tous G. Biochemistry. 1985;24:7711–7717. doi: 10.1021/bi00347a031. [DOI] [PubMed] [Google Scholar]
- 25.Bycroft M, Fersht A R. Biochemistry. 1988;27:7390–7394. doi: 10.1021/bi00419a033. [DOI] [PubMed] [Google Scholar]
- 26.Markley J L, Porubcan M A. J Mol Biol. 1976;102:487–509. doi: 10.1016/0022-2836(76)90330-2. [DOI] [PubMed] [Google Scholar]
- 27.Rawlings N D, Barrett A J. Methods Enzymol. 1994;244:19–61. doi: 10.1016/0076-6879(94)44004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J H. Proc Natl Acad Sci USA. 1970;66:874–881. doi: 10.1073/pnas.66.3.874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Polgar L. Acta Biochim Biophys. 1972;7:29–34. [PubMed] [Google Scholar]
- 30.Satterthwait A C, Jencks W P. J Am Chem Soc. 1974;96:7018–7031. doi: 10.1021/ja00829a034. [DOI] [PubMed] [Google Scholar]
- 31.Bachovchin W W, Wong W Y, Farr J S, Shenvi A B, Kettner C A. Biochemistry. 1988;27:7689–7697. doi: 10.1021/bi00420a018. [DOI] [PubMed] [Google Scholar]
- 32.Tsilikounas E, Kettner C A, Bachovchin W W. Biochemistry. 1993;32:12651–12655. doi: 10.1021/bi00210a013. [DOI] [PubMed] [Google Scholar]
- 33.Day R M. Ph.D. thesis. Boston, MA: Tufts University; 1995. [Google Scholar]
- 34.Kettner C A, Shenvi A B. J Biol Chem. 1984;259:15106–15114. [PubMed] [Google Scholar]
- 35.Vincent M V. Ph.D. thesis. Boston: Tufts University; 1992. [Google Scholar]
- 36.Kettner C A, Bone R, Agard D A, Bachovchin W W. Biochemistry. 1988;27:7682–7688. doi: 10.1021/bi00420a017. [DOI] [PubMed] [Google Scholar]
- 37.Torchilin E V. Ph.D. thesis. Boston, MA: Tufts University; 1999. [Google Scholar]
- 38.Sachs D H, Schechter A N, Cohen J S. J Biol Chem. 1971;246:6576–6580. [PubMed] [Google Scholar]
- 39.Porubcan M A, Neves D E, Rausch S K, Markley J L. Biochemistry. 1978;17:4640–4647. doi: 10.1021/bi00615a009. [DOI] [PubMed] [Google Scholar]
- 40.Porubcan M A, Westler W M, Ibanez I B, Markley J L. Biochemistry. 1979;18:4108–4116. doi: 10.1021/bi00586a008. [DOI] [PubMed] [Google Scholar]
- 41.Bao D, Cheng J-T, Kettner C, Jordan F. J Am Chem Soc. 1998;120:3485–3489. [Google Scholar]
- 42.Lin J, Westler W M, Cleland W W, Markley J L, Frey P A. Proc Natl Acad Sci USA. 1998;95:14664–14668. doi: 10.1073/pnas.95.25.14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bao D, Huskey W P, Kettner C A, Jordan F. J Am Chem Soc. 1999;121:4684–4689. [Google Scholar]
- 44.Consonni R, Molinari H, Greco F, Zannoni G, Zetta L, Carrea G, Riva S. Biochim Biophys Acta. 1992;1119:39–44. doi: 10.1016/0167-4838(92)90231-2. [DOI] [PubMed] [Google Scholar]
- 45.Sudmeier J L, Evelhoch J L, Jonsson N B H. J Magn Reson. 1980;40:377–390. [Google Scholar]
- 46.Bachovchin W W, Switzman S. Spectrosc Int J. 1983;2:219–226. [Google Scholar]
- 47.Bachovchin W W. Biochemistry. 1986;25:7751–7759. doi: 10.1021/bi00371a070. [DOI] [PubMed] [Google Scholar]
- 48.Ösapay K, Case D A. J Am Chem Soc. 1991;113:9436–9444. [Google Scholar]
- 49.Herranz J, Gonzalez C, Rico M, Nieto J L, Santoro J, Jimenez M A, Bruix M, Neira J L, Blanco F J. Magn Reson Chem. 1992;30:1012–1018. [Google Scholar]
- 50.Williamson M P, Asakura T. J Magn Reson B. 1993;101:63–71. [Google Scholar]
- 51.Asakura T, Taoka K, Demura M, Williamson M P. J Biomol NMR. 1995;6:227–236. doi: 10.1007/BF00197804. [DOI] [PubMed] [Google Scholar]
- 52.Fujinaga M, Delbaere L T J, Brayer G D, James M N G. J Mol Biol. 1985;183:479–502. doi: 10.1016/0022-2836(85)90296-7. [DOI] [PubMed] [Google Scholar]
- 53.Jencks W P. Catalysis in Chemistry and Enzymology. New York: Dover; 1987. , Ch. 2. [Google Scholar]
- 54.Kraut J. Annu Rev Biochem. 1977;46:331–358. doi: 10.1146/annurev.bi.46.070177.001555. [DOI] [PubMed] [Google Scholar]
- 55.Kidd R D, Sears P, Huang D, Witte K, Wong C, Farber G K. Protein Sci. 1999;8:410–417. doi: 10.1110/ps.8.2.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gerlt J A, Gassman P G. J Am Chem Soc. 1993;115:11552–11568. [Google Scholar]
- 57.Cleland W W, Kreevoy M M. Science. 1994;264:1887–1890. doi: 10.1126/science.8009219. [DOI] [PubMed] [Google Scholar]
- 58.Frey P A, Whitt S A, Tobin J B. Science. 1994;264:1927–1930. doi: 10.1126/science.7661899. [DOI] [PubMed] [Google Scholar]
- 59.Shan S O, Loh S, Hershlag D. Science. 1996;272:97–101. doi: 10.1126/science.272.5258.97. [DOI] [PubMed] [Google Scholar]
- 60.Warshel A, Papazyan A, Kollman P A. Science. 1995;269:102–104. doi: 10.1126/science.7661987. [DOI] [PubMed] [Google Scholar]
- 61.Warshel A, Papazyan A. Proc Natl Acad Sci USA. 1996;93:13665–13670. doi: 10.1073/pnas.93.24.13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}