

Abstract
Interleukin-8α (IL-8α) is an antimicrobial peptide derived from the chemokine IL-8. Solution NMR was used to determine the atomic resolution structure of IL-8α in SDS micelles. Solid state NMR and tryptophan fluorescence were used to probe the interaction of IL-8α with model membranes. The peptide interacted differently with anionic versus purely zwitterionic micelles or bilayers. Tryptophan fluorescence demonstrated a deeper position of Trp 4 in SDS micelles and POPC/POPG bilayers compared to pure POPC bilayers, consistent with 2H order parameters, which also indicated a deeper position of the peptide in POPC/POPG bilayers compared to POPC bilayers. Paramagnetic probe data showed that IL-8α was situated roughly parallel to the SDS micelle surface, with a slight tilt that positioned the N-terminus more deeply in the micelle compared to the C-terminus. 15N solid-state NMR spectra indicated a similar, nearly parallel position for the peptide in POPC/POPG bilayers. 31P and 2H solid-state NMR demonstrated that the peptide did not induce the formation of any non-lamellar phases and did not significantly disrupt bilayer orientation in aligned model membranes composed of POPC or POPC/POPG.
Keywords: α-helix, solution structure, micelle, solid-state NMR, aligned lipid bilayers
Antimicrobial peptides (AMPs) have been studied for several years due to their ability to kill microbes, often by targeting and disrupting their membranes (1). More recently, it has been recognized that AMPs can have dual functions that involve direct and indirect host defense roles. There are antimicrobial peptides that act directly against microbes, but are also able to modulate the host immune response (2). Moreover, there are well-characterized components of the immune system, such as chemokines, for which direct antimicrobial properties have now been identified (3; 4). The present study concerns the structure-activity relationships of one such peptide, IL-8α, derived from the helical region of the chemokine interleukin-8 (IL-8). IL-8 is a 72 residue dimeric chemokine produced primarily by macrophages, in response to an inflammatory stimulus. NMR (5; 6) and crystallography (7) structural studies have demonstrated that its two monomers associate to form two antiparallel α-helices sitting on a six-stranded β-sheet.
There has been much recent interest in AMPs largely due to the possibility that they could be useful as adjunctives or alternatives to traditional antibiotics. In this respect, AMPs have broad antimicrobial spectra, are active against organisms refractory to many classical antibiotics, and may exert one or more mechanisms of action that differ from traditional antibiotics (8). Antimicrobial peptides of human origin are of particular interest as they represent highly attractive templates for novel anti-infective therapeutics in humans. A number of AMPs present in mammals have been studied, including lactoferrin, cathelicidin and defensins (9). α-defensins (10) share a conserved CXC motif, which is also found in chemokines. This similarity suggested that chemokines could exert antibacterial activity, and this was indeed shown to be the case for platelet basic peptide / CXCL7 and its derivatives CTAP-3 and NAP-2 (11), for platelet factor-4 / CXCL4 family polypeptides and RANTES / CCL5 (12), for MIG/CXCL9, IP-10/CXCL10 and I-TAC/CXCL11 (3; 13) and for the kinocidins GRO-α / CXCL1, I-309 / CCL1, MCP-2 / CCL2, and lymphotactin / CL1 (14). Direct antimicrobial activity has also been detected for additional chemokines, including macrophage inflammatory protein-3α/CCL20 (15), CCL28 (16), granulocyte chemotactic protein 2/CXCL6 (17) and CXCL14 (18). Yang et al identified an in vitro antimicrobial activity for 17 of the 30 human chemokines they tested (15). Several peptides derived from the C-terminal helical region of chemokines have also been shown to exert antimicrobial activity, such as thrombocidins TC-1 or TC-2, derived from CXCL7 (19) and CDAP-4, derived from CCL13 (20) and peptides with antimicrobial activity are also generated by the thrombin cleavage of human platelets CXCL4, CCL5 and CTAP-3 (21). Some antimicrobial peptides have been developed as analogs of chemokine domains, like platelet microbicidal protein 1, which is analogous to the C-terminal regions of human platelet factor-4/CXCL4 and IL-8 (22-25).
In common with many previously characterized AMPs, several chemokines and chemokine-derived peptides present large positive patches on their surface, leading to the suggestion that they might permeabilize bacterial membranes via interactions with negatively charged bacterial membrane components (15). Indeed, the chemokine-derived peptide CDAP-4 has been shown to affect the surface of Pseudomonas aeruginosa and might thus insert in the bacterial membrane (20). However, for TC-1 and TC-2, membrane potential measurement experiments showed that these proteins do not dissipate the bacterial membrane potential. It was thus postulated that their microbial target might be intracellular (19). The antibacterial activity of thombin-induced platelet microbicidal protein-1, which is an orthologue of the chemokine hPF-4, involves both the interaction with cell membrane and the interference with intramolecular targets (26; 27). Hence, further studies to elucidate the molecular mechanism(s) of chemokine-derived AMPs are necessary to better understand these important host defense molecules.
The peptide IL-8α consists of the C-terminal α-helix of IL-8 and has been shown to exert antimicrobial activity (14; 28). Although there are, as yet, no published reports detailing that IL-8α or a similar peptide is produced in vivo, this does not seem unlikely given that acid hydrolysis experiments generate a peptide which consists of the IL-8α sequence plus an extra proline at its N-terminal end (28). Moreover, there are many examples of antimicrobial peptides that are produced by cleavage of a precursor protein (29). Indeed, C-terminal domains of IL-8, PF-4 and other kinocidins are deployed from their respective holoproteins in vitro when subjected to proteases relevant to settings of tissue injury or infection (Yeaman et al. unpublished data; (30). IL-8α exerts antimicrobial activity against both Gram-negative (Escherichia coli, Salmonella enterica MR10, Klebsiella pneumoniae, Helicobacter pylori) and Gram-positive (Streptococcus pyogenes) bacteria (14; 28). Some of these bacteria, such as E. coli and K. pneumoniae, have been shown to be resistant to many conventional antibiotics and thus developing new therapeutics targeting these and other multi-drug-resistant pathogens is especially important. Yount et al have shown that IL-8α retains its antimicrobial properties in human blood and blood matrices ex vivo (14), an important characteristic for a therapeutic molecule. Solid-phase assays, performed by measuring the diameter of inhibition zones, have been used to determine a lethal concentration of ∼60μM for IL-8α, which is comparable with the ∼55μM lethal concentration of the cecropin-like peptide Hp(2-20). Activity in the presence of salt is another important characteristic in a therapeutic molecule. Like many antibacterial peptides such as indolicidins(31), gramicidins and magainins (32), IL-8α had a reduced activity at higher NaCl concentrations and completely lost its antimicrobial activity at 350mM NaCl. However, at 255mM NaCl, IL-8α appeared to be relatively salt-resistant as it still retained ∼60% of its activity (28), compared to the 65% of activity of CXCL14 at 100mM NaCl (18) and GCP-2/CXCL6, which showed complete abrogation of antimicrobial activity at 100mM NaCl (17). When the pH was decreased from 7.4 to 5.0, IL-8α exerted a higher antibacterial activity. In inhibition zone assays performed by Björstad (28) and Cole (3) or in colony-forming assays (15) no measurable antimicrobial activity could be observed for full length IL-8 against Escherichia coli, Listeria monocytogenes or Staphylococcus aureus at pH=7.4-7.5. On the other hand, in solid-phase assays performed at pH 5.5, Yount et al (14) showed full-length IL-8 exerts not only an antibacterial activity against Salmonella typhimurium and modest activity against S. aureus, but also antifungal efficacy equivalent to that of defensin hNP-1 against Candida albicans. Importantly, in solution phase studies at pH 5.5 and 7.5, the IL-8α peptide exerted strong candidacidal activity, whereas its anti-bacterial efficacy was comparably less (24). These activity profiles were conferred by the IL-8α peptide, autonomous from the N-terminal regions of the IL-8 holoprotein.
We have previously studied the structure of RP-1, a synthetic antimicrobial peptide designed based on the C-terminal α-helix of the chemokine platelet factor-4/CXCL4 (33). Similar structures were obtained for RP-1 in complex with micelles composed of SDS, which roughly mimic anionic bacterial membranes, and DPC, which approximate zwitterionic eukaryotic membranes. This observation lead to the suggestion that RP-1's preferential specificity for bacterial versus mammalian membranes is likely due to differences in the mode of lipid-peptide interaction rather than differences in peptide structure in the two environments. Thus, for the IL-8α peptide, we chose to limit the solution NMR structural studies to a single micelle system, SDS, and focus on its interactions with bilayers using solid state NMR of oriented lipid bilayers. Additionally, tryptophan fluorescence studies were performed to elucidate the role of the tryptophan residue in the bilayer interactions, as well as to allow the direct comparison of IL-8α's interactions with micelles and bilayers, as this technique can be used on both contexts.
Materials and Methods
Materials
The IL-8α peptide (NH2-K1ENWV5QRVVE10KFLKR15AENS-COOH) was produced by solid phase synthesis using Fmoc (O-fluorenylmethyl-oxycarbonyl) chemistry. 15N labels were incorporated at positions V5, V8, L13 and A16. The peptide was purified using reversed-phase HPLC, and its mass was authenticated by MALDI-TOF spectrometry. Purity of the resulting sample was determined to be ≥ 95% by analytical HPLC.
Sodium-dodecylsulfate-d25, was obtained from Cambridge Isotope Laboratories (Andover, MA). Sodium-dodecylsulfate, 16-Doxyl-stearic acid (16-DSA) and sodium azide were from Sigma-Aldrich. 1-Palmitoyl-2-Oleoyl-sn-Glycero-3-Phosphocholine (POPC), 1-Palmitoyl(D31)-2-Oleoyl-sn-Glycero-3-Phosphocholine (POPC-d31), 1-Palmitoyl-2-Oleoyl-sn-Glycero-3-[Phospho-rac-(1-glycerol)] (Sodium Salt) (POPG), 1-Palmitoyl(D31)-2-Oleoyl-sn-Glycero-3-[Phospho-rac-(1-glycerol)] (Sodium Salt) (POPG-d31) were purchased from Avanti Polar Lipids, Inc.
Circular dichroism
The sample of IL-8α in pure water was used to run circular dichroism experiments. These were performed on a Jasco J-810 spectropolarimeter. The mean of 4 spectra acquired at 308K over a range of 180-300nm was analyzed.
Solution NMR
NMR structure determination
Spectra were acquired on a Bruker Avance 500MHz spectrometer equipped with z-gradients and a triple-resonance TXI probe, processed with NMRPipe (34) and analyzed using SPARKY (35). Two IL-8α samples were prepared in pure water and in SDS micelles. For both samples, 1.5mM peptide was dissolved in 90% H2O, 10% D2O, pH=5.0. Additionally, 0.2mM 2,2-dimethyl-2-silapentane-5-sulfonate (sodium salt, DSS) and 0.2mM sodium azide were added. 150mM deuterated SDS was added to one of the samples for the experiments in micelles. For structure analysis in water, 2D 15N-1H HSQC spectra, 2D TOCSY spectra and 2D NOESY spectra were run at 35°C. For the study in micelles, samples were analyzed by 2D 15N-1H HSQC, 2D TOCSY and 2D NOESY with mixing times varying from 100 to 200ms and at temperatures of 25 and 35°C. The chemical shifts were referenced with respect to DSS (0.0 ppm). Resonance frequency assignments were determined using the 2D TOCSY and NOESY spectra recorded at 35°C (36). All peptide resonances expected to be observable except K1 Hε* and Hγ*, K11 Hε* and F12 Hζ, were assigned. The N-terminal group was not observed and it was not possible to make stereospecific assignments.
NOEs were quantified using peak height from the 2D NOESY spectrum with a mixing time of 150 ms, at 35°C. The NOEs were divided into three classes corresponding to strong, medium and weak intensities and assigned target distances of 1.8-2.5, 1.8-3.5 and 1.8-5 Å respectively (37). For structure calculations, hydrogen bond (Hi+4•••Oi = 2.5 Å and Ni+4•••Oi = 3.5 Å) and dihedral angle (φ= -60 ± 30° and φ= -40 ± 40°) restraints were defined for the segment where NOE patterns clearly indicated an α-helix (residues 3-16). Ambiguous NOEs were used when two to six possible inter-residues assignments existed for a cross peak. Fifty structures were calculated using the simulated annealing algorithm within CNS 1.1 (38). Analysis of the final structures was performed using the program Procheck-NMR (39; 40). Structures were visualized in MOLMOL (41) and Swiss-Pdb Viewer (42).
Paramagnetic probe experiments
Spectra were acquired on a Bruker Avance 600MHz. For paramagnetic line-broadening experiments, IL-8α was made up at a concentration of 1.5mM in 90% H2O, 10% D2O, 150mM SDS-d25, pH=5.0, 0.2mM DSS and 0.2mM sodium azide. A 0.4M stock solution of 16-DSA in deuterated methanol was prepared. It was then mixed with the protein/micelle sample to concentrations of 0.6mM, 1.2mM and 2.4mM. The maximal amount of deuterated methanol in the sample was 5μL for a total sample volume of 600μL. 15N-1H HSQC and TOCSY spectra were recorded at 35°C, before and after the addition of 16-DSA. The pH of the sample was adjusted after each addition of the 16-DSA.
DOSY experiments
In order to determine the hydrodynamic radius (RH) of IL-8α and SDS alone and in complex, diffusion experiments were performed. Three samples were prepared: 1) 1.5mM IL-8α in H2O/D2O, 2) 1.5mM IL-8α and 150mM SDS-d25 in H2O/D2O and 3) 150mM SDS in H2O/D2O. For all of the three samples, 0.4mM DSS and 0.4mM NaN3 were added and the pH was adjusted to 5.0. DOSY (Diffusion-ordered spectroscopy) experiments were acquired with pulsed field gradient (PFG) NMR (43) on a Bruker Avance 600MHz spectrometer, using stimulated echo with bipolar gradient pulses (44), followed by 3-9-19 pulse for water suppression (45). The spectra were collected in 32 steps, attenuating the signal to about 5% of the initial value by increasing the gradient strength from 2% to 95% of the maximum amplitude, for a constant diffusion time of 100ms and an optimized gradient pulse length of 1 ms.
The spectra were processed by Bruker Topspin 2.0. Diffusion constants were derived from the following equation:
where I is the observed intensity, I0 the unattenuated signal intensity, D the diffusion coefficient, γ the gyromagnetic ratio of 1H, g the gradient strength, δ the gradient length and Δ is the diffusion time.
Solid-state NMR
Sample preparation
The lipids and peptide were dissolved in a mix of CH3OH/CDCl3 (1/1 in volume). The lipids used were 4-10mg of POPC/POPC-d31, POPC-d31/POPC/POPG or POPC/POPG/POPG-d31, with a molar ratio of 3/1 of POPC/POPG and 30% of the sample (by weight) was deuterated. The solution (a total volume of 200μL) was spread homogeneously on 12 mica plates. They were then dried for approximately 2 hrs in a fume hood before being placed in a vacuum chamber overnight. Each plate was then spread with 5μL of double distilled water and put in hydration chamber with a saturated ammonia phosphate solution at 4°C for 2 days. The plates were then stacked together, wrapped with plastic film and sealed by polystyrene plastic. Samples were stored at 4°C before the NMR experiments.
NMR
The spectra were acquired on a Bruker Avance solid state NMR spectrometer operated at frequencies of 243.02 MHz, 92.15MHz and 60.83 MHz for nuclei 31P, 2H and 15N respectively. The oriented samples were positioned with the normal to the bilayer parallel to the magnetic field. For 31P and 15N experiments, the sample was inserted into a Bruker dual tuned 4mm flat coil probe and for the 2H experiments a Bruker triple tuned 3mm flat-coil probe was used. For 31P experiments, a proton-decoupled experiment was carried out with a 90° pulse length of 12μs. Other acquisition parameters were: recycle delay of 5s; SPINAL-64 proton decoupling at strength 65kHz; spectral width of 200ppm; and a total of 2888 scans. The chemical shift was externally referenced to 85% H3PO4 as 0 ppm. For 15N experiments, a cross polarization (CP) pulse sequence was used, with TPPM proton decoupling (46). Using 1024 data points, 65498 scans were acquired with a CP contact time of 6ms and a recycle delay of 5s. For 2H experiments, a quadrupole echo pulse sequence (π/2-τ-7π/2-τ-acq) was used with π/2 and τ set at 2.5μs and 15μs respectively. 10240 scans were acquired and the recycle delay was 1s. The spectral width was 200kHz. The experiments were performed at 20 °C.
Order parameter calculation
The deuterium spectra enabled us to calculate the order parameters for the carbon acyl chains. The peaks in these spectra were assigned with the appropriate carbon positions of the (sn1) acyl chain. The center pair is attributed to the deuterons on the methyl group, which have the more flexible motion. Then, the following pair is contributed by deuterons next to the methyl group (C15) and so on for deuterons on C14, C13, C12 and C11, which have larger splittings due to their position closer to the headgroup and consequent more constrained motion. The order parameter changes slowly along the chain, which is characterized by an overlap of the doublets on the 2H NMR spectrum. The dependence of orientational order on position can be estimated by the smoothed order parameter profile approach (47). In this unresolved region of the spectrum, called the orientational order parameter plateau, the peak positions are approximated by dividing the spectrum corresponding area into 9 equal parts and assuming that the quadrupole splitting changes monotonically with position along the chain. The order parameters can then be calculated using the following formula
where ΔνCDi is the quadrupole splitting of deuterons at the ith carbon position of the acyl chain, νCD is the quadrupole coupling constant of deuterium (167kHz in deuterated paraffin hydrocarbons (48)) and SCDi is the order parameter.
Fluorescence experiments
Small unilamellar vesicles (SUVs) were prepared by mixing the lipids in chloroform (10mg/mL), and drying off the solvent under a stream of nitrogen gas and then under vacuum overnight. The samples were then resuspended in H2O to create a 0.1M lipid stock solution. The lipid suspension was freeze-thawed five times before being sonicated in a water-bath sonicator until the solution became opalescent.
Steady-state fluorescence spectra were obtained using a Shimadzu RF-540 spectrofluorometer. Measurements were performed with an excitation wavelength of 295 nm to avoid the fluorescence of other aromatic residues and to reduce absorbance by acrylamide. The emission wavelength was scanned from 300 to 450 nm with 0.5 nm increments, in a 3 by 10mm quartz cell at room temperature. Both the excitation and emission slit widths were set to 5 nm. Spectrum baselines were corrected by subtracting the blank spectra of the detergent or lipid solution without peptide. The concentration of peptide was 2μM and experiments were performed in the presence or absence of 25mM SDS or 5 mM lipid vesicles. Quenching experiments were accomplished by adding increasing concentrations of acrylamide (from 10 to 200mM). The emission was monitored at the peak obtained from the spectra in the absence of quencher. The effect of acrylamide on the peptide was analyzed using the Stern-Volmer equation (49)
where F0 and F are the fluorescence intensity in the absence and presence of the quencher, respectively, KSV is the Stern-Volmer quenching constant and Q is the quencher concentration.
Results
Structure of IL-8α in aqueous solution
Circular dichroism experiments were run for IL-8α in aqueous solution. Overall, the CD spectra were characteristic of random coil conformation (data not shown), but an inflection point near 222nm suggested a propensity to structure as α-helix (50). This α-helical propensity was further substantiated by the NMR experiments. The 1D proton and 2D 15N-1H HSQC spectra (not shown) displayed more dispersion of the peptide amide proton chemical shifts than expected for a peptide in random coil conformation. In the NOESY spectrum, we observed strong HN-HN correlations, which are indicative of α-helical structure.
Structure of IL-8α in SDS micelles
The 3D structure of IL-8α was determined in SDS micelles using solution NMR. 1D proton and 15N-1H HSQC spectra were acquired at 25, 30 and 35°C. 35°C was chosen for the structural analysis as the peptide showed the least conformational inhomogeneity at that temperature. 2D-TOCSY and 2D-NOESY experiments were thus recorded at 35°C. The strong HNi/HNi+1 and the HNi/HNi+2 NOEs, added to the various Hαi/HN1+3 and Hαi/Hβ1+3 correlations indicated an α-helical conformation for residues 3 to 16. This was supported by the chemical shift index (CSI) values, the difference between the Hα proton chemical shifts in SDS environment and the chemical shifts in a random coil (51) (Fig. 1). The CSI values indicate a helical structure for residues 3 to 17 with a weakening of the helix toward the C-terminal end.
Figure 1.
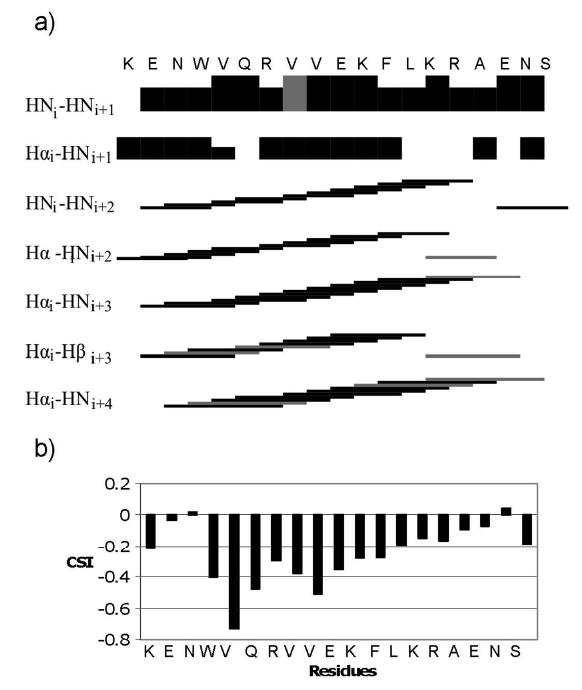
Secondary structure indicators for IL-8α. a) Black bars indicate unambiguously assigned NOEs and gray bars represent ambiguous NOEs. For sequential NOEs, the height of the bars corresponds to the intensity of the NOE. b) Chemical shift indices (CSI) for IL-8α (51).
Structure calculations were performed using the NOE-derived distances. α-helical hydrogen bond and dihedral angle restraints were also added for residues 3 to 16. An ensemble of 50 structures were calculated using 86 sequential, 113 medium range and 42 ambiguous NOEs. Of this ensemble, the 10 lowest energy structures were retained for analysis. An average of ∼12 restraints per residue allowed for good definition of the structure, with a backbone RMSD (Root Mean Square Deviation) of 0.89Å. Ninety-two percent of the residues fell within the most favored region of the Ramachandran plot, while no residues were found in the disallowed region (Table 1). The calculated structure displays a well-defined α-helix for residues 3 to 16 (Fig. 2). The N- and C-terminal residues are more flexible due to their position at the end of the peptide. The sidechains orient in a way that makes the helix amphipathic.
Table 1.
NMR structure calculation parameters
| Restraints used in structure calculation | |
|---|---|
| Total number of NOE restraints | 242 |
| Sequential [(i-j)=1] | 86 |
| Medium-range (|i-j|≤4) | 114 |
| Ambiguous | 42 |
| Hydrogen-bond restraints | 24 |
| Dihedral angle restraints | 31 |
| CNS energy (kcal/mol) for the ensemble of 10 lowest energy structures | |
| Eoverall | 58.27 ± 7.9 |
| Ebond | 2.75 ± 0.5 |
| Eangle | 25.92 ± 3.37 |
| Eimproper | 4.22 ± 1.42 |
| Evdw | 14.24 ± 1.86 |
| Enoe | 11.06 ± 1.51 |
| Ecdih | 0.07 ± 0.15 |
| there were no violations >0.4 Å | |
| Ramachandran plot summary (%) | |
| Most favoured | 91.8 |
| Additionally allowed | 3.5 |
| Generously allowed | 4.7 |
| Disallowed | 0 |
| Average RMSD from mean structure (Å) | |
| Backbone atoms | 0.899 |
| All heavy atoms | 1.416 |
The default parameters and force constants of protein-allhdg.param and anneal.inp in CNS 1.1 were used for calculation.
Figure 2.
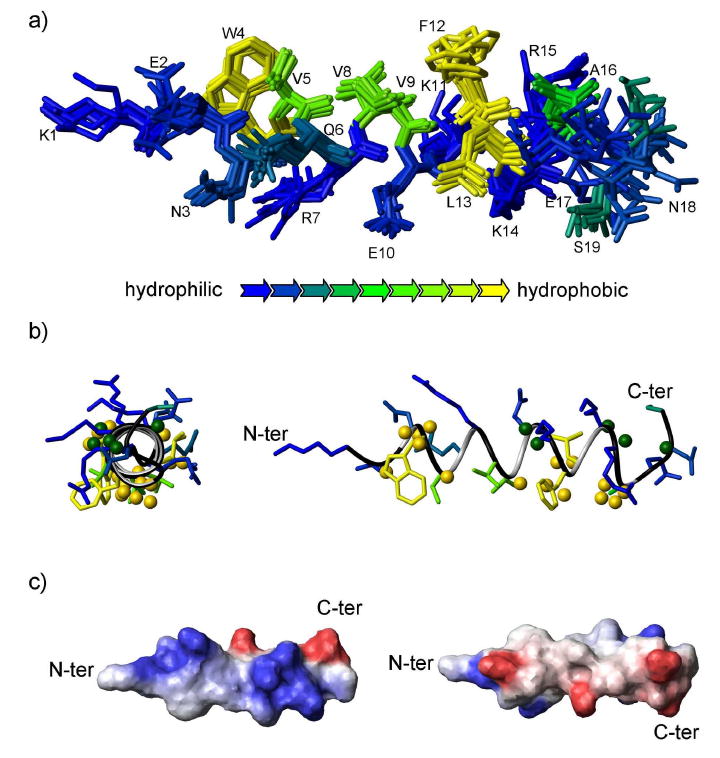
Structure of IL-8α. a) Ensemble of the ten lowest energy structures of IL-8α. The structures were superimposed using the backbone atoms from residues 3 to 16. The amino acids are coloured according to hydrophobicity (see key for color scheme). b) Ribbon representation of the structure of IL-8α with the lowest energy (sideview on the left). Spheres represent the results of the paramagnetic probe experiments: Hα and Hβ atoms located closer to the micelle interior are represented as yellow spheres, whereas Hα and Hβ atoms located closer to the micelle exterior are represented as green spheres. c) Electrostatic potential surface representation of the two opposite faces of the IL-8α representative structure with blue and red for positive and negative charge, respectively, and white for non-polar.
Peptide-micelle interactions
To further probe the IL-8α/SDS micelle interaction, diffusion NMR experiments were performed to determine the translational diffusion coefficient of the SDS micelles and IL-8α peptide alone and in complex (see supporting data). When alone in solution, the values were 0.98×10-10m2/s and 3.23×10-10m2/s for SDS micelles and IL-8α, respectively. The results for SDS alone in solution are consistent with the known size of SDS micelles (52; 53). The diffusion constant for the complex was found to be 2.46×10-10m2/s as measured using the IL-8α peaks. The reason the complex appears to diffuse faster than an SDS micelle alone may be a result of fast exchange of the peptide between bound and unbound states, making the peptide's observed diffusion constant a weighted average of the bound and unbound diffusion constants (54; 55).
To assess the position of IL-8α within the SDS micelles, 16-DSA was added to the peptide/micelle sample. This paramagnetic probe incorporates into the micelles with its free radical group positioned close to the center of the micelle, inducing a broadening and thus a height reduction of signals for the protons that are close to the micelle center (56; 57). A linewidth broadening was observed for the entire spectrum of the peptide after 16-DSA addition, as expected for a peptide bound close to the surface of micelles. The intensity reduction of Hα-HN and Hβ-HN correlations was monitored in TOCSY spectra and compared to the average intensity reduction (Fig. 3). The data for Hα-HN from K1, Q6, V8 and E17 and for Hβ-HN from K1, Q6, R7, V8, E10, K14, E17 and S19 were not retained in the analysis because they exhibited signal overlap. The protons experiencing a reduction in signal intensity close to the average were not plotted on the structure. The remaining residues were grouped as either exhibiting a larger or a smaller reduction in intensity than the average value. The former are thus located facing inside the micelles while the latter face the exterior of the micelle. This information was mapped on the structure displayed in Fig. 2b. Most of the residues closest to the DSA probe localize to one face of the helix, consistent with IL-8α positioning roughly parallel to the micelle-water interface. However, the N-terminal residues were more affected by the 16-DSA than the C-terminal residues. Thus, it seems that the peptide inserts itself in the SDS micelle with a slight angle, leaving the C-terminus and the exterior face of the C-terminal end of the helix outside the micelle.
Figure 3.
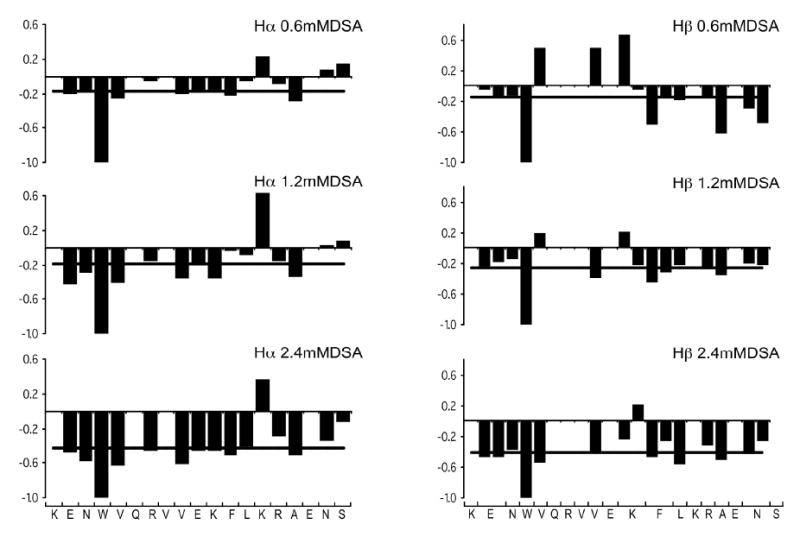
Effect of 16-DSA on IL-8α. The ratio of 2D-TOCSY crosspeak intensity with and without 16-DSA is plotted along the amino acid sequence at 0.6mM 16-DSA (top), 1.2mM 16-DSA (middle), 2.4mM 16-DSA (bottom), together with the average values at each 16-DSA concentration, represented as a black line, for Hα protons on the left and Hβ protons on the right.
Tryptophan fluorescence quenching in micelles and vesicles
The aromatic ring of the tryptophan at position 4 is located on the hydrophobic face of IL-8α, but near the juncture between the hydrophobic and hydrophilic faces (Fig. 2b) and is thus likely to position at the interfacial region between the micelle and the water. Tryptophan fluorescence experiments were carried out to characterize the interaction of this residue with micelles, as well as bilayers.
Emission spectra of IL-8α were recorded in the presence and absence of detergents or lipids. In pure H2O, the maximum fluorescence emission was observed at 350nm. When POPC SUVs were added, only a small intensity increase was observed and the maximum emission remained close to 350nm (data not shown). This indicates the tryptophan remains in a polar environment when the peptide is bound to POPC SUVs (58). When SDS micelles or POPC/POPG SUVs were added, the fluorescence intensity increased markedly and we observed a blue-shift of the tryptophan fluorescence (maximum peak at 336nm in SDS and 332nm in POPC/POPG) (59). This indicates a more hydrophobic environment surrounding the tryptophan in SDS micelles and POPC/POPG SUVs as compared to pure POPC SUVs, consistent with a deeper insertion of the tryptophan into the anionic micelles and SUVs.
To get further insight into the peptide insertion, we added increasing concentrations of acrylamide to the samples and measured the reduction in tryptophan fluorescence intensity. Acrylamide is a polar and uncharged compound, and thus it remains in solution and does not extensively affect the fluorescence of tryptophan if it is buried in the lipid phase. Figure 4 shows the Stern-Volmer plots of fluorescence quenching and the Stern-Volmer constants calculated from these plots. The tryptophan was readily quenched in pure water, indicating that it is easily accessible in the solution. When the peptide was in complex with SDS detergent micelles or lipid SUVs, the tryptophan fluorescence was less affected by the acrylamide addition. KSV values, which indicate the extent of Trp's exposure to solution, were calculated for IL-8α in the various environments. The KSV values calculated for IL-8α is in the presence of SDS micelles and POPC/POPG SUVs are significantly lower compared to the KSV in the presence of POPC SUVs. In the former conditions, the tryptophan is more protected from the acrylamide quenching than in the latter conditions. These results show that the tryptophan inserts deeply into anionic micelles or lipid SUVs, whereas in the POPC SUVs, it remains either more accessible to the solvent or in a very hydrophilic environment. The KSVs are almost the same in SDS micelles and in POPC/POPG SUVs, indicating that the tryptophan is equally protected in both environments.
Figure 4.
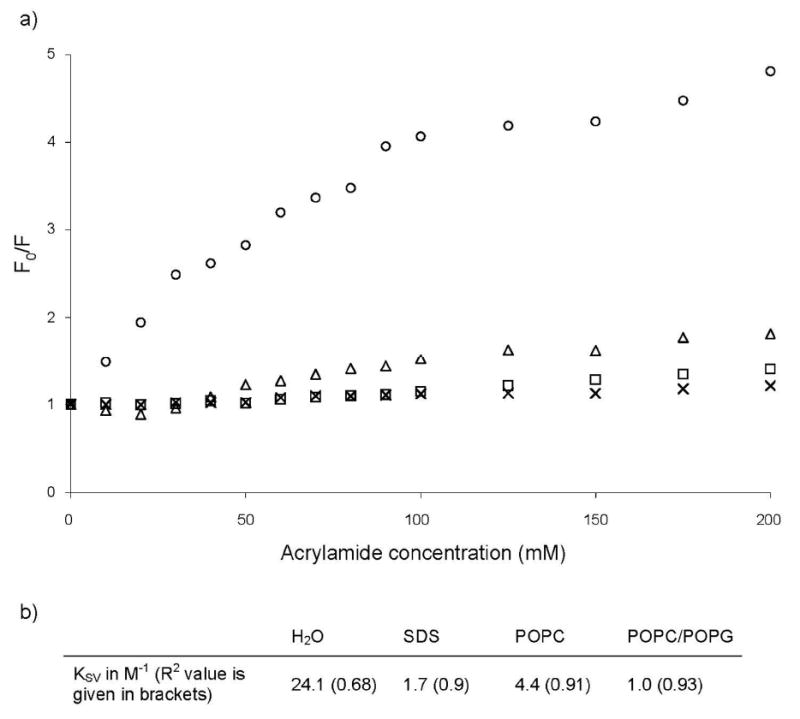
Tryptophan fluorescence quenching by acrylamide. Stern-Volner plots (a) and constants (b) of IL-8α for acrylamide quenching experiments in pure H2O (○) and bound to SDS micelles (⌈), POPC SUVs (Δ) and POPC/POPG SUVs (×).
Solid-state NMR of IL-8α in oriented lipid bilayers
Interactions with membranes are critical aspects of AMP function, whether they act by disrupting membranes or merely pass through membranes on their way to an intracellular target. Hence, we used solid state NMR to assess the impact of IL-8α on lipid bilayers composed of POPC, to mimic mammalian membranes, or POPC/POPG, to mimic bacterial membranes. The effects of the peptide on lipid bilayers were examined by acquiring 31P and 2H NMR spectra of oriented lipids without peptide and with 3% and 6% (by mole) of peptide.
Firstly, to study the interaction of IL-8α with the lipid headgroups, as well as the effect of the peptide on overall bilayer orientation, 31P experiments were performed. For lipids alone, the major peaks were observed at 36.4 and 39 ppm for the POPC and POPC/POPG samples, respectively, with minimal signal from 0 to -12ppm (Fig. 5), indicating that in the absence of peptide, the lipid bilayers were well aligned. Addition of 3 or 6 mol% peptide to the POPC bilayer did not appear to increase the ratio of unaligned species, indicating that the peptide did not perturb the bilayer alignment. There was a small decrease in the chemical shift of the major 31P peak with peptide addition. 31P MAS experiments indicated no peptide-induced change in isotropic chemical shift or linewidth, which would suggest a change in headgroup dynamics (Supplementary Figure 2). The MAS data imply that the source of the small shift seen in the oriented, static spectra is likely a change in headgroup tilt, which is consistent with a peripheral association of the peptide with the bilayers such that headgroup tilt is affected without modification of bilayer alignment. When peptide was added to the POPC/POPG bilayers, some increase in unaligned lipid species was seen in most samples, however the peptide-induced perturbation of the bilayer alignment was minimal, even at relatively high peptide concentrations.
Figure 5.
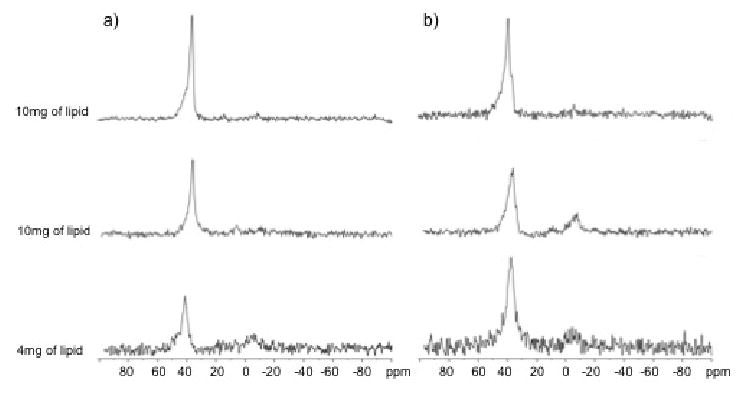
31P spectra of mechanically oriented bilayers of (a) POPC/POPC-d31=7/3, w/w and (b) POPC-d31/POPG=3/1, mol/mol in the absence (at the top) and in the presence of 3 mol% (in the middle) and 6 mol% (at the bottom) of IL-8α. The vertical scale of each spectrum was corrected according to the relative amount of lipids that were used to prepare the sample. The bilayers were oriented with the bilayer normal along the magnetic field. Spectra of POPC/POPG-d31 were also acquired but are not shown since they appear very similar to those of POPC-d31/POPG.
Deuterium NMR was employed to study the effects of IL-8α peptide on the lipid chains. The 2H spectra of POPC-d31, POPC-d31/POPG and POPC/POPG-d31 are shown in Fig. 6 in the presence and absence of IL-8α. In the absence of peptide, a symmetric spectrum with well-resolved peaks characteristic of well-aligned bilayers was obtained. When IL-8α was included in the sample, the amount of rapidly reorienting material remained very small, as indicated by the absence of strong signals at the center of the spectrum. The peaks broaden slightly as the concentration of IL-8α increases. In POPC, the inclusion of 3 mol% peptide induces a reduction in the quadrupole splitting, indicating a less restricted motion of lipid acyl chains in the presence of IL-8α. When 6mol% of peptide was added, the spectral resolution decreases dramatically (data not shown), precluding order parameter calculation. In POPC/POPG samples, IL-8α significantly reduces the quadrupolar coupling only at the highest concentration (6 mol%).
Figure 6.
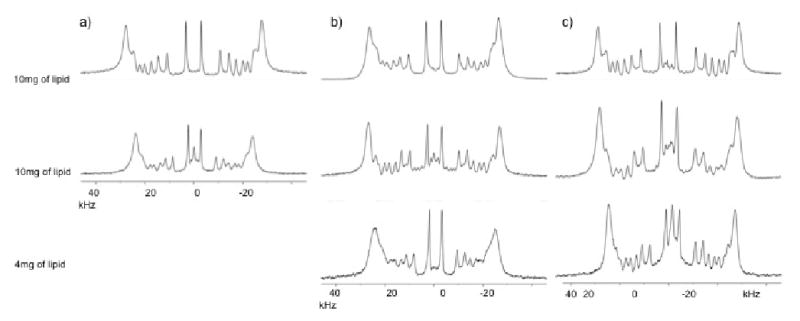
2H spectra of mechanically oriented bilayers composed of (a) POPC/POPC-d31=7/3, w/w, (b) POPC-d31/POPG=3/1, mol/mol and (c) POPC/POPG-d31=3/1, mol/mol in the absence (at the top) and in the presence of 3% (in the middle) and 6% (at the bottom) of IL-8α. The vertical scale of each spectrum has been corrected according to the relative amount of lipids that were used to prepare the sample. The bilayers were oriented with the bilayer normal along the magnetic field.
To ascertain the effect of the peptide along the lipid chain, order parameter profiles were calculated from the quadrupolar splittings in the 2H spectra (Fig. 7). In POPC, the addition of IL-8α resulted in a decrease in the order parameters for the entire lipid chain length. For the POPC/POPG mix, the degree of order did not change when 3% of IL-8α was added, using either POPC or POPG as the deuterium labeled species. In POPC/POPG, a slight decrease of the order parameters could be observed once 6% of IL-8α was included.
Figure 7.
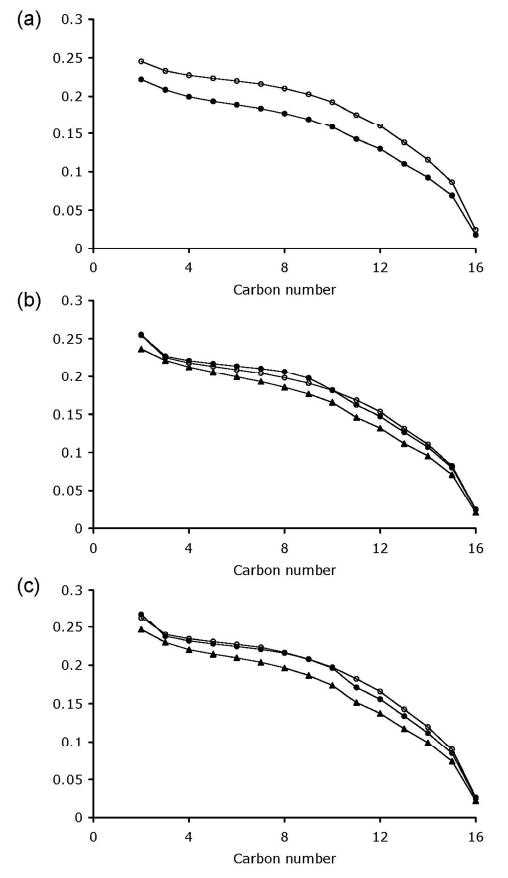
Order parameters profile of the sn1 chains of POPC-d31 in POPC/POPC-d31 samples (a), of POPC-d31 in POPC-d31/POPG samples (b) and of POPG-d31 in POPC/POPG-d31 samples (c), in the absence (empty circles) and in the presence of 3% (filled circles) and 6% (triangles) of IL-8α. Data derived from Figure 6.
As there are 4 residues enriched with 15N in the peptide (V5, V8, L13 and A16), it was possible to run static 15N cross polarization spectra of IL-8α in oriented bilayers (Fig. 8). This experiment gives the anisotropic chemical shits for these atoms, which depend on the orientation of the helical axis with respect to the bilayer normal. Chemical shifts <100ppm correspond to an in-plane alignment while chemical shifts in the 200ppm region indicate a transmembrane orientation (60). In the 15N spectrum of the sample with 10mg POPC-d31/POPG and 3% IL-8α there are three peaks in the ∼60-70 ppm range and a fourth close to 120 ppm. It may be that the 120 ppm peak corresponds to A16 since this is the terminal residue of the helix and thus may deviate more from the canonical angles than the other three residues that are in the more regular part of the helix. In any case, that most of the 15N signal is less than 100 ppm indicates a predominantly in-plane alignment for the helix.
Figure 8.
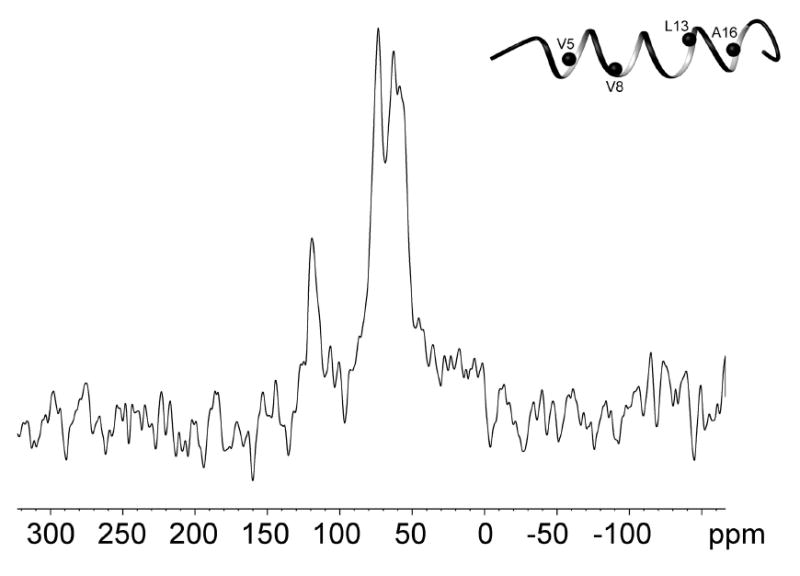
15N spectrum of mechanically oriented 3:1 POPC-d31/POPG, with 3% (mol) of IL-8α. The inset shows a ribbon representation of the representative structure of SDS-bound structure of IL-8α with 15N atoms as black spheres.
Discussion
Compared to most antimicrobial peptides found in the Antimicrobial Peptide Database, IL-8α has a rather modest positive charge (+2 as compared to the average of +4.4) and hydrophobic content (36% as compared to the average of 48%) (61). These distinctive physicochemical attributes provide a special context in which to contemplate the molecular mechanisms of this peptide, which has been shown to exert an antibacterial activity against both Gram-negative and Gram-positive bacteria (14; 28). We started by assessing the structure of IL-8α in water and in complex with SDS micelles that approximate prokaryotic membranes. In pure water, circular dichroism and NMR spectra indicated that IL-8α is largely random coil in conformation, but with an apparent propensity for α-helical conformation. Upon interaction with SDS micelles, IL-8α assumes a well-defined, highly amphipathic α-helix spanning residues 3 to 16 (Fig. 2). These results are in line with previous studies of antimicrobial peptides that exhibit disorder in aqueous solution but structure upon binding to detergent micelles (33; 62; 63). Comparison of the structure of IL-8α in SDS micelles with the structure of this fragment within the context of the entire IL-8 protein (5-7) demonstrates that this region structures similarly in both cases, with only small differences in sidechain orientation.
It was also of interest to investigate the spatial orientation through which IL-8α interacts with target lipid systems. The paramagnetic probe data indicate that IL-8α orients with its helical axis roughly parallel to the SDS micelle/water interface, but with a slight angle that positions the N-terminal residues more deeply into the micelles than the C-terminal residues (Fig. 2b). This finding is consistent with the extended structure adopted by Lys1 and Arg7, which may allow the positively charged termini of their side chains to position close to the micelle surface, while their backbone and aliphatic chain atoms remain buried within the micelle. This so-called “snorkeling” phenomenon has been observed previously (64; 65) and is hypothesized to be important for the interaction between protein and lipids (66). Based on the position of IL-8α within the micelle, combined with the dispensation of the sidechains in the structure (Fig. 2b), it seems reasonable to speculate that the tryptophan at position 4 may be the entity responsible for drawing the N-terminus of the peptide further in towards the micelle interior.
Tryptophan itself is an amphipathic residue (67), having a sidechain with both polar and hydrophobic groups and thus a tendency to position at interfaces (68). Tryptophan residues have been shown to be critical for the activity of several antimicrobial peptides (69; 70), related to their role in anchoring peptides in membranes and stabilizing peptide structure in bilayers (71; 72). Some antimicrobial peptides are rich in tryptophan, a composition which enables them to disrupt bacterial membranes (73; 74). In bovine lactoferricin, the substitution of Trp6 or Trp8 by alanine leads to a dramatic decrease of antibacterial activity (75). However, even if tryptophan facilitates membrane insertion, it is likely that other processes (e.g. electrophoretic) contribute to transmigration of such peptides across the membrane if they are to reach intracellular targets (8).
To gain insight into the role of tryptophan in IL-8α/target interactions, we performed intrinsic tryptophan fluorescence experiments in the presence of detergents and lipids. This approach was additionally helpful in providing one measure of the peptide position in both micelles and lipid bilayers, thus directly linking the solution and solid-state NMR work. The peptide penetrated to a very similar extent all the anionic membrane mimetics, i.e. the SDS micelles and POPC/POPG SUVs, with the tryptophan well buried and inaccessible to the solvent. In contrast, the tryptophan residue penetrated less deeply into the zwitterionic POPC SUVs, remaining in a polar environment. These differences in penetration with model membrane composition are in keeping the apparent preferential specificity of IL-8α for bacterial over mammalian membranes.
In addition to the tryptophan residue, IL-8α has one additional aromatic sidechain, a phenylalanine at position 12. Like tryptophan, phenylalanine residues are known to be key residues in antimicrobial activity of some peptides. For example, the phenylalanine location is critical for antibacterial activity in aurein 1.2 analogs (76). In tachyplesin I derivatives, the replacement of cysteines by phenylalanines leads to an increase of antimicrobial activity against fungi and E coli (77). In the Pis-1 PG Piscidin 1 analogue, the insertion of phenylalanines in the membrane surface contributes to the anchoring of the peptide to the cell membrane (78). Likewise, in the IL-8α/SDS micelle complex, the phenylalanine 12 sidechain orients itself in the same direction as the tryptophan sidechain, and is thus in a key position for strong interactions with the lipid acyl chains of bacterial cell membranes. Given the position of these two aromatic sidechains within the peptide-micelle complex, (i.e. facing the interior of the micelle and at the end of the helix that is tilted more deeply into the micelle), it seems likely that these two residues may compensate in-part for the reduced hydrophobicity and charge of IL-8α as compared to the average values for AMPs.
The current solution NMR studies provided detailed information on the peptide structure and its interactions with SDS micelles. Moreover, the tryptophan fluorescence experiments showed that the tryptophan residue adopts a similar position in both SDS micelles and POPC/POPG SUVs. Following from these observations, it was of interest to characterize in more detail the interaction of IL-8α peptide with lipid bilayers, which are more biologically relevant than micelles. Two model membranes were used. The first one consisted of POPC alone and was studied because this zwitterionic lipid is abundant in mammalian cell membranes. The second model membrane consisted of POPC/POPG at a 3:1 molar ratio, as POPG is an anionic lipid commonly found in bacterial plasma membranes, and the POPC/POPG mix has a membrane-charge density similar to that of bacterial cytoplasmic membranes (79). The orientation of an antimicrobial peptide within the bilayer offers further insights into potential mechanisms of peptide membrane interaction and disruption that may contribute to AMP efficacy. Thus the 15N NMR spectrum of IL-8α was determined using mechanically oriented POPC/POPG bilayers. Most of the intensity in the 15N spectrum was between 50 and 90 ppm (Fig. 8), consistent with a largely in-plane orientation of the peptide within the bilayer, with a tilt angle in the realm of 0-15° (80). This orientation is consistent with the position of the peptide in SDS micelles, which was also found to be largely in-plane, but with a slight tilt of the N-terminus towards the micelle centre. Such a parallel orientation of IL-8α tends to rule out the toroid pore and barrel stave mechanisms of membrane disruption (81; 82), since these are thought to require a transmembrane orientation. On the contrary, if membrane disruption is a significant mechanistic determinant of IL-8α antimicrobial activity, the present findings point more towards carpet mechanism (83) or perhaps one of the mechanisms, such as the sinking raft, that have been proposed to involve only transient formation of distortions (84). However, it is possible that IL-8α and similar peptides execute one or more antimicrobial effects through mechanisms that do not require significant initial or direct membrane perturbation (Yeaman et al., unpublished data).
As a complement to the above studies, which focused on peptide structure and orientation, 2H and 31P solid-state NMR studies were used to assess the impact of IL-8α on the lipids in the oriented bilayers. The order parameter profiles derived from the 2H spectra indicated a more peripheral interaction of the peptide with zwitterionic POPC bilayers and a deeper penetration into the anionic containing POPC/POPG bilayers. The addition of 3 mol% IL-8α to POPC-only bilayers lead to a decrease in order parameters over the entire lipid chain length, consistent with a peripheral interaction of the peptide that increases the spacing between lipid head groups and thus increases the motional freedom of the chains (85; 86). These results are consistent with the fluorescence experiments, which show an only peripheral interaction of IL-8α with POPC. Similar observations have been made for the antimicrobial peptide KIGAKI (87), in which fluorescence was not affected by POPC addition although there were decreases in the lipid order (88). On the other hand, the 2H order parameter profiles for POPC/POPG in the presence of 0 and 3% of IL-8α were very similar, suggesting that the motions of the lipid acyl chains remain the same in the absence or the presence of the peptide. This type of behavior has been interpreted as indicative of a relatively deep location of the peptide in the lipid bilayer, where the peptide would constrain the motion of the lipid chains more than if it were located more peripherally within the bilayers (87; 89; 90). At 6 mol% IL-8α, the POPC/POPG 2H order parameter profile is affected, perhaps indicating the acyl chain position might be saturated and the peptide might thus start to interact more peripherally with the lipid headgroups. The differences in IL-8α's interactions with the two model membranes are consistent with this peptide's putative preferential targeting of bacterial cells as compared to mammalian cells, as well as the tryptophan fluorescence results.
In neither POPC nor POPC/POPG with up to 6% peptide was there any evidence for non-lamellar phase structures, such as cubic or hexagonal phases (Fig. 5), or fragmentation of the bilayers to rapidly tumbling species such as micelles (Fig. 6). The 2H spectra (Fig. 6) also provided no evidence for the formation of toroidal pores, which would give rise to a superposed spectrum at half the splitting observed, or significant membrane thinning, which could result in distortions to the observed spectra (91). These observations argue for a carpet-like mechanism of membrane disruption, rather than a pore or detergent type disruption. In fact, overall bilayer alignment was not significantly perturbed by the presence of IL-8α, as judged by the 2H or 31P NMR spectra (Figs. 5 & 6). POPC bilayer alignment was not perturbed at all, even at 6 mol% peptide and POPC/POPG bilayers were only very slightly perturbed at this peptide concentration. Additionally, the position of the 31P peak did not shift significantly in the presence of peptide, indicating that IL-8α had no major effects on lipid head group tilt angle. Six mol% is quite a high peptide:lipid ratio and comparable to the higher concentrations employed in many studies of AMPs that use oriented solid state NMR, but since the antimicrobial assays with IL-8α (28) were performed in the solid phase, it is not possible to directly compare the peptide/lipid ratios between the functional and NMR studies, and thus we do not know for certain that 6 mol% is a “functional” concentration.
There are at least three potential reasons why an antimicrobial peptide might not perturb model lipid bilayers, as probed by solid-state NMR studies of oriented bilayers. Firstly, its mechanism may involve little bilayer disruption, either because it only perturbs the membrane enough to cross it and gain access to an intracellular target, or its membrane-disrupting mechanism does not involve large-scale disruptions (92-95). Secondly, it is likely that at least some antimicrobial peptides act via mechanisms that involve only superficial interactions that trigger intracellular responses leading to target cell inhibition or death (8; 96). Thirdly, the membrane-disrupting activities of an AMP may be highly dependent on membrane composition and thus not apparent in all model lipid systems. For example, Cheng et al (97) found very modest effects with three aurein peptides on 3:1 POPC/POPG bilayers, versus much larger effects on bilayer disruption with DMPC/DMPG bilayers, which are thinner. Similarly, a Pardaxin peptide did not greatly disrupt POPC/POPG bilayers, but significantly perturbed a number of other model lipid bilayers (98). Finally, a peptide based on granulysin, termed G15, was shown to have little effect on 3:1 POPC/POPG, but much more of an effect on bilayers made from E. coli lipid extract (99). This latter study is of particular interest in comparison to the present findings, as the G15 peptide has a very similar charge and hydrophobicity to IL-8α and, like IL-8α, contains a Trp and Phe residue positioned two helix-turns apart from each other. Similarly to IL-8α, G15 did not promote the formation of non-lamellar phase structures or rapidly tumbling lipid structures, such as micelles. Hence, it is possible that IL-8α and G15 share a similar molecular mechanism of action that derives from their characteristic modest charge and hydrophobicity, as well as their shared Trp/Phe motif.
On balance, it is important to recognize the limitations of the observations reported herein. Studies employing model membranes like those presented in the current work provide a necessary first stage to understanding mechanisms of antimicrobial peptides such as IL-8α. However, further work performed in the context of even more physiologically relevant systems, such as actual microbial and host cell membranes, is needed to more thoroughly elucidate the biological mechanism(s) of antimicrobial action of IL-8α or any AMP.
Supplementary Material
Supplementary Figure 1: Curve fitting for DOSY experiments.
Supplementary Figure 2: 31P MAS spectra of POPC in the absence and presence of 3 mol% IL-8α.
Acknowledgments
We thank Stéphan Dubrau for his help in preparing Figure 1, Michael Hayley for suggesting the tryptophan fluorescence experiments, and Celine Schneider for NMR help, especially with the MAS experiments.
Funding: This work was supported by an NSERC grant to V.B. and by NIH / NIAID research grants 5RO1AI39001 to M.R.Y and A.J.W., and 5RO1AI48031 to M.R.Y.
Abbreviations
- AMP
antimicrobial peptide
- CFU
colony forming unit
- CP
cross polarization
- CSI
chemical shift index
- 16-DSA
16-Doxyl-stearic acid
- DOSY
Diffusion-ordered spectroscopy
- DSS
0.2mM 2,2-dimethyl-2-silapentane-5-sulfonate (sodium salt)
- PFG
pulsed field gradient
- POPC
Palmitoyl-2-Oleoyl-sn-Glycero-3-Phosphocholine
- POPE
phosphatidyl ethanolamine
- POPG
1-Palmitoyl-2-Oleoyl-sn-Glycero-3-[Phospho-rac-(1-glycerol)] (Sodium Salt)
- SUV
small unilamellar vesicle
Footnotes
The chemical shifts for IL-8α in SDS micelles can be found on the BMRB database, ID:20044.
Supporting Information is available
This material is available free of charge via the Internet at http://pubs.acs.orgmonths after publication.
Transparency Statement: M.R.Y. is a shareholder of NovaDigm Therapeutics, Inc. He has received past research funding from Pfizer Inc., Amgen Inc., Cubist Pharmaceuticals, and Novozymes Pharmaceuticals. None of these entities provided support for the current studies.
References
- 1.Shai Y. Mode of action of membrane active antimicrobial peptides. Biopolymers. 2002;66:236–248. doi: 10.1002/bip.10260. [DOI] [PubMed] [Google Scholar]
- 2.Mookherjee N, Hancock RE. Cationic host defence peptides: innate immune regulatory peptides as a novel approach for treating infections. Cell Mol Life Sci. 2007;64:922–933. doi: 10.1007/s00018-007-6475-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cole AM, Ganz T, Liese AM, Burdick MD, Liu L, Strieter RM. Cutting edge: IFN-inducible ELR- CXC chemokines display defensin-like antimicrobial activity. J Immunol. 2001;167:623–627. doi: 10.4049/jimmunol.167.2.623. [DOI] [PubMed] [Google Scholar]
- 4.Durr M, Peschel A. Chemokines meet defensins: the merging concepts of chemoattractants and antimicrobial peptides in host defense. Infect Immun. 2002;70:6515–6517. doi: 10.1128/IAI.70.12.6515-6517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clore GM, Appella E, Yamada M, Matsushima K, Gronenborn AM. Three-dimensional structure of interleukin 8 in solution. Biochemistry. 1990;29:1689–1696. doi: 10.1021/bi00459a004. [DOI] [PubMed] [Google Scholar]
- 6.Rajarathnam K, Clark-Lewis I, Sykes BD. 1H NMR solution structure of an active monomeric interleukin-8. Biochemistry. 1995;34:12983–12990. doi: 10.1021/bi00040a008. [DOI] [PubMed] [Google Scholar]
- 7.Baldwin ET, Weber IT, St Charles R, Xuan JC, Appella E, Yamada M, Matsushima K, Edwards BF, Clore GM, Gronenborn AM, et al. Crystal structure of interleukin 8: symbiosis of NMR and crystallography. Proc Natl Acad Sci U S A. 1991;88:502–506. doi: 10.1073/pnas.88.2.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yeaman MR, Yount NY. Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev. 2003;55:27–55. doi: 10.1124/pr.55.1.2. [DOI] [PubMed] [Google Scholar]
- 9.De Smet K, Contreras R. Human antimicrobial peptides: defensins, cathelicidins and histatins. Biotechnol Lett. 2005;27:1337–1347. doi: 10.1007/s10529-005-0936-5. [DOI] [PubMed] [Google Scholar]
- 10.Pazgier M, Li X, Lu W, Lubkowski J. Human defensins: synthesis and structural properties. Curr Pharm Des. 2007;13:3096–3118. doi: 10.2174/138161207782110381. [DOI] [PubMed] [Google Scholar]
- 11.Tang YQ, Yeaman MR, Selsted ME. Purification, characterization, and antimicrobial properties of peptides released from thrombin-induced human platelets. Blood. 1995;86:910a. [Google Scholar]
- 12.Yeaman MR, Tang YQ, Shen AJ, Bayer AS, Selsted ME. Purification and in vitro activities of rabbit platelet microbicidal proteins. Infect Immun. 1997;65:1023–1031. doi: 10.1128/iai.65.3.1023-1031.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crawford MA, Zhu Y, Green CS, Burdick MD, Sanz P, Alem F, O'Brien AD, Mehrad B, Strieter RM, Hughes MA. Antimicrobial effects of interferon-inducible CXC chemokines against Bacillus anthracis spores and bacilli. Infect Immun. 2009;77:1664–1678. doi: 10.1128/IAI.01208-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yount NY, Waring AJ, Gank KD, Welch WH, Kupferwasser D, Yeaman MR. Structural correlates of antimicrobial efficacy in IL-8 and related human kinocidins. Biochim Biophys Acta. 2007;1768:598–608. doi: 10.1016/j.bbamem.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 15.Yang D, Chen Q, Hoover DM, Staley P, Tucker KD, Lubkowski J, Oppenheim JJ. Many chemokines including CCL20/MIP-3alpha display antimicrobial activity. J Leukoc Biol. 2003;74:448–455. doi: 10.1189/jlb.0103024. [DOI] [PubMed] [Google Scholar]
- 16.Hieshima K, Ohtani H, Shibano M, Izawa D, Nakayama T, Kawasaki Y, Shiba F, Shiota M, Katou F, Saito T, Yoshie O. CCL28 has dual roles in mucosal immunity as a chemokine with broad-spectrum antimicrobial activity. J Immunol. 2003;170:1452–1461. doi: 10.4049/jimmunol.170.3.1452. [DOI] [PubMed] [Google Scholar]
- 17.Linge HM, Collin M, Nordenfelt P, Morgelin M, Malmsten M, Egesten A. The human CXC chemokine granulocyte chemotactic protein 2 (GCP-2)/CXCL6 possesses membrane-disrupting properties and is antibacterial. Antimicrob Agents Chemother. 2008;52:2599–2607. doi: 10.1128/AAC.00028-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maerki C, Meuter S, Liebi M, Muhlemann K, Frederick MJ, Yawalkar N, Moser B, Wolf M. Potent and broad-spectrum antimicrobial activity of CXCL14 suggests an immediate role in skin infections. J Immunol. 2009;182:507–514. doi: 10.4049/jimmunol.182.1.507. [DOI] [PubMed] [Google Scholar]
- 19.Krijgsveld J, Zaat SA, Meeldijk J, van Veelen PA, Fang G, Poolman B, Brandt E, Ehlert JE, Kuijpers AJ, Engbers GH, Feijen J, Dankert J. Thrombocidins, microbicidal proteins from human blood platelets, are C-terminal deletion products of CXC chemokines. J Biol Chem. 2000;275:20374–20381. doi: 10.1074/jbc.275.27.20374. [DOI] [PubMed] [Google Scholar]
- 20.Martinez-Becerra F, Silva DA, Dominguez-Ramirez L, Mendoza-Hernandez G, Lopez-Vidal Y, Soldevila G, Garcia-Zepeda EA. Analysis of the antimicrobial activities of a chemokine-derived peptide (CDAP-4) on Pseudomonas aeruginosa. Biochem Biophys Res Commun. 2007;355:352–358. doi: 10.1016/j.bbrc.2007.01.188. [DOI] [PubMed] [Google Scholar]
- 21.Tang YQ, Yeaman MR, Selsted ME. Antimicrobial peptides from human platelets. Infect Immun. 2002;70:6524–6533. doi: 10.1128/IAI.70.12.6524-6533.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yeaman MR, Gank KD, Bayer AS, Brass EP. Synthetic peptides that exert antimicrobial activities in whole blood and blood-derived matrices. Antimicrob Agents Chemother. 2002;46:3883–3891. doi: 10.1128/AAC.46.12.3883-3891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yount NY, Gank KD, Xiong YQ, Bayer AS, Pender T, Welch WH, Yeaman MR. Platelet microbicidal protein 1: structural themes of a multifunctional antimicrobial peptide. Antimicrob Agents Chemother. 2004;48:4395–4404. doi: 10.1128/AAC.48.11.4395-4404.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yount NY, Andrés MT, Fierro JF, Yeaman MR. The gamma-core motif correlates with antimicrobial activity in cysteine-containing kaliocin-1 originating from transferrins. Biochim Biophys Acta. 2007;1768:2862–2872. doi: 10.1016/j.bbamem.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 25.Yeaman MR, Yount NY, Waring AJ, Gank KD, Kupferwasser D, Wiese R, Bayer AS, Welch WH. Modular determinants of antimicrobial activity in platelet factor-4 family kinocidins. Biochim Biophys Acta. 2007;1768:609–619. doi: 10.1016/j.bbamem.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiong YQ, Yeaman MR, Bayer AS. In vitro antibacterial activities of platelet microbicidal protein and neutrophil defensin against Staphylococcus aureus are influenced by antibiotics differing in mechanism of action. Antimicrob Agents Chemother. 1999;43:1111–1117. doi: 10.1128/aac.43.5.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yeaman MR, Bayer AS, Koo SP, Foss W, Sullam PM. Platelet microbicidal proteins and neutrophil defensin disrupt the Staphylococcus aureus cytoplasmic membrane by distinct mechanisms of action. J Clin Invest. 1998;101:178–187. doi: 10.1172/JCI562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Björstad A, Fu H, Karlsson A, Dahlgren C, Bylund J. Interleukin-8-derived peptide has antibacterial activity. Antimicrob Agents Chemother. 2005;49:3889–3895. doi: 10.1128/AAC.49.9.3889-3895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gallo RL, Murakami M, Ohtake T, Zaiou M. Biology and clinical relevance of naturally occurring antimicrobial peptides. J Allergy Clin Immunol. 2002;110:823–831. doi: 10.1067/mai.2002.129801. [DOI] [PubMed] [Google Scholar]
- 30.Yeaman MR, Yount NY. Code among chaos: Immunorelativity and the AEGIS model of antimicrobial peptides. Asm News. 2005;71 [Google Scholar]
- 31.Lee DG, Kim HK, Kim SA, Park Y, Park SC, Jang SH, Hahm KS. Fungicidal effect of indolicidin and its interaction with phospholipid membranes. Biochem Biophys Res Commun. 2003;305:305–310. doi: 10.1016/s0006-291x(03)00755-1. [DOI] [PubMed] [Google Scholar]
- 32.Lee IH, Cho Y, Lehrer RI. Effects of pH and salinity on the antimicrobial properties of clavanins. Infect Immun. 1997;65:2898–2903. doi: 10.1128/iai.65.7.2898-2903.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bourbigot S, Dodd E, Horwood C, Cumby N, Fardy L, Welch WH, Ramjan Z, Sharma S, Waring AJ, Yeaman MR, Booth V. Antimicrobial peptide RP-1 structure and interactions with anionic versus zwitterionic micelles. Biopolymers. 2009;91:1–13. doi: 10.1002/bip.21071. [DOI] [PubMed] [Google Scholar]
- 34.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 35.Goddard TD, Kneller DG. Sparky. Vol. 3 [Google Scholar]
- 36.Wuthrich K. NMR of Proteins and Nucleic Acids. Book 1986 [Google Scholar]
- 37.Clore GM, Nilges M, Sukumaran DK, Brunger AT, Karplus M, Gronenborn AM. The three-dimensional structure of alpha1-purothionin in solution: combined use of nuclear magnetic resonance, distance geometry and restrained molecular dynamics. Embo J. 1986;5:2729–2735. doi: 10.1002/j.1460-2075.1986.tb04557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 39.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 40.Morris AL, MacArthur MW, Hutchinson EG, Thornton JM. Stereochemical quality of protein structure coordinates. Proteins. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 41.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–5. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 42.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 43.Morris KF, Johnson CS., Jr Diffusion-ordered two-dimensional nuclear magnetic resonance spectroscopy. Journal of the American Chemical Society. 1992;114:3139–3141. [Google Scholar]
- 44.Tanner JE. Use of the Stimulated Echo in NMR Diffusion Studies. J Chem Phys. 1970;52:2523–2526. [Google Scholar]
- 45.Sklenar V, Piotto M, Leppik R, Saudek V. Gradient-Tailored Water Suppression for 1H-15N HSQC Experiments Optimized to Retain Full Sensitivity. Journal of Magnetic Resonance, Series A. 1993;102:241–245. [Google Scholar]
- 46.Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. Heteronuclear decoupling in rotating solids. J Chem Phys. 1995;103:6951–6958. [Google Scholar]
- 47.Sternin E, Fine B, Bloom M, Tilcock CP, Wong KF, Cullis PR. Acyl chain orientational order in the hexagonal HII phase of phospholipid-water dispersions. Biophysical Journal. 1988;54:689–694. doi: 10.1016/S0006-3495(88)83004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burnett LJ, Muller BH. Deuteron Quadrupole Coupling Constants in Three Solid Deuterated Paraffin Hydrocarbons: C2D6, C4D10, C6D14. J Chem Phys. 1971;55:5829–5831. [Google Scholar]
- 49.Eftink MR, Ghiron CA. Exposure of tryptophanyl residues in proteins. Quantitative determination by fluorescence quenching studies. Biochemistry. 1976;15:672–680. doi: 10.1021/bi00648a035. [DOI] [PubMed] [Google Scholar]
- 50.Kelly SM, Jess TJ, Price NC. How to study proteins by circular dichroism. Biochim Biophys Acta. 2005;1751:119–139. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 51.Wishart DS, Bigam CG, Holm A, Hodges RS, Sykes BD. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J Biomol NMR. 1995;5:67–81. doi: 10.1007/BF00227471. [DOI] [PubMed] [Google Scholar]
- 52.Hayter JB, Penfold J. Self-consistent structural and dynamic study of concentrated micelle solutions. Journal of the Chemical Society, Faraday Transactions 1. 1981;77:1851–1863. [Google Scholar]
- 53.Itri R, Amaral LQ. Distance distribution function of sodium dodecyl sulfate micelles by x-ray scattering. The Journal of Physical Chemistry. 1991;95:423–427. [Google Scholar]
- 54.Whitehead TL, Jones LM, Hicks RP. Effects of the incorporation of CHAPS into SDS micelles on neuropeptide-micelle binding: separation of the role of electrostatic interactions from hydrophobic interactions. Biopolymers. 2001;58:593–605. doi: 10.1002/1097-0282(200106)58:7<593::AID-BIP1033>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 55.Stilbs P. Fourier transform NMR pulsed-gradient spin-echo (FT-PGSE) self-diffusion measurements of solubilization equilibria in SDS solutions. J Colloid Interface Sci. 1982;87:385–394. [Google Scholar]
- 56.Papavoine CH, Konings RN, Hilbers CW, van de Ven FJ. Location of M13 coat protein in sodium dodecyl sulfate micelles as determined by NMR. Biochemistry. 1994;33:12990–12997. doi: 10.1021/bi00248a007. [DOI] [PubMed] [Google Scholar]
- 57.Zamoon J, Mascioni A, Thomas DD, Veglia G. NMR solution structure and topological orientation of monomeric phospholamban in dodecylphosphocholine micelles. Biophys J. 2003;85:2589–2598. doi: 10.1016/s0006-3495(03)74681-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Surewicz WK, Epand RM. Role of peptide structure in lipid-peptide interactions: a fluorescence study of the binding of pentagastrin-related pentapeptides to phospholipid vesicles. Biochemistry. 1984;23:6072–6077. doi: 10.1021/bi00320a026. [DOI] [PubMed] [Google Scholar]
- 59.Lakowicz JR, Masters BR. Principles of fluorescence spectroscopy. Journal of Biomedical Optics. 2008;13:029901. [Google Scholar]
- 60.Bechinger B, Sizun C. Alignment and structural analysis of membrane polypeptides by 15 N and 31 P solid-state NMR spectroscopy. Concepts in Magnetic Resonance. 2003;18:130–145. [Google Scholar]
- 61.Wang G, Li X, Wang Z. APD2: the updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 2009;37:D933–7. doi: 10.1093/nar/gkn823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bello J, Bello HR, Granados E. Conformation and aggregation of melittin: dependence on pH and concentration. Biochemistry. 1982;21 doi: 10.1021/bi00532a007. [DOI] [PubMed] [Google Scholar]
- 63.Dathe M, Wieprecht T. Structural features of helical antimicrobial peptides: their potential to modulate activity on model membranes and biological cells. Biochim Biophys Acta. 1999;1462:71–87. doi: 10.1016/s0005-2736(99)00201-1. [DOI] [PubMed] [Google Scholar]
- 64.de Planque MR, Kruijtzer JA, Liskamp RM, Marsh D, Greathouse DV, Koeppe RE, de Kruijff B, Killian JA. Different membrane anchoring positions of tryptophan and lysine in synthetic transmembrane alpha-helical peptides. J Biol Chem. 1999;274:20839–20846. doi: 10.1074/jbc.274.30.20839. [DOI] [PubMed] [Google Scholar]
- 65.Johnston JM, Cook GA, Tomich JM, Sansom MS. Conformation and environment of channel-forming peptides: a simulation study. Biophys J. 2006;90:1855–1864. doi: 10.1529/biophysj.105.069625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Planque MR, Boots JW, Rijkers DT, Liskamp RM, Greathouse DV, Killian JA. The effects of hydrophobic mismatch between phosphatidylcholine bilayers and transmembrane alpha-helical peptides depend on the nature of interfacially exposed aromatic and charged residues. Biochemistry. 2002;41:8396–8404. doi: 10.1021/bi0257686. [DOI] [PubMed] [Google Scholar]
- 67.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 68.Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 69.Schibli DJ, Epand RF, Vogel HJ, Epand RM. Tryptophan-rich antimicrobial peptides: comparative properties and membrane interactions. Biochemistry and Cell Biology. 2002;80:667–677. doi: 10.1139/o02-147. [DOI] [PubMed] [Google Scholar]
- 70.Schibli DJ, Montelaro RC, Vogel HJ. The membrane-proximal tryptophan-rich region of the HIV glycoprotein, gp41, forms a well-defined helix in dodecylphosphocholine micelles. Biochemistry. 2001;40:9570–9578. doi: 10.1021/bi010640u. [DOI] [PubMed] [Google Scholar]
- 71.Schiffer M, Chang CH, Stevens FJ. The functions of tryptophan residues in membrane proteins. Protein Eng. 1992;5:213–214. doi: 10.1093/protein/5.3.213. [DOI] [PubMed] [Google Scholar]
- 72.Stopar D, Spruijt RB, Hemminga MA. Anchoring mechanisms of membrane-associated M13 major coat protein. Chem Phys Lipids. 2006;141:83–93. doi: 10.1016/j.chemphyslip.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 73.Rezansoff AJ, Hunter HN, Jing W, Park IY, Kim SC, Vogel HJ. Interactions of the antimicrobial peptide Ac-FRWWHR-NH(2) with model membrane systems and bacterial cells. J Pept Res. 2005;65:491–501. doi: 10.1111/j.1399-3011.2005.00263.x. [DOI] [PubMed] [Google Scholar]
- 74.Hunter HN, Jing W, Schibli DJ, Trinh T, Park IY, Kim SC, Vogel HJ. The interactions of antimicrobial peptides derived from lysozyme with model membrane systems. Biochim Biophys Acta. 2005;1668:175–189. doi: 10.1016/j.bbamem.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 75.Strom MB, Rekdal O, Svendsen JS. Antibacterial activity of 15-residue lactoferricin derivatives. J Pept Res. 2000;56:265–274. doi: 10.1034/j.1399-3011.2000.00770.x. [DOI] [PubMed] [Google Scholar]
- 76.Li X, Li Y, Peterkofsky A, Wang G. NMR studies of aurein 1.2 analogs. Biochim Biophys Acta. 2006;1758:1203–1214. doi: 10.1016/j.bbamem.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 77.Rao AG. Conformation and antimicrobial activity of linear derivatives of tachyplesin lacking disulfide bonds. Arch Biochem Biophys. 1999;361:127–134. doi: 10.1006/abbi.1998.0962. [DOI] [PubMed] [Google Scholar]
- 78.Lee SA, Kim YK, Lim SS, Zhu WL, Ko H, Shin SY, Hahm KS, Kim Y. Solution structure and cell selectivity of piscidin 1 and its analogues. Biochemistry. 2007;46:3653–3663. doi: 10.1021/bi062233u. [DOI] [PubMed] [Google Scholar]
- 79.Ratledge C, Wilkinson SG. Microbial Lipids. 1988. [Google Scholar]
- 80.Bechinger B, Aisenbrey C, Bertani P. The alignment, structure and dynamics of membrane-associated polypeptides by solid-state NMR spectroscopy. Biochim Biophys Acta. 2004;1666:190–204. doi: 10.1016/j.bbamem.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 81.Ludtke SJ, He K, Heller WT, Harroun TA, Yang L, Huang HW. Membrane pores induced by magainin. Biochemistry. 1996;35:13723–13728. doi: 10.1021/bi9620621. [DOI] [PubMed] [Google Scholar]
- 82.Matsuzaki K, Murase O, Fujii N, Miyajima K. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry. 1996;35:11361–11368. doi: 10.1021/bi960016v. [DOI] [PubMed] [Google Scholar]
- 83.Pouny Y, Rapaport D, Mor A, Nicolas P, Shai Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry. 1992;31:12416–12423. doi: 10.1021/bi00164a017. [DOI] [PubMed] [Google Scholar]
- 84.Chan DI, Prenner EJ, Vogel HJ. Tryptophan- and arginine-rich antimicrobial peptides: structures and mechanisms of action. Biochim Biophys Acta. 2006;1758:1184–1202. doi: 10.1016/j.bbamem.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 85.Mecke A, Lee DK, Ramamoorthy A, Orr BG, Banaszak Holl MM. Membrane thinning due to antimicrobial peptide binding: an atomic force microscopy study of MSI-78 in lipid bilayers. Biophys J. 2005;89:4043–4050. doi: 10.1529/biophysj.105.062596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.K AHW, G VM, M FB, R A. Perturbation of the hydrophobic core of lipid bilayers by the human antimicrobial peptide LL-37. Biochemistry. 2004;43:8459–8469. doi: 10.1021/bi036284s. [DOI] [PubMed] [Google Scholar]
- 87.Lu JX, Damodaran K, Blazyk J, Lorigan GA. Solid-state nuclear magnetic resonance relaxation studies of the interaction mechanism of antimicrobial peptides with phospholipid bilayer membranes. Biochemistry. 2005;44:10208–10217. doi: 10.1021/bi050730p. [DOI] [PubMed] [Google Scholar]
- 88.Jin Y, Mozsolits H, Hammer J, Zmuda E, Zhu F, Zhang Y, Aguilar MI, Blazyk J. Influence of tryptophan on lipid binding of linear amphipathic cationic antimicrobial peptides. Biochemistry. 2003;42:9395–9405. doi: 10.1021/bi034338s. [DOI] [PubMed] [Google Scholar]
- 89.Antharam VC, Farver RS, Kuznetsova A, Sippel KH, Mills FD, Elliott DW, Sternin E, Long JR. Interactions of the C-terminus of lung surfactant protein B with lipid bilayers are modulated by acyl chain saturation. Biochim Biophys Acta. 2008;1778:2544–2554. doi: 10.1016/j.bbamem.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Koenig BW, Ferretti JA, Gawrisch K. Site-specific deuterium order parameters and membrane-bound behavior of a peptide fragment from the intracellular domain of HIV-1 gp41. Biochemistry. 1999;38:6327–6334. doi: 10.1021/bi982800g. [DOI] [PubMed] [Google Scholar]
- 91.Wi S, Kim C. Pore structure, thinning effect, and lateral diffusive dynamics of oriented lipid membranes interacting with antimicrobial peptide protegrin-1: 31P and 2H solid-state NMR study. J Phys Chem B. 2008;112:11402–11414. doi: 10.1021/jp801825k. [DOI] [PubMed] [Google Scholar]
- 92.Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 93.Jenssen H, Hamill P, Hancock RE. Peptide antimicrobial agents. Clin Microbiol Rev. 2006;19:491–511. doi: 10.1128/CMR.00056-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bechinger B. The structure, dynamics and orientation of antimicrobial peptides in membranes by multidimensional solid-state NMR spectroscopy. Biochim Biophys Acta. 1999;1462:157–183. doi: 10.1016/s0005-2736(99)00205-9. [DOI] [PubMed] [Google Scholar]
- 95.Oren Z, Hong J, Shai Y. A repertoire of novel antibacterial diastereomeric peptides with selective cytolytic activity. J Biol Chem. 1997;272:14643–14649. doi: 10.1074/jbc.272.23.14643. [DOI] [PubMed] [Google Scholar]
- 96.Yeaman MR, Yount NY. Unifying themes in host defence effector polypeptides. Nat Rev Microbiol. 2007;5:727–740. doi: 10.1038/nrmicro1744. [DOI] [PubMed] [Google Scholar]
- 97.Cheng JT, Hale JD, Elliot M, Hancock RE, Straus SK. Effect of membrane composition on antimicrobial peptides aurein 2.2 and 2.3 from Australian southern bell frogs. Biophys J. 2009;96:552–565. doi: 10.1016/j.bpj.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hallock KJ, Lee DK, Omnaas J, Mosberg HI, Ramamoorthy A. Membrane composition determines pardaxin's mechanism of lipid bilayer disruption. Biophys J. 2002;83:1004–1013. doi: 10.1016/S0006-3495(02)75226-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ramamoorthy A, Thennarasu S, Tan A, Lee DK, Clayberger C, Krensky AM. Cell selectivity correlates with membrane-specific interactions: a case study on the antimicrobial peptide G15 derived from granulysin. Biochim Biophys Acta. 2006;1758:154–163. doi: 10.1016/j.bbamem.2006.02.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Curve fitting for DOSY experiments.
Supplementary Figure 2: 31P MAS spectra of POPC in the absence and presence of 3 mol% IL-8α.