

Abstract
A novel algorithm, PILOT_PTM, has been developed for the untargeted identification of post-translational modifications (PTMs) on a template sequence. The algorithm consists of an analysis of an MS/MS spectrum via an integer linear optimization model to output a rank-ordered list of PTMs that best match the experimental data. Each MS/MS spectrum is analyzed by a preprocessing algorithm to reduce spectral noise and label potential complimentary, offset, isotope, and multiply charged peaks. Postprocessing of the rank-ordered list from the integer linear optimization model will resolve fragment mass errors and will reorder the list of PTMs based on the cross-correlation between the experimental and theoretical MS/MS spectrum. PILOT_PTM is instrument-independent, capable of handling multiple fragmentation technologies, and can address the universe of PTMs for every amino acid on the template sequence. The various features of PILOT_PTM are presented, and it is tested on several modified and unmodified data sets including chemically synthesized phosphopeptides, histone H3-(1–50) polypeptides, histone H3-(1–50) tryptic fragments, and peptides generated from proteins extracted from chromatin-enriched fractions. The data sets consist of spectra derived from fragmentation via collision-induced dissociation, electron transfer dissociation, and electron capture dissociation. The capability of PILOT_PTM is then benchmarked using five state-of-the-art methods, InsPecT, Virtual Expert Mass Spectrometrist (VEMS), Modi, Mascot, and X!Tandem. PILOT_PTM demonstrates superior accuracy on both the small and large scale proteome experiments. A protocol is finally developed for the analysis of a complete LC-MS/MS scan using template sequences generated from SEQUEST and is demonstrated on over 270,000 MS/MS spectra collected from a total chromatin digest.
Identification of the types of post-translational modifications (PTMs)1 of various organisms is currently a major challenge in the field of proteomics. MS/MS has shown to be an excellent tool for de novo peptide sequence prediction and database peptide identification and is indispensable in determining PTMs (1, 2). Many research groups (3–28) have incorporated modification discovery into their respective identification algorithms and utilize multiple databases, including UniMod (29), RESID (30), and Delta Mass,2 to build a list of variable modifications that can exist on a candidate peptide. To date, there exist two types of algorithms for identification of PTMs: (a) hybrid sequence tag/database approaches (3–9, 28), which develop a sequence tag and subsequently compare this tag with a database to extract a candidate peptide sequence and determine the set of PTMs that best explain the MS/MS spectrum and (b) pure database-based approaches (10–15, 23–27), which directly compare the experimental peak list with a theoretical peak list derived from candidate peptides in a database. Both approaches have had success both in validation of known modifications and discovery of novel ones. To our knowledge, there is no de novo approach for identification of PTMs using a comprehensive variable modification list.
The hybrid methods, denoted as (a), are beneficial because the derivation of the sequence tag may limit the size of the database to proteins that contain that sequence tag. This approach (3) can allow for a richer set of variable modifications to be considered on candidate peptide sequences due to the database size reduction. For example, the InsPecT algorithm (4) will generate de novo sequence tags of a fixed length and scan a trie-based database for all instances of the tag. Each distinct variable modification combination, or decoration, is entered in a mass-ordered list prior to database searching. When a peptide matching the tag is found, the algorithm will attempt to increase the length of the tag with an amino acid sequence if the mass of the sequence plus the mass of one decoration is equal to the predetermined mass gap. The Virtual Expert Mass Spectrometrist (VEMS) (6) uses both a database-independent search for generation of sequence tags and a database-dependent search to determine possible peptides. Any sequence tag that is not validated by a peptide found in the database-dependent search is compared with the list of proteins containing peptides found in the database-dependent search to generate candidate amino acid sequences. All combinations of variable modifications that equal the difference between the parent mass and the candidate sequence mass are tested to derive the best possible modification (6). The Modi algorithm (7) assumes that the database has been reduced a priori to a candidate subset of 20 proteins. After filtering the MS/MS spectrum, the algorithm generates a list of sequence tags derived from the spectrum and attempts to explain the mass gaps using any of the modifications from the UniMod database.
Although sequence tags have proven to be very capable in determining the candidate peptide sequence, the success of these methods relies greatly on the accurate prediction of the sequence tag. Pure database methods, denoted earlier as (b), remove this need by directly obtaining the peptide sequence (with or without modifications) from a database. These approaches are also beneficial because they use all of the MS/MS spectrum peak information at once when analyzing a candidate peptide. That is, for each candidate sequence in the database, a full set of theoretical ion fragments may be compared with the experimental MS/MS spectrum peaks to derive a score for the candidate sequence. The potential drawback of the pure database algorithms is the limitation of variable modifications that can be analyzed. Each variable modification will create an additional copy of the amino acid that must be analyzed when developing a theoretical candidate peptide from the database.
When analyzing the entire database with a small modification set, these algorithms have been very effective in identifying modified spectra. The SEQUEST algorithm (10) uses a technique known as cross-correlation to mathematically compare the overlap between the theoretical spectrum from a candidate database peptide and the experimental spectrum. Mascot (11) incorporates probability-based searching to locate a candidate peptide sequence that scores above a certain expectation threshold dependent on the size of the database. X!Tandem (14) also uses a probabilistic search method to determine the best peptide match to a spectrum.
A major limitation of many of the preceding algorithms is the inability to interpret electron transfer dissociation (ETD) (32–34) or electron collision dissociation (ECD) (35, 36) spectra. ECD and ETD both involve the reaction of an electron with a protonated cation to form an odd electron peptide. This process induces large amounts of backbone cleavage to yield c-ions and z•-ions (32, 35) that are analogous to the b-ions and y-ions produced from CID. Although the c-ions and z•-ions are often the most abundant ions present, both ETD and ECD spectra have been known to show b-ions, y-ions, and their neutral losses as well (37). ECD and ETD enhance the diversity of peptides that can be fragmented because they can analyze bigger peptides with higher charge state. In fact, a recent decision tree model (38) was developed to differentiate which parent mass and charge states are most appropriate for CID and ETD. Generally, CID will provide the most fragmentation for peptides of charge 2 or high mass peptides of charge 3. Low mass peptides of charge 3 and all peptides of charge greater than 3 may have better fragmentation using ETD or ECD (38). Unlike CID, ECD and ETD cleavage is very weakly affected by the amino acid sequence and generally provides more complete coverage than CID alone when used on peptides with higher charge density. Depending on the precursor charge and basic residue location, one can expect a large fraction of complementary c-ions and z•-ions to be present in the spectral data. Additionally, both ECD and ETD also prevent cleavage of labile modifications (33, 36). Although the mechanism of cleavage during ECD and ETD is still debated, PTMs are fully present on the c-ions and z•-ions produced during cleavage. As ETD/ECD fragmentation techniques become more readily available, they will serve as a complement for CID technology, and hence it is desirable that computational algorithms be able to handle inputs from all three techniques.
A further limitation of most of the preceding algorithms is the inability to search for a large amount of variable modifications. Enumerating all combinations of the variable modifications will lead to an exponential increase in the search time and can pose a significant problem when the database size is large. This may be reduced by implementing a two-pass approach (39–41) where the database is initially scanned either with no modifications or a small subset of variable modifications to eliminate proteins that did not score above a given threshold (based on the peptide hits). Mascot (40), X!Tandem (39), and InsPecT (41) will run a first pass search with a small set of variable modifications to analyze spectra that are either unmodified or contain the queried modifications. Because of the reduced database size, additional variable modifications as well as missed cleavages and other unusual digestion/fragmentation information can be incorporated into the search.
Several groups (16, 17, 19, 21–23, 28, 40, 42) have developed untargeted algorithms to assign integer mass modifications to candidate peptide sequences. These algorithms place a restriction on the number of modification sites to enhance computational efficiency and reduce the false detection of low mass modifications. Alternatively, the Modi algorithm (7) currently uses the entire UniMod (29) database as a variable modification list, allows a user to input as many additional modifications as necessary, and does not place an upper bound on the amount of modification sites. A recent approach involving selective mass screening (18) has also been developed to identify low abundance modifications using the Modi (7) algorithm. To aid in the development of de novo identification algorithms for modified peptides, MS-Profile (43) was recently developed to generate spectral profiles of tandem mass spectra. Using the forward-backward algorithm, MS-Profile is able to determine the probability that a spectral peak corresponds to a peptide prefix mass without explicitly enumerating the complete spectral dictionary for the MS/MS spectrum.
We have developed a novel method, PILOT_PTM (Fig. 1), for untargeted PTM prediction via integer linear optimization (ILP) and tandem mass spectrometry. ILP has been an integral tool in the de novo sequencing algorithm PILOT (44, 45) and the hybrid algorithm PILOT_SEQUEL (46). Similar to these previous methods, our objective function seeks to maximize the sum of intensity contributions from theoretical peak matches to the experimental spectrum given a set of logical constraints. We expand on the previous methodology by directly incorporating the intensity contribution from both sets of complementary ion peaks as well as all corresponding offsets in the objective function. Given a template sequence of amino acids, the model will seek to determine the optimal set of modifications among a “universal” list based on the MS/MS spectral data assuming that all template positions can contain a PTM. This universal list (supplemental Table 1) consists of 912 known PTMs, chemical derivatives, amino acid substitutions, non-enzymatic modifications, isotopic labels, and artifacts in the UniMod (29), RESID (30), and Delta Mass2 databases. No upper bound is placed on the amount of modification types or modification sites that can exist on the template. The method rigorously guarantees the optimal set of modifications without having to enumerate all combinations of possible modifications.
Fig. 1.

a, overall framework for PILOT_PTM. b, identification of globally and locally significant peaks. The highest intensity filtered peaks are labeled as globally significant. Any other filtered peak is labeled as locally significant if the peak intensity is greater than all other peaks within a 5.0-Da mass window. c, a set of singly charged support peaks (red) for a candidate ion peak (blue). Each peak labeled as globally or locally significant will be assigned a set of supporting peaks based on the filtered MS/MS. d, output from an optimal solution of PILOT_PTM. The blue peaks on the left represent all MS/MS peaks that are activated for the optimal solution. The sets of blue peaks on the right represent all candidate ion peak sets that will activate the optimal combination of MS/MS peaks. e, cross-correlation example for the template sequence KSTGGKAPR with N-terminal propionylation, Lys-1 dimethylation, and Lys-6 acetylation.
EXPERIMENTAL PROCEDURES
Sample Preparation and Annotation
This section will detail the preparation of each of the data sets and the annotation procedure for determination of PTMs. Annotated spectra for all test data sets are provided as supplemental material. The fragmentation methods, MS/MS instruments, and total scans for each data set are given in Table I.
Table I. Annotated test set information.
Fragmentation method, MS/MS instruments, and total MS/MS spectra are presented for each test data set.
| Subset | MS instrument | MS/MS instrument | No. of CID scans | No. of ETD scans | No. of ECD scans |
|---|---|---|---|---|---|
| A1 | Ion trap | Ion trap | 0 | 102 | 0 |
| A2 | Ion trap | Ion trap | 31 | 17 | 0 |
| A3 | Orbitrap | Ion trap | 13 | 55 | 0 |
| B | FTICR | FTICR | 0 | 0 | 58 |
| C | Orbitrap | Ion trap | 553 | 0 | 0 |
| D1 | Orbitrap | Ion trap | 525 | 0 | 0 |
| D2 | Orbitrap | Ion trap | 6,025 | 0 | 0 |
| E1 | Ion trap | Ion trap | 36 | 0 | 0 |
| E2 | Q-TOF | Q-TOF | 37 | 0 | 0 |
| E3 | Orbitrap | Orbitrap | 401 | 0 | 0 |
Test Set A: Phosphopeptides
Three sets of chemically synthesized phosphopeptides (AnaSpec) were prepared for mass spectrometry by using 50% acetonitrile. The peptides were analyzed using a data-dependent mode setting measuring the parent mass followed by MS/MS fragmentation using alternating CID/ETD scans (Table I). All peptides were isolated based on parent mass, and a total of 218 spectra were manually validated.
Test Set B: Histone H3-(1–50) N-terminal Tail
Histone H3 was isolated from HeLa cells and prepared for mass spectrometry as described previously (47). The H3-(1–50) N-terminal tail was analyzed using an 8.5-tesla quadrupole FTMS instrument. The 8+ charge state was enhanced using the quadrupole and second octopole of the instrument for selective ion accumulation. Selected species were selected for fragmentation by ECD, and 58 spectra were manually validated (47). Although multiple modified forms may be present in each MS/MS spectrum, the annotation assigned corresponds to the most abundant modified form.
Test Set C: Propionylated Histone Fragments
Histone H3 was isolated from mouse embryonic fibroblast cells and prepared for mass spectrometry as described previously (48). Propionylated H3 peptides were analyzed by nanoflow reverse-phase HPLC-MS/MS using a linear quadrupole ion trap-Orbitrap mass spectrometer (ThermoFisher, San Jose, CA) operated in the data-dependent mode with one full MS acquired in the Orbitrap followed by seven data-dependent MS/MS spectra acquired via CID in the ion trap. A representative set of parent masses was selected using the five peptide fragments associated with the H3-(1–50) N-terminal tail and modifications that are commonly found on histone H3 (Lys methylation, Lys dimethylation, and Lys acetylation) or are artifacts of the propionylation procedure (Lys propionylation, Lys methylated propionylation, N-terminal propionylation, N-terminal acetylation, and C-terminal methylation), and a total of 553 spectra were isolated and manually annotated.
Test Set D: Total Chromatin Fraction
HeLa S3 cells were cultured and harvested as described recently (47). Chromatin fractions from the HeLa cells were roughly prepared according to published procedures (49). Extracted protein was separated using one-dimensional SDS-PAGE and in-gel digested by trypsin following treatment with iodoacetamide. Peptide digests were then analyzed by nanoflow LC-MS/MS on an Orbitrap mass spectrometer as described previously (50).
To develop an annotated test set, we initially utilized the SEQUEST algorithm (10) with a set of eight variable modifications (Met oxidation, Lys methylation, Lys dimethylation, Lys acetylation, Arg methylation, Ser phosphorylation, N-terminal acetylation, and C-terminal amidation). We scanned the NCBInr database with human taxonomy and allowed up to three missed cleavages. The fragment ion tolerance was set to 0.5 Da, and the parent ion tolerance was set to 0.1 Da. The cutoff XCorr for an annotation was 2.0 for a charge 1 precursor, 2.2 for charge 2, 2.50 for charge 3, and 3.0 for charge 4. All such assignments were then analyzed using Mascot (11) with the same variable modification list and search parameters. The assignments that were validated by Mascot with an expectation value of at most 0.1 were retained.
The annotations were then manually examined to remove assignments that appeared to correspond to low quality spectra. Each peptide annotation was theoretically fragmented to develop a list of predicted b- and y-ions. Using an appropriate noise threshold (51), we remove all MS/MS spectra that do not contain at least 50% (Quality < 0.5) of the theoretical b- and y-ions (Equation 1).
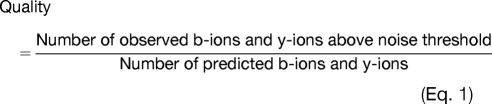 |
This procedure produced 525 modified peptides corresponding to 193 different proteins and 6,025 unmodified peptides corresponding to 2,123 different proteins.
Test Set E: Additional Unmodified Peptides
Ion Trap Peptides
These spectra from the organism Mycobacterium smegmatis are available from the Open Proteomics Database (52). A test set of 36 spectra were verified by Mascot (11) and SEQUEST (10) and further filtered based on the amount of b-ions and y-ions above the noise level as described previously (44).
Q-TOF Peptides
These spectra were derived from a publicly available data set (53). The spectra were collected with Q-TOF2 and Q-TOF-Global mass spectrometers using a mixture of alcohol dehydrogenase (yeast), myoglobin (horse), albumin (bovine; BSA), and cytochrome c (horse). A test set of 37 spectra was obtained using only “acceptable spectra” as defined previously (44).
Orbitrap Peptides
Stock solutions of a 16-peptide mixture were prepared containing equal amounts of each protein as described previously (46). The proteins were digested with trypsin and analyzed by automated microcapillary liquid chromatography and an LTQ-Orbitrap hybrid mass spectrometer (Thermo Finnigan, San Jose, CA). Both MS and MS/MS spectra were recorded on the instrument, and a test set of 401 spectra was annotated using the SEQUEST algorithm (10).
Novel PILOT_PTM Algorithm
The framework for PILOT_PTM (Fig. 1a) begins with a preprocessing algorithm that filters the raw spectrum to extract all globally and locally significant peaks based on their intensity (Fig. 1b). The preprocessor is capable of handling inputs from multiple fragmentation methods including CID, ETD, and ECD and will label candidate b-ion (CID) or c-ion peaks (ETD/ECD), the appropriate complementary y-ion (CID) or z•-ion (ETD/ECD), and any supporting peaks (isotopes, neutral offsets, etc.) that may exist (Fig. 1c). The ILP model will derive a rank-ordered list of activated globally significant peaks for the template peptide sequence based on one or more sets of candidate ion peaks (Fig. 1d). A complete list of candidate modified sequences that satisfy the appropriate mass conservation constraints for each candidate ion peak set is then constructed. The postprocessing algorithm section uses a cross-correlation function to mathematically verify the overlap between the experimental MS/MS and the theoretical spectrum created by a candidate sequence (Fig. 1e). Each candidate sequence is assigned a cross-correlation score and placed in a rank-ordered list. The modified sequence that best explains the experimental data will have the highest cross-correlation score.
ILP Model
Given a template amino acid sequence of length K, each amino acid is assigned an index, k, corresponding to the position in the template sequence. Without loss of generality, the N-terminal amino acid will correspond to k = 1, and the C-terminal amino acid will correspond to k = K. During the preprocessing stage, a list of candidate ion peaks, j, is generated that represent possible b-ions (for CID) or c-ions (for ETD/ECD). The peak masses will correspond to singly charged ions, and the choice of ion type is arbitrary as y-ions or z•-ions could easily be used in the formulation of the problem.
Sets
The set CSk (Equation 2) consists of all candidate ion peaks j that are valid peaks for the template amino acid sequence at position k. Given the universal list of modifications, the theoretical lower (mkL) and upper (mkU) bounds on the masses of the ion peaks used to construct the candidate sequence can be easily calculated. We can then efficiently construct each set CSk by enumerating all j subject to mkL ≤ mj ≤ mkU, and there exists an amino acid path from j to both the N-terminal and C-terminal boundary conditions (N-term and C-term B.C.) (54). The set Posj is simply a list of all template positions for which j can be a candidate ion peak (Equation 3).
 |
 |
For each candidate ion peak j, we construct the set of supporting MS/MS spectrum peaks, Supportj (Equation 4) using the globally and locally significant peaks (indexed over i) determined from the preprocessor (Fig. 1b). Supportj is intended to detail as much information about the candidate ion peak j and is dependent on the fragmentation method used. For ETD/ECD spectra, Supportj consists of c2+-ions, z•-ions, z•2+-ions, b-ions, y-ions, and their corresponding +1 and +2 isotopes. For CID spectra, the appropriate ions are b2+-ions, y-ions, y2+-ions, their corresponding +1 and +2 isotopes, and their corresponding offsets (i.e. −H2O, −NH3, and −CO). The y-ion or z•-ion series can be calculated from the modified parent mass by the formula c-ion + z•-ion = mP + mH + 2·mH+ for ETD/ECD spectra and y-ion + b-ion = mP + 2·mH+ for CID spectra, respectively. The set Multi is the set of all j such that i is a supporting peak for j (Equation 5).
Binary Variables
We use binary variables (Equations 6 and 7) to model the logical use of a candidate ion peak j at a template position k (pj,k) as well as the logical use of an MS/MS spectrum ion peak i as supporting information (yi). These variables are defined as follows.
 |
 |
Mathematical Model
The constraints of the problem are chosen to ensure proper use of the logical binary variables. At most, one candidate ion peak j is able to be assigned to a template position k (Equation 9). Additionally, we allow for missing candidate ion peaks j associated with a template position k but require that there can be no more than three consecutive missing candidate ion peaks (Equation 10). We can also enforce the constraint that a candidate ion peak j can be used at most once in the construction of a modified sequence (Equation 11). Constraints are also introduced to ensure that an MS/MS spectrum peak i is used properly as supporting information. An MS/MS spectrum peak i can only be activated if at least one of the corresponding candidate ion peaks j in the set Multi is activated for any valid template position k in the set Posj (Equation 12). We also ensure that a candidate ion peak j is not activated if the corresponding MS/MS spectrum ion peak i is not activated (Equation 13). The objective of the problem is to maximize the intensity of the MS/MS spectrum peaks i used to construct the modified sequence (Equation 8).
 |
subject to
 |
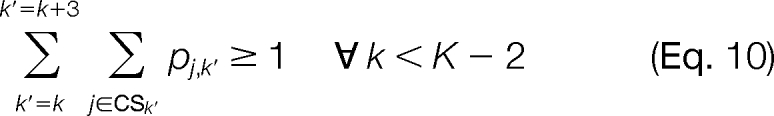 |
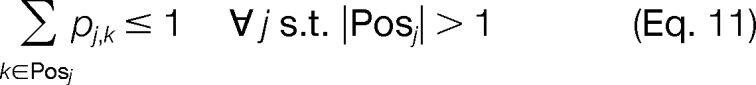 |
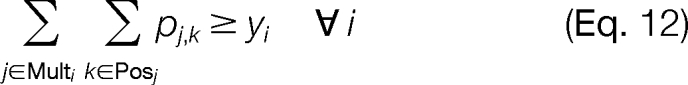 |
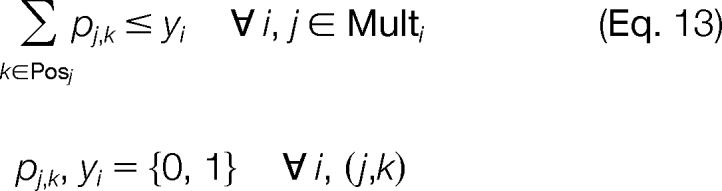 |
This ILP model can be solved to global optimality using CPLEX (55) to obtain a set of MS/MS spectrum peaks that correspond to one or more modified sequences. Using integer cuts (56), a rank-ordered list of the top 10 sets of MS/MS spectrum peak variables will be generated. CPLEX uses a branch-and-cut algorithm (57) where a subset of the integer variables is fixed and the remaining integer variables are relaxed so they can take on continuous values, and a linear programming (LP) relaxation is formed. A branch-and-bound tree keeps track of which integer variables are fixed in a given relaxation and stores them in a “node” in the tree. The algorithm then parses through the tree and solves an LP relaxation at each node to get a theoretical upper bound on the optimal solution of the original problem. The traversal of the tree is dependent on the search techniques being used, some of which include depth-first search, breadth-first search, and best-bound search. Complete enumeration is avoided using fathoming criteria after each LP relaxation is solved. For further detail, the reader is directed to Refs. 56 and 57. A complete description of the ILP model and detailed solution strategies can be found in the supplemental material. Note that the ILP model can be formulated using network-based constraints (44, 45, 58–62).
Cutting Plane Constraints
When incorporating all of the previous constraints, it is still possible to obtain linear programming relaxations that consider a set of pj,k at adjacent template positions that do not correspond to the mass difference of a modified or unmodified amino acid. For each pj,k, we determine Invj,k,k′L and Invj,k,k′U, the set of candidate ion peaks j′ at template position k′ (k′ < k and k′ > k, respectively) such that no jump exists between j and j′.
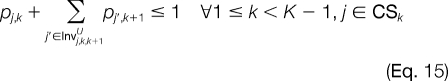 |
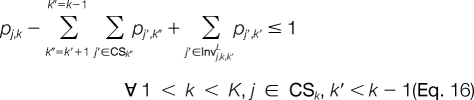 |
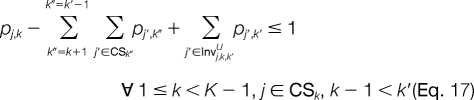 |
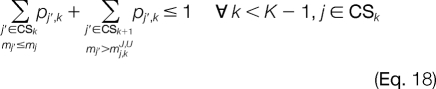 |
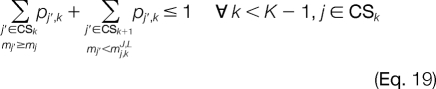 |
The improper assignment of any invalid peak combination at adjacent template positions is prevented with Equations 14 and 15. For candidate ion peaks j and j′ at template positions k and k′, respectively, where |k′ − k| > 1, we would like to prevent an invalid combination only if a candidate peak is not activated at any template position k" between k and k′. This is illustrated using Equations 16 and 17. The next set of constraints will be added when the linear relaxation activates candidate ion peaks at adjacent template positions where the mass difference between them is less than the smallest modified amino acid or greater than the largest modified amino acid. Thus, for each candidate ion peak j at template position k, we establish mj,kJ,L and mj,kJ,U, which are the maximum and minimum masses that can be reached from j, respectively. All j′ ϵ CSk + 1 for which mj′ > mj,kJ,U and mj′ < mj,kJ,L correspond to candidate ion peaks outside the minimum and maximum possible mass peak boundaries. The improper assignment of peak variables can be prevented by Equations 18 and 19.
Incorporating all of these equations in the initial formulation of the problem results in a large number of constraints, many of which are not activated for the optimal solution. To circumvent this computational burden, we apply them dynamically as cuts. That is, for a given ILP relaxation, the violations of Equations 14–19 are checked, and cuts are then added when needed.
Preprocessing Algorithm
The preprocessing algorithm begins by removing all peaks that are associated with the precursor ion. For CID spectra, this includes the precursor ion, its +1 and +2 isotopes, and any neutral losses (i.e. −H2O, −NH3, and −CO) (63). For ETD/ECD spectra, we must remove all peaks that correspond to distinct charge states of the precursor ion and their isotopes. Additionally, all peaks that correspond to a common neutral loss of a charge-reduced form of the precursor ion (37) are removed. The MS/MS spectrum is then filtered to remove any peak that is within an appropriate tolerance of another peak of higher intensity. The filtered MS/MS spectrum is scanned to extract the peaks with the highest intensity (Fig. 1b). All locally significant peaks are then extracted if the peak intensity is greater than all other peaks within an appropriate mass window (Fig. 1b).
The preprocessor scans and removes all peaks that are determined to be +1 or +2 isotopes. If any doubly or triply charged peaks are found based on isotopic offsets, the appropriate singly charged peak of the same intensity is constructed. For CID spectra, all neutral offsets are removed if the offset does not have a complementary peak. The preprocessor then queries all candidate peaks and determines a full list of supporting peaks (Fig. 1c) for each candidate ion peak. For CID spectra, this will include +1 and +2 isotopic offsets, neutral losses (i.e. −H2O, −NH3, and −CO), and doubly charged peaks. For ETD/ECD spectra, this will include isotopic offsets and doubly charged peaks.
Postprocessing Algorithm
A postprocessing algorithm is used to score the candidate modified peptide sequences that are derived from the peak sets in the ILP rank-ordered list. A cross-correlation technique (10) is used to measure the mathematical overlap between the theoretical ions produced from the candidate PTM set and the experimental spectrum. A generalized model is established that is similar to that used in PILOT (44, 45) and PILOT_SEQUEL (46). A mathematical overlap between the theoretical and experimental spectrum is then calculated based on monoisotopic masses for each candidate modified peptide (Fig. 1e). Each candidate modified peptide is assigned a cross-correlation score and inserted into a rank-ordered list. Similar to SEQUEST, this score is a measure of how well the “expected” fragmentation pattern of a particular modified peptide matches the experimental data and is not a probabilistic metric (10). The modified peptide thought to best explain the experimental data is given the highest cross-correlation score.
Once all peak intensities are assigned, the postprocessor scans each set of candidate ion peaks j output from the ILP model. If the mass difference between two candidate ion peaks j and j′ that are at least two template positions apart is equal to the sum of the intermediate unmodified residue masses, but the activated candidate ion peaks in between j and j′ indicate a possible modification, then these intermediate candidate ion peak assignments are checked by looking for the presence of peaks in the MS/MS spectrum that indicate unmodified residues. If enough supporting information exists, then the intermediate candidate ion peaks are reassigned to that of the unmodified sequence and subsequently rescored.
Algorithm Scoring
The accuracy of an algorithm is measured using three metrics: residue prediction accuracy, peptide prediction accuracy, and subsequence accuracy. The definitions of each accuracy metric for PILOT_PTM and all compared algorithms are given below.
Residue Prediction Accuracy
For a given template amino acid, we will define the residue prediction accuracy for PILOT_PTM as 1 for the assignment of a modification (or lack thereof) with mass within 0.1 Da (0.01 Da for ECD spectra) of the proper annotated modification mass and 0 otherwise. For alternative algorithms, the residue prediction accuracy will be equal to 1 if the algorithm assigned any amino acid (modified or unmodified) with mass within 0.1 Da (0.01 Da for ECD spectra) of the proper modified residue and 0 otherwise. When the size of the peptide predicted by a competing algorithm is not equal to that of the annotated peptide, we look for the alignment between the predicted peptide and the annotated peptide that will yield the highest amount of correct residues. If multiple peptides report the same “best” score for an algorithm, then the peptide that has the highest amount of correct residues is selected for accuracy quantitation.
Peptide Prediction Accuracy
The complete peptide prediction accuracy is set to 1 if all residues have been correctly annotated with the proper modification (or lack thereof) and 0 otherwise. The complete prediction accuracy within N residues is set to 1 if at most N residues are assigned incorrectly and 0 otherwise. When the size of the peptide predicted by a competing algorithm is not equal to that of the annotated peptide, the peptide prediction accuracy is calculated using the alignment found during calculation of the residue prediction accuracy.
Subsequence Length Accuracy
The subsequence length of an MS/MS spectrum is the longest string of residues that were annotated correctly. That is, if an MS/MS spectrum is assigned a subsequence with length L, then there exists L consecutive amino acids that were assigned a residue prediction accuracy of 1. The subsequence accuracy for a given length across a data set is then determined by dividing all peptides that contain a properly annotated subsequence with at least that length by the total number of peptides with at least that length.
RESULTS
Algorithm Validation
The proposed method was tested on four modified test data sets, including 218 phosphopeptides fragmented via ETD and CID (A1–A3), 58 histone H3-(1–50) N-terminal tail spectra fragmented via ECD (47) (B), 553 propionylated histone H3-(1–50) peptides fragmented via CID (48) (C), and 525 peptides from a total chromatin fraction fragmented via CID. (D1) PILOT_PTM was able to accurately identify 100% of the modified residues from data set A1, 93.8% from A2, 89.7% from A3, 97.9% from B, 98.6% from C, and 96.5% from D1 (Table II). The decrease in accuracy between data set A1 and data sets A2 and A3 was thought to be due to the lack of fragmentation of the MS/MS spectrum in these data sets. We note that the prediction accuracy for modified residues is the highest for data sets A1 and C. These data sets generally had the best fragmentation and thus contained many of their singly charged ion peaks (supplemental annotations). For all modified data sets, PILOT_PTM was able to accurately identify 2,339 of the 2,393 modified residues (97.7%) and 15,752 of the 15,864 unmodified residues (99.3%).
Table II. PILOT_PTM residue prediction accuracy.
A correctly predicted amino acid residue (modified or unmodified) is assigned a value of 1 if the correct modification (or lack thereof) was assigned to the residue and 0 otherwise. The accuracy for a data set is simply the total residue prediction accuracy for all peptides in the data set. The percent of correct annotations is given in parenthesis next to the number of correct annotations. N/A, not applicable.
| Data set | All residues | Modified residues | Unmodified residues |
|---|---|---|---|
| Modified | |||
| A1 | 943/943 (1.000) | 102/102 (1.000) | 841/841 (1.000) |
| A2 | 747/772 (0.968) | 45/48 (0.938) | 702/724 (0.970) |
| A3 | 947/960 (0.986) | 61/68 (0.897) | 886/892 (0.993) |
| B | 2,888/2,900 (0.996) | 284/290 (0.979) | 2,604/2,610 (0.998) |
| C | 5,716/5,790 (0.987) | 1,295/1,313 (0.986) | 4,421/4,477 (0.987) |
| D1 | 6,850/6,892 (0.994) | 552/572 (0.965) | 6,298/6,320 (0.997) |
| Total | 18,091/18,257 (0.991) | 2,339/2,393 (0.977) | 15,752/15,864 (0.993) |
| Unmodified | |||
| D2 | 84,498/84,521 (0.999) | N/A | 84,498/84,521 (0.999) |
| E1 | 402/408 (0.982) | N/A | 402/408 (0.982) |
| E2 | 417/418 (0.997) | N/A | 417/418 (0.997) |
| E3 | 3,632/3,638 (0.998) | N/A | 3,632/3,638 (0.998) |
| Total | 88,985/88,949 (0.999) | N/A | 88,985/88,949 (0.999) |
PILOT_PTM was also tested on two unmodified data sets, including the 6,025 unmodified spectra identified from the total chromatin fraction (D2) and 474 unmodified spectra fragmented via CID using ion trap, Q-TOF, and Orbitrap instruments (44–46) (E1–E3). PILOT_PTM achieves a residue prediction accuracy of 99.9% for the total chromatin data set, 98.2% for the ion trap data set, 99.7% for the Q-TOF data set, and 99.8% for the Orbitrap data set. These results are evidence both of the ability to properly identify no modifications on unmodified peptides for various types of spectral instruments and the potential for enhanced accuracy with better spectral resolution (Table II).
The peptide prediction accuracy for a data set is defined as the total amount of peptides with the correct modification (or lack thereof) assigned to all residues in the peptide and is displayed in Table III. PILOT_PTM reports a complete peptide prediction accuracy of 100% for data set A1, 93.8% for data set A2, 89.7% for data set A3, 89.7% for data set B, 94.4% for data set C, and 95.2% for data set D1. Note that the accuracies for data sets A1, A2, and A3 will be exactly equal to the modified residue prediction accuracy (Table II) because each of the peptides contain exactly one modified phosphorylation residue. The high prediction accuracies of data sets C and D1 show the ability of PILOT_PTM to fully annotate peptides that are either highly modified (data set C) or part of a very complex sample (data set D1). We also note that PILOT_PTM was able to fully annotate 52 of the 58 histone N-terminal tail peptides in data set B. This is an important result because these peptides are the longest in all test data sets with 50 amino acids, the fragmentation near the middle of the peptide is not nearly as strong as it is near the termini (supplemental annotations), and these spectra contain additional modified peptides at lower stoichiometric amounts that could reduce the ability of an algorithm to identify the most prevalent form (47). The local inclusion of high resolution peaks in the PILOT_PTM preprocessor as well as the accurate identification of peaks of charge 2+ and charge 3+ from isotopic information allow for the proper assignment of the lysine modifications near the middle of the peptide. Table III also shows the improvement in the peptide prediction accuracy when allowing for up to one or two incorrect residues. When allowing for two incorrect modifications, PILOT_PTM was able to annotate 6,497 of the 6,499 unmodified peptides (100%) and 1,323 of the 1,354 modified peptides (97.7%), showing that PILOT_PTM was still able to annotate a majority of the peptide even when some residues are incorrectly identified (Table III).
Table III. PILOT_PTM peptide prediction accuracy.
Peptide prediction accuracy is defined as the total amount of peptides with the correct modification (or lack thereof) assigned to all residues. The accuracy within one or two residues represents the total amount of peptides with at most one or two incorrect residues, respectively. The percent of correct annotations is given in parenthesis next to the number of correct annotations.
| Data set | Total | Completely | Within 1 | Within 2 |
|---|---|---|---|---|
| Modified | ||||
| A1 | 102 | 102 (1.000) | 102 (1.000) | 102 (1.000) |
| A2 | 48 | 45 (0.938) | 45 (0.938) | 48 (1.000) |
| A3 | 68 | 61 (0.897) | 61 (0.897) | 68 (1.000) |
| B | 58 | 52 (0.897) | 52 (0.896) | 58 (1.000) |
| C | 553 | 522 (0.944) | 522 (0.944) | 536 (0.969) |
| D1 | 525 | 500 (0.952) | 500 (0.952) | 511 (0.973) |
| Total | 1,354 | 1,282 (0.947) | 1,282 (0.947) | 1,323 (0.977) |
| Unmodified | ||||
| D2 | 6,025 | 6,011 (0.998) | 6,011 (0.998) | 6,023 (0.999) |
| E1 | 36 | 33 (0.917) | 33 (0.917) | 36 (1.000) |
| E2 | 37 | 36 (0.973) | 37 (1.000) | 37 (1.000) |
| E3 | 401 | 398 (0.993) | 398 (0.992) | 401 (1.000) |
| Total | 6,499 | 6,478 (0.997) | 6,479 (0.997) | 6,497 (1.000) |
Comparative Studies
To benchmark the capability of the method, PILOT_PTM was compared with five state-of-the-art algorithms using the modified data sets B, C, and D1. The compared algorithms include three hybrid sequence tag/database approaches (InsPecT (4), Modi (7), and VEMS (6)) and two pure database approaches (Mascot (11) and X!Tandem (14)). Data sets A1, A2, and A3 were not used because all spectra are chemically synthesized and thus did not necessarily correspond to a peptide that would be found in a database as a result of a tryptic digest. These data sets were instead analyzed with phosphopeptide site assignment software Phosida (31). Details about the algorithm parameters used for each data set are given in the supplemental methods.
Test Set A: Chemically Synthesized Phosphopeptides
As a large majority of the phosphopeptides used in this data set did not correspond to a tryptically digested peptide found in the NCBInr database, a comparison with the five software packages listed above could not be done. A comparison can be made with current phosphopeptide site assignment software such as Phosida (31), which attempts to localize a phosphorylation modification on a template amino acid sequence. Phosida is only capable of predicting serine and threonine phosphorylations on peptides that contain at least 13 amino acids. Of the four peptides meeting this criteria (P1, DLDVPIPGRFDRRVpSVAAE; P2, FQpSEEQQQTEDELQDK; P3, RPVSSAApSVYAGAC; and P4, SFVLNPTNIGMpSKSSQGHVTK), Phosida was only able to assign the correct phosphorylation residue to P1 and P3. The serine at position 15 was incorrectly assigned the modification for P4, and no phosphorylation was assigned for P1.
Test Set B: Histone H3-(1–50) N-terminal Tail
Mascot was the only compared algorithm for data set B because it is the only algorithm of the five that is specifically capable of handling the highly modified ECD spectra. Although X!Tandem is able to search for c- and z•-ions, the algorithm imposes an upper bound of one modification type for each amino acid. As all of the histone MS/MS spectra in data set B contain more than one type of lysine modification, it was expected that X!Tandem would not be able to accurately identify the modifications in this data set. In fact, when tested, X!Tandem was unable to assign a sequence to any of the 58 spectra.
Of the 58 MS/MS spectra in data set B, Mascot was not able to completely annotate any of the peptides and only correctly annotated 1 (1.7%) when allowing for up to two incorrect modifications (Table IV). Alternatively, PILOT_PTM was able to completely annotate 52 peptides (89.7%). Of the six peptides that PILOT_PTM did not completely annotate, the acetylation on lysine 14 was improperly assigned to lysine 18, indicating that PILOT_PTM was still able to assign the proper modification type. The total number of correct residues is also higher for PILOT_PTM (2,888; 99.6%) than for Mascot (2,595; 89.5%) even though Mascot only allows for modifications on lysine, arginine, serine, threonine, and the termini (supplemental methods), whereas PILOT_PTM utilizes the universal list. This is clear evidence of the ability of PILOT_PTM to accurately predict modification types when given a high resolution MS/MS spectrum. The authors note that Mascot is able to search using the entire list of modifications found in the UniMod database (29) in an error-tolerant search (40). The results of the error-tolerant search were slightly worse than the original search. Mascot retained 99.2% of the original residue annotations with some of the previously unmodified residues now containing small mass shifts associated with deamidation or amidation.
Table IV. Comparison results for H3-(1–50) spectra (data set B).
There exist 464 lysine residues and 2,900 total residues for the 58 spectra. Lysine residues 9, 14, 23, 27, and 36 correspond to the modified residues for each spectra. The percent of correct annotations is given in parenthesis next to the number of correct annotations. PILOT_PTM data is reported in boldface font.
| PILOT_PTM | Mascot | |
|---|---|---|
| Lysine modifications | ||
| Lys-4 | 58 (1.000) | 51 (0.879) |
| Lys-9 | 58 (1.000) | 18 (0.310) |
| Lys-14 | 52 (0.966) | 48 (0.828) |
| Lys-18 | 52 (0.966) | 1 (0.017) |
| Lys-23 | 58 (1.000) | 2 (0.034) |
| Lys-27 | 58 (1.000) | 31 (0.534) |
| Lys-36 | 58 (1.000) | 39 (0.672) |
| Lys-37 | 58 (1.000) | 56 (0.966) |
| Modified | 452 (0.974) | 246 (0.530) |
| Total | 452 (0.974) | 246 (0.530) |
| Overall accuracy | ||
| Correct peptides | 52 (0.897) | 0 (0.000) |
| Within 1 residue | 52 (0.897) | 0 (0.000) |
| Within 2 residues | 52 (0.897) | 1 (0.017) |
| Correct residues | 2,888 (0.996) | 2,595 (0.895) |
It is speculated that lysine methylation, dimethylation, trimethylation, and acetylation interact together on the histone H3-(1–50) N-terminal tail to give rise to a potential histone “code” (47). It is highly essential that a PTM prediction algorithm be capable of accurately identifying not only the types of modifications but also the appropriate residues. Thus, we focused on the annotation of the eight lysine residues in the H3-(1–50) N-terminal tail (Table IV). PILOT_PTM was able to accurately identify 452 of the 464 (97.4%) lysine residues (modified or unmodified), whereas Mascot was only able to identify 246 (53.0%) residues. Moreover, PILOT_PTM was able to correctly annotate the lysines at positions 9, 14, 23, 27, 36, and 37 for all 58 spectra. In fact, the highest scoring lysine residues for Mascot are at positions closest to the termini (4, 14, and 37) where the fragmentation is most prevalent for the annotated modified form (supplemental annotations). Mascot scored very poorly for the lysine residues at positions 18 and 23, possibly resulting from the weaker fragmentation and the likely presence of other modified forms of lower abundance (Table IV).
Test Sets C and D1: Algorithm Comparison Protocol
To compare the capability of PILOT_PTM against alternative prediction algorithms for test data sets C and D1, a testing protocol was developed for those algorithms that place an upper bound on the number of modification sites or types. We begin by constructing the set SAnn, which is the set of modifications that was used to create the annotated spectra. Note that SAnn will be different for data sets C and D1. For each data set, we then create a superset of common modifications, STest, from the set SAnn by adding additional modifications that have been reported on the peptides (data set C) (30) or are commonly found (data set D1) (11). A set of modifications is chosen for a trial as follows. 1) Select a set of modifications, SKnown, from the annotated set SAnn that are known to be in the sample. This reflects the user's knowledge of the sample in question and the PTMs thought to be present. The set SKnown was fixed to be the four most prevalent modifications in the annotated data set. 2) Select at random a set of unknown modifications, SUnk, from the remaining modifications in STest until the total amount of known and unknown modifications totals nine, which is the upper bound for variable modification types for Mascot. This reflects the user's uncertainty about the additional test modifications that may or may not be present. The set of nine modifications extracted from steps 1 and 2 represents the variable modifications that will be checked. The modification list used in the protocol is presented in Table V. The modifications that comprise STest for each data set will be marked as either present (P) in the test set only, present in the annotated set (A), or present in the annotated set and the known set (C). The modifications in SAnn will either be marked as A or C, and the modifications in SKnown are marked as C. All remaining modifications are marked as not present in the data set (N).
Table V. Testing protocol modification list.
Each modification is either marked as “N,” not present in the test set STest, “P,” present in the test set only, “A,” present in the test set and in the annotated set SAnn, or “C,” present in the test and annotated sets and kept constant during the testing protocol (present in SKnown).
| Residue | Modification | Mass (Da) | Data set C | Data set D1 |
|---|---|---|---|---|
| C terminus | Amidation | −0.9841 | P | A |
| Asn | Deamidation | 0.9841 | N | P |
| Gln | Deamidation | 0.9841 | N | P |
| Arg | Citrullination | 0.9841 | P | N |
| Asp | Methylation | 14.0157 | N | P |
| Glu | Methylation | 14.0157 | N | P |
| Lys | Methylation | 14.0157 | A | A |
| Arg | Methylation | 14.0157 | P | A |
| C terminus | Methylation | 14.0157 | A | N |
| Met | Oxidation | 15.9949 | N | C |
| Trp | Oxidation | 15.9949 | N | P |
| Lys | Dimethylation | 28.0313 | A | C |
| Arg | Dimethylation | 28.0313 | P | P |
| Met | Dioxidation | 31.9898 | N | P |
| Lys | Acetylation | 42.0106 | C | C |
| N terminus | Acetylation | 42.0106 | A | C |
| Lys | Trimethylation | 42.0470 | P | N |
| N terminus | Propionylation | 56.0262 | C | N |
| Lys | Propionylation | 56.0262 | C | N |
| Lys | Methylated propionylation | 70.0419 | C | N |
| Ser | Phosphorylation | 79.9663 | P | A |
| Thr | Phosphorylation | 79.9663 | P | P |
| Tyr | Phosphorylation | 79.9663 | P | P |
To estimate the expected prediction accuracy, we create multiple modification lists using the above methodology to be tested with the methods Mascot (11), X!Tandem (14), InsPecT (4), and VEMS (6). Note that multiple modification lists are not needed for PILOT_PTM or Modi (7) because these methods place no restriction on the number of variable modifications. For each variable modification list, a separate trial was conducted for Mascot, X!Tandem, InsPecT, and VEMS. We report both the average results and aggregate results over all trials. Average results are calculated by first calculating the accuracy of an algorithm for each trial and then determining the average result over all trials. The aggregate result is calculated by first finding the highest scoring peptide for each spectra over all trials and then performing the accuracy calculations on these peptides.
For data set D1, the protocol is slightly modified for the Modi algorithm to account for the fact that Modi can handle the universal list of modifications but requires a database of at most 20 proteins. Thus, we first determine the 10 proteins that correspond to the largest total amount of modified peptides and then randomly select 10 additional proteins that contain at least one modified or unmodified spectrum in data sets D1 or D2. Average and aggregate results are calculated in a way similar to that for the above methods. Note that this procedure is not necessary for data set C because all of the test peptides come from a single histone H3 protein. The remaining 19 proteins are randomly selected from the NCBInr database with human taxonomy.
Test Sets C and D1
The peptide and residue prediction accuracy for all algorithms is presented in Table VI. We first note that the aggregate score for any algorithm is higher than the corresponding average score. This is not surprising because the aggregate accuracy comprises the highest scoring results from each trial, but it should be noted that multiple trials are needed to determine the aggregate score. Thus, there is clearly an added cost in terms of the number of trials that need to be run to achieve the aggregate score. We look to the average score for an indication of how accurate an algorithm is on any given trial and expect the accuracy to improve to the aggregate accuracy as more trials are run. Note that PILOT_PTM, Modi (data set C only), and InsPecT in unrestricted mode (data set D1 only) do not require more than one trial. Although the average and aggregate scores are reported for the residue prediction accuracy (Table VI), the peptide prediction accuracy (Table VI), and the subsequence accuracy (Table VII and Fig. 2), the following discussion will solely focus on the aggregate results.
Table VI. Peptide and residue accuracies for comparison using propionylated histone fragments (data set C) and total chromatin peptides (data set D1).
Data set C contained 553 spectra with a total of 5,790 residues. Data set D1 contained 525 spectra with a total of 6,892 residues. Parameters for each of the algorithms were chosen to reflect the quality of the spectra as well as the possibility for multiple modifications and missed cleavages. The results for Mascot, InsPecT, VEMS, X!Tandem, and Modi contain both averaged (Avg.) and aggregated (Agg.) results based on the protocol described in the text. For data set D1, InsPecT was also run in unrestricted search mode (Unr.) while allowing up to two modifications. The percent of correct annotations is given in parenthesis next to the number of correct annotations. PILOT_PTM data is reported in boldface font.
| Algorithm | Peptide | Within 1 | Within 2 | Residue |
|---|---|---|---|---|
| Data set C: propionylated histone fragments | ||||
| PILOT_PTM | 522 (0.944) | 522 (0.944) | 536 (0.969) | 5,716 (0.987) |
| Mascot (Avg.) | 449.8 (0.813) | 449.8 (0.813) | 473.2 (0.856) | 5,115.3 (0.883) |
| Mascot (Agg.) | 474 (0.857) | 474 (0.857) | 501 (0.906) | 5,337 (0.922) |
| InsPecT (Avg.) | 464.3 (0.840) | 464.3 (0.840) | 490.5 (0.887) | 5,289.8 (0.914) |
| InsPecT (Agg.) | 484 (0.875) | 484 (0.875) | 524 (0.948) | 5,492 (0.949) |
| VEMS (Avg.) | 107.6 (0.195) | 107.6 (0.195) | 180.3 (0.326) | 2,196.2 (0.379) |
| VEMS (Agg.) | 127 (0.230) | 127 (0.230) | 216 (0.391) | 2,391 (0.413) |
| X!Tandem (Avg.) | 186.6 (0.337) | 225.3 (0.407) | 253.0 (0.458) | 2,976.1 (0.514) |
| X!Tandem (Agg.) | 213 (0.385) | 251 (0.454) | 301 (0.544) | 3,365 (0.581) |
| Modi | 146 (0.264) | 146 (0.264) | 206 (0.373) | 2,333 (0.403) |
| Data set D1: total chromatin peptides | ||||
| PILOT_PTM | 500 (0.952) | 500 (0.952) | 511 (0.973) | 6,850 (0.994) |
| InsPecT (Avg.) | 468.8 (0.893) | 471.5 (0.898) | 486.6 (0.927) | 6,603.4 (0.958) |
| InsPecT (Agg.) | 482 (0.918) | 485 (0.924) | 493 (0.939) | 6,693 (0.971) |
| InsPecT (Unr.) | 274 (0.522) | 290 (0.552) | 385 (0.733) | 5,490 (0.797) |
| VEMS (Avg.) | 377.6 (0.719) | 377.9 (0.720) | 400.6 (0.763) | 5,474.8 (0.794) |
| VEMS (Agg.) | 390 (0.743) | 391 (0.745) | 441 (0.840) | 5,639 (0.818) |
| X!Tandem (Avg.) | 455.4 (0.867) | 455.4 (0.867) | 464.0 (0.884) | 6,271.7 (0.910) |
| X!Tandem (Agg.) | 471 (0.897) | 471 (0.897) | 493 (0.939) | 6,420 (0.932) |
| Modi (Avg.) | 73.1 (0.139) | 73.1 (0.139) | 101.5 (0.193) | 1,263.8 (0.183) |
| Modi (Agg.) | 295 (0.562) | 295 (0.562) | 327 (0.623) | 4,299 (0.624) |
Table VII. Subsequence accuracy results for comparison using propionylated histone fragments (data set C) and total chromatin peptides (data set D1).
The results for Mascot, InsPecT, VEMS, X!Tandem, and Modi contain both averaged (Avg.) and aggregated (Agg.) results based on the protocol described in the text. For data set D1, InsPecT was also run in unrestricted search mode (Unr.) while allowing up to two modifications. The percent of correct annotations is given in parenthesis next to the number of correct annotations. PILOT_PTM data is reported in boldface font.
| L = 3 | L = 4 | L = 5 | L = 6 | L = 7 | L = 8 | L = 9 | |
|---|---|---|---|---|---|---|---|
| Data set C: propionylated histone fragments | |||||||
| Total peptides | 553 | 553 | 553 | 553 | 514 | 514 | 514 |
| PILOT_PTM | 553 (1.000) | 553 (1.000) | 548 (0.991) | 548 (0.991) | 501 (0.975) | 501 (0.975) | 495 (0.963) |
| Mascot (Avg.) | 490.9 (0.888) | 489.1 (0.884) | 489.0 (0.884) | 480.2 (0.868) | 458.3 (0.892) | 458.2 (0.891) | 458.2 (0.891) |
| Mascot (Agg.) | 526 (0.951) | 525 (0.949) | 521 (0.942) | 521 (0.942) | 483 (0.940) | 483 (0.940) | 482 (0.938) |
| InsPecT (Avg.) | 501.6 (0.907) | 472.9 (0.855) | 456.5 (0.825) | 425.7 (0.770) | 403.7 (0.785) | 402.7 (0.783) | 402.1 (0.782) |
| InsPecT (Agg.) | 525 (0.949) | 520 (0.940) | 511 (0.924) | 502 (0.908) | 431 (0.839) | 430 (0.837) | 430 (0.837) |
| X!Tandem (Avg.) | 308.4 (0.558) | 307.3 (0.556) | 297.7 (0.538) | 294.5 (0.533) | 265.4 (0.516) | 262.1 (0.510) | 221.1 (0.430) |
| X!Tandem (Agg.) | 342 (0.618) | 339 (0.613) | 331 (0.599) | 328 (0.593) | 301 (0.586) | 301 (0.586) | 286 (0.556) |
| VEMS (Avg.) | 332.8 (0.602) | 203.3 (0.368) | 193.9 (0.351) | 189.2 (0.342) | 175.6 (0.342) | 175.6 (0.342) | 175.6 (0.342) |
| VEMS (Agg.) | 363 (0.656) | 287 (0.519) | 259 (0.468) | 243 (0.439) | 212 (0.412) | 209 (0.407) | 209 (0.407) |
| Modi | 231 (0.418) | 219 (0.396) | 211 (0.382) | 211 (0.382) | 210 (0.409) | 210 (0.409) | 210 (0.409) |
| Data set D1: total chromatin peptides | |||||||
| Total peptides | 525 | 525 | 525 | 525 | 523 | 515 | 505 |
| PILOT_PTM | 525 (1.000) | 522 (0.994) | 521 (0.992) | 521 (0.992) | 519 (0.992) | 511 (0.992) | 495 (0.980) |
| InsPecT (Avg.) | 503.3 (0.959) | 499.2 (0.951) | 491.4 (0.936) | 487.5 (0.929) | 483.7 (0.925) | 480.5 (0.933) | 472.5 (0.936) |
| InsPecT (Agg.) | 518 (0.987) | 509 (0.970) | 505 (0.962) | 499 (0.950) | 497 (0.950) | 491 (0.953) | 485 (0.960) |
| InsPecT (Unr.) | 515 (0.981) | 502 (0.956) | 486 (0.926) | 471 (0.897) | 453 (0.866) | 431 (0.837) | 412 (0.816) |
| X!Tandem (Avg.) | 465.9 (0.887) | 465.9 (0.887) | 464.0 (0.884) | 463.4 (0.883) | 463.4 (0.886) | 463.3 (0.900) | 456.4 (0.904) |
| X!Tandem (Agg.) | 481 (0.916) | 481 (0.916) | 479 (0.912) | 478 (0.910) | 478 (0.914) | 478 (0.928) | 471 (0.933) |
| VEMS (Avg.) | 457.8 (0.872) | 451.7 (0.860) | 445.6 (0.849) | 443.4 (0.845) | 440.8 (0.843) | 439.4 (0.853) | 424.8 (0.841) |
| VEMS (Agg.) | 471 (0.897) | 463 (0.882) | 461 (0.878) | 457 (0.870) | 457 (0.874) | 455 (0.883) | 441 (0.873) |
| Modi (Avg.) | 328.2 (0.625) | 319.1 (0.608) | 319.1 (0.608) | 317.9 (0.606) | 267.2 (0.511) | 107.3 (0.208) | 70.1 (0.139) |
| Modi (Agg.) | 332 (0.632) | 330 (0.629) | 328 (0.625) | 328 (0.625) | 301 (0.576) | 298 (0.579) | 278 (0.550) |
Fig. 2.

Subsequence accuracy results from comparisons using data sets C and D1. The results from Mascot (data set C), InsPecT (restricted), VEMS, X!Tandem, and Modi (data set D1) are calculated using many trials, each consisting of a distinct variable modification list. The results for PILOT_PTM, Modi (data set C), and InsPecT in blind search mode (unrestricted (Unr.); data set D1) are reported for only one trial because these algorithms do not place a restriction on the types of variable modifications considered. a, aggregate (Agg.) subsequence accuracies for the histone fragment test data set. b, average (Avg.) subsequence accuracies for the histone fragment test data set. c, aggregate subsequence accuracies for the chromatin test data set. d, average subsequence accuracies for the chromatin test data set.
PILOT_PTM is able to fully predict 522 (94.4%) of the peptides from data set C and 500 (95.2%) from data set D1. Alternatively, Mascot is only able to fully identify 474 (85.7%) peptides from data set C, whereas InsPecT fully identifies 484 (87.5%) from data set C and 482 (91.8%) from data set D1 (Table VI). The accuracy of the remaining algorithms for data set C was significantly lower than that for Mascot with X!Tandem reporting the highest of the remainder (213 peptides). X!Tandem was also able to fully identify 471 (89.7%) of the peptides in data set D1 followed by VEMS with a total of 390 (74.3%). The blind search of InsPecT reported only 274 (52.2%) fully annotated peptides from data set D1, the lowest of all algorithms (Table VI, aggregate scores only). When allowing for up to two incorrect residues, PILOT_PTM is able to predict 536 peptides (96.9%) from data set C and 511 (97.3%) from data set D1. InsPecT is the next highest in data set C with 524 identified peptides (94.8%) and ties X!Tandem with 493 peptides (93.9%) for data set D1. Mascot scores the next highest in data set C with 501 peptides (90.6%), and VEMS follows InsPecT and X!Tandem in data set D1 with 441 peptides (84.0%). We also see that the blind search of InsPecT accuracy improves to 73.3% (385 peptides) when allowing for two incorrect modifications, although it is still 10.7% lower than VEMS.
The ability to predict a subsequence of a given length gives insight into the effectiveness of an algorithm to sequence a portion of the peptide using appropriate spectral information (Fig. 2 and Table VII). PILOT_PTM reports a subsequence accuracy of 100% for all L ≤ 4 in data set C and for all L ≤ 3 in data set D1. This implies that PILOT_PTM was able to correctly annotate four consecutive amino acids for all 553 spectra in data set C and three consecutive amino acids for all 525 spectra in data set D1. Additionally, PILOT_PTM outperforms all competing algorithms for each listed length for both data set C and set D1 and maintains an accuracy that is at least 3.5% greater than the next highest scoring algorithm for data set C and at least 1.3% greater for data set D1 (Table VII).
Complete MS Analysis
Complete LC-MS/MS Untargeted Modification Search Protocol
To run an untargeted modification search with PILOT_PTM on a complete MS scan, we must first generate a set of candidate template sequences for use with the algorithm. The sequences will be generated by initially scanning the data using a peptide sequencing algorithm to uncover all spectra that are either unmodified or contain oxidized methionine. Using the SEQUEST algorithm (10), a protein list (PL; probability >5e−5) was generated, and a superset of candidate template sequences (TS) is then defined as the non-redundant list of peptide sequences found in the search. We augment PL with a dummy “No match” protein and then map all peptides in TS to their corresponding proteins in PL. Any peptide that was not assigned a protein from the SEQUEST search is assigned No match.
The spectra not annotated by SEQUEST were subject to filtering where those that did not contain at least 50 ion peaks were removed. For each remaining spectra, we run the following sequence of steps. 1) Determine a three-amino acid unmodified sequence tag based on the experimental data. 2) Search all peptides in TS to derive a list of template sequences that exactly contain the sequence tag. 3) Run the PILOT_PTM algorithm for each template sequence. 4) Compare the cross-correlation score of the top modified peptide for each template sequence and select the peptide that has the highest score. Because of the large number of template sequences generated during step 2 of the above procedure, we imposed a window on the possible mass gaps for possible modifications (step 3). We set the lower bound to be −50 Da and the upper bound to be 250 Da.
Case Study: Total Chromatin Extraction
The above protocol was tested on several data sets generated from a total chromatin extraction. All spectra that had a minimum XCorr value (1.5 for z = 1, 2.0 for z = 2, 2.5 for z = 3, and 3.0 for z = 4) were annotated with the associated peptide and oxidized methionine modifications (if applicable), and PL and TS were generated as described above. A total of 466,905 spectra were initially analyzed with the SEQUEST algorithm. A total of 81,961 unmodified spectra and 4,838 modified spectra were found, yielding 19,250 distinct peptides and 1,913 distinct proteins. After applying the ion peak filtering, a total of 273,733 spectra were analyzed with PILOT_PTM. Prior to searching, all isotopic labels were removed from the universal list of modifications as these modifications will not be present in the chromatin data.
The sequence tag generation step reduced the number of template sequences per MS/MS spectrum to ∼10 on average. Some of these sequences will not be able to generate appropriate sets of valid candidate ion peaks (CSk) for several consecutive amino acids because of the inability to connect candidate ion peaks with an appropriate series of jumps. The total number of template sequences fully analyzed by PILOT_PTM is ∼6 per MS/MS spectrum on average (46,610 sequences).
PILOT_PTM assigned a sequence with modifications or amino acid substitutions to 7,641 spectra, including a total of 6,356 modifications and 11,391 substitutions. We report the histogram of modifications for all annotated spectra in Fig. 3. Oxidized methionine (1,957 PTMs) was removed from Fig. 3 to show the counts of the other modifications in higher detail. C-terminal methylation and N-terminal acetylation appeared 829 and 330 times, respectively. However, these modifications are likely the result of sample preparation and not post-translational modification. Methylation is the most prevalent modification appearing on the N terminus (257 PTMs), lysine (179 PTMs), arginine (110 PTMs), aspartic acid (109 PTMs), asparagine (88 PTMs), threonine (67 PTMs), and glutamine (33 PTMs). Dimethylation is the next most abundant modification appearing on the N terminus (247 PTMs), lysine (184 PTMs), arginine (102 PTMs), and asparagine (35 PTMs). Acetylation is annotated on serine (110 PTMs) and lysine (46 PTMs); deamidation is annotated on glutamine (163 PTMs), asparagine (83 PTMs), and arginine (15 PTMs); formylation is annotated on serine (65 PTMs) and threonine (62 PTMs); and hydroxylation is annotated on proline (121 PTMs), valine (103 PTMs), and aspartic acid (39 PTMs).
Fig. 3.

Modification histogram for untargeted analysis of a chromatin fraction. Each modification is given an abbreviation (Abb.) of the form AA-M where AA is the name of the amino acid and M is the modification type. Note that CT and NT refer to the C terminus and N terminus, respectively. a, histogram of the total amount of modification counts present in all 7,668 annotated spectra. b, numerical table for the modifications in a.
DISCUSSION
A novel integer linear framework for the assignment of PTMs on a template sequence was developed. PILOT_PTM utilizes the universal list of modifications while placing no restrictions on the amount of modification types or modification sites for a given peptide. The case studies presented above demonstrate the high accuracy of the PILOT_PTM algorithm when analyzing modified spectra that come from different mass spectrometers as well as different fragmentation patterns. The superior ability of PILOT_PTM when compared with five current PTM prediction algorithms is demonstrated using highly modified histone H3-(1–50) peptides and peptides from a large scale chromatin-enriched fraction. The performance of PILOT_PTM may be due to the amount of peaks selected from the MS/MS spectrum for analysis. Database and hybrid methods may use fewer peaks to discriminate between correct and incorrect results, but it is often necessary to utilize lower abundance peaks to properly assign the modification type and modification site when a large variable modification list is considered. To maintain the efficiency of PILOT_PTM when the MS/MS spectrum contains many peaks, a strict filtering algorithm is used during the preprocessing stage (supplemental methods) to eliminate all possible isotopes, neutral losses, and multiply charged ions from consideration in the candidate peak list. In fact, the preprocessing stage is crucial for the ECD data where the spectral resolution often enables proper assignment of many charge states that can be converted into the appropriate singly charged peak or removed from consideration.
The computational run time for a completely automated run of PILOT_PTM for a single template sequence is shown in detail for the data sets in Table VIII. The time is reported for both the single thread and parallelized version of CPLEX (55) (eight threads) on average on a Intel Pentium 4 3.0-GHz Linux-based computer. For each data set, we calculate the average number of residues, R̄, per peptide by dividing the total number of residues (Table II) by the total number of peptides (Table III). The average CPU time for all data sets ranged from 8.7 to 18.3 CPU s with ranges from 9.1 to 16.1 for all data sets except set B. The increase in CPU time for this data set is due to the large number of peaks present in the ECD data set (R̄ = 50) that were retained by PILOT_PTM. The average run time is reduced on average by a factor of 4.85 if a parallelized version of CPLEX is used. With an average computational time of 2.8 CPU s per spectrum, the total time required to run all stages of PILOT_PTM and output results for all 7,853 spectra is 6.1 CPU h. Furthermore, addition of modifications to the universal list does not result in an increase in PILOT_PTM run time because the amount of binary variables and constraints will remain unchanged in the ILP. The raw spectral data and source code for the PILOT_PTM algorithm are available upon request.
Table VIII. Average PILOT_PTM computational time per spectrum for each data set.
The average time to process a spectrum was measured using CPLEX version 11.1 on a Pentium 4 3.0-GHz Linux-based computer. The parallel time utilized the parallel CPLEX software package on an eight-thread unit. The reported time is taken as the average over all spectrum in that data set. The average number of residues per peptide, R̄, is calculated for each data set as the total number of residues divided by the total number of peptides.
| Data set | Avg. time | Avg. parallel time | R̄ |
|---|---|---|---|
| s | s | ||
| A1 | 8.7 | 1.7 | 9.2 |
| A2 | 18.3 | 3.5 | 16.1 |
| A3 | 16.9 | 3.5 | 14.1 |
| B | 98.3 | 20.1 | 50.0 |
| C | 17.3 | 3.4 | 10.5 |
| D1 | 15.2 | 3.0 | 13.1 |
| D2 | 12.6 | 2.6 | 14.0 |
| E1 | 11.8 | 2.4 | 11.3 |
| E2 | 10.9 | 2.1 | 11.3 |
| E3 | 9.8 | 2.0 | 9.1 |
| Total | 13.6 | 2.8 | 13.7 |
Supplementary Material
* This work was supported, in whole or in part, by National Institutes of Health Grant R01LM009338 (to C. A. F.). This work was also supported by the United States Environmental Protection Agency Science to Achieve Results Program through Grant R 832721-010.
 This article contains supplemental Table 1, methods, and annotations.
This article contains supplemental Table 1, methods, and annotations.
2 K. Mitchelhill, Delta Mass: a Database of Protein Post Translational Modifications.
1 The abbreviations used are:
- PTM
- post-translational modification
- VEMS
- Virtual Expert Mass Spectrometrist
- ETD
- electron transfer dissociation
- ECD
- electron collision dissociation
- ILP
- integer linear optimization
- LP
- linear programming
- PL
- protein list
- TS
- template sequences
- CPU
- central processing unit.
REFERENCES
- 1.Nesvizhskii A. I., Vitek O., Aebersold R. (2007) Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat. Methods 4, 787–797 [DOI] [PubMed] [Google Scholar]
- 2.Witze E. S., Old W. M., Resing K. A., Ahn N. G. (2007) Mapping protein post-translational modifications with mass spectrometry. Nat. Methods 4, 798–806 [DOI] [PubMed] [Google Scholar]
- 3.Mann M., Wilm M. (1994) Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal. Chem 66, 4390–4399 [DOI] [PubMed] [Google Scholar]
- 4.Tanner S., Shu H., Frank A., Wang L. C., Zandi E., Mumby M., Pevzner P. A., Bafna V. (2005) InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem 77, 4626–4639 [DOI] [PubMed] [Google Scholar]
- 5.Searle B. C., Dasari S., Wilmarth P. A., Turner M., Reddy A. P., David L. L., Nagalla S. R. (2005) Identification of protein modifications using MS/MS de novo sequencing and the OpenSea alignment algorithm. J. Proteome Res 4, 546–554 [DOI] [PubMed] [Google Scholar]
- 6.Matthiesen R., Trelle M. B., Højrup P., Bunkenborg J., Jensen O. N. (2005) VEMS 3.0: algorithms and computational tools for tandem mass spectrometry based identification of post-translational modifications in proteins. J. Proteome Res 4, 2338–2347 [DOI] [PubMed] [Google Scholar]
- 7.Kim S., Na S., Sim J. W., Park H., Jeong J., Kim H., Seo Y., Seo J., Lee K. J., Paek E. (2006) Modi: a powerful and convenient web server for identifying multiple post-translational peptide modifications from tandem mass spectra. Nucleic Acids Res 34, W258–W263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savitski M. M., Nielsen M. L., Zubarev R. A. (2006) ModifiComb, a new proteomic tool for mapping substoichiometric post-translational modifications, finding novel types of modifications, and fingerprinting complex protein mixtures. Mol. Cell. Proteomics 5, 935–948 [DOI] [PubMed] [Google Scholar]
- 9.Zamdborg L., LeDuc R. D., Glowacz K. J., Kim Y. B., Viswanathan V., Spaulding I. T., Early B. P., Bluhm E. J., Babai S., Kelleher N. L. (2007) Prosight PTM 2.0: improved protein identification and characterization for top down mass spectrometry. Nucleic Acids Res 35, W701–W706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yates J. R., 3rd, Eng J. K., McCormack A. L., Schieltz D. (1995) Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem 67, 1426–1436 [DOI] [PubMed] [Google Scholar]
- 11.Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 12.Lu B., Chen T. (2003) A suffix tree approach to the interpretation of tandem mass spectra: applications to peptides of non-specific digestion and post-translational modifications. Bioinformatics 19, ii113–ii121 [DOI] [PubMed] [Google Scholar]
- 13.Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 14.Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 15.Shadforth I., Xu W., Crowther D., Bessant C. (2006) GAPP: a fully automated software for the confident identification of human peptides from tandem mass spectra. J. Proteome Res 5, 2849–2852 [DOI] [PubMed] [Google Scholar]
- 16.Baumgartner C., Rejtar T., Kullolli M., Akella L. M., Karger B. L. (2008) SeMoP: a new computational strategy for the unrestricted search for modified peptides using LC-MS/MS data. J. Proteome Res 7, 4199–4208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu C., Yan B., Song Y., Xu Y., Cai L. (2006) Peptide sequence tag-based blind identification of post-translational modifications with point process model. Bioinformatics 22, e307–e313 [DOI] [PubMed] [Google Scholar]
- 18.Seo J., Jeong J., Kim Y. M., Hwang N., Paek E., Lee K. J. (2008) Strategy for Comprehensive identification of post-translational modifications in cellular proteins, including low abundant modifications: application to glyceraldehyde-3-phosphate dehydrogenase. J. Proteome Res 7, 587–602 [DOI] [PubMed] [Google Scholar]
- 19.Hansen B. T., Davey S. W., Ham A. J., Liebler D. C. (2005) P-Mod: an algorithm and software to map modifications to peptide sequences using tandem MS data. J. Proteome Res 4, 358–368 [DOI] [PubMed] [Google Scholar]
- 20.DiMaggio P. A., Jr., Young N. L., Baliban R. C., Garcia B. A., Floudas C. A. (2009) A mixed-integer linear optimization framework for the identification and quantification of targeted post-translational modifications of highly modified proteins using multiplexed ETD tandem mass spectrometry. Mol. Cell. Proteomics 8, 2527–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Havilio M., Wool A. (2007) Large-scale unrestricted identification of post-translational modifications using tandem mass spectrometry. Anal. Chem 79, 1362–1368 [DOI] [PubMed] [Google Scholar]
- 22.Chen Y., Chen W., Cobb M. H., Zhao Y. (2009) PTMap—a sequence alignment software for unrestricted, accurate, and full-spectrum identification of post-translational modification sites. Proc. Natl. Acad. Sci. U.S.A 106, 761–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chalkley R. J., Baker P. R., Medzihradszky K. F., Lynn A. J., Burlingame A. L. (2008) In-depth analysis of tandem mass spectrometry data from disparate instrument types. Mol. Cell. Proteomics 7, 2386–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang N., Aebersold R., Schwikowski B. (2002) ProbID: a probabilistic algorithm to identify peptides through sequence database searching using tandem mass spectrometry. Proteomics 2, 1406–1412 [DOI] [PubMed] [Google Scholar]
- 25.Agilent (2004) Spectrum Mill for MassHunter Workstation, Agilent, Santa Clara, CA [Google Scholar]
- 26.Colinge J., Masselot A., Giron M., Dessingy T., Magnin J. (2003) OLAV: towards high-throughput tandem mass spectrometry data identification. Proteomics 3, 1454–1463 [DOI] [PubMed] [Google Scholar]
- 27.Tabb D. L., Fernando C. G., Chambers M. C. (2007) MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res 6, 654–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hernandez P., Gras R., Frey J., Appel R. D. (2003) Popitam: towards new heuristic strategies to improve protein identification from tandem mass spectrometry data. Proteomics 3, 870–878 [DOI] [PubMed] [Google Scholar]
- 29.Creasy D. M., Cottrell J. S. (2004) Unimod: protein modifications for mass spectrometry. Proteomics 4, 1534–1536 [DOI] [PubMed] [Google Scholar]
- 30.Garavelli J. S. (2004) The RESID database of protein modifications as a resource and annotation tool. Proteomics 4, 1527–1533 [DOI] [PubMed] [Google Scholar]
- 31.Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 32.Syka J. E., Coon J. J., Schroeder M. J., Shabanowitz J., Hunt D. F. (2004) Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U.S.A 101, 9528–9533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mikesh L. M., Ueberheide B., Chi A., Coon J. J., Syka J. E., Shabanowitz J., Hunt D. F. (2006) The utility of ETD mass spectrometry in proteomic analysis. Biochim. Biophys. Acta 1764, 1811–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Udeshi N. D., Shabanowitz J., Hunt D. F., Rose K. L. (2007) Analysis of proteins and peptides on a chromatographic timescale by electron-transfer dissociation MS. FEBS J 274, 6269–6276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zubarev R. A., Kelleher N. L., McLafferty F. W. (1998) Electron capture dissociation of multiply charged protein cations. A nonergodic process. J. Am. Chem. Soc 120, 3265–3266 [Google Scholar]
- 36.Bakhtiar R., Guan Z. (2006) Electron capture dissociation mass spectrometry in characterization of peptides and proteins. Biotechnol. Lett 28, 1047–1059 [DOI] [PubMed] [Google Scholar]
- 37.Good D. M., Wenger C. D., McAlister G. C., Bai D. L., Hunt D. F., Coon J. J. (2009) Post-acquisition ETD spectral processing for increased peptide identifications. J. Am. Soc. Mass Spectrom 20, 1435–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swaney D. L., McAlister G. C., Coon J. J. (2008) Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat. Methods 5, 959–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Craig R., Beavis R. C. (2003) A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun. Mass Spectrom 17, 2310–2316 [DOI] [PubMed] [Google Scholar]
- 40.Creasy D. M., Cottrell J. S. (2002) Error tolerant searching of uninterpreted tandem mass spectrometry data. Proteomics 2, 1426–1434 [DOI] [PubMed] [Google Scholar]
- 41.Tanner S., Pevzner P. A., Bafna V. (2006) Unrestrictive identification of post-translational modifications through peptide mass spectrometry. Nat. Protoc 1, 67–72 [DOI] [PubMed] [Google Scholar]
- 42.Tsur D., Tanner S., Zandi E., Bafna V., Pevzner P. A. (2005) Identification of post-translational modifications by blind search of mass spectra. Nat. Biotechnol 23, 1562–1567 [DOI] [PubMed] [Google Scholar]
- 43.Kim S., Bandeira N., Pevzner P. A. (2009) Spectral profiles, a novel representation of tandem mass spectra and their applications for de novo peptide sequencing and identification. Mol. Cell. Proteomics 8, 1391–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dimaggio P. A., Floudas C. A. (2007) A mixed-integer optimization framework for de novo peptide identification. AIChE J 53, 160–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DiMaggio P. A., Jr., Floudas C. A. (2007) De novo peptide identification via tandem mass spectrometry and integer linear optimization. Anal. Chem 79, 1433–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DiMaggio P. A., Jr., Floudas C. A., Lu B., Yates J. R., 3rd (2008) A hybrid method for peptide identification using integer linear optimization, local database search, and quadrupole time-of-flight or orbitrap tandem mass spectrometry. J. Proteome Res 7, 1584–1593 [DOI] [PubMed] [Google Scholar]
- 47.Garcia B. A., Pesavento J. J., Mizzen C. A., Kelleher N. L. (2007) Pervasive combinatorial modification of histone H3 in human cells. Nat. Methods 4, 487–489 [DOI] [PubMed] [Google Scholar]
- 48.Garcia B. A., Mollah S., Ueberheide B. M., Busby S. A., Muratore T. L., Shabanowitz J., Hunt D. F. (2007) Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc 2, 933–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shiio Y., Eisenman R. N., Yi. E. C., Donohoe S., Goodlett D. R., Aebersold R. (2003) Quantitative proteomic analysis of chromatin-associated factors. J. Am. Soc. Mass Spectrom 14, 696–703 [DOI] [PubMed] [Google Scholar]
- 50.El Gazzar M., Yoza B. K., Chen X., Garcia B. A., Young N. L., McCall C. E. (2009) Chromatin-specific remodeling by HMGB1 and linker histone H1 silences proinflammatory genes during endotoxin tolerance. Mol. Cell. Biol 29, 1959–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Purvine S., Kolker N., Kolker E. (2004) Spectral quality assessment for high throughput tandem mass spectrometry proteomics. OMICS 8, 255–265 [DOI] [PubMed] [Google Scholar]
- 52.Prince J. T., Carlson M. W., Wang R., Lu P., Marcotte E. M. (2004) The need for a public proteomics repository. Nat. Biotechnol 22, 471–472 [DOI] [PubMed] [Google Scholar]
- 53.Ma B., Zhang K., Hendrie C., Liang C., Li M., Doherty-Kirby A., Lajoie G. (2003) PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom 17, 2337–2342 [DOI] [PubMed] [Google Scholar]
- 54.Hubler S. L., Jue A., Keith J., McAlister G. C., Craciun G., Coon J. J. (2008) Valence parity renders z(*)-type ions chemically distinct. J. Am. Chem. Soc 130, 6388–6394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.IBM ILOG (2008) ILOG CPLEX C++ API 11.1 Reference Manual, IBM ILOG, Armonk, NY [Google Scholar]
- 56.Floudas C. A. (1995) Nonlinear and Mixed-Integer Optimization, Oxford University Press, New York [Google Scholar]
- 57.Nemhauser G. L., Wolsey L. A. (1988) Integer and Combinatorial Optimization, John Wiley and Sons, Inc., New York [Google Scholar]
- 58.Floudas C. A., Paules G. E., 4th (1988) A mixed-integer nonlinear programming formulation for the synthesis of heat-integrated distillation sequences. Comp. Chem. Eng 12, 531–546 [Google Scholar]
- 59.Kokossis A. C., Floudas C. A. (1991) Synthesis of isothermal reactor-separator-recycle systems. Chem. Eng. Sci 46, 1361–1383 [Google Scholar]
- 60.Kokossis A. C., Floudas C. A. (1994) Optimization of complex reactor networks—II: nonisothermal operation. Chem. Eng. Sci 49, 1037–1051 [Google Scholar]
- 61.Floudas C. A., Anastasiadis S. H. (1988) Synthesis of general distillation sequences with several multicomponent feeds and products. Chem. Eng. Sci 43, 2407–2419 [Google Scholar]
- 62.Ciric A. R., Floudas C. A. (1989) A retrofit approach for heat exchanger networks. Comp. Chem. Eng 13, 703–715 [Google Scholar]
- 63.Kinter M., Sherman N. E. (2000) Protein Sequencing and Identification Using Tandem Mass Spectrometry, John Wiley & Sons, Inc., New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.