

Abstract
Mutations of the tumor suppressor PTEN, a phosphatase with specificity for 3-phosphorylated inositol phospholipids, accompany progression of brain tumors from benign to the most malignant forms. Tumor progression, particularly in aggressive and malignant tumors, is associated with the induction of angiogenesis, a process termed the angiogenic switch. Therefore, we tested whether PTEN regulates tumor progression by modulating angiogenesis. U87MG glioma cells stably reconstituted with PTEN cDNA were tested for growth in a nude mouse orthotopic brain tumor model. We observed that the reconstitution of wild-type PTEN had no effect on in vitro proliferation but dramatically decreased tumor growth in vivo and prolonged survival in mice implanted intracranially with these tumor cells. PTEN reconstitution diminished phosphorylation of AKT within the PTEN-reconstituted tumor, induced thrombospondin 1 expression, and suppressed angiogenic activity. These effects were not observed in tumors reconstituted with a lipid phosphatase inactive G129E mutant of PTEN, a result that provides evidence that the lipid phosphatase activity of PTEN regulates the angiogenic response in vivo. These data provide evidence that PTEN regulates tumor-induced angiogenesis and the progression of gliomas to a malignant phenotype via the regulation of phosphoinositide-dependent signals.
The reversible phosphorylation of proteins and lipids is critical to the control of signal transduction in mammalian cells and is regulated by kinases and phosphatases (1). The product of the tumor suppressor gene PTEN/MMAC (hereafter termed PTEN) was identified as a dual specificity phosphatase and has been shown to dephosphorylate inositol phospholipids (2–9). The PTEN gene is mutated in 40–50% of high-grade gliomas as well as prostate, endometrial, breast, lung, and other tumors (2, 3, 10). In addition, PTEN is mutated in several rare autosomal dominant cancer predisposition syndromes, including Cowden disease, Lhermitte-Duclos disease, and Bannayan-Zonana syndrome (11–14). The phenotype of PTEN-knockout mice reveal a requirement for this phosphatase in normal development and confirm its role as a tumor suppressor (15–17).
PTEN is a 55-kDa protein comprising an N-terminal catalytic domain, identified as a segment with homology to the cytoskeletal protein tensin and containing the sequence HC(X)5R, which is the signature motif of members of the protein tyrosine phosphatase family, and a C-terminal C2 domain with lipid-binding and membrane-targeting functions (18). The sequence of the extreme C terminus of PTEN suggests a function in binding PDZ domain-containing proteins. PTEN is a dual specificity phosphatase that displays a pronounced preference for acidic substrates (5). Importantly, PTEN possesses lipid phosphatase activity, preferentially dephosphorylating phosphoinositides at the D3 position of the inositol ring. It is the only enzyme known to dephosphorylate the D3 position in inositol phospholipids, suggesting that PTEN may function as a direct antagonist of phosphatidylinositol 3-kinase (PI3-kinase) and phosphatidylinositol 3,4,5-trisphosphate [PtdIns(3,4,5)P3]-dependent signaling (7, 13). Reconstitution of PTEN in tumor cells that carry mutations in the PTEN gene have established that this phosphatase regulates the PI3-kinase-dependent activation of AKT, a major player in cell survival (6, 14). However, despite progress in understanding the biochemistry of PTEN, the role of this phosphatase in tumor progression, as it relates to its diverse effects on cell growth, angiogenesis, and/or survival, remains unclear.
Mutations in PTEN accompany progression of brain tumors from grade I/II to malignant grade III and IV in vivo (19, 20). Tumor progression is associated with angiogenesis, the formation of new blood vessels from existing vascular structures, with increases in microvessel density (MVD) and increased invasion of tumor cells into brain parenchyma (21–23). For tumor growth to occur, tumor dormancy must be broken, an event termed the angiogenic switch. During angiogenesis endothelial cells are induced to degrade the basement membrane of existing vessels, break away, and migrate to the site of the tumor, where they proliferate to form linear structures that differentiate to form blood vessels. Factors that control angiogenesis include growth factors, matrix metalloproteinases, plasminogen activators, thrombospondins, integrins αvβ3, αvβ5, and α5β1, etc. (23–25). The angiogenic switch involves a shift in the balance of angiogenic stimulators and angiogenic inhibitors. Stimulators include the growth factors, vascular endothelial growth factor and basic fibroblast growth factor, and the induction of matrix remodeling via matrix metalloproteinases (26). Inhibitors include thrombospondin 1 (TSP-1), angiostatin, endostatin, tissue inhibitors of metalloproteinases, and others (24, 27). It has been observed that neovascularization and PTEN mutations are associated with high-grade gliomas and are not observed in low-grade glial tumors, leading to the hypothesis that these two events may be causally linked.
Regulation of PI3-kinase-dependent signals, including activation of AKT by vascular endothelial growth factor and its receptors, the protein tyrosine kinases Flt-1 and KDR, have been implicated in brain tumor angiogenesis (28). Data generated in the chicken chorioallantoic membrane model suggests that PI3-kinase-dependent pathways may regulate angiogenesis and vascular endothelial growth factor expression in endothelial cells (29). Furthermore, correlative studies in prostate tumor specimens have demonstrated that tumors containing PTEN mutations have higher microvessel counts than tumors expressing wild-type (WT) PTEN (30). However, whether PTEN is causally linked to induction of angiogenesis by the tumor cell remains unproven. These and other observations led us to hypothesize that PTEN may control tumor-induced angiogenesis and contribute to the high mortality associated with malignant brain tumors.
Materials and Methods
Cell Culture, Constructs, and Reagents.
WT PTEN or mutant PTEN (G129E, R130 M) cDNAs were subcloned into the pBabe-puro retroviral expression vector. Stable clones of U87MG cells expressing WT PTEN (WT.E1, WT.C7) or mutant PTEN (G129E, R130 M) were established under puromycin selection (2 μg/ml) (6). Muristirone-induced expression of PTEN in U87MG cells was performed as described by J. Stolarov (31). Antibodies were obtained specific for PTEN (6), AKT and phospho-S473-AKT (New England Biolabs, #9270), TSP-1 (Calbiochem, #605230), and anti-BrdUrd mAb clone BU33 (Sigma, #B9285).
Tumor Implantation.
Cells were cultured in fresh medium for 24 h and harvested, adjusting the cell concentration to 1 × 106 in 10 μl of RPMI medium. Mice, under general anesthesia were placed into the stereotactic device (model 963, Kopf Instruments, Tujunga, CA). Stereotactically controlled drill assembly was used to provide a hole 0.3 mm deep and of 0.8 mm diameter in cranium at a position 0.5 mm anterior and 1.2 mm lateral to the bregmal anatomical landmark. Tumor cells (1 × 106) were introduced slowly through a 10-μl Hamilton syringe at a depth of 2.5 mm at a rate of 2 μl/min. We then slowly removed the needle at a rate of 0.5 mm/min. After needle removal we sealed the hole with bone wax and closed the incision with a wound clip. In some of the mice, 5 × 106 tumor cells were implanted s.c. into the right flank to monitor tumor volume and to perform biochemical and immunohistochemical analysis of tumor tissue. All animal experiments performed were approved by the Animal Care Committee at Indiana University School of Medicine.
Biochemical Analysis.
Immunoblots were performed on cell lysates obtained from U87 cells grown in tissue culture or from multiple cryostat sections of s.c. tumor tissues. A Bradford assay was performed to determine protein concentration of each lysate. Equivalent amounts of protein were resolved by SDS/PAGE and transferred to nitrocellulose. Membranes were probed with antisera specific for PTEN, AKT, phospho-S473-AKT, or TSP-1. The RNase protection assay was performed by using a RPA III kit from (Ambion) according to the manufacturer's specifications. Briefly, 20 μg of total RNA was precipitated and resuspended in 10 μl of hybridization buffer containing the radioactive probe. The RNA then was heated to 95°C for 10 min and hybridized for 16 h at 42°C. A total of 150 μl of this mixture was treated with 1:100 dilution of RNase in RNase buffer for 30 min. RNase was inactivated, and RNA was reprecipitated and resolved on 5% acrylamide gel. RNA probes were synthesized by using maxi script using PCR templates and T7 polymerase. The glyceraldehyde-3-phosphate dehydrogenase probe was provided in the kit, and TSP-1 probe represents a 590-nt sequence in the 3′ untranslated region of TSP-1 sequence. All probes were sequenced.
Immunohistochemical Analysis.
MVD was determined for each s.c. and brain tumors by CD31 staining, and a proliferative index was determined by using anti-BrdUrd mAb staining performed on cryostat sections (7 μm), fixed in acetone, blocked in 1% goat serum, and stained with anti-CD31 antibody (PharMingen, #01951D). Antibody staining was visualized with peroxidase-conjugated anti-mouse and counterstained with hematoxylin. A negative control was performed on each tumor tissue stained with mouse IgG. Two sections from each tumor were scanned under low-power magnification (×40) to identify areas of highest CD31-positive vessel density (32), followed by digitization of three fields from this area. For in vivo BrdUrd labeling, mice received 100 μl of BrdUrd in PBS (10 mM) injected into the tail vein 1 h before tumor harvesting. The digitized images representing one ×20 field were counted for the number of CD31-positive vascular elements and number of BrdUrd-positive cells/field. The number of cells positive for BrdUrd staining ranged between 5% and 8% of total number of tumor cells. Data were collected from two independent observers without knowledge of which tumors were viewed. The average number of microvessels or cells positive for BrdUrd staining per digitized field was determined for five tumors per experimental group and analyzed by Student's t test.
Results
Effect of PTEN Reconstitution on in Vitro Growth of Tumor.
To determine whether PTEN exerts control over angiogenesis and the growth of glial tumors, we developed an orthotopic brain tumor model in which PTEN-deficient tumor cells were genetically manipulated in vitro and then stereotactically injected into the skin or frontal cerebral cortex of nude mice. The U87MG cell line is derived from a patient diagnosed with glioblastoma multiforme, a highly malignant and uniformly fatal brain tumor. This tumor and other human glioblastomas and glioblastoma cell lines contain a mutation in both PTEN alleles, resulting in a null genotype. In light of these observations, we reconstituted the PTEN gene in the parental U87MG (U87) cells.
Stable derivatives of the parental U87 cells were generated after transduction with retroviruses encoding cDNA for WT PTEN or specific mutants of this phosphatase. In particular, we used missense mutations in the PTP signature motif to ascertain the importance of the enzymatic activity of PTEN to its tumor suppressor function. This included R130 M, in which all phosphatase activity is abrogated (5, 33), and G129E, which has been identified in Cowden disease and endometrial cancer and in which the activity toward inositol phospholipids is severely attenuated but protein phosphatase activity is essentially intact.
Tumor cells were characterized biochemically for levels of activated AKT (phospho-S473-AKT), growth in vitro, and PTEN expression (Fig. 1). Anti-PTEN blots confirmed that parental U87 cells do not express PTEN and that after reconstitution of mutant or WT PTEN expression, U87 cells express amounts comparable to WT physiological levels. Expression of WT PTEN, to levels similar to those observed in a mouse brain lysate and primary human astrocytes, suppressed the activated state of AKT observed in PTEN-deficient U87 cells (Fig. 1A, lanes 3 and 5). After expression of the R130 M and G129E mutant forms of PTEN, the levels of phospho-AKT were similar to those observed in the parental U87 cells (Fig. 1A, lanes 1, 2, and 4), suggesting that the lipid phosphatase activity of PTEN was essential for the effects on the PtdIns(3,4,5)P3-dependent activation of AKT. Importantly, the growth of the different PTEN-expressing U87 cell lines in vitro was similar in 2%, 5%, and 10% FBS (data not shown and Fig. 1B). Therefore, we compared these cell lines further in our in vivo models.
Figure 1.

Stable expression of PTEN and PTEN mutants in U87MG cells regulates AKT. (A) Cell lysates from the U87MG (U87) cell line and U87 cells infected with a retroviral vector encoding PTEN (pBabe-Puro-PTEN) or mutants of PTEN (pBabe-Puro-PTEN-G129E or R130 M) were resolved by SDS/PAGE, and equal amounts of proteins were loaded per lane, immunoblotted with antisera to PTEN, phospho-AKT, and total AKT, and visualized by enhanced chemiluminescence. The basal levels of PTEN (Top), phosphorylated AKT (Ser-473) (Middle), and total AKT (Bottom) are shown. The status of the PTEN gene in each stable cell line was designated as: WT.E1 and WT.C7, two separate clones expressing WT PTEN; R130 M and G129E are mutated PTEN, R130 M is inert as both a protein and a lipid phosphatase. The G129E PTEN can dephosphorylate acidic phosphopeptides, but cannot dephosphorylate lipid substrate, PtdIns(3,4,5)P3. The U87MG (U87) cell line is the parental cell line isolated from a human glioblastoma multiforme patient. (B) Comparison of in vitro growth of U87MG cells transduced with mutants of PTEN. Equal number of cells (1 × 105) were incubated in RPMI + 10% FBS for different times, and cell numbers were quantitated by direct cell counting.
Effect of PTEN Reconstitution on in Vivo Growth and Proliferation.
We implanted athymic nude mice s.c. and by intracranial injection. Production of s.c. tumors allowed us to monitor the size of the tumor and perform direct biochemical analysis of tumor tissue for PTEN expression and levels of AKT activation without significant contamination from other tissues. Tumor tissue blocks were processed for hematoxylin/eosin staining. Greater than 95% of tissue analyzed was tumor. We compared the levels of PTEN in tumor tissue and numerous normal tissues within the athymic nude mouse. Using anti-PTEN antisera, we detected the expression of PTEN in all tissues, with the exception of skeletal and heart muscle (data not shown), but no PTEN was detected in parental U87-derived tumor tissue (Fig. 2C, lane 4). These results indicate that the tumor tissue sampled represents predominantly tumor cell-derived proteins. As observed in the cell lines grown in vitro, s.c. tumors, derived from U87 cells reconstituted with mutant or WT PTEN, displayed similar levels of PTEN expression (Fig. 2C, lanes 1–3 and 5). Phospho-AKT activity was higher in PTEN-null U87 cells and U87 cells reconstituted with R130 M and to a lesser extent in U87 cells expressing the G129E mutant (Fig. 2C, lane 1) as compared with the WT PTEN-transduced cells (Fig. 2C, compare lanes 1, 2, and 4 to lanes 3 and 5). The pattern of phosphorylated AKT was similar when the different U87 mutant expressing cell lines were assayed in vitro or in vivo (compare Fig. 1A to 2C).
Figure 2.

Effects of PTEN on growth of U87MG cells in vivo. (A) Cell growth in vivo. To determine the rate of cell growth in vivo, equal amounts of cells (5 × 106) from each cell line were implanted at the right ventral flank by s.c. injection. The formation and growth of the s.c. tumor was monitored, and the volume of the tumor was determined by a three-dimensional measurement at the times indicated (day 0, the date of implantation, no tumor is detected). Data were analyzed by Student's t test, and differences were significant comparing the PTEN deficient (U87MG, R130 M, G129E) to the WT PTEN (WT.E1, WT.C7), n = 5, number of mice; P < 0.0001. (B) Stereophotography of s.c. tumor sites in mice implanted with the parental U87 tumor, PTEN minus (Left) versus WT PTEN reconstituted tumor cells (Right). These tumors represent 25 and 42 days after implantation for PTEN minus versus WT PTEN reconstituted tumors, respectively. (Magnification, ×40.) (C) Immunoblot of cryostat tissue sections from s.c. tumor for the expression pattern of PTEN, AKT, and phosphorylated AKT. Frozen tissue sections were solubilized in Laemmli sample buffer, total protein was quantitated, and equal protein was loaded on SDS/PAGE. The data shown are representative of tissue analysis from five animals per experimental group.
Despite the similar in vitro growth rate, there was a dramatic difference in the growth of tumors derived from parental U87 cells compared with cells reconstituted with WT PTEN (Fig. 2 A and B). The average volume of U87-derived tumors on day 25 after implantation was 848 ± 203 mm3, compared with 91 ± 27 mm3 for tumors derived from WT PTEN-reconstituted cells (n = 5, P < 0.0001). Interestingly, the reconstitution of U87 cells with catalytically impaired PTEN (G129E or R130 M mutants) (Fig. 2A) shows an intermediate level of growth suppression, a result that suggests some residual function of these mutants in vivo.
Despite dramatic differences in the size of these tumors, in vivo BrdUrd labeling of tumor cells revealed no significant difference in number of cells in S phase: 72 ± 6 BrdUrd-positive cells per field in parental U87MG tumor mass versus 68.5 ± 3 in WT PTEN reconstituted tumors (data not shown). The percentage of TUNEL-positive nuclei within the U87MG and WT PTEN reconstituted tumors was similar. These results suggested that something other than the proliferative or apoptotic rate of the U87MG versus U87MG reconstituted with WT PTEN accounted for the difference in growth potential. We observed that the loss of inositol phospholipid phosphatase activity results in deregulated tumor growth comparable to the total ablation of catalytic activity and that the despite differences in overall growth in vivo, the proliferative rate of these tumors is similar. These findings are consistent with a role for PTEN in the regulation of another aspect of tumor biology, such as angiogenesis. This led us to assess the effect of PTEN reconstitution on the induction of angiogenesis in this model.
PTEN Suppresses Tumor-Induced Angiogenesis.
To examine the effect of PTEN on angiogenesis, we compared parental U87 cells to cells reconstituted with WT or mutant PTEN. We stained cryostat sections from s.c. tumors for CD31 (PECAM). CD31 is an endothelial marker used to measure the MVD of these tumors. MVD was assessed from multiple digitized images of CD31-stained tumor tissue at ×100 magnification (three fields were evaluated per tumor) and counted blindly for the number of CD31-positive microvessels per unit surface area as described (32). Reconstitution of PTEN expression in U87 cells dramatically suppressed the angiogenic response in vivo (Fig. 3 A and B). Quantitation of MVD in tumors derived from parental U87 cells (77 ± 13) and U87 cells expressing WT PTEN (38 ± 7) revealed an ≈50% suppression of angiogenesis (Fig. 3C) (n = 5, P < 0.001). The MVD of tumors derived from U87 cells reconstituted with catalytically impaired PTEN (R130 M, 84 ± 15 or G129E, 69 ± 16) were not significantly different (P > 0.05) from the parental U87 cell line (Fig. 3C). The levels of phospho-AKT detected within the tumor mass in vivo provide a correlation between the loss of the inositol lipid phosphatase function of PTEN, the phosphorylation status AKT, and the angiogenic phenotype within the tumor.
Figure 3.
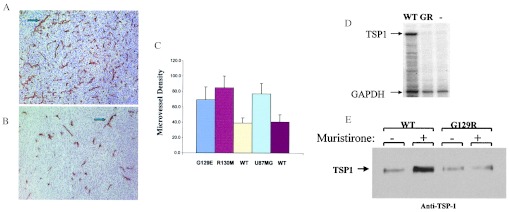
PTEN suppresses angiogenesis. Immunohistochemical analysis of staining with CD31 antibody to evaluate the angiogenesis response within the parental U87MG tumor (A) and PTEN reconstituted tumors (B), implanted into the s.c. tissue. In PTEN minus and tumors expressing mutants of PTEN, there are more new vessels formed (angiogenesis) (Upper, arrow indicated) than in WT PTEN reconstituted tumor (Lower), indicating the PTEN has direct influence on angiogenesis during tumor growth. (C) MVD counts were performed on tumor tissue stained with anti-CD31 antibody as described (32) to determine the effect of expression of PTEN and specific PTEN mutants (G129E or R130 M) on tumor-induced angiogenesis. Bars represent SD, five animals per group. Statistical analysis by Student's t test demonstrateed significant difference between MVD of PTEN null and PTEN catalytic mutants as compared with WT PTEN reconstituted tumors, n = 5, number of mice; P < 0.001. (D) PTEN regulates the expression of TSP-1 in U87MG cells. RNase protection assay was used to measure levels of TSP-1 mRNA in WT PTEN expressing U87 cells or cells transduced with a mutant catalytically dead PTEN (G129R). U87MG cells were infected with retrovirus encoding WT PTEN (WT), the catalytically dead, G129R mutant (GR), or empty vector retrovirus (−) and selected for 10 days in puromycin. RNA was harvested and RNase protection assays were carried out by using probes for TSP-1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). A probe for GAPDH was used as a normalization control. (E) Thrombospondin immunoblot analysis. U87MG transduced with WT PTEN (WT) or a catalytic mutant PTEN (G129R) in an ecdysone inducible expression system (36) were induced (48 h) with 0.5 μM muristirone or assayed without induction to determine the effect of PTEN expression on the induction of TSP-1 by Western blotting. Supernates from cells were prepared and proteins were resolved on SDS/PAGE and probed with anti-TSP-1 antibody. There is clear up-regulation of TSP-1 in WT PTEN-transduced U87 cultures compared with U87 cells expressing the lipid phosphatase-deficient G129R mutant PTEN.
PTEN Induction of Thrombospondin.
Recent in vitro data suggest a link between PTEN and downstream targets including AKT, HIF1α, and vascular endothelial growth factor in the potential control of angiogenesis (34, 35). We used the RNase protection assay to confirm an observation we initially made during a cDNA microarray analysis comparing the effect of PTEN reconstitution on U87MG TSP-1 expression. RNase protection assay was performed with a TSP-1 specific probe in the same U87MG cells constitutively expressing WT PTEN or G129R PTEN (Fig. 3D). The data demonstrate that WT but not mutant PTEN expression induces TSP-1 in U87 cells. To confirm these results, we performed Western blot analysis for TSP-1 expression with a retroviral-based ecdysone-inducible PTEN expression system (36). Inducible and dose-dependent expression of PTEN was confirmed in U87 cells, and we noted that the induced expression of WT PTEN and not G129R PTEN (not shown) resulted in augmented TSP-1 expression (Fig. 3E), and WT PTEN suppressed the activation of phospho-AKT without affecting total AKT (data not shown). The induced expression of mutant G129R had no effect on phospho-AKT. Therefore, our data demonstrate that PTEN positively modulates the expression of TSP-1, a negative regulator of angiogenesis (Fig. 3 D and E) (37, 38). These data identify one potential mechanism by which PTEN may regulate angiogenesis through the control of TSP-1.
PTEN Is a Determinant of Survival in Orthotopic Brain Tumor Model.
Brain tumor-induced angiogenic responses occur in the context of brain-specific stromal and extracellular matrix interactions. This fact led us to determine whether the expression of the PTEN affects the survival of mice in an orthotopic brain tumor model. For these experiments, U87 cells expressing either WT or mutant forms of PTEN were implanted under stereotactic control into the right frontal lobe of nude mice (Fig. 4B, see arrow for site of implantation). The results indicate that reconstitution of WT PTEN in U87 cells suppressed the malignant potential of these cells in an orthotopic animal model. Thus, there was 90% survival at 40 days in animals implanted with the WT PTEN-reconstituted U87 cells compared with 100% mortality of mice implanted with the parental cells at 27 days (Fig. 4C) (n = 15, P < 0.0001). PTEN reconstituted tumor cells grew more slowly when implanted in the frontal lobe (Fig. 4, compare A and B) and remained circumscribed to that area of brain (data not shown). U87 cells reconstituted with mutants of PTEN, either ablated in inositol lipid phosphatase activity (G129E) or catalytically inactive (R130 M), displayed a phenotype similar to the PTEN-negative, parental U87 cells (Fig. 4C). Animals with tumors derived from U87 cells reconstituted with PTEN-G129E displayed slightly prolonged survival (50% at day 30) compared with those implanted with parental U87 cells; however, all animals died by day 40. Immunohistochemical analysis for CD31-positive neovascular structures within the brain tumor demonstrated marked suppression of angiogenesis within the brain tumor by WT PTEN reconstitution (compare CD31-positive MVD in Fig. 4 D to F). These combined data suggest that ablation of the inositol lipid phosphatase activity of PTEN alone is sufficient to yield an angiogenic (Fig. 3) and malignant glioma (Fig. 4C) after implantation of U87 tumor cells.
Figure 4.

Effects of PTEN reconstitution on survival in an orthotopic brain tumor model. Equivalent number of parental U87 cells or U87 cells reconstituted with WT or mutant alleles of PTEN (1 × 106 cells) were implanted in right frontal lobe of nude mice. Stereophotography of whole brains from mice implanted with U87MG tumor cells (day 25) (A) or PTEN reconstituted (day 42) (B). The implantation site is shown by position of arrow in the WT PTEN reconstituted tumor (B). (Magnification: ×20.) (C) Survival plots for mice implanted with PTEN minus or parental U87 cells transduced with mutants of PTEN as shown. Survival data represents 15 animals per experimental group. P < 0.0001 for difference observed between the PTEN+ and PTEN− groups for survival. (D) Immunohistochemical analysis for anti-CD31 staining (brown staining) for microvessels within a U87MG parental PTEN-deficient brain tumor on day 22 after stereotactic implantation. (E) Anti-CD31 staining of microvessels within a WT PTEN reconstituted U87MG brain tumor evaluated on day 42 after implantation. (F) MVD counts were performed on brain tumor tissue stained with anti-CD31 antibody as described (32) to determine the effect of expression of PTEN and specific PTEN mutants (G129E or R130 M) on tumor-induced angiogenesis. Bars represent SD, four animals per group. Statistical analysis by n = 5 (number of mice), P < 0.01.
Discussion
In this report we have investigated the effects of expressing WT and mutant forms of PTEN on the tumorigenicity of the U87 malignant glioblastoma cell line. We have observed that expression of WT PTEN did not affect the growth of the cell line in vitro. Furthermore, the proliferative rate of cell lines expressing WT PTEN was indistinguishable from the parental U87MG line when the cells were injected either s.c. or intracranially in nude mice. Although the proliferative rates were indistinguishable, there was a marked difference in tumor volume that was accompanied by a dramatic difference in the vascularization of the tumor. Even when tumors of the same size were assayed, tumors derived from cells expressing WT PTEN had fewer blood vessels than those derived from the parental cell line or cell lines expressing mutant forms of PTEN. Importantly, use of a specific point mutation in PTEN (PTEN G129E), in which lipid phosphatase activity is impaired but protein phosphatase activity remains essentially intact (5), revealed that it is the lipid phosphatase activity of PTEN that is required for inhibition of angiogenesis. Because the percentage of terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling positive nuclei within the U87MG and WT PTEN reconstituted tumors was similar, the difference in tumor size is likely to be the result of changes in PI3-kinase-dependent angiogenesis, rather than effects on proliferative or apoptotic rates.
Other groups have noted that reconstitution of PTEN to supraphysiological levels in the U87 cell line can suppress cell proliferation (33, 39–44). In those experiments, PTEN was noted to effect growth in vitro through the induction of a G1 cell cycle arrest. We have been unable to detect a G1 arrest in our cell lines. Significantly, our system was designed to express PTEN in the U87 cells at levels similar to those observed in normal brain or primary astrocytes. PTEN is catalytically active and may be susceptible to regulation, for example, through interaction with potential regulatory/targeting proteins (45–49) or phosphorylation-dependent control at the level of proteolysis (50). Therefore, we reasoned that even small deviations from physiological levels could have profound repercussions on the maintenance of the normal function and regulation of the phosphatase. It is possible that the G1 arrest detected by many groups is a result of the high levels of overexpression of PTEN. This conclusion is supported by the finding that most cell lines and tissues express PTEN without undergoing a G1 arrest.
As shown in our work, PTEN controls the expression and secretion of TSP-1, a negative regulator of angiogenesis. The work of Lawler et al. (51) and Hsu et al. (38) have clearly established that TSP-1 exerts a negative control over endothelial cell physiology and suppresses angiogenesis. A considerable body of literature has established which domains of this complex matrix protein exert its antiangiogenic effects and worked out some of the mechanisms which underlie TSP-1 effects on endothelial cells survival and angiogenesis (52, 53). Our data are similar to the observation that p53 regulates angiogenesis and provide evidence for one potential mechanism by which PTEN may negatively regulate the angiogenic response by the induction of TSP-1 expression. However, it seems unlikely that this is the sole mechanism. Induction of TSP-1 expression by PTEN likely reflects the recruitment of a larger program of gene expression, one that is inhibitory to angiogenesis and tissue remodeling. Our preliminary studies suggest that PTEN expression also induces TIMP3, an inhibitor of matrix metalloproteinases, and suppresses matrix degradation in vivo, an important component of angiogenesis in vivo. Further analysis of the role of PTEN in the regulation of angiogenesis will increase our understanding of how PtdIns(3,4,5)P3 pathways interface with the angiogenic switch mechanisms.
PTEN was first characterized as a dual specificity phosphatase that displayed selectivity for acidic substrates, including both protein and nonprotein targets. Therefore, a question remains as to which activity is essential to the tumor suppressor effects of PTEN. There have been suggestions that a primary target of PTEN may be the protein tyrosine kinase, FAK, a regulator of integrin-dependent signaling and cell migration (54). However, the properties of U87 cells reconstituted with the PTEN-G129E mutant suggest that it is the lipid phosphatase activity of PTEN that is a critical determinant of its function in this brain tumor model, in particular the induction of tumor-induced angiogenesis in vivo. The observations of Holland et al. (55) are consistent with this hypothesis: they reported that the introduction of activated AKT, a downstream effector of PI3-kinase, into glial cells of the mouse brain results in the development of glioblastomas. Thus, our data and the data of Holland et al. suggest a potential therapeutic benefit of inhibitors of PI3-kinase and downstream targets such as AKT in treatment of malignant gliomas. Preliminary work from our laboratory demonstrates that the treatment of mice with the PI3-kinase inhibitor LY294002 blocks angiogenesis and prevents the intracranial growth of U87MG tumors (unpublished observation). Further analysis should be directed at determining how LY294002 suppresses the angiogenic response of gliomas in this model. Based on our combined data we suggest that PI3-kinase inhibitors may have efficacy in the treatment of malignant gliomas.
Acknowledgments
We dedicate this work to the memory of our patients Ross Feikls and Kandra Grzesik for their bravery and the inspiration they gave us. We acknowledge the excellent technical assistance of Lee Ann Balderidge and the immunohistochemical core facility. We thank Drs. Carrie Phillips and Biagio Azzarelli for their support. We thank Drs. Anat Epstein and Mary C. Dinauer for review of this manuscript before submission. Funding for this work was from National Institutes of Health Grants RO1-CA75637 and PO1 CA81403–01A1 (to D.L.D.), National Institutes of Health Grants CA53840 and GM55989 and a grant from the Mellam Family Foundation (to N.K.T.), a grant from the V Foundation (to M.P.M.), and grants from the U. S. Department of the Army (9DAMD-17–94-14247), the National Cancer Institute, 5R35 CA39829, Tularik Inc. and 1 in 9: The Long Island Breast Cancer Action Coalition (to M.H.W.). M.H.W. is an American Cancer Society Research Professor. D.L.D. is a member of the Children's Oncology Group and the New Agents Neuroblastoma Treatment consortium for neuroblastoma research.
Abbreviations
- PI3-kinase
phosphatidylinositol 3-kinase
- PtdIns(3,4,5)P3
phosphatidylinositol 3,4,5-trisphosphate
- TSP-1
thrombospondin 1
- WT
wild type
- MVD
microvessel density
References
- 1.Hunter T. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- 2.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang S I, Puc J, Miliaresis C, Rodgers L, McCombie R, et al. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 3.Steck P A, Pershouse M A, Jasser S A, Yung W K, Lin H, Ligon A H, Langford L A, Baumgard M L, Hattier T, Davis T, et al. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 4.Li D M, Sun H. Cancer Res. 1997;57:2124–2129. [PubMed] [Google Scholar]
- 5.Myers M P, Stolarov J P, Eng C, Li J, Wang S I, Wigler M H, Parsons R, Tonks N K. Proc Natl Acad Sci USA. 1997;94:9052–9057. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Myers M P, Pass I, Batty I H, Van der Kaay J, Stolarov J P, Hemmings B A, Wigler M H, Downes C P, Tonks N K. Proc Natl Acad Sci USA. 1998;95:13513–13518. doi: 10.1073/pnas.95.23.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maehama T, Dixon J E. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 8.Stambolic V, Suzuki A, de la Pompa J L, Brothers G M, Mirtsos C, Sasaki T, Ruland J, Penninger J M, Siderovski D P, Mak T W. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 9.Wu X, Senechal K, Neshat M S, Whang Y E, Sawyers C L. Proc Natl Acad Sci USA. 1998;95:15587–15591. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maier D, Zhang Z, Taylor E, Hamou M F, Gratzl O, Van Meir E G, Scott R J, Merlo A. Oncogene. 1998;16:3331–3335. doi: 10.1038/sj.onc.1201832. [DOI] [PubMed] [Google Scholar]
- 11.Liaw D, Marsh D J, Li J, Dahia P L, Wang S I, Zheng Z, Bose S, Call K M, Tsou H C, Peacocke M, et al. Nat Genet. 1997;16:64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 12.Myers M P, Tonks N K. Am J Hum Genet. 1997;61:1234–1238. doi: 10.1086/301659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maehama T, Dixon J E. Trends Cell Biol. 1999;9:125–128. doi: 10.1016/s0962-8924(99)01519-6. [DOI] [PubMed] [Google Scholar]
- 14.Cantley L C, Neel B G. Proc Natl Acad Sci USA. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Podsypanina K, Ellenson L H, Nemes A, Gu J, Tamura M, Yamada K M, Cordon-Cardo C, Catoretti G, Fisher P E, Parsons R. Proc Natl Acad Sci USA. 1999;96:1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki A, de la Pompa J L, Stambolic V, Elia A J, Sasaki T, del Barco Barrantes I, Ho A, Wakeham A, Itie A, Khoo W, et al. Curr Biol. 1998;8:1169–1178. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- 17.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi P P. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 18.Lee J O, Yang H, Georgescu M M, Di Cristofano A, Maehama T, Shi Y, Dixon J E, Pandolfi P, Pavletich N P. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 19.Rasheed B K, Stenzel T T, McLendon R E, Parsons R, Friedman A H, Friedman H S, Bigner D D, Bigner S H. Cancer Res. 1997;57:4187–4190. [PubMed] [Google Scholar]
- 20.Raffel C, Frederick L, O'Fallon J R, Atherton-Skaff P, Perry A, Jenkins R B, James C D. Clin Cancer Res. 1999;5:4085–4090. [PubMed] [Google Scholar]
- 21.Folkman J. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 22.Brem S, Cotran R, Folkman J. J Natl Cancer Inst. 1972;48:347–356. [PubMed] [Google Scholar]
- 23.Hanahan D, Folkman J. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 24.Dameron K M, Volpert O V, Tainsky M A, Bouck N. Science. 1994;265:1582–1584. doi: 10.1126/science.7521539. [DOI] [PubMed] [Google Scholar]
- 25.Brooks P C, Montgomery A M, Rosenfeld M, Reisfeld R A, Hu T, Klier G, Cheresh D A. Cell. 1994;79:1157–1164. doi: 10.1016/0092-8674(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 26.Fang J, Shing Y, Wiederschain D, Yan L, Butterfield C, Jackson G, Harper J, Tamvakopoulos G, Moses M A. Proc Natl Acad Sci USA. 2000;97:3884–3889. doi: 10.1073/pnas.97.8.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Good D J, Polverini P J, Rastinejad F, Le Beau M M, Lemons R S, Frazier W A, Bouck N P. Proc Natl Acad Sci USA. 1990;87:6624–6628. doi: 10.1073/pnas.87.17.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plate K H, Breier G, Weich H A, Risau W. Nature (London) 1992;359:845–848. doi: 10.1038/359845a0. [DOI] [PubMed] [Google Scholar]
- 29.Jiang B H, Zheng J Z, Aoki M, Vogt P K. Proc Natl Acad Sci USA. 2000;97:1749–1753. doi: 10.1073/pnas.040560897. . (First Published February 4, 2000; 10.1073/pnas.040560897) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giri D, Ittmann M. Hum Pathol. 1999;30:419–424. doi: 10.1016/s0046-8177(99)90117-x. [DOI] [PubMed] [Google Scholar]
- 31.Stolarov J. Thesis Dissertation. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2001. [Google Scholar]
- 32.Weidner N, Semple J P, Welch W R, Folkman J. N Engl J Med. 1991;324:1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- 33.Furnari F B, Lin H, Huang H S, Cavenee W K. Proc Natl Acad Sci USA. 1997;94:12479–12484. doi: 10.1073/pnas.94.23.12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E, Gottschalk A R, Ryan H E, Johnson R S, Jefferson A B, et al. Genes Dev. 2000;14:391–396. [PMC free article] [PubMed] [Google Scholar]
- 35.Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu M M, Simons J W, Semenza G L. Cancer Res. 2000;60:1541–1545. [PubMed] [Google Scholar]
- 36.No D, Yao T P, Evans R M. Proc Natl Acad Sci USA. 1996;93:3346–3351. doi: 10.1073/pnas.93.8.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheibani N, Frazier W A. Histol Histopathol. 1999;14:285–294. doi: 10.14670/HH-14.285. [DOI] [PubMed] [Google Scholar]
- 38.Hsu S C, Volpert O V, Steck P A, Mikkelsen T, Polverini P J, Rao S, Chou P, Bouck N P. Cancer Res. 1996;56:5684–5691. [PubMed] [Google Scholar]
- 39.Cheney I W, Johnson D E, Vaillancourt M T, Avanzini J, Morimoto A, Demers G W, Wills K N, Shabram P W, Bolen J B, Tavtigian S V, Bookstein R. Cancer Res. 1998;58:2331–2334. [PubMed] [Google Scholar]
- 40.Li D M, Sun H. Proc Natl Acad Sci USA. 1998;95:15406–15411. doi: 10.1073/pnas.95.26.15406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Furnari F B, Huang H J, Cavenee W K. Cancer Res. 1998;58:5002–5008. [PubMed] [Google Scholar]
- 42.Ramaswamy S, Nakamura N, Vazquez F, Batt D B, Perera S, Roberts T M, Sellers W R. Proc Natl Acad Sci USA. 1999;96:2110–2115. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun H, Lesche R, Li D M, Liliental J, Zhang H, Gao J, Gavrilova N, Mueller B, Liu X, Wu H. Proc Natl Acad Sci USA. 1999;96:6199–6204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adachi J, Ohbayashi K, Suzuki T, Sasaki T. J Neurosurg. 1999;91:822–830. doi: 10.3171/jns.1999.91.5.0822. [DOI] [PubMed] [Google Scholar]
- 45.Georgescu M M, Kirsch K H, Akagi T, Shishido T, Hanafusa H. Proc Natl Acad Sci USA. 1999;96:10182–10187. doi: 10.1073/pnas.96.18.10182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adey N B, Huang L, Ormonde P A, Baumgard M L, Pero R, Byreddy D V, Tavtigian S V, Bartel P L. Cancer Res. 2000;60:35–37. [PubMed] [Google Scholar]
- 47.Wu X, Hepner K, Castelino-Prabhu S, Do D, Kaye M B, Yuan X J, Wood J, Ross C, Sawyers C L, Whang Y E. Proc Natl Acad Sci USA. 2000;97:4233–4238. doi: 10.1073/pnas.97.8.4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leslie N R, Gray A, Pass I, Orchiston E A, Downes C P. Biochem J. 2000;346:827–833. [PMC free article] [PubMed] [Google Scholar]
- 49.Marfatia S M, Byron O, Campbell G, Liu S C, Chishti A H. J Biol Chem. 2000;275:13759–13770. doi: 10.1074/jbc.275.18.13759. [DOI] [PubMed] [Google Scholar]
- 50.Vazquez F, Ramaswamy S, Nakamura N, Sellers W R. Mol Cell Biol. 2000;20:5010–5018. doi: 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lawler J, Sunday M, Thibert V, Duquette M, George E L, Rayburn H, Hynes R O. J Clin Invest. 1998;101:982–992. doi: 10.1172/JCI1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dawson D W, Pearce S F, Zhong R, Silverstein R L, Frazier W A, Bouck N P. J Cell Biol. 1997;138:707–717. doi: 10.1083/jcb.138.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dawson D W, Volpert O V, Pearce S F, Schneider A J, Silverstein R L, Henkin J, Bouck N P. Mol Pharmacol. 1999;55:332–338. doi: 10.1124/mol.55.2.332. [DOI] [PubMed] [Google Scholar]
- 54.Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada K M. Science. 1998;280:1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- 55.Holland E C, Celestino J, Dai C, Schaefer L, Sawaya R E, Fuller G N. Nat Genet. 2000;25:55–57. doi: 10.1038/75596. [DOI] [PubMed] [Google Scholar]