

Abstract
Although pioneering sequencing projects have shed light on the boxer and poodle genomes, a number of challenges need to be met before the sequencing and annotation of the dog genome can be considered complete. Here, we present the DNA sequence of the Jindo dog genome, sequenced to 45-fold average coverage using Illumina massively parallel sequencing technology. A comparison of the sequence to the reference boxer genome led to the identification of 4 675 437 single nucleotide polymorphisms (SNPs, including 3 346 058 novel SNPs), 71 642 indels and 8131 structural variations. Of these, 339 non-synonymous SNPs and 3 indels are located within coding sequences (CDS). In particular, 3 non-synonymous SNPs and a 26-bp deletion occur in the TCOF1 locus, implying that the difference observed in cranial facial morphology between Jindo and boxer dogs might be influenced by those variations. Through the annotation of the Jindo olfactory receptor gene family, we found 2 unique olfactory receptor genes and 236 olfactory receptor genes harbouring non-synonymous homozygous SNPs that are likely to affect smelling capability. In addition, we determined the DNA sequence of the Jindo dog mitochondrial genome and identified Jindo dog-specific mtDNA genotypes. This Jindo genome data upgrade our understanding of dog genomic architecture and will be a very valuable resource for investigating not only dog genetics and genomics but also human and dog disease genetics and comparative genomics.
Keywords: genome sequencing, Jindo dog, massively parallel sequencing
1. Introduction
The Korean Jindo Dog (Jindotgae in Korean) is a breed of hunting dog that originated on Jindo Island in South Korea. The Jindo dog breed was recognized by the United Kennel Club on 1 January 1998 and by the Fédération Cynologique Internationale in 2005, and, owing to its fierce loyalty to humans and brave nature, it has spread far beyond the Korean peninsula. The Jindo dog has notably acute hearing and scenting ability, a medium-sized body, erect ears and a sickle-shaped tail.
The genome of the domestic dog is of great interest not only to biologists and animal breeders but also to medical scientists. Dogs and humans share many highly prevalent diseases, including cancers, epilepsy, cataracts, diabetes, blindness, heart disease, hip dysplasia and deafness,1,2 and the clinical manifestations of these diseases in the two species are often similar.3 Understanding the genetic bases of behavioural traits and morphological variations4 in physical characteristics, such as size, skull shape and coat colour and texture in domestic dogs, could help animal breeders to breed dogs that are better suited to human requirements. In addition, correctly positioning the dog within the mammalian evolutionary tree will provide important insight into the human genome.5
Pioneering genome sequencing projects5,6 using the traditional Sanger method to sequence the boxer and poodle genomes shed the first light on the dog genome sequence and structure, genetic evolution, haplotype structure, linkage disequilibrium patterns, single nucleotide polymorphism (SNP) map and evolutionary phylogeny.7 Along with major advances in canine genomics, great progress was made in identifying the olfactory receptor genes8–11 that are associated with dogs' excellent smell-discriminating ability. Dog mitochondrial genome data were also used in these studies12–14 to provide insight into the phylogenetic origins of diverse kinds of dog breeds.
Despite these achievements in the fields of dog genomics and genetics, there are still issues that remain to be resolved before the sequencing and annotation of the dog genome can be considered complete. In contrast to the cell-free system used with next-generation sequencing (NGS) techniques, the in vivo cloning steps used with the Sanger method of whole-genome shot-gun sequencing could result in gap regions.15 In addition, the coverage generated by the Sanger method is low (7.5-fold for the boxer genome5 and 1.5-fold for the poodle genome)6 owing to the high cost of this method, which could prevent dog population-level genome sequencing. Recently, next-generation massively parallel sequencing platforms, including the Illumina Genome Analyser, the Roche 454 Genome Sequencer FLX Instrument and the ABI SOLiD System, have revolutionized the genome sequencing process by providing high through-put, high speed and cost-effective high coverage.15–18
In this study, we applied the next-generation massively parallel sequencing technique (Illumina HiSeq 2000) to the sequencing and analysis of a dog genome for the first time. We generated 45-fold coverage sequence data, performed complete sequence annotation of the Jindo dog mitochondrial genome and provided deep insight into the SNP map, gap regions in the reference boxer genome, the olfactory gene family responsible for a dog's ability to smell and evolutionary phylogeny.
2. Materials and methods
2.1. DNA extraction, library preparation and Illumina massively parallel sequencing platform
Genomic DNA was isolated from blood cells collected from a male Korean Jindo dog using a QIAGEN genomic DNA isolation kit. DNA sample preparation kits were used to prepare DNA libraries with template insert sizes of 300–400 bp for single and paired-end sequencing (Illumina® sequencing manual). All data were generated on an Illumina HiSeq2000 using sequencing protocols provided by the manufacturer.
2.2. Alignment of short reads onto the reference boxer genome
After filtering the artificial reads from the total collection of short reads, we aligned a clean, usable read set to the reference boxer genome (canFam2) using a fast short-read alignment program, BWA (ver. 0.5.9), with the default parameters. This program efficiently uses the information obtained from paired-end reads to correct alignments and accurately map short reads to repetitive sequences.19
2.3. SNP identification
The short reads were aligned to the reference boxer genome by the BWA program under conditions that allowed two-base pair mismatches for the detection of SNPs. We then performed statistical calculations based on Samtools and the Illumina quality system to judge whether a mismatched base was an error or an SNP. The utilized criteria were as follows: minimum read depth of four (−d 4), maximum depth of 100 (−D 100) to filter out randomly placed repetitive hits, consensus quality score ≥20 or error rate <1% (Q20), adjacent sequence quality (Q20) and no indel within a 3-bp flanking region. The filter criteria used here included a Q20 quality cut-off, estimated copy number of flanking sequences (<2), minimum distance between any two SNPs (≥5 bp) and overall depth (≤100) at a given position in the reference. For the homozygous SNPs, at least four reads should be observed. For the heterozygous SNPs, each allele should be supported by at least four reads.
2.4. Detection of small indels
We identified small indels using indel detection methods based on the information generated from paired-end reads (BWA and Samtools). We used standard criteria stating that each indel should be confirmed by a minimum of three reads and that it should also be observed in both strands. Multiple indels occurring within a 20-bp window were filtered out from the BWA results because closely spaced indels can be caused by alignment errors.
2.5. Detection of structural variants
We detected structural variants (SVs) by using information about the span size and orientation of each paired-end read. Unusually long or short distances (more than twice the median insert size of each DNA library or much less than that) between two paired-end reads were identified as harbouring SVs only when they were clustered with a read depth of more than two. We also dismissed cases where clusters of such paired-end reads were present in repeat regions of the genome. Finally, genomic deletions with a size of >10 kb were filtered out.
2.6. Data sources
The NCBI dog reference genome, NCBI reference gene information and dbSNP (ver. CanFam2.0) were obtained from the Ensembl database (http://www.ensembl.org/), which provides information about genes mapped onto the NCBI build 2.
2.7. SNP annotation
SNPs in the Jindo dog genome were compared with ENSEMBL dbSNP (ver. CanFam2.0) to distinguish between known and novel SNPs after using BWA to identify SNPs that were confirmed by more than four reads. Each SNP was mapped onto the regions corresponding to the genomic features of the NCBI gene structure, such as introns, UTRs and coding sequences (CDS). Information about non-synonymous SNPs was extracted by comparison with the NCBI reference gene information.
2.8. Mitochondrial genome assembly
All reads corresponding to the Jindo dog mitochondrial genome sequence were searched using Bowtie, and we found a total of 234 817 such reads. The number of mitochondrial genome reads was reduced to 217 988 at the error correction stage. The reads were assembled using SOAPdenovo, generating 43 large contigs (>200 bp in size).
2.9. Data access
The whole-genome sequencing short-read data for the Korean Jindo dog have been deposited in the DDBJ under the accession number DRA000473.
3. Results
3.1. Massively parallel sequencing of the Jindo dog genome
Genomic DNA was isolated from blood samples collected from a male Korean Jindo dog. Using the Illumina HiSeq2000 platform, we performed massively parallel sequencing, generating 1 102 978 656 high-quality reads with an average read length of 100 bp (Table 1). Of these reads, 1 090 271 942 (98.84%) were mapped to the reference boxer genome5 by a fast short-read alignment program, BWA19 (version 0.5.9; see Section 2). The total number of sequenced nucleotides was 110.2 Gb, corresponding to an average depth coverage of 45-fold over the reference boxer genome (estimated genome size: 2.445 Gb). The proportion of the regions in which our mapped reads were matched to the reference boxer genome corresponded to 94.02% of the genome. The relationship between the short-read depth coverage and the GC content throughout the whole dog genome shows that our Illumina short reads cover regions in the Jindo dog genome corresponding to nearly entire GC content range, and especially, the read depth coverage ranges from 15- to 47-fold in the genomic regions with the GC contents ranging from 11 to 77% (Supplementary Fig. S1A and B). These results suggest that massively parallel sequencing using the Illumina HiSeq 2000 platform is very cost-effective and accurate for the generation of high-quality reads.
Table 1.
Summary of the Jindo dog genome sequencing results
| Overview of sequencing | |
|---|---|
| Average genomic DNA insert size (bp) | 340 |
| Average read length (bp) | 100 |
| Number of reads | 1 102 978 656 |
| Number of mapped reads | 1 090 271 942 (98.84%) |
| Number of sequenced nucleotides (Gb) | 110.2 |
| Average depth coverage relative to the reference boxer genome (x) | 45 |
| Total percentage of matched reference genome regions | 94.02% |
3.2. Single nucleotide polymorphisms
Based on the standard criteria that each homozygous SNP must be corroborated by at least four reads, and every heterozygous SNP must be supported by at least four reads for each corresponding allele, we identified SNPs and corrected errors (see Section 2) in the sequence read data. Given that a previous study15 showed that the existence of two short reads for each SNP site, together with SNP error correction, can guarantee an accuracy of 99% of identified SNPs in the case of genome read data with a 13-fold average coverage, our criteria should enable almost perfect accuracy in the identification of SNPs in Jindo dog genome short-read data with an average depth coverage of 45-fold. We detected a total of 4 675 437 SNPs, of which 1 976 029 and 2 699 408 corresponded to homozygous and heterozygous SNPs, respectively (Fig. 1A). Compared with the reference boxer genome, the Jindo dog genome showed 3 346 058 novel and 1 329 379 shared SNPs (Fig. 1B). We also determined that 1065 Jindo dog SNPs were located within intragenic regions, of which 46 (in 35 genes), 812 (in 503 genes) and 207 (in 133 genes) were positioned within the 5′untranslated region (UTR), CDS and 3′UTR, respectively (Fig. 1C and Supplementary Table S1).
Figure 1.

SNPs in the Jindo dog genome. (A) Homozygous and heterozygous SNPs. (B) A comparison between Jindo and boxer SNPs. (C) Intragenic SNPs in the Jindo dog genome.
Importantly, of the 812 CDS SNPs, 339 (in 222 genes) were non-synonymous. We used the NCBI OMIM (Online Mendelian Inheritance in Man) database to identify which of the 339 non-synonymous CDS SNPs are located within genes that are associated with known disease phenotypes. We confirmed that 89 of the 222 genes have been reported to be associated with diseases in humans, dogs and other species (mice, rats, drosophila, zebra fish, rabbits and cattle; Supplementary Table S2). Notably, a majority of the genes (58 genes) were associated with disease phenotypes that are known to be shared by humans and other species. In particular, 18 of the 58 genes were associated with mutational phenotypes shared mainly between humans and dogs (Table 2). Of these 18 genes, TCOF120 (OMIM accession: 606847, associated with Treacher Collins syndrome 1), HLA-DQB1 (OMIM accession: 604305, associated with Creutzfeldt–Jakob disease), AMN (OMIM accession: 605799, associated with megaloblastic anaemia-1), COL4A5 (OMIM accession: 303630, associated with Alport syndrome), COL7A1 (OMIM accession: 120120, associated with EBD inversa), DBH (OMIM accession: 609312, associated with dopamine beta-hydroxylase deficiency) and GUSB (OMIM accession: 611499, associated with mucopolysaccharidosis VII) harbour more than one non-synonymous CDS SNP.
Table 2.
Non-synonymous SNPs within CDS regions of known dog and human disease-associated genes
| Gene namea | Dog chromosome | OMIM accession number | Number of non-synonymous SNPsb | Disease phenotype in humans | Disease-sharing animals |
|---|---|---|---|---|---|
| AGL | chr6 | 610860 | 1 | Glycogen storage disease III a and b | Dog |
| AMN | chr8 | 605799 | 2 | Megaloblastic anaemia-1 | Dog |
| ATP7B | chr22 | 606882 | 1 | Wilson disease | Dog, rat, mouse |
| COL4A5 | chrX | 303630 | 2 | Alport syndrome | Dog |
| COL7A1 | chr20 | 120120 | 2 | EBD inversa, epidermolysis bullosa dystrophica, AD | Dog, mouse |
| CUBN | chr2 | 602997 | 1 | Megaloblastic anaemia-1, Finnish type | Dog |
| DBH | chr9 | 609312 | 2 | Dopamine beta-hydroxylase deficiency | Dog |
| DES | chr37 | 125660 | 1 | Cardiomyopathy, dilated, 1I, myopathy | Dog |
| DMD | chrX | 300377 | 1 | Duchenne muscular dystrophy | Dog |
| DNASE1 | chr6 | 125505 | 1 | Systemic lupus erythematosus susceptibility | Dog |
| FLCN | chr5 | 607273 | 1 | Birt-Hogg-Dube syndrome, colorectal cancer | Dog |
| GUSB | chr6 | 611499 | 2 | Mucopolysaccharidosis VII | Dog |
| HLA-DQB1 | chr12 | 604305 | 3 | Creutzfeldt–Jakob disease | Dog |
| NEFH | chr26 | 162230 | 1 | Amyotrophic lateral sclerosis | Dog |
| NHLRC1 | chr35 | 608072 | 1 | Epilepsy, progressive myoclonic 2B (Lafora) | Dog |
| RPGR | chrX | 312610 | 1 | Cone-rod dystrophy-1, macular degeneration | Dog |
| TCOF1 | chr4 | 606847 | 3 | Treacher Collins syndrome 1 | Dog |
| TNF | chr12 | 191160 | 1 | Asthma, dementia, susceptibility to malaria | Dog |
aThe gene name is common in dogs and humans.
bNon-synonymous SNPs in the Jindo dog genome compared with the boxer genome.
The TCOF1 gene also plays an important role in cranial facial development in dogs.21 Even a single amino acid change affects the cranial and facial shape.7 Among the 3 non-synonymous SNPs and a 26-bp deletion within the TCOF1 gene locus, which have been experimentally confirmed by genomic PCR and capillary-based Sanger sequencing in this study (Supplementary Fig. S2A–D), the SNP (G in Jindo versus A in boxer at the nucleotide 61 932 564 in the chromosome 4) exactly corresponded to the SNP (a C396T variant, leading to a Pro117Ser substitution), which had been previously reported to have a decisively influential effect in making the differences in dog breed-unique craniofacial characteristics among other dog breeds.21 In this regard, the newly verified non-synonymous SNP (at nucleotide 61 932 564 in chromosome 4) with the 2 other non-synonymous SNPs and the 26-bp deletion within the Jindo dog TCOF1 gene, compared with the boxer, could make a crucial contribution to the developmental difference in skull and face shapes between the Jindo and boxer breeds.
3.3. Indels
We identified a total of 71 642 indels (insertion and deletion variations) present within the short-read sequences, of which 27 517 and 44 125 corresponded to homozygous and heterozygous indels, respectively (Fig. 2A). In particular, nine indels occurred within intragenic regions; three indels were present within the CDS regions of the genes KRT1 (a 3-bp insertion: GGC), TCOF1 (a 26-bp deletion: GGGCACCTGCAGCCTCACCTGAACAG, as shown in Supplementary Fig. S2D) and SET (a 9-bp deletion: GATGATGAT; Table 3). TCOF1 (OMIM accession: 606847) is known to be associated with the Treacher-Collins–Franceschetti syndrome in humans and dogs, and KRT1 (OMIM accession: 139350) is known to be associated with Curth–Macklin Palmoplantar keratoderma in humans.
Figure 2.

Indels and structural variations in the Jindo dog genome. (A) Indels; (B) structural variations and (C) structural deletion variations.
Table 3.
Jindo dog indels in intragenic regionsa
| Gene name | Accession number | Chromosome | Orientation | Genic region | INDEL sequence |
|---|---|---|---|---|---|
| CYP4A11 | NM_001048034 | chr15 | + | 3′UTR | +T |
| MS4A1 | NM_001048028 | chr21 | + | 3′UTR | −C |
| DZIP1 | NM_001166008 | chr22 | − | 3′UTR | −AT |
| KRT1 | NM_001003392 | chr27 | + | CDS | +GGC |
| BHLHE41 | NM_001002973 | chr27 | + | 3′UTR | −GGTGCC |
| CRYAA | NM_001080898 | chr31 | + | 3′UTR | +T |
| TCOF1 | NM_001003057 | chr4 | + | CDS + intron | −GGGCACCTGCAGCCTCACCTGAACAG |
| TRAF5 | NM_001197118 | chr7 | + | 5′UTR | −AC |
| SET | NM_001003031 | chrX | + | CDS | −GATGATGAT |
aIndels in the Jindo dog genome compared with the boxer genome.
3.4. Structural variation
Next, we searched for structural variations using the paired-end short-read data. We identified a total of 8131 structural variations (7780 deletions, 231 insertions and 120 inversions) in the Jindo genome relative to the boxer reference genome (Fig. 2B). Among the structural deletion variations (7780), 1086 and 6694 events were caused by non-retrotransposons and retrotransposons, respectively, and of the 6694 retrotransposon-induced deletion variations, 2068, 2033, 389, 242 and 1962 events were mediated by LINE, SINE, LTR and DNA elements and other retrotransposons, respectively (Fig. 2C and Supplementary Fig. S3).
Interestingly, our analysis of the SINE-mediated deletions is consistent with a recent study22 that reported that there are >10 000 loci that are bimorphic for the SINEs in the dog genome and that 3–5% of them could vary between dog breeds as a result of deletion or insertion. Such differences in the absence or presence of SINEs (especially within intragenic regions) between dog breeds appear to be significantly associated with breed-specific novel exon creation (owing to the donation of splice acceptor sites by SINEs).22
3.5. Identification of novel dog genome sequences corresponding to gap regions in the boxer reference genome
Another important finding of this study was the discovery of novel dog genome sequences that correspond to the gap regions in the boxer breed reference genome. By performing de novo assembly of the 12 706 714 (1.15%) reads that were unmapped to the reference genome, we acquired 36 900 contigs and 25 805 scaffolds. We identified a significant match for 2517 (≥300 bp in length) of these contigs in the NR (non-redundant) database using BlastN; 3855 (≥300 bp in length) of these contigs did not have a match. Of the matched contigs, 734 (Supplementary Table S3) and 991 (Supplementary Table S4) had a match in the NR database (protein database) based on BlastX and in the expressed sequence tag (EST) database (human, mouse and others) based on BlastN, respectively. Of the unmatched contigs, 862 (Supplementary Table S5) and 42 (Supplementary Table S6) had a match in the NR (protein database) and EST databases, respectively. We matched 2438 (≥300 bp in length) of the scaffolds to the NR database using BlastN, and 3792 scaffolds (≥300 bp in length) were unmatched. Of these, 700 (matched; Supplementary Table S7) and 863 (unmatched; Supplementary Table S8) scaffolds were matched to the NR database (protein database), respectively, and 958 (matched; Supplementary Table S9) and 42 (unmatched; Supplementary Table S10) were matched to the EST database, respectively.
During the painstaking and time-consuming manual annotation [in the UCSC boxer reference genome (Broad/CanFam2) browser] of each contig or scaffold (unmapped to the reference genome) showing a match to the EST and NR protein databases, we realized that the unmapped reads mainly corresponded to cDNA or EST sequences that could be classified into three groups.
The first group includes EST sequences (dog, human and others) that span a known genomic region and a partial unknown sub-region within a gap region in the reference genome. Intriguingly, many unmapped scaffolds matched only to the parts (of the EST sequences) corresponding to the unknown sub-regions in the gaps, which explains why those reads and scaffolds could not be mapped to the reference genome. The many unmapped contigs and scaffolds that belonged to this first group included the contigs C64934 6.0 (corresponding gap location: chr9: 22362967–22363440), C72210 10.0 (chr25: 21595815–21596814), C61340 8.0 (chr1:5236 4890–52366081), C57026 6.0 (chr28:17167026 –17167309), C61208 6.0 (chr11:27745205–27745630) and C59020 6.0 (chr2:87164377–87165300) and the scaffolds 190 7.6 (chr26: 19087754–19088523) and 263 6.6 (chr1:383 95670–38396089). This paper is the first analysis of the dog genome using massively parallel next generation sequencing techniques, as opposed to the previously published boxer reference genome, which was based on cloning in bacterial cells, Sanger sequencing and whole genome shot-gun assembly. Similar to our results, the first human genome resequencing15 using NGS (next-generation sequencing) techniques added novel human genome sequences to the gaps in the reference human genome, which, at that time, was based on Sanger sequencing. In contrast to the cell-free system used with the NGS techniques, the traditional Sanger technique-based whole-genome sequencing method involves cloning steps in E. coli cells that are likely to cause the loss of sequence regions that are unsuited for in vivo cloning.15 Therefore, our Jindo dog short-read genome sequence data could make a significant contribution towards partially filling the gap regions in the reference boxer genome.
The second group of reads includes dog EST and cDNA sequences, the genomic locations of which are unknown at present. In addition, the third group consists of EST and cDNA sequences (from humans, bears, mice and others) for which the homologous dog partners (ESTs and cDNAs) are absent from dog EST libraries and their genomic locations in the dog genome are still obscure. Thus, our unmapped contigs and scaffolds that belong to the second and third groups appear to correspond to the middle regions of large gaps, which are so long that a known EST sequence flanking a gap, if it exists, cannot cover the middle region.
3.6. Olfactory receptor gene family
In general, dogs have much keener olfactory abilities than humans, and the Korean Jindo dog is known to have a particularly acute olfactory sense. Previously, ∼900 and 1094 olfactory receptor genes have been reported in humans and dogs, respectively, and 63% (567) and 20.3% (222) of the respective human and dog olfactory receptor gene repertoires have been annotated as pseudo genes, providing evidence that more functional genes are involved in the olfactory process in dogs than in humans.8,11,23
Using the recently updated boxer olfactory receptor gene repertoire (1179 genes, including predicted and experimentally invalidated genes; http://genome.weizmann.ac.il/horde/organism/index/organism:Dog),24 we identified the corresponding olfactory receptor genes in our Jindo dog genome short-read data and analysed SNPs within their sequences. We detected a total of 2299 SNPs in those genes, of which 773 and 1526 are homozygous and heterozygous SNPs, respectively (Supplementary Tables S11 and S12). The 773 homozygous SNPs are located within 265 coding genes and 108 pseudo genes and the 1526 heterozygous SNPs are located within 369 coding genes and 152 pseudo genes. In the case of the heterozygous SNP-containing Jindo olfactory genes, their Jindo alleles with no SNP are also present in the boxer genome. Therefore, we focused on further analysis of the homozygous SNP-containing Jindo olfactory genes for which the genotypes are present only in the Jindo dog genome and not in the boxer genome. Such analysis showed that, of the homozygous SNP-containing Jindo olfactory receptor genes (373), 236 and 137 had non-synonymous (Supplementary Table S13) and synonymous SNPs, respectively. The existence of the non-synonymous, homozygous SNPs within the 236 Jindo olfactory genes and their absence from the corresponding genes in the boxer genome implies that the smelling capabilities of the Jindo and boxer dog breeds might be significantly different. This finding provides strong evidence of such differences from a new molecular level perspective and is consistent with the recent discovery25 that the domestication of dogs resulted in changes in dog brain morphologies, accompanied by the reorientation of olfactory lobes and bulbs and consequent changes in olfactory sensory abilities among diverse domesticated dog breeds.
Another interesting result of this study is our discovery of two unique dog olfactory receptor genes (named cOR5AS1 and cOR14C36) in the Jindo breed dog genome. These receptors had not previously been reported. Importantly, the unique dog olfactory receptor gene cOR5AS1 has a single exon encoding a deduced amino acid sequence with seven transmemebrane domains, which is typical of the olfactory receptor proteins (Fig. 3A). Interestingly, using the UCSC Affymetrix exon array chip database track, we identified that the human predicted olfactory receptor gene OR5AS1, which had the highest sequence identity (90%) at the amino acid level to the unique dog gene cOR5AS1, was expressed at low levels in the human cerebellum, thyroid and other tissues (Fig. 3B). In addition, not only the nucleotide sequence of the cOR5AS1 gene was highly conserved among a variety of mammalian species, but also its amino acid sequence with the seven transmembrane domain regions showed high homology (human: 90%, chimpanzee: 88%, macaque: 89%, pig: 88%, mouse: 81% and rat: 82%) with orthologous protein sequences from humans, chimpanzees, macaques, pigs, mice and rats (Fig. 3C). These results imply that the protein encoded by this unique gene might be functionally involved in the olfactory process in humans and dogs and that its function could be evolutionarily preserved in mammalian species.26
Figure 3.

The unique olfactory receptor gene cOR5AS1. (A) The proteins encoded by the unique dog olfactory receptor gene and its human homologue are both membrane-embedded proteins with seven transmembrane domains. (B) The unique dog olfactory receptor gene and its human homologue, both of which consist of a single exon, are closely aligned in the UCSC genome browser. The human homologue shows expression on the UCSC Affymetrix exon chip array track and also exhibits high evolutionary conservation among mammalian species on the UCSC conservation track. (C) Sequence identities among protein sequences (homologous to the unique dog olfactory receptor) from seven species.
We found that the other unique dog olfactory gene discovered in this study, cOR14C36, is a pseudo gene harbouring stop codons in its CDS, unlike its human homolog (OR14C36), which has a CDS encoding a deduced amino acid sequence uninterrupted by any mid-stop codons. This finding suggests that the second unique olfactory receptor gene has been conserved evolutionarily between humans and dogs, but functionally degenerated in dog species during the evolutionary process. It remains to be seen whether the pseudo olfactory receptor genes corresponding to ∼20% of all olfactory genes represent evolutionary remnants or wasteful rubbish in the dog genome or why these apparently useless genes are evolutionarily preserved in the human (54%), dog (20%), mouse (20%) and rat (19.5%) genomes. In light of the recent breakthrough discovery27 that there are huge regulatory interaction networks connecting pseudo genes and their homologous coding genes (via microRNAs) that are mediated by the MRE (microRNA response element), the so-called ‘Rosetta stone28 of a hidden RNA language’, now would be the time to reassess the real value of the olfactory pseudo genes.
3.7. Mitochondrial genome sequence reveals Jindo dog-specific genotypes
We determined the mitochondrial genome sequence of the Jindo dog by performing de novo assembly of mitochondrial reads extracted from our sequence read data (see Section 2). The mitochondrial genome of the Jindo dog consists of 13 protein-coding genes, 22 tRNA genes and 2 rRNAs genes (16 and 12S; Fig. 4). The sequence length of the Jindo dog mitochondrial genome determined in this study was 16 100 bp, but we estimated that the correct length of this mitochondrial DNA genome could be ∼16.7 kb because there might be a gap (corresponding to a deficiency in a control region between tRNAPro and tRNAPhe) spanning ∼600 bp.29
Figure 4.

Annotation of the Jindo dog mitochondrial genome sequence. Red arrows and numbers indicate the base positions corresponding to the Jindo dog-specific SNPs in the mitochondrial genome sequence.
A comparative analysis of mitochondrial genomic DNA sequences from 80 dog breeds, including the Jindo dog, showed Jindo dog-specific genotypes at 9 sequence positions (Table 4). Of these nine positions, two were present in intergenic regions and the other seven in the COX2 and ND5 genes. This result suggests that these nine genotypes could be used as genetic markers for identification of the Jindo dog breed.
Table 4.
Jindo dog-specific mitochondrial DNA genotypes
| MT DNA position | Genotype in Jindo dog | Genotype in 79 dog breeds | Gene name |
|---|---|---|---|
| 3760 | T | A | Intergenic region |
| 5234 | A | G | Intergenic region |
| 7649 | T | C | COX2 |
| 7658 | G | A | COX2 |
| 11 626 | T | C | ND5 |
| 11 694 | T | C | ND5 |
| 11 775 | A | G | ND5 |
| 11 797 | A | T | ND5 |
| 12 693 | A | G | ND5 |
3.8. Phylogenetic analysis suggests that Jindo dog is a purebred dog breed
To elucidate the phylogenetic origin of the domesticated Jindo dog breed, we drew a phylogenetic tree using mitochondrial DNA sequences from the grey wolf and 80 dog breeds including the Jindo dog and the mtDNA sequence from a coyote as an out-group (Fig. 5). We determined that these dog breeds could be phylogenetically classified into five relatively large groups (A–E) and two small groups (F and G). Surprisingly, the Jindo dog breed did not belong to any of these groups, providing evidence that the Jindo dog breed could have a maternally unique phylogenetic history during the domestication processes from grey wolves to dogs. Moreover, the Sapsari dog breed,29 which also originated in Korea, is not grouped with the Jindo dog breed in our phylogenetic tree. This result indicates that the Jindo dog breed is an independent purebred that originated in Korea, separate from the Sapsari dog breed.
Figure 5.
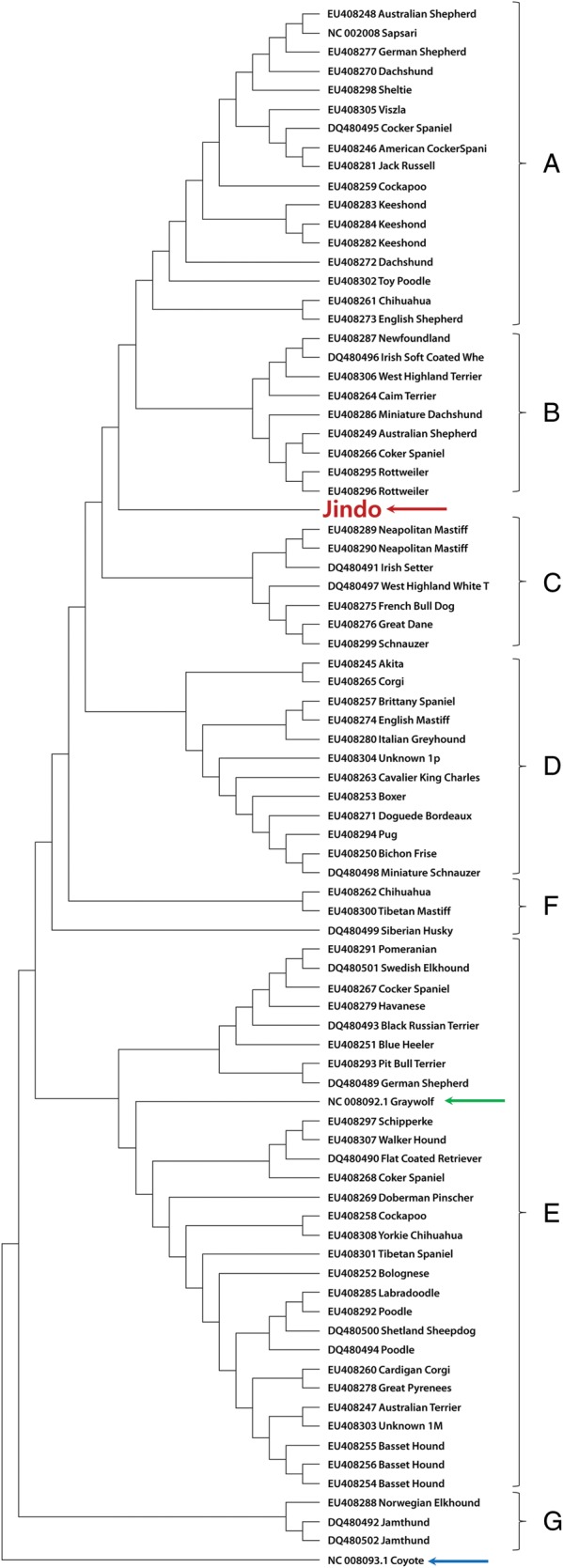
Phylogenetic tree generated using mitochondrial genome sequences from 80 dog breeds. A coyote mitochondrial genome sequence was used as an out-group. Red, green and blue arrows indicate where the Jindo dog, grey wolf and coyote are phylogenetically located, respectively. The letters A–F and G indicate the names of the groups to which each of the dog breeds belong phylogenetically.
Our phylogenetic tree also shows that dog breeds and grey wolf are included together in a large group (E), but all of the dog breeds are clearly split from coyote, supporting the evidence that the grey wolf could be an ancestor of domesticated dogs.7,30,31
4. Discussion
Our analysis of the Jindo dog genome using Illumina massively parallel sequencing technology is, to our knowledge, the first application of next-generation sequencing techniques to a dog genome since the traditional Sanger method-based sequencing of the boxer and poodle genomes.
We show that short reads derived from a cell-free system could partially cover the gap regions in the reference boxer genome sequence, which might not have been sequenced originally because of difficulty in in vivo cloning of these regions during the Sanger sequencing and assembly of the boxer genome. This finding strongly suggests that a significant portion of the remaining gaps in the reference genome could be partially or completely filled in the near future by the more cost-effective NGS read data (Illumina short reads and 454 reads).
The establishment of a correct genome-wide SNP map is very important for distinguishing between dog breeds and between individual dogs with regard to disease susceptibility, haplotype, gene expression and allele types. Using the 45-fold coverage short-read data, we identified 3 346 058 novel SNPs in the Jindo genome compared with boxer SNPs that had previously been identified based on the 7.5-fold coverage reference genome data. In addition, based on the non-synonymous SNPs in the Jindo dog genome, we indicated the probabilities of differences in disease susceptibility among the Jindo, boxer and other dog breeds. This result demonstrates that the Illumina NGS technology can cost-effectively generate high fold coverage read data that can be aligned with most (94.02%) of the reference boxer genome regions, undoubtedly guaranteeing a much higher accuracy of SNP identification than the low-coverage (7.5) boxer genome data based on the traditional Sanger method.
There is growing evidence32 that the synonymous SNPs located within CDS of the protein-coding genes cannot be considered as silent or insignificant with regard to genetic disease causing. The synonymous SNPs (sSNPs) within CDS can cause a change in codon usage that could result in a change in a ribosome movement speed during protein translation, subsequently causing mis-folding in protein conformation and consequently affecting protein function.32,33 The sSNPs within exonic spicing enhancer sequences in the coding sequences could also change splicing processes and consequently mRNA structures.32,34 In light of these previous studies, the 473 CDS sSNPs (Fig. 1C and Supplementary Table S1) identified in this study could not be ignorable targets in future researches.
The in-depth annotation of the dog genome could have a significant impact on human and medical biology. More than 360 genetic diseases found in humans are known to also be found in dogs.2,3 This fact suggests that the dog is a suitable model animal for identifying the loci of human disease-associated genes and for studying the causative mechanisms of human diseases. Through the annotation of the Jindo dog short-read genome data, we identified unique olfactory receptor genes, SNPs located in olfactory receptor genes and CDS regions of genes that are associated with genetic diseases and morphological development and breed-specific Jindo SNP genotypes in the mitochondrial genome sequence. Such information could be valuable for genome-wide association studies aimed at locating disease genes and SNPs in the dog genome. In conclusion, our Jindo genome data help gain deeper insight into dog genome sequence, structure and architecture and comparative genomics.
Supplementary Data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This research was supported by grant 2009-0084206 from the Ministry of Education, Science and Technology (MEST) and grant KGM5411011 from KRIBB.
Supplementary Material
Footnotes
Edited by Masahira Hattori
References
- 1.Ostrander E.A., Galibert F., Patterson D.F. Canine genetics comes of age. Trends Genet. 2000;16:117–24. doi: 10.1016/s0168-9525(99)01958-7. [DOI] [PubMed] [Google Scholar]
- 2.Patterson D.F. Companion animal medicine in the age of medical genetics. J. Vet. Intern. Med. 2000;14:1–9. [PubMed] [Google Scholar]
- 3.Sargan D.R. IDID: inherited diseases in dogs: web-based information for canine inherited disease genetics. Mamm. Genome. 2004;15:503–6. doi: 10.1007/s00335-004-3047-z. [DOI] [PubMed] [Google Scholar]
- 4.Wayne R.K. Limb morphology of domestic and wild canids: the influence of development on morphologic change. J. Morphol. 1986;187:301–19. doi: 10.1002/jmor.1051870304. [DOI] [PubMed] [Google Scholar]
- 5.Lindblad-Toh K., Wade C.M., Mikkelsen T.S., et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–19. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- 6.Kirkness E.F., Bafna V., Halpern A.L., et al. The dog genome: survey sequencing and comparative analysis. Science. 2003;301:1898–903. doi: 10.1126/science.1086432. [DOI] [PubMed] [Google Scholar]
- 7.Ostrander E.A., Wayne R.K. The canine genome. Genome Res. 2005;15:1706–16. doi: 10.1101/gr.3736605. [DOI] [PubMed] [Google Scholar]
- 8.Quignon P., Kirkness E., Cadieu E., et al. Comparison of the canine and human olfactory receptor gene repertoires. Genome Biol. 2003;4:R80. doi: 10.1186/gb-2003-4-12-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tacher S., Quignon P., Rimbault M., Dreano S., Andre C., Galibert F. Olfactory receptor sequence polymorphism within and between breeds of dogs. J. Hered. 2005;96:812–6. doi: 10.1093/jhered/esi113. [DOI] [PubMed] [Google Scholar]
- 10.Olender T., Fuchs T., Linhart C., et al. The canine olfactory subgenome. Genomics. 2004;83:361–72. doi: 10.1016/j.ygeno.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 11.Quignon P., Giraud M., Rimbault M., et al. The dog and rat olfactory receptor repertoires. Genome Biol. 2005;6:R83. doi: 10.1186/gb-2005-6-10-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gundry R. L., Allard M.W., Moretti T.R., et al. Mitochondrial DNA analysis of the domestic dog: control region variation within and among breeds. J. Forensic. Sci. 2007;52:562–72. doi: 10.1111/j.1556-4029.2007.00425.x. [DOI] [PubMed] [Google Scholar]
- 13.Eichmann C., Parson W. Molecular characterization of the canine mitochondrial DNA control region for forensic applications. Int. J. Legal Med. 2007;121:411–6. doi: 10.1007/s00414-006-0143-5. [DOI] [PubMed] [Google Scholar]
- 14.Pang J.F., Kluetsch C., Zou X.J., et al. mtDNA data indicate a single origin for dogs south of Yangtze River, less than 16,300 years ago, from numerous wolves. Mol. Biol. Evol. 2009;26:2849–64. doi: 10.1093/molbev/msp195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wheeler D.A., Srinivasan M., Egholm M., et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–6. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 16.Wang J., Wang W., Li R., et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–5. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ju Y.S., Kim J.I., Kim S., et al. Extensive genomic and transcriptional diversity identified through massively parallel DNA and RNA sequencing of eighteen Korean individuals. Nat. Genet. 2011;43:745–52. doi: 10.1038/ng.872. [DOI] [PubMed] [Google Scholar]
- 18.Metzker M.L. Sequencing technologies—the next generation. Nat. Rev. Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 19.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Splendore A., Fanganiello R.D., Masotti C., Morganti L.S., Passos-Bueno M.R. TCOF1 mutation database: novel mutation in the alternatively spliced exon 6A and update in mutation nomenclature. Hum. Mutat. 2005;25:429–34. doi: 10.1002/humu.20159. [DOI] [PubMed] [Google Scholar]
- 21.Haworth K.E., Islam I., Breen M., et al. Canine TCOF1; cloning, chromosome assignment and genetic analysis in dogs with different head types. Mamm. Genome. 2001;12:622–9. doi: 10.1007/s00335-001-3011-0. [DOI] [PubMed] [Google Scholar]
- 22.Wang W., Kirkness E.F. Short interspersed elements (SINEs) are a major source of canine genomic diversity. Genome Res. 2005;15:1798–808. doi: 10.1101/gr.3765505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malnic B., Godfrey P.A., Buck L.B. The human olfactory receptor gene family. Proc. Natl. Acad. Sci. USA. 2004;101:2584–9. doi: 10.1073/pnas.0307882100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aloni R., Olender T., Lancet D. Ancient genomic architecture for mammalian olfactory receptor clusters. Genome Biol. 2006;7:R88. doi: 10.1186/gb-2006-7-10-r88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roberts T., McGreevy P., Valenzuela M. Human induced rotation and reorganization of the brain of domestic dogs. PLoS One. 2010;5:e11946. doi: 10.1371/journal.pone.0011946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mainland J.D., Johnson B.N., Khan R., Ivry R.B., Sobel N. Olfactory impairments in patients with unilateral cerebellar lesions are selective to inputs from the contralesional nostril. J. Neurosci. 2005;25:6362–71. doi: 10.1523/JNEUROSCI.0920-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poliseno L., Salmena L., Zhang J., Carver B., Haveman W.J., Pandolfi P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–8. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P.P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–8. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim K.S., Lee S.E., Jeong H.W., Ha J.H. The complete nucleotide sequence of the domestic dog (Canis familiaris) mitochondrial genome. Mol. Phylogenet. Evol. 1998;10:210–20. doi: 10.1006/mpev.1998.0513. [DOI] [PubMed] [Google Scholar]
- 30.Savolainen P., Zhang Y.P., Luo J., Lundeberg J., Leitner T. Genetic evidence for an East Asian origin of domestic dogs. Science. 2002;298:1610–3. doi: 10.1126/science.1073906. [DOI] [PubMed] [Google Scholar]
- 31.Vonholdt B.M., Pollinger J.P., Lohmueller K.E., et al. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature. 2010;464:898–902. doi: 10.1038/nature08837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sauna Z.E., Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 2011;12:683–91. doi: 10.1038/nrg3051. [DOI] [PubMed] [Google Scholar]
- 33.Sauna Z.E., Kimchi-Sarfaty C., Ambudkar S.V., Gottesman M.M. Silent polymorphisms speak: how they affect pharmacogenomics and the treatment of cancer. Cancer Res. 2007;67:9609–12. doi: 10.1158/0008-5472.CAN-07-2377. [DOI] [PubMed] [Google Scholar]
- 34.Chamary J.V., Parmley J.L., Hurst L.D. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 2006;7:98–108. doi: 10.1038/nrg1770. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.