

Abstract
The prediction of prostate cancer clinical outcome remains a major challenge after the diagnosis, even with improved early detection by prostate-specific antigen (PSA) monitoring. To evaluate whether copy number variation (CNV) of the genomes in prostate cancer tumor, in benign prostate tissues adjacent to the tumor (AT), and in the blood of patients with prostate cancer predicts biochemical (PSA) relapse and the kinetics of relapse, 241 samples (104 tumor, 49 matched AT, 85 matched blood, and 3 cell lines) were analyzed using Affymetrix SNP 6.0 chips. By using gene-specific CNV from tumor, the genome model correctly predicted 73% (receiver operating characteristic P = 0.003) cases for relapse and 75% (P < 0.001) cases for short PSA doubling time (PSADT, <4 months). The gene-specific CNV model from AT correctly predicted 67% (P = 0.041) cases for relapse and 77% (P = 0.015) cases for short PSADT. By using median-sized CNV from blood, the genome model correctly predicted 81% (P < 0.001) cases for relapse and 69% (P = 0.001) cases for short PSADT. By using median-sized CNV from tumor, the genome model correctly predicted 75% (P < 0.001) cases for relapse and 80% (P < 0.001) cases for short PSADT. For the first time, our analysis indicates that genomic abnormalities in either benign or malignant tissues are predictive of the clinical outcome of a malignancy.
Prostate cancer is one of the most common and lethal malignancies for men. The annual mortality rate reached 32,000 in the United States in 2009.1–3 Although active monitoring of the serum prostate-specific antigen (PSA) level for men >50 years has greatly improved early detection of prostate cancer, the mortality rate from prostate cancer does not significantly improve.4,5 Several treatment options are available for patients with prostate cancer, including watchful waiting, radiation, hormone therapy/chemotherapy, and radical prostatectomy. Gleason grading alone or in combination with other clinical indicators, such as serum PSA levels and pathological or clinical staging, has been the guiding tool in selecting these treatment options. However, many patients with prostate cancer experienced relapse after surgical resection of the prostate gland. There is clearly a need for better prediction of the behavior of prostate cancer. Previous cytogenetic and other genome studies6–11 suggest a clear link between genome abnormalities and the prostate cancer. To detect genome abnormalities in prostate cancer, a comprehensive genome analysis on 241 prostate cancer samples (104 prostate cancer, 85 matched blood samples, 49 matched benign prostate tissues adjacent to cancer, and 3 cell lines) was performed using SNP 6.0 chips (Affymetrix, Santa Clara, CA). Our analyses indicate that genome copy number variation (CNV) occurred in both cancer and noncancer tissues and that the CNV of these tissues predicts prostate cancer progression. Specifically, prediction models of prostate cancer relapse or short PSA doubling time (PSADT) were generated from specific CNV patterns in tumor or benign prostate tissues adjacent to cancer samples. In addition, mean and median sizes of CNV from patients' blood, benign prostate tissues adjacent to the tumor (AT), and tumor samples are also predictive of these clinical outcomes, independent of specific genes and regions.
Materials and Methods
Tissue Processing, DNA Extraction, Amplicon Generation, Labeling, Hybridization, Washing, and Scanning of SNP 6.0 Chips
Prostate cancer samples were obtained from the University of Pittsburgh Medical Center Tissue Bank, Pittsburgh, PA. These samples were collected from 1998 to 2009. To make the analysis balance, samples of short PSADT (<4 months), long PSADT (>15 months), and no relapse (cancer free for >5 years after radical prostatectomy) each were made to constitute approximately one third of the total number. Whenever possible, nonrelapse samples were chosen to match pathological stages and Gleason grades of relapse samples. A total of 214 samples were from whites, whereas 5 samples were from African Americans and 19 samples were from patients with an unknown race. The patients whom these samples were obtained from either experienced relapse or had no relapse for at least 5 years, based on chemical (serum PSA) and radiological evidence. Frozen tissues were used for blood, prostate cancer, and benign prostate tissue adjacent to cancer. Clinical follow-up was conducted by office examination record, blood PSA survey, and radiographical follow-up. These follow-up visits were performed for up to a 10-year period after the patient underwent a radical prostatectomy. The protocol was approved by the Institutional Review Board. For prostate cancer, microdissection was performed to achieve tumor purity >80%. For benign prostate tissues adjacent to cancer, benign tissues away from prostate cancer (at least 3 mm) were microdissected. Whenever available, whole blood or buffy coat from the same patients was used as a normal control. PC3, DU145, and LNCaP cells were obtained from American Type Culture Collection Inc. (Manassas, VA) in 2000, 2001, and 2007, respectively. The genomes of these cell lines were tested for short tandem repeat DNA profiling on eight different loci (CSF1PO, D13S317, D16S539, D5S818, D7S820, THO1, TPOX, and vWA) of the genomes by PCR using the following sets of primers: CSF1PO, 5′-AACCTGAGTCTGCCAAGGACTAGC-3′ and 5′-TTCCACACACCACTGGCCATCTTC-3′; D13S317, 5′-ACAGAAGTCTGGGATGTGGA-3′ and 5′-GCCCAAAAAGACAGACAGAA-3′; D16S539, 5′-GATCCCAAGCTCTTCCTCTT-3′ and 5′-ACGTTTGTGTGTGCATCTGT-3′; D5S818, 5′-GGGTGATTTTCCTCTTTGGT-3′ and 5′-TGATTCCAATCATAGCCACA-3′; D7S820, 5′-TGTCATAGTTTAGAACGAACTAACG-3′ and 5′-CTGAGGTATCAAAAACTCAGAGG-3′; TH01, 5′-GTGGGCTGAAAAGCTCCCGATTAT-3′ and 5′-ATTCAAAGGGTATCTGGGCTCTGG-3′; TPOX, 5′-ACTGGCACAGAACAGGCACTTAGG-3′ and 5′-GGAGGAACTGGGAACCACACAGGT-3′; and vWA, 5′-CCCTAGTGGATGATAAGAATAATCAGTATG-3′ and 5′-GGACAGATGATAAATACATAGGATGGATGG-3′. These cell lines were authenticated because the short tandem repeat profiles of the cell lines have a perfect match with those published by American Type Culture Collection Inc. DNA was then extracted using a Qiagen tissue kit (Qiagen, Valencia, CA). Detailed case information is shown in Tables 1, 2, and 3 and Supplemental Table S1 (available at http://ajp.amjpathol.org). Genome DNA (500 ng), was digested with StyI and Nsp1 for 2 hours at 37°C. The digested DNA was purified and ligated with primer/adaptors at 16°C for 12 to 16 hours. Amplicons were generated by performing PCR using primers provided by the manufacturer (Affymetrix, Santa Clara, CA) on the ligation products using the following program: 94°C for 3 minutes and then 35 cycles of 94°C for 30 seconds, 60°C for 45 seconds, and 65°C for 1 minute. This was followed by extension at 68°C for 7 minutes. The PCR products were then purified and digested with DNaseI for 35 minutes at 37°C to fragment the amplified DNA. The fragmented DNA was then labeled with biotinylated nucleotide through terminal deoxynucleotide transferase for 4 hours at 37°C. Fragmented DNA, 250 μg, was hybridized with a pre-equilibrated Affymetrix SNP 6.0 chip at 50°C for 18 hours. Procedures of washing and scanning of SNP 6.0 chips followed the manuals provided by Affymetrix.
Table 1.
Clinical and Pathological Characteristics of Prostate Cancer Samples
| Relapse status | None (n = 28) | Long PSADT (n = 42) | Short PSADT (n = 33) |
|---|---|---|---|
| Mean age (P = 0.0783) | 56.07 | 59.29 | 56.06 |
| Cancer stage (P = 0.0224) | |||
| pT1 | 3 (2.9) | 1 (1.0) | 0 (0) |
| pT2 | 10 (9.7) | 9 (8.7) | 7 (6.8) |
| pT3a | 7 (6.8) | 18 (17.5) | 6 (5.8) |
| pT3b | 8 (7.8) | 14 (13.6) | 20 (19.4) |
| Gleason grade (P = 0.6569) | |||
| 6 | 6 (5.8) | 8 (7.8) | 3 (2.9) |
| 7 | 16 (15.5) | 26 (25.2) | 21 (20.4) |
| 8–10 | 6 (5.8) | 8 (7.8) | 9 (8.7) |
| Race (P = 0.2349) | |||
| Black | 1 | 2 | 0 |
| Unknown | 4 | 1 | 2 |
| White | 23 | 39 | 31 |
| Median follow-up (months) | 154 | 124.8 | 54.8 |
| Median time to progression (months) | NA | 47.355 | 1.87 |
| Median PSADT (months) | NA | 23.2 | 3.21 |
| Mean preoperative PSA (P = 0.42) | 8.61 | 12.31 | 10.79 |
Data are given as number (percentage) of the 103 samples unless otherwise indicated.
NA, not applicable.
Table 2.
Clinical and Pathological Characteristics of Prostate Tissues Adjacent to Tumor
| Relapse status | None (n = 28) | Long PSADT (n = 13) | Short PSADT (n = 8) |
|---|---|---|---|
| Mean age (P = 0.554) | 55 | 57.69 | 56.06 |
| Cancer stage (P = 0.541) | |||
| pT1 | 3 (6.1) | 0 (0) | 0 (0) |
| pT2 | 11 (22.4) | 4 (8.2) | 3 (6.1) |
| pT3a | 7 (14.3) | 6 (12.2) | 1 (2.0) |
| pT3b | 7 (14.3) | 3 (6.1) | 4 (8.2) |
| Gleason grade (P = 0.9849) | |||
| 5 | 1 (2.0) | 0 (0) | 0 (0) |
| 6 | 7 (14.3) | 3 (6.1) | 2 (4.1) |
| 7 | 16 (32.7) | 7 (14.3) | 5 (10.2) |
| 8–10 | 4 (8.2) | 3 (6.1) | 1 (2.0) |
| Race (P = 0.08387) | |||
| Black | 0 | 1 | 0 |
| Unknown | 6 | 0 | 0 |
| White | 22 | 12 | 8 |
| Median follow-up (months) | 155 | 149 | 29.205 |
| Median time to progression (months) | NA | 54.6 | 3.09 |
| Median PSADT (months) | NA | 26.9 | 2.46 |
| Mean preoperative PSA (P = 0.074) | 8.23 | 12.98 | 6.3 |
Data are given as number (percentage) of the 49 samples unless otherwise indicated.
NA, not applicable.
Table 3.
Clinical and Pathological Characteristics of Blood Samples from Patients with Prostate Cancer
| Relapse status | None (n = 18) | Long PSADT (n = 35) | Short PSADT (n = 31) |
|---|---|---|---|
| Mean age (P = 0.268) | 58.33 | 59.43 | 56.77 |
| Cancer stage (P = 0.003893) | |||
| pT1 | 5 (6.0) | 1 (1.2) | 0 (0) |
| pT2 | 6 (7.1) | 12 (14.3) | 6 (7.1) |
| pT3a | 1 (1.2) | 10 (11.9) | 5 (6.0) |
| pT3b | 6 (7.1) | 12 (14.3) | 20 (23.8) |
| Gleason grade (P = 0.2248) | |||
| 6 | 5 (6.0) | 8 (9.5) | 3 (3.6) |
| 7 | 7 (8.3) | 21 (25.0) | 18 (21.4) |
| 8–10 | 6 (7.1) | 6 (7.1) | 10 (11.9) |
| Race (P = 0.08387) | |||
| Black | 0 | 1 | 0 |
| Unknown | 3 | 1 | 2 |
| White | 15 | 33 | 29 |
| Median follow-up (months) | 152 | 109.14 | 54.8 |
| Median time to progression (months) | NA | 47.27 | 3.23 |
| Median PSADT (months) | NA | 26 | 3.21 |
| Mean preoperative PSA (P = 0.868) | 10.48 | 12.17 | 10.87 |
Data are given as number (percentage) of the 84 samples unless otherwise indicated.
NA, not applicable.
SYBR Green Real-Time qPCR
A LightCycler FastStart DNA Master SYBR Green I Kit (Hoffman-La Roche, Inc., Nutley, NJ) was used for real-time PCR amplification. The reaction was performed in a MasterCycler Realplex (Eppendorf, Hauppauge, NY). A quantitation standard curve of normal male DNA, from 50,000 to 500,000 copies of genome, was generated using known amounts of template copies. Genomic DNA, 20 ng, was used for all of the experimental and control samples. TaqDNA polymerase was activated with a 2-minute pre-incubation step at 94°C. Amplification of the following primers was performed: ARL17B, 5′-ACTGTCATAGCAGTGCTGAGG-3′ and 5′-ACTTACCTACTGTAGGGACGG-3′; SCAPER, 5′-AGGAAGGCCTATTCGTTCTCG-3′ and 5′-GAACAGTATGGGAGGAGTTCG-3′; WWOX, 5′-GCCAGTTGATGTGACAACTGC-3′ and 5′-CAGCTGAGAGTGGTTTCTTTGC-3′; EPHA3, 5′-ATCAGGACTTACCAGGTGTGC-3′ and 5′-ACCGTGTCTGGAAACATAGCC-3′; and ERBB4, 5′-AGTGGCCTGTCCTTGCTTATC-3′ and 5′-CAGAGCAACAATTCTGACCGG-3′. There were 35 cycles of the following program: 94°C for 30 seconds, 62°C for 30 seconds, and 68°C for 3 minutes. Realplex data software (Eppendorf, Inc., Hauppauge, NY) was used to quantify and fit the data with a standard curve. A separate β-actin (5′-TCTTTGCACTTTCTGCATGTCCCC-3′ and 5′-GTCCATCACGATGCCAGTGGTAC-3′) DNA quantification was also performed as an internal control for each analysis.
Statistical Analysis
A total of 241 cel files were analyzed with Genotyping console (Affymetrix) for quality control analysis. Samples with a quality control call of >80% and a quality control contrast ratio of >0.4 were admitted into the analysis. To analyze CNV, cel files were imported into Partek Genome Suite 6.6 (Partek, Inc., St. Louis, MO) to generate copy number from raw intensity. To plot the histograms, deletion or amplification of genomes was analyzed by first limiting to the regions with P < 0.05/total number of regions detected (ie, the familywise error rate is controlled using Bonferroni's correction).12 The selected regions were subsequently filtered by limiting to the regions with at least 100 markers and 10 kb. The regions were then mapped to known genes. The frequencies of amplification and deletions were plotted to the genome corresponding to the gene locations.
Prediction Analysis and ROC Curve
The following prediction analysis for the comparison of nonrelapse versus fast relapse plus slow relapse and nonrelapse plus slow relapse versus fast relapse was performed. A test sample was first left out from prediction model construction. The remaining samples were used as the training set. Loci with more than r% amplification or r% deletion in the case group but no locus aberration in the control group were selected as predictive loci. To predict the left-out test sample, the percentage of locus aberration (amplification or deletion) among the identified predictive loci was calculated. The test sample was predicted as a case if the percentage of aberration is greater than p% threshold, and control otherwise. The leave-one-out cross validation was repeated until each sample was left out and predicted. In this prediction scheme, r is a parameter that determines the number of predictive loci used in the model. For a given r, the threshold p% was varied to locus rate an receiver operating characteristic (ROC) curve with sensitivity/specificity trade-off. We selected r that produced the best area under the curve (AUC).13 To report the best sensitivity and specificity trade-off and overall accuracy rate, we chose the threshold p% such that the Youden index (sensitivity + specificity − 1) is maximized. This criterion gave equal importance to sensitivity and specificity. To further evaluate whether the prediction result is better than obtained by random, AUC was used as a test statistic, and permutation analysis was performed to assess the statistical significance. Specifically, class labels (case and control) were randomly shuffled and AUC calculation was performed. Such permutations were repeated for 1000 times to generate the null distribution. The P value was calculated as the percentage that the 1000 null AUCs from permutation are greater than the observed AUC. The genes that are overlapped with the loci used in the test and the frequency of use are listed in Supplemental Tables S2 through S5 (available at http://ajp.amjpathol.org). For Gleason score prediction, the ROC curve was generated by varying the Gleason score threshold. AUC and its associated P value were similarly calculated. For CNV size prediction, CNV was limited to >2 kb, P < 0.001, and >10 markers. The ROC curve was generated by varying sizes of CNV threshold. AUC and its associated P value were similarly calculated.
Prediction Analysis for Blood versus Tumor
To predict blood versus tumor, the total number of aberrations in each sample was counted instead of the predictive locus selection previously described. The ROC curve, AUC, and the associated P value were similarly generated.
Results
The SNP 6.0 chip hybridization results were analyzed using Partek Genome Suite 6.6, with blood samples as normal references. As shown in the histograms of Figure 1A, abnormalities of genome in copy number can be found in all chromosomes in prostate cancer. An average of 91.6 loci (minimum, 10 kb) per sample involving 1092 genes were identified as either amplified or deleted in prostate cancer genomes, as determined by >100 markers (P < 5.5 × 10−9; Bonferroni correction, Figure 1B). Deletions of large segments, more than three megabases, of chromosomes 8p, 13p, 16q, and 17p occurred with high frequencies, whereas amplification of 8q and X chromosomes occurred in a subset of prostate cancer samples. Similar amplification and deletion of the same regions also occurred in benign prostate tissues adjacent to cancer, albeit with smaller sizes and a lower frequency. The blood of patients with prostate cancer contains significant CNVs in genomes (1329 genes total, or 4.4 loci and 32.6 genes/sample). Most of these CNVs are not unique and are overlapped with those of prostate cancer samples (Figure 1C). Prostate cancers were then subdivided based on clinical behavior: those with no relapse after prostatectomy, those with a relapse and a slow increase in serum PSA level (doubling in >15 months), and those with a relapse and a rapid increase in serum PSA level (doubling in <4 months). The kinetics of PSA increase after prostatectomy are predictive of prostate cancer-specific death, with rapid increases highly associated with lethal prostate cancer.14 The spectrum of CNVs increases from blood to prostate cancer in an incremental manner: the least in blood to the most in rapidly progressive prostate cancer (doubling in <4 months) (Figure 1D).
Figure 1.

Deletion and amplification of segments of genomes in blood, AT, and prostate cancer samples. A: Histograms of genome deletion (blue) or amplification (red) of blood (B), AT, and tumor (T) in 23 pairs of human chromosomes. B: Box plot of number of genes overlapping with CNV per sample. Outliers in blood and AT samples are indicated. C: Venn diagram of deleted or amplified genes occurring in at least one sample overlapping between blood, AT, and tumor. D: The spectrum of genes that are amplified or deleted in blood, AT, and tumors that did not relapse (Tnone), tumors that relapsed and had a PSADT at or after 15 months of radical prostatectomy (Tslow), and tumors that relapsed and had a PSADT within 4 months of radical prostatectomy (Tfast).
To assess the reproducibility of these analyses, a large set of reference normal samples (n = 800) available to the public through Partek, Inc., was used. This re-analysis showed that genome segment CNVs of blood, AT, and tumor overlapped by at least 93% between these two analyses (see Supplemental Figure S1A at http://ajp.amjpathol.org). In addition, a third analysis using a different set of normal samples (GeneSpring GX11, n = 265) was performed, showing that 94% to 99% of the amplified or deleted genome segments from blood, AT, and tumor overlap with those obtained from Partek Genome Suite analyses using blood as baseline (see Supplemental Figure S1A at http://ajp.amjpathol.org). Affymetrix SNP 6.0 contains separate probe sets for SNP and CNV detection. Most large genome deletions are accompanied by loss of heterozygosity (LOH). Profiles of LOH for blood, AT, and tumor samples were generated to validate the deletions detected by CNV analysis. Genome deletion frequently accompanied LOH (91% to 98%), with average matches for blood, AT, and tumor ranging from 93% to 96% (see Supplemental Figure S1B at http://ajp.amjpathol.org). This suggests that the analyses are reproducible and robust.
Five loci from chromosomes 2, 3, and 15 through 17, with deletions of at least 10 kb and overlap with nearby genes, were selected for quantitative PCR (qPCR) analysis. As shown in Supplemental Figure S1C (available at http://ajp.amjpathol.org), a deletion by qPCR was found in four of five samples predicted to have a deletion in the region overlapping with ARL17B, a gene homologous to ADP-ribosylation factor located at 17q21.15 Similar confirmation was found in qPCR of SCAPER, the S-phase cyclin A–associated protein in the endoplasmic reticulum located at 15q2416 (five of five samples); of WWOX, WW domain containing oxidoreductase located at 16q2317–19 (five of five samples); of EPHA3 or ephrin receptor 3, a protein tyrosine receptor frequently mutated in a variety of human cancers20–22 (four of five samples); and of ERBB4 or v-erb-a erythroblastic leukemia viral oncogene homolog 423,24 (five of five samples) in blood samples from patients with prostate cancer. The concordance rate of qPCR and copy number analysis was 92%. Our analysis indicates that CNV is not limited to prostate cancer or benign prostate tissue adjacent to cancer but is also found in blood from patients with prostate cancer.
To investigate whether the CNV profiles of blood, AT, and tumor are distinct from each other, classification analysis was performed to predict genomes of blood versus those of prostate cancer, by aggregating genome loci that have differential amplification or deletion proportion between blood and prostate cancer (see Materials and Methods for more detail). The prediction accuracy under unbiased leave-one-out cross validation25 was 89% (76/85) for blood and 94% (98/104) for prostate cancer. The overall accuracy was 92% (174/189; see Supplemental Figure S1D at http://ajp.amjpathol.org). To investigate whether AT is genetically more related to cancer or normal tissues, the CNV profiles of blood and tumor samples were constructed into a logistic regression model as normal and prostate cancer training sets, respectively. This model was then used to classify each of the 49 AT samples as either normal or prostate cancer. Such analysis predicts 42 (86%) of 49 benign prostate tissue as cancer, whereas only seven AT tissues were classified as normal. All prostate cancer cell lines were classified as cancer. These analyses clearly indicate that most AT samples have copy number profiles similar to those of prostate cancer, rather than those of normal, resembling a field effect similarly found for gene expression profiling.26
Most prostate cancers are not lethal.27 Prediction analysis with leave-one-out cross validation, based on loci that have a significant proportion of amplification or deletion in the group of relapse but none in the nonrelapsed group, was performed. The resulting ROC curves were generated by varying sensitivity-specificity trade-off (Figure 2, A and B). The cutoff that generates the best Youden index (ie, sensitivity + specificity − 1) has an accuracy of 73% (74/102; ROC P = 0.003), a positive prediction of 76% (57/75), and a negative prediction of 63% (17/27) for relapse prediction. Gleason grading has been a strong predictor of recurrence; however, in this analysis, it was statistically insignificant from baseline (ROC P = 0.32) and much worse than CNV analysis. When stratifying CNV prediction rate on each Gleason category, it appears that CNV prediction for relapse is statistically significant at Gleason grade 7 (Table 4, Fisher's exact test P = 0.002).
Figure 2.

Genome copy variation in prostate cancer predicts relapse. A: Histograms of genome deletion (blue) or amplification (red) of tumors that did not relapse (Tnone), tumors that relapsed and had a PSADT at or after 15 months of radical prostatectomy (Tslow), and tumors that relapsed and had a PSADT within 4 months of radical prostatectomy (Tfast) in 23 pairs of human chromosomes. B: ROC curves of predicting prostate cancer relapse. The prostate cancer was separated into a group that relapsed within 5 years of prostatectomy (n = 75) and a group that did not relapse (n = 27). Prediction using gene deletions or amplifications unique to the relapsing group, generated through leave-one-out analysis, was performed to produce the ROC chart. C: ROC curves of predicting prostate cancer short PSADT. The prostate cancer was separated into a group that had a PSADT within 4 months of prostatectomy (n = 33) and a group that did not (n = 69). Prediction using gene deletions or amplifications unique to the fast-relapsing group, generated through leave-one-out analysis, was performed to produce the ROC chart. Dotted line, random prediction baseline; broken line, prediction generated from Gleason grading.
Table 4.
Prediction of Relapse or Short PSADT of Prostate Cancer by CNV in Each Gleason Grade
| Gleason grade | Correct for relapse status | Correct for short PSADT status |
|---|---|---|
| 6 | 70.6 (12/17) | 88.2 (15/17) |
| 7 | 75.8 (47/62)⁎ | 71.0 (44/62)⁎ |
| 8 | 76.9 (10/13) | 84.6 (11/13) |
| 9 | 42.9 (3/7) | 71.4 (5/7) |
| 10 | 66.7 (2/3) | 33.3 (1/3) |
Data are given as percentage (number/total). The analysis excluded samples that did not have clinical follow-up.
P < 0.01.
Prostate cancers with rapid progression, as defined by rates of PSA increase, are lethal.14,28 Those with a PSADT of <4 months after relapse and those who died of prostate cancer were compared with those with PSADT of >15 months or having no relapse. A similar prediction with leave-one-out cross-validation analysis was performed to examine the accuracy of CNV profiling in predicting rapidly progressing prostate cancer. As shown in Figure 2C, the accuracy of predicting rapid progression was 75% (P < 0.001), with positive and negative predictive values of 58% and 83%, respectively. In contrast, the histological characteristics of the cancer, as defined by Gleason grading, failed to achieve >50% predictive values simultaneously on positive and negative predictions (ROC P = 0.074). When stratifying the CNV prediction rate on each Gleason category, it appears that CNV prediction is statistically significant at Gleason grade 7 (Table 4, P = 0.007) and combined Gleason grades 8 through 10 (P = 0.042).
Because the genome alterations in AT are most similar to those of tumor, the CNV of AT to predict relapse was examined using cross validation. As shown in Figure 3, A and C, the CNV profile of AT is moderately predictive of prostate cancer relapse: sensitivity of 76% and specificity of 56% (ROC P = 0.041). Surprisingly, the CNV profile of AT is more accurate in predicting short PSADT (88% sensitivity and 75% specificity, P = 0.015; Figure 3, B and D). By using the same approach, the CNV profiles from blood failed to generate an ROC statistically different from baseline in predicting relapse or short PSADT. However, our analysis showed that the average and median sizes of CNV are significantly larger in blood samples (70 and 23 kb, respectively) from patients with relapse than those without relapse (40 and 17 kb, respectively). Based on the sizes of CNV, highly statistically significant ROCs were generated (Figure 4, A and B), predicting 81% (P < 0.001) relapse and 69% (P = 0.001) short PSADT correctly through median CNV sizes. The CNV size correlation with relapse was also found in tumor (817-kb mean and 647-kb median for relapse versus 385-kb mean and 185-kb median for nonrelapse) and AT (246-kb mean and 18-kb median for relapse versus 95-kb mean and 16-kb median for nonrelapse) samples, suggesting that a larger CNV size is a common feature for prostate cancer relapse regardless of tissues. Both median and mean sizes of CNV from tumor and blood, and mean size of CNV from AT, predict prostate cancer relapse, whereas mean and median sizes of CNV from tumor and blood predict short PSADT (Figure 4, C and D; see also Supplemental Figure S2, A–D, and Supplemental Figure S3, A and B, at http://ajp.amjpathol.org). Interestingly, similar relapse prediction results were also replicated using the sizes of either amplified or deleted loci of blood (see Supplemental Figure S4, A–D, and Supplemental Figure S5, A–D, at http://ajp.amjpathol.org). To exclude aging being a factor in our analysis, correlation analyses between our gene-specific or size-based model and the patient age were performed; these analyses revealed no significant correlation between age and our prediction methods. Age did not predict relapse or short PSADT (see Supplemental Figure S6, A and B, at http://ajp.amjpathol.org).
Figure 3.

Genome copy variation in benign prostate tissues adjacent to cancer predicts prostate cancer relapse. A: Histograms of genome deletion (blue) or amplification (red) of AT no relapse, AT relapse, AT not short PSADT, and AT short PSADT in 23 pairs of human chromosomes. B: ROC curves of AT predicting prostate cancer relapse. The AT samples were separated into a group that relapsed within 5 years of prostatectomy (n = 21) and a group that did not relapse (n = 28). Prediction using gene deletions or amplifications unique to the relapsing group, generated through leave-one-out analysis, was performed to produce the ROC chart. ROC curves of AT predicting prostate cancer short PSADT (C and D). The AT samples were separated into a group that had PSADT within 4 months of prostatectomy (n = 8) and a group that did not (n = 41). Prediction using gene deletions or amplifications unique to the fast-relapsing group, generated through leave-one-out analysis, was performed to produce the ROC chart. Dotted line, random prediction baseline.
Figure 4.
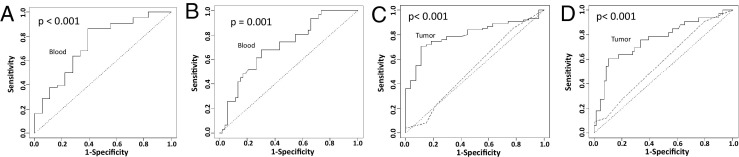
Median size variation of CNV of blood and tumor samples predicts prostate cancer relapse and short PSADT. A: ROC curves of CNV median size of blood samples predicting prostate cancer relapse. The blood samples were separated as described in A. The optimal prediction rates for CNV median size of blood model are 86% (57/66) sensitivity and 61% (11/18) specificity. B: ROC curves of CNV median size of blood predicting prostate cancer short PSADT. The blood samples were separated into a group that had PSADT within 4 months of prostatectomy (n = 31) and a group that did not (n = 53). The optimal prediction rates for CNV median size of blood model are 68% (21/31) sensitivity and 70% (37/53) specificity. C: ROC curves of predicting prostate cancer relapse using median sizes of CNV from tumor samples. The prostate cancer was separated into a group that relapsed within 5 years of prostatectomy (n = 75) and a group that did not relapse (n = 27). The optimal prediction rates for CNV median size of tumor model are 71% (53/75) sensitivity and 89% (24/27) specificity. D: ROC curves of predicting prostate cancer short PSADT using CNV median sizes from tumor samples. The prostate cancer was separated into a group that had PSADT within 4 months of prostatectomy (n = 33) and a group that did not (n = 69). The optimal prediction rates for CNV median size of tumor model are 61% (20/33) sensitivity and 90% (62/69) specificity. Prediction using various CNV median sizes was performed to produce the ROC charts. Dotted line, random prediction baseline; broken line, prediction generated from Gleason grading.
To investigate the reproducibility of our prediction models, we collected an additional 25 samples, including 10 tumors, 10 benign tissues adjacent to tumors, and 5 blood samples from patients with prostate cancer. These experiments and analyses were performed in a separate time period and by different personnel. By using a gene-specific model, we correctly predicted 7 of 10 relapse and 8 of 10 short PSADT from tumor samples, whereas we correctly predicted 7 of 10 for both relapse and short PSADT from AT samples. By using mean size of CNV from tumor, we correctly predicted 7 of 10 cases of both relapse and short PSADT, 7 of 10 for relapse from AT, and 4 of 5 for relapse and 4 of 5 for short PSADT from blood. By using median size of CNV from tumors, we correctly predicted 6 of 10 for relapse and 7 of 10 for short PSADT, whereas from blood, we correctly predicted 5 of 5 for relapse and 4 of 5 for short PSADT. Taken together, the gene-specific CNV model has an overall prediction rate of 72.5% in the replication data set (see Supplemental Table S6 at http://ajp.amjpathol.org), similar to those found in the first set of data. The mean CNV sizes of blood, tumor, and benign prostate tissues have an overall prediction rate of 72% for relapse, and the mean CNV sizes of blood and tumor samples have an overall prediction rate of 73% for short PSADT, whereas the median CNV sizes of blood and tumor have overall prediction rates of 73% for relapse and 80% for short PSADT (see Supplemental Table S6 at http://ajp.amjpathol.org). These results are also similar to those found in the original study, reflecting good consistency and reproducibility of our prediction models.
Discussion
Genome-wide analyses of prostate cancer and matched tissues using other methods were previously performed.29–34 However, there was no attempt to construct a model to predict the prognosis of prostate cancer. Although a tiny amount (<0.1% of the blood cell population) of circulating tumor cells may exist in the blood sample,35,36 the stringency of CNV analysis (>30% contamination to be detected) excluded contamination of tumor cells in the blood as a contending interpretation for CNV in the blood. Similarly, the similarity of AT and tumor samples in CNV profiling is unlikely because of the small amount of contaminated tumor cells in normal tissues; it will be well beyond the sensitivity of CNV analysis. An analysis of some of the previously published matched normal samples of other malignancies37,38 also reveals significant CNV. This suggests that CNV is widely present in tissues of patients carrying malignancies. However, it is unclear whether healthy individuals carry these CNVs. The CNV of blood may be somatic and acquired through aging; this alteration would tend to be random and spontaneous. Alternatively, genome copy number abnormalities may occur at the germ-line level. To distinguish these two possibilities, longitudinal blood samples of the same aging individual could identify if CNV is accumulated. Independent of the mechanism, however, genome CNV correlates with the eventual behavior of prostate cancer: This is observed in the primary prostate cancer, in the histologically normal tissue from a prostate gland containing cancer, and in the blood of a patient with prostate cancer.
Conceivably, CNV analysis offers a better option than Gleason grading in predicting the behavior of prostate cancer because of a better prediction rate on the tumor samples and its applicability to nontumor tissues. There are several salient potentials for clinical application using the CNV tests. For a patient being diagnosed as having prostate cancer, CNV analysis performed on the blood or perhaps other normal tissues from the patient would eliminate the need for an additional invasive procedure to decide a treatment mode. For a patient already undergoing a radical prostatectomy, the CNV analysis on a tumor or blood sample may help to decide whether additional treatment is warranted to prevent relapse. When the morphological characteristics become indeterminate in a biopsy sample, the gene-specific CNV field effect in benign prostate tissues may help to obtain a firmer diagnosis. The main limitation of the genome CNV analysis for a clinical test is its requirement of high-quality genome DNA. Formalin-fixed, paraffin-embedded tissues may not be suitable. When gene-specific CNV prediction is performed, a training set containing samples with known outcome is required for the prediction (there is no need for a training set when the size of CNV analysis is performed). Despite these limitations, CNV analysis on the genome of blood, normal prostate, or tumor tissues of the patients with prostate cancer holds promise to become a more efficient and accurate way to predict the behavior of prostate cancer.
Acknowledgments
We thank Jeffrey Romoff for his constructive comments and support for the project and Chia-Yueh Yen for her technical support.
Footnotes
Supported by a grant from the American Cancer Society (RSG-08-137-01-CNE to Y.P.Y.), a grant from the National Cancer Institute (RO1 CA098249 to J.-H.L.), and a grant from the University of Pittsburgh Cancer Institute.
Y.P.Y. and C.S. contributed equally to this work.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at http://dx.doi.org/10.1016/j.ajpath.2012.03.008.
Supplementary data
Validation analysis of genome CNV of blood (B), AT, and prostate cancer samples. A: Loci overlaps of deleted or amplified segments of genomes using our 85 blood samples (/b), 265 normal reference samples from GeneSpring (/gs), or 800 normal reference samples from Partek, Inc. (/p), as baselines in analyzing genome CNV of B, AT, and tumor (T) samples. B: Overlaps of deleted loci of genomes in B, AT, and T samples, with loci detected as LOH in these samples. C: Matching of predicted deletion with qPCR. The qPCRs were performed on five selected blood samples predicted to have loci of deletion overlapping with ARL17b, SCAPER, EPHA3, ERBB4, and WWOX. The average copy number and SD are shown for CNV prediction (P) and measured by qPCR (M). D: ROC curves of predicting prostate cancer genome versus normal (blood) genome. P < 0.001. Dotted line, random chance baseline.
Mean size variation of CNV of blood (B) and tumor (T) samples predicts prostate cancer relapse and fast relapse. A: ROC curves of CNV mean size of B predicting prostate cancer relapse. The B samples were separated into a group that relapsed within 5 years of prostatectomy (n = 66) and a group that did not relapse (n = 18). The optimal prediction rates for CNV mean size of blood sample model are 95% (63/66) sensitivity and 56% (10/18) specificity. B: ROC curves of CNV mean size of B predicting prostate cancer fast relapse. The B samples were separated into a group that had PSADT within 4 months of prostatectomy (n = 31) and a group that did not (n = 53). The optimal prediction rates for CNV mean size of blood model are 100% (31/31) sensitivity and 30% (16/53) specificity. C: ROC curves of predicting prostate cancer relapse using mean sizes of CNV from T samples. The prostate cancer was separated into a group that relapsed within 5 years of prostatectomy (n = 75) and a group that did not relapse (n = 27). The optimal prediction rates for CNV mean size of T model are 81% (61/75) sensitivity and 44% (12/27) specificity. D: ROC curves of predicting prostate cancer fast relapse using CNV mean sizes from T samples. The prostate cancer was separated into a group that had PSADT within 4 months of prostatectomy (n = 33) and a group that did not (n = 69). The optimal prediction rates for CNV mean size of T model are 45% (15/33) sensitivity and 86% (59/69) specificity. Prediction using various CNV mean sizes was performed to produce the ROC charts. Dotted line, random prediction baseline.
Mean size variation of genomes from AT predicts prostate cancer relapse. A: ROC curves of CNV mean size of AT predicting prostate cancer relapse. The AT samples were separated into a group that relapsed within 5 years of prostatectomy (n = 21) and a group that did not relapse (n = 28). The optimal prediction rates for CNV mean size of AT model are 57% (12/21) sensitivity and 78% (21/27) specificity. B: ROC curves of CNV mean sizes of AT predicting prostate cancer fast relapse. The AT samples were separated into a group that had a PSADT within 4 months of prostatectomy (n = 8) and a group that did not (n = 40). The optimal prediction rates for CNV mean size of AT model are 88% (7/8) sensitivity and 45% (18/40) specificity. Prediction using various CNV mean sizes was performed to produce the ROC charts. Dotted line, random prediction baseline.
Mean size variation of deletion or amplification of genomes from blood (B) cells predicts prostate cancer relapse and fast relapse. A: ROC curves of amplification mean size of B predicting prostate cancer relapse. The B samples were separated into a group that relapsed within 5 years of prostatectomy (n = 66) and a group that did not relapse (n = 18). The optimal prediction rates for amplification mean size of B model are 97% (64/66) sensitivity and 39% (7/18) specificity. B: ROC curves of amplification mean size of B predicting prostate cancer fast relapse. The B samples were separated into a group that had a PSADT within 4 months of prostatectomy (n = 31) and a group that did not (n = 53). The optimal prediction rates for amplification mean size of B model are 61% (19/31) sensitivity and 57% (30/53) specificity. C: ROC curves of deletion mean size of B predicting prostate cancer relapse. The B samples were separated as described in A. The optimal prediction rates for deletion mean size of B model are 73% (48/66) sensitivity and 61% (11/18) specificity. D: ROC curves of deletion mean size of B predicting prostate cancer fast relapse. The B samples were separated as described in B. The optimal prediction rates for deletion mean size of B model are 84% (26/31) sensitivity and 34% (18/53) specificity. Prediction using various deletion mean sizes was performed to produce the ROC charts. Dotted line, random prediction baseline.
Median size variation of deletion or amplification of genomes from blood (B) cells predicts prostate cancer relapse and fast relapse. A: ROC curves of amplification median size of B predicting prostate cancer relapse. The B samples were separated into a group that relapsed within 5 years of prostatectomy (n = 66) and a group that did not relapse (n = 18). The optimal prediction rates for amplification median size of B model are 89% (59/66) sensitivity and 44% (8/18) specificity. B: ROC curves of amplification median size of B predicting prostate cancer fast relapse. The B samples were separated into a group that had a PSADT within 4 months of prostatectomy (n = 31) and a group that did not (n = 53). The optimal prediction rates for amplification median size of B model are 77% (24/31) sensitivity and 47% (25/53) specificity. C: ROC curves of deletion median size of B predicting prostate cancer relapse. The B samples were separated as described in A. The optimal prediction rates for deletion median size of B model are 58% (38/66) sensitivity and 89% (16/18) specificity. D: ROC curves of deletion median size of B predicting prostate cancer fast relapse. The B samples were separated as described in B. The optimal prediction rates for deletion median size of B model are 58% (18/31) sensitivity and 72% (38/53) specificity. Prediction using various deletion median sizes was performed to produce the ROC charts. Dotted line, random prediction baseline.
Age does not predict relapse or fast relapse of prostate cancer. A: ROC curves of ages (40 to 70 years) of tumor (T), AT, and blood (B) predicting prostate cancer relapse. B: ROC curves of ages (40 to 70 years) of T, AT, and B predicting prostate cancer fast relapse.
References
- 1.Jemal A., Siegel R., Ward E., Hao Y., Xu J., Murray T., Thun M.J. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A., Siegel R., Ward E., Hao Y., Xu J., Thun M.J. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A., Siegel R., Xu J., Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2011;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 4.Andriole G.L., Crawford E.D., Grubb R.L., 3rd, Buys S.S., Chia D., Church T.R., Fouad M.N., Gelmann E.P., Kvale P.A., Reding D.J., Weissfeld J.L., Yokochi L.A., O'Brien B., Clapp J.D., Rathmell J.M., Riley T.L., Hayes R.B., Kramer B.S., Izmirlian G., Miller A.B., Pinsky P.F., Prorok P.C., Gohagan J.K., Berg C.D. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med. 2009;360:1310–1319. doi: 10.1056/NEJMoa0810696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schroder F.H., Hugosson J., Roobol M.J., Tammela T.L., Ciatto S., Nelen V., Kwiatkowski M., Lujan M., Lilja H., Zappa M., Denis L.J., Recker F., Berenguer A., Maattanen L., Bangma C.H., Aus G., Villers A., Rebillard X., van der Kwast T., Blijenberg B.G., Moss S.M., de Koning H.J., Auvinen A. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med. 2009;360:1320–1328. doi: 10.1056/NEJMoa0810084. [DOI] [PubMed] [Google Scholar]
- 6.Pang S.T., Weng W.H., Flores-Morales A., Johansson B., Pourian M.R., Nilsson P., Pousette A., Larsson C., Norstedt G. Cytogenetic and expression profiles associated with transformation to androgen-resistant prostate cancer. Prostate. 2006;66:157–172. doi: 10.1002/pros.20328. [DOI] [PubMed] [Google Scholar]
- 7.Matsui S., LaDuca J., Rossi M.R., Nowak N.J., Cowell J.K. Molecular characterization of a consistent 4.5-megabase deletion at 4q28 in prostate cancer cells. Cancer Genet Cytogenet. 2005;159:18–26. doi: 10.1016/j.cancergencyto.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 8.Bettendorf O., Schmidt H., Eltze E., Gockel I., Semjonow A., Burger H., Bocker W., Brandt B. Cytogenetic changes and loss of heterozygosity in atypical adenomatous hyperplasia, in carcinoma of the prostate and in non-neoplastic prostate tissue using comparative genomic hybridization and multiplex-PCR. Int J Oncol. 2005;26:267–274. [PubMed] [Google Scholar]
- 9.Teixeira M.R., Ribeiro F.R., Eknaes M., Waehre H., Stenwig A.E., Giercksky K.E., Heim S., Lothe R.A. Genomic analysis of prostate carcinoma specimens obtained via ultrasound-guided needle biopsy may be of use in preoperative decision-making. Cancer. 2004;101:1786–1793. doi: 10.1002/cncr.20527. [DOI] [PubMed] [Google Scholar]
- 10.Macoska J.A., Paris P., Collins C., Andaya A., Beheshti B., Chaib H., Kant R., Begley L., MacDonald J.W., Squire J.A. Evolution of 8p loss in transformed human prostate epithelial cells. Cancer Genet Cytogenet. 2004;154:36–43. doi: 10.1016/j.cancergencyto.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 11.Kraus J., Pantel K., Pinkel D., Albertson D.G., Speicher M.R. High-resolution genomic profiling of occult micrometastatic tumor cells. Genes Chromosomes Cancer. 2003;36:159–166. doi: 10.1002/gcc.10160. [DOI] [PubMed] [Google Scholar]
- 12.Strassburger K., Bretz F. Compatible simultaneous lower confidence bounds for the Holm procedure and other Bonferroni-based closed tests. Stat Med. 2008;27:4914–4927. doi: 10.1002/sim.3338. [DOI] [PubMed] [Google Scholar]
- 13.Hanczar B., Hua J., Sima C., Weinstein J., Bittner M., Dougherty E.R. Small-sample precision of ROC-related estimates. Bioinformatics. 2010;26:822–830. doi: 10.1093/bioinformatics/btq037. [DOI] [PubMed] [Google Scholar]
- 14.Hanks G.E., Hanlon A.L., Lee W.R., Slivjak A., Schultheiss T.E. Pretreatment prostate-specific antigen doubling times: clinical utility of this predictor of prostate cancer behavior. Int J Radiat Oncol Biol Phys. 1996;34:549–553. doi: 10.1016/0360-3016(95)02154-x. [DOI] [PubMed] [Google Scholar]
- 15.Strausberg R.L., Feingold E.A., Grouse L.H., Derge J.G., Klausner R.D., Collins F.S. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci U S A. 2002;99:16899–16903. doi: 10.1073/pnas.242603899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsang W.Y., Wang L., Chen Z., Sanchez I., Dynlacht B.D. SCAPER, a novel cyclin A-interacting protein that regulates cell cycle progression. J Cell Biol. 2007;178:621–633. doi: 10.1083/jcb.200701166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang J., Cogdell D., Yang D., Hu L., Li H., Zheng H., Du X., Pang Y., Trent J., Chen K., Zhang W. Deletion of the WWOX gene and frequent loss of its protein expression in human osteosarcoma. Cancer Lett. 2010;291:31–38. doi: 10.1016/j.canlet.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 18.Nunez M.I., Rosen D.G., Ludes-Meyers J.H., Abba M.C., Kil H., Page R., Klein-Szanto A.J., Godwin A.K., Liu J., Mills G.B., Aldaz C.M. WWOX protein expression varies among ovarian carcinoma histotypes and correlates with less favorable outcome. BMC Cancer. 2005;5:64. doi: 10.1186/1471-2407-5-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yakicier M.C., Legoix P., Vaury C., Gressin L., Tubacher E., Capron F., Bayer J., Degott C., Balabaud C., Zucman-Rossi J. Identification of homozygous deletions at chromosome 16q23 in aflatoxin B1 exposed hepatocellular carcinoma. Oncogene. 2001;20:5232–5238. doi: 10.1038/sj.onc.1204674. [DOI] [PubMed] [Google Scholar]
- 20.Blackford A., Parmigiani G., Kensler T.W., Wolfgang C., Jones S., Zhang X., Parsons D.W., Lin J.C., Leary R.J., Eshleman J.R., Goggins M., Jaffee E.M., Iacobuzio-Donahue C.A., Maitra A., Klein A., Cameron J.L., Olino K., Schulick R., Winter J., Vogelstein B., Velculescu V.E., Kinzler K.W., Hruban R.H. Genetic mutations associated with cigarette smoking in pancreatic cancer. Cancer Res. 2009;69:3681–3688. doi: 10.1158/0008-5472.CAN-09-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clifford N., Smith L.M., Powell J., Gattenlohner S., Marx A., O'Connor R. The EphA3 receptor is expressed in a subset of rhabdomyosarcoma cell lines and suppresses cell adhesion and migration. J Cell Biochem. 2008;105:1250–1259. doi: 10.1002/jcb.21926. [DOI] [PubMed] [Google Scholar]
- 22.Bae H.J., Song J.H., Noh J.H., Kim J.K., Jung K.H., Eun J.W., Xie H.J., Ryu J.C., Ahn Y.M., Kim S.Y., Lee S.H., Yoo N.J., Lee J.Y., Park W.S., Nam S.W. Low frequency mutation of the Ephrin receptor A3 gene in hepatocellular carcinoma. Neoplasma. 2009;56:331–334. doi: 10.4149/neo_2009_04_331. [DOI] [PubMed] [Google Scholar]
- 23.Koutras A.K., Fountzilas G., Kalogeras K.T., Starakis I., Iconomou G., Kalofonos H.P. The upgraded role of HER3 and HER4 receptors in breast cancer. Crit Rev Oncol Hematol. 2010;74:73–78. doi: 10.1016/j.critrevonc.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 24.Lee S.Y., Kim M.J., Jin G., Yoo S.S., Park J.Y., Choi J.E., Jeon H.S., Cho S., Lee E.B., Cha S.I., Park T.I., Kim C.H., Jung T.H., Park J.Y. Somatic mutations in epidermal growth factor receptor signaling pathway genes in non-small cell lung cancers. J Thorac Oncol. 2010;5:1734–1740. doi: 10.1097/JTO.0b013e3181f0beca. [DOI] [PubMed] [Google Scholar]
- 25.Golub T.R., Slonim D.K., Tamayo P., Huard C., Gaasenbeek M., Mesirov J.P., Coller H., Loh M.L., Downing J.R., Caligiuri M.A., Bloomfield C.D., Lander E.S. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 26.Yu Y.P., Landsittel D., Jing L., Nelson J., Ren B., Liu L., McDonald C., Thomas R., Dhir R., Finkelstein S., Michalopoulos G., Becich M., Luo J.H. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22:2790–2799. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 27.Isaacs J.T. Molecular markers for prostate cancer metastasis: developing diagnostic methods for predicting the aggressiveness of prostate cancer. Am J Pathol. 1997;150:1511–1521. [PMC free article] [PubMed] [Google Scholar]
- 28.Stephenson A.J., Shariat S.F., Zelefsky M.J., Kattan M.W., Butler E.B., Teh B.S., Klein E.A., Kupelian P.A., Roehrborn C.G., Pistenmaa D.A., Pacholke H.D., Liauw S.L., Katz M.S., Leibel S.A., Scardino P.T., Slawin K.M. Salvage radiotherapy for recurrent prostate cancer after radical prostatectomy. JAMA. 2004;291:1325–1332. doi: 10.1001/jama.291.11.1325. [DOI] [PubMed] [Google Scholar]
- 29.Kim J.H., Dhanasekaran S.M., Mehra R., Tomlins S.A., Gu W., Yu J., Kumar-Sinha C., Cao X., Dash A., Wang L., Ghosh D., Shedden K., Montie J.E., Rubin M.A., Pienta K.J., Shah R.B., Chinnaiyan A.M. Integrative analysis of genomic aberrations associated with prostate cancer progression. Cancer Res. 2007;67:8229–8239. doi: 10.1158/0008-5472.CAN-07-1297. [DOI] [PubMed] [Google Scholar]
- 30.Zhao H., Kim Y., Wang P., Lapointe J., Tibshirani R., Pollack J.R., Brooks J.D. Genome-wide characterization of gene expression variations and DNA copy number changes in prostate cancer cell lines. Prostate. 2005;63:187–197. doi: 10.1002/pros.20158. [DOI] [PubMed] [Google Scholar]
- 31.Ren B., Yu G., Tseng G.C., Cieply K., Gavel T., Nelson J., Michalopoulos G., Yu Y.P., Luo J.H. MCM7 amplification and overexpression are associated with prostate cancer progression. Oncogene. 2006;25:1090–1098. doi: 10.1038/sj.onc.1209134. [DOI] [PubMed] [Google Scholar]
- 32.Liu W., Laitinen S., Khan S., Vihinen M., Kowalski J., Yu G., Chen L., Ewing C.M., Eisenberger M.A., Carducci M.A., Nelson W.G., Yegnasubramanian S., Luo J., Wang Y., Xu J., Isaacs W.B., Visakorpi T., Bova G.S. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat Med. 2009;15:559–565. doi: 10.1038/nm.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taylor B.S., Schultz N., Hieronymus H., Gopalan A., Xiao Y., Carver B.S., Arora V.K., Kaushik P., Cerami E., Reva B., Antipin Y., Mitsiades N., Landers T., Dolgalev I., Major J.E., Wilson M., Socci N.D., Lash A.E., Heguy A., Eastham J.A., Scher H.I., Reuter V.E., Scardino P.T., Sander C., Sawyers C.L., Gerald W.L. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Penney K.L., Pyne S., Schumacher F.R., Sinnott J.A., Mucci L.A., Kraft P.L., Ma J., Oh W.K., Kurth T., Kantoff P.W., Giovannucci E.L., Stampfer M.J., Hunter D.J., Freedman M.L. Genome-wide association study of prostate cancer mortality. Cancer Epidemiol Biomarkers Prev. 2010;19:2869–2876. doi: 10.1158/1055-9965.EPI-10-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cristofanilli M., Budd G.T., Ellis M.J., Stopeck A., Matera J., Miller M.C., Reuben J.M., Doyle G.V., Allard W.J., Terstappen L.W., Hayes D.F. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004;351:781–791. doi: 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]
- 36.Moreno J.G., Croce C.M., Fischer R., Monne M., Vihko P., Mulholland S.G., Gomella L.G. Detection of hematogenous micrometastasis in patients with prostate cancer. Cancer Res. 1992;52:6110–6112. [PubMed] [Google Scholar]
- 37.Green M.R., Monti S., Rodig S.J., Juszczynski P., Currie T., O'Donnell E., Chapuy B., Takeyama K., Neuberg D., Golub T.R., Kutok J.L., Shipp M.A. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116:3268–3277. doi: 10.1182/blood-2010-05-282780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parkin B., Erba H., Ouillette P., Roulston D., Purkayastha A., Karp J., Talpaz M., Kujawski L., Shakhan S., Li C., Shedden K., Malek S.N. Acquired genomic copy number aberrations and survival in adult acute myelogenous leukemia. Blood. 2010;116:4958–4967. doi: 10.1182/blood-2010-01-266999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Validation analysis of genome CNV of blood (B), AT, and prostate cancer samples. A: Loci overlaps of deleted or amplified segments of genomes using our 85 blood samples (/b), 265 normal reference samples from GeneSpring (/gs), or 800 normal reference samples from Partek, Inc. (/p), as baselines in analyzing genome CNV of B, AT, and tumor (T) samples. B: Overlaps of deleted loci of genomes in B, AT, and T samples, with loci detected as LOH in these samples. C: Matching of predicted deletion with qPCR. The qPCRs were performed on five selected blood samples predicted to have loci of deletion overlapping with ARL17b, SCAPER, EPHA3, ERBB4, and WWOX. The average copy number and SD are shown for CNV prediction (P) and measured by qPCR (M). D: ROC curves of predicting prostate cancer genome versus normal (blood) genome. P < 0.001. Dotted line, random chance baseline.
Mean size variation of CNV of blood (B) and tumor (T) samples predicts prostate cancer relapse and fast relapse. A: ROC curves of CNV mean size of B predicting prostate cancer relapse. The B samples were separated into a group that relapsed within 5 years of prostatectomy (n = 66) and a group that did not relapse (n = 18). The optimal prediction rates for CNV mean size of blood sample model are 95% (63/66) sensitivity and 56% (10/18) specificity. B: ROC curves of CNV mean size of B predicting prostate cancer fast relapse. The B samples were separated into a group that had PSADT within 4 months of prostatectomy (n = 31) and a group that did not (n = 53). The optimal prediction rates for CNV mean size of blood model are 100% (31/31) sensitivity and 30% (16/53) specificity. C: ROC curves of predicting prostate cancer relapse using mean sizes of CNV from T samples. The prostate cancer was separated into a group that relapsed within 5 years of prostatectomy (n = 75) and a group that did not relapse (n = 27). The optimal prediction rates for CNV mean size of T model are 81% (61/75) sensitivity and 44% (12/27) specificity. D: ROC curves of predicting prostate cancer fast relapse using CNV mean sizes from T samples. The prostate cancer was separated into a group that had PSADT within 4 months of prostatectomy (n = 33) and a group that did not (n = 69). The optimal prediction rates for CNV mean size of T model are 45% (15/33) sensitivity and 86% (59/69) specificity. Prediction using various CNV mean sizes was performed to produce the ROC charts. Dotted line, random prediction baseline.
Mean size variation of genomes from AT predicts prostate cancer relapse. A: ROC curves of CNV mean size of AT predicting prostate cancer relapse. The AT samples were separated into a group that relapsed within 5 years of prostatectomy (n = 21) and a group that did not relapse (n = 28). The optimal prediction rates for CNV mean size of AT model are 57% (12/21) sensitivity and 78% (21/27) specificity. B: ROC curves of CNV mean sizes of AT predicting prostate cancer fast relapse. The AT samples were separated into a group that had a PSADT within 4 months of prostatectomy (n = 8) and a group that did not (n = 40). The optimal prediction rates for CNV mean size of AT model are 88% (7/8) sensitivity and 45% (18/40) specificity. Prediction using various CNV mean sizes was performed to produce the ROC charts. Dotted line, random prediction baseline.
Mean size variation of deletion or amplification of genomes from blood (B) cells predicts prostate cancer relapse and fast relapse. A: ROC curves of amplification mean size of B predicting prostate cancer relapse. The B samples were separated into a group that relapsed within 5 years of prostatectomy (n = 66) and a group that did not relapse (n = 18). The optimal prediction rates for amplification mean size of B model are 97% (64/66) sensitivity and 39% (7/18) specificity. B: ROC curves of amplification mean size of B predicting prostate cancer fast relapse. The B samples were separated into a group that had a PSADT within 4 months of prostatectomy (n = 31) and a group that did not (n = 53). The optimal prediction rates for amplification mean size of B model are 61% (19/31) sensitivity and 57% (30/53) specificity. C: ROC curves of deletion mean size of B predicting prostate cancer relapse. The B samples were separated as described in A. The optimal prediction rates for deletion mean size of B model are 73% (48/66) sensitivity and 61% (11/18) specificity. D: ROC curves of deletion mean size of B predicting prostate cancer fast relapse. The B samples were separated as described in B. The optimal prediction rates for deletion mean size of B model are 84% (26/31) sensitivity and 34% (18/53) specificity. Prediction using various deletion mean sizes was performed to produce the ROC charts. Dotted line, random prediction baseline.
Median size variation of deletion or amplification of genomes from blood (B) cells predicts prostate cancer relapse and fast relapse. A: ROC curves of amplification median size of B predicting prostate cancer relapse. The B samples were separated into a group that relapsed within 5 years of prostatectomy (n = 66) and a group that did not relapse (n = 18). The optimal prediction rates for amplification median size of B model are 89% (59/66) sensitivity and 44% (8/18) specificity. B: ROC curves of amplification median size of B predicting prostate cancer fast relapse. The B samples were separated into a group that had a PSADT within 4 months of prostatectomy (n = 31) and a group that did not (n = 53). The optimal prediction rates for amplification median size of B model are 77% (24/31) sensitivity and 47% (25/53) specificity. C: ROC curves of deletion median size of B predicting prostate cancer relapse. The B samples were separated as described in A. The optimal prediction rates for deletion median size of B model are 58% (38/66) sensitivity and 89% (16/18) specificity. D: ROC curves of deletion median size of B predicting prostate cancer fast relapse. The B samples were separated as described in B. The optimal prediction rates for deletion median size of B model are 58% (18/31) sensitivity and 72% (38/53) specificity. Prediction using various deletion median sizes was performed to produce the ROC charts. Dotted line, random prediction baseline.
Age does not predict relapse or fast relapse of prostate cancer. A: ROC curves of ages (40 to 70 years) of tumor (T), AT, and blood (B) predicting prostate cancer relapse. B: ROC curves of ages (40 to 70 years) of T, AT, and B predicting prostate cancer fast relapse.