

Abstract
Gene transfer to eukaryotic cells requires the uptake of exogenous DNA into the cell nucleus. Except during mitosis, molecular access to the nuclear interior is limited to passage through the nuclear pores. Here we demonstrate the nuclear uptake of extended linear DNA molecules by a combination of fluorescence microscopy and single-molecule manipulation techniques, using the latter to follow uptake kinetics of individual molecules in real time. The assays were carried out on nuclei reconstituted in vitro from extracts of Xenopus eggs, which provide both a complete complement of biochemical factors involved in nuclear protein import, and unobstructed access to the nuclear pores. We find that uptake of DNA is independent of ATP or GTP hydrolysis, but is blocked by wheat germ agglutinin. The kinetics are much slower than would be expected from hydrodynamic considerations. A fit of the data to a simple model suggests femto-Newton forces and a large friction relevant to the uptake process.
A common component in many types of viral infection (1, 2), strategies for genetic therapy (3), and direct DNA vaccination (4) is the accumulation of exogenous DNA in a host cell nucleus. While much attention has been paid to viral invasion mechanisms for cellular entry, relatively little is known about the uptake of DNA into the nucleus itself, particularly as it is not a part of normal cellular physiology. In nondividing cells, molecular exchange across the nuclear envelope normally takes place through the nuclear pore complexes (NPCs). These large channels, whose physiological role is primarily in regulating traffic of proteins and protein-RNA complexes between the nucleus and cytoplasm (5–8), exhibit distinct modes of passive exchange for small molecules and peptide signal-mediated selective transport for larger ones (9). Several pathways for the latter have been identified, based on affinity for receptors of the karyopherin β/importin β (10, 11) and related families (12). Less is known about the biochemistry related to DNA uptake, even whether such exists at all. Indeed, a prominent feature of the viral mechanisms is their ability to co-opt the native protein import machinery by expression of appropriate nuclear localization signals (NLS; ref. 1).
The size scales of the NPC make it an unlikely transporter for DNA. The upper cutoff for diffusive passage of colloidal gold particles is ≈9 nm, whereas the NLS-mediated mechanism displays an apparent cutoff at ≈25 nm diameter (13, 14). This scale easily encompasses the size of most protein transport substrates, for which the molecular weight of the cargo–receptor complex will be in the low hundreds of kDa. A recent study of transport rates finds the passage of several molecules per second in the NLS-mediated mode (15). Tightly compacted messenger ribonucleoprotein (mRNP) particles have been observed to unwind during their export through NPCs, conforming to the restricted space within the pore (16). The virulence proteins VirD2 and VirE2 of Agrobacterium tumefaciens mediate the import of single-stranded DNA to plant nuclei. VirE2 winds the DNA into a telephone-coil structure of ≈13 nm outer diameter, presumably in steric preparation for nuclear entry (17, 18). Concentrations of multivalent salts, as found in sperm cells, can condense double-stranded DNA (dsDNA) into compact toroidal configurations with a typical diameter of ≈100 nm (19). Under normal physiological salt conditions, however, dsDNA is a polymeric molecule with a persistence length of roughly 50 nm and the form of a random coil (20).
Here we report on a physical study of nuclear uptake of dsDNA. To focus as closely as possible on translocation through the NPC itself, we make use of the cell-free nuclear reconstitution system based on extracts of Xenopus laevis eggs (21). One of the classic assays for protein import (22), it provides at once a full complement of biochemical transport factors and unhindered access to the nuclear envelope. The latter is essential for the experiments to be described here. As a linear, double-stranded transport substrate we use bacteriophage λ DNA, both for convenience in single-molecule manipulation and for the unlikelihood of specific sequence recognition in the vertebrate cell extract. We show first that fluorescent DNA molecules accumulate in the reconstituted nuclei. We then present a study of the uptake kinetics for individual DNA molecules, using optical tweezers (23) for molecular manipulation and length measurements.
Materials and Methods
In Vitro Nuclear Reconstitution.
Interphase extracts of X. laevis eggs were prepared according to the protocol of Newmeyer and Wilson (24), including centrifugation at 200,000 × g to separate the crude extract to membrane-containing and clarified cytosol components, and kept frozen at −80°C. Nuclei were reconstituted using demembranated Xenopus sperm as the source of chromatin and a 1:10 to 1:20 ratio of membranes to cytosol. In some experiments the preparation of reconstituted nuclei was further diluted between 1:1 and 4:1 with cytosol. This reduced the concentration of free membrane and vesicles in the final extract. Under such conditions the formation of new nuclear membrane was prevented, even on freshly added demembranated sperm. Cytochalasin D (0.6 μg/ml) and nocodozole (15 μM) were added to depolymerize F-actin and microtubules, respectively. An energy regenerating system (1 mM ATP/1 mM GTP/50 μg/ml creatine phosphokinase/10 mM phosphocreatine) was added for assembly. Apyrase (100 units/ml) was used when required for ATP depletion. Reagents were purchased from Sigma.
Preparation of Transport Substrates.
Fluorescent human serum albumin (HSA) and bovine serum albumin (BSA) (Sigma) were linked to a synthetic NLS peptide of the SV40 large T antigen (CTPVKKKRKV) at a ratio of ≈20 peptides/protein by using the maleimido-bis-succinimide heterobifunctional crosslinking reagent (MBS from Pierce). DNA was labeled covalently with Cy3 fluorophores by using the LabelIt kit (Mirus, Madison, WI), following the manufacturer's instructions. Intercalant dyes were not suitable because of their tendency to stain the nuclear chromatin.
Transport Assays with Fluorescent Substrates.
Following nuclear assembly, fluorescent albumin-NLS (final concentration 50 μg/ml) or DNA (final concentration of ≈15 μg/ml) was mixed with nuclei in an Eppendorf tube and turned for 5 min on a gentle end-to-end mixer, followed by incubation at room temperature for 1 h. For ATP depletion assays, apyrase was added for 30 min before addition of transport substrates. For inhibition by wheat germ agglutinin (WGA), the lectin was also added 30 min before addition of transport substrates. For assays at 4°C, the nuclei were held on ice while a precooled solution of DNA was added and incubated for 1 h on ice. Similarly, cold protein-NLS was added and incubated for 10 min. Glutaraldehyde was then added to final concentration of 2% as fixative. Inhibition assays were done in parallel to compare between protein and DNA uptake using the same preparations of nuclei. Fluorescein-labeled dextran of 167 kDa molecular weight (Sigma, final concentration 50 μg/ml) was added along with Cy3-labeled DNA to check for integrity of the nuclear membranes. Images in Fig. 1 were recorded with an Olympus Fluoview confocal microscope, using a PlanApo ×60/1.4 objective.
Figure 1.

λ Phage DNA is concentrated in reconstituted nuclei, but responds differently than NLS-protein transport substrate to inhibitors of protein import. Each panel shows a single nucleus imaged by scanning confocal microscopy, with fluorescent molecules as marked: protein [human serum albumin (HSA)-NLS or BSA-NLS] in column 1, DNA in column 2, and 167-kDa dextran in column 3. Each row in columns 2 and 3 show the same nuclei in two color channels. (A–C) Both DNA and protein are accumulated in intact nuclei. (D–I) Protein import is sensitive to ATP depletion and cold incubation, whereas DNA uptake is not. (J–L) Wheat germ agglutinin blocks both protein and DNA accumulation. (M–O) the fluorescent labeling reagent (see Materials and Methods) is not specifically accumulated in the nucleus: (M) differential interference contrast image; (N and O) fluorescence of Cy3 and dextran, respectively. (Scale bars, 5 μm.)
Linking Beads to DNA and Testing.
Polystyrene spheres of 1 or 3 μm diameter with amine surface functionalization (Polysciences) were coupled covalently to anti-digoxygenin Fab fragments (Roche Molecular Biochemicals) by glutaraldehyde crosslinker, following the bead manufacturer's recommended procedure. λ Phage DNA was prepared for coupling by one of two methods: (i) Following the method of Cluzel et al. (25), AT-rich adhesive segments of 732 bp length were prepared by PCR from the pUX13 plasmid reported previously, incorporating U-biotin or U-digoxygenin at 1:9 U:T ratio. This leads to an average of ten bio or dig sites per segment. Side-specific linkers were ligated to the λ and to the overhangs on adhesive segments left by digestion of the PCR products with EcoRI restriction enzyme. (ii) λ Molecules were ligated directly to synthetic 12-mer complements to the natural 5′ overhangs, synthesized with 3′ biotin or digoxygenin labels (Operon Technologies, Alameda, CA). Coupling was carried out by incubating the beads with prepared DNA molecules at a 1:1 ratio for 3 h. Efficiency of the coupling was checked first by the block of penetration into an agarose gel, and second by the direct molecular stretching assay described below. For experiments in which the biotin end was linked to the SV40 NLS, the peptide was first reacted with biotin-maleimide (Sigma) through the cysteine residue, and then coupled to the DNA via a streptavidin bridge. Bead constructs were isolated from the reagents by centrifugation.
Final testing of the bead-DNA constructs was performed by anchoring the distal end of the DNA molecules to the glass coverslip and extending them directly by means of the optical tweezers. A solution of biotinylated BSA (Sigma, 5 mg/ml) was left overnight to adsorb to the glass at room temperature, then rinsed with PBS buffer. Streptavidin was added and incubated at room temperature for several hours and rinsed with PBS. A suspension of bead-DNA constructs was then injected and the slide mounted on the microscope. The beads settled by gravity, allowing for binding of the biotin to the surface-adsorbed streptavidin. Beads were drawn in at least three directions, using the optical tweezers to measure the length of the DNA. Equal extensions with a radius of 16 μm indicate attachment by a single molecule, the measured length being consistent with the weak stretching force. In a typical successful preparation, 80% of the beads were connected to the glass surface by a single molecule.
For testing the effect of the egg extract on single DNA molecules, a perfusion chamber was prepared by punching an 8-mm hole in a small sheet of Parafilm wax and cutting it in half along the hole diameter. The two halves were separated by 1 to 2 mm and used to adhere a clean, #1.5 glass coverslip, size 22 × 40 mm, crosswise to a standard glass microscope slide. Solutions were exchanged by wicking on one side with tissue. Complete exchange of the contents was verified by using measured volumes of fluorescein and water.
Optical Tweezers and Imaging.
Optical tweezers were based on a near-infrared laser diode [model 5432 (830 nm); SDL, San Jose, CA] and a Zeiss Axiovert 35 inverted microscope, with a Fluar 100/1.3 objective and differential interference contrast accessories. The beam could be steered by means of galvanometer-driven mirrors (model 6450, Cambridge Technology, Cambridge, MA), using the mouse of a PC, or with a function generator for scanning. The maximum trapping force applied on a 3-μm bead, 5 μm above the chamber surface was 12 pN, measured by the escape against Stokes drag. Deeper into the sample, or using smaller beads, the escape force is reduced. Analysis of the rate at which nuclei follow beads indicates a typical force of ≈4 pN in experiments.
Experiments were recorded by video camera to S-VHS video cassettes. Images were digitized to a PC and analyzed by using Matrox INSPECTOR software (Matrox, Dorval, QC, Canada). Particle tracking in frame-by-frame sequence analysis, for bead recoil and viscosity measurements, was performed using a crosscorrelation algorithm in the Matrox MIL software package.
Curve Fitting for Uptake Measurements.
In the single-molecule uptake kinetic measurements, the moment of initial association between the DNA and the nuclear pore is unknown and must be deduced to assemble the data from individual runs into the plot of Fig. 3B. Each measurement set was fit to a three-parameter function that describes the curve shape well, using a least-squares algorithm in mathematica (Wolfram Research, Champaign, IL). A common master curve was defined by using the median values of the parameters. The fit from each individual run was then refit to the master curve by matching the length of the first measurement. This procedure aligns the remaining points with the master curve, using the estimate of initial time (i.e., shift along the horizontal axis) as a single free parameter.
Figure 3.

Kinetic measurements of DNA uptake. (A) A reconstituted nucleus being dragged after a 3-μm-diameter bead, linked by a molecule of DNA. The time interval between measurements in the first and second images is 532 sec, between the second and third, 302 sec. Note the shortening of the maximum distance between bead and nucleus. (B) measurements of maximum extension vs. time with conditions as marked. The solid line shows a fit to Eq. 4.
Results
dsDNA Accumulates in Reconstituted Nuclei.
Nuclear accumulation and concentration of fluorescent DNA and NLS-labeled albumin proteins are compared in Fig. 1. As expected, the protein substrate was concentrated in the reconstituted nuclei (Fig. 1A). Fluorescently labeled λ phage DNA accumulates in these nuclei and reaches a concentration higher than that on the outside (Fig. 1B). (It is imperative in the uptake assay to verify that DNA molecules remain individually dispersed in suspension, which we did by direct observation in a fluorescence microscope. Partly aggregated DNA often clustered around the nuclear membrane, erroneously suggesting that DNA could not pass into the nuclei.) Exclusion of dextran (167 kDa) demonstrates completeness of the nuclear envelope (Fig. 1C). Depletion of ATP from the extract by apyrase treatment blocks the accumulation of protein (Fig. 1D), where transport substrate binds to the nuclear pores, but its release is hindered (26). [The blockage is actually attributed to loss of GTP (27, 28).] DNA accumulation is not blocked by ATP depletion, as seen in Fig. 1E. In each observation of nonblockage of DNA uptake, the effectiveness of ATP depletion was confirmed by a protein import assay. Incubation at 4°C also inhibited protein import (Fig. 1G), whereas again DNA uptake continued (Fig. 1H).
In contrast to ATP depletion and cold incubation, treatment of the nuclei with wheat germ agglutinin (WGA) did hinder the uptake of both protein-NLS and DNA (Fig. 1 J and K, respectively). Again, 167-kDa dextran was excluded (Fig. 1L). Finally, we checked that the fluorescent reagent used to label the DNA transport substrate did not accumulate in the nuclei by reaction with the chromatin (Fig. 1 M–O), ruling out the possibility that staining of the native chromatin by traces of unreacted reagent could account for the above observations.
Single Molecule Study of DNA Stability in the Egg Extract.
To set the stage for single-molecule studies of uptake kinetics, we prepared constructs of λ phage DNA with the ends labeled by biotin and digoxygenin moieites (as described in Materials and Methods and depicted in Fig. 2A). Beads were coupled to the glass coverslip surface in perfusion chambers, and attachment via single molecules was verified by stretching in several directions. The PBS buffer in which beads were originally suspended was exchanged for the cytosolic fraction of the egg extract. A selected bead was then subjected to stretches, roughly every 10 min, to test for shortening of the DNA link that would indicate its condensation, or alternately its release by lysis. After 30 to ≈60 min, another bead was selected and subjected to repeated stretches, having spent the first period since solution exchange unperturbed. Fig. 2B summarizes these observations, showing that the measured relative length between the first and last stretch changes by less than 15% during the exposure to the interphase extract. In four cases, the membrane fraction used in nuclear assembly was introduced at a 1:10 ratio together with the cytosolic fraction. Although we could sometimes observe adsorption of lipids to the DNA, again there was no evidence of its collapse or condensation over more than 1 h. We note that these glycogen-depleted, high-speed centrifuged extracts are not efficient for nuclear assembly on naked DNA substrates (29).
Figure 2.

(A) Schematic of attachment linking beads to DNA. For some of the single-molecule nuclear uptake experiments, streptavidin was added to the free biotin moieties, and then to biotinylated NLS peptide. (B and C) The free biotin was used to bridge the bead-DNA construct to the microscope slide, which was precoated with biotinylated-BSA. (B) Test for DNA condensation or lysis in egg extract. The plot shows the relative extended length of DNA molecules vs. time in extract. Lines of a given color represent first and last stretches, whereas the horizontal axis shows the time since exchange of buffer by cytosol. (C) Recoil kinetics of beads stretched from an anchor point on the glass surface in buffer and in extract (red points, in PBS buffer; blue points, in extract).
The DNA is not likely to remain naked in the protein-rich extract. Dense protein adsorption could be expected to alter the elastic properties of the molecule, as has been shown for RecA binding (30–32). To test for this, we stretched the DNA and followed its recoil toward the equilibrium position by using video-based particle-tracking analysis. The kinetics of the recoil were analyzed in buffer and in the egg extract cytosol; typical results appear in Fig. 2C. Equating the Stokes drag on the bead with the worm-like chain force-extension curve appropriate to DNA (33),
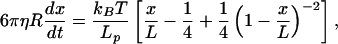 |
1 |
shows that the recoil velocity scales inversely with the product ηLp, where R is the bead radius, Lp is the persistence length of the DNA and x/L its fractional extension, and η is the viscosity of the medium. The viscosities of buffer and extract were measured independently by direct analysis of the diffusion of 3 μm diameter beads, using the particle-tracking facility (34) and the expression for the mean square displacement in two dimensions:
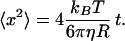 |
2 |
Comparison of the recoil kinetics in the extract and in buffer shows a ratio in the product ηLp of 2.5, equal within 20% to the measured ratio of viscosities alone. Thus, we find no substantial changes in the elastic properties of the DNA in the interphase egg extract.
Nuclear Uptake Kinetics of Individual DNA Molecules.
To test the nuclear uptake kinetics of individual DNA molecules, we prepared bead-DNA constructs in a variety of configurations to be detailed below. These were added to a preparation of reconstituted nuclei and turned for 5 min on an end-to-end mixer. They were then transferred to an observation chamber consisting of a rectangular coverslip fixed crosswise to a microscope slide by using parallel strips of Parafilm. The chamber was sealed with paraffin and quickly placed on the microscope stage to search for beads near the nuclei. (Microtubules and F-actin were depolymerized to permit free passage of the large beads, which were otherwise constrained in a gel of cytoskeletal filaments.) On searching through the sample, most beads were simply floating in Brownian motion. Beads lying nearby a nucleus were trapped with the optical tweezers and drawn away. In some cases no interaction was observed (i.e., on release the bead returned to Brownian motion in its new location). In other cases, dragging the bead dragged the whole nucleus behind it at a distance. If the nucleus was stuck to a surface, the bead pulled out of the optical trap after being drawn to some maximum distance, after which it recoiled toward the nuclear membrane. In both cases this maximum extension was less than the fully extended length of the DNA molecule, and once released, the bead returned to Brownian motion near its initial location. Dynamics of the recoil were analyzed to confirm that the object connecting between the bead and the nucleus had similar elastic properties to the DNA stretched from the glass surface (Fig. 2C). Repeating such stretches on the same bead, normally at two minutes intervals, the maximum length decreased with each measurement. (Repeated stretches in immediate succession, however, showed similar extensions.) Within the imaging resolution, the bead was always connected to the same point on the nuclear membrane. We were not able to induce a lateral interaction between the DNA and the membrane surface even by deliberately wrapping it around the nucleus. In combination with the data of Fig. 1, we conclude that the measured shortening of individual DNA molecules outside the nucleus signifies their irreversible passage through a nuclear pore.
Results of the single-molecule experiments in several variations are summarized in Fig. 3. Measurements were performed using beads of 3 μm and 1 μm diameter. The distal end of the DNA may be attached to streptavidin-NLS for specific targeting to the NPC, or left with unconjugated biotin linkers (Fig. 2A). Experiments were also conducted with DNA lacking NLS under conditions of ATP depletion, where protein-import assays in the same preparations showed blockage (Fig. 1D). Deliberately holding the bead so as to extend the DNA imposed a counterforce to the uptake on the order of 1 pN. The uptake made no progress during such periods, but once released the subsequent kinetics proceeded as normal. Twenty-eight series of shortening kinetics were collected. For clarity, only ten are shown. Because the initial moment of association between the DNA and the nuclear pore is unknown, each series was fit to a common master curve to shift the different initial lengths appropriately along the time axis (as described in Materials and Methods). Following this procedure, the shapes of all of the measurements are remarkably consistent. The series not shown are also consistent, but start lower on the vertical axis.
Discussion
The NPC Is Not a Barrier to Uptake of DNA.
First, the data presented here show that the NPC does not in itself represent a barrier to nuclear uptake of DNA. By contrast to observations in digitonin-permeabilized cells and by microinjection (35, 36), we observe uptake of very long DNA molecules. The difference is presumably due to the free access to the nuclear envelope afforded by the cell-free reconstitution system. Indeed, one can estimate the typical size scale for dsDNA of 50,000 base pairs from the rms end-to-end separation: d = 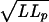 . Taking L = 16.5 μm and Lp = 50 nm, we obtain a size of ≈1 μm for an uncondensed random coil. It is easy to appreciate that such a large object would have difficulty in navigating the many membrane and filamentous barriers in the cytoplasm (37, 38). Even in the present experiments, where such barriers were absent, the initial association between the DNA end and a nuclear pore was apparently a slow process. The attachment of NLS peptides to the DNA end did qualitatively improve the probability of association, however.
. Taking L = 16.5 μm and Lp = 50 nm, we obtain a size of ≈1 μm for an uncondensed random coil. It is easy to appreciate that such a large object would have difficulty in navigating the many membrane and filamentous barriers in the cytoplasm (37, 38). Even in the present experiments, where such barriers were absent, the initial association between the DNA end and a nuclear pore was apparently a slow process. The attachment of NLS peptides to the DNA end did qualitatively improve the probability of association, however.
Given that the long DNA molecules accumulate by passage through nuclear pores, we may consider several hypotheses concerning the mechanism. The physics of polymer translocation through a small pore has received considerable attention recently (39–44). In the present context, one possibility is that spontaneous adsorption of NLS-bearing proteins ferries the DNA through the pores by invoking the protein import mechanism. A second one is that some unknown motor protein activity is responsible for “reeling in” the DNA, acting either at the pore or perhaps by targeting the DNA or attached NLS to specific sites within the nucleus. A third possibility, which we favor according to the evidence presented here, is that the uptake process is essentially a passive ratchet, governed by retention of the segment that has already entered the nucleus. This retention may be due to some degree of chromatinization in the nuclear environment, as has been proposed (45), to adsorption of nuclear-retained proteins, or to interaction with nuclear structures such as lamins or native chromatin. A conceptually similar mechanism has been implicated in polypeptide translocation through endoplasmic reticulum channels (46, 47).
Nuclear Uptake of DNA Is Distinct from NLS-Mediated Protein Import.
Three classic tests were applied to check whether the observed uptake of DNA is mediated by the biochemistry associated with nuclear protein import, either directly or by adsorption of NLS-bearing proteins. As seen in Fig. 1, the two processes are distinct. DNA uptake is uninhibited by ATP depletion and cold, both of which halt active protein import. Insensitivity to these inhibitors also argues strongly against the involvement of ATPase mechanoenzymes. DNA uptake is blocked, however, by wheat germ agglutinin (WGA). Although we cannot deduce from this whether the block was due to loss of a specific biochemical interaction or to plugging of the pores by shuttling proteins and transport complexes, this does indicate that the DNA translocation takes place through the NPCs.
Evidence for Serpentine Passage Through the Pore.
The evidence presented in Fig. 1 could be explained by compaction of the DNA to a condensed structure sufficiently small to pass the NPC as a unit, or by passage of the uncondensed oligomer along its own contour (i.e., threading through the pore). A third possibility is that the DNA is degraded in the cytosol and the resulting fluorescent nucleotides accumulate in the nuclei by diffusion. These hypotheses are addressed by the single-molecule manipulation studies. First, we have shown in Fig. 2B that individual DNA molecules are neither condensed nor lysed on the time scale of the fluorescent uptake measurements in Fig. 1. In Fig. 3, we have shown a monotonic shortening of the DNA remaining available for extension by using the optical tweezers. In combination with the evidence in Fig. 1, this shortening is most consistent with a gradual disappearance of the DNA within the nucleus, and its irreversible retention there.
Measured Kinetics Suggest a Passive Mechanism of Uptake.
The kinetics of DNA uptake as seen in Fig. 3 show first a linear rate of shortening, followed by a slowing down for lengths shorter than ≈4 μm. The curves are similar for all of the tested permutations of bead size, NLS presence or absence, and ATP depletion. They are remarkably repeatable once aligned to account for the length of the initial measurement. The initial linear rate of uptake is measured at 28 nm/sec. As in Fig. 1, insensitivity to ATP depletion argues against the involvement of mechanoenzymes in the uptake process.
The spontaneous nuclear accumulation of DNA indicates an exothermic process (i.e., uptake is driven by a chemical potential difference between nuclear and cytoplasmic segments). A polymer in transit will feel a net inward force F0, which we presume is constant during the entry process. The persistence length of dsDNA in physiological salt conditions (20) is of the same order as the transit length through the pore (5–8). This conformity suggests that the oligomer would be able to pass the channel without a large penalty for stretching it out. The width of a bare dsDNA molecule is ≈2 nm. Of course this may be increased by protein adsorption in the egg extract, but it remains quite reasonable that the oligomer could pass the constricted channel of the NPC by linear diffusion, without the need for conformational changes or specific biochemical interaction with pore components.
We ask next what generates the unusual kinetics. Four elements might be rate-limiting: hydrodynamic drag, length-dependent kinetics of the retention process (e.g., crowding), friction in passage through the pore itself, and resistance to entry of the segment remaining outside. From the first effect we would expect a time scale determined by diffusion of the whole polymer across its hydrodynamic radius (41), roughly the rms end-to-end length: t = x2/2D = (LLp)3πη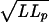 /kBT, where D is the diffusion constant (other variables are defined as above). For numerical values appropriate to λ DNA, this time is on the order of 1 sec, much faster than the observed time scale. Moreover we could expect an acceleration of the uptake as the remaining length decreases. Were the rate limited by hydrodynamic drag on the bead, we should have seen a significant difference between the rates for attached beads of 1 and 3 μm diameter. The second possibility is more difficult to ascertain, but it is hard to envision a reason for the consistency of the measurements, or for the coincidence leading to the observed slowing-down after 12 μm length of uptake. If the kinetics were dominated by friction in passing the pore, on the other hand, we would expect a linear rate of uptake under constant force, as is indeed observed during the first stage of the process. We propose a simple model based on this and the fourth consideration, that is, that the segment of DNA remaining outside the nucleus applies a resistance to entry whose magnitude depends on its instantaneous length.
/kBT, where D is the diffusion constant (other variables are defined as above). For numerical values appropriate to λ DNA, this time is on the order of 1 sec, much faster than the observed time scale. Moreover we could expect an acceleration of the uptake as the remaining length decreases. Were the rate limited by hydrodynamic drag on the bead, we should have seen a significant difference between the rates for attached beads of 1 and 3 μm diameter. The second possibility is more difficult to ascertain, but it is hard to envision a reason for the consistency of the measurements, or for the coincidence leading to the observed slowing-down after 12 μm length of uptake. If the kinetics were dominated by friction in passing the pore, on the other hand, we would expect a linear rate of uptake under constant force, as is indeed observed during the first stage of the process. We propose a simple model based on this and the fourth consideration, that is, that the segment of DNA remaining outside the nucleus applies a resistance to entry whose magnitude depends on its instantaneous length.
In our experiment, one end of the DNA chain remaining outside the nucleus is attached to a bead, while the other is pinned at the nuclear pore. As the bead is excluded from the volume occupied by the polymer coil, the average separation between the ends is roughly 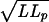 and this imposes a non-zero static tension in the polymer that opposes its passage through the pore. When the DNA is long the effect is negligible, but as it shortens the restoring force becomes progressively more significant. An important consideration is that the time scale for uptake is much slower than any of the polymer time scales, so that hydrodynamic effects are negligible and the DNA is subject to quasistatic conditions.
and this imposes a non-zero static tension in the polymer that opposes its passage through the pore. When the DNA is long the effect is negligible, but as it shortens the restoring force becomes progressively more significant. An important consideration is that the time scale for uptake is much slower than any of the polymer time scales, so that hydrodynamic effects are negligible and the DNA is subject to quasistatic conditions.
The model is expressed as a balance between the uptake force F0, the opposing tension developed in the DNA, and the frictional force where the latter is given, in linear response, by the translocation velocity times an effective friction coefficient φ:
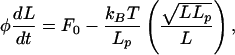 |
3 |
where we have used the Gaussian chain model to estimate the stretch force in the second term. The constancy of φ reflects our presumption that the effective friction is dominated by passage of the polymer through the pore. The equation can be integrated analytically to yield:
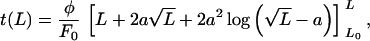 |
4 |
where a = kBT/F0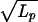 and L0 is the initial extended length. This function is fit to the data in Fig. 3. Note that the ratio φ/F0 has units of an inverse velocity, and that a fit of the initial uptake rate and the length at which the uptake stalls (or the uptake time diverges) determine both parameters. Numerically we obtain φ = 3.8 × 10−4 pN sec/nm and F0 = 1.5 × 10−2 pN. For comparison, the coefficient of viscous drag on a 1-μm-radius object in water, 6πηR = 1.9 × 10−5 pN sec/nm, is one order of magnitude smaller. Similarly, the effective force F0 is two orders of magnitude smaller than the pico-Newton force scale associated with motor protein activity, and rather close to the scale of Brownian motion on μm-sized objects: kBT/1μm ≈ 4 × 10−3 pN. In such a model, the slowing-down appears as a consequence of the attached bead, but one that provides an important insight to the mechanism in the regime where its effect is not felt. In the absence of the static tension, we can expect the natural velocity of the DNA uptake to be F0/φ = 39 nm/sec. While this is faster than the measured rate of 28 nm/sec, we note that the second and third terms in Eq. 4 make a significant numerical contribution over the range from 5 to 16 μm, although they introduce negligible curvature. Therefore, the model is consistent with the apparently linear initial rate of uptake in the measurements.
and L0 is the initial extended length. This function is fit to the data in Fig. 3. Note that the ratio φ/F0 has units of an inverse velocity, and that a fit of the initial uptake rate and the length at which the uptake stalls (or the uptake time diverges) determine both parameters. Numerically we obtain φ = 3.8 × 10−4 pN sec/nm and F0 = 1.5 × 10−2 pN. For comparison, the coefficient of viscous drag on a 1-μm-radius object in water, 6πηR = 1.9 × 10−5 pN sec/nm, is one order of magnitude smaller. Similarly, the effective force F0 is two orders of magnitude smaller than the pico-Newton force scale associated with motor protein activity, and rather close to the scale of Brownian motion on μm-sized objects: kBT/1μm ≈ 4 × 10−3 pN. In such a model, the slowing-down appears as a consequence of the attached bead, but one that provides an important insight to the mechanism in the regime where its effect is not felt. In the absence of the static tension, we can expect the natural velocity of the DNA uptake to be F0/φ = 39 nm/sec. While this is faster than the measured rate of 28 nm/sec, we note that the second and third terms in Eq. 4 make a significant numerical contribution over the range from 5 to 16 μm, although they introduce negligible curvature. Therefore, the model is consistent with the apparently linear initial rate of uptake in the measurements.
Mechanistically we envisage the translocation process as a passive ratchet, in which no energy-consuming motor is required. A beautiful example of such a process has been seen in the uptake to synthetic vesicles of T5 phage DNA when the former are loaded with condensing concentrations of multivalent ions (48). The relevant energy scale is that of deforming the polymeric molecule, leading to force scales of a magnitude associated with thermal diffusion. As such, the translocation could be considered a process of biased or rectified diffusion, rather than driven motion.
Conclusions
We have established that nuclear uptake of DNA can take place by linear passage through nuclear pores, and that this import depends on a biochemistry distinct from that governing active protein import. Using single-molecule manipulation techniques, we have measured the rate of uptake for long, linear dsDNA molecules of bacteriophage λ. Together, the observations indicate an exothermic process consistent with the picture of a passive ratchet governed by linear diffusion through the NPC and irreversible retention within the nucleus. A simple model reproduces the kinetic data. Quantitative estimates of its two parameters, the effective uptake force and the relevant coefficient of drag, are consistent with the very slow kinetics observed.
Given the very weak forces involved, and the improbability of the first recognition between a DNA end and a nuclear pore, it is no surprise that DNA uptake to the nuclei of nondividing somatic cells is a rare process. Judicious labeling of linear plasmids by NLS has been shown to enhance protein expression in transfection assays (35, 45), whereas for plasmid DNA a sequence requirement is observed for nuclear uptake (49). Both suggest that suitably modified DNA may be ferried through the NPC by invoking the protein import machinery, thus in a way mimicking viral import strategies. Our findings contribute a direct measure of the DNA uptake kinetics in a model system, and indicate the physical mechanism by which long DNA molecules may pass through the nuclear pore complex.
Acknowledgments
The authors are indebted to D. Forbes for encouragement and advice concerning nuclear reconstitution, to D. Frank for help in Xenopus handling, and to Y. Gruenbaum, K. Wilson, Z. Reich, D. Kessler, and R. Granek for helpful discussions. This work was supported in part by the Minerva Research Foundation, the Gerhard M. J. Schmidt Minerva Center for Supramolecular Architecture, the Arc-en-Ciel-Keshet program for France–Israel cooperation, and the Gabriel Alhadeff Research Fund. M.E. is incumbent of the Delta Career Development Chair.
Abbreviations
- NPC
nuclear pore complex
- NLS
nuclear localization signal
- dsDNA
double-stranded DNA
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Whittaker G R, Helenius A. Virology. 1998;246:1–23. doi: 10.1006/viro.1998.9165. [DOI] [PubMed] [Google Scholar]
- 2.Kasamatsu H, Nakanishi A. Annu Rev Microbiol. 1998;52:627–686. doi: 10.1146/annurev.micro.52.1.627. [DOI] [PubMed] [Google Scholar]
- 3.Luo D, Saltzman W M. Nat Biotechnol. 2000;18:33–37. doi: 10.1038/71889. [DOI] [PubMed] [Google Scholar]
- 4.Felgner P L. Curr Biol. 1998;8:551–553. doi: 10.1016/s0960-9822(07)00358-2. [DOI] [PubMed] [Google Scholar]
- 5.Pante N, Aebi U. J Cell Biol. 1993;122:977–984. doi: 10.1083/jcb.122.5.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akey C W, Radermacher M. J Cell Biol. 1993;122:1–19. doi: 10.1083/jcb.122.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pante N, Aebi U. Curr Opin Cell Biol. 1996;8:397–406. doi: 10.1016/s0955-0674(96)80016-0. [DOI] [PubMed] [Google Scholar]
- 8.Kiseleva E, Goldberg M W, Cronshaw J, Allen T D. Crit Rev Eukaryotic Gene Expression. 2000;10:101–112. [PubMed] [Google Scholar]
- 9.Gorlich D, Kutay U. Annu Rev Cell Dev Biol. 1999;15:607–660. doi: 10.1146/annurev.cellbio.15.1.607. [DOI] [PubMed] [Google Scholar]
- 10.Rexach M, Blobel G. Cell. 1995;83:683–692. doi: 10.1016/0092-8674(95)90181-7. [DOI] [PubMed] [Google Scholar]
- 11.Gorlich D, Kostka S, Kraft R, Dingwall C, Laskey R A, Hartmann E, Prehn S. Curr Biol. 1995;5:383–392. doi: 10.1016/s0960-9822(95)00079-0. [DOI] [PubMed] [Google Scholar]
- 12.Pollard V W, Michael W M, Nakielny S, Siomi M C, Wang F, Dreyfuss G. Cell. 1996;86:985–994. doi: 10.1016/s0092-8674(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 13.Dworetzky S I, Landford R E, Feldherr C M. J Cell Biol. 1988;107:1279–1287. doi: 10.1083/jcb.107.4.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feldherr C M, Akin D. J Cell Biol. 1990;111:1–8. doi: 10.1083/jcb.111.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keminer O, Siebrasse J-P, Zerf K, Peters R. Proc Natl Acad Sci USA. 1999;96:11842–11847. doi: 10.1073/pnas.96.21.11842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kiseleva E, Goldberg M W, Allen T D, Akey C W. J Cell Sci. 1998;111:223–236. doi: 10.1242/jcs.111.2.223. [DOI] [PubMed] [Google Scholar]
- 17.Citovsky V, Guralnick B, Simon M N, Wall J S. J Mol Biol. 1997;271:718–727. doi: 10.1006/jmbi.1997.1230. [DOI] [PubMed] [Google Scholar]
- 18.Ziemienowicz A, Görlich D, Lanka E, Hohn B, Rossi L. Proc Natl Acad Sci USA. 1999;96:3729–3733. doi: 10.1073/pnas.96.7.3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bloomfield V A. Biopolymers. 1998;44:269–282. doi: 10.1002/(SICI)1097-0282(1997)44:3<269::AID-BIP6>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 20.Baumann C G, Smith S B, Bloomfield V A, Bustamante C. Proc Natl Acad Sci USA. 1997;94:6185–6190. doi: 10.1073/pnas.94.12.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lohka M J, Masui Y. Science. 1983;220:719–721. doi: 10.1126/science.6601299. [DOI] [PubMed] [Google Scholar]
- 22.Newmeyer D D, Finlay D R, Forbes D J. J Cell Biol. 1986;103:2091–2102. doi: 10.1083/jcb.103.6.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Svoboda K, Block S M. Annu Rev Biophys Biomol Struct. 1994;23:247–285. doi: 10.1146/annurev.bb.23.060194.001335. [DOI] [PubMed] [Google Scholar]
- 24.Newmeyer D D, Wilson K L. Methods Cell Biol. 1991;36:607–631. doi: 10.1016/s0091-679x(08)60299-x. [DOI] [PubMed] [Google Scholar]
- 25.Cluzel P, Lebrun A, Heller C, Lavery R, Viovy J L, Chatenay D, Caron F. Science. 1996;271:792–794. doi: 10.1126/science.271.5250.792. [DOI] [PubMed] [Google Scholar]
- 26.Newmeyer D D, Forbes D J. Cell. 1988;52:641–653. doi: 10.1016/0092-8674(88)90402-3. [DOI] [PubMed] [Google Scholar]
- 27.Schwoebel E D, Talcott B, Cushman I, Moore M S. J Biol Chem. 1998;273:35170–35175. doi: 10.1074/jbc.273.52.35170. [DOI] [PubMed] [Google Scholar]
- 28.Englmeier L, Olivo J C, Mattaj I W. Curr Biol. 1999;9:30–41. doi: 10.1016/s0960-9822(99)80044-x. [DOI] [PubMed] [Google Scholar]
- 29.Hartl P, Olson E, Dang T, Forbes D J. J Cell Biol. 1994;124:235–248. doi: 10.1083/jcb.124.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leger J F, Robert J, Bourdieu L, Chatenay D, Marko J F. Proc Natl Acad Sci USA. 1998;95:12295–12299. doi: 10.1073/pnas.95.21.12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hegner M, Smith S B, Bustamante C. Proc Natl Acad Sci USA. 1999;96:10109–10114. doi: 10.1073/pnas.96.18.10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shivashankar G V, Feingold M, Krichevsky O, Libchaber A. Proc Natl Acad Sci USA. 1999;96:7916–7921. doi: 10.1073/pnas.96.14.7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bustamante C, Marko J F, Siggia E D, Smith S. Science. 1994;265:1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 34.Caspi A, Elbaum M, Granek R, Lachish A, Zbaida D. Phys Rev Lett. 1998;80:1106–1109. [Google Scholar]
- 35.Hagstrom J E, Ludtke J J, Bassik M C, Sebestyén M G, Adam S, A, Wolff J A. J Cell Sci. 1997;110:2323–2331. doi: 10.1242/jcs.110.18.2323. [DOI] [PubMed] [Google Scholar]
- 36.Ludtke J J, Zhang G, Sebestyen M G, Wolff J A. J Cell Sci. 1999;112:2033–2041. doi: 10.1242/jcs.112.12.2033. [DOI] [PubMed] [Google Scholar]
- 37.Lukacs G L, Haggie P, Seksek O, Lechardeur D, Freedman N, Verkman A S. J Biol Chem. 2000;275:1625–1629. doi: 10.1074/jbc.275.3.1625. [DOI] [PubMed] [Google Scholar]
- 38.Luby-Phelps K, Castle P E, Taylor D L, Lanni F. Proc Natl Acad Sci USA. 1987;84:4910–4913. doi: 10.1073/pnas.84.14.4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sung W, Park P J. Phys Rev Lett. 1996;77:783–786. doi: 10.1103/PhysRevLett.77.783. [DOI] [PubMed] [Google Scholar]
- 40.Park P J, Sung W. J Chem Phys. 1998;108:3013–3018. [Google Scholar]
- 41.De Gennes P-G. Proc Natl Acad Sci USA. 1999;96:7262–7264. doi: 10.1073/pnas.96.13.7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lubensky D K, Nelson D R. Biophys J. 2000;77:1824–1838. doi: 10.1016/S0006-3495(99)77027-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meller A, Nivon L, Brandin E, Goluvchenko J, Branton D. Proc Natl Acad Sci USA. 2000;97:1079–1084. doi: 10.1073/pnas.97.3.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kasianowicz J J, Brandin E, Branton D, Deamer D W. Proc Natl Acad Sci USA. 1996;93:13770–13773. doi: 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zanta M A, Belguise-Valladeir P, Behr J. Proc Natl Acad Sci USA. 1999;96:91–94. doi: 10.1073/pnas.96.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simon S M, Peskin C S, Oster G F. Proc Natl Acad Sci USA. 1992;89:3770–3374. doi: 10.1073/pnas.89.9.3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matlack K E S, Misselwitz B, Plath K, Rapoport T A. Cell. 1999;97:553–564. doi: 10.1016/s0092-8674(00)80767-9. [DOI] [PubMed] [Google Scholar]
- 48.Lambert O, Letellier L, Gelbart W M, Rigaud J-L. Proc Natl Acad Sci USA. 2000;97:7248–7253. doi: 10.1073/pnas.130187297. . (First Published June 6, 2000; 10.1073/pnas.130187297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson G L, Dean B S, Wang G, Dean D A. J Biol Chem. 1999;274:22025–22032. doi: 10.1074/jbc.274.31.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]