

Abstract
Cancer associated fibroblasts (CAF) are abundant in the stroma of desmoplastic cancers where they promote tumor progression. CAF are `activated' and as such may be uniquely susceptible to apoptosis. Using cholangiocarcinoma (CCA) as a desmoplastic tumor model, we investigated the sensitivity of liver CAF to the cytotoxic drug navitoclax, a BH3 mimetic. Navitoclax induced apoptosis in CAF and in myofibroblastic human hepatic stellate cells, but lacked similar effects in quiescent fibroblasts or CCA cells. Unlike CCA cells, niether CAF nor quiescent fibroblasts expressed Mcl-1, a known resistance factor for navitoclax cytotoxicity. Explaining this paradox, we found that mitochondria isolated from CAF or cells treated with navitoclax both released the apoptogenic factors Smac and cytochrome c, suggesting that they are primed for cell death. Such death priming in CAF appeared to be due in part to upregulation of the pro-apoptotic protein Bax. shRNA-mediated attenuation of Bax repressed navitoclax-mediated mitochondrial dysfunction, release of apoptogenic factors and apoptotic cell death. In a syngeneic rat model of CCA, navitoclax treatment triggered CAF apoptosis, diminishing expression of the desmoplastic extracellular matrix protein tenascin C, suppressing tumor outgrowth and improving host survival. Together, our findings argue that navitoclax may be useful for destroying CAF in the tumor microenvironment as a general strategy to attack solid tumors.
Keywords: Bcl-2 proteins, BH3 mimetic, carcinoma associated fibroblasts, hepatic stellate cells, tumor stroma
INTRODUCTION
Cancer progression is a complex, multifaceted process that involves not only intrinsic genetic changes unique to the malignant cell but also dynamic reciprocal communication between the cancer cell and stromal cells within the tumor microenvironment (1). As cancers progress, the stromal cells become reactive, phenotypically mimicking tissues undergoing wound healing or chronic inflammation. Indeed, many solid human cancers are characterized by desmoplasia, with high numbers of myofibroblasts (2). These myofibroblasts often termed cancer associated fibroblasts (CAF), express tenascin C (Ten C), periostin, seprase, matrix metalloproteinases and other proteins promoting tumor procession, invasion and metastases (3–5). Notably, Ten C has been implicated in tumor progression by facilitating metastasis (6).
In liver, CAF likely originate from hepatic stellate cells, although CAF may also be derived from portal fibroblasts or potentially even from bone-marrow derived precursor cells (3). Activation of hepatic stellate cells not only converts these cells into matrix producing myofibroblasts, but also enhances their susceptibility to apoptosis analogous to the activation-induced cell death observed in T-lymphocytes (7). For example, resolution of the wound healing response in acute liver injury is associated with enhanced myofibroblast apoptosis, thereby reducing their absolute numbers in the liver (8). By analogy, one may predict that CAF may also be uniquely sensitive to pro-apoptotic stimuli, suggesting that selective deletion of CAF with pro-apoptotic therapies could potentially abrogate their support of cancer cells. This in turn, would be expected to negatively affect tumor survival, growth and progression. However, selectively targeting CAF as anticancer strategy remains largely unexplored.
Apoptosis is regulated by members of the Bcl-2 family of proteins, which control mitochondrial outer membrane permeabilization (MOMP) (9). A pro-apoptotic subset of these proteins displays only a single Bcl-2 homology domain termed the BH3 domain. BH3-only proteins act as initiators of cell death by either promoting activation of the multi-domain pro-apoptotic Bcl-2 proteins Bax and Bak, (10) or by neutralizing the anti-apoptotic Bcl-2 proteins (9). Given the ability of the BH3-only proteins to initiate cell death pathways, the pharmaceutical industry has developed BH3-only protein mimetics such as navitoclax (ABT- 263) (11). This small organic molecule may exert single-agent activity in selected cells despite its inability to directly activate Bax or Bak. The direct cytotoxicity by a sensitizing BH3 mimetic can be explained if the mitochondria of these cells are “primed” for cell death (12). In one model of priming, the mitochondrial anti-apoptotic proteins are nearly saturated with activator BH3-only proteins. In this model, the BH3 mimetic may displace and liberate preexisting bound activators, thus allowing them to trigger Bax or Bak oligomerization and MOMP. In an alternative model termed the “embedded together” model, anti-apoptotic proteins inhibit not only BH-3 only proteins, but also Bax or Bak within the outer mitochondrial membrane (13, 14). In this latter model, BH3 mimetics may alter the competing equilibrium between the various Bcl-2 binding partners permitting Bax or Bak activation and MOMP.
Herein, we used cholangiocarcinoma as a model of a highly desmoplastic cancer to examine the role of pro-apoptotic signaling in targeting CAF. Our results suggest that compared to quiescent fibroblasts and CCA cells, CAF are significantly more susceptible to cell death by navitoclax in vitro and in vivo. Moreover, our data indicate that mitochondria from CAF appear to be primed for cell death. These mechanistic insights expand the role of pro-apoptotic therapies in cancer by suggesting they may target the tumor microenvironment as well as the cancer cells.
MATERIALS AND METHODS
Cell lines and culture
The human cholangiocarcinoma cell lines HuCCT-1 (15), Mz-ChA-1 (16), KMCH (17) and KMBC (18), the human hepatic myofibroblast LX-2 (19), quiescent human fibroblasts (hFB) kindly provided by V.H. Shah (Division of Gastroenterology and Hepatology, Mayo Clinic Rochester) and R.S. Bahn (Division of Endocrinology), the rat cholangiocarcinoma cell line BDEneu (20), BDEneu tumor associated myofibroblasts (rCAF)(21) and quiescent rat fibroblasts (rFB) were cultured in Dulbecco's modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, penicillin (100 U/mL) and streptomycin (100 μg/mL) under standard conditions. Cell lines were authenticated in June (HuCCT-1, MzChA-1, KMCH. KMBC) and July 2011 (LX-2) by Genetica DNA Laboratories (Cincinnati, OH) using an AmpF/STR® Identifiler® kit and GeneMapper v3.2 software. Three primary human cholangiocarcinoma associated myofibroblasts cell lines (hCAF) were isolated and cultured. These cell lines were obtained in full compliance and approval of the Mayo Institutional Review Board (IRB). Briefly, liver cancer tissue was processed with the BD MediMachine (BD Biosciences, San Jose CA). Disaggregated cells were cultured in RPMI medium supplemented with 5% PLTMax (Mill Creek Life Sciences, Rochester, MN) (22). The hCAF cell lines were karyotyped to confirm a normal karyotype. Expansion media consisted of DMEM with 10% fetal bovine serum, 100 U/ml penicillin, 100 g/ml streptomycin.
Immunoprecipitation of Bcl-XL and associated BH3 only proteins
LX-2 cells were grown on 20 cm tissue culture dishes to subconfluency and treated with navitoclax (1 μM) for 24 hrs. Cells were subsequently lysed in cold CHAPS lysis buffer [1% CHAPS, 150 mM NaCl, 20 mM HEPES, 1% Glycerol, 3% Thiodiglycol, 1 mM EGTA, 1 mM sodium orthovanadate, 10 mM sodium pyrophosphate, 1 mM PMSF, 1× protease inhibitor mix, 100 mM sodium fluoride, 25 nM Microcystin]. Lysates were centrifuged for 15 min at 15000 × g to pellet cellular debris. Protein concentration was determined via Bradford assay. 400 μg of total protein were pre-cleared by incubation with 40 μl agarose A/G beads (Invitrogen, Camarillo, CA) for 1 hrs at 4°C. Bcl-XL (clone N-20, Santa Cruz Biotechnology (Santa Cruz, CA) and control rabbit IgG (BD Pharmingen, Franklin Lakes, NJ) crosslinked beads were prepared as previously described (23) 40μl of control IgG or Bcl-XL cross-linked beads were added to the precleared 400 μg of total lysate protein and incubated rotating over night at 4°C. The beads were then pelleted by 2 min centrifugation at 8000 × g, washed 4 times in CHAPS lysis buffer, resuspended in sample buffer [4 M urea, 2% SDS, 62.5 mM Tris-HCl (pH 6.8), 1 mM EDTA and 5% 2-mercaptoethanol] and boiled for 20 min to release immunoprecipitated proteins. The samples were centrifuged 2 min at 8000 × g and the supernatant was subject to SDS-PAGE and transfer to PVDF membrane as previously described.
Bax activation by navitoclax
Cells were grown on glass slides and treated with navitoclax or vehicle. Slides were then washed with PBS fixed for 30 min with 4% paraformaldehyde and permeabilized with 0.01% CHAPS in PBS. After a further washing, slides were incubated with a conformation specific antibody against activated Bax 6A7 (1:100) over night at 4°C, washed and incubated with secondary antibody (goat-anti mouse Alexa 488 1:1000) (24).
Generation of stable transfectants
HEK 293T cells were transfected with pCMV-VSV-G (Addgene, Cambridge, MA), pCMV-dR8.2 dvpr (Addgene, Cambridge, MA) and the lentiviral shBax or shBak constructs (Open Biosystems, V2LHS_240441 and V2LHS_94682), respectively, using Lipofectamine LTX reagent (Invitrogen, Camarillo, CA) to amplify the shBax or shBak containing lentivirus. Target LX-2 cells were grown to 50% confluency and infected with lentivirus containing medium from the HEK 293T cells. Medium was previously passed through a 0.45 μm pore filter polybrene (Sigma Aldrich, St. Louis, MO) was then added to a final concentration of 8 μg/ml. LX-2 cells were incubated with infectious medium for 3 hrs before medium was replaced with fresh non-infectious medium. Infection was again repeated 24 hrs after the initial exposure. Infected LX-2 cells were split into selection medium containing 10 ug/ml puromycin. Cell lysates were prepared from shBax and shBak cells to confirm knockdown of Bax or Bak protein by Western blot. Stably transfected shMcl-1 KMCH clones were generated as previously described in detail (25). LX-2 cells were stably transfected with an S-peptide tagged Mcl-1 construct as previously described for HuH-7 cells (26). The S-tag results in a slight increase in molecular weight for Mcl-1 which can readily be identified by immunoblot analysis to verify stable expression of the protein.
Syngeneic rodent model of intrahepatic cholangiocarcinoma
All animal experimentation described in this study was performed in accordance with and approved by the Institutional Animal Care and Use Committee. Syngeneic, in vivo cell transplantation was performed in adult Fischer 344 male rats (Harlan, Indianapolis, IN) with initial mean body weights ranging between 180 and 230 g, as we have previously described in detail (27). Buprenorphine (0.05 mg/kg SubQ) was used for postoperative analgesia. For apoptosis studies, navitoclax (5 mg/kg) or vehicle was given i.p. once daily for 2 consecutive days starting 7 d after tumor implantation. Twenty-four hours after receiving the second treatment, the rats were euthanized and the livers were removed for analysis. For tumor size and metastasis analysis, navitoclax (5 mg/kg) or vehicle was given i.p. once daily for ten consecutive days starting 7 d after tumor implantation. Animals were sacrificed at day 18 and the livers were removed for analysis. Survival studies were performed as regulated by the Institutional Animal Care and Use Committee and animals were euthanized according to defined endpoints (weight loss > 25% of original body weight, disabilitating tumor mass, inability to reach food or water, moribund appearance).
Statistical analysis
Data represent at least three independent experiments using cells from a minimum of three separate isolations and are expressed as means ± SEM. Differences between groups were compared using two-tailed Student's t tests or χ2 tests. Survival data were analyzed and Kaplan-Meier graphs were generated using GraphPad Prism software 6 (GraphPad Software, La Jolla, CA).
Other Materials and Methods
Other materials and methods for PCR, cell death assays, immunoblot analysis, mitochondrial membrane depolarization, Smac release and immunofluorescence are described in the Supplementary Materials and Methods.
RESULTS
Navitoclax selectively induces myofibroblast apoptosis
CAF are characterized by the expression of α-smooth muscle actin (α-SMA) as a hallmark of the myofibroblast phenotype, and expression plus secretion of Ten C protein (4, 28). We confirmed α-SMA and Ten C and mRNA expression in hCAFs, human liver derived myofibroblastic LX-2 cell line and their shBAX and shBAK modified clones, as well as the absence of these markers in quiescent human fibroblasts (hFB) and human CCA cell lines (Supplemental. Fig. 1). Next we examined the potential, single-agent, pro-apoptotic effects of navitoclax on hCAF, LX-2, and hFB (Fig. 1A). As assessed by both morphologic and biochemical criteria, navitoclax markedly induced apoptotic cell death in CAF and LX-2 cells, while quiescent fibroblasts were resistant to navitoclax cytotoxicity. To confirm that CAF were in fact undergoing caspase-dependent apoptosis, the pan-caspase inhibitor QVD was employed. QVD (5 mM) effectively reduced navitoclax induced apoptosis in CAF cells (<5% apoptotic cells after 48 hrs of 1 μM navitoclax treatment; data not shown). In contrast to the myofibroblasts, human CCA cells were relatively resistant to navitoclax-mediated cell death (Fig. 1B upper and lower panel), although the HuCCT-1 cell line displayed a moderate increase in caspase 3/7 activity following exposure to navitoclax. The partial sensitivity of HuCCT-1 cells to navitoclax-induced apoptosis may be due to their enhanced Bax plus reduced Bcl-2 and Bcl-XL expression altering the complex balance of anti- and pro-apototic regulators (Supplemental Figure 2). Finally, treatment of quiescent fibroblasts with TGF-β not only resulted in their activation, as observed by induction of α-SMA expression, but also sensitized these cells to navitoclax induced apoptosis (Supplemental Fig. 3). Taken together, these data suggest navitoclax selectively induces apoptosis in CAF as compared to CCA cells.
Figure 1. Navitoclax induces apoptosis in human CAF, but not CCA cells.

Quiescent human fibroblast (hFB), human primary cancer associated fibroblast from three different CCA patients (hCAF 1, 2 and 3) and activated hepatic myofibroblasts (LX-2) were plated onto multiwell plates and grown to approximately 70% confluency. Cells were treated as indicated with increasing doses of navitoclax for 48 hrs. Cells were analyzed for apoptotic nuclear morphology by DAPI-staining and quantitation of apoptotic nuclei by fluorescence microscopy (panel A; mean ± SEM; n=3; ** p≤0.01). hFB, hCAF, LX2 as well as the human CCA cell lines HuCCT-1, Mz-ChA-1, KMCH and KMBC were treated with navitoclax (1μM) or vehicle for 48 hrs. Apoptosis was measured by DAPI-staining with quantitation of apoptotic nuclei by fluorescence microscopy (panel B, upper graph; mean ± SEM; n=3; ** p≤0.01) or fluorometric analysis of caspase 3/7 activity displayed as fold change compared to vehicle control (panel B, lower graph; mean ± SEM; n≥5; * p≤0.05).
CAF do not express Mcl-1 and upregulate Bax
We next explored the potential mechanisms resulting in selective CAF sensitivity to navitoclax-mediated apoptosis. We first profiled CAF for antiapoptotic multidomain Bcl-2 proteins (Bcl-2, Bcl-XL, and Mcl-1), pro-apoptotic Bcl-2 proteins (Bax and Bak), and BH3-only proteins (Bad, Bim, Bid, Noxa, and PUMA) (Fig. 2A). Expression of these proteins was quite variable between the different cell lines. Interestingly, Mcl-1, a known resistance factor for navitoclax cytotoxicity (29), was not expressed by CAF or even quiescent fibroblasts, although it was expressed by CCA, as previously described (25) (Fig. 2B). Bax was consistently up-regulated in the myofibroblasts as compared to quiescent fibroblasts (Fig. 2A), which is likely due to post-translational regulatory differences as Bax mRNA levels were similar between quiescent fibroblasts and the activated phenotype (data not shown). LX-2 cells also displayed an increase of activated Bax compared to quiescent fibroblasts following navitoclax treatment (Fig. 2C). Finally, shRNA targeted knockdown of Bax but not Bak reduced navitoclax-induced cytotoxicity of myofibroblastic LX-2 cells (Fig. 2D). Although Mcl-1 has been previously identified as a resistance factor for navitoclax cytotoxicity (30), we further verified the effect of Mcl-1 on navitoclax sensitivity in our paradigms (Fig. 3). shRNA knockdown of Mcl-1 in KMCH cells sensitized the cancer cells to navitoclax-mediated apoptosis. Conversely, enforced expression of Mcl-1 in LX-2 cells rendered these cells resistant to navitoclax-induced cell killing. Collectively, these observations suggest that the absence of Mcl-1 expression and the increase in Bax protein levels contribute to the sensitivity of CAFs to navitoclax-mediated apoptosis.
Figure 2. Activated hepatic myofibroblasts exhibit alterations in their Bcl-2 protein profile and are sensitized to navitoclax by Bax.

Whole cell lysates were prepared from quiescent hFB, hepatic myofibroblasts LX-2 and hCAF as well as quiescent rat fibroblasts (rFB), rat cancer associated fibroblasts (rCAF) and the malignant erbB-2/neu transformed rat cholangiocyte cell line (BDEneu) that is employed in the described in-vivo model of cholangiocarcinoma. Cell lysates were subject to immunoblot analysis of Bcl-2 proteins. Except where indicated by white lines, all lanes were adjacent on the membranes; in some cases, additional lanes originally run between those shown are omitted for clarity (panels A and B). All full-length blots/gels are presented in Supplementary Figure 6. hFB, LX-2 and hCAF cells were grown to approximately 50% confluency on glass chamber slides and treated with navitoclax (1μM) for the indicated time. Cells were then analyzed by fluorescence microscopy using a conformation-specific antibody (6A7) against activated Bax. Bax positive cells are plotted as percentage of all cells (panel C; mean ± SEM; n=4; ** p≤0.01). Wild-type LX-2 cells as well as stably transfected shBax and shBak LX-2 cells were treated with navitoclax (1μM, 24h) and apoptosis was assessed by DAPI staining and fluorescence microscopy (panel D upper graph; mean ± SEM; n=4; ** p≤0.01) as well as fluorometric measurement of caspase 3/7 activity (panel D lower graph; mean ± SEM; n=6; ** p≤0.01)
Figure 3. Knockdown of Mcl-1 in CCA cells induces navitoclax sensitivity while Mcl-1 overexpression in LX-2 cells confers resistance to navitoclax induced apoptosis.
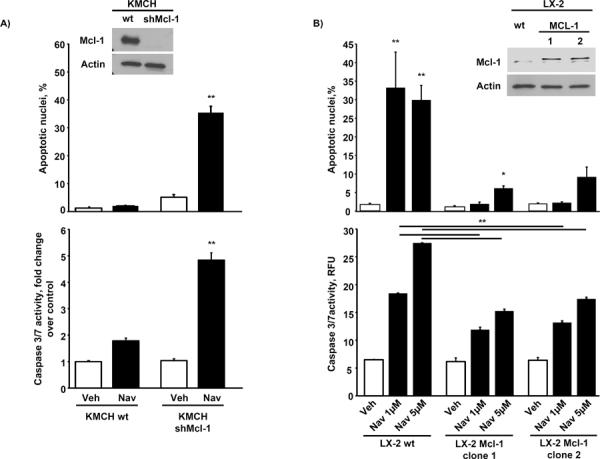
Mcl-1 was knocked down in the human CCA cell line KMCH by shRNA technique (Western blot for Mcl-1, see panel A inset) and cells were treated with navitoclax (1 μM) or vehicle for 48 hrs. Apoptosis was measured by DAPI-staining and fluorescence microscopy (panel A, upper graph; mean ± SEM; n=3; ** p≤0.01) or fluorometric analysis of caspase 3/7 activity displayed as fold change compared to vehicle control (panel A, lower graph; mean ± SEM; n≥5; ** p≤0.001). Mcl-1 was overexpressed in the activated hepatic myofibroblast LX-2 (Western blot for Mcl-1, see panel B inset) and cells were treated with navitoclax (1 μM or 5 μM) or vehicle for 48 hrs. Apoptosis was assessed by DAPI-staining and measurement of caspase 3/7 activity as described above ( mean ± SEM; n=3; ** p≤0.01)
Liver myofibroblasts display mitochondrial priming for cell death
Navitoclax is incapable of directly inducing Bax activation (11, 31). Moreover, quiescent fibroblasts were also resistant to navitoclax cell killing despite failure to express Mcl-1. To reconcile these observations, we postulated that CAF mitochondria were primed for cell death (32). Mitochondrial priming refers to the empiric observation that activated or transformed cells have a reduced threshold for BH3 peptide– or BH3 mimetic-induced mitochondrial release of apoptogenic factors such as Smac and cytochrome c (33). For these studies we employed the LX-2 cells, given their ability to be easily transfected. Upon navitoclax treatment, we observed substantial Smac release from isolated cellular heavy membranes enriched in mitochondria (Fig. 4A) as well as into the cytoplasm of whole cells (Fig. 4B). However, Smac release was not observed in shBax cells (Fig. 4C). Mitochondria from CCA cells did not display release of apoptogenic factors upon exposure to navitoclax (Fig. 4D). Finally, consistent with being primed for cell death, the LX-2 cells but not quiescent fibroblasts nor CCA cells underwent mitochondrial membrane depolarization following navitoclax treatment and this effect was abolished in shBax LX-2 cells (Fig. 4E). To further examine mechanisms responsible for this observed priming, mitochondrial enriched heavy membrane fractions obtained from CCA, LX-2 and hCAF cells were profiled for activator BH3-only proteins (Bid, Bim and Puma), and Bax and Bak by immunoblot analysis (Fig. 5A). Increased Bax, Bid and Bim were identified in the mitochondrial fraction of myofibroblasts but not CCA cells. In accordance with other studies of priming (30), navitoclax treatment of LX-2 cells decreased Bim binding to Bcl-XL as examined by immunoprecipitation studies (Fig. 5B). These observations support the concept that the activated state of CAF is associated with translocation of Bax and BH3 only proteins to mitochondria. The presence of Bim association with Bcl-XL in the presence of Bax on mitochondria supports an embedded together model which primes the cells for cell death by navitoclax (13).
Figure 4. Navitoclax selectively induces release of mitochondrial proapoptotic factors in activated myofibroblasts.

Heavy membranes enriched in mitochondria were isolated from LX-2 cells and incubated with navitoclax (10 μM) or vehicle. Mitochondria were separated from supernatant by centrifugation and fractions were subject to immunoblot analysis to assay for Smac release from the mitochondria into the supernatant (panel A). LX-2 cells were treated with navitoclax (1 μM) and separated into cytoplasmic and mitochondria-containing fractions (pellet - loading control) by differential centrifugation after selective digitonin permeabilization. Fractions were analyzed for Smac and cytochrome C by immunoblot (panel B). Cytoplasmic and mitochondria-containing fractions (pellet) of shBax and shBak LX-2 cells and human CCA cell lines MzChA-1 and HuCCT-1 treated with navitoclax (1 μM) are shown in panels C and D, respectively. Note in these short-term incubation studies, the pool of cytochrome C and/or Smac released was limited and did not significantly influence the amount of these proteins in the pellet. hFB, LX-2, shBax LX-2 and Mz-ChA-1 CCA cells were treated with navitoclax or vehicle for 24 hrs. After tetramethylrhodamine methyl ester (TMRM, 100 nM) loading, cells were analyzed for mitochondrial depolarization and subsequent loss of fluorescence by flow cytometry. TMRM fluorescence (excitation wavelength 544 nM) intensity was measured in 50,000 cells (panel E).
Figure 5. Activated myofibroblasts display increased localization of proapoptotic BH3 only proteins to mitochondria and binding to anti-apoptotic Bcl-2 proteins.
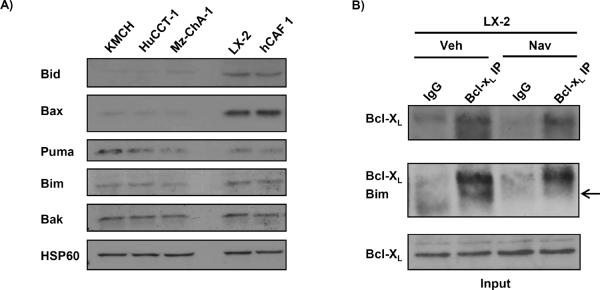
Mitochondria from LX-2, hCAF and human CCA cells were isolated by nitrogen cavitation and subject to immunoblot analysis for pro-apoptotic Bcl-2 family proteins (panel A). Immunoprecipitation of Bcl-XL from cell lysates of LX-2 cells and subsequent immunoblot analysis for the pro-apoptotic BH3 only protein Bim is shown in panel B. non-specific rabbit IgG was used as negative control during the immunoprecipitation (panel B).
Navitoclax reduces tumor burden and metastasis in vivo
To ascertain whether the effects of navitoclax on CAF from CCA observed in vitro may be translated to an in vivo model, we employed a syngeneic, orthotopic rodent model of CCA (34). First, cytotoxicity assays for the employed BDEneu tumor cells and rCAF previously isolated from orthotopic rat tumors were performed analogous to the previous assays with human cells (Suppl. Fig. 4A and B). These studies demonstrated that rat CAF, similar to human CAF, are more sensitive to navitoclax than the corresponding cancer cells. To also confirm that the stromal composition in this animal model was comparable to human CCA (28), we performed immunofluorescence for CAF-derived Ten C, an extracellular matrix protein produced by myofibroblasts, and α-SMA, a marker of the activated myofibroblast phenotype (3). Cytokeratin 7 was employed to identify CCA cells and distinguish them from the CAF. Indeed, the rodent CCA was associated with extensive tumor desmoplasia which strongly expressed CAF markers (Figure 6A). We then asked, whether navitoclax treatment induces CAF cytotoxicity in vivo. Animals were treated with navitoclax for 48 h, 7 days after tumor implantation. TUNEL assays were then performed with dual labeling for α-SMA and cytokeratin 7 to assess for TUNEL positive/α-SMA (CAF) and/or TUNEL positive/CK7 cells (CCA cells). The number of α-SMA positive cells which were TUNEL positive was increased compared to tumors from vehicle treated animals (Fig. 6B) and was greater than the TUNEL positive/CK7 cells. Consistent with this observation, there was also a net reduction in quantitative immunofluorescence staining for the tumor stroma markers α-SMA and Ten C (Fig. 6C). This observation was also confirmed by quantitative analysis of α-SMA and Ten C mRNA expression in the tumor samples (Fig. 6D). Finally, we examined the potential tumor suppressing effect of navitoclax treatment in this model. In vehicle and navitoclax treated rats, tumor weight and metastases were examined after 10 days of treatment, initiated 7 days after tumor implantation. Mean gross tumor wet weight (Fig. 7A) and mean tumor-liver weight ratio (Fig. 7B) were reduced in the navitoclax-treated animals. Peritoneal metastases were also reduced in this group (Fig. 7C). The navitoclax treatment associated tumor suppressive effects also translated into an improved animal survival (Fig. 7D). The morphometric analysis revealed an altered tumor composition after 10 days of treatment, namely a reduction in the ratio of α-SMA and Ten C positive stroma to tumor area (Fig. 7E). Histologically the tumors appeared less desmoplastic with larger contiguous areas of tumor cells rather than isolated tumor glands typical for CCA (Fig. 7F). Finally, we were also able to demonstrate Bax activation and loss of mitochondrial Smac in CAF following navitoclax treatment for 48 hrs, thereby, suggesting the mechanisms of navitoclax-mediated CAF cytotoxicity in vivo parallels that observed in vitro (Supplemental Fig. 5). Taken together, these observations suggest navitoclax impairs tumor growth and metastasis by depleting the tumor stroma of CAF, thereby reducing the extent of growth supporting CCA tumor stroma.
Figure 6. Navitoclax depletes CAF from BDEneu tumors and reduces characteristic tumor stroma ECM.

Immunofluorescence double stainings for α-SMA (red) and cytokeratin 7 (CK-7, green; panel A, upper image, 40×) as well as tenascin C (Ten C, green) and cytokeratin 7 (CK-7 red; panel A, lower image, 40×) were performed on representative sections of BDEneu tumors harvested 17 days after tumor cell implantation. Frozen sections of BDEneu tumors from animals treated with two doses of vehicle or navitoclax (5 mg/kg) were stained for α-SMA or cytokeratin 7 and co-labeled with the TUNEL staining. The number of α-SMA/TUNEL and CK-7/TUNEL positive cells was assessed and plotted as percentage of all α-SMA or CK-7 positive cells, respectively (panel B; mean ± SEM; n=4; * p≤0.05). BDEneu tumors from animals treated with two doses of navitoclax (5 mg/kg) were stained for α-SMA, tenascin C and cytokeratin 7. Confocal microphotographs of representative tumor areas were analyzed by digital morphometry and α-SMA and tenascin C positive areas were expressed as percent of total tumor area (panel C; mean ± SEM; n=8; ** p≤0.001). α-SMA, Ten C and CK-7 mRNA expression was quantified by real-time PCR and normalized to 18S rRNA. Expression of α-SMA and tenascin c mRNA was plotted as ratio to CK-7 to control for the quantity of tumor-stroma in the sample (panel D; mean ± SEM; n=7; * p≤0.05)
Figure 7. Navitoclax treatment reduces BDEneu tumor size and metastasis, improves survival and alters tumor composition.

BDEneu tumors from animals treated with navitoclax (5 mg/kg) or vehicle for 10 days were carefully excised, remaining liver tissue was removed, then tumors were measured and weighted (panel A; mean ± SEM; n=8; * p≤0.05) and tumor-liver weight ratios were calculated (panel B; mean ± SEM; n=8; * p≤0.05). After removal of the liver, the peritoneal cavity was carefully examined for metastases (panel C; mean ± SEM; n=8; * p≤0.05). To assess effects of navitoclax on survival, animals were treated with navitoclax as described for 21 days and survival was documented (panel D; mean ± SEM; n=18; p≤0.05). Frozen tumor sections were stained for α-SMA, tenascin C and cytokeratin 7. Digital morphometry was performed on confocal microphotographs and ratio of α-SMA and tenascin C positive stroma to tumor area was calculated (panel E; mean ± SEM; n=4; ** p≤0.01). Representative photomicrographs of hematoxylin-eosin stained tumor sections are shown in panel F. Characteristic tumor areas are outlined.
DISCUSSION
The results of this study provide new mechanistic insights regarding therapeutic targeting of CAF within the tumor microenvironment as an anticancer strategy. These data indicate that: (a) liver CAF: display sensitivity to single agent cell killing by the BH3 mimetic navitoclax; (b) the sensitivity of CAF to navitoclax is associated with enhanced Bax expression and mitochondrial priming for cell death; and (c) targeting CAF for cell death in the tumor microenvironment is therapeutic in a syngeneic rodent in vivo model of CCA. These findings are discussed in greater detail below.
Navitoclax mimics the binding characteristics of the BH3-only protein Bad, and like Bad does not bind to the anti-apoptotic protein Mcl-1 (14); indeed, Mcl-1 expression is a well-documented resistance factor for navitoclax (29). Given that CCA express Mcl-1 (25), their resistance to navitoclax is not surprising. However, resistance to navitoclax is unlikely to be mediated solely by a single Bcl-2 protein given the complexity of the interactions between pro- and anti-apoptotic members of this family. For example, the HuCCT cells displayed partial sensitivity to navitoclax despite Mcl-1 expression, likely due to their increased Bax plus reduced Bcl-2 and Bcl-XL expression. Alternatively, lack of Mcl-1 phosphorylation at Ser-64 in these cells could potentially explain the insufficiency of Mcl-1 in preventing navitoclax cytotoxicity (35).
The absence of Mcl-1 expression by CAF was unexpected, but, partially explains their sensitivity to navitoclax-mediated apoptosis. The mechanism for silencing Mcl-1 expression by CAF is beyond the scope of this study, given the complexity of Mcl-1 regulation (36). Because navitoclax cannot directly activate Bax or Bak, the absence of Mcl-1 expression is not sufficient to explain the sensitivity of CAF to navitoclax killing. Indeed, quiescent fibroblasts were also resistant to navitoclax cytotoxicity despite their lacking of Mcl-1 expression. These observations suggest CAF may also be primed for cell death.
In one model, mitochondrial priming is considered to be a state in which the capacity of anti-apoptotic Bcl-2 proteins to sequester pro-apoptotic BH3 only proteins or bind Bax/Bak is almost completely exhausted (32). In this situation minimal shifts of the Bcl-2 equilibrium can cause cells to rapidly enter apoptosis (37). By binding to the anti-apoptotic Bcl-2 proteins Bcl-2 and Bcl-XL, navitoclax displaces sequestered BH3 proteins like Bim from these antiapoptotic proteins and enables them to activate Bax and Bak, inducing apoptosis (14). Indeed, we were able to demonstrate Bim displacement from Bcl-XL following navitoclax treatment in our current studies. In an alternative model, coined the “embedded together” model, anti-apoptotic proteins inhibit not only BH-3 only proteins, but also Bax or Bak all embedded within the outer mitochondrial membrane (13, 14) In this model, BH3 mimetics may alter binding between the various Bcl-2 binding polypeptides allowing Bax or Bak activation and MOMP. Our data are perhaps most consistent with an embedded together model as we observed that Bim and Bax were already associated with mitochondria under basal conditions in myofibroblasts. Following navitoclax treatment, Bim was released from Bcl-XL suggesting the BH3 mimetic altered the complex binding dynamics in the membrane in such a manner as to favor Bax activation and MOMP.
In our in vivo studies, despite the more prominent cytotoxic effects on CAF as compared to CCA cells, navitoclax treatment was sufficient to markedly reduce tumor size. These observations support the concept that tumor stroma is crucial for tumor development and even metastasis (38). Quantitative depletion of CAF from the stroma by navitoclax also reduced the expression of typical components of the tumor extracellular matrix, such as Ten C, a protein recently found to support cancer growth (6, 39, 40). Thus, therapeutic induction of CAF apoptosis can be coupled to a decrease in tumor promoting components of the extracellular matrix. The noticeable reduction in metastasis might be a result of this depletion particularly of Ten C from the microenvironment, which has just recently been described as important matrix component of the metastatic niche (6, 41).
In this study we employed CCA as a model of a highly desmoplastic cancer containing abundant CAF and tumor-specific extracellular matrix (4). This cancer is similar to other desmoplastic cancers, such as prostate, breast and pancreatic cancer, where tumor stroma contributes to tumor development and progression (42, 43). Thus, the results of this study are not only germane to CCA, but may also provide information relevant to desmoplastic cancers in general (2, 44). While cancer therapy targeting the cancer cells becomes increasingly more individualized, reflecting the multitude of genetic and cell biological alterations an individual tumor may present, the stromal reaction of the organism appears more uniform across many different types of malignancies (45). This suggests the tumor stroma to be a viable and attractive target for combination anti-cancer therapy. Our results further suggest that CAF priming to select pro-apoptotic therapies targeting this cell population represents a novel anticancer strategy. In this regard, navitoclax may be a clinically relevant therapeutic agent.
Supplementary Material
ACKNOWLEDEMENTS
We are indebted to helpful scientific discussions and suggestions by Drs. Scott H. Kaufmann and Haiming Dai. We thank Courtney Hoover for her outstanding secretarial assistance.
GRANT SUPPORT This work was supported by grants NIH DK59427 (GJG), NIH R01 CA 39225 (AES), the optical microscopy core of NIH DK84567, and the Mayo Foundation. J.C. Mertens is a scholar of the Swiss National Science Foundation (Schweizerischer Nationalfond SNF; grant PBSKP3_130612/1).
Abbreviations
- CCA
cholangiocarcinoma
- CAF
carcinoma associated fibroblast
- hFB
human fibroblasts
- rFB
rat fibroblasts
- CK7
cytokeratin 7
- α-SMA
α-smooth muscle actin
- Ten C
tenascin C
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
Footnotes
Conflict of Interest statement: Michael P. Gustafson and Allan B. Dietz have patent applications regarding culture of primary human cells, and Allen Dietz holds shares in Millcreek Life Sciences.
REFERENCES
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 3.Sirica AE. The role of cancer-associated myofibroblasts in intrahepatic cholangiocarcinoma. Nat Rev Gastro Hepat. 2011;9:44–54. doi: 10.1038/nrgastro.2011.222. [DOI] [PubMed] [Google Scholar]
- 4.Sirica AE, Dumur CI, Campbell DJ, Almenara JA, Ogunwobi OO, Dewitt JL. Intrahepatic cholangiocarcinoma progression: prognostic factors and basic mechanisms. Clin Gastroenterol Hepatol. 2009;7:S68–78. doi: 10.1016/j.cgh.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rasanen K, Vaheri A. Activation of fibroblasts in cancer stroma. Exp Cell Res. 2010;316:2713–22. doi: 10.1016/j.yexcr.2010.04.032. [DOI] [PubMed] [Google Scholar]
- 6.Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med. 2011;17:867–74. doi: 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brenner D, Krammer PH, Arnold R. Concepts of activated T cell death. Crit Rev Oncol Hematol. 2008;66:52–64. doi: 10.1016/j.critrevonc.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S, et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. 1998;102:538–49. doi: 10.1172/JCI1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30:3667–83. doi: 10.1038/emboj.2011.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, et al. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010;330:1390–3. doi: 10.1126/science.1190217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walensky LD. From mitochondrial biology to magic bullet: navitoclax disarms BCL-2 in chronic lymphocytic leukemia. J Clin Oncol. 2012;30:554–7. doi: 10.1200/JCO.2011.37.9339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–33. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leber B, Lin J, Andrews DW. Embedded together: the life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis. 2007;12:897–911. doi: 10.1007/s10495-007-0746-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, et al. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell. 2011;44:517–31. doi: 10.1016/j.molcel.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishimura N, Isomoto H, Bronk SF, Gores GJ. Trail induces cell migration and invasion in apoptosis-resistant cholangiocarcinoma cells. Am J Physiol-Gastr L. 2006;290:G129–36. doi: 10.1152/ajpgi.00242.2005. [DOI] [PubMed] [Google Scholar]
- 16.Knuth A, Gabbert H, Dippold W, Klein O, Sachsse W, Bitter-Suermann D, et al. Biliary adenocarcinoma. Characterisation of three new human tumor cell lines. J Hepatol. 1985;1:579–96. doi: 10.1016/s0168-8278(85)80002-7. [DOI] [PubMed] [Google Scholar]
- 17.Anan A, Baskin-Bey ES, Bronk SF, Werneburg NW, Shah VH, Gores GJ. Proteasome inhibition induces hepatic stellate cell apoptosis. Hepatology. 2006;43:335–44. doi: 10.1002/hep.21036. [DOI] [PubMed] [Google Scholar]
- 18.Yano H, Maruiwa M, Iemura A, Mizoguchi A, Kojiro M. Establishment and characterization of a new human extrahepatic bile duct carcinoma cell line (KMBC) Cancer. 1992;69:1664–73. doi: 10.1002/1097-0142(19920401)69:7<1664::aid-cncr2820690705>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 19.Xu L, Hui AY, Albanis E, Arthur MJ, O'Byrne SM, Blaner WS, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54:142–51. doi: 10.1136/gut.2004.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sirica AE, Zhang Z, Lai GH, Asano T, Shen XN, Ward DJ, et al. A novel “patient-like” model of cholangiocarcinoma progression based on bile duct inoculation of tumorigenic rat cholangiocyte cell lines. Hepatology. 2008;47:1178–90. doi: 10.1002/hep.22088. [DOI] [PubMed] [Google Scholar]
- 21.Campbell DJ, Dumur CI, Lamour NF, Dewitt JL, Sirica AE. Novel organotypic culture model of cholangiocarcinoma progression. Hepatol Res. 2012 doi: 10.1111/j.1872-034X.2012.01026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crespo-Diaz R, Behfar A, Butler GW, Padley DJ, Sarr MG, Bartunek J, et al. Platelet lysate consisting of a natural repair proteome supports human mesenchymal stem cell proliferation and chromosomal stability. Cell Transplant. 2011;20:797–811. doi: 10.3727/096368910X543376. [DOI] [PubMed] [Google Scholar]
- 23.Dai H, Meng XW, Lee SH, Schneider PA, Kaufmann SH. Context-dependent Bcl-2/Bak interactions regulate lymphoid cell apoptosis. J Biol Chem. 2009;284:18311–22. doi: 10.1074/jbc.M109.004770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mott JL, Bronk SF, Mesa RA, Kaufmann SH, Gores GJ. BH3-only protein mimetic obatoclax sensitizes cholangiocarcinoma cells to Apo2L/TRAIL-induced apoptosis. Mol Cancer Ther. 2008;7:2339–47. doi: 10.1158/1535-7163.MCT-08-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taniai M, Grambihler A, Higuchi H, Werneburg N, Bronk SF, Farrugia DJ, et al. Mcl-1 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in human cholangiocarcinoma cells. Cancer Res. 2004;64:3517–24. doi: 10.1158/0008-5472.CAN-03-2770. [DOI] [PubMed] [Google Scholar]
- 26.Masuoka HC, Mott J, Bronk SF, Werneburg NW, Akazawa Y, Kaufmann SH, et al. Mcl-1 degradation during hepatocyte lipoapoptosis. J Biol Chem. 2009;284:30039–48. doi: 10.1074/jbc.M109.039545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smoot RL, Blechacz BR, Werneburg NW, Bronk SF, Sinicrope FA, Sirica AE, et al. A Bax-mediated mechanism for obatoclax-induced apoptosis of cholangiocarcinoma cells. Cancer Res. 2010;70:1960–9. doi: 10.1158/0008-5472.CAN-09-3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aishima S, Taguchi K, Terashi T, Matsuura S, Shimada M, Tsuneyoshi M. Tenascin expression at the invasive front is associated with poor prognosis in intrahepatic cholangiocarcinoma. Modern Pathol. 2003;16:1019–27. doi: 10.1097/01.MP.0000086860.65672.73. [DOI] [PubMed] [Google Scholar]
- 29.Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115:3304–13. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Certo M, Moore Vdel G, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–65. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 31.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 32.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–65. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 33.Letai AG. Diagnosing and exploiting cancer's addiction to blocks in apoptosis. Nat Rev Cancer. 2008;8:121–32. doi: 10.1038/nrc2297. [DOI] [PubMed] [Google Scholar]
- 34.Fingas CD, Bronk SF, Werneburg NW, Mott JL, Guicciardi ME, Cazanave SC, et al. Myofibroblast-derived PDGF-BB promotes Hedgehog survival signaling in cholangiocarcinoma cells. Hepatology. 2011;54:2076–88. doi: 10.1002/hep.24588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mazumder S, Choudhary GS, Al-Harbi S, Almasan A. Mcl-1 Phosphorylation defines ABT-737 resistance that can be overcome by increased NOXA expression in leukemic B cells. Cancer Res. 2012;72:3069–79. doi: 10.1158/0008-5472.CAN-11-4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gores GJ, Kaufmann SH. Selectively targeting Mcl-1 for the treatment of acute myelogenous leukemia and solid tumors. Genes Dev. 2012;26:305–11. doi: 10.1101/gad.186189.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–21. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Utispan K, Thuwajit P, Abiko Y, Charngkaew K, Paupairoj A, Chau-in S, et al. Gene expression profiling of cholangiocarcinoma-derived fibroblast reveals alterations related to tumor progression and indicates periostin as a poor prognostic marker. Mol Cancer. 2010;9:13. doi: 10.1186/1476-4598-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paron I, Berchtold S, Voros J, Shamarla M, Erkan M, Hofler H, et al. Tenascin-C enhances pancreatic cancer cell growth and motility and affects cell adhesion through activation of the integrin pathway. PloS one. 2011;6:e21684. doi: 10.1371/journal.pone.0021684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brellier F, Chiquet-Ehrismann R. How do tenascins influence the birth and life of a malignant cell? J Cell Mol Med. 2012;16:32–40. doi: 10.1111/j.1582-4934.2011.01360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS, et al. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. P Natl Acad Sci USA. 2011;108:16002–7. doi: 10.1073/pnas.1109493108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–11. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Carey L, et al. Reconstruction of functionally normal and malignant human breast tissues in mice. P Natl Acad Sci USA. 2004;101:4966–71. doi: 10.1073/pnas.0401064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erez N, Truitt M, Olson P, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell. 2010;17:135–47. doi: 10.1016/j.ccr.2009.12.041. [DOI] [PubMed] [Google Scholar]
- 45.Engels B, Rowley DA, Schreiber H. Targeting stroma to treat cancers. Semin Cancer Biol. 2012;22:41–9. doi: 10.1016/j.semcancer.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.