

Abstract
Proteins have evolved to use water to help guide folding. A physically motivated, nonpairwise-additive model of water-mediated interactions added to a protein structure prediction Hamiltonian yields marked improvement in the quality of structure prediction for larger proteins. Free energy profile analysis suggests that long-range water-mediated potentials guide folding and smooth the underlying folding funnel. Analyzing simulation trajectories gives direct evidence that water-mediated interactions facilitate native-like packing of supersecondary structural elements. Long-range pairing of hydrophilic groups is an integral part of protein architecture. Specific water-mediated interactions are a universal feature of biomolecular recognition landscapes in both folding and binding.
Water is intimately involved in protein folding (1–4). That proteins denature both on heating and cooling strongly implicates the involvement of water degrees of freedom. Kauzmann (5) correctly inferred from thermodynamics the hydrophobic layering characteristic of protein structure before protein structures were determined crystallographically. The kinetics of water exclusion is often considered in discussing mechanisms of protein folding, but again it is the avoidance of water in the final folded structure that is emphasized (1). Hydrophobicity patterns have long been a dominant consideration in predicting protein structure by using sequence data (6) and are basic in synthetic protein design (7). Nevertheless, the structured character of water has not been a paramount factor in most existing algorithms for structure prediction (8). These usually rely on effective pair potentials (9) or buried surface area terms to account for the free energy of burying hydrophobic residues (10).
In this article, we hypothesize that specific water-mediated interactions help guide the folding process even before native contacts form. Using this idea we develop a bioinformatic, nonpairwise-additive interaction model accounting for water and show that it greatly improves the efficiency and accuracy of structure prediction for α-helical proteins. Analysis of folding trajectories with this potential strongly implicates the guiding role of long-range water-mediated interactions. Interestingly, we find here that long-range hydrophilic interactions, as distinct from hydrophobic interactions, also take center stage.
The bioinformatic route to water-mediated potentials is difficult in several ways (for more directly physical approaches see ref. 11). Although bound water is visible in structures, localizing waters is more difficult than localizing main-chain atoms. Monomeric protein structures also have relatively few visible water-mediated interactions. Our path to a water-mediated potential started with an energy landscape analysis of protein–protein interactions and a bioinformatic survey of interfaces in dimer structures (12, 13). We found that the often-used contact potentials (9) worked well to describe hydrophobic binding interfaces; however, hydrophilic interfaces were poorly recognized (13). This finding suggests that longer-range interresidue contacts, mediated by water, play an important role in stabilizing these interfaces (13). To test this hypothesis, we derived both direct and water-mediated binding potentials (13). When these two potentials were used simultaneously (13), smooth recognition of diverse binding interfaces was achieved (in contrast to the direct contact potential). Here, we show that water-mediated interactions play an important role not only in binding interfaces but in folding of monomeric proteins.
We use the associative memory (AM) Hamiltonian molecular dynamics model as a starting point (14–16). This Hamiltonian has two principal components: general polymer physics-based terms that are sequence independent, collectively called “backbone,” and sequence-dependent knowledge-based distance-dependent additive potentials, collectively denoted as AM/C (AM/contact). The AM part describes interactions between all pairs of residues that are separated in sequence between 3 and 12 residues. It uses a set of nonhomologous memory proteins to build a funneled energy landscape by matching fragments. The C part applies to tertiary contacts between residues separated by >12 residues in sequence. All parameters in the potential have been optimized with a self-consistent procedure based on the energy landscape theory as described (15) (see Appendix: Computational Details and Supporting Text, which is published as supporting information on the PNAS web site).
The C part of the AM/C potential describes effective interactions between Cβ (Cα for Gly) atoms in each residue pair. It consists of three wells covering the 4.5- to 8.5-Å, 8.5- to 10.0-Å, and 10.0- to 15.0-Å distance intervals. Similarly, the potentials used in this study contain a first well for the 4.5- to 6.5-Å interval, whereas the second well is replaced by a local density-dependent potential (discussed below). They contain no third well, because it is unlikely that specific interresidue interactions are mediated by water to such a long distance (10–15 Å). There is also a residue-specific many-body burial profile potential describing coordination preferences of all 20 amino acids. The parameters for the resulting potential, which we call AM/W (W for water), were optimized by using our earlier sequence-based approach (13). We have further refined these parameters by using the self-consistent structurally based optimization scheme from energy landscape theory (15). We denote the original potential, AM/W-0, and the more refined one, AM/W-1 (see Appendix for Computational Details and Supporting Text).
For the coarse-grained models considered in our study, the definition of water-mediated contacts naturally becomes somewhat more indirect than, for example, in full-atom simulations. Because direct contacts are defined as occurring between residues that have a distance between Cβ (Cα for Gly) atoms of <6.5 Å, a similar constraint for water-mediated contacts places them in the 6.5- to 9.5-Å distance interval. A more extensive discussion of the rationale for this choice is given in ref. 13 where the results for protein binding recognition also was found to be robust with respect to various alternative definitions of water-mediated contact range.
To interact through water, we require that both residues are sufficiently exposed to water, or equivalently, neither residue should be buried in the protein interior (hydrophobic core). To model this we use a highly nonadditive local density-dependent potential: when either of the residues in the pair attains a local neighbor density above a critical threshold value (i.e., becomes buried), the potential switches smoothly but quickly from water mediated to protein mediated.
Results and Discussion
Physical Interpretation of the Interaction Potentials. Before discussing the simulation results, we briefly analyze the main qualitative differences between the AM/C and AM/W interaction potentials. The interactions within the range of the first well and the protein-mediated interactions of the second well of the AM/W potential are qualitatively similar to their corresponding AM/C counterparts (see Fig. 1 A and B). The main difference lies in the interactions between hydrophilic residues in the second-well water-mediated interactions (see Fig. 1C). Whereas very polar second-well interactions are destabilized on average for the AM/C potential, they are highly stabilized for the AM/W potential when two residues are in a low-density environment, i.e., when residues interact through water (Fig. 1C).
Fig. 1.

The detailed interactions in the bioinformatic prediction energy functions are compared. We partition 210 interresidue pairs into 165 pairs having at least one hydrophobic partner (hydrophobic group) and 45 pairs having only charged and polar residues in the pair (polar group). More positive values for the matrix elements indicate more favorable interactions. (A) The 165 interaction matrix elements for first-well interactions among residue pairs having at least one hydrophobic partner are shown. The hydrophobic group first-well interactions show very similar profiles among the AM/C and AM/W potentials. (B) The 45 interactions for first-well interactions among all charged and polar residue pairs are shown. The first-well polar group contact interactions are rather similar in each potential, except for the most charged pairs, which are more destabilized in the AM/W-0 and AM/W-1 potentials. (C) The 45 interactions for second-well protein-mediated (filled symbols) and second-well water-mediated (dotted symbols) interactions among all charged and polar residue pairs are shown. Again the second-well protein-mediated interactions in AM/W are similar to those in AM/C, but the AM/W water-mediated interactions stand out as being different (see the text for discussion).
Although these potentials are knowledge-based in origin, examining their details gives interesting physical insight into the nature of biomolecular forces (13). Perusing of charged residue interactions in Fig. 1 B and C suggests that a large desolvation penalty must be paid when a fully direct contact is formed, and, therefore, charged and highly polar residues prefer to avoid complete desolvation by interacting through one or two water layers. Even more interestingly, not only do oppositely charged residues attract each other when interacting through water, but so do residues of the same charge (Fig. 1C). This finding either indicates that residues of the same charge alter their mutual pKa so only one residue is really charged (i.e., one has in fact a charged-polar interaction) or that correlated fluctuations of the counterion cloud (17) and the perturbation of the water hydrogen-bonding network bind the like-charged residues together.
General Trends. Given the differences among the potentials outlined above, we anticipate that the AM/W potential would significantly improve the AM/C potential results for those proteins that contain explicit water-bridged interactions in their native state. As we shall see, these water-mediated interactions also appear transiently during collapse and folding of the chain and help guide the heteropolymer into a correct topology. For each protein of 14 chosen for study (discussed below), we have carried out five distinct annealing runs (7.2 × 105 time steps) with each of the three potentials (AM/C, AM/W-0, and AM/W-1), starting from a randomly generated extended-coil conformation at high temperature (we have not optimized the annealing protocol for the AM/W potentials nor used other minimization techniques; ref. 18). For each run we have taken 240 snapshots at equal time intervals, monitoring the progress toward achieving a native-like conformation by using a contact overlap measure Q. Our Q measure is more stringent than the usual contact Q, because it takes into account not only the correctness of contacts that occur in the native structure but also the correctness of distances between all pairs of residues even when they are far apart in the native state. In addition to Q, when discussing various structures, we use other structural similarity measures, such as rms displacement (RMSD) and the combinatorial extension (CE) method, which makes sequence-independent alignment of two conformations (19). It is perhaps not surprising that comparing protein structures is a tricky business, involving several means of similarity as discussed (20).
The validation of any knowledge-based potential must be done on an unrelated set of test proteins because of the risk of parameter overlearning. Nine of the 14 α-helical proteins used are “training” proteins for the AM/C potential, i.e., they were used to derive the parameters for the AM and C parts of the potential (15). The W part of AM/W-0 was optimized by using a sequence-based technique for an unrelated set of proteins. Thus for the AM/W-0 tertiary contact potential these nine proteins serve partially as test proteins. On the other hand, the W part of the AM/W-1 potential was refined by using the same training set of nine proteins. We emphasize these relationships to be attentive to the possibility of overlearning. It is necessary to apply the potential to an unrelated set of test proteins for confirmation.
The performance of the AM/W potentials is well documented by five α-helical test proteins that we discuss in detail below (see also Supporting Text). Two test proteins [Protein Data Bank (PDB) codes 1BG8 (21) and 1JWE (22)] were targets taken from the CASP3 (Critical Assessment of Techniques for Protein Structure Prediction) event (ref. 20; a detailed compilation of all CASP results may be found at http://predictioncenter.llnl.gov), and three, target T0170 [PDB code 1H40 (23)], target T172b [PDB code 1N2X (24)], and target T129a [PDB code 1IZM (A. Galkin, E. Sarikaya, C. Lehmann, A. Howard, and O. Herzberg, personal communication)], were taken from the CASP5 event (http://predictioncenter.llnl.gov and ref. 25). Our results compare favorably with the top CASP predictions for these proteins (http://predictioncenter.llnl.gov), but have nevertheless been obtained a posteriori (although in a fairly automatic manner) and should not be regarded as new CASP entries. Nevertheless, because numerous prediction groups participated in CASP, the CASP experiment has generated valuable statistical data that may be used to calibrate progress.
When the best Q scores obtained for each protein during all five annealing runs are compared across all 14 proteins (Fig. 2), the following trends becomes evident. First, AM/C and AM/W potentials show similar performance for small (<115 residues) training proteins. As for small test proteins, 1BG8 (21) is greatly improved by both AM/W-0 and AM/W-1 potentials, whereas T0170 is improved only by AM/W-0. The most significant trend, one that is highly desirable, comes when the largest proteins (>115 residues) are considered. A methodical improvement in the prediction of both training and test proteins is achieved by both AM/W-0 and AM/W-1 potentials, the latter showing a more uniform trend (Fig. 2). For large proteins, an improvement of 0.05–0.10 in Q is very significant, typically improving global RMSD by a few Å and significantly improving other measures of fold recognition, such as CE Z score.
Fig. 2.

Structure prediction performance and the comparison of AM/C, AM/W-0, and AM/W-1 potentials. The maximum Q scores versus chain length attained during five annealing runs for each of 14 proteins using three different potentials are shown. PDB codes for the training proteins are in violet, and the test proteins are in green.
Specific Targets. Having achieved substantial progress in protein structure prediction by using the tertiary contact potential incorporating long-range water-mediated interactions, we next investigate the cause of the improved structural recognition. We focus on three proteins: (i) PDB code 2FHA (26), a training protein, the largest one in the protein set; (ii) PDB code 1BG8 (21), a small test protein for which both AM/W-0 and AM/W-1 show very significant improvements; and (iii) CASP5 target T129a (A. Galkin, E. Sarikaya, C. Lehmann, A. Howard, and O. Herzberg, personal communication), the largest test protein in the protein set, that has two interacting domains.
Human iron storage protein, ferritin [PDB code 2FHA (26)], is the largest protein studied (172 residues). Although it was a training protein for AM/C, there was a large improvement in structure prediction when using both AM/W-0 and AM/W-1 (Fig. 3A). During the cooling schedule the divergence between the trajectories in nativeness occurs around T = 1.05. To further evaluate the difference between the potentials, we carried out free energy calculations as a function of Q by using the histogramming technique (16). These calculations show that the free energy minimum shifts toward more native-like structure for AM/W-0 vs. AM/C (data not shown). In addition, the thermodynamic energy as a function of Q (Fig. 3B) indicates that AM/W-0 energy landscape is slightly more funneled than AM/C. The AM/C potential generates a rougher energy landscape than the AM/W potentials. This is even more exaggerated in the E(Q) plot of Fig. 3B caused by the statistical noise caused by slow exploration of deeper traps of the AM/C potential energy surface. These thermodynamic observations at least partially explain the improved prediction from the AM/W potentials.
Fig. 3.

Structure predictions for ferritin, PDB code 2FHA. (A) The best (of five for each potential) Q-score annealing trajectories are shown for three different potentials. (B) The average thermodynamic energy vs. Q.(C) Superposition of the AM/W-0 best Q-score structure (blue) and the native structure (red) is indicated. Spheres indicate charged residue Cα atoms. (D) The distance plot for the AM/W-0 best Q-score structure (blue, upper triangle) and the native structure (red, lower triangle). (E) Distance plot for the AM/C best Q-score structure (blue, upper triangle) and the native structure (red, lower triangle) are compared. In the AM/W-0 structure (D), only a small number of contacts are missing and a small registry shift near residue 70 occurs. In the AM/C structure (E) the C-terminal half misses on a major interhelical interface.
The highest Q structure from AM/W-0 annealing is superimposed on the native ferritin structure in Fig. 3C. The CE structure alignment method gives a Z score of 5.7 for the alignment of 134 residues with an RMSD of 3.4 Å (a Z score >3.5 is considered as significant fold recognition). The first and last 15 residues were not aligned by CE, as evident in Fig. 3C. Because of this mispacking of these two relatively small fragments, the global RMSD of 10.2 Å turns out to be somewhat misleading. The distance plots for the native and the predicted structure (Fig. 3D) indicate that the overall fold and the tertiary interactions are captured quite accurately. The best AM/C snapshot, on the other hand, describes correctly only a smaller part of the overall structure (118 residues are aligned with RMSD of 5.0 Å and Z score of 3.9). When we partition the Q-score data into native contacts that are short and medium range in sequence (between 3 and 12 residues) as opposed to long range in sequence (tertiary contacts, >12 residues), then the resulting Qshort and Qmedium are rather similar for AM/C and AM/W potentials (Qs = 0.74 and Qm = 0.59 vs. Qs = 0.79 and Qm = 0.64). The main performance gain comes from improved packing of supersecondary structural elements (Qlong = 0.20 vs. Qlong = 0.36). Although expected, because the AM potential describing short- and medium-range contacts is shared by all potentials, this finding directly shows that the AM/W potentials improve nativeness by better treatment of tertiary interactions.
The small test (CASP3) protein for which we have observed large enhancement in native structure recognition is Escherichia coli stress-response protein HdeA [PDB code 1BG8 (21)]. The superposition of best predicted structure with AM/W-1 and the native is shown in Fig. 4A. At the overall Q score of 0.47, the CE alignment of 70 residues of total 76 residues produces an RMSD of 4.2 Å and Z score of 3.7. The global RMSD was 5.1 Å. The best AM/C prediction (Fig. 4B) again captures correctly large chunks of the structure (CE alignment of residues 7–62 produces a Z score of 3.3 and RMSD of 5.7 Å), but fails to pack them globally (overall Q = 0.31, global RMSD = 12.0 Å). Free energy calculations show that the minimum in F(Q) is shifted substantially toward the native for the AM/W potentials (Fig. 4C), rationalizing annealing results.
Fig. 4.

Structure predictions for 1HdeA, PDB code 1bg8 (CASP3). (A) A superposition of the best Q-score structure from the AM/W-1 potential (blue) and the native structure (red) is shown. Spheres indicate charged residue Cα atoms. (B) The superposition of the best Q-score structure from the AM/C potential (blue) and the native structure (red) is shown. (C) Free energy vs. Q as computed with a histogramming technique. (D) Annealing trajectories of individual fragment Q scores, large N-terminal fragment containing residues 1–61, and small C-terminal domain containing residues 62–76 are shown as a function of the instantaneous temperature through the run. (E) Annealing trajectories of interfragment Q scores are indicated. (F) Annealing trajectories of interfragment Q scores partitioned into the first-well and second-well contributions are shown.
Closer examination of the HdeA sequence reveals that both N- and C-terminal 11-residue fragments are highly charged (four charged residues in the N-terminal fragment and seven charged residues in the C-terminal fragment). Fig. 4B shows that it is these terminal fragments that are packed incorrectly by the AM/C potential. For analysis, we thus partitioned the protein into two fragments: a larger N-terminal fragment consisting of residues 1–61, and a smaller C-terminal terminal fragment consisting of residues 62–76. The annealing trajectories for the corresponding fragment Q scores (Fig. 4D) indicate that AM/C and AM/W-1 produce qualitatively similar fragment structures down to T = 0.8, at which temperature the larger fragment experiences a jump in the nativeness for the AM/W-1 potential. This event is immediately preceded by a jump in the interfragment Q value (Fig. 4E), suggesting that native interfragment interface formation nucleates the folding of the larger fragment. We have additionally partitioned the interfragment Q into first-well and second-well contributions (Fig. 4F). This analysis shows the major improvement in the interface recognition comes from the second-well interactions. Because protein-mediated second-well interactions are greatly diminished in AM/W-1 (see Fig. 1C) and we notice the charged nature of the C-terminal fragment (Fig. 4A), we see that it is AM/W water-mediated interactions that greatly facilitate correct packing of secondary structure elements in HdeA.
When the HdeA crystal structure was originally published, Yang et al. (21) could not find any sequence or structural similarity to any other known protein. Its functional role was also unknown (21). In a subsequent study (27), it was demonstrated that HdeA provides acid resistance in bacterial pathogens (HdeA is stable under extreme acidic conditions). It was suggested that in neutral pH HdeA forms a dimer (the dimer interface is formed mainly by hydrophobic residues), which dissociates to become an active monomer when pH is significantly lowered (27), the exact mechanism of dissociation being unclear. Gajiwala and Burley (27) hypothesized that perhaps pH-induced conformational changes of unknown nature lead to dissociation. In light of our analysis, it indeed seems plausible that a change in the protonation state of terminal fragments would lead to large structural rearrangement, perhaps causing the dimer dissociation.
The final test protein for analysis is a two-domain CASP5 target, T129a [PDB code 1IZM; the structure has not been released at the time of writing (A. Galkin, E. Sarikaya, C. Lehmann, A. Howard, and O. Herzberg, personal communication)]. The distance plot comparing the best Q (0.36) predicted structure and the crystal structure (Fig. 5A) shows that the major features of the protein fold are well captured (global RMSD was found at 8.7 Å). Interestingly enough, the same snapshot also has the best interdomain Q score for the same trajectory. However, there exist snapshots with somewhat better individual domain structures that are docked incorrectly (Fig. 5B). The individual domain II Q scores between the best AM/C and AM/W-1 trajectories are of similar quality, but the AM/W-1 potential produces more native-like structures for domain I (Fig. 5C). As in the case of HdeA, the interdomain Q (Fig. 5D) shows the most improvement for AM/W-1 compared with AM/C. Partitioning the interdomain Q into first- and second-well contributions (Fig. 5 E and F), again leads to the conclusion that water-mediated interactions enhance native-like packing of supersecondary structure elements.
Fig. 5.

Structure predictions for CASP5 target protein T129a (PDB code 1IZM, structural information not yet officially released at the time of writing). (A) The distance plot for AM/W-1 best Q-score structure (blue, upper triangle) and the native structure (red, lower triangle) is shown. (B) The distance plot for AM/W-1 structure with the best sum of individual domain Q scores (blue, upper triangle) and the native structure (red, lower triangle) is shown. (C) Annealing trajectories of individual domain Q scores, N-terminal domain containing residues 1–75, and C-terminal domain containing residues 76–170 are indicated. (D) Annealing trajectories of interdomain Q scores are shown. (E) Annealing trajectories of interdomain first-well Q scores are plotted. (F) Annealing trajectories of interdomain second-well Q scores are plotted.
At a coarse-grained level, the interplay between direct contact interactions and longer-range water-mediated interactions, both guiding the folding process, suggests some new protein physics. Direct contact potentials are crudely equivalent to surface tension between the protein and its solvent environment, whereas longer-range water-mediated interactions depend on the curvature of the protein–water interface. The complex solvation physics of polar and charged species in the presence of counterions shapes the curvature landscape. Our results imply that, at least in the cases studied, evolution has tuned both the surface tension and curvature contributions to be consistent with the principle of minimal frustration (28).
Conclusions
In summary, specific water-mediated interactions are a universal feature of biomolecular recognition, both in folding of monomers and binding of many dimers. We have shown that the inclusion of long-range water-mediated interactions, through a nonpairwise-additive switching potential, in structure prediction Hamiltonians leads to systematically improved predictions for protein structures. Detailed analysis of annealing trajectories for the model reveals explicitly that water-mediated interactions indeed help to correctly assemble supersecondary structure elements into the global native fold. We hope that the water model presented in this article will also help advance the important ongoing efforts toward building an accurate coarsegrained representation of water for self-assembly of both biological and nonbiological systems.
Supplementary Material
Acknowledgments
We thank Michael Prentiss for help with setting up CASP5 target calculations and useful discussions; Dr. Yaakov Levy for helpful comments when preparing the manuscript; Dr. Osnat Herzberg for giving us permission to show distance plots for CASP5 target T129a in Fig. 5 before publication of the crystallographic information; and W. M. Keek Foundation for providing access to computing resources at the W. M. Keek Laboratory for Integrated Biology. G.A.P. thanks the National Institutes of Health for its generous support of this work through a Postdoctoral Fellowship Award. J.U. thanks the Swedish Research Council and the San Diego Supercomputing Center for providing postdoctoral fellowships. The efforts of P.G.W. in concepts of protein folding are supported through National Institutes of Health Grant 5R01GM44557. Additional support was received from National Science Foundation Grants PHY-0216556 and PHY-0225630.
Appendix: Computational Details
The AM/C Hamiltonian. The AM/C Hamiltonian has been discussed at great length in the literature (14–16, 29–32). The Hamiltonian, HAM/C = Hbb + HAM + Hcontact, consists of a general polymer physics-based backbone potential (see refs. 15, 16, and 29 for details), an AM term defining an energy funnel for residues separated by <12 residues (15, 16, 31), and a contact term that describes tertiary interactions. The contact Hamiltonian, Hcontact, has three wells covering the 4.5- to 8.5-Å, 8.5- to 10.0-Å, and 10.0-to 15.0-Å intervals. Supporting Text provides additional details about the AM/C Hamiltonian.
The AM/W Hamiltonian. The AM/W Hamiltonian is a modification of the AM/C Hamiltonian, where the tertiary contact part of AM/C Hamiltonian is replaced by a potential based on water-mediated interactions, HAM/W = Hbb + HAM + HRg + Hcontact + Hwater + Hburial, where Hbb and HAM are the same as in the AM/C potential, HRg is a quadratic potential that helps to collapse the chains (Hrg = C * [Rg({r}) - Rg(N)]2, based on work from ref. 33), Hcontact keeps the same functional form as in AM/C, but it contains only a single, direct contact, defined between 4.5 and 6.5 Å, Hwater is a nonpairwise additive second-well switching potential (defined below), and Hburial is a many-body potential indicating the burial preferences for each amino acid (defined below). The water-mediated second-well potential is, 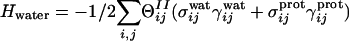 , where switching functions
, where switching functions 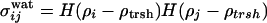 and
and 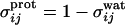 are used, that depend on local density environment of residues i and j
are used, that depend on local density environment of residues i and j 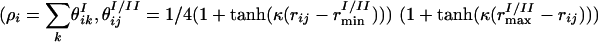 , and
, and 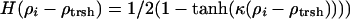 . In these expressions rij is the distance between residues i and j, rmin and rmax indicate the endpoints of corresponding wells (4.5–6.5 Å for the first well, 6.5–9.5 Å for the second well), and κ is a parameter that describes the sharpness of the switching tanh functions (κ was set to 5.0). The σ switching functions are constructed so that when the local density ρ for each residue increases beyond a threshold value of ρtrsh [chosen to be 2.6 from a structural survey of the monomer database (34), see below], the σwat switches smoothly from 1 to 0, whereas σprot switches from 0 to 1.
. In these expressions rij is the distance between residues i and j, rmin and rmax indicate the endpoints of corresponding wells (4.5–6.5 Å for the first well, 6.5–9.5 Å for the second well), and κ is a parameter that describes the sharpness of the switching tanh functions (κ was set to 5.0). The σ switching functions are constructed so that when the local density ρ for each residue increases beyond a threshold value of ρtrsh [chosen to be 2.6 from a structural survey of the monomer database (34), see below], the σwat switches smoothly from 1 to 0, whereas σprot switches from 0 to 1.
The burial profile term, Hburial, is a many-body local density based on three-well potential, which indicates amino acid preferences for a particular coordination density,  , where
, where 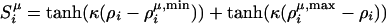 , where (μ = 1, 2, or 3), are indicative whether the particular residue i is found in low, medium, or high local density environment. The intervals for these three wells were defined from zero to three, three to six, and six to nine residues (i.e., the potentials becomes zero when the coordination number is increased beyond nine).
, where (μ = 1, 2, or 3), are indicative whether the particular residue i is found in low, medium, or high local density environment. The intervals for these three wells were defined from zero to three, three to six, and six to nine residues (i.e., the potentials becomes zero when the coordination number is increased beyond nine).
Optimization of the AM/W-0 Potential. The training set consists of 156 proteins from a database of Banavar and colleagues (34). In AM/W-0 the 210 parameters for the first-well interactions, the second-well through-protein interactions, and the second-well through-water interactions were found with a sequence shuffling protocol as described (13). These were scaled to the magnitude of the AM/C interactions.
The 60 parameters (20 amino acids × 3 wells) of the many-body burial profile were obtained from the same database of monomeric proteins (34) by calculating the frequency of occurrence of a particular coordination number within a 3-Å interval for each of the 20 amino acids, and the logarithms of the corresponding frequencies were taken.
Optimization of the AM/W-1 Potential. Our self-consistent structural-based optimization strategy maximizes the TF/TG (folding temperature/glass transition temperature) by using ideas from the energy landscape theory (30). For each of the nine training proteins, molten globules and native ensembles were generated with the AM/W-0 potential by using long constant temperature runs (1.26 × 106 time steps) at the corresponding equilibration temperatures with a Q-constrained potential.
The Hamiltonian is linear H = Σiγiεi, where εis are real interaction terms in the Hamiltonian described previously. The γs are parameters that scale these terms. Our previously described variational optimization procedure based on the energy landscape theory is used to maximize TF/TG (30). We introduce two auxiliary mathematical objects, a vector  , which is indicative of the stability gap, and a matrix
, which is indicative of the stability gap, and a matrix  , which characterizes the excess ruggedness of the molten globule ensemble compared with the native ensemble. When TF and TG are computed in terms of A and B, then TF/TG optimization leads to γ ≈ γ0 + T * B-1A, where T is the simulation temperature (15, 30, 35).
, which characterizes the excess ruggedness of the molten globule ensemble compared with the native ensemble. When TF and TG are computed in terms of A and B, then TF/TG optimization leads to γ ≈ γ0 + T * B-1A, where T is the simulation temperature (15, 30, 35).
In the current work, the standard procedure was modified by using each training protein to generate its own Bγ ≈ Bγ0 + T* A equation. We used least-square singular value decomposition solution of an overdetermined set of equations for all proteins simultaneously (36). Robustness of the solution was tested by using Poisson noise and repeatedly (20 times) solving for γ with different random seeds. To prevent overlearning from the small training set we allowed only a modulation of the AM/W-0 potential with an hydrophobic/polar coarse-grained grouping of amino acid interactions (37).
All parameters that are new in AM/W-1 as compared with AM/C are given in Supporting Text, Fig. 6, and Tables 1–6, which are published as supporting information on the PNAS web site.
Training Proteins for AM/C and AM/W-1 Parameter Optimization. Nine α-helical proteins (PDB codes 1R69, 1UTG, 3ICB, 256BA, 4CPV, 1CCR, 2MHR, 1MBA, and 2FHA) were used for training.
Abbreviations: AM, associative memory; AM/C, AM/contact; AM/W, AM/water; RMSD, rms displacement; CE, combinatorial extension; PDB, Protein Data Bank; CASP, Critical Assessment of Techniques for Protein Structure Prediction.
See Commentary on page 3325.
References
- 1.Cheung, M. S., Garcia, A. E. & Onuchic, J. N. (2002) Proc. Natl. Acad. Sci. USA 99, 685-690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Head-Gordon, T. & Brown, S. (2003) Curr. Opin. Struct. Biol. 13, 160-167. [DOI] [PubMed] [Google Scholar]
- 3.Kaya, H. & Chan, H. S. (2003) J. Mol. Biol. 326, 911-931. [DOI] [PubMed] [Google Scholar]
- 4.Van der vaart, A., Bursulaya, B. D., Brooks, C. L. & Merz, K. M. (2000) J. Phys. Chem. B 104, 9554-9563. [Google Scholar]
- 5.Kauzmann, W. (1959) Adv. Protein Chem. 14, 1-59. [DOI] [PubMed] [Google Scholar]
- 6.Eisenberg, D., Weiss, R. M. & Terwilliger, T. C. (1984) Proc. Natl. Acad. Sci. USA 81, 140-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeGrado, W. F., Wasserman, Z. R. & Lear, J. D. (1989) Science 243, 622-628. [DOI] [PubMed] [Google Scholar]
- 8.Hardin, C., Pogorelov, T. V. & Luthey-Schulten, Z. (2002) Curr. Opin. Struct. Biol. 12, 176-181. [DOI] [PubMed] [Google Scholar]
- 9.Miyazawa, S. & Jernigan, R. L. (1996) J. Mol. Biol. 256, 623-644. [DOI] [PubMed] [Google Scholar]
- 10.Hummer, G., Garde, S., Garcia, A. E. & Pratt, L. R. (2000) Chem. Phys. 258, 349-370. [Google Scholar]
- 11.Pertsemlidis, A., Soper, A. K., Sorenson, J. M. & Head-Gordon, T. (1999) Proc. Natl. Acad. Sci. USA 96, 481-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papoian, G. A. & Wolynes, P. G. (2003) Biopolymers 68, 333-349. [DOI] [PubMed] [Google Scholar]
- 13.Papoian, G. A., Ulander, J. & Wolynes, P. G. (2003) J. Am. Chem. Soc. 125, 9170-9178. [DOI] [PubMed] [Google Scholar]
- 14.Friedrichs, M. S. & Wolynes, P. G. (1989) Science 246, 371-373. [DOI] [PubMed] [Google Scholar]
- 15.Hardin, C., Eastwood, M. P., Luthey-Schulten, Z. & Wolynes, P. G. (2000) Proc. Natl. Acad. Sci. USA 97, 14235-14240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eastwood, M. P., Hardin, C., Luthey-Schulten, Z. & Wolynes, P. G. (2001) IBM J. Res. Dev. 45, 475-497. [Google Scholar]
- 17.Evans, D. F. & Wennerström, H. (1999) The Colloidal Domain Where Physics, Chemistry, Biology, and Technology Meet (Wiley, New York).
- 18.Wales, D. J. & Scheraga, H. A. (1999) Science 285, 1368-1372. [DOI] [PubMed] [Google Scholar]
- 19.Shindyalov, I. N & Bourne, P. E. (1998) Protein Eng. 11, 739-747. [DOI] [PubMed] [Google Scholar]
- 20.Murzin, A. G. (1999) Proteins Struct. Funct. Genet. 37, Suppl. 3, 88-103. [DOI] [PubMed] [Google Scholar]
- 21.Yang, F., Gustafson, K. R., Boyd, M. R. & Wlodawer, A. (1998) Nat. Struct. Biol. 5, 763-764. [DOI] [PubMed] [Google Scholar]
- 22.Weigelt, J., Brown, S. E., Miles, C. S., Dixon, N. E. & Otting, G. (1999) Struct. Fold. Design 7, 681-690. [DOI] [PubMed] [Google Scholar]
- 23.Allen, M., Friedler, A., Schon, O. & Bycroft, M. (2002) J. Mol. Biol. 323, 411-416. [DOI] [PubMed] [Google Scholar]
- 24.Miller, D. J., Ouellette, N., Evdokimova, E., Savchenko, A., Edwards, A. & Anderson, W. F. (2003) Protein Sci. 12, 1432-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tramontano, A. (2003) Nat. Struct. Biol. 10, 87-90. [DOI] [PubMed] [Google Scholar]
- 26.Lawson, D. M., Artymiuk, P. J., Yewdall, S. J., Smith, J. M. A., Livingstone, J. C., Treffry, A., Luzzago, A., Levi, S., Arosio, P., Cesareni, G., et al. (1991) Nature 349, 541-544. [DOI] [PubMed] [Google Scholar]
- 27.Gajiwala, K. S. & Burley, S. K. (2000) J. Mol. Biol. 295, 605-612. [DOI] [PubMed] [Google Scholar]
- 28.Bryngelson, J. D. & Wolynes, P. G. (1987) Proc. Natl. Acad. Sci. USA 84, 7524-7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardin, C., Luthey-Schulten, Z. & Wolynes, P. G. (1999) Proteins 34, 281-294. [PubMed] [Google Scholar]
- 30.Eastwood, M. P., Luthey-Schulten, Z. & Wolynes, P. G. (2003) J. Chem. Phys. 118, 8500-8512. [Google Scholar]
- 31.Hardin, C., Eastwood, M. P., Prentiss, M., Luthey-Schulten, Z. & Wolynes, P. G. (2002) J. Comput. Chem. 23, 138-146. [DOI] [PubMed] [Google Scholar]
- 32.Eastwood, M. P., Hardin, C., Luthey-Schulten, Z. & Wolynes, P. G. (2002) J. Chem. Phys. 117, 4602-4615. [Google Scholar]
- 33.Kolinski, A., Skolnick, J., Godzik, A. & Hu, W. P. (1997) Proteins Struct. Funct. Genet. 27, 290-308. [PubMed] [Google Scholar]
- 34.Chang, I., Cieplak, M., Dima, R. I., Maritan, A. & Banavar, J. R. (2001) Proc. Natl. Acad. Sci. USA 98, 14350-14355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koretke, K. K., Luthey-Schulten, Z. & Wolynes, P. G. (1998) Proc. Natl. Acad. Sci. USA 95, 2932-2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Press, W. H., Teukolsky, S. A., Vetterling, W. T. & Flannery, B. P. (2002) Numerical Recipes in C++ (Cambridge Univ. Press, Cambridge, U.K.).
- 37.Wang, J. & Wang, W. (1999) Nat. Struct. Biol. 6, 1033-1038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.