

Abstract
Background
Familial British and Familial Danish dementias (FBD and FDD, respectively) are associated with mutations in the BRI2 gene. Processing of the mutated BRI2 protein leads to the accumulation in the brain of the 34-mer amyloid Bri (ABri) and amyloid Dan (ADan) peptides, accompanied by neurofibrillary tangles. Recently, transgenic mice successfully reproduced different aspects of FDD, while modeling of FBD in vivo has been more difficult. In this work we have modeled FBD and FDD in Drosophila and tested the hypothesis that ABri and ADan are differentially neurotoxic.
Results
By using site-directed insertion, we generated transgenic lines carrying ABri, ADan, Bri2-23 (the normal product of wild-type BRI2 processing) and amyloid-β (Aβ) 1–42 as a well-characterized neurotoxic peptide, alone or with a His-tag. Therefore, we avoided random insertion effects and were able to compare levels of accumulation accurately. Peptides were expressed with the GAL4-Upstream Activating Sequence (UAS) system using specific drivers. Despite low levels of expression, toxicity in the eye was characterized by mild disorganization of ommatidia and amyloid peptides accumulation. The highest toxicity was seen for ADan, followed by Aβ42 and ABri. Pan-neuronal expression in the CNS revealed an age-dependent toxicity of amyloid peptides as determined by the ability of flies to climb in a geotaxis paradigm when compared to Bri2-23. This effect was stronger for ADan, detected at 7 days post-eclosion, and followed by ABri and Aβ42, whose toxicity became evident after 15 and 21 days, respectively. Histological analysis showed mild vacuolization and thioflavine-S-negative deposits of amyloid peptides. In contrast, the over-expression of amyloid peptides in the specific subset of lateral neurons that control circadian locomotor activity showed no toxicity.
Conclusions
Our results support the differential neurotoxicity of ADan and ABri in the Drosophila eye and CNS at low expression levels. Such differences may be partially attributed to rates of aggregation and accumulation. In the CNS, both peptides appear to be more neurotoxic than wild-type Aβ42. These Drosophila models will allow a systematic and unambiguous comparison of differences and similarities in the mechanisms of toxicity of diverse amyloid peptides associated with dementia.
Keywords: Alzheimer’s disease, Familial British dementia, Familial Danish dementia, ABri, ADan, Neurotoxicity, Drosophila
Background
Familial British Dementia (FBD) and Familial Danish Dementia (FDD) are autosomal dominant neurodegenerative disorders associated with mutations in the BRI2 gene on chromosome 13 (also named ITM2B) [1,2]. FBD usually starts in the fifth decade of life with progressive dementia, spasticity and ataxia leading to death in ~9 years [3]. FDD starts earlier, before 30 years of age with cataracts, visual loss followed by impaired hearing, progressive cerebellar ataxia and late dementia. Patients die within the sixth-seventh decade of life [4]. A prominent neuropathological finding in FBD and FDD patients is the accumulation of amyloid proteins in the walls of small arteries with a widespread distribution. This includes the cerebral cortex, leptomeninges, cerebellum, brain stem and white matter [5,6]. In addition, parenchymal amyloid deposits and neurofibrillary tangles are consistently found with a notorious and severe compromise of the hippocampus. In this regard, FBD and FDD (together with some cases of hereditary prionoses) are closely similar to Alzheimer’s disease (AD), the major cause of dementia in aging populations. A further similarity between FBD, FDD and AD is that amyloid deposits are made of short peptides generated in the brain by internal proteolysis of larger transmembrane precursor proteins. These peptides of ~4 kDa, are: amyloid β (Aβ) in AD [7,8], ABri in FBD and ADan in FDD [1,2]. In FBD, a missense mutation at the BRI2 stop codon leads to the generation of the ABri peptide sequence [1]. In FDD, a 10-nucleotide duplication insertion causes a frame shift and the generation of the ADan sequence [2]. Both 34-residue peptides (ABri and ADan) and the normal peptide product of wild-type BRI2 (Bri2-23) are released by furin and other subtilisin/kexin-like proprotein convertases (PCs) by cleavage of the BRI2 carboxyl-terminus along the secretory pathway [9].
Processing of BRI2 seems to be complex, involving several proteases in addition to PCs. The pro-protein and mature BRI2 (m-BRI2) protein may also be cleaved by a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10), releasing the Brichos domain [10] and an N-terminal fragment (NTF). The NTF is also the subject of additional proteolysis by signal peptide peptidase-like 2 (SPPL2), releasing an intracellular domain (ICD) and the BRI2 C-peptide (Figure 1) [11]. To date, the biological roles of BRI2 and the pro-peptide Bri2-23 have not been elucidated. In vitro studies showed that oligomers of ABri and ADan are toxic to neuronal cell lines [12,13].
Figure 1.
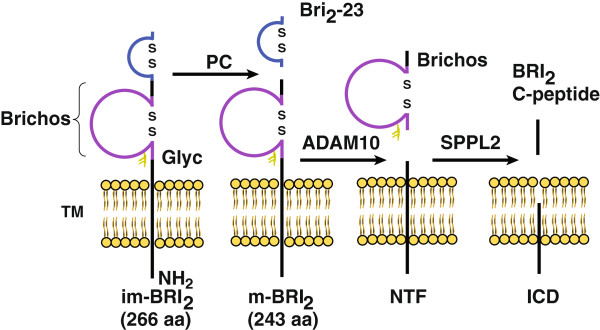
Proteolytic processing of BRI2. Schematic diagram representing the proteolytic events of BRI2 processing. Proprotein convertases (PC) release a C-terminal 23-residue peptide (Bri2-23). Cleavage by ADAM10 of the BRI2 ectodomain releases the Brichos domain to the extracellular space and leaves an N-terminal fragment (NTF) attached to the membrane. NTF is further processed by signal peptide peptidase-like 2 (SPPL2) generating an intracellular domain (ICD) and an extracellular BRI2 –C-terminal peptide (BRi2-C peptide). Instead of Bri2-23, cleavage of the mutant BRI2 by PC releases ABri in FBD or ADan in FDD, respectively (not depicted). im-BRI2, immature BRI2; m-BRI2, mature BRI2. Glyc, glycosylation; TM, trans-membrane; ICD, intracellular domain.
Recently, transgenic mouse models for FDD have been generated. The first reported line carries a mutant BRI2 under the mouse prion protein promoter and after 6 months of age shows extensive vascular deposition, parenchymal ADan accumulation, gliosis and an increase of phosphorylated-tau immunoreactivity [14]. When this transgenic animal was crossed with tau-P301S transgenic mice (Tg-Tau P301S), there was an increase of tau accumulation, phosphorylation and caspase cleavage of tau at Asp421 [15]. A knock-in (KI) mouse, carrying the FDD mutation in endogenous BRI2 has also been generated although it did not show detectable brain abnormalities [16]. In addition, two other transgenic lines that overexpress BRI2 containing the FDD mutation have been produced [17]. The FDD-like line with higher expression displays ADan accumulation in the hippocampus and meningeal vessels after 2 months of age with a marked age-dependent increase in amyloid deposition, particularly in the microvasculature. In addition, when crossed with Tg-tau P301S mice, these animals show a significant increment in the accumulation of hyperphosphorylated tau as compared to the Tg-tau P301S alone. Therefore, mouse models of FDD that overexpress mutant BRI2 in the brain recapitulate several key features of the human disease.
Regarding FBD, the development of a transgenic animal model reproducing basic lesions of the disease has been more elusive. Lines of transgenic mice carrying the FBD mutation have been generated and despite high levels of mutant BRI2 expression, no brain pathology was detected. Moreover, ABri peptide did not accumulate in the brain and only minimal amounts were detected by immunoprecipitation even after exogenous furin overexpression [18]. A second approach, using the KI strategy, showed that mice carrying the FBD mutation in one mouse BRI2 allele developed a significant deficit in hippocampal-dependent memory tasks by the age of 9 months without neuropathology or ABri deposition [19]. However, a reduction in BRI2 was detected in synaptic vesicles as compared to wild-type mice. These results together with the finding of similar memory deficits in BRI2 haplo-deficient mice (Bri2+/-) and lower levels of BRI2 in FBD brains has led to the hypothesis that loss of function of BRI2 may be a contributing factor to the development of dementia in FBD patients [19]. Other studies have reported that BRI2 may modulate amyloid β precursor protein (AβPP) processing in transfected cells and in transgenic mice leading to a reduced Aβ production and aggregation [20,21].
Drosophila melanogaster has been extensively used to reproduce basic aspects of several neurodegenerative diseases including AD, Parkinson disease, frontotemporal dementia, polyglutamine diseases, non-coding trinucleotide repeat expansion disease, amyotrophic lateral sclerosis and prion diseases [22-25]. The first fly model for AD was designed to over-express human AβPP, human β-secretase and presenilin1 mutants associated with familial AD. These animals showed Aβ accumulation and shortened life span [26]. Other groups have generated transgenic flies targeting Aβ directly to the secretory pathway. These flies developed age-dependent amyloid accumulation and reduced life span accompanied by defects in locomotion, memory and learning [27-30]. Further studies of these lines showed that Aβ caused defects in axonal transport, synaptic integrity and mitochondrial mislocalization [31-33].
In this study we report the generation and characterization of transgenic flies that over-express ABri, ADan and the normal product of BRI2, Bri2-23, by site-directed insertion. For comparison, a line expressing Aβ1-42 was also generated. Although the toxicity of wild-type Aβ and Aβ mutants in flies has been extensively described [26-41], the strategy used in this study included Aβ as a reference for comparison with ABri and ADan, for which no Drosophila models have been reported. Our results support the neurotoxicity of both FBD and FDD-associated peptides in contrast to Bri2-23. In addition, important differences were appreciated in terms of degree of toxicity and vulnerability of neuronal types. Moreover, this is the first report of the toxic effects of ABri expression in an animal model.
Results and discussion
Generation of transgenic lines
To examine and compare the effects of ABri, ADan and Aβ42 over-expression in Drosophila, we generated transgenic lines carrying the sequence of each amyloid peptide fused to the signal peptide of Drosophila Necrotic (Nec) that targets the protein to the secretory pathway This strategy has been used successfully with Aβ [29] and circumvents possible difficulties due to an insufficient processing of BRI2 (particularly in the case of ABri, as suggested by the mouse models). To avoid positional effects associated with random insertion in the genome, we used the φ-recombinase-based system [42] to direct insertion of transgenes at a specific site on chromosome 3. Since there are no specific antibodies to Bri2-23, we generated a set of constructs with a 6 x His-tag at their amino-termini (Additional file 1). This allowed us to compare the levels of accumulation of each peptide by using the same antibody. All the peptides designed in this study are schematically shown in Figure 2A. PCRs with specific primers were used to assess the insertion of the correct cDNAs (Figure 2B). The “site-directed” strategy allows a better comparison of the effect caused by each peptide because the levels of mRNA expression should be similar. Accordingly, Quantitative Real-Time PCR (QRT-PCR) using the same set of primers showed no significant differences in the expression levels of Bri2-23, ABri and ADan mRNAs (Figure 2C).
Figure 2.
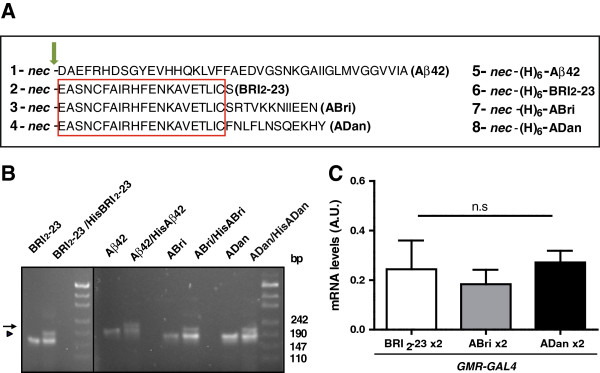
Generation of transgenic lines with similar expression levels. A, schematic representation of the fusion peptides used to generate transgenic flies. Amino-acid single-letter code is used. Nec, the signal peptide of necrotic (MASKVSILLLLTVHLLAAQTFAQ); (H)6 indicates the 6 × His-tag. The arrow indicates the site of cleavage in the secretory pathway. The rectangle encloses the sequence shared by Bri2-23, ABri and ADan. B, agarose gels showing the PCR products obtained from genomic DNA of transgenic lines carrying one copy of untagged (arrowheads) or two copies: one untagged and the other with a 6 × His-tagged peptide DNAs (arrows). On the right, molecular markers in base-pairs (bp). C, similar levels of mRNA expression of Bri2-23, ABri and ADan in the eye as determined by QRT-PCR normalized with tubulin mRNA. Bars represent the mean ± SEM of 3 independent experiments. A.U., arbitrary units.
Expression and processing of Bri2-23, Aβ42, ABri and ADan
To examine whether the fusion peptides were correctly targeted and cleaved to release two copies of Bri2-23, Aβ42, ABri and ADan, peptides were expressed in the eye using the Glass Multiple Reporter (GMR-GAL4) driver and the GAL4-UAS bipartite system. Ten days post-eclosion (p.e), homogenates from transgenic and control fly heads were analyzed by Western blots. In the GMR-GAL4/HisBri2-23 line, anti-His showed a very faint band of ~3 kDa (at the limit of detection) compatible with a properly processed Bri2-23 (Figure 3A). The Aβ42 monomer was clearly detected with monoclonal 6E10 antibody (Figure 3B). Regarding ABri, a specific band with the size of a monomer was detected with anti-ABri antibody (Figure 3C). The processing of ADan was assessed with a specific antibody directed to the peptide carboxyl-terminus, which showed a 4 kDa band consistent with ADan monomer (Figure 3D). It is of note that differently from other reports using Aβ-transgenic Drosophila, mouse models of ADan or brain tissue from FBD and FDD patients, no SDS-resistant high order oligomeric species were detected in our transgenic lines (Additional file 2). In addition, the bulk of amyloid peptides were solubilized in buffer containing 1% Triton X-100 and 1% SDS, while only traces of Aβ42 and ADan remained as a formic-soluble fraction (Additional file 2). These results suggest that the proteins are correctly processed along the secretory pathway, generating mostly soluble peptides with small amounts of insoluble aggregates.
Figure 3.

Amyloid peptides are properly processed in Drosophila eye. RIPA-soluble proteins extracted from flies expressing two copies of each peptide in the eye using the GMR-GAL4 driver. A, immunoreactivity with anti-His showing a very faint 3 kDa band consistent with HisBri2-23 monomer (arrowhead). B, Western blot with 6E10 showing the 4.5 kDa monomeric Aβ42 in transgenic line (arrowhead). Synthetic (sp) Aβ42 was added to wild type Drosophila protein extract to compare molecular masses. C, immunoreactivity using anti-ABri revealed a specific band with the expected sizes of a monomer (arrowhead); on the right, synthetic (sp) ABri peptide added to the protein extract from a non-transgenic fly. D, Western blot using anti-ADan show the presence of a band (arrowhead) consistent with ADan monomer in the transgenic line. Wild type Drosophila protein extract was spiked with sp ADan peptide for comparison. M, monomers. CNT, control flies (GMR-GAL4/+). In all lanes, RIPA-soluble proteins from 15 head homogenates (150 μg) were loaded. Representative gels from more than 3 independent experiments for each genotype are shown.
Toxicity of Aβ42, ABri and ADan in the eye
At 10 days p.e, eyes were morphologically examined for toxicity by light microscopy and scanning electron microscopy (ESEM). When one or two copies of Aβ42, ABri and ADan were expressed in the eye using GMR-GAL4 and flies maintained at 25°C, no toxic effects were detected (not shown). By raising temperature to 28°C, an expected mild toxicity was seen in heterozygous GMR-GAL4/+ lines, as reported [43]. However, such effect was greatly reduced in the GMR-GAL4/Bri2-23 line, likely due to titration of free GAL4 to the UAS repeats available for GAL4 binding (Additional file 3). Therefore, we used GMR-GAL4/Bri2-23 as a control to compare the effects of Aβ42, ABri and ADan expression. The eyes from GMR-GAL4/Bri2-23 flies showed few scattered ommatidia of different sizes, but this defect was much lower than the disorganization seen in the amyloid-expressing lines. Light microscopy (not shown) and ESEM micrographs revealed a pattern of ommatidia disarray in the amyloid- expressing eyes related to BRI2-23 (Figure 4A). The penetrance of these phenotypes was 90-100% in all cases. Since the phenotype was mild, the extent of toxicity was assessed by counting the number of ommatidia, ommatidia fusions and number of bristles in a representative central area of the eye for each genotype. The number of fused ommatidia was significantly higher for ADan, Aβ42 and ABri as compared to BRI2-23 (Figure 4B) while the number of bristles per ommatidia was significantly lower in flies expressing the amyloid peptides as compared to control (Figure 4C). In addition, there was a significant loss of ommatidia in Aβ42 and ADan-expressing lines as compared to BRI2-23 (Additional file 3). Overall, the degree of toxicity reflected as ommatidia size heterogeneity and fusion tended to be higher for ADan and Aβ42 relative to ABri. These phenotypes obtained with Aβ42, ABri and ADan in our system were subtle as compared to the typical full-rough eye as reported for flies expressing high levels of Aβ [37]. When we compared our Aβ42 line with the reference line Aβ42 Hj2.12, the latter presented ~2.5-fold higher Aβ42 accumulation as measured by ELISA (7.5 vs 17.5 ng/mg total protein, respectively) and a more severe eye toxicity (Additional file 3), indicating that the “site-directed lines” used here have relatively low levels of expression, and yet, a toxic effect in the eye can be detected.
Figure 4.

Differential toxicity of Aβ42, ABri and ADan in the eye. A, morphological analysis of eye integrity from flies expressing Bri2-23, Aβ42, ABri and ADan. Top panel, ESEM microphotographs indicating the selected area (135 × 135 μm) used to count the number of ommatidia and bristles.The area used for quantification of fused ommatidia was slightly larger (195 × 170 μm) and it is not shown for clarity. Bottom panel, higher magnification of the selected regions as delineated in the top panel. Fused ommatidia are indicated with arrows. Bar scales, 100 μm and 50 μm for top and bottom panels, respectively. B, quantification of fused ommatidia in the selected area for each genotype. C, quantification of bristles in the selected area for each genotype. One- way ANOVA followed by Dunnett’s comparison test analysis was performed. Eight eyes per genotype, randomly taken from 5 independent biological experiments were analyzed. Data are expressed as mean ± SEM. Asterisks indicate statistically significant differences (*p < 0.05, **p < 0.01).
In order to examine the accumulation levels of amyloid peptides in vivo in a comparable way, two copies of the His-tagged constructs were expressed in the eye and assessed by Western blots using anti-His. As shown in Figure 5, the reactivity of HisADan and HisAβ42 were ~4 fold and ~3 fold higher than HisABri, respectively.
Figure 5.

Analysis of His-tagged amyloid protein levels. A, Western blots of fly head homogenates expressing two copies of His-tagged peptides in the eye. Top panel, anti-His shows different levels of monomeric HisAβ42, HisABri and HisADan accumulation in transgenic flies after 10 days p.e. In this experiment, the expression levels of HisBri2-23 were below the limit of detection. Bottom panel, Western blot of the same membrane re-probed with anti-actin antibody for loading normalization. B, quantification of HisAβ42 and HisADan levels relative to HisABri. At least 3 independent experiments were performed. A.U., arbitrary units.
To evaluate the possible effect of the His tag upon amyloid peptides, transgenic lines carrying two copies of tagged or untagged peptides were analyzed by Western blots using amyloid-specific antibodies (Additional file 4). In the case of HisAβ42, there was a ~3 fold increase as compared to Aβ42 while there were no differences between ADan and HisADan. The effect upon ABri could not be accurately measured due to the very low signal and high background of the blots. These results raise the possibility that, at least for ADan (for which the His tag had no effect) its levels of accumulation may correlate with toxicity, as suggested for Aβ42. Further in vivo and in vitro studies may help to clarify the mechanisms by which the His tag imposes differential effects on amyloid peptides accumulation.
Aβ42, ABri and ADan show different patterns of non-fibrillar deposition in the eye
Histological analysis of retina paraffin sections stained with hematoxilin and eosin (H&E) were consistent with the eye external phenotype, showing mild disorganization and heterogeneity in the sizes of ommatidia in GMR-GAL4/Aβ42, GMR-GAL4/ABri and GMR-GAL4/ADan as compared to GMR-GAL4/ Bri2-23 (Figure 6A).
Figure 6.

Accumulation of amyloid peptides in the retina. A, paraffin sections of the retina stained with H&E showing disorganization of ommatidia (arrows). Scale bar = 25 μm. B, horizontal sections from the retina of transgenic lines: GMR-GAL4/Aβ42 × 2, GMR-GAL4/ABri × 2 andGMR-GAL4/ADan x 2 were immunostained with 6E10, anti-ABri and anti-ADan, respectively and sections from GMR-GAL4/Bri2-23 x 2 control lines probed with the amyloid peptide-specific antibodies. Arrows indicate immunoreactivity consistent with amyloid peptide deposits. Images represent the results of at least 2 independent biological experiments. Scale bar = 20 μm.
The accumulation of Aβ42, ABri and ADan in the eye (expressing two copies of each peptide under GMR-GAL4) was studied by immunohistochemistry with specific antibodies on paraffin sections of the fly heads (Figure 6B). Specificity of staining was assessed by using 6E10, anti-ABri and anti-ADan antibodies upon eye sections of GMR-GAL4/Bri2-23 lines and unrelated IgG as primary antibodies. Aβ42 staining showed deposits in the retina following rhabdomers topology that were more abundant toward the basal ommatidial region. ABri immunoreactivity was scattered, showing discrete deposits in ommatidia close to the lamina different from ADan distribution that was mainly along the inner ommatidial rhabdomers with a stronger immunoreactivity. Thioflavine-S (ThS) staining of similar sections was negative for all peptides (not shown). These findings suggest that in the Drosophila eye, and despite similar levels of mRNA transcription, non-fibrillar Aβ42, ABri and ADan induced different degrees of toxicity, possibly related to different levels of accumulation and distribution.
Toxicity of Aβ42, ABri and ADan in the CNS
To study Aβ42, ABri and ADan neurotoxicity in the CNS, peptides were expressed with the pan-neuronal driver elav-GAL4. Lines expressing Bri2-23 were used as negative controls and two behavioral paradigms were tested; negative geotaxis (to evaluate locomotor coordination) and positive phototaxis (to evaluate response to light). In the geotaxis assay, flies expressing one copy of each transgene and kept at 25°C showed no climbing defects (not shown). However, flies expressing two copies of amyloid peptides and kept at 28°C after eclosion showed behavioral defects that worsened with age (Figure 7A). After 7 days p.e, ADan-expressing flies displayed a significant impairment in climbing ability (in cm) compared to Bri2-23 (2.5 ± 0.7 vs 4.6 ± 0.2, respectively, p < 0.001) while in Aβ42 (4.6 ± 0.2) and ABri (3.8 ± 0.2) there was no detectable toxicity. After 15 days, flies expressing ABri showed a significant toxicity compared to Bri2-23 (2.3 ± 0.3 vs 3.8 ± 0.1 respectively, p < 0.01), while Aβ42 effect was only evident after 21 days as compared to Bri2-23 (1.5 ± 0.3 vs 3 ± 0.2, respectively, p < 0.01). Therefore, the three amyloid peptides were capable of causing a significant worsening of climbing ability in the geotaxis assay. However, Aβ42 and ABri neurotoxicity was rather mild and strongly age-dependent, as reported for other Aβ Drosophila models [32,33]. ADan, in turn, was highly toxic at the first time-point analyzed. In the phototaxis assay, flies expressing one copy of each peptide and kept at 25°C showed that only ADan was capable of exerting a significant impairment in 25 days-old flies (p < 0.05) reinforcing the higher toxicity of this peptide (Figure 7B and Additional file 5). These results were unlikely due to locomotor defects since flies expressing one copy of amyloid peptides under the same conditions showed no deficits in climbing assays, as mentioned above. Next, a specific group of neurons was tested for their vulnerability to amyloid peptides. The small and large lateral ventral neurons is a key subgroup of neurons that produce the neuropeptide pigment dispersing factor (pdf) and control circadian locomotor activity. We used the pdf-GAL4 driver to direct expression of two copies of Bri2-23, ABri, ADan and Aβ42 to these cells. Neurodegeneration of pdf-neurons results in a dysregulation of the circadian rhythm with aging [44]. The analysis of circadian locomotor activity in flies expressing amyloid peptides showed normal rhythm and period both in young (5-days old) and aged (21-days old) individuals as compared to pdf-GAL4/ Bri2-23 or pdf-GAL4/+ (Additional file 6). This result was interesting although not unexpected, according to a previous report in which no circadian behavioral phenotype was observed in pdf neurons expressing the Arctic Aβ mutant [45]. Taken together, our behavioral experiments point at a different vulnerability of neuronal populations to amyloid peptides toxicity.
Figure 7.
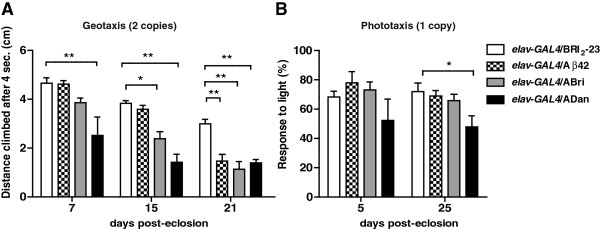
Age and amyloid peptide-dependent toxicity in the CNS. Pan-neuronal transgene expression was induced using the elav-GAL4 driver. A, negative geotaxis assays were performed at the indicated times with flies expressing two copies of the transgenes. The slight decline in climbing performance with age in non-transgenic flies has been reported [reviewed in 36]. ADan expression was associated with reduced climbing at 7 days of age while the toxic effect of ABri and Aβ42 was seen at 15 and 21 days, respectively. Climbing was expressed in cm as the mean ± SEM. Data from 5 technical replicates from each of 3 independent biological experiments per genotype were analyzed. B, phototaxis assays were performed at the indicated times with flies expressing one copy of each transgene. At 25 days p.e, flies expressing ADan had a lower light response in comparison to Bri2-23, Aβ42 or ABri. Results from 2 technical replicates from each of 3 independent biological experiments per genotype are shown. Phototaxis was expressed as the percentage (mean ± SEM) of flies that responded to light. Asterisks indicate statistically significant differences, (*p < 0.05; ** p < 0.01; *** p < 0.001). Geotaxis and Phototaxis data were statistically analyzed by two-way repeated measures ANOVA, followed by Bonferroni’s comparison test for all genotypes and time points which are shown in Additional file 5.
Histological analysis and immunostaining in the CNS
To assess the effect of amyloid accumulation on the integrity of the CNS, two copies of each peptide were expressed under the pan-neuronal driver elav-GAL4 and brain paraffin sections stained with H&E were examined (Figure 8A) under light microscopy. Specifically, we looked at the degree of vacuolization, a widely used parameter of neuronal death [46,47]. Amyloid peptides expression increased vacuolization, quantified as the loss of tissue area in cortical neurons and neuropil that was significantly higher for Aβ42 as compared to Bri2-23 (p < 0.001) while ABri and ADan showed a non-significant tendency (Figure 8B). Next, the accumulation of amyloid peptides in the brain was assessed by immunohistochemistry with specific antibodies using elav-GAL4/Bri2-23 flies as negative controls. In the three cases, immunostaining was detected in the cortex (Figure 9). Aβ42 showed a highly discrete and specific labeling that was consistent with intracellular accumulation, as described [29]. ABri and ADan distribution was much more widespread, compatible with intra and extracellular accumulation. These features may explain the apparent contradiction between the extent of tissue loss and the degree of neurotoxicity among the peptides. ABri and ADan soluble species, with a more diffused distribution than Aβ42, may induce an earlier synaptic dysfunction (reflected in behavioral defects) in contrast to localized neuronal death. Moreover, brain paraffin sections showing a negative staining with ThS support the non-fibrillar form of deposition of the three amyloid peptides (Additional file 7).
Figure 8.
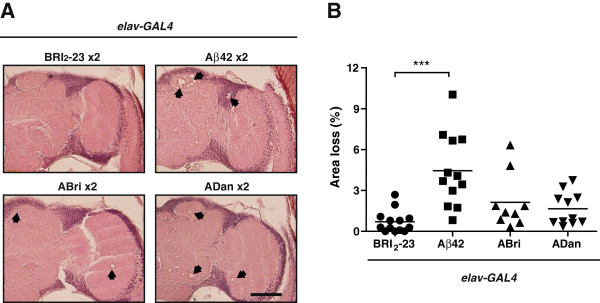
Histological analysis of fly brains. A, frontal sections of 21-day-old flies were stained with H&E and examined by bright-field microscopy. Vacuolization (>3 μm) of brain tissue was significantly higher in lines expressing Aβ42 as compared to Bri2-23, ABri and ADan. B, percentage of the area of tissue loss expressed as mean ± SEM. All lines expressed 2 copies of each peptide and 8–10 hemispheres per genotype were used for quantification. Asterisks indicate significant differences (One-way ANOVA, followed by Dunnett’s multiple comparison test). Scale bar = 50 μm.
Figure 9.
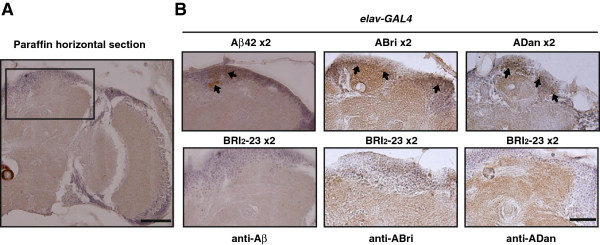
Specific accumulation of amyloid peptides in the Drosophila CNS. A, low magnification micrograph of brain section indicating the selected area from which immunohistochemistry is shown. B, amyloid deposits in the CNS were assessed with specific anti-amyloid peptide antibodies in lines expressing 2 copies of each transgene. Microphotographs of frontal paraffin sections showed discrete aggregates of Aβ42 and more widespread ABri and ADan deposits which were detected mainly in the supraesophagical brain, both in neurons and neuropil (arrows). No staining was detected in Bri2-23 line, used as a negative control for antibody specificity. Representative images from 3 independent biological experiments are shown. Scale bar = 50 μm.
Conclusions
Our results describe a novel Drosophila model of mutant peptides associated with human dementias, in which the major neuropathological hallmarks are amyloid deposits and neurofibrillary tangles. In the case of ADan, the fly model may contribute to recently reported studies in transgenic mice by increasing our understanding of the molecular mechanism associated with ADan toxicity and the pathogenesis of FDD. Regarding ABri, this is the first animal model in which neurotoxicity is directly tested in vivo.
Although low levels of expression of the transgenes may be a drawback for an easy detection of amyloid peptide accumulation, they allow a more reliable measure of toxicity without artifacts associated with acute and high over-expression. Importantly, despite the relatively low levels of expression of our transgenes, we were able to detect toxicity in the eye and in the CNS of flies. The phenotypes, as expected, were mild as compared to published data on wild-type Aβ and Aβ mutants, and yet, sufficient to detect important similarities and differences regarding the degree of toxicity and accumulation among Aβ42, ABri and ADan. Both in the CNS and in the eye, ADan was more toxic than Aβ42 and ABri, possibly due to a faster rate of aggregation. In the CNS, ABri appeared to be more toxic than Aβ42, but the relationship between peptide levels was not straightforward, suggesting additional factors involved such as conformation or differential neuronal vulnerability. The solubility, the pattern of deposition, and the fact that all deposits in the brain and the eye were not stained by ThS, strongly suggest that toxicity of the three amyloid peptides was likely related to the accretion of soluble species. Our data on the Drosophila models correlate well with recent work on a double transgenic mouse model for FDD, in which enhanced tauopathy, tau truncation, and synaptic loss occurs prior to any detectable ADan deposition [15].
In summary, the Drosophila lines presented in this paper support the neurotoxicity of ABri and confirm the toxicity of ADan and Aβ42, suggesting that this effect may be important in the pathogenesis of human dementia associated with the slow and relentless accumulation of amyloid peptides.
Methods
Generation of transgenic lines and stocks
The Bri2-23, ABri and ADan cDNAs were obtained by PCR from BRI2 mutant templates [14,48]. Aβ42 cDNAs was amplified from human AβPP-CT99 fragment cloned in pGEX. The cDNA of necrotic protein signal peptide MASKVSILLLLTVHLLAAQTFAQ was obtained from the pOT Nec (GH10112). The primers’ sequences and overall strategy to generate fusion constructs are described in Additional file 1. Constructs were subcloned in the pUAST- attB expression vector. Transgenic flies were obtained by injection of embryos from line ΦX-86Fb (y w M {eGFP.vas-int. Dm} ZH-2A; +; M {RFP. attP} ZH-86Fb; +) with constructs cloned in pUAST- attB, and therefore integration took place at a specific site on chromosome 3 [42]. The GMR-GAL4 and elav c155 -GAL4 lines were obtained from Bloomington Stock Center. The Hj2.12 line was kindly provided by Dr. Mary Konsolaki. This line has several UAS-Aβ42 randomly inserted on chromosome 2 (personal communication). All crosses and maintenance of flies were done at 25°C or 28°C as indicated.
Peptides and antibodies
Synthetic Aβ1-42 was obtained from American Peptide Co. Synthetic ABri was a generous gift of Prof. Jorge Ghiso, New York University. Monoclonal anti-Aβ 6E10 was from Signet. Monoclonal antibodies anti-His were obtained from Abcam, Cambridge, MA and Qiagen, Valencia, CA. Polyclonal antisera anti-ADan and anti-ABri were generated as reported before [14].
DNA extraction and PCR
Thirty flies were homogenized in 400 μl of Buffer A (100 mM Tris–HCl, pH 7.5, 100 mM EDTA,100 mM NaCl, 0.5% SDS) and incubated at 65°C for 30 min. Eight hundred μl of a 5 M KOAc: 6 M LiCl (1:2.5) solution were added and left 10 min on ice. After centrifugation at 14,000 rpm for 15 min, 700 μl of isopropanol were added per ml of supernatant. Samples were centrifuged at 14,000 rpm for 15 min, the pellet washed with cold ethanol and resuspended in water. PCR reactions were done using KAPA Taq (Kapa Biosystems, Woburn, MA) and run on a Bio-Rad cycler (Bio-Rad, Hercules, CA). The following primers, which are described in Additional file 1, were used: forward, Br_F1b and reverse Br_R2 for BRI2-23, Br_R3 for ABri, Br_R5 for ADan and R-Aβ42 for Aβ42.
RNA extraction and quantitative real time PCR analysis
For each experiment, 30–40 flies carrying two copies of each transgene were collected and frozen. Heads were mechanically isolated and total RNA extracted using Trizol (Invitrogen Carlsbad, USA) according to the manufacturer’s protocol. An additional centrifugation step at 11,000 × g for 10 min was used to remove the cuticles prior to the addition of chloroform. The concentration of total RNA purified from each sample was measured with a Nanodrop 2000 spectrophotometer (Thermo Scientific Walthman, MA). One to 2.5 μg of total RNA were digested with DNAse I (Promega Madison, WI) immediately followed by reverse transcription using the Superscript II system (Invitrogen, Carlsbad, USA) with oligo (dT) primers. PCR reactions were done using KAPA SYBR* FAST qPCR Master Mix (2×) from KAPA BIOSYSTEMS (Woburn, MA) following the manufacturers’ instructions. Reactions were run in an Mx3005P Cycler (Stratagene, Santa Clara, CA). Data were analyzed using MxPRO-Mx3005P software. The primers used for Nec-Bri2-23, Nec-ABri and Nec-ADan transgenes were directed to: 1) the Nec sequence (forward: 5′-ATA CGA ATT CAT GGC GAG CAA AG-3′); and 2) a common sequence for the three peptides, C-terminal of Bri2-23 (reverse 5′-GTT TCCACG GCA AAT TTG TT-3′). For tubulin cDNA amplification, the following primers were used: forward 5′-GCC TGA ACA TAG CGG TGA AC-3′ and reverse 5′-ATC CCC AAC AAC GTG AAG AC-3′.
Tissue processing, SDS-PAGE and Western blots
Fly heads were homogenized in RIPA buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, containing 0.5% sodium deoxycholate, 1% Triton X-100), 1% SDS, 5 mM EDTA, 5 mM EGTA, 5 μg/ml leupeptin, 10 μg/ml aprotinin, 1 μg/ml pepstatin, 50 mM sodium fluoride and 5 mM sodium orthovanadate. After sonication, homogenates were centrifuged at 14,000 rpm for 1 h. RIPA insoluble pellets were homogenized in 90% formic acid (FA) followed by centrifugation as described above. FA was evaporated by Speed Vac (Savant, SC100) and RIPA and FA-soluble proteins were re-suspended in sample buffer containing 0.1 M dithiothreitol. Protein extracts were separated on 12.5 or 15% Tris-Tricine gels and transferred onto PVDF membranes (GE Bioscience, Piscataway, NJ). Membranes were incubated with 6E10 monoclonal antibody (1/1000), anti-ABri (1/500) anti-ADan (1/1000) or a mixture of both monoclonal anti-His (1/500 each Qiagen and Abcam). Anti-actin (Sigma) was used for protein loading normalization. Immunoreactivity was detected with anti-rabbit or anti-mouse horseradish peroxidase-labeled IgG (Dako Denmark) and enhanced chemiluminescence ECL Plus (GE Bioscience Piscataway, NJ). Immunoblots were scanned with Storm 840 and band intensities quantified with ImageQuant 5.1 software (GE Bioscience Piscataway, NJ).
Eye toxicity
Flies expressing one or two copies of the peptides in the eye (GMR-GAL4) were raised at 25°C or 28°C and aged for 10 days before examination under light microscope. At least 100 eyes per genotype were analyzed from 5 independent biological experiments. Due to the high penetrance of phenotypes (90-100%), 8–10 eyes per genotype were randomly selected for Environmental Scanning Electron Microscopy (ESEM) analysis. Adult flies were immobilized on the ESEM mount using water-based colloidal carbon glue for proper orientation. The electro-scan was performed with an ESEM microscope model XL30 (Philips) at 20.0 kV and 0.9 Torr in the auxiliary mode. This technology did not require metal coating of the specimen. To analyze toxicity in a quantitative manner, eyes were properly oriented and a comparable area for each genotype was selected. The number of ommatidia, fused ommatidia and bristles were also counted in the selected areas. Eight fly eyes were analyzed per genotype. All the counting and measurements were done as blind experiments.
Histology and immunostaining
Fly heads were fixed in Carnoy solution (60% ethanol, 30% chloroform, 10% acetic acid) at 4°C overnight and dehydrated in ethanol. Samples were treated for 30 min with butanol:ethanol (1:1), butanol:toluene (1:1), toluene, and finally soaked in toluene: paraffin (1:1) for 30 min at 65°C. After incubation for 2 h in pure paraffin, heads were embedded and cut in 7-μm serial frontal sections. After H&E staining, images were captured using an OLYMPUS B × 50 Microscope and analyzed with the Image Pro Plus software (Media Cybernetics). Neurodegeneration was quantified and reported as the percentage of area lost in the tissue. The ratio was calculated by dividing the area of vacuolization (each vacuole with a diameter of at least 3 μm) by the total area of the brain section. At least 8 brains per genotype were analyzed. ThS staining was used for the detection of amyloid fibrillar deposits. The tissue was deparaffinized and incubated in 50% ethanol containing 1% ThS (Sigma St. Louis, MO) for 20 min. After washing in 50% ethanol and PBS, tissue sections were analyzed using a confocal Zeiss LSM510 microscope. Immunostaining was performed by incubating sections with anti-Aβ monoclonal 6E10 (1/500) and protein A-sepharose affinity-purified anti-ADan and anti-ABri antibodies (1/100). Immunoreactivity was detected with biotinylated anti-mouse or anti-rabbit IgG followed by incubation with avidin–biotin complex (Vector Laboratories, Burlingame, CA). The reaction products were visualized with 0.05% diaminobenzidine tetrahydrochloride, 0.01% hydrogen peroxide and, in some cases with nickel enhancement. Negative controls included incubation with primary unrelated antibodies and immunostainig of GAL4/Bri2-23 fly sections with amyloid peptides-specific antibodies.
Climbing assay (rapid iterative negative geotaxis, RING)
Flies expressing one or two copies of Bri2-23, Aβ42, ABri and ADan peptides in the CNS (with elav-GAL4) were raised at 25°C and incubated at 28°C in groups of 40 males in 4-inch glass vials with food replacement every 3–4 days. Vertical mobility was tested using the RING assay as described [49]. Briefly, the day before the assay 10 males per genotype were randomly selected under CO2. The following day, each group was transferred into empty vials without anesthesia and the vials were loaded into the RING apparatus. The apparatus was tapped three times in rapid succession to initiate a negative geotaxis response. After 4 sec, digital images were taken. The climbed distance in cm was measured for each fly and the average height per genotype calculated using the Scion Image software. Data from 5 technical replicates from each of 3 independent biological experiments per genotype were analyzed.
Phototaxis assay
Flies expressing one copy of Bri2-23, Aβ42, ABri and ADan in the CNS (with elav-GAL4) were raised and kept at 25°C. Phototaxis was performed as described [44]. Briefly, the day before the assay, 30 males per genotype were randomly selected under CO2. The following day, 15 min before testing, flies were transferred to darkness. Further manipulations were performed under red light. For the assay, flies were transferred to empty tubes and for 2 min they were able to move towards a collecting tube which had a white cold light at the end of it. The number of flies that moved to the collecting tube, towards the light (defined as a positive response) was counted and the results were expressed as the percentage of flies that responded to light of the total number of animals per assay. Data from 2 technical replicates from each of 3 independent biological experiments per genotype were analyzed.
Circadian locomotor activity
Fly activity was monitored as described [44]. Briefly, newly eclosed male flies were trained under 12 h light-darkness (LD) cycles until the beginning of the experiment. Then, young (5 days-old) and aged (21 days-old) flies were placed in glass tubes and monitored for activity with infrared detectors. Fly activity was monitored under LD conditions for 3 days and then released into constant darkness (DD) for at least one week employing commercially available activity monitors (TriKinetics, Waltham, MA). Period and rhythmicity were estimated using the ClockLab software (Actimetrics, Evanston, IL) from data collected under DD. Each experiment was repeated at least two times from two independent biological replicates.
ELISA
Five fly heads from our line GMR/Aβ42 carrying two copies of the transgene and 5 heads from the GMR/Hj2.12 line were homogenized and proteins extracted in 50 mM Tris–HCl, pH 8 containing 5 M guanidine and a cocktail of proteases inhibitors (Sigma St. Louis, MO). After centrifugation at 10,000 × g, proteins in the supernatant were quantified using a bicinchoninic acid kit (Thermo Scientific, Rockford, IL). Aβ42 concentration was determined using a commercial capture ELISA kit (Invitrogen Frederick, MD) following the manufacturer’s instructions.
Statistical analysis
Data were analyzed by Student’s t-test, One-way ANOVA, and Two-way repeated measures ANOVA, followed by post-hoc tests. Graph Pad Prism v.5 software was used. Statistical results were presented as means ± SEM. Asterisks indicate levels of significance (*p <0.05, **p <0.01 and ***p < 0.001).
Abbreviations
Aβ: Amyloid β; ABri: Amyloid Bri; ADan: Amyloid Dan; AD: Alzheimer’s disease; ADAM10: A disintegrin and metalloproteinase domain-containing protein 10; AβPP: Amyloid β-precursor protein; DD: Constant darkness; ELISA: Enzyme-linked immuno-sorbent assay; ESEM: Environmental scanning electron microscopy; FBD: Familial British dementia; FDD: Familial Danish dementia; GMR: Glass-multiple-reporter; H&E: Hematoxylin-eosin; ITM2B: Integral trans-membrane protein 2B; LD: Light-darkness; Nec: Necrotic; Bri2-23: The BRI2 wild-type derived peptide of 23 residues; PBS: Phosphate buffered saline; PBS-T: PBS containing 1% Triton X-100; PC: Pro-protein convertases; PCR: Polymerase chain reaction; pdf: Pigment-dispersing factor; p. e: Post-eclosion; PVDF: Polyvinylidene fluoride; QRT: Quantitative real time; RING: Rapid iterative negative geotaxis; SDS: Sodium dodecyl sulfate; sp: Synthetic peptide; ThS: Thioflavine S; UAS: Upstream-activator sequence.
Competing interests
The authors have no competing interests.
Authors’ contributions
MSM analyzed toxicity, performed histological analysis, Western blots and wrote the paper; AFG performed immunohistochemistry and Western blots; LAA generated and maintained transgenic lines, CR and OLP performed tissue sections and histological data analysis; LM, RV, MFC were involved in the experimental design; EMC designed experiments and wrote the paper. All authors read and approved the final manuscript.
Supplementary Material
Primers used for constructs generation. Table with the sequences of all primers used in this study and PCR strategy.
Western blots of amyloid peptides in SDS-insoluble factions from Drosophila eyes. Accumulation of Aβ42 and ADan in the formic acid-soluble fraction. Non-specific immunoreactive bands in Western blots from RIPA-soluble homogenates.
Toxicity in the Drosophila eye. Toxicity of GMR-GAL/+ as compared to GMR-GAL4/Bri2-23 lines at 28°C for 10 days; dose dependent Aβ42 toxicity in the eye and number of ommatidia in transgenic vs non-transgenic lines.
Accumulation of untagged and His-tagged amyloid peptides. Western blot anti specific peptides from fly heads expressing tag and untagged peptides.
Geotaxis and Phototaxis statistical analysis. Two-way repeated measures ANOVA, followed by comparison tests for all genotypes and time points.
pdf -neurons are resistant to amyloid peptides toxicity. Figure showing the circadian locomotor activity of transgenic flies.
Thioflavine S-negative accumulation of Aβ, ABri and ADan in the CNS of Drosophila. Figure showing negative Thioflavine-S staining of brain sections from Drosophila transgenic lines.
Contributor Information
María S Marcora, Email: mmarcora@leloir.org.ar.
Agata C Fernández-Gamba, Email: afernandezg@leloir.org.ar.
Luz A Avendaño, Email: lavazquez@leloir.org.ar.
Cecilia Rotondaro, Email: crotondaro@leloir.org.ar.
Osvaldo L Podhajcer, Email: opodhajcer@leloir.org.ar.
Rubén Vidal, Email: rvidal@iupui.edu.
Laura Morelli, Email: lmorelli@leloir.org.ar.
María F Ceriani, Email: fceriani@leloir.org.ar.
Eduardo M Castaño, Email: ecastano@leloir.org.ar.
Acknowledgements
This work was supported by a grant from the Alzheimer’s Association (IIRG-11-205127), a fellowship from the John Simon Guggenheim Memorial Foundation (to EMC), a grant PICT2007-678 (to MFC) and grants from the National Institute on Neurological Disorders and Stroke NS050227 and NS063056 (to RV).
LM, OLP, MFC and EMC are independent investigators of CONICET. MSM is supported by a CONICET fellowship. We thank Bloomington Stock Center for flies stocks.
References
- Vidal R, Frangione B, Rostagno A, Mead S, Révész T, Plant G. et al. A stop-codon mutation in the BRI gene associated with familial British dementia. Nature. 1999;24:776–781. doi: 10.1038/21637. [DOI] [PubMed] [Google Scholar]
- Vidal R, Revesz T, Rostagno A, Kim E, Holton JL, Bek T. et al. A decamer duplication in the 3′ region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc Natl Acad Sci U S A. 2000;97:4920–4925. doi: 10.1073/pnas.080076097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead S, James-Galton M, Revesz T, Doshi RB, Harwood G, Pan EL. et al. Familial British dementia with amyloid angiopathy: early clinical, neuropsychological and imaging findings. Brain. 2000;123:975–991. doi: 10.1093/brain/123.5.975. [DOI] [PubMed] [Google Scholar]
- Strömgren E, Dalby A, Dalby MA, Ranheim B. Cataract, deafness, cerebellar ataxia, psychosis and dementia: a new syndrome. Acta Neurol Scand. 1970;46:261–262. doi: 10.1111/j.1600-0404.1970.tb02219.x. [DOI] [PubMed] [Google Scholar]
- Holton JL, Lashley T, Ghiso J, Braendgaard H, Vidal R, Guerin CJ. et al. Familial Danish dementia: a novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta. J Neuropathol Exp Neurol. 2002;61:254–267. doi: 10.1093/jnen/61.3.254. [DOI] [PubMed] [Google Scholar]
- Holton JL, Ghiso J, Lashley T, Rostagno A, Guerin CJ, Gibb G. et al. Regional distribution of amyloid-Bri deposition and its association with neurofibrillary degeneration in familial British dementia. Am J Pathol. 2001;158:515–526. doi: 10.1016/S0002-9440(10)63993-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;210:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creemers JWM, Chu S, Thinakaran G, Sisodia SS. Proteolytic processing of familial British dementia-associated BRI variants: evidence for enhanced intracellular accumulation of amyloidogenic peptides. J Biol Chem. 2002;277:1872–1877. doi: 10.1074/jbc.M108739200. [DOI] [PubMed] [Google Scholar]
- Sánchez-Pulido L, Devos D, Valencia A. BRICHOS: a conserved domain in proteins associated with dementia, respiratory distress and cancer. Trends Biochem Sci. 2002;27:329–332. doi: 10.1016/S0968-0004(02)02134-5. [DOI] [PubMed] [Google Scholar]
- Garringer HJ, Murrell J, D’Adamio L, Ghetti B, Vidal R. Modeling familial British and Danish dementia. Brain Struct Funct. 2010;214:235–244. doi: 10.1007/s00429-009-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Agnaf OM, Nagala S, Patel BP, Austen BM. Non-fibrillar oligomeric species of the amyloid ABri peptide, implicated in familial British dementia, are more potent at inducing apoptotic cell death than protofibrils or mature fibrils. J Mol Biol. 2001;310:157–168. doi: 10.1006/jmbi.2001.4743. [DOI] [PubMed] [Google Scholar]
- Gibson G, Gunasekera N, Lee M, Lelyveld V, El-Agnaf OM, Wright A, Austen BM. Oligomerization and neurotoxicity of the amyloid ADan peptide implicated in familial Danish dementia. J Neurochem. 2004;88:281–290. doi: 10.1046/j.1471-4159.2003.02134.x. [DOI] [PubMed] [Google Scholar]
- Vidal R, Barbeito AG, Miravalle L, Ghetti B. Cerebral amyloid angiopathy and parenchymal amyloid deposition in transgenic mice expressing the Danish mutant form of human BRI2. Brain Pathol. 2009;19:58–68. doi: 10.1111/j.1750-3639.2008.00164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garringer HJ, Murrell J, Sammeta N, Gnezda A, Ghetti B, Vidal R. Increased tau phosphorylation and tau truncation, and decreased synaptophysin levels in mutant BRI2/tau transgenic mice. PLoS ONE. 2013;8:e56426. doi: 10.1371/journal.pone.0056426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giliberto L, Matsuda S, Vidal R, D’Adamio L. Generation and initial characterization of FDD knock in mice. PLoS ONE. 2009;4:e7900. doi: 10.1371/journal.pone.0007900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coomaraswamya J, Kilgera E, Wölfinga H, Schäfera C, Kaesera SA, Wegenast-Brauna BM. et al. Modeling familial Danish dementia in mice supports the concept of the amyloid hypothesis of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2010;107:7969–7974. doi: 10.1073/pnas.1001056107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickford F, Coomaraswamy J, Jucker M, McGowan E. Modeling familial British dementia in transgenic mice. Brain Pathol. 2006;16:80–85. doi: 10.1111/j.1750-3639.2006.tb00564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamayev R, Giliberto L, Li W, d’Abramo C, Arancio O, Vidal R. et al. Memory deficits due to familial British dementia BRI2 mutation are caused by loss of BRI2 function rather than amyloidosis. J Neurosci. 2010;30:14915–14924. doi: 10.1523/JNEUROSCI.3917-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotinopoulou A, Tsachaki M, Vlavaki M, Poulopoulos A, Rostagno A, Frangione B. et al. BRI2 interacts with amyloid precursor protein (APP) and regulates amyloid beta (Aβ) production. J Biol Chem. 2005;280:30768–30772. doi: 10.1074/jbc.C500231200. [DOI] [PubMed] [Google Scholar]
- Kim J, Miller VM, Levites Y, West KJ, Zwizinski CW, Moore BD. et al. BRI2 (ITM2b) inhibits Aβ deposition in vivo. J Neurosci. 2008;28:6030–6036. doi: 10.1523/JNEUROSCI.0891-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran J, Lewis P. Mad fly disease. J Neurosci. 2007;27:971–972. doi: 10.1523/JNEUROSCI.5427-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Vogel H. Drosophila models of neurodegenerative diseases. Annu Rev Pathol. 2009;4:315–342. doi: 10.1146/annurev.pathol.3.121806.151529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanson NA Jr, Pandey UB. FUS-related proteinopathies: lessons from animal models. Brain Res. 2012;1462:44–60. doi: 10.1016/j.brainres.2012.01.039. [DOI] [PubMed] [Google Scholar]
- Jaiswal M, Sandoval H, Zhang K, Bayat V, Bellen HJ. Probing mechanisms that underlie human neurodegenerative diseases in Drosophila. Annu Rev Genet. 2012;46:371–396. doi: 10.1146/annurev-genet-110711-155456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greeve I, Kretzschmar D, Tschäpe JA, Beyn A, Brellinger C, Schweizer M. et al. Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J Neurosci. 2004;24:3899–3906. doi: 10.1523/JNEUROSCI.0283-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finelli A, Kelkar A, Song HJ, Yang H, Konsolaki M. A model for studying Alzheimer’s Aβ42-induced toxicity in Drosophila melanogaster. Mol Cell Neurosci. 2004;26:365–375. doi: 10.1016/j.mcn.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolakis M, Zhong Y. Dissecting the pathological effects of human Aβ40 and Aβ42 in Drosophila: a potential model for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101:6623–6628. doi: 10.1073/pnas.0400895101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, Duthie FA. et al. Intraneuronal Aβ, non-amyloid aggregates and neurodegenertion in a Drosophila model of Alzheimer’s disease. Neuroscience. 2005;132:123–135. doi: 10.1016/j.neuroscience.2004.12.025. [DOI] [PubMed] [Google Scholar]
- Iijima K, Chiang H, Hearn S, Hakker I, Gatt A, Shenton C. et al. Aβ42 mutants with different aggregation profiles induce distinct pathologies in Drosophila. PLoS One. 2008;3:e1703. doi: 10.1371/journal.pone.0001703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima AK, Hearn SA, Shenton C, Gatt A, Zhao LJ, Iijima K. Mitochondrial mislocalization underlies Aβ42-induced neuronal dysfunction in a Drosophila model of Alzheimer’s disease. PLoS One. 2009;4:e8310. doi: 10.1371/journal.pone.0008310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XL, Wang WA, Tan JX, Huang JK, Zhang X, Zhang BZ. et al. Expression of beta-amyloid induced age-dependent presynaptic and axonal changes in Drosophila. J Neurosci. 2010;30:1512–1522. doi: 10.1523/JNEUROSCI.3699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JK, Ma PL, Ji SY, Zhao XL, Tan JX, Sun XJ, Huang FD. Age-dependent alterations in the presynaptic active zone in a Drosophila model of Alzheimer’s. Neurobiol Dis. 2013;51:161–167. doi: 10.1016/j.nbd.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Luheshi LM, Tartaglia GG, Brorsson AC, Pawar AP, Watson IE, Chiti F. et al. Systematic in vivo analysis of the intrinsic determinants of amyloid β pathogenicity. PLoS Biol. 2007;5:e290. doi: 10.1371/journal.pbio.0050290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima-Ando K, Hearn SA, Granger L, Shenton C, Gatt A, Chiang HC. et al. Overexpression of neprilysin reduces alzheimer amyloid-β42 (Aβ42)-induced neuron loss and intraneuronal Aβ42 deposits but causes a reduction in cAMP-responsive element-binding protein-mediated transcription, age-dependent axon pathology, and premature death in Drosophila. J Biol Chem. 2008;283:19066–19076. doi: 10.1074/jbc.M710509200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijimaa K, Kanae Iijima-Ando K. Drosophila models of Alzheimer’s amyloidosis: the challenge of dissecting the complex mechanisms of toxicity of amyloid-β 42. J Alz Dis. 2008;15:523–540. doi: 10.3233/jad-2008-15402. [DOI] [PubMed] [Google Scholar]
- Cao W, Song HJ, Gangi T, Kelkar A, Antani I, Garza D. et al. Identification of novel genes that modify phenotypes induced by Alzheimer’s β-amyloid overexpression in Drosophila. Genetics. 2008;178:1457–1471. doi: 10.1534/genetics.107.078394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L, Schedl P, Song HJ, Garza D, Konsolaki M. The Toll → NFkB signaling pathway mediates the neuropathological effects of the human Alzheimer’s Aβ42 polypeptide in Drosophila. PLoS One. 2008;3:e3966. doi: 10.1371/journal.pone.0003966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang HC, Wang L, Xied Z, Yaua A, Zhonga Y. PI3 kinase signaling is involved in Aβ-induced memory loss in Drosophila. Proc Natl Acad Sci U S A. 2010;107:7060–7065. doi: 10.1073/pnas.0909314107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tare M, Modi RM, Nainaparampil JJ, Puli OR, Bedi S, Fernandez-Funez P. et al. Activation of JNK signaling mediates amyloid-β-dependent cell death. PLoS One. 2011;6:e24361. doi: 10.1371/journal.pone.0024361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers I, Kerr F, Martinez P, Hardy J, Lovestone S, Partridge L. Ageing increases vulnerability to Aβ42 toxicity in Drosophila. PLoS One. 2012;7:e40569. doi: 10.1371/journal.pone.0040569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific-C31 integrases. Proc Natl Acad Sci U S A. 2007;104:3312–3317. doi: 10.1073/pnas.0611511104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer JM, Staveley BE. GAL4 causes developmental defects and apoptosis when expressed in the developing eye of Drosophila melanogaster. Genet Mol Res. 2003;2:43–47. [PubMed] [Google Scholar]
- Rezával C, Berni J, Gorostiza EA, Werbajh S, Fagilde MM, Fernández MP. et al. A functional misexpression screen uncovers a role for enabled in progressive neurodegeneration. PLoS One. 2008;3:e3332. doi: 10.1371/journal.pone.0003332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiAngelo JR, Erion R, Crocker A, Sehgal A. The central clock neurons regulate lipid storage in Drosophila. PLoS One. 2011;6:e19921. doi: 10.1371/journal.pone.0019921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan RL, Benzer S. Defective glia in the drosophila brain degeneration mutant drop-dead. Neuron. 1993;10:839–850. doi: 10.1016/0896-6273(93)90200-B. [DOI] [PubMed] [Google Scholar]
- Lang M, Wang L, Fan Q, Xiao G, Wang X, Zhong Y. et al. Genetic inhibition of solute-linked carrier 39 family transporter 1 ameliorates Aβ pathology in a Drosophila model of Alzheimer’s disease. PLoS Genet. 2012;8:e1002683. doi: 10.1371/journal.pgen.1002683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SI, Vidal R, Frangione B, Levy E. Axonal transport of British and Danish amyloid peptides via secretory vesicles. FASEB J. 2004;18:373–375. doi: 10.1096/fj.03-0730fje. [DOI] [PubMed] [Google Scholar]
- Gargano JW, Martin I, Bhandari P, Grotewiel MS. Rapid iterative negative geotaxis (RING): a new method for assessing age-related locomotor decline in Drosophila. Exp Gerontol. 2005;40:386–395. doi: 10.1016/j.exger.2005.02.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers used for constructs generation. Table with the sequences of all primers used in this study and PCR strategy.
Western blots of amyloid peptides in SDS-insoluble factions from Drosophila eyes. Accumulation of Aβ42 and ADan in the formic acid-soluble fraction. Non-specific immunoreactive bands in Western blots from RIPA-soluble homogenates.
Toxicity in the Drosophila eye. Toxicity of GMR-GAL/+ as compared to GMR-GAL4/Bri2-23 lines at 28°C for 10 days; dose dependent Aβ42 toxicity in the eye and number of ommatidia in transgenic vs non-transgenic lines.
Accumulation of untagged and His-tagged amyloid peptides. Western blot anti specific peptides from fly heads expressing tag and untagged peptides.
Geotaxis and Phototaxis statistical analysis. Two-way repeated measures ANOVA, followed by comparison tests for all genotypes and time points.
pdf -neurons are resistant to amyloid peptides toxicity. Figure showing the circadian locomotor activity of transgenic flies.
Thioflavine S-negative accumulation of Aβ, ABri and ADan in the CNS of Drosophila. Figure showing negative Thioflavine-S staining of brain sections from Drosophila transgenic lines.