

ABSTRACT
Poliovirus (PV), a model for interactions of picornaviruses with host cells, replicates its genomic RNA in association with cellular membranes. The origin of PV replication membranes has not been determined. Hypotheses about the origin of replication membranes, based largely on localization of viral proteins, include modification of coat protein complex I (COPI) and/or COPII secretory pathway vesicles and subversion of autophagic membranes. Here, we use an antibody against double-stranded RNA (dsRNA) to identify replication complexes by detection of dsRNA replication intermediates. dsRNA signal is dependent on virus genome replication and colocalizes with the viral integral membrane protein 3A, which is part of the RNA replication complex. We show that early in infection, dsRNA does not colocalize with a marker for autophagic vesicles, making it unlikely that autophagosomes contribute to the generation of PV RNA replication membranes. We also find that dsRNA does not colocalize with a marker of the COPII coat, Sec31, and, in fact, we demonstrate proteasome-dependent loss of full-length Sec31 during PV infection. These data indicate that COPII vesicles are an unlikely source of PV replication membranes. We show that the Golgi resident G-protein Arf1 and its associated guanine nucleotide exchange factor (GEF), GBF1, transiently colocalize with dsRNA early in infection. In uninfected cells, Arf1 nucleates COPI coat formation, although during infection the COPI coat itself does not colocalize with dsRNA. Phosphatidylinositol-4-phosphate, which is associated with enterovirus-induced vesicles, tightly colocalizes with Arf1/GBF1 throughout infection. Our data point to a noncanonical role for some of the COPI-generating machinery in producing unique replication surfaces for PV RNA replication.
IMPORTANCE
Picornaviruses are a diverse and major cause of human disease, and their genomes replicate in association with intracellular membranes. There are multiple hypotheses to explain the nature and origin of these membranes, and a complete understanding of the host requirements for membrane rearrangement would provide novel drug targets essential for viral genome replication. Here, we study the model picornavirus, poliovirus, and show that some, but not all, components of the cellular machinery required for retrograde traffic from the Golgi apparatus to the endoplasmic reticulum are transiently present at the sites of viral RNA replication. We also show that the full-length Sec31 protein, which has been suggested to be present on PV RNA replication membranes, is lost during infection in a proteasome-dependent manner. This study helps to reconcile multiple hypotheses about the origin of poliovirus replication membranes and points to known host cell protein complexes that would make likely drug targets to inhibit picornavirus infections.
INTRODUCTION
Poliovirus (PV), like all positive-strand RNA viruses, replicates its RNA genome in association with cellular membranes (1). The intracellular sites of poliovirus genomic RNA replication have been studied for many years, and these studies have resulted in multiple hypotheses about their origin. One hypothesis involves subversion of the autophagic degradation pathway by the virus, resulting in the cytoplasmic accumulation of double-membraned autophagosome-like vesicles. These vesicles are marked with the viral 3A and 2C proteins, which are both essential components of the RNA replication complex (2–4). Treatment of cells with 3-methyladenine (3-MA), an inhibitor of phosphatidylinositol-3 (PI3) kinases that has been well documented to prevent the formation of autophagosomes, attenuates viral RNA replication (5, 6). These data have led us, and others, to hypothesize that autophagosome-like vesicles are sites of PV RNA replication (7–9).
However, another hypothesis was proposed based on data showing that the viral 2B protein localizes to single-membraned vesicles containing Sec13 and Sec31, both components of the cellular coat protein complex II (COPII) (10). COPII is a set of highly conserved proteins responsible for creating small membrane vesicles that originate from the endoplasmic reticulum (ER) (11, 12). During the final stage of COPII-coated vesicle formation, the Sec13-Sec31 complex is recruited to ER membranes, where it polymerizes the COPII complex into a coat, which brings about vesicle budding (13–16). It was recently shown that infection with PV results in a transient increase in COPII vesicle budding from the ER (17). Taken together, these data led to a hypothesis that the PV genome was replicating on vesicles with a COPII secretory pathway origin.
A third hypothesis was based on the sensitivity of PV RNA replication to the fungal metabolite brefeldin A (BFA) (18). BFA inhibits the activation of the small GTPase Arf1 by interacting with specific guanine nucleotide exchange factors (GEFs) (19). These GEFs recycle Arf1 from its inactive GDP-bound form to an active GTP bound form. In its activated form, Arf1-GTP binds to Golgi membranes, where it recruits coat protein complex I (COPI) proteins (20). COPI vesicles have been shown to participate in the retrieval of proteins from the Golgi back to the ER (21–23). BFA specifically inhibits the activity of the Arf1 GEFs GBF1, BIG1, and BIG2 (24). The PV 3A and 2C proteins colocalize with GBF1 and Arf1, respectively (25, 26). These data led to a model in which PV RNA replication membranes are generated by the subversion of the host cell COPI secretory pathway (26–28).
GBF1 and Arf1 also play important roles in the production of phosphatidylinositol-4-phosphate (PI4P) lipids (29–31). During infection with another member of the enterovirus family, coxsackievirus B3 (CVB3), Arf1 and phosphatidylinositol-4-kinase III β (PI4KIIIβ) colocalize at sites of viral RNA replication (32). PI4 kinases, such as PI4KIIIβ, catalyze the phosphorylation of phosphatidylinositol at the D-4 position of the inositol ring, which generates PI4P (33). The activity of PI4KIIIβ is absolutely required for both CVB3 and PV RNA replication, leading to speculation that PI4KIIIβ may represent a conserved host requirement among enteroviruses (32). In addition, the PV RNA-dependent RNA polymerase preferentially binds to PI4P, supporting a role for these lipids in the replication membrane.
From the GBF1 and PI4P data, a model was generated that PV, like CVB3, replicates its RNA genome on PI4P-rich replication organelles harboring components of the COPI secretory machinery (32). Genomic replication of CVB3, as well as another enterovirus, human rhinovirus type 2 (HRV-2), has been shown to occur on membranes enriched for both PI4P as well as cholesterol (34). Poliovirus replication complex proteins colocalize with both cholesterol and PI4P, and the replication of both CVB3 and PV is attenuated when free cholesterol is depleted within the cell (34). This has led to a revised model, in which enterovirus RNA replication relies on the production of both PI4P and cholesterol to generate membranes capable of supporting viral RNA replication. Recent work has indicated a role for the Golgi protein ACBD3 in poliovirus replication (35, 36). ACBD3, which interacts with PV 3A and double-stranded RNA, is important for normal levels of PV replication. These data further the idea that Golgi resident proteins may be crucial for the creation of PV RNA replication vesicles.
Electron microscopy (EM) tomography has been used to observe host cell membranes at discrete points postinfection (p.i.) (9). At 3 h p.i., PV RNA replication can be observed on single-membraned vesicles. At 4 h p.i., RNA replication was observed on extended convoluted membranes, some of which appear similar to the crescent-shaped precursors to autophagosomes. At 7 h p.i., RNA replication was observed on double-membraned vesicles. These snapshots of individual time points led to a suggestion that the early single-membraned vesicles morph into late double-membraned vesicles, a possibility which could unify multiple hypotheses in the field.
With evidence in the literature for at least three models, we wanted to ask if the virus replicates on a variety of different membranes during infection, some originating from the secretory pathways, others originating from the autophagic pathway. Alternatively, the virus may generate a single replication organelle, unique to infection, containing markers from multiple cellular membrane pathways, with the shape and nature of this organelle changing as infection progresses. By employing an antibody that recognizes double-stranded RNA (dsRNA), which is formed during the synthesis of nascent positive-strand RNA genomes, we were able to avoid relying on viral proteins for detection of RNA replication complexes. Viral proteins can be unreliable markers of replication sites, as these proteins are generally multifunctional and may be present at cellular locales that are not sites of RNA replication. We show that early in infection the autophagic marker LC3 does not significantly colocalize with active RNA replication. Arf1, GBF1, and PI4P are, in agreement with recent reports, found at replication sites, although we find that components of the COPI and COPII coats are not themselves recruited to PV RNA replication sites. Furthermore, levels of the full-length COPII coat protein Sec31 are reduced, in a proteasome-dependent manner, during infection. We suggest a model in which a few select components of the COPI machinery generate PI4P-rich vesicles for PV RNA replication.
RESULTS
Visualization of PV RNA replication using an antibody directed against dsRNA.
To visualize the sites of PV RNA replication, we employed a monoclonal antibody against dsRNA. This antibody recognizes all double-stranded RNAs longer than 40 bp, independent of sequence (37). Poliovirus generates double-stranded RNA intermediates twice during replication of the positive-sense genome: first during production of the negative-sense strand generated using the incoming genome as a template, and then again when generating nascent positive-strand genomes from the negative-strand template (38). Our first goal was to determine if the dsRNA antibody signal was specific to viral RNA replication and to identify levels of background signal from rare dsRNA events in uninfected cells. Cells were either mock infected or infected with PV at a multiplicity of infection (MOI) of 50 PFU/cell, fixed at 3, 5, and 7 h p.i., and then incubated with the dsRNA antibody, followed by secondary antibody. Signal was observed in all PV-infected cells, while no signal was observed in mock-infected cells at 7 h p.i. (Fig. 1A). To test if the observed signal was specific to cells with active PV RNA replication, we infected cells following treatment with guanidine HCl (GHCl). GHCl is a potent, specific inhibitor of PV RNA replication (39). We observed no significant dsRNA signal in cells treated with GHCl.
FIG 1 .

Visualization of poliovirus RNA replication sites. (A) H1-HeLa cells were infected at an MOI of 50 PFU/cell and fixed and permeabilized with −20°C methanol at the indicated times postinfection. Cells were stained with a monoclonal antibody against dsRNA. Guanidine HCl (GHCl) was added at a final concentration of 20 mM at the start of infection. (B) H1-Hela cells were infected at an MOI of 50, fixed and permeabilized with −20°C methanol at the indicated times postinfection, and stained by indirect immunofluorescence using a monoclonal antibody against dsRNA and a polyclonal antibody against the poliovirus 3A protein. Blue staining in this and subsequent figures is nuclear staining with DAPI. The white box in the field image denotes the cell shown in the panels to the right.
To further confirm that the dsRNA signal we observed was marking viral RNA replication complexes, we investigated the localization of the signal with respect to the viral 3A protein. 3A is a transmembrane protein thought to nucleate the assembly of the replication complex, so we would expect the dsRNA signal to colocalize with 3A at membranes associated with active replication sites (40–42). When cells were incubated with antibodies directed against dsRNA and 3A, the two signals colocalized throughout infection (Fig. 1B). The Pearson’s coefficients of dsRNA and 3A signal were calculated as described in Materials and Methods, and the coefficients remain high throughout infection (Pearson correlation coefficient Rr = 0.84 to 0.89). These results led us to conclude that the dsRNA antibody could be successfully used to detect the sites of PV RNA replication.
PV RNA replication does not colocalize with LC3-positive structures early in infection.
To determine if active viral RNA replication complexes form on membranes derived from the autophagic pathway, we investigated the localization of the dsRNA signal with respect to the autophagosome marker protein LC3. In resting cells, the majority of LC3 is cytosolic and referred to as LC3-I. Following induction of autophagy, LC3 becomes lipidated, which confers its association with the autophagosome membrane. This lipidated form of the protein is referred to as LC3-II (43, 44). PV infection dramatically increases cellular levels of LC3-II (45). Association of LC3-II with the autophagosome membrane is absolutely required for formation of the double-membraned vesicles, making it a reliable protein marker for autophagosomes (46, 47). LC3 was viewed by expression of an LC3 protein with an amino-terminal fusion to green fluorescent protein (GFP) (7). By this method, autophagosomes appear as discrete, GFP-positive puncta (47). In order to determine whether active viral RNA replication occurred on LC3-positive membranes, cells transfected with GFP-LC3 were infected with PV 24 h after transfection. Cells were then fixed and incubated with the dsRNA antibody. As shown in Fig. 2, the dsRNA signal did not significantly overlap the punctate GFP-LC3 signal at early time points (Rr = 0.13 to 0.26). We also examined the localization of the dsRNA signal with respect to the punctate LC3 signal at both 5 and 7 h postinfection, time points after the peak of viral RNA replication (Fig. 3). At these later time points, the two signals appeared to be in close proximity to one another, which was reflected in a slight increase in the Pearson’s coefficients (Rr = 0.23 to 0.42). However, we did not observe a direct, significant overlap in the immunofluorescence signals of GFP-LC3 and dsRNA.
FIG 2 .
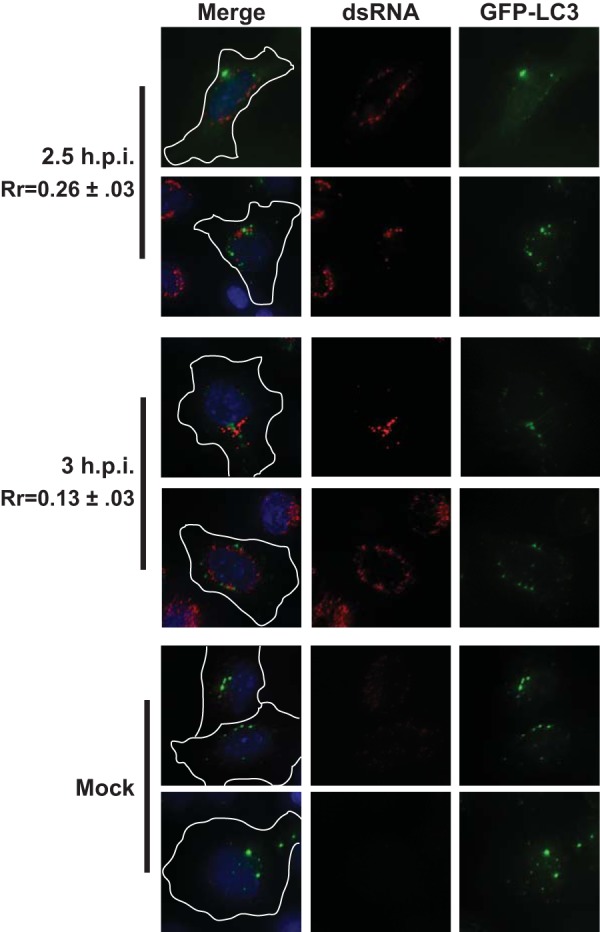
Poliovirus RNA replication does not colocalize with GFP-LC3 early in infection. H1-HeLa cells expressing a GFP-tagged form of the autophagosome membrane protein LC3 were infected at an MOI of 50 PFU/cell and fixed and permeabilized with −20°C methanol at the indicated times postinfection. Cells were then stained with a monoclonal antibody against dsRNA. An outline of the cell was generated from a differential inference contrast (DIC) image and is shown in white on the merged images. Two fields are shown from a representative experiment.
FIG 3 .
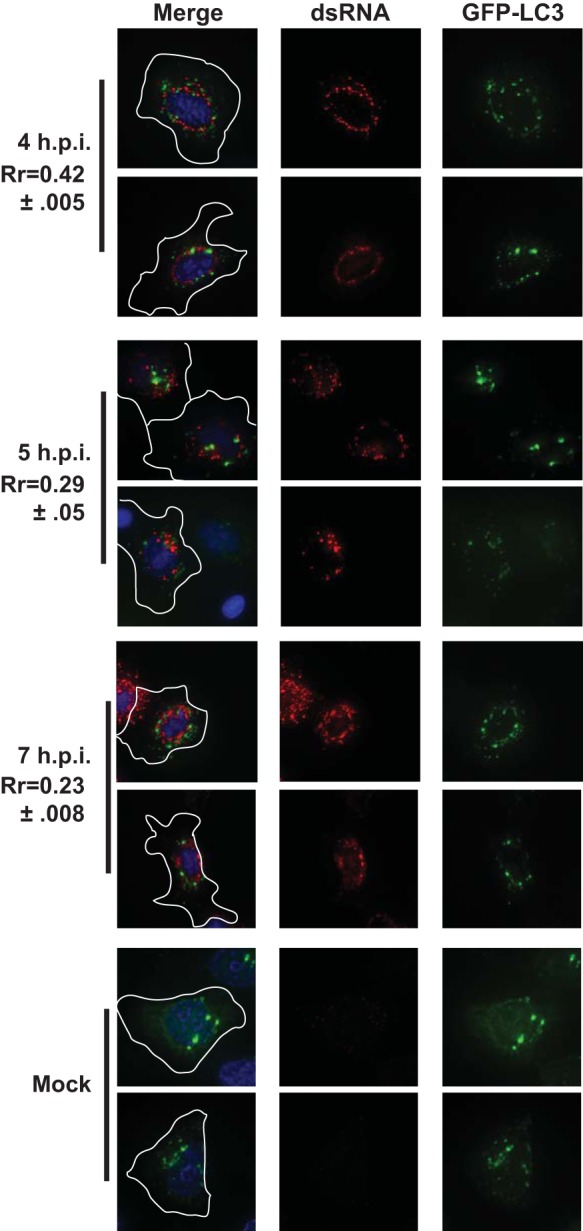
Poliovirus RNA replication does not colocalize with GFP-LC3 late in infection. H1-HeLa cells expressing a GFP-tagged LC3 protein were infected at an MOI of 50 PFU/cell and fixed and permeabilized with −20°C methanol at the indicated times postinfection. Cells were then stained with a monoclonal antibody against dsRNA. An outline of the cell was generated from a DIC image and is shown in white on the merged images. Two fields are shown from a representative experiment.
We found this result surprising, as previous reports had reported that dsRNA had localized with double-membraned vesicles late in infection (9). We considered the possibility that the dsRNA-associated membranes in the previous report were not LC3-containing double-membraned vesicles. The previous report used immuno-EM with DAB (3,3′-diaminobenzidine) peroxidase staining, a technique which does not allow colocalization studies. Therefore, that study may have been detecting another type of vesicle. To understand the late-infection phenomenon at an ultrastructural level, we performed scanning EM with gold particle-labeled secondary antibodies so we could simultaneously detect LC3 and dsRNA. LC3 was detected using an anti-GFP antibody in cells transfected with a GFP-LC3 construct (tested LC3 antibodies were not successful in immuno-EM experiments.) Our controls indicated low dsRNA or GFP background in cells lacking the antigens (data not shown.) As seen in Fig. 4, we found that at 2.5 h, the two signals did not colocalize. However, at 5 h p.i., we found regular pockets of strong colocalization of the two signals, often associated with apparent double- or multimembraned bodies.
FIG 4 .
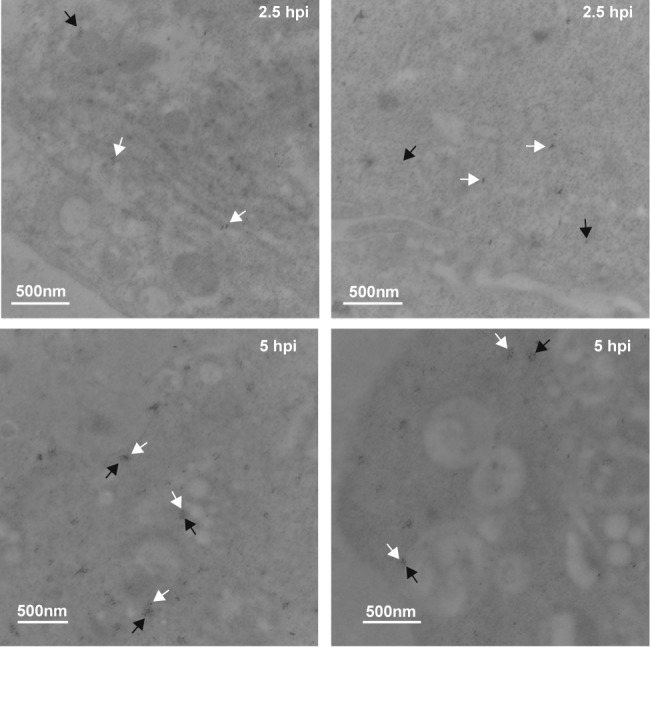
Electron microscopy indicates colocalization of dsRNA and GFP-LC3 late, but not early, in infection. H1-HeLa cells were transfected with GFP-LC3 vector, and 24 h later cells were infected with PV at an MOI of 50 PFU/cell and fixed for electron microscopy analysis at 2.5 and 5 h postinfection, as described in Materials and Methods. Two images are shown for each time point. Anti-goat secondary antibody conjugated to 6-nm gold particles recognizes anti-GFP antibody. Anti-mouse secondary antibody conjugated to 10-nm gold particles recognizes anti-dsRNA antibody. Bars (500 nm) are shown for scale in each image. Black arrowheads, 6 nm, indicating LC3-GFP; white arrowheads, 10 nm, indicating dsRNA.
Therefore, our immunofluorescence microscopy (IF) and immuno-EM data are both compelling but provide opposite conclusions. It is possible that the amplification of the signal achieved by using the anti-GFP antibody may allow us to visualize LC3 protein that is below the level of detection of IF. Alternatively, secondary detection of the anti-GFP antibody may provide an artifact, and direct observance of the GFP signal by IF may be a better method. We have compared GFP-LC3 fluorescence signal to indirect immunofluorescence detection of LC3 using our GFP antibody and find essentially complete overlap of the signals (data not shown). Ultimately, we find the immuno-EM result convincing enough to conclude that by 5 h p.i., at least some RNA replication is occurring in association with LC3-containing structures. This is in agreement with the previous report showing dsRNA associated with double-membraned vesicles (9). We present all data here so that the scientific record will show the different conclusions that might be drawn from IF and immuno-EM experiments with GFP-LC3. However, it is important to point out that using either method, at earlier time points, when RNA replication is at its peak, RNA replication sites do not colocalize with LC3.
PV RNA replication localizes with GBF1 and Arf1 early in infection.
To test the hypothesis that components of the COPI secretory pathway are recruited to the PV replication membrane, we investigated the localization of dsRNA with respect to both Arf1 and GBF1. Arf1 was visualized by expression of an Arf1 protein with a carboxy-terminal fusion to GFP protein (48). GBF1 was viewed by indirect immunofluorescence using a polyclonal antibody. In mock-infected cells, both Arf1 and GBF1 partially colocalize with the cis-Golgi marker GM130 in a juxtanuclear pattern, which is expected given their roles in COPI vesicle formation (see Fig. S1 in the supplemental material). By 2.5 h p.i., Arf1 relocalizes from a Golgi-like staining pattern to a punctate perinuclear pattern. This relocalization of Arf1 following PV infection has been previously demonstrated (25). Early in infection (2.5 h p.i.), the Arf1 signal partially colocalized with the dsRNA signal, although a portion of the Arf1 signal remained distinct from the dsRNA signal (Rr = 0.77). At later time points (3 and 4 h p.i.), the Arf1 signal and the dsRNA signal remained in close proximity to one another, although the regions of tight colocalization of the two signals diminished (Rr = 0.54 to 0.63) (Fig. 5).
FIG 5 .
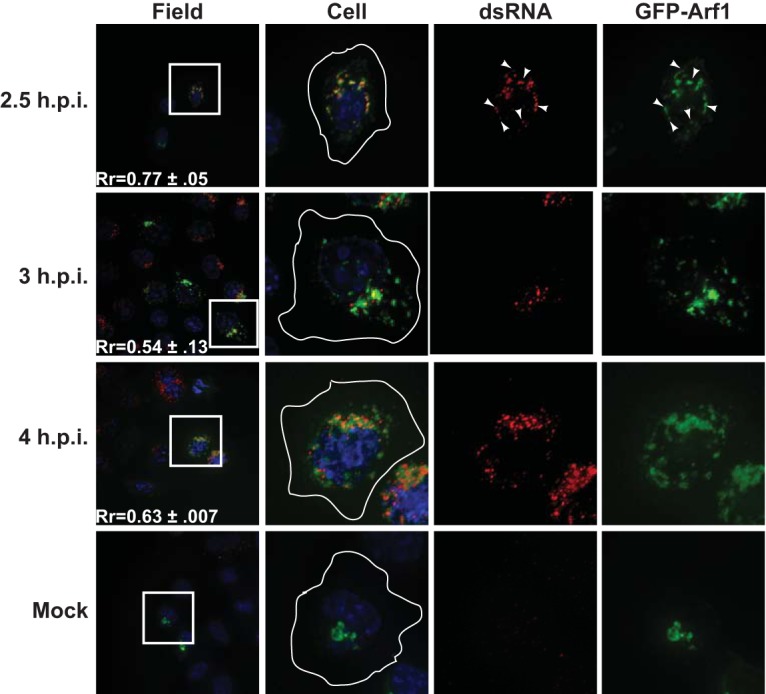
Poliovirus RNA replication transiently colocalizes with Arf1 early in infection. H1-HeLa cells expressing a GFP-tagged Arf1 protein were infected at an MOI of 50 PFU/cell and fixed and permeabilized with −20°C methanol at the indicated times postinfection. Cells were then stained with a monoclonal antibody against dsRNA. An outline of the cell was generated from a DIC image and is shown in white on the cell images. The white box in the field image denotes the cell shown in the panels to the right. An outline of the cell was generated from a DIC image and is shown in white on the merged images. Arrowheads denote areas of colocalization.
As was observed with Arf1, by 2.5 h p.i., the GBF1 signal was relocalized to a punctate perinuclear pattern during PV infection (Fig. 6). This relocalization has also previously been reported (26). Early in infection (2.5 h p.i.), the GBF1 signal colocalized with the dsRNA signal (Rr = 0.62). As the infection proceeded, the GBF1 and dsRNA signals remained in close proximity to one another, although as was observed with Arf1, the tight colocalization diminished (Rr = 0.48 to 0.52).
FIG 6 .
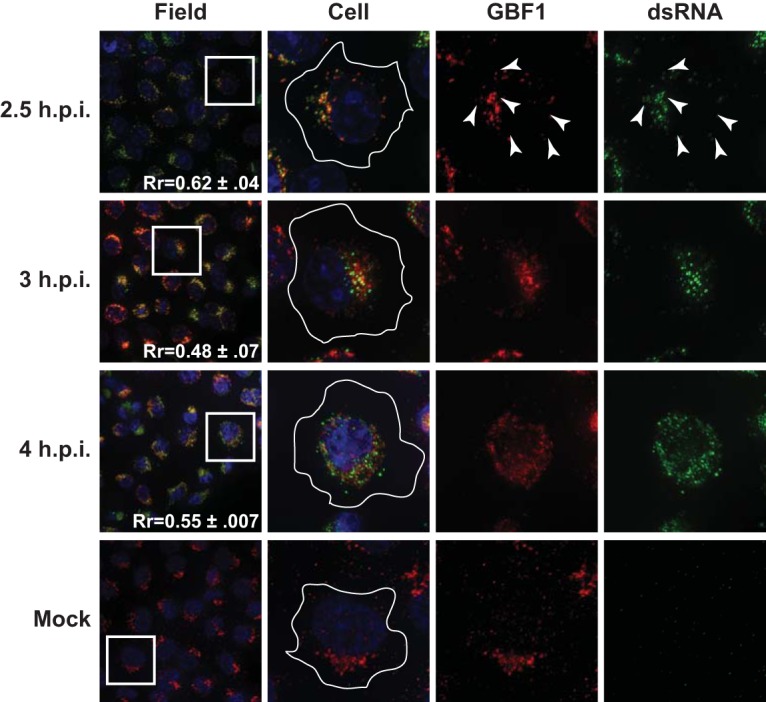
Poliovirus RNA replication transiently colocalizes with GBF1 early in infection. H1-Hela cells were infected at an MOI of 50 PFU/cell and fixed and permeabilized with −20°C methanol at the indicated times postinfection. Cells were stained both with a monoclonal antibody against dsRNA and a polyclonal antibody against the endogenous GBF1. An outline of the cell was generated from a DIC image and is shown in white on the cell images. The white box in the field image denotes the cell shown in the panels to the right. An outline of the cell was generated from a DIC image and is shown in white on the merged images. Arrowheads denote areas of colocalization.
GBF1 localizes with PI4P throughout PV infection.
We also examined the localization of GBF1 with respect to PI4P during infection. In mock-infected cells, the signals from both GBF1 and PI4P localized to the same juxtanuclear region of the cell. Following PV infection, both signals relocalized to a punctate perinuclear pattern. The signals from both antibodies demonstrated a high degree of colocalization throughout the infection (Rr = 0.82 to 0.72) (Fig. 7).
FIG 7 .

PI4P and GBF1 colocalize throughout poliovirus infection. H1-Hela cells were infected at an MOI of 50 PFU/cell and fixed and permeabilized with −20°C methanol at the indicated times postinfection. Cells were stained both with a monoclonal antibody against PI4P and a polyclonal antibody against the endogenous GBF1. An outline of the cell was generated from a DIC image and is shown in white on the cell images. The white box in the field image denotes the cell shown in the panels to the right. An outline of the cell was generated from a DIC image and is shown in white on the merged images. Arrowheads denote areas of colocalization.
The sensitivity of viral RNA replication to inhibitors of PI4KIIIβ, combined with data showing that the viral RNA polymerase has a high affinity for PI4P lipids, has led to a hypothesis that PV may require a membrane that is rich in PI4P on which to assemble its viral RNA replication complexes (32). We visualized PI4P during infection using indirect immunofluorescence using a monoclonal antibody against PI4P. We did not simultaneously observe the localization of dsRNA and PI4P due to the lack of compatible, IF-competent antibodies. We therefore relied on 3A as a marker for the location of active PV RNA replication (Fig. 1). Following PV infection, PI4P and 3A relocalized to a punctate perinuclear pattern. The signals from both antibodies demonstrated a high degree of colocalization throughout the infection (Rr = 0.84) (see Fig. S2 in the supplemental material).
COPI coat proteins are not recruited to the sites of PV RNA replication.
The localization of Arf1 and GBF1 with respect to active RNA replication complexes prompted us to investigate whether or not components of the COPI coat protein complex were recruited to replication sites. In uninfected cells, COPI coat proteins are recruited to Golgi membranes by activated Arf1 (20). Expression of the PV 3A protein in isolation has been shown to prevent the membrane association of COPI coat proteins, although the localization of COPI proteins during infection has never been investigated (49). β-COP is one of seven proteins that make up the COPI coat complex that facilitates vesicle budding from the Golgi, and it has been shown to be absolutely essential for the assembly of the coat complexes (50, 51). We examined the localization of β-COP during PV infection using a polyclonal antibody directed against endogenous β-COP. To determine if β-COP was recruited to replication sites, we compared the localization of the β-COP signal to the signal from our dsRNA antibody. In mock-infected cells, β-COP signal is concentrated in a juxtanuclear region of the cell. Following PV infection, β-COP appeared to be redistributed throughout the cytoplasm. The β-COP signal did not show significant colocalization with the dsRNA signal at any of the time points we investigated during infection (Rr = 0.18 to 0.26) (Fig. 8).
FIG 8 .

β-COP is not recruited to the sites of poliovirus RNA replication. H1-Hela cells were infected at an MOI of 50 PFU/cell and fixed and permeabilized with −20°C methanol at the indicated times postinfection. Cells were stained using a monoclonal antibody against dsRNA and a polyclonal antibody against the endogenous β-COP. An outline of the cell was generated from a DIC image and is shown in white on the cell images. The white box in the field image denotes the cell shown in the panels to the right. An outline of the cell was generated from a DIC image and is shown in white on the merged images.
Sec31 is not recruited to replication membranes.
The PV 2C protein has been shown to localize to vesicles whose membranes contain components of the COPII coat protein complex, specifically Sec13 and Sec31 (10). Given the essential roles of 2BC and 2C in viral RNA replication, we hypothesized that COPII proteins might be recruited to the sites of active viral RNA replication. We investigated the localization of Sec31 throughout infection using indirect immunofluorescence using a monoclonal antibody directed against endogenous Sec31. Initially, we relied on GBF1 as a marker for the location of active PV RNA replication, as the dsRNA antibody is not compatible with the available GBF1 antibody. While GBF1 staining revealed the expected perinuclear localization, Sec31 appeared to be redistributed throughout the cytoplasm of infected cells. The relocalization was evident as early as 3 h p.i., and the signal remained relocalized throughout infection (Fig. 9). As the experiment progressed, Sec31 signal became difficult to detect; while the Pearson’s coefficients are relatively high at 0.43 to 0.5, we suspect this may be due to an increase in the nonpunctate cytosolic signal.
FIG 9 .
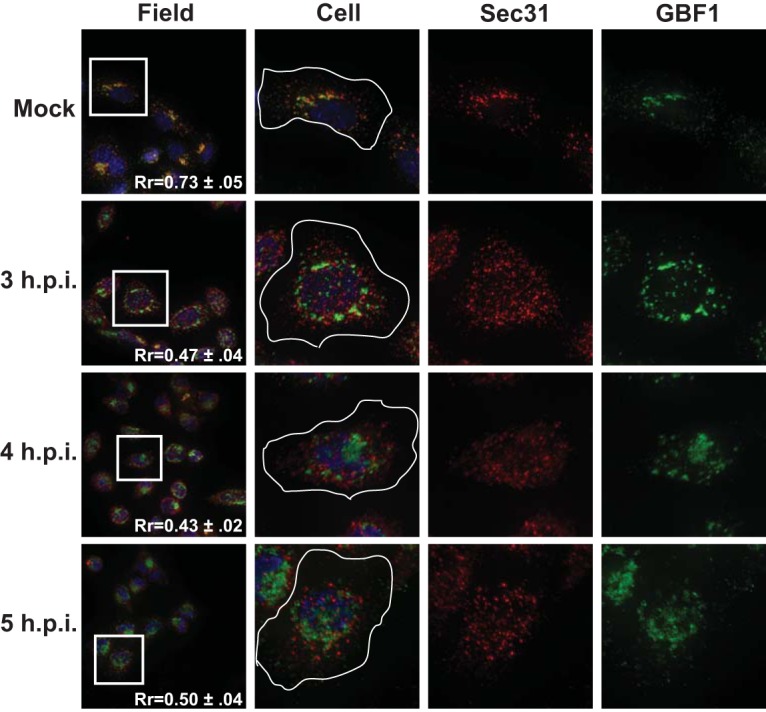
Sec31 is not recruited to the sites of poliovirus RNA replication. H1-Hela cells were infected at an MOI of 50 PFU/cell and fixed and permeabilized with −20°C methanol at the indicated times postinfection. Cells were stained both with a monoclonal antibody against Sec31 and a polyclonal antibody against the endogenous GBF1. An outline of the cell was generated from a DIC image and is shown in white on the cell images. The white box in the field image denotes the cell shown in the panels to the right. An outline of the cell was generated from a DIC image and is shown in white on the merged images. The cell panels represent the single cell in the white boxes shown in the left panels.
Due to the somewhat faint detection signal for Sec31 using the available antibody, we obtained a Sec31-GFP fusion protein and repeated these experiments. This construct allowed us to codetect dsRNA and Sec31. Cells transfected with Sec31-GFP were infected with PV 24 h after transfection. Cells were then fixed and incubated with the dsRNA antibody. As shown in Fig. S4, the dsRNA signal did not significantly overlap the Sec31-GFP signal at any of the time points we investigated during infection (Rr = 0.02 to 0.22).
Steady-state levels of full-length Sec31, but not β-COP, decrease during PV infection.
The change in immunofluorescence staining observed for Sec31 and β-COP could be due to the proteins being relocalized during infection or it could be due to a change in the protein levels. Therefore, we investigated the effect of PV infection on the steady-state levels of each protein by Western blot analysis. We observed a decrease in the level of full-length Sec31 beginning at 3 h p.i. By 6 h p.i., full-length Sec31 was almost undetectable (Fig. 10A). Concurrent with the loss of full-length Sec31, we noted the appearance of a higher mobility band which also reacted with the Sec31 antibody. The levels of β-COP remained unchanged throughout PV infection (Fig. 10B).
FIG 10 .

Steady-state levels of Sec31 but not β-COP decrease during poliovirus infection. (A) H1-Hela cells were mock infected or infected with poliovirus at an MOI of 50 PFU/cell. Cells were lysed at the indicated hours postinfection. Western blots were performed for calnexin and Sec31. Arrow indicates a lower-molecular-weight band observed following PV infection. (B) H1-Hela cells were mock infected or infected with poliovirus at an MOI of 50 PFU/cell. Cells were lysed at the indicated hours postinfection. Western blots were performed for calnexin and β-COP. (C) Six-hour infections, as described for panels A and B; MG132 was used at a final concentration of 5 µM and added to the media at the time of infection. Leupeptin was used at 20 µM, and cells were pretreated for 14 h prior to infection and then kept under leupeptin throughout infection. Cycloheximide was added at 50 µg/ml 30 min prior to infection and kept on cells after infection. Western blotting was performed for Sec31, β-COP, PV 3A protein, and calnexin.
We next wanted to understand the nature of the loss of Sec31 protein levels we observed during PV infection. The intensity of the higher mobility band increased as the infection proceeded, leading us to speculate that it may represent a degradation product of Sec31 (Fig. 10A). To test if the loss of full-length Sec31 and appearance of the higher mobility band is dependent on the activity of the proteasome, we treated cells with MG132, a specific inhibitor of the 26S proteasome (52). This treatment prevented both the reduction in the level of full-length Sec31 and the appearance of the higher mobility band. MG132 has no effect on PV replication at 5 µM as determined by 3A blot analysis (Fig. 10C; see also Fig. S5 in the supplemental material) and plaque assays for viral yield (data not shown.). To test if lysosomal proteases were responsible for degradation of Sec31, we treated cells with leupeptin, a thiol protease inhibitor, and this had no effect on Sec31 degradation during PV infection (53). To determine if viral protein synthesis was required for the loss of full-length Sec 31 during infection, cells were treated with cycloheximide throughout infection. In treated cells, levels of Sec31 were unchanged from those observed in mock-infected cells, indicating that the observed decrease in Sec31 levels is not due to protein turnover (Fig. 10C). None of these treatments had an effect on β-COP levels during PV infection.
DISCUSSION
Studies of poliovirus and other positive-strand RNA viruses have for years identified various cellular membrane-associated proteins that colocalize with viral RNA replication complex proteins, but few studies have directly identified active RNA replication (7, 10, 25, 26). In situ RNA hybridization to the negative viral RNA strand, which our lab has previously used as a reliable marker for active RNA replication, can be difficult to use for antibody colocalization studies, presumably due to the RNases present in essentially all antibody preparations (54). However, some RNA replication proteins can be unreliable markers of replication sites, as viral proteins are generally multifunctional and may be present at cellular locales that are not sites of RNA replication.
We show here, with essential controls, that active PV RNA replication complexes can be detected by an antibody directed against dsRNA. We recognize that a portion of the dsRNA signal we observe may be stalled double-stranded intermediates in which the positive strand and negative strand have not separated, but active replication is no longer occurring. While we cannot provide a quantitative value to the proportion of the signal in which this is the case, our data indicate that we are detecting most active replication complexes, with the possibility of detecting some that are halted in a double-stranded intermediate. It is unlikely that the dsRNA signal observed is due to detection of cellular double-stranded RNA products, as the signal is dependent on active PV RNA replication and colocalizes with 3A, a viral replication complex protein. The localization of PV minus- and plus-strand RNA has been previously investigated by fluorescent in situ hybridization (55). The authors show that a probe specific for the minus strand of viral RNA is present in distinct, regularly sized, round structures throughout the viral replication cycle. A probe recognizing the plus strand of viral RNA is present in two distinct structures: large round bodies, which also harbor minus-strand RNA, and small granules which lack a recognizable minus-strand-specific signal. The localization of the viral RNA with respect to cellular proteins was not investigated.
With the ability to detect active RNA replication in immunofluorescence experiments, we show for the first time that, early in infection, viral RNA replication occurs on membranes positive for Arf1 and its activating GEF, GBF1 (Fig. 11B). These data are in agreement with previous studies showing that GBF1 and Arf1 colocalize with viral proteins (25–28). The colocalization of GBF1 and Arf1 with active replication complexes is the most extensive early in infection, between 2 and 3 h p.i., although the proteins remain in proximity to dsRNA as the infection proceeds. We interpret these observations to mean that Arf1/GBF1 may be essential for formation of the replication vesicles but that they do not need to remain associated with the membrane after it has been generated. A “formation only” role for these proteins during infection would be consistent with the way these proteins function in uninfected cells. Arf1 and GBF1 are essential for the formation of COPI vesicles, but both are dispensable once the vesicles have formed (56, 57).
FIG 11 .

Relocalization of cellular lipids and proteins during poliovirus infection. (A) In uninfected cells, Sec31 is an essential component of the COPII protein coat complex. COPII-coated vesicles bud from the ER and traffic toward the cis-Golgi complex. Arf1 is activated by GBF1 at the cis-Golgi membrane. Activated Arf1 recruits COPI coat proteins, including β-COP, to the cis-Golgi. COPI coat proteins facilitate the budding of COPI-coated vesicles from the Golgi. COPI vesicles traffic from the cis-Golgi to the ER. Golgi membranes are rich in PI4P as a result of several Golgi-localized PI4 kinases. (B) In poliovirus-infected cells, the Golgi is fragmented and Arf1 and GBF1 localize to viral replication membranes. The replication membrane is enriched for PI4P, although the Golgi membrane protein giantin does not associate with the replication membranes. Unlike Arf1 and GBF1, β-COP is redistributed throughout the cytoplasm of infected cells. Sec31 is also redistributed during infection, and levels of full-length Sec31 decrease during infection, possibly due to proteasome-mediated cleavage or degradation.
The colocalization of 3A and GBF1 with PI4P leads us to conclude, in agreement with previously suggested hypotheses, that replication membranes contain elevated levels of PI4P (Fig. 7B; see also Fig. S2 in the supplemental material). PI4P in the Golgi membrane has been shown to recruit GBF1 to these Golgi membranes (58). Therefore, it is conceivable that PI4P present in replication membranes is responsible for the recruitment of GBF1 to these membranes. While Arf1 and GBF1 are normally associated with the formation of COPI-coated vesicles, the replication membranes generated by poliovirus appear to be distinct from COPI vesicles, as we found that the COPI complex protein β-COP is not present at sites of viral RNA replication. Interestingly, infection with BFA-resistant PV can proceed in the absence of activated Arf1, but the mutant virus still requires GBF1. The inhibitory effect of BFA on PV RNA replication can be rescued by expression of the N-terminal domain of GBF1, which lacks the catalytic domain required for Arf1 activation. These data hint that PV may be utilizing GBF1 for a purpose other than Arf1 activation (59).
PV RNA replication has been shown to require the activity of GBF1, PI4KIIIβ, and ACBD3, all of which are associated with the Golgi in uninfected cells (24, 60, 61). This is particularly intriguing, as PV infection results in disruption of the Golgi complex (see Fig. S3 in the supplemental material) (62). In addition to inhibiting host protein secretion during infection, this disassembly of the Golgi apparatus may provide the virus access to Golgi resident proteins it requires for RNA replication. One future direction of this work will be to determine if disruption of the Golgi by PV is required for Golgi resident proteins to function in viral RNA replication. Although the Golgi-associated proteins Arf1 and GBF1 are recruited to replication membranes, we saw no evidence that Golgi integral membrane proteins were incorporated into replication membranes, as giantin staining did not colocalize with the dsRNA signal during infection (see Fig. S3).
Autophagosome-like vesicles have been proposed to be sites of PV RNA replication by our group and others (6, 7, 9). However, we did not observe LC3, an essential autophagosome membrane protein, at the sites of viral RNA replication at times corresponding to the peak of viral RNA replication. We conclude that it is unlikely that cellular autophagosomes are the primary sites of viral genome replication. We previously demonstrated that pharmacological inhibition of autophagy reduces viral RNA replication. This could be due to an effect of anti-autophagy drugs on late RNA replication, which seems to occur on autophagosomes. Alternatively, anti-autophagy drugs could alter the formation of early viral replication vesicles, independent of their effects on the autophagic pathway.
Previously, we have shown that autophagosomes primarily play a post-RNA replication role in PV infection (6, 7). However, in our hands and others, electron microscopy has led to the observation of RNA replication in association with LC3-labeled, double-membraned structures (9). We suggest two explanations for these data. One possibility is that LC3 is incorporated into replication membranes late in infection. Alternatively, due to the increased presence of cytoplasmic autophagosomes late in infection, replication complexes formed late in infection may utilize double-membraned vesicles as minor sites of genome replication.
The membranes of vesicles carrying the essential COPII coat protein, Sec31, have been shown to also contain the viral 2BC protein. 2BC and 2C are essential parts of the viral RNA replication complex, leading to speculation that the COPII-like vesicles observed are primary sites of viral RNA replication. Our data provide no evidence that the COPII coat protein, Sec31, was present at sites of viral RNA replication. Instead, the Sec31 signal became faint and was redistributed to the cytosol in the infected cell. Following up with Western analysis, we found that full-length Sec31 levels are reduced during PV infection accompanied by the appearance of a faster-migrating band. This effect appears to be dependent on the activity of the 26S proteasome, leading us to speculate that Sec31 is cleaved or degraded during infection. To the best of our knowledge, this is the first report of Sec31 degradation during viral infection. Since it is known that PV inhibits ER-to-Golgi trafficking through the action of the 3A protein, we hypothesize that degradation of Sec31 may be a redundant mechanism for ensuring that bulk protein flow from the ER is blocked during infection. Sec31 localization during PV infection has been previously investigated in normal rat kidney (NRK) cells expressing the poliovirus receptor (17). The authors did not report a significant change in the localization of Sec31 at 2 h postinfection. We suggest that the differences between these data and our own are the result of the earlier time point examined in the previous work. Our Western blot analysis of Sec31 during infection showed no significant change in the level of full-length Sec31 at 2 h postinfection (Fig. 10). Since we hypothesize that the loss of full-length Sec31 is responsible for the redistribution we observed during infection, we would not expect to see a change in localization at 2 h p.i. While there may be a burst of COPII traffic early in infection, our data do not support the hypothesis that COPII traffic provides precursors for viral replication membranes. We suggest that the transient COPII trafficking is a cellular response to infection, possibly an attempt to warn the immune system, and one which is halted by the virus-induced reduction in levels of full-length Sec31.
We feel it is important to note that our data do not contradict any of the published reports on the localization of viral proteins during infection. Rather, we have demonstrated that although these proteins have essential functions at the viral RNA replication complex, they may be acting outside the replication complex, likely performing essential functions related to converting the host cell into an environment capable of robust virus production. We have only begun to scratch the surface of the many host factors recruited to the viral RNA replication factories, and many questions remain on how the proteins and lipids coordinate to generate the unique architecture of the viral replication membrane.
MATERIALS AND METHODS
Cell culture and transfection.
H1-HeLa (human cervical adenocarcinoma cells) were maintained in minimum essential medium (MEM) (Invitrogen) supplemented with 10% calf serum and 1% penicillin-streptomycin-glutamine (100×) (Invitrogen) and grown at 37°C, 5% CO2. The GFP-tagged Arf1 and GFP-tagged Sec31 constructs were obtained from Addgene (Cambridge, MA). The GFP-LC3 plasmid was previously described (7). Expression of either GFP fusion constructs was by transient transfection of H1-HeLa cells with Effectene (Qiagen) according to the manufacturer’s instructions. Cells were infected with PV 24 h after transfection.
Immunofluorescence microscopy.
H1-HeLa cells were grown on glass coverslips in a 24-well TC plate. For all experiments, approximately 1.5 × 105 cells were infected at an MOI of 50 PFU/cell. Following infection, cells were washed three times with phosphate-buffered saline (PBS) and permeabilized with −20°C methanol at 4°C for 10 min. Cells were then washed three times with PBS and incubated overnight with blocking buffer (20% goat serum, 0.05% saponin in PBS) at 4°C. Cells were then incubated with the primary antibody for 3 h at room temperature. All primary antibodies were diluted in blocking buffer. Cells were washed three times with PBS and then incubated with the appropriate secondary antibody for 1 h at room temperature. All secondary antibodies were diluted in blocking buffer. Cells were next washed three times with PBS and mounted using ProLong Gold antifade reagent with DAPI (4′,6-diamidino-2-phenylindole; Invitrogen). For all images, a Z-stack of approximately 30 slices was taken. A deconvolved maximum projection was generated using the AutoQuant X3 software (MediaCybernetics). Deconvolution was performed using AutoQuant X3’s three-dimensional blind (adaptive point spread functional [PSF]) deconvolution algorithm. Colocalization analysis was performed by ImageJ software using the Manders coefficient plug in. Rr values represent the Pearson’s correlation coefficient. Correlation coefficients were calculated for two or three random fields of multiple cells, and the average values are displayed in the figures. For colocalization of dsRNA with GFP-tagged proteins, correlation coefficients were determined for three images containing only transfected cells.
Antibodies.
The J2 monoclonal antibody was obtained from English and Scientific Consulting. Polyclonal antibodies against GBF1 and β-COP were obtained from Abcam. The monoclonal antibody against PI4P was obtained from Echelon. The Sec31 antibody was obtained from BD Transduction Laboratories. Anti-GFP (goat) antibody was obtained from Rockland Immunochemicals. The rabbit PV 3A antibody was generated by Biomatik USA (Wilmington, DE) by using the peptide KDLKIDIKTSPPPEC, which corresponds to amino acids 6 to 21 of the PV 3A protein, and was affinity purified.
Immunoelectron microscopy.
Transfected cell cultures (2.5 h and 5 h) were vitrified using an EMPact2 high-pressure freezer (Leica) and freeze substituted in a temperature-controlling device (Reichert AFS) into Lowicryl HM20 resin (Electron Microscopy Sciences, Pennsylvania) following the protocol of Hawes et al. (63). The freeze substitution medium contained 2% (wt/vol) uranyl acetate, 0.25% glutaraldehyde, 10% (vol/vol) dry methanol, 89% dry acetone, and 1% water. Double labeling was performed on ultratyin sections mounted on Formvar-coated copper grids using anti-GFP antibody labeled with rabbit anti-goat 6-nm gold particles (EMS) and anti-J2 antibody labeled with donkey anti-mouse (10 nm; EMS).
SUPPLEMENTAL MATERIAL
Colocalization of GBF1 and GM130 in uninfected cells. H1-Hela cells were mock infected and fixed and permeabilized with −20°C methanol at 6 h postinfection. Cells were stained using a monoclonal antibody against the Golgi protein GM130 and a polyclonal antibody against the endogenous GBF1. Download
PV 3A colocalizes with phosphatidylinositol-4-phosphate (PI4P) during PV infection. H1-Hela cells were infected at an MOI of 50 PFU/cell and fixed and permeabilized with −20°C methanol at the indicated times postinfection. Cells were stained both with a monoclonal antibody against PI4P and a polyclonal antibody against the poliovirus 3A protein. Download
dsRNA does not colocalize with the Golgi marker giantin. H1-Hela cells were infected at an MOI of 50 or mock infected and then fixed and permeabilized with −20°C methanol at the indicated times postinfection. Cells were stained using a monoclonal antibody against dsRNA and a polyclonal antibody against the Golgi protein giantin. Download
GFP-Sec31 is not recruited to the sites of poliovirus RNA replication. H1-Hela cells were transfected with GFP-Sec31 and 24 h later infected at an MOI of 50 PFU/cell and fixed and permeabilized with −20°C methanol at the indicated times postinfection. Cells were stained using dsRNA antibody for visualization of RNA replication sites. Download
Effect of MG132 on Sec31 levels during PV infection. H1-Hela cells were infected at an MOI of 50 PFU/cell. MG132 was used at various concentrations and added to the medium from the time of infection until collection, unless otherwise indicated. The rightmost lane used 20 µM MG132, but otherwise the protocol of N. Neznanov, E. M. Dragunsky, K. M. Chumakov, L. Neznanova, R. C. Wek, A. V. Gudkov, and A. K. Banerjee (PLoS One 3:e1887, 2008, doi:10.1371/journal.pone.0001887). was followed by pretreating cells with MG132 for two hours, infecting cells, and removing MG132 at 1 h p.i. Western blottings were performed for PV-3A, Sec31, and GAPDH. Download
ACKNOWLEDGMENT
This work was supported by grant AI104928 from the National Institutes of Health.
Footnotes
Citation Richards AL, Soares-Martins JAP, Riddell GT, Jackson WT. 2014. Generation of unique poliovirus RNA replication organelles. mBio 5(2):e00833-13. doi:10.1128/mBio.00833-13.
REFERENCES
- 1. Whitton JL, Cornell CT, Feuer R. 2005. Host and virus determinants of picornavirus pathogenesis and tropism. Nat. Rev. Microbiol. 3:765–776. 10.1038/nrmicro1284 [DOI] [PubMed] [Google Scholar]
- 2. Hope DA, Diamond SE, Kirkegaard K. 1997. Genetic dissection of interaction between poliovirus 3D polymerase and viral protein 3AB. J. Virol. 71:9490–9498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xiang W, Cuconati A, Hope D, Kirkegaard K, Wimmer E. 1998. Complete protein linkage map of poliovirus P3 proteins: interaction of polymerase 3Dpol with VPg and with genetic variants of 3AB. J. Virol. 72:6732–6741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bienz K, Egger D, Troxler M, Pasamontes L. 1990. Structural organization of poliovirus RNA replication is mediated by viral proteins of the P2 genomic region. J. Virol. 64:1156–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Seglen PO, Gordon PB. 1982. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. U. S. A. 79:1889–1892. 10.1073/pnas.79.6.1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Richards AL, Jackson WT. 2012. Intracellular vesicle acidification promotes maturation of infectious poliovirus particles. PLoS Pathog. 8:e1003046. 10.1371/journal.ppat.1003046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jackson WT, Giddings TH, Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. 2005. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3:e156. 10.1371/journal.pbio.0030156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Suhy DA, Giddings TH, Jr, Kirkegaard K. 2000. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J. Virol. 74:8953–8965. 10.1128/JVI.74.19.8953-8965.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Belov GA, Nair V, Hansen BT, Hoyt FH, Fischer ER, Ehrenfeld E. 2012. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 86:302–312. 10.1128/JVI.05937-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rust RC, Landmann L, Gosert R, Tang BL, Hong W, Hauri HP, Egger D, Bienz K. 2001. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J. Virol. 75:9808–9818. 10.1128/JVI.75.20.9808-9818.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee MC, Miller EA, Goldberg J, Orci L, Schekman R. 2004. Bi-directional protein transport between the ER and Golgi. Annu. Rev. Cell Dev. Biol. 20:87–123. 10.1146/annurev.cellbio.20.010403.105307 [DOI] [PubMed] [Google Scholar]
- 12. Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R. 1994. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 77:895–907. 10.1016/0092-8674(94)90138-4 [DOI] [PubMed] [Google Scholar]
- 13. Aridor M, Weissman J, Bannykh S, Nuoffer C, Balch WE. 1998. Cargo selection by the COPII budding machinery during export from the ER. J. Cell Biol. 141:61–70. 10.1083/jcb.141.1.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Salama NR, Chuang JS, Schekman RW. 1997. Sec31 encodes an essential component of the COPII coat required for transport vesicle budding from the endoplasmic reticulum. Mol. Biol. Cell 8:205–217. 10.1091/mbc.8.2.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tang BL, Zhang T, Low DY, Wong ET, Horstmann H, Hong W. 2000. Mammalian homologues of yeast sec31p. An ubiquitously expressed form is localized to endoplasmic reticulum (ER) exit sites and is essential for ER-Golgi transport. J. Biol. Chem. 275:13597–13604. 10.1074/jbc.275.18.13597 [DOI] [PubMed] [Google Scholar]
- 16. Schekman R, Orci L. 1996. Coat proteins and vesicle budding. Science 271:1526–1533. 10.1126/science.271.5255.1526 [DOI] [PubMed] [Google Scholar]
- 17. Trahey M, Oh HS, Cameron CE, Hay JC. 2012. Poliovirus infection transiently increases COPII vesicle budding. J. Virol. 86:9675–9682. 10.1128/JVI.01159-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maynell LA, Kirkegaard K, Klymkowsky MW. 1992. Inhibition of poliovirus RNA synthesis by brefeldin A. J. Virol. 66:1985–1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Niu TK, Pfeifer AC, Lippincott-Schwartz J, Jackson CL. 2005. Dynamics of GBF1, a brefeldin A-sensitive Arf1 exchange factor at the Golgi. Mol. Biol. Cell 16:1213–1222. 10.1091/mbc.E04-07-0599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Popoff V, Adolf F, Brügger B, Wieland F. 2011. COPI budding within the Golgi stack. Cold Spring Harb. Perspect. Biol. 3:a005231. 10.1101/cshperspect.a005231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Letourneur F, Gaynor EC, Hennecke S, Démollière C, Duden R, Emr SD, Riezman H, Cosson P. 1994. Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell 79:1199–1207. 10.1016/0092-8674(94)90011-6 [DOI] [PubMed] [Google Scholar]
- 22. Cosson P, Letourneur F. 1994. Coatomer interaction with di-lysine endoplasmic reticulum retention motifs. Science 263:1629–1631. 10.1126/science.8128252 [DOI] [PubMed] [Google Scholar]
- 23. Elazar Z, Orci L, Ostermann J, Amherdt M, Tanigawa G, Rothman JE. 1994. ADP-ribosylation factor and coatomer couple fusion to vesicle budding. J. Cell Biol. 124:415–424. 10.1083/jcb.124.4.415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Manolea F, Claude A, Chun J, Rosas J, Melançon P. 2008. Distinct functions for Arf guanine nucleotide exchange factors at the Golgi complex: GBF1 and BIGs are required for assembly and maintenance of the Golgi stack and trans-Golgi network, respectively. Mol. Biol. Cell 19:523–535. 10.1091/mbc.E07-04-0394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Belov GA, Fogg MH, Ehrenfeld E. 2005. Poliovirus proteins induce membrane association of GTPase ADP-ribosylation factor. J. Virol. 79:7207–7216. 10.1128/JVI.79.11.7207-7216.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Belov GA, Altan-Bonnet N, Kovtunovych G, Jackson CL, Lippincott-Schwartz J, Ehrenfeld E. 2007. Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. J. Virol. 81:558–567. 10.1128/JVI.01820-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Belov GA, Feng Q, Nikovics K, Jackson CL, Ehrenfeld E. 2008. A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLoS Pathog. 4:e1000216. 10.1371/journal.ppat.1000216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Belov GA, Ehrenfeld E. 2007. Involvement of cellular membrane traffic proteins in poliovirus replication. Cell Cycle 6:36–38. 10.4161/cc.6.1.3683 [DOI] [PubMed] [Google Scholar]
- 29. De Matteis MA, Godi A. 2004. Protein-lipid interactions in membrane trafficking at the Golgi complex. Biochim. Biophys. Acta 1666:264–274. 10.1016/j.bbamem.2004.07.002 [DOI] [PubMed] [Google Scholar]
- 30. Levine TP, Munro S. 2002. Targeting of Golgi-specific pleckstrin homology domains involves both PtdIns 4-kinase-dependent and -independent components. Curr. Biol. 12:695–704. 10.1016/S0960-9822(02)00779-0 [DOI] [PubMed] [Google Scholar]
- 31. Godi A, Pertile P, Meyers R, Marra P, Di Tullio G, Iurisci C, Luini A, Corda D, De Matteis MA. 1999. ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat. Cell Biol. 1:280–287. 10.1038/12993 [DOI] [PubMed] [Google Scholar]
- 32. Hsu NY, Ilnytska O, Belov G, Santiana M, Chen YH, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T, Cameron CE, Ehrenfeld E, van Kuppeveld FJ, Altan-Bonnet N. 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141:799–811. 10.1016/j.cell.2010.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. De Matteis MA, D’Angelo G. 2007. The role of the phosphoinositides at the Golgi complex. Biochem. Soc. Symp. 74:107–116 [DOI] [PubMed] [Google Scholar]
- 34. Ilnytska O, Santiana M, Hsu NY, Du WL, Chen YH, Viktorova EG, Belov G, Brinker A, Storch J, Moore C, Dixon JL, Altan-Bonnet N. 2013. Enteroviruses harness the cellular endocytic machinery to remodel the host cell cholesterol landscape for effective viral replication. Cell Host Microbe 14:281–293. 10.1016/j.chom.2013.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Téoulé F, Brisac C, Pelletier I, Vidalain PO, Jégouic S, Mirabelli C, Bessaud M, Combelas N, Autret A, Tangy F, Delpeyroux F, Blondel B. 2013. The Golgi protein ACBD3, an interactor for poliovirus protein 3A, modulates poliovirus replication. J. Virol. 87:11031–11046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Greninger AL, Knudsen GM, Betegon M, Burlingame AL, Derisi JL. 2012. The 3A protein from multiple picornaviruses utilizes the Golgi adaptor protein ACBD3 to recruit PI4KIIIbeta. J. Virol. 86:3605–3616. 10.1128/JVI.06778-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schönborn J, Oberstrass J, Breyel E, Tittgen J, Schumacher J, Lukacs N. 1991. Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res. 19:2993–3000. 10.1093/nar/19.11.2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Agol VI, Paul AV, Wimmer E. 1999. Paradoxes of the replication of picornaviral genomes. Virus Res. 62:129–147. 10.1016/S0168-1702(99)00037-4 [DOI] [PubMed] [Google Scholar]
- 39. Caliguiri LA, Tamm I. 1968. Action of guanidine on the replication of poliovirus RNA. Virology 35:408–417. 10.1016/0042-6822(68)90219-5 [DOI] [PubMed] [Google Scholar]
- 40. Lama J, Sanz MA, Rodríguez PL. 1995. A role for 3AB protein in poliovirus genome replication. J. Biol. Chem. 270:14430–14438. 10.1074/jbc.270.24.14430 [DOI] [PubMed] [Google Scholar]
- 41. Strauss DM, Glustrom LW, Wuttke DS. 2003. Towards an understanding of the poliovirus replication complex: the solution structure of the soluble domain of the poliovirus 3A protein. J. Mol. Biol. 330:225–234. 10.1016/S0022-2836(03)00577-1 [DOI] [PubMed] [Google Scholar]
- 42. Xiang W, Cuconati A, Paul AV, Cao X, Wimmer E. 1995. Molecular dissection of the multifunctional poliovirus RNA-binding protein 3AB. RNA 1:892–904 [PMC free article] [PubMed] [Google Scholar]
- 43. Tanida I. 2011. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 14:2201–2214. 10.1089/ars.2010.3482 [DOI] [PubMed] [Google Scholar]
- 44. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720–5728. 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taylor MP, Kirkegaard K. 2007. Modification of cellular autophagy protein LC3 by poliovirus. J. Virol. 81:12543–12553. 10.1128/JVI.00755-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, Ohsumi M, Takao T, Noda T, Ohsumi Y. 2000. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 151:263–276. 10.1083/jcb.151.2.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, Aleo MF, Alirezaei M, Almasan A, Almonte-Becerril M, Amano A, Amaravadi R, Amarnath S, Amer AO, Andrieu-Abadie N, Anantharam V, Ann DK, Anoopkumar-Dukie S, Aoki H, Apostolova N, Auberger P, Baba M, Backues SK, Baehrecke EH, Bahr BA, Bai XY, Bailly Y, Baiocchi R, Baldini G, Balduini W, Ballabio A, Bamber BA, Bampton ET, Banhegyi G, Bartholomew CR, Bassham DC, et al. 2012. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8:445–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chun J, Shapovalova Z, Dejgaard SY, Presley JF, Melançon P. 2008. Characterization of class I and II ADP-ribosylation factors (Arfs) in live cells: GDP-bound class II Arfs associate with the ER-Golgi intermediate compartment independently of GBF1. Mol. Biol. Cell 19:3488–3500. 10.1091/mbc.E08-04-0373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wessels E, Duijsings D, Lanke KH, van Dooren SH, Jackson CL, Melchers WJ, van Kuppeveld FJ. 2006. Effects of picornavirus 3A proteins on protein transport and GBF1-dependent COP-I recruitment. J. Virol. 80:11852–11860. 10.1128/JVI.01225-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rothman JE. 1996. The protein machinery of vesicle budding and fusion. Protein Sci. 5:185–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peter F, Plutner H, Zhu H, Kreis TE, Balch WE. 1993. Beta-COP is essential for transport of protein from the endoplasmic reticulum to the Golgi in vitro. J. Cell Biol. 122:1155–1167. 10.1083/jcb.122.6.1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Han YH, Moon HJ, You BR, Park WH. 2009. The effect of MG132, a proteasome inhibitor on HeLa cells in relation to cell growth, reactive oxygen species and GSH. Oncol. Rep. 22:215–221 [PubMed] [Google Scholar]
- 53. Schnellmann RG, Williams SW. 1998. Proteases in renal cell death: calpains mediate cell death produced by diverse toxicants. Ren. Fail. 20:679–686. 10.3109/08860229809045162 [DOI] [PubMed] [Google Scholar]
- 54. Quiner CA, Jackson WT. 2010. Fragmentation of the Golgi apparatus provides replication membranes for human rhinovirus 1A. Virology 407:185–195. 10.1016/j.virol.2010.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bolten R, Egger D, Gosert R, Schaub G, Landmann L, Bienz K. 1998. Intracellular localization of poliovirus plus- and minus-strand RNA visualized by strand-specific fluorescent in situ hybridization. J. Virol. 72:8578–8585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Szul T, Garcia-Mata R, Brandon E, Shestopal S, Alvarez C, Sztul E. 2005. Dissection of membrane dynamics of the ARF-guanine nucleotide exchange factor GBF1. Traffic 6:374–385. 10.1111/j.1600-0854.2005.00282.x [DOI] [PubMed] [Google Scholar]
- 57. Presley JF, Ward TH, Pfeifer AC, Siggia ED, Phair RD, Lippincott-Schwartz J. 2002. Dissection of COPI and Arf1 dynamics in vivo and role in Golgi membrane transport. Nature 417:187–193. 10.1038/417187a [DOI] [PubMed] [Google Scholar]
- 58. Alvarez C, Garcia-Mata R, Brandon E, Sztul E. 2003. COPI recruitment is modulated by a Rab1b-dependent mechanism. Mol. Biol. Cell 14:2116–2127. 10.1091/mbc.E02-09-0625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Belov GA, Kovtunovych G, Jackson CL, Ehrenfeld E. 2010. Poliovirus replication requires the N-terminus but not the catalytic Sec7 domain of ArfGEF GBF1. Cell. Microbiol. 12:1463–1479. 10.1111/j.1462-5822.2010.01482.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wong K, Meyers ddR, Cantley LC. 1997. Meyers d, Cantley LC. Subcellular locations of phosphatidylinositol 4-kinase isoforms. J. Biol. Chem. 272:13236–13241. 10.1074/jbc.272.20.13236 [DOI] [PubMed] [Google Scholar]
- 61. Fan J, Liu J, Culty M, Papadopoulos V. 2010. Acyl-coenzyme A binding domain containing 3 (ACBD3; PAP7; GCP60): an emerging signaling molecule. Prog. Lipid Res. 49:218–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Beske O, Reichelt M, Taylor MP, Kirkegaard K, Andino R. 2007. Poliovirus infection blocks ERGIC-to-Golgi trafficking and induces microtubule-dependent disruption of the Golgi complex. J. Cell Sci. 120:3207–3218. 10.1242/jcs.03483 [DOI] [PubMed] [Google Scholar]
- 63. Hawes P, Netherton CL, Mueller M, Wileman T, Monaghan P. 2007. Rapid freeze-substitution preserves membranes in high-pressure frozen tissue culture cells. J. Microsc. 226:182–189. 10.1111/j.1365-2818.2007.01767.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Colocalization of GBF1 and GM130 in uninfected cells. H1-Hela cells were mock infected and fixed and permeabilized with −20°C methanol at 6 h postinfection. Cells were stained using a monoclonal antibody against the Golgi protein GM130 and a polyclonal antibody against the endogenous GBF1. Download
PV 3A colocalizes with phosphatidylinositol-4-phosphate (PI4P) during PV infection. H1-Hela cells were infected at an MOI of 50 PFU/cell and fixed and permeabilized with −20°C methanol at the indicated times postinfection. Cells were stained both with a monoclonal antibody against PI4P and a polyclonal antibody against the poliovirus 3A protein. Download
dsRNA does not colocalize with the Golgi marker giantin. H1-Hela cells were infected at an MOI of 50 or mock infected and then fixed and permeabilized with −20°C methanol at the indicated times postinfection. Cells were stained using a monoclonal antibody against dsRNA and a polyclonal antibody against the Golgi protein giantin. Download
GFP-Sec31 is not recruited to the sites of poliovirus RNA replication. H1-Hela cells were transfected with GFP-Sec31 and 24 h later infected at an MOI of 50 PFU/cell and fixed and permeabilized with −20°C methanol at the indicated times postinfection. Cells were stained using dsRNA antibody for visualization of RNA replication sites. Download
Effect of MG132 on Sec31 levels during PV infection. H1-Hela cells were infected at an MOI of 50 PFU/cell. MG132 was used at various concentrations and added to the medium from the time of infection until collection, unless otherwise indicated. The rightmost lane used 20 µM MG132, but otherwise the protocol of N. Neznanov, E. M. Dragunsky, K. M. Chumakov, L. Neznanova, R. C. Wek, A. V. Gudkov, and A. K. Banerjee (PLoS One 3:e1887, 2008, doi:10.1371/journal.pone.0001887). was followed by pretreating cells with MG132 for two hours, infecting cells, and removing MG132 at 1 h p.i. Western blottings were performed for PV-3A, Sec31, and GAPDH. Download