

Abstract
We describe a synthesis strategy for the preparation of lysine isotopologues that differ in mass by as little as 6 mDa. We demonstrate that incorporation of these molecules into the proteomes of actively growing cells does not affect cellular proliferation, and we discuss how to use the embedded mass signatures (neutron encoding (NeuCode)) for multiplexed proteome quantification by means of high-resolution mass spectrometry. NeuCode SILAC amalgamates the quantitative accuracy of SILAC with the multiplexing of isobaric tags and, in doing so, offers up new opportunities for biological investigation. We applied NeuCode SILAC to examine the relationship between transcript and protein levels in yeast cells responding to environmental stress. Finally, we monitored the time-resolved responses of five signaling mutants in a single 18-plex experiment.
Large-scale technologies for the comparative analysis of proteomes have become essential for modern biology and medicine (1, 2). To satisfy this increasing demand and boost statistical power, parallel processing of proteomes (i.e. multiplexing) is key. The ground was broken in this field about two decades ago by advances in both MS and stable isotope labeling (3–5). Since then, two distinct strategies have emerged, each with its own strengths and weaknesses. The first approach, stable isotope labeling by amino acids in cell culture (SILAC),1 metabolically incorporates labeled amino acids into proteins and is considered the gold standard (6–8). SILAC quantification provides unmatched accuracy, but simultaneous comparison of more than three proteomes, although possible, is not practical for most global studies (9, 10). A second, and increasingly popular, method is to chemically modify peptides originating from up to 10 different sources (a 3- to 5-fold boost in throughput over SILAC) with isobaric reagents (e.g. TMT or iTRAQ) (11–14). The escalated throughput afforded by this strategy is, for many applications, essential; however, multiplexing via isobaric tagging comes at the cost of quantitative accuracy (15–17). Furthermore, because each sample is handled independently prior to labeling, systematic and random variation that occurs during sample processing cannot be accounted for as it is with metabolic labeling. Thus, experimenters designing a quantitative proteomics experiment must choose between accuracy and throughput.
Recently we described a new approach that blends the SILAC and isobaric tagging methods (18). The strategy, neutron encoding (NeuCode), relies on the mass defects of atoms and their isotopes (19). In studies using two isotopologues of lysine, differing by 36 mDa, NeuCode SILAC quantified proteins as well as traditional SILAC, but it allowed deeper proteome coverage. NeuCode harnesses the exceptional resolving power of modern FT-MS systems so that quantitative information is only revealed by high-resolution scanning when desired, in either MS or tandem MS scans (20, 21).
Owing to the lack of suitable lysine isotopologues, our initial work with NeuCode SILAC offered only duplex quantification; consequently, we could only predict the utility of NeuCode SILAC (18). To test our supposition that NeuCode SILAC has the potential to combine the benefits of traditional SILAC and isobaric tagging, we began the synthesis of novel lysine isotopologues. Here we present a synthetic route that allows for precise tuning of C, H, and N stable isotopes in lysine. With this strategy we generated four new lysine isotopologues that, when combined with existing lysines, deliver 6-plex NeuCode SILAC quantification. We determined that yeast cell growth is not affected by the isotopic composition, or “flavor,” of the lysine—a prerequisite for metabolic labeling. Next, we used the six closely spaced lysine isotopologues (6 mDa) to test theoretical calculations that peptides bearing these signatures could indeed be detected (i.e. resolved) with current commercially available FT-MS systems.
With the synthetic and theoretical assertions of NeuCode SILAC performance vetted, we benchmarked the new approach against traditional SILAC, presenting on a variety of figures of merit, including sampling depth, quantitative precision and accuracy, throughput, and plexing capacity. Having achieved favorable performance in this comparison, we then used NeuCode SILAC to generate a quantitative sketch of the yeast proteome during environmental stress. We first examined the relationship between protein levels (measured via NeuCode SILAC and TMT isobaric tags, separately) and their corresponding transcripts in cells exposed to salt stress. Here, using quantitative measurements for over 4,000 proteins, we reveal that a wider dynamic range equips NeuCode SILAC with greater sensitivity and discovery potential. We then monitored the temporal responses of an additional five signaling yeast mutants by coupling NeuCode SILAC with nominal mass difference tagging. The 18 plexes of quantification offered by this approach greatly expand the scale of comparative proteome analysis.
EXPERIMENTAL PROCEDURES
Theoretical Calculations
A library of 71,499 yeast Lys-C peptides identified via mass spectrometry was compiled. The theoretical full width at 10% maximum (FWTM) peak height for each library peptide is calculated by
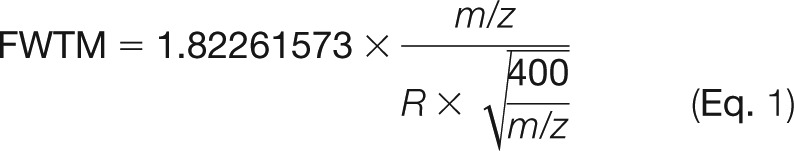 |
where the resolving power R is defined as the minimum m/z difference that can be resolved at 400 m/z, and the coefficient (1.82261573) is derived from Gaussian peak shape modeling. The m/z difference (Δm/z) for each theoretical isotope set, assuming lysine isotopologue mass differences (ΔI) of 6, 12, 18, and 36 mDa, is given by
 |
where n is the number of lysines in the peptide sequence and z is the charge of the peptide. An isotopologue set is considered resolvable only if Δm/z > FWTM at the given resolving power and isotopologue mass difference.
Synthesis of Lysine Isotopologues
Four isotopologues of lysine with eight additional neutrons (M + 8) were targeted for synthesis carrying various ratios of 13C, 2H, and 15N incorporation (K#13C#2H#15N): K422, K521, K341, and K440. A common synthetic route to these compounds was developed based on the protected glutamic acid derivatives carrying the 13C and 15N substitutions required at C1–C5 of the target lysine. Reduction of the free acid moiety with either sodium borohydride or sodium borodeuteride allowed the introduction of two deuterium atoms into the target, if required. The reduced alcohol was then converted to the iodide via standard chemistry. Displacement of the iodide with labeled potassium cyanide provided the means to introduce further labels at the 6-position of the final product, depending on the labeling pattern of the cyanide selected. Finally, reduction of the cyanide under catalytic conditions with either deuterium or hydrogen gas provided the option of adding two further deuterium atoms, if required for the final target. The products were purified by means of ion exchange chromatography and recrystallized. The overall purity was typically 97% to 99%, as determined via HPLC analysis against an authentic standard, and enrichments were ≥98%. The overall yield for the process was 40% to 50%.
Yeast Cell Growth and Salt Stress
All Saccharomyces cerevisiae strains were of the BY4742 background (supplemental Table S3) and, except for BY MATα hog1Δ::KANMX lys2Δ0 and BY MATα dot6Δ::KANMX tod6Δ::HYGMX lys2Δ0, were acquired from Open Biosystems, Lafayette, CO (22, 23). To control for effects of the KANMX cassette in this histidine-auxotrophic strain background, BY4742 his3Δ::KANMX was used as the WT reference. Deletion strains were verified by means of diagnostic PCR. The hog1Δ::KANMX lys2Δ0 MATα and dot6Δ::KANMX tod6Δ::HYGMX lys2Δ0 MATα strains were generated by mating BY4741-hog1Δ::KANMX (AGY260) and BY4741-dot6Δ::KANMX tod6Δ::HYGMX (AGY594) (24) with BY4742. Following sporulation, tetrads were dissected (25) and cells were subjected to diagnostic PCR to verify the loss of LYS2 and maintenance of the original deletion(s). Subsequently, a halo mating assay was performed, and MATα variants were used for all experiments (26).
For the mixed ratio experiments, S. cerevisiae strain BY4742 was grown in defined, synthetic dropout (SC-lys, Sunrise Science, San Diego, CA) medium supplemented with one of eight lysines (supplemental Table S1): K000 (Sigma Aldrich), K040 (Cambridge Isotopes, Andover, MA), K602 (Cambridge Isotopes), K422 (Cambridge Isotopes), K521 (Cambridge Isotopes), K341 (Cambridge Isotopes), K440 (Cambridge Isotopes), or K080 (Cambridge Isotopes). Cells were allowed to propagate for a minimum of 10 doublings at 30 °C to ensure complete lysine incorporation. Upon reaching mid-log phase, the cells were harvested via centrifugation at 3,000 rpm for 3 min, washed three times with chilled double distilled H20, flash-frozen in liquid nitrogen, and stored at −80 °C until use.
For the 4-plex salt stress experiments, S. cerevisiae strain BY4742-his3Δ::KANMX was grown in SC-lys medium supplemented with K000, K602, K521, K440, or K080. Upon reaching mid-log phase, cells were either immediately harvested (time 0, unstressed reference) or collected 60 min after exposure to 0.7 m NaCl (time 60). For the higher plexing salt stress experiments, S. cerevisiae strains BY4742-his3Δ::KANMX, BY4742-sto1Δ::KANMX (AGY0877), hog1Δ::KANMX lys2Δ0 MATα (AGY0863), BY4742-rck2Δ::KANMX (AGY0866), cka2Δ::KANMX (AGY0867), and dot6Δ::KANMX tod6Δ::HYGMX lys2Δ0 MATα (AGY0865) were grown in SC-lys medium supplemented with K602, K422, K521, K341, K440, and K080, respectively. Upon reaching mid-log phase, a sample of unstressed cells was removed from each of the six cultures. Pre-warmed medium was added for a final concentration of 0.7 m NaCl. Samples were removed at 60 and 240 min post-NaCl treatment. In order to maintain log-phase growth at the 240-min collection, the cultures were diluted with pre-warmed medium to a final concentration of 0.7 m NaCl following the 60-min collection.
Cell pellets were resuspended in lysis buffer (50 mm Tris, pH 8.0, 8 m urea, 75 mm sodium chloride, 100 mm sodium butyrate, 1 mm sodium orthovanadate, protease and phosphatase inhibitor tablet), and total protein was extracted via glass-bead milling (Retsch, Haan, Germany).
Microarray Sample Preparation and Data Analysis
BY4742- his3Δ::KANMX was grown in batch at 30 °C in SC medium for at least seven generations, reaching mid-log phase before an unstressed reference sample was removed. Cultures were then subjected to a final concentration of 0.7 m NaCl, and samples were removed 30 (T30) and 60 (T60) minutes after salt addition. Cell collection, RNA preparation, cDNA synthesis, and labeling were performed as previously described (27), using cyanine dyes (Flownamics, Madison, WI), amino-allyl-dUTP (Ambion, Grand Island, NY), and Superscript III (Invitrogen). In order to determine NaCl-dependent gene expression changes, we mixed cDNA from the unstressed reference sample with cDNA from either the T30 or the T60 salt-treated sample and hybridized to whole-genome tiled DNA microarrays (Roche Nimblegen). Arrays were scanned on a GenePix4000 scanner (Molecular Devices, Sunnyvale, CA), and data normalization was performed according to a previously described method (28). Gene expression differences were determined by taking the log2 of the green/red signals. Genes exhibiting NaCl-induced differential expression were defined as having a greater than 1.5-fold change between the reference and salt-treated samples. Genes not meeting this cutoff were considered to be non–salt responsive. Arrays were performed in biological singlet. Array data are available through the NIH at GEO accession # GSE53549.
Proteomic Sample Preparation
Lysate protein concentration was measured via BCA (Pierce). Protein disulfide bonds were reduced by the addition of 5 mm dithiothreitol and incubation for 45 min at 55 °C. Free thiols were alkylated by the addition of 15 mm iodoacetamide and incubation in the dark, at ambient temperature, for 45 min. The alkylation reaction was quenched by the addition of 5 mm dithiothreitol. The urea concentration was diluted to 4 m with 50 mm Tris, pH 8.0. Proteolytic digestion was accomplished by Lys-C (Wako, Richmond, VA) added at a 1:50 enzyme-to-protein ratio and incubation at ambient temperature for 16 h. For the last hour, another aliquot of Lys-C (1:50 enzyme:protein) was added. The digestion reaction was quenched by the addition of TFA, and then samples were desalted with tC18 Sep-Pak cartridges (Waters, Milford, MA).
Traditional SILAC known ratios were prepared by mixing labeled yeast peptides 1:1, 1:10, 1:1:1, and 1:10:2 by mass. NeuCode SILAC known ratios were prepared by mixing labeled yeast peptides 1:1, 1:10, 1:1:1, 1:10:2, 1:1:1:1, 1:10:2:5, 1:1:1:1:1:1, and 1:10:2:5:1:10 by mass. For the 4-plex yeast salt stress experiment, NeuCode SILAC peptides were mixed in equal amounts by mass. Unlabeled peptides were labeled with four TMT tags (Pierce) according to the manufacturer's protocol and mixed in equal amounts. NeuCode SILAC and TMT samples were each fractionated via strong cation exchange chromatography. For each method, a total of 12 fractions were collected, lyophilized, and desalted on tC18 Sep-Pak cartridges (Waters). For the higher plexing yeast salt stress experiments, NeuCode SILAC peptides from each time point were mixed in equal amounts by mass for duplex, triplex, 4-plex, and 6-plex configurations. Mixtures from each time point were differentially labeled using reductive dimethylation to create a maximum of three distinct isotope clusters that each contained up to six isotopologues (29).
Nano-LC-MS/MS
Reversed phase columns were prepared in-house. Briefly, a 75–360 μm inner–outer diameter bare-fused silica capillary with a laser pulled electrospray tip was packed in 1.7-μm diameter, 130-Å pore size Bridged Ethylene Hybrid C18 particles (Waters) to a final length of 30 cm. The column was installed on a nanoAcquity UPLC (Waters) using a stainless steel ultra-high-pressure union (IDEX, Oak Harbor, WA) and heated to 60 °C for all runs. Mobile phase A was composed of water, 0.2% formic acid, and 5% dimethyl sulfoxide. Mobile phase B was composed of acetonitrile and 0.2% formic acid. The known ratio and 4-plex salt stress experiments used a 70-min gradient, whereas the higher plexing salt stress experiments used a 210-min gradient.
Eluting peptide cations were converted to gas-phase ions by electrospray ionization and analyzed on an Orbitrap Elite mass spectrometer (Thermo Scientific). For NeuCode SILAC analyses, a survey scan was performed by the Orbitrap at 30,000 resolving power to identify precursors to sample for data-dependent, top-20 ion trap CAD MS/MS (rapid scan analysis). An additional, quantitative 480,000 or 960,000 resolving power scan immediately followed the survey scan. Preview mode was enabled. Precursors with unknown charge or a charge of +1 were excluded from MS/MS. MS1 and MS/MS target-ion accumulation values were set to 1 × 106 and 3 × 103, respectively. Monoisotopic precursor selection was turned on. Dynamic exclusion was set to 45 s for −25 ppm and +15 ppm around the selected precursor and its isotopes. All other analyses were conducted in the same manner but utilized survey scans at 60,000 resolution for both precursor sampling and peptide quantification and excluded a 10-ppm tolerance around the selected precursor and its isotopes. For TMT analyses, data-dependent, top-15 MS/MS (5 × 104 target-ion accumulation value) was performed via HCD fragmentation with a normalized collision energy of 35 and MS analysis in the Orbitrap at 15,000 resolving power. All scan sequences employed can be configured using the standard method editor, Xcalibur (Thermo Scientific).
Data Processing
MS raw files were converted to searchable text files using DTA Generator (v. 1.1) and searched against a target-decoy (30) database (Saccharomyces Genome Database (yeast), February 3, 2011) using the Open Mass Spectrometry Search Algorithm (v. 2.1.8) (31, 32). For all samples, cysteine carbamidomethylation and methionine oxidation were searched as fixed and variable modifications, respectively. In addition, all samples were searched with LysC specificity and up to three missed cleavages. Traditional SILAC samples were searched independently with fixed lysine modifications of +0.0000 Da (K000), +4.0251 Da (K040), and +8.0142 Da (K602) and were later combined during false discovery rate (FDR) filtering. NeuCode SILAC samples were searched with a single fixed lysine modification (+8.0322 Da) that represented the average mass increase of the K602 and K080 isotopologues. Dimethyl-labeled samples were searched independently with fixed N terminus and lysine modifications of +28.0313 Da (“light dimethyl”), +32.0564 Da (“medium dimethyl”), and +36.0757 Da (“heavy dimethyl”) and were later combined during FDR filtering. TMT-labeled samples were searched with fixed N terminus and lysine modifications and variable tyrosine modification of +229.1629 Da. The precursor mass tolerance was defined as 150 ppm, and the fragment ion mass tolerance was set at 0.35 Da (ion trap MS/MS) and 0.015 Da (FT-MS/MS). Search results were filtered to a 1% FDR based on precursor mass error and E-value within the COMPASS environment (32). Peptides were grouped into proteins and filtered to a 1% FDR according to rules previously described (33).
Quantification
TMT-labeled peptides were quantified using the TagQuant module of COMPASS (32). All forms of MS1-based peptide quantification (e.g. traditional SILAC, NeuCode SILAC, reductive dimethylation) were performed using NeuQuant, a module developed in-house to fit within the COMPASS workflow. After database searching, the FDR-filtered list of peptide spectral matches was first used to calculate the systematic mass error associated with the dataset and to adjust precursor masses accordingly. Then, for every peptide deemed theoretically resolvable at FWTM at the given spacing and resolving power, a list containing all high-resolution MS1 scans within ±30 s of a peptide spectral match was compiled. This retention window extracted quantitative data throughout a peptide's elution to accommodate any shifts in chromatographic retention due to deuterium-containing labels. If the proper number of peaks, each with a signal-to-noise ratio greater than 3, were found within the specified tolerance (5 ppm below the lightest isotopologue's m/z to 5 ppm above the heaviest isotopologue's m/z for NeuCode SILAC; ±5 ppm around each m/z for traditional SILAC and dimethyl labeling), that particular isotope and MS1 scan were considered for peptide quantification. Any peaks below the noise level simply contributed a noise-based intensity to the appropriate missing channel. Once sets of quantitative peaks were assembled, they were discarded if they didn't meet the spacing criteria (<20% error for NeuCode SILAC mDa spacings; <0.5% error for traditional SILAC and dimethyl labeling Da spacings). Quantitative peaks were isolated and evaluated in this manner for the mono- and first two isotopes of the isotopic cluster and for each relevant MS1 spectrum. If a peptide had three or more quantitative measurements (i.e. isotopes or MS1 scans), it was quantified by summing the channel intensities across all measurements. Any peak intensities below a threshold relative to the maximum channel intensity were excluded; typically a threshold of 1/(2e) times the maximum intensity is employed, where e is the mathematical constant, which represents 1.75 standard deviations (90% area) of the standard normal distribution. Protein quantification was accomplished by averaging the ratios of all corresponding peptides and manually normalizing to a median fold change of 1 to account for unequal mixing.
RESULTS
Synthesis of Novel Lysine Isotopologues
With M + 8 lysine as our scaffold, and considering all non-exchangeable atoms—six carbons, nine hydrogens, and two nitrogens—as potential neutron recipients, there were 21 distinct entities (supplemental Fig. S1). These isotopologues span a 36-mDa range and can be coupled to match instrument resolution. The extreme isotopologues, for example, require a resolving power of 120,000 (m/z 400, Orbitrap analyzer) for quantification (i.e. ∼70% of peptides are resolved, FWTM), permitting duplex NeuCode SILAC quantification (Fig. 1A). 3-, 4-, and 7-plex NeuCode SILAC can be achieved by reducing isotopologue spacing to 18, 12, and 6 mDa, respectively. As spacing is decreased, MS resolving power must increase, up to 960,000 for 6 mDa (Fig. 1A). The utility of NeuCode SILAC for highly plexed proteome quantification therefore hinges on two factors: MS resolving power and reagent availability. Both ion cyclotron resonance and Orbitrap analyzers achieve resolving powers greater than 106, with commercial Orbitrap systems reaching at least 480,000 (20, 21). We conclude that the expansion of NeuCode SILAC plexing is principally limited by lysine isotopologue availability.
Fig. 1.

Synthesis of novel lysine isotopologues for NeuCode SILAC. A, theoretical calculation depicting the percentage of peptides that are resolved (full width at 10% maximum peak height (FWTM)) when spaced 6, 12, 18, or 36 mDa apart for resolving powers from 15,000 to 1 million (m/z 400, Orbitrap analyzer). B, four synthetic targets of lysine M + 8. C, the developed common synthetic route to the target compounds. D, synthetic lysine products are easily resolved in Orbitrap MS1 scans collected at 480,000 resolving power.
Of the 21 theoretical M + 8 lysine isotopologues, only two have been synthesized (supplemental Fig. S1, black). We developed a common synthetic route to generate four additional isotopologues (Fig. 1B; supplemental Fig. S1, red): protected glutamic acid derivatives carried the 13C and 15N substitutions required at C1–C5 of the target lysine; reduction of the free acid moiety introduced two deuterium atoms, if required; and conversion of the reduced alcohol to the iodide and subsequent displacement of the iodide introduced further labels at the 6-position of the final product. Finally, reduction of the cyanide inserted two more deuterium atoms, if required. The products were purified to 97% to 99% for final use. Note that this synthetic route provides a template from which to build additional lysine isotopologues for further NeuCode SILAC development (Fig. 1C).
Following synthesis, we verified that the experimental masses of all six M + 8 lysine isotopologues were within 1 ppm of their theoretical values (Fig. 1D). Next, we grew yeast separately on media supplemented with each of eight different lysines (supplemental Table S1) to assess defects in cell growth. Regardless of isotopologue, we found that the various stable isotopes did not affect rates of cellular proliferation in this biological system (one-way analysis of variance, p > 0.5).
Characterizing NeuCode SILAC Quantitative Performance
Using these novel lysine isotopologues, we characterized quantitative figures of merit for NeuCode SILAC. Proteins encoded with each of the eight lysines were LysC-digested, and the resulting peptides were mixed in equal ratios by mass for various combinations (Table I). For the unlabeled and traditional SILAC control samples, a 60,000 resolving power survey MS scan preceded 20 data-dependent MS/MS ion trap scans. NeuCode SILAC analysis utilized a medium-resolution (30,000) survey scan to dictate MS/MS sampling, and while the ion trap handled the tandem MS events, the Orbitrap collected a high-resolution (480,000) MS1 to reveal the embedded NeuCode SILAC labels permitting quantification (Fig. 2A). When conducted in this parallelized manner, the high-resolution scan minimally affects the overall duty cycle (Fig. 2B).
Table I. Lysine combinations for multiplexed traditional SILAC and NeuCode SILAC experiments.
| Label type | Number of channels | Lysines used |
|---|---|---|
| Unlabeled | 1 | K000 |
| Traditional SILAC | 2 | K000, K602 |
| Traditional SILAC | 3 | K000, K040, K602 |
| NeuCode SILAC | 2 | K602, K080 |
| NeuCode SILAC | 3 | K602, K341, K080 |
| NeuCode SILAC | 4 | K602, K521, K440, K080 |
| NeuCode SILAC | 6 | K602, K422, K521, K341, K440, K080 |
Fig. 2.
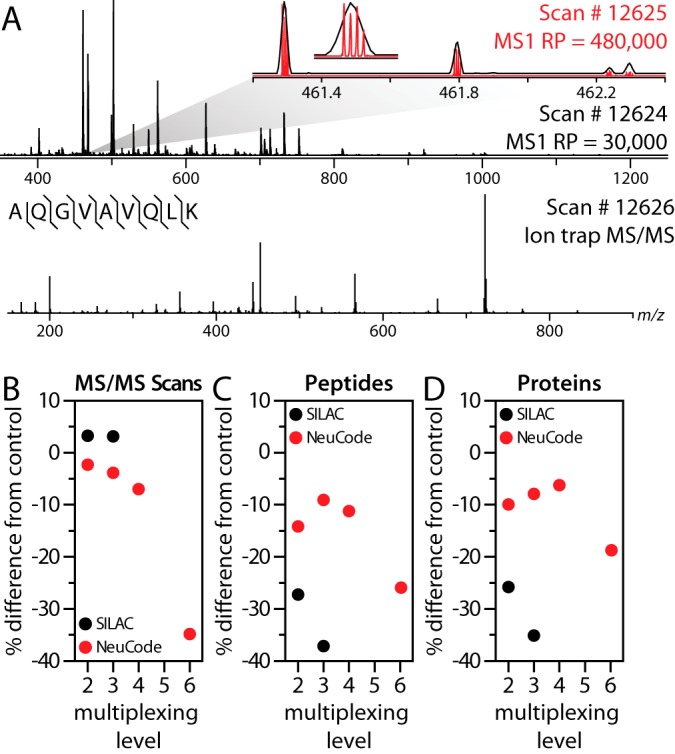
Resolving power conceals and reveals NeuCode-embedded signatures for peptide identification and quantification. A, MS1 scan collected with 30,000 resolving power from a nano-LC-MS/MS analysis of yeast LysC peptides and (inset) of a selected precursor at 461 m/z (black trace). The signal recorded in a subsequent high-resolution MS1 scan (480,000 resolving power; red trace) reveals the quantitative data. Presented below the MS1 scan is an MS/MS spectrum following CAD and ion trap m/z analysis of the NeuCode peptide. SILAC (2-plex and 3-plex) and NeuCode (2-plex, 3-plex, 4-plex, and 6-plex) at equal mixing ratios are compared with an unlabeled sample for (B) MS/MS spectra acquired, (C) peptide identifications, and (D) protein identifications. Traditional SILAC analyses utilize 60,000 resolving power MS1 scans for quantification. All NeuCode SILAC analyses utilize 480,000 resolving power, except for 6-plex, which utilizes 960,000.
One unique asset of the NeuCode SILAC label is that its visibility is resolution-dependent, providing a substantial benefit in proteome coverage. High resolving powers authorize quantification, but under low to medium resolving powers NeuCode SILAC peptides appear unlabeled, which leaves other important processes, such as MS/MS sampling, unaffected. Relative to an unlabeled control, NeuCode SILAC samples (up to 4-plex) incur only a 5% to 15% reduction in peptide and protein identifications (Figs. 2C and 2D). As a result of redundant sampling, traditional SILAC labeling generates at least 25% fewer identifications. Strikingly, identifications obtained from 6-plex NeuCode SILAC requiring 960,000 resolving power for quantification still surpassed both traditional SILAC analyses.
Encouraged by the significant boost in sampling depth attainable with NeuCode SILAC, we next benchmarked the quantitative accuracy and precision of NeuCode SILAC against that of traditional SILAC. For both approaches, we mixed peptides encoded with the appropriate lysine isotopologues in 1:10 (duplex) and 1:10:2 (triplex) ratios by mass (Table I). Peptide and protein measurements afforded by traditional SILAC and NeuCode SILAC were similar in terms of accuracy and precision (Figs. 3A and 3B). For example, 10-fold peptide/protein ratios were computed as 8.8/8.2 (duplex traditional SILAC), 9.3/9.3 (duplex NeuCode SILAC), 7.3/7.3 (triplex traditional SILAC), and 11.0/9.9 (triplex NeuCode SILAC). When measured via 4-plex and 6-plex NeuCode SILAC, median ratios still were within 20% of the expected values (Figs. 3C and 3D), indicating that quantitative accuracy and precision are maintained at higher levels of multiplexing.
Fig. 3.

Peptide and protein quantification via traditional SILAC and NeuCode SILAC. Boxplots showing the measured (box and whiskers) and true (dashed line) values for traditional SILAC (black) and NeuCode SILAC (red) (A) peptides and (B) proteins at mixing ratios of 1:10 (2-plex) and 1:10:2 (3-plex). Boxplots demarcate the median (stripe), the 25th to 75th percentile (interquartile range; box), 1.5 times the interquartile range (whiskers), and outliers (open circles). Boxplots showing the measured and true values for NeuCode (C) peptides and (D) proteins at mixing ratios of 1:10:2:5 (4-plex; 480,000 MS1 resolving power) and 1:10:2:5:1:10 (6-plex; 960,000 MS1 resolving power).
Benchmarking Quantitative Dynamic Range in Yeast Stress Response
Capturing the dynamics of complex proteomes demands sensitive methods for quantification. Although isobaric tagging strategies excel in multiplexing proteome analysis, they suffer from low accuracy (15–17). We hypothesized that NeuCode SILAC would deliver improved sensitivity in detecting protein changes at similar levels of sample throughput. To test this, we compared 4-plex NeuCode SILAC and TMT labeling in the context of salt stress in yeast (supplemental Fig. S2). Both multiplexing approaches afforded deep, quantitative coverage of the NaCl-treated yeast proteome (supplemental Table S2). The 3,715 proteins quantified via both methods exhibited a strong correlation in terms of measured relative abundance (R2 = 0.72, Fig. 4A). NeuCode SILAC labeling produced a considerably wider dynamic range in protein abundance measurements, as indicated by the slope (m) of the linear regression curve (TMT versus NeuCode SILAC m = 0.53, Fig. 4A), enabling more sensitive detection of protein changes and affording novel biological insight. For example, the maximum change captured by NeuCode SILAC was a 432-fold up-regulation of the aldehyde dehydrogenase Ald3 (34). Osmotic shock induced the corresponding transcript 123-fold, indicating that post-transcriptional regulation influences Ald3 protein abundance. In contrast, TMT reported only a 7-fold change in Ald3, underestimating the NeuCode SILAC measurement and obscuring the implicated post-transcriptional regulation. NeuCode SILAC's increased accuracy substantially improved discovery potential: half of the proteins changing at least 1.5-fold were detected only by NeuCode SILAC (214 of the 438). We conclude that NeuCode SILAC provides a major advantage over TMT labeling.
Fig. 4.
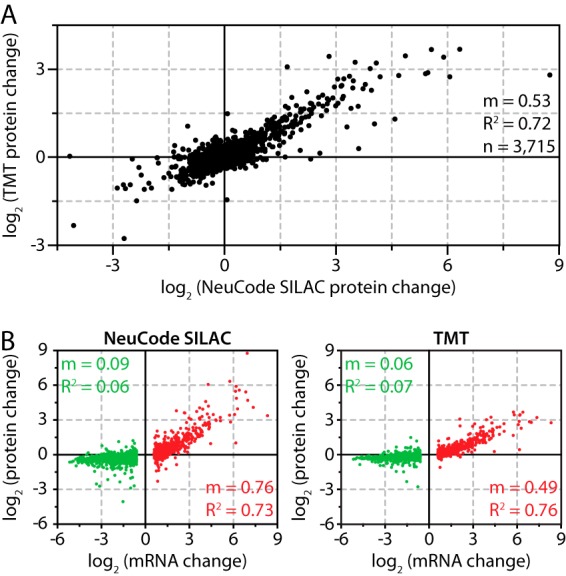
Yeast response to salt stress measured via NeuCode SILAC and TMT labeling. A, salt-induced protein changes were reproducibly quantified via both methods (R2 = 0.72), but NeuCode SILAC achieved a greater dynamic range (slope m = 0.53). B, regression of log2 changes in mRNA and protein for 771 induced transcript–protein pairs (red) and 1,196 repressed transcript–protein pairs (green).
We used this advance in quantitative accuracy to critically examine how well changes in mRNA levels predict changes in protein abundance. We previously used TMT labeling to compare salt-induced transcript and protein changes and found a high correlation between induced transcripts and their proteins (24). Like others, we observed that the fold change in transcript abundance was significantly higher than the fold-change in protein levels (24, 35). Using NeuCode SILAC, we confirmed a high correlation between induced transcripts and their corresponding proteins (R2 = 0.73; Fig. 4B, red). However, NeuCode SILAC revealed that the magnitude of protein change was closer to the magnitude of transcript changes (NeuCode SILAC m = 0.76, TMT m = 0.49; Fig. 4B). With the broader dynamic range, we reinvestigated our previous finding that transcript repression upon NaCl stress does not affect corresponding protein levels. NeuCode SILAC confirmed our previous conclusions (Fig. 4B, green), indicating that transcript repression during the salt response serves a different purpose than mediating protein abundance change (24).
NeuCode SILAC Multiplexing Diversifies Proteomic Experimental Design
The quantification approach presented here expands the number of biological variables that can be interrogated with a single metabolic labeling experiment. Combining NeuCode SILAC with dimethyl labeling (29), we compared changes in the yeast proteome over 18 different conditions (three isotopic clusters of 6-plex NeuCode SILAC) (supplemental Figs. S3A and S3B). These 18 channels permitted simultaneous characterization of five mutants (deletions of the regulatory kinases Hog1 (36) and CK2 (37), proteins involved in translation Sto1 (38) and Rck2 (39), and the transcriptional repressors Dot6/Tod6 (40)) and the WT responding over three time points (supplemental Table S3).
We first tested in a biological context how multiplexing affects quantitative performance. This malleable experimental set-up facilitates duplex (1 × 2) up to 18-plex (3 × 6) NeuCode SILAC proteome quantification (a × b, where a = 1, 2, or 3 dimethyl-labeled isotope clusters and b = 2, 3, 4, or 6 lysine isotopologues). We characterized the proportion of identified peptides quantified, not quantified, or excluded from quantification at all levels of multiplexing (Fig. 5A). The number of unique proteins quantified declined as a function of multiplexing level (Fig. 5B). Sample throughput, the total number of proteins quantified across all channels, escalated to a maximum in the 9-plex (3 × 3) experiment, however, demonstrating an advantage of quantitative multiplexing (Fig. 5C).
Fig. 5.
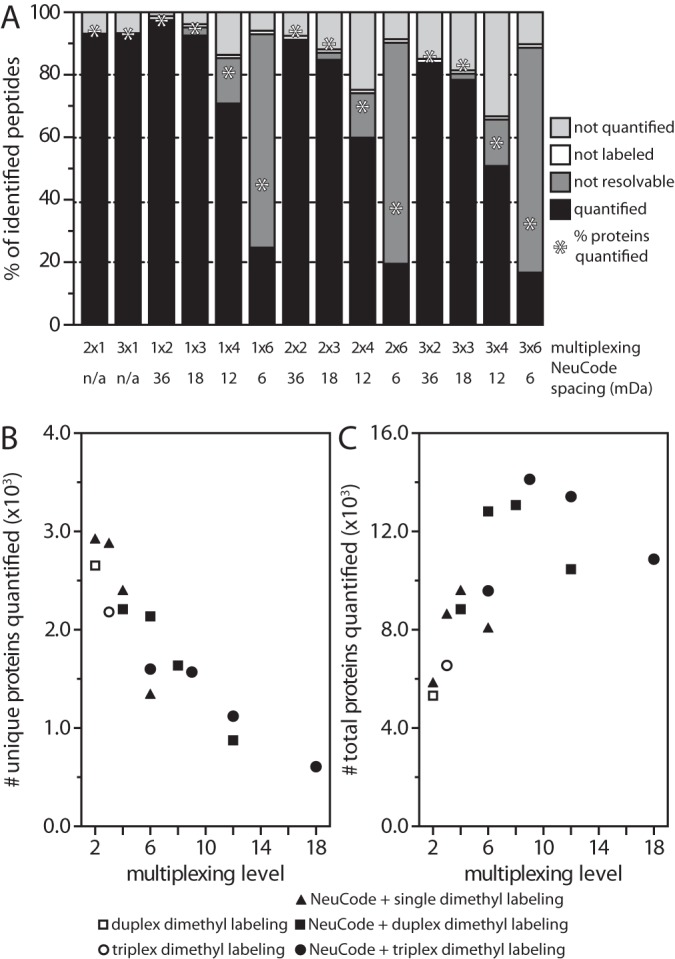
Quantitative performance of strategies combining NeuCode SILAC and dimethyl labeling. A resolving power of 480,000 was used for all analyses. A, the distribution of identified peptides that were quantified, excluded from quantification (not resolvable or not labeled), or not quantified changed with multiplexing level. B, the depth of proteome coverage and NeuCode multiplexing level were inversely related. C, the total number of proteins quantified (quantified unique proteins multiplied by number of channels) increased with multiplexing level to a maximum at the 3 × 3 (9-plex) configuration.
To assess global reproducibility as a function of multiplexing, we compared the proteome of unstressed WT yeast cells as measured in all 14 experimental configurations. We found a strong correlation between all configurations (Spearman rank r > 0.80, Fig. 6A), indicating that protein quantification retained integrity as NeuCode SILAC quantitative capacity was expanded. Quantitative precision was also confirmed at the level of individual proteins (Fig. 6B). For example, the Hbt1 protein drastically increased (≥20-fold) after salt stress in both WT and dot6Δtod6Δ mutant yeast strains. Increased Rtc3 abundance was also salt-dependent in both strains, but it was consistently stunted in cells lacking Dot6/Tod6 across eight different experiments (p < 10−14). Quantitative measurements made via NeuCode SILAC provided the reproducibility and dynamic range needed to isolate strain-specific defects as the proteome responds to a given stimulus.
Fig. 6.
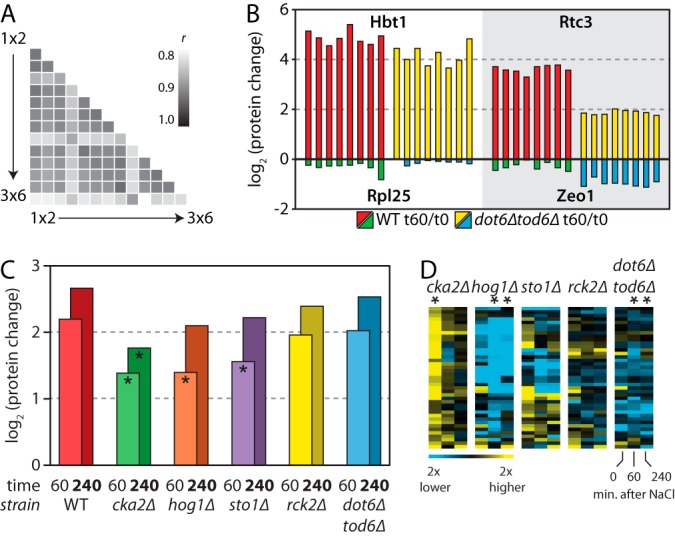
18-plex NeuCode quantification of yeast salt stress response. A, Spearman rank correlation coefficients (r) were calculated to compare proteome measurements (wild-type, untreated) across 14 experiments with levels of multiplexing ranging from duplex (1 × 2) to 18-plex (3 × 6). B, reproducible measurement of protein dynamics across eight different experiments (2 × 2, 2 × 3, 2 × 4, 2 × 6, 3 × 2, 3 × 3, 3 × 4, and 3 × 6). Hbt1 and Rpl25 proteins were induced (red, yellow) or repressed (green, blue) similarly in both wild-type and mutant strains (left), whereas Rtc3 and Zeo1 displayed salt-dependent differences (right). C, average fold-change in protein abundance across 41 proteins induced in response to NaCl in each of six strains. Each bar shows the average fold change at the denoted time point (tx) relative to the unstressed 0-min sample (t0) for that strain (*p < 10−5, relative to WT tx/t0). D, the log2 differences in 41 proteins (rows) at three time points (columns) for five mutant strains relative to WT (*p < 10−5). Yellow and blue colors indicate increased and decreased abundance, respectively, relative to WT.
NeuCode SILAC multiplexing requires high mass-resolving powers (more isotopologues, b in a × b) and/or increased spectral complexity (more isotopic clusters, a in a × b), both of which reduce coverage of the quantified proteome. Fewer proteins simultaneously analyzed across many conditions, however, does not preclude meaningful biological interpretation. For example, over half of the 603 proteins quantified via 18-plex NeuCode SILAC were differentially expressed in at least 1 of the 15 strain and time-point comparisons enabled by this labeling scheme (supplemental Table S4). We hierarchically clustered these proteome changes and focused on 41 stress-induced proteins showing interesting interstrain patterns (Fig. 6C). The most prominent defects in NaCl-induced protein changes were displayed by the hog1Δ and cka2Δ mutants (p < 10−5), whereas the defects exhibited by cells without Sto1, Rck2, and Dot6/Tod6 were surprisingly subtle. Considering the major transcriptional defect in the hog1Δ mutant responding to salt, our results suggest that NaCl-dependent protein changes are likely due primarily to defective transcript changes (41–43). However, a subset of the 41 proteins exhibited defective increase in the sto1Δ mutant responding to salt exposure, implicating Sto1-dependent translational effects (Fig. 6D). The apparent defect in protein increase in the CK2 mutant might alternatively be due to an elevated basal abundance under non-stress conditions (Fig. 6D). We conclude that NeuCode SILAC affords unprecedented multiplexing of global proteome quantification from cell culture systems.
DISCUSSION
In this work we transformed NeuCode SILAC from an interesting theoretical proposition to a bona fide method that outperformed the current cutting-edge technologies, in both plexing scale and performance metrics. Development of a synthesis strategy to fine-tune lysine isotopic content was the critical first step—an approach we validated by generating four novel lysine isotopologues. Combining these amino acids with existing ones, we achieved duplex, triplex, 4-plex, and 6-plex NeuCode SILAC quantification within a single isotopic cluster. We show that plexing capacity scaled with mass spectrometer resolving power (Fig. 1A). Doubtless this instrument performance metric will continue to improve, and with it, so too will the plexing capacity of NeuCode. To achieve the highest levels of plexing, we combined this approach with chemical labels to generate multiple isotopic clusters of two, three, four, or six NeuCode SILAC channels. We anticipate that additional lysine molecules will be generated, using the synthetic routes we describe here, that offer NeuCode SILAC quantification in other offset masses of lysine. For example, multiple isotopologues of lysine can be generated at M + 4, M + 8 (those we describe here), and M + 12. The availability of these molecules will ameliorate the need for additional chemical labels, simplifying the protocol. Beyond that, comparable NeuCode versions of arginine can be used in conjunction with the lysine isotopologues so that most tryptic peptides bear a NeuCode signature. In fact, owing to its greater nitrogen content, arginine offers even greater multiplexing capacity than lysine.
That said, NeuCode SILAC does have certain limitations. Similar to traditional SILAC, the method requires custom amino acid availability, cells that are auxotrophic for the selected amino acid(s), and compatibility for growth on supplemented minimal media. Commercial development of these lysine isotopologues, and others, is currently underway and should resolve availability concerns. Mammalian tissues and biofluids, however, are not as easily metabolically labeled and seemingly are not amenable to the strategy (44–46). For such samples we envision the development of mass defect-bearing chemical tags (47–50). Moreover, these NeuCode labels, unlike the amino acid reagents used herein, could be designed without the use of deuterium. Peptides bearing lysine isotopologues differing most in deuterium content (i.e. K602 and K080) vary in chromatographic retention by ∼2 s and demonstrate a subtle decrease in quantitative precision (standard deviation of 0.7 for traditional SILAC with deuterium versus 0.6 without deuterium; data not shown). The performance metrics outlined here should be entirely transferred for that work, as data collection methods would be virtually identical.
We note that NeuCode does require the collection of high-resolution mass spectra—so high that only FT-MS systems are currently compatible. Orbitrap mass analyzers are ubiquitous, however, and constitute a major percentage of the MS systems currently conducting proteome analysis. Given that, we suppose most researchers in the field presently have access to a NeuCode-compatible system. We demonstrate here that current commercial FT-MS instruments incur only a subtle duty cycle penalty when analyzing NeuCode samples (Fig. 2B). Doubtless future systems with boosted parallelization capabilities will mitigate this concern altogether (51, 52). Lastly, peak coalescence is a phenomenon that can cause two closely spaced m/z peaks to be detected as one (53, 54). We did observe coalescence on the most abundant m/z features in a spectrum, typically those with a signal-to-noise exceeding 2,500. These cases occur relatively infrequently (for ∼1% of identified peptides) and do not thwart quantification (i.e. >90% of intense peptides are still quantified), as quantitative information can be extracted from less intense isotopic peaks and/or from other spectra collected lower in the peptide's elution profile, as previously described (18).
NeuCode SILAC is a new and promising tool for biologists seeking to map the quantitative landscape of proteins in cell culture systems. When linked to mRNA levels, accurate protein abundance measurements provide insight on post-transcriptional regulation mechanisms that might be critical for cells adapting to environmental stress. High levels of plexing facilitate comparisons among strains, treatments, and/or time points within a single experiment, greatly diversifying experimental design options. The versatility of NeuCode SILAC, as depicted by this collection of vignettes, will greatly enhance the quantitative study of proteomes throughout biology.
Supplementary Material
Footnotes
Author contributions: A.E.M., A.S.H., M.E.M., C.M.R., A.P.G., and J.J.C. designed research; A.E.M., A.S.H., and M.E.M. performed research; J.C.B., W.W.W., and M.E. contributed new reagents or analytic tools; A.E.M., A.S.H., M.E.M., C.M.R., D.J.B., M.S.W., and A.P.G. analyzed data; A.E.M., M.E.M., A.P.G., and J.J.C. wrote the paper.
* This work was supported by NIH Grant No. R01 GM080148 to J.J.C. A.E.M. gratefully acknowledges support from an NIH-funded Genomic Sciences Training Program (5T32HG002760). C.M.R. was funded by an NSF Graduate Research Fellowship and NIH Trainee-ship (T32GM008505).
 This article contains supplemental material. Data available at ChorusProject.org under project name NeuCode SILAC or with the following links: https://chorusproject.org/anonymous/download/experiment/-1068165243734741551, https://chorusproject.org/anonymous/download/experiment/-2868352629863054252, and https://chorusproject.org/anonymous/download/experiment/4869941559505874158.
This article contains supplemental material. Data available at ChorusProject.org under project name NeuCode SILAC or with the following links: https://chorusproject.org/anonymous/download/experiment/-1068165243734741551, https://chorusproject.org/anonymous/download/experiment/-2868352629863054252, and https://chorusproject.org/anonymous/download/experiment/4869941559505874158.
Competing financial interests: A.S.H. and J.J.C. are co-inventors on a patent application (U.S. 13/660677) related in part to the material presented here.
1 The abbreviations used are:
- SILAC
- stable isotope labeling by amino acids in cell culture
- NeuCode
- neutron encoding
- TMT
- tandem mass tags
- FT
- Fourier transform
- FWTM
- full width at 10% maximum
- WT
- wild type
- FDR
- false discovery rate
- CAD
- collisionally activated dissociation
- HCD
- higher-energy collisional dissociation.
REFERENCES
- 1. Altelaar A. F., Munoz J., Heck A. J. (2013) Next-generation proteomics: towards an integrative view of proteome dynamics. Nat. Rev. Genet. 14, 35–48 [DOI] [PubMed] [Google Scholar]
- 2. Ahmad Y., Lamond A. I. (2014) A perspective on proteomics in cell biology. Trends Cell Biol. 24, 257–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gygi S. P., Rist B., Gerber S. A., Turecek F., Gelb M. H., Aebersold R. (1999) Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 17, 994–999 [DOI] [PubMed] [Google Scholar]
- 4. Washburn M. P., Wolters D., Yates J. R., 3rd (2001) Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 [DOI] [PubMed] [Google Scholar]
- 5. Syka J. E., Marto J. A., Bai D. L., Horning S., Senko M. W., Schwartz J. C., Ueberheide B., Garcia B., Busby S., Muratore T., Shabanowitz J., Hunt D. F. (2004) Novel linear quadrupole ion trap/FT mass spectrometer: performance characterization and use in the comparative analysis of histone H3 post-translational modifications. J. Proteome Res. 3, 621–626 [DOI] [PubMed] [Google Scholar]
- 6. Jiang H., English A. M. (2002) Quantitative analysis of the yeast proteome by incorporation of isotopically labeled leucine. J. Proteome Res. 1, 345–350 [DOI] [PubMed] [Google Scholar]
- 7. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 8. Zhu H., Pan S., Gu S., Bradbury E. M., Chen X. (2002) Amino acid residue specific stable isotope labeling for quantitative proteomics. Rapid Commun. Mass Spectrom. 16, 2115–2123 [DOI] [PubMed] [Google Scholar]
- 9. Molina H., Yang Y., Ruch T., Kim J. W., Mortensen P., Otto T., Nalli A., Tang Q. Q., Lane M. D., Chaerkady R., Pandey A. (2009) Temporal profiling of the adipocyte proteome during differentiation using a five-plex SILAC based strategy. J. Proteome Res. 8, 48–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tzouros M., Golling S., Avila D., Lamerz J., Berrera M., Ebeling M., Langen H., Augustin A. (2013) Development of a 5-plex SILAC method tuned for the quantitation of tyrosine phosphorylation dynamics. Mol. Cell. Proteomics 12, 3339–3349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thompson A., Schafer J., Kuhn K., Kienle S., Schwarz J., Schmidt G., Neumann T., Johnstone R., Mohammed A. K., Hamon C. (2003) Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–1904 [DOI] [PubMed] [Google Scholar]
- 12. Ross P. L., Huang Y. N., Marchese J. N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D. J. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 13. McAlister G. C., Huttlin E. L., Haas W., Ting L., Jedrychowski M. P., Rogers J. C., Kuhn K., Pike I., Grothe R. A., Blethrow J. D., Gygi S. P. (2012) Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem. 84, 7469–7478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Werner T., Becher I., Sweetman G., Doce C., Savitski M. M., Bantscheff M. (2012) High-resolution enabled TMT 8-plexing. Anal. Chem. 84, 7188–7194 [DOI] [PubMed] [Google Scholar]
- 15. Ow S. Y., Salim M., Noirel J., Evans C., Rehman I., Wright P. C. (2009) iTRAQ underestimation in simple and complex mixtures: “the good, the bad and the ugly.” J. Proteome Res. 8, 5347–5355 [DOI] [PubMed] [Google Scholar]
- 16. Wenger C. D., Lee M. V., Hebert A. S., McAlister G. C., Phanstiel D. H., Westphall M. S., Coon J. J. (2011) Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat. Methods 8, 933–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ting L., Rad R., Gygi S. P., Haas W. (2011) MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hebert A. S., Merrill A. E., Bailey D. J., Still A. J., Westphall M. S., Strieter E. R., Pagliarini D. J., Coon J. J. (2013) Neutron-encoded mass signatures for multiplexed proteome quantification. Nat. Methods 10, 332–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sleno L. (2012) The use of mass defect in modern mass spectrometry. J. Mass Spectrom. 47, 226–236 [DOI] [PubMed] [Google Scholar]
- 20. Schaub T. M., Hendrickson C. L., Horning S., Quinn J. P., Senko M. W., Marshall A. G. (2008) High-performance mass spectrometry: Fourier transform ion cyclotron resonance at 14.5 Tesla. Anal. Chem. 80, 3985–3990 [DOI] [PubMed] [Google Scholar]
- 21. Denisov E., Damoc E., Lange O., Makarov A. (2012) Orbitrap mass spectrometry with resolving powers above 1,000,000. Int. J. Mass Spectrom. 325–327, 80–85 [Google Scholar]
- 22. Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., Boeke J. D. (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132 [DOI] [PubMed] [Google Scholar]
- 23. Giaever G., Chu A. M., Ni L., Connelly C., Riles L., Veronneau S., Dow S., Lucau-Danila A., Anderson K., Andre B., Arkin A. P., Astromoff A., El-Bakkoury M., Bangham R., Benito R., Brachat S., Campanaro S., Curtiss M., Davis K., Deutschbauer A., Entian K. D., Flaherty P., Foury F., Garfinkel D. J., Gerstein M., Gotte D., Guldener U., Hegemann J. H., Hempel S., Herman Z., Jaramillo D. F., Kelly D. E., Kelly S. L., Kotter P., LaBonte D., Lamb D. C., Lan N., Liang H., Liao H., Liu L., Luo C., Lussier M., Mao R., Menard P., Ooi S. L., Revuelta J. L., Roberts C. J., Rose M., Ross-Macdonald P., Scherens B., Schimmack G., Shafer B., Shoemaker D. D., Sookhai-Mahadeo S., Storms R. K., Strathern J. N., Valle G., Voet M., Volckaert G., Wang C. Y., Ward T. R., Wilhelmy J., Winzeler E. A., Yang Y., Yen G., Youngman E., Yu K., Bussey H., Boeke J. D., Snyder M., Philippsen P., Davis R. W., Johnston M. (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391 [DOI] [PubMed] [Google Scholar]
- 24. Lee M. V., Topper S. E., Hubler S. L., Hose J., Wenger C. D., Coon J. J., Gasch A. P. (2011) A dynamic model of proteome changes reveals new roles for transcript alteration in yeast. Mol. Syst. Biol. 7, 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sherman F. (2002) Getting started with yeast. Methods Enzymol. 350, 3–41 [DOI] [PubMed] [Google Scholar]
- 26. Julius D., Blair L., Brake A., Sprague G., Thorner J. (1983) Yeast alpha factor is processed from a larger precursor polypeptide: the essential role of a membrane-bound dipeptidyl aminopeptidase. Cell 32, 839–852 [DOI] [PubMed] [Google Scholar]
- 27. Lewis J. A., Gasch A. P. (2012) Natural variation in the yeast glucose-signaling network reveals a new role for the Mig3p transcription factor. G3 2, 1607–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wohlbach D. J., Kuo A., Sato T. K., Potts K. M., Salamov A. A., Labutti K. M., Sun H., Clum A., Pangilinan J. L., Lindquist E. A., Lucas S., Lapidus A., Jin M., Gunawan C., Balan V., Dale B. E., Jeffries T. W., Zinkel R., Barry K. W., Grigoriev I. V., Gasch A. P. (2011) Comparative genomics of xylose-fermenting fungi for enhanced biofuel production. Proc. Natl. Acad. Sci. U.S.A. 108, 13212–13217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boersema P. J., Raijmakers R., Lemeer S., Mohammed S., Heck A. J. (2009) Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 4, 484–494 [DOI] [PubMed] [Google Scholar]
- 30. Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 31. Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 32. Wenger C. D., Phanstiel D. H., Lee M. V., Bailey D. J., Coon J. J. (2011) COMPASS: a suite of pre- and post-search proteomics software tools for OMSSA. Proteomics 11, 1064–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nesvizhskii A. I., Aebersold R. (2005) Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics 4, 1419–1440 [DOI] [PubMed] [Google Scholar]
- 34. Aranda A., del Olmo Ml M. (2003) Response to acetaldehyde stress in the yeast Saccharomyces cerevisiae involves a strain-dependent regulation of several ALD genes and is mediated by the general stress response pathway. Yeast 20, 747–759 [DOI] [PubMed] [Google Scholar]
- 35. Cox B., Kislinger T., Wigle D. A., Kannan A., Brown K., Okubo T., Hogan B., Jurisica I., Frey B., Rossant J., Emili A. (2007) Integrated proteomic and transcriptomic profiling of mouse lung development and Nmyc target genes. Mol. Syst. Biol. 3, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. de Nadal E., Posas F. (2010) Multilayered control of gene expression by stress-activated protein kinases. EMBO J. 29, 4–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Meggio F., Pinna L. A. (2003) One-thousand-and-one substrates of protein kinase CK2? FASEB J. 17, 349–368 [DOI] [PubMed] [Google Scholar]
- 38. Garre E., Romero-Santacreu L., De Clercq N., Blasco-Angulo N., Sunnerhagen P., Alepuz P. (2012) Yeast mRNA cap-binding protein Cbc1/Sto1 is necessary for the rapid reprogramming of translation after hyperosmotic shock. Mol. Biol. Cell 23, 137–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Teige M., Scheikl E., Reiser V., Ruis H., Ammerer G. (2001) Rck2, a member of the calmodulin-protein kinase family, links protein synthesis to high osmolarity MAP kinase signaling in budding yeast. Proc. Natl. Acad. Sci. U.S.A. 98, 5625–5630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lippman S. I., Broach J. R. (2009) Protein kinase A and TORC1 activate genes for ribosomal biogenesis by inactivating repressors encoded by Dot6 and its homolog Tod6. Proc. Natl. Acad. Sci. U.S.A. 106, 19928–19933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alepuz P. M., Jovanovic A., Reiser V., Ammerer G. (2001) Stress-induced map kinase Hog1 is part of transcription activation complexes. Mol. Cell 7, 767–777 [DOI] [PubMed] [Google Scholar]
- 42. Proft M., Mas G., de Nadal E., Vendrell A., Noriega N., Struhl K., Posas F. (2006) The stress-activated Hog1 kinase is a selective transcriptional elongation factor for genes responding to osmotic stress. Mol. Cell 23, 241–250 [DOI] [PubMed] [Google Scholar]
- 43. Rep M., Krantz M., Thevelein J. M., Hohmann S. (2000) The transcriptional response of Saccharomyces cerevisiae to osmotic shock. Hot1p and Msn2p/Msn4p are required for the induction of subsets of high osmolarity glycerol pathway-dependent genes. J. Biol. Chem. 275, 8290–8300 [DOI] [PubMed] [Google Scholar]
- 44. Kruger M., Moser M., Ussar S., Thievessen I., Luber C. A., Forner F., Schmidt S., Zanivan S., Fassler R., Mann M. (2008) SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell 134, 353–364 [DOI] [PubMed] [Google Scholar]
- 45. Geiger T., Cox J., Ostasiewicz P., Wisniewski J. R., Mann M. (2010) Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat. Methods 7, 383–385 [DOI] [PubMed] [Google Scholar]
- 46. Monetti M., Nagaraj N., Sharma K., Mann M. (2011) Large-scale phosphosite quantification in tissues by a spike-in SILAC method. Nat. Methods 8, 655–658 [DOI] [PubMed] [Google Scholar]
- 47. Hebert A. S., Merrill A. E., Stefely J. A., Bailey D. J., Wenger C. D., Westphall M. S., Pagliarini D. J., Coon J. J. (2013) Amine-reactive neutron-encoded labels for highly plexed proteomic quantitation. Mol. Cell. Proteomics 12, 3360–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ulbrich A., Merrill A. E., Hebert A. S., Westphall M. S., Keller M. P., Attie A. D., Coon J. J. (2014) Neutron-encoded protein quantification by peptide carbamylation. J. Am. Soc. Mass Spectrom. 25, 6–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou Y., Shan Y., Wu Q., Zhang S., Zhang L., Zhang Y. (2013) Mass defect-based pseudo-isobaric dimethyl labeling for proteome quantification. Anal. Chem. 85, 10658–10663 [DOI] [PubMed] [Google Scholar]
- 50. Bamberger C., Pankow S., Park S. K., Yates J. R. (2014) Interference-free proteome quantification with MS/MS-based isobaric isotopologue detection. J. Proteome Res. 13, 1494–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hebert A. S., Richards A. L., Bailey D. J., Ulbrich A., Coughlin E. E., Westphall M. S., Coon J. J. (2013) The one hour yeast proteome. Mol. Cell. Proteomics 13, 339–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Senko M. W., Remes P. M., Canterbury J. D., Mathur R., Song Q., Eliuk S. M., Mullen C., Earley L., Hardman M., Blethrow J. D., Bui H., Specht A., Lange O., Denisov E., Makarov A., Horning S., Zabrouskov V. (2013) Novel parallelized quadrupole/linear ion trap/Orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal. Chem. 85, 11710–11714 [DOI] [PubMed] [Google Scholar]
- 53. Mitchell D. W., Smith R. D. (1995) Cyclotron motion of two Coulombically interacting ion clouds with implications to Fourier-transform ion cyclotron resonance mass spectrometry. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics 52, 4366–4386 [DOI] [PubMed] [Google Scholar]
- 54. Gorshkov M. V., Fornelli L., Tsybin Y. O. (2012) Observation of ion coalescence in Orbitrap Fourier transform mass spectrometry. Rapid Commun. Mass Spectrom. 26, 1711–1717 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.