

Abstract
The m-AAA protease subunit AFG3L2 is involved in degradation and processing of substrates in the inner mitochondrial membrane. Mutations in AFG3L2 are associated with spinocerebellar ataxia SCA28 in humans and impair axonal development and neuronal survival in mice. The loss of AFG3L2 causes fragmentation of the mitochondrial network. However, the pathogenic mechanism of neurodegeneration in the absence of AFG3L2 is still unclear. Here, we show that depletion of AFG3L2 leads to a specific defect of anterograde transport of mitochondria in murine cortical neurons. We observe similar transport deficiencies upon loss of AFG3L2 in OMA1-deficient neurons, indicating that they are not caused by OMA1-mediated degradation of the dynamin-like GTPase OPA1 and inhibition of mitochondrial fusion. Treatment of neurons with antioxidants, such as N-acetylcysteine or vitamin E, or decreasing tau levels in axons restored mitochondrial transport in AFG3L2-depleted neurons. Consistently, tau hyperphosphorylation and activation of ERK kinases are detected in mouse neurons postnatally deleted for Afg3l2. We propose that reactive oxygen species signaling leads to cytoskeletal modifications that impair mitochondrial transport in neurons lacking AFG3L2.
Keywords: m-AAA protease, mitochondrial transport, N-acetylcysteine, Neurodegeneration, tau
Introduction
Mitochondria are essential organelles, whose functional integrity is ensured by efficient quality control mechanisms, including maintenance of mitochondrial proteostasis by chaperones and proteases, ongoing mitochondrial fusion and fission, and removal of dysfunctional mitochondria by mitophagy (Rugarli & Langer, 2012). Neurons are particularly susceptible to defects of any of these systems, which fail in several neurodegenerative conditions (Schon & Przedborski, 2011). Since neurons are highly polarized and need to traffic mitochondria to sites very distant from where mitochondrial biogenesis takes place, mechanisms are likely to exist to couple transport of mitochondria to their functionality. For instance, PINK1 and parkin target the mitochondrial motor adaptor miro for degradation, thus preventing transport of dysfunctional mitochondria (Wang et al, 2011).
The mitochondrial protease AFG3L2 is a central component of the intra-mitochondrial quality control system. AFG3L2 forms homo-oligomeric complexes or hetero-oligomeric hexamers with the homologous subunit paraplegin to constitute the m-AAA (matrix-ATPase associated with various cellular activities) protease in the inner mitochondrial membrane (Atorino et al, 2003; Koppen et al, 2007). This complex exerts ATP-dependent proteolytic activity, leading either to degradation or processing of specific substrates (Gerdes et al, 2012). Quality control substrates of the m-AAA protease include misfolded polypeptides in the inner membrane, such as subunits of the respiratory chain (Hornig-Do et al, 2012), while the best characterized processing substrate is the mitoribosome subunit MRPL32 (Nolden et al, 2005; Bonn et al, 2011). Mitochondria lacking AFG3L2 show defective assembly of the mitoribosome and reduced levels of mitochondrial protein synthesis (Almajan et al, 2012).
Mutations in AFG3L2 have been linked to two neurodegenerative conditions in humans, a dominant form of spinocerebellar ataxia (SCA28), and a severe recessive form of spastic-ataxia with early-onset, and rapid progression (SPAX5; Di Bella et al, 2010; Pierson et al, 2011). In contrast, mutations in SPG7, encoding paraplegin, are found in patients with a recessive form of hereditary spastic paraplegia (HSP; Casari et al, 1998). Given the pleiotropic roles of m-AAA proteases in mitochondria, the pathogenic cascade in these diseases is still unclear.
Constitutive Afg3l2 knockout mice are affected by a severe form of spasticity and muscle weakness, and die before 3 weeks of age (Maltecca et al, 2008). Strikingly, these mice display a characteristic neurodevelopmental phenotype, characterized by maintenance of neuronal cell number, but failure to develop spinal and peripheral large caliber axons (Maltecca et al, 2008). Neurons from Afg3l2-deficient mice contain abnormal mitochondria in the cell body and a paucity of neurofilaments in their processes (Maltecca et al, 2008). This phenotype led to the proposal that impaired trafficking of mitochondria and other cargos underlies the developmental failure of the axons. AFG3L2 is also required cell-autonomously for survival of adult neurons. Deletion of the gene in Purkinje cells following development provokes dramatic cell loss associated with secondary inflammation (Almajan et al, 2012). Notably, the first abnormalities observed in neurons lacking AFG3L2 are mitochondrial fragmentation and clustering, further hinting to impaired mitochondrial dynamics and transport (Almajan et al, 2012).
Studies in cell lines have shown that mitochondria lacking AFG3L2 are fragmented and fusion-incompetent owing to activation of OMA1, a mitochondrial protease that cleaves the GTPase fusion molecule OPA1 (Ehses et al, 2009; Head et al, 2009). OPA1 is essential for inner membrane fusion (Song et al, 2007). Various stress conditions activate the peptidase OMA1 in the inner membrane that cleaves OPA1, converting long OPA1 forms into short forms and inhibiting mitochondrial fusion (Duvezin-Caubet et al, 2006; Ishihara et al, 2006; Griparic et al, 2007; Ehses et al, 2009; Head et al, 2009; Baker et al, 2014). It is therefore conceivable that mitochondrial fragmentation impairs the axonal transport of mitochondria in Afg3l2-deficient neurons leading to neurodegeneration.
Here, we use primary murine cortical neurons to examine this possibility. We show that depletion of AFG3L2 leads to a specific defect of anterograde transport of mitochondria. Surprisingly, these transport defects are independent of OMA1 activation and mitochondrial fragmentation, but can be rescued by treatment of neurons with antioxidants and by modulation of tau levels. In vivo, deletion of Afg3l2 in cortical and hippocampal neurons leads to tau hyperphosphorylation and activation of ERK kinases. Our data suggest a cross talk between dysfunctional mitochondria and the cytoskeleton, which may have implications for other neurodegenerative conditions characterized by tau aggregations.
Results
Depletion of AFG3L2 in primary neurons leads to fragmentation and defective anterograde transport of mitochondria
To investigate whether mitochondrial transport is affected in neurodegenerative diseases caused by mutations in AFG3L2, we established primary cultures of mouse cortical neurons and downregulated Afg3l2 using RNA interference while plating the neurons. A construct expressing mCherry targeted to mitochondria (mito-mCherry) was co-transfected together with different Afg3l2-specific or control siRNA oligonucleotides (Ehses et al, 2009). We imaged morphology and motility of mitochondria in axons with a length of at least 200 μm. Downregulation of Afg3l2 resulted in mitochondrial fragmentation in comparison with control neurons (Fig1A, Supplementary Fig S1A). A significantly different distribution of mitochondrial length was observed in neurons with depleted levels of AFG3L2 (Fig1B, Supplementary Fig S1B). Furthermore, the percentage of axonal length occupied by mitochondrial mass, hereafter referred to as mitochondrial occupancy, was reduced upon depletion of AFG3L2 when compared to control (Fig1C, Supplementary Fig S1C). Mitochondrial fragmentation often precedes mitophagy that in turn could account for the reduced occupancy (Twig et al, 2008). The best characterized mitophagic pathway involves recruitment of parkin to depolarized mitochondria (Narendra et al, 2008). However, we found no recruitment of parkin to the mitochondrial surface in neurons downregulated for Afg3l2 (Supplementary Fig S2A). Moreover, mitochondrial membrane potential was not significantly affected in most neurons by AFG3L2 depletion (Supplementary Fig S2B and C).
Figure 1. Depletion of AFG3L2 causes mitochondrial defects.

- Representative kymographs of axons (shown above respective kymographs) from neurons co-transfected with mito-mCherry and control or Afg3l2 siRNAs. Schematic of different color-coded transport types is shown below. Scale bars, 10 μm.
- The percentage of mitochondria belonging to a given length bin was averaged from three independent experiments (120–150 mitochondria were measured in each experiment). Bars represent SEM. Chi-square test: P = 0.002.
- Quantification of mitochondrial occupancy per axon (n = 3 experiments; 7–8 axons per experiment).
- Quantification of mitochondrial transport types in the axon. Data represent mean ± SEM of three independent experiments. 7–8 axons from each experiment were analyzed. P-value was determined with Student's t-test.
- Average mitochondrial velocity in the anterograde and retrograde direction. Data represent mean ± SEM of three independent experiments. The velocity of mitochondria from at least 5 axons per experiment was analyzed.
To analyze mitochondrial transport, we scored the percentage of mitochondria moving in the anterograde direction, in the retrograde direction, changing their directionality (defined as oscillatory), or remaining stationary during the time of recording. Strikingly, depletion of AFG3L2 resulted in a selective defect of anterograde movement of mitochondria into the axons (Fig1D, Supplementary Fig S1D). The number of stationary mitochondria slightly increased, in some cases to statistically significant values (Supplementary Table S1). Finally, we determined whether downregulation of Afg3l2 affects the velocity of mitochondrial movement. We found that the average speed of both anterogradely and retrogradely moving mitochondria was not significantly reduced in Afg3l2-downregulated neurons (Fig1E).
Mitochondrial transport defects in Afg3l2 knockdown axons are independent of OMA1 activation
It is conceivable that mitochondrial fragmentation causes transport defects. To test this possibility, we downregulated Opa1 in cortical neurons using two different siRNA oligonucleotides and performed live-imaging experiments. As expected, Opa1 downregulation led to mitochondrial fragmentation (Supplementary Fig S3A and B). It is noteworthy that mitochondrial occupancy was also significantly decreased with siRNA-B (Supplementary Fig S3C). However, mitochondria were normally transported into the axons, as previously observed (Misko et al, 2010). This experiment suggests that mitochondrial fragmentation by itself does not impair mitochondrial movement (Supplementary Fig S3D).
To substantiate these findings, we investigated whether Oma1 deletion could alleviate defects in mitochondrial morphology, occupancy, and transport in primary cortical neurons depleted for AFG3L2. In Afg3l2-deficient mouse embryonic fibroblasts (MEFs), long OPA1 forms are processed by the protease OMA1, inhibiting fusion (Ehses et al, 2009). In Oma1 knockout neurons, mitochondrial length was not significantly affected by downregulation of Afg3l2, and mitochondrial occupancy was only slightly reduced (Fig2A-C). Strikingly, anterograde mitochondrial transport was still impaired upon depletion of AFG3L2 (Fig2D), indicating that the mitochondrial transport defect is independent of OMA1 activation and mitochondrial fragmentation.
Figure 2. OMA1 ablation does not rescue mitochondrial transport defect of AFG3L2-depleted neurons.

- Representative kymographs of axons (shown above respective kymographs) from neurons isolated from Oma1 knockout mice co-transfected with mito-mCherry and control or Afg3l2 siRNAs. Schematic of different color-coded transport types is shown below. Scale bars, 10 μm.
- The percentage of mitochondria belonging to a given length bin was averaged from three independent experiments (105–200 mitochondria were measured in each experiment). Bars represent SEM.
- Quantification of mitochondrial occupancy per axon (n = 3 experiments; at least 8 axons per experiment).
- Quantification of mitochondrial transport types in the axon. Data represent mean ± SEM of three independent experiments. At least 8 axons from each experiment were analyzed. P-value was determined with Student's t-test.
Afg3l2 deficiency leads to disruption of the microtubule network and tau hyperphosphorylation in vivo
The previous data suggest that dysfunctional mitochondria in Afg3l2 knockdown neurons might be sensed in the cell body leading to inhibition of the anterograde transport. To elucidate the underlying mechanism, we examined the cerebral cortex of mice constitutively lacking Afg3l2 (Afg3l2Emv66/Emv66 mice, from now on referred to as Afg3l2 KO; Maltecca et al, 2008). Nissl staining of serial coronal sections of the brain at postnatal day 14, shortly before Afg3l2 KO mice die, showed a reduction in the thickness of the cerebral cortex, while the organized six-layered structure was preserved (Fig3A). Cortical neurons in layer V appeared shrunken and lacked processes, a hallmark of degeneration (Fig3A).
Figure 3. Constitutive Afg3l2 deletion leads to microtubule fragmentation and tau hyperphosphorylation.

- Nissl staining of coronal sections across the cerebral cortex of Afg3l2 knockout (KO) and wild-type (WT) show normal lamination but degenerating neurons in layer V (arrows).
- In situ hybridization of brain coronal sections of WT and KO using the indicated probes.
- Immunofluorescence analysis of coronal sections across the cerebral cortex of Afg3l2 KO and WT mice with the indicated antibodies shows dramatic loss of myelinated axons. cc: corpus callosum.
- Electron micrographs of cortical neuronal processes show microtubule fragmentation and disorganization, and abnormal mitochondria in KO mice. Arrows: mitochondria; asterisks: microtubules.
- Cerebral cortex sections were stained with AT8 antibody. Phosphorylated tau accumulates in cell bodies and dendrites of KO mice.
- Western blot analysis of brain lysates from Afg3l2 KO and WT littermates using the indicated antibodies. KO brains display increased levels of phosphorylated tau species.
Data information: Scale bar in (A) 200 μm, in (C) 20 μm, in (D) 0.5 μm.
Source data are available online for this figure.
We performed in situ hybridization experiments to evaluate the expression of laminar-specific genes, such as Cux2 for layers II–IV, Rorβ for layer IV, and Er81 for layer V (Molyneaux et al, 2007). These genes were expressed in the respective layer, indicating normal migration and specification of cortical neurons during development (Fig3B). However, Er81-positive neurons were decreased, pointing to pronounced secondary degeneration of neurons in layer V.
We then examined neuronal projections by staining brain sections with antibodies against phosphorylated neurofilaments (SMI31) to detect axons, microtubule-associated protein 2 (SMI52) to label dendrites, and myelin basic protein (MBP) to decorate myelin sheaths. A drastic reduction of myelinated axonal projections containing neurofilaments was found in Afg3l2 KO mice, while dendrites were affected to a lesser degree (Fig3C). These findings are consistent with previous observations in the spinal cord of these mice (Maltecca et al, 2008). To further characterize neuronal alterations in the absence of AFG3L2, we performed ultrastructural studies of the cerebral cortex. Neurons demonstrated pronounced mitochondrial abnormalities. Mitochondria appeared swollen, lost their tubular morphology, and had disrupted cristae pushed to the organelle periphery (Fig3D). Microtubules in control dendrites and axons were aligned along the major axis, while they were fragmented and disorganized in Afg3l2-deficient neuronal processes (Fig3D).
The microtubule binding protein tau plays an important role in microtubule stabilization (Morris et al, 2011). Tau hyperphosphorylation detaches tau from the microtubules, ultimately leading to their instability and fragmentation, similar to our observations in Afg3l2-deficient neuronal projections. We therefore assessed the levels of phosphorylated tau species in the brain of Afg3l2 KO mice. Hyperphosphorylation of tau can be detected with the AT8 antibody, which recognizes epitopes (serine 202 and threonine 205) that are pathologically hyperphosphorylated in Alzheimer's disease and other tauopathies. Immunoreactivity for AT8 antibodies was greatly enhanced in neurons of Afg3l2 KO mice compared with wild-types (Fig3E). Western blot analyses confirmed these results and further indicated increased levels of phosphorylation of tau on threonine 181 and serines 199 and 396 (Fig3F).
In conclusion, constitutive loss of AFG3L2 affects development of long projection axons in the cerebral cortex and is associated with microtubule disorganization and tau hyperphosphorylation.
Deletion of Afg3l2 in adult cortical neurons causes tau hyperphosphorylation and activation of PKA and ERK1/2 kinases
Tau is highly phosphorylated during embryonic development and in the early postnatal periods at several residues. However, after the majority of axons have reached their target, tau phosphorylation is downregulated (Wang & Liu, 2008). Tau hyperphosphorylation in Afg3l2 null mice may therefore reflect the axonal developmental failure. To test whether lack of AFG3L2 can also lead to tau hyperphosphorylation in adult neurons, we crossed Afg3l2fl/fl mice (Almajan et al, 2012) with a transgenic line expressing the Cre recombinase under the control of the CamKIIα promoter, leading to efficient depletion of AFG3L2 in postnatal neurons of the cortex, and the hippocampus (Minichiello et al, 1999; Supplementary Fig S4A). Deletion of Afg3l2 in neurons (Afg3l2NKO) leads to prominent neurodegeneration. At 8 weeks of age, Afg3l2NKO mice display evident loss of neurons in both the cortex and the hippocampus, as shown by Nissl staining (Fig4A). Remarkably, tau hyperphosphorylation was prominent in degenerating neurons and involved similar residues as in the constitutive Afg3l2 KO mice (Fig4B and C). Thus, tau phosphorylation is activated during the neurodegenerative process that is triggered by the loss of AFG3L2.
Figure 4. Deletion of Afg3l2 in adult forebrain neurons causes tau hyperphosphorylation and activation of PKA and ERK1/2 kinases.

- Brain coronal sections of Afg3l2 forebrain neuronal specific knockout (Afg3l2fl/fl; CamKIIα-Cre positive; NKO) and littermate control (Afg3l2fl/fl; CamKIIα-Cre negative, WT) at 8 weeks of age were stained with Nissl solution. Pronounced neuronal degeneration is observed in the cerebral cortex and hippocampus.
- AT8 immunohistochemistry staining of adjacent sections from (A) shows accumulation of hyperphosphorylated tau in neuronal cell bodies in both cerebral cortex and hippocampus of Afg3l2NKO.
- Western blot analysis of hippocampus and cerebral cortex lysates from Afg3l2 NKO and WT littermates using the indicated antibodies detects increased phosphorylation of tau.
- Western blot analysis of hippocampus and cerebral cortex lysates from Afg3l2 NKO and WT littermates using the indicated antibodies show activation of PKA and ERK1/2 kinases.
Data information: Scale bar in (A) 50 μm (cortex) and 200 μm (hippocampus), in (B) 50 μm (cortex) and 200 μm (hippocampus).
Source data are available online for this figure.
To address the mechanism underlying tau phosphorylation, we examined the activation of potential tau kinases in the Afg3l2NKO model. The main tau kinase GSK3β was found to be inhibited by phosphorylation at serine 9, and activation of MARK, CDK5, or its activator p35 was not observed (Supplementary Fig S4B). In contrast, deletion of Afg3l2 in adult neurons increased the levels of the phosphorylated active forms of mitogen-activated protein kinase (MAPK) ERK1/2 and of cAMP–dependent protein kinase A (PKA; Fig4D). Both kinases have been previously implicated in tau phosphorylation in pathological conditions (Martin et al, 2013). MAP kinases are activated by oxidative stress, which in turn can lead to tau hyperphosphorylation (Chu et al, 2004; Petersen et al, 2007). We therefore examined the brains of Afg3l2 null and NKO mice for signs of oxidative damage, but could not find accumulation of carbonylated proteins or increased levels of the antioxidant SOD2 (Supplementary Fig S5).
We conclude from these experiments that the MAP kinases ERK1/2 and PKA are activated upon loss of AFG3L2 in adult neurons of the cortex and hippocampus leading to tau hyperphosphorylation in vivo.
Tau downregulation affects mitochondrial transport in AFG3L2-depleted neurons
Tau binds selectively to anterograde-directed motors and can therefore regulate anterograde transport of mitochondria (Dixit et al, 2008). The finding of tau hyperphosphorylation in the absence of AFG3L2 in vivo prompted us to examine whether tau levels in primary cortical neurons can affect the mitochondrial transport and morphology defects, associated with acute Afg3l2 downregulation. Mapt-specific siRNA oligonucleotides were tested for their ability to downregulate overexpressed tau in cultured MEFs (Supplementary Fig S6). Afg3l2 siRNA oligonucleotides were transfected together with a control siRNA or Mapt siRNA in cortical neurons, and mitochondrial morphology and transport were evaluated. Notably, tau depletion did not affect mitochondrial length, occupancy, transport, and velocity in wild-type neurons (Fig5A–E, Table S1). However, mitochondria were less fragmented, and a partial but significant rescue of the anterograde transport defect was detected upon concomitant downregulation of Afg3l2 and Mapt when compared to AFG3L2 depletion alone (Fig5). Thus, modulation of tau levels affects mitochondrial transport in pathological conditions.
Figure 5. Tau levels affect mitochondrial transport in AFG3L2-depleted neurons.

A The percentage of mitochondria belonging to a given length bin was averaged from three independent experiments (120–210 mitochondria were measured in each experiment). Bars represent SEM. P-values were calculated with the chi-square test. Control vs. Afg3l2/control: P = 7 × 10−4, control vs. Afg3l2/Mapt: P = 9 × 10−3, Afg3l2/control vs. Afg3l2/Mapt: P = 5 × 10−5.
B Quantification of mitochondrial occupancy per axon (n = 3 experiments; 7–9 axons per experiment).
C Quantification of mitochondrial transport types in the axon. Data represent mean ± SEM of three independent experiments. 7–9 neurons from each experiment were analyzed.
D, E Average mitochondrial velocity in the anterograde (D) and retrograde (E) direction. Data represent mean ± SEM of three independent experiments. The velocity of mitochondria from 3 axons per experiment was analyzed.
Data information: P-values in (B) and (C) were determined with Student's t-test.
Antioxidants suppress mitochondrial transport defects in AFG3L2-depleted neurons
As oxidative stress triggers tau hyperphosphorylation, we explored the possibility that increased reactive oxygen species (ROS) production affects mitochondrial motility. Firstly, we tested whether primary neurons downregulated for Afg3l2 show signs of increased ROS by staining them with CellROX green. This dye becomes highly fluorescent upon oxidation and labels the DNA of both nucleus and mitochondria. However, we did not observe consistent, statistically significant differences in the fluorescence between control and AFG3L2-depleted neurons (Supplementary Fig S7A and B). This result could be due to limited sensitivity of the dye that is suitable to detect massive changes in ROS levels, such as those induced by treating neurons with menadione (Supplementary Fig S7C and D). We therefore asked whether interfering with basal ROS levels by culturing neurons in the presence of antioxidants, such as N-acetylcysteine (NAC) or vitamin E, has an impact on mitochondrial motility. NAC is a drug that affects the redox state of cells and indirectly acts as antioxidant by increasing the pool of reduced glutathione (Kelly, 1998), while vitamin E scavenges the peroxyl radicals and prevents the propagation of free radicals (Niki & Traber, 2012). Both drugs efficiently reduce the levels of ROS induced in primary neurons by menadione (Supplementary Fig S7C and D). Moreover, both NAC and vitamin E reduce the levels of phospho-tau in cultured neurons in vitro (Supplementary Fig S7E and F). Strikingly, NAC treatment of primary neurons leads to a global change in cytoskeletal components, by reducing the levels of β-tubulin, tyrosinated and acetylated tubulin, actin, and total tau (Supplementary Fig S7E).
The presence of NAC in the culture medium did not affect the distribution of mitochondrial length or the occupancy of mitochondria in control neurons. Surprisingly, the percentage of mitochondria moving in the anterograde direction and their velocity was significantly increased in control neurons when cultured in medium supplemented with NAC (Fig6A–F, Supplementary Table S1). Moreover, NAC treatment significantly suppressed the defects observed upon Afg3l2 downregulation: mitochondrial length, mitochondrial occupancy, and the percentage of mitochondria moving in anterograde direction increased to levels comparable to control (Fig6A–D).
Figure 6. Mitochondrial defects in AFG3L2-depleted neurons are rescued by NAC.

A Representative kymographs of axons (shown above respective kymographs) from neurons co-transfected with mito-mCherry and Afg3l2 siRNAs in the absence or presence of NAC. Schematic of different color-coded transport types is shown below. Scale bars, 10 μm.
B The percentage of mitochondria belonging to a given length bin was averaged from four independent experiments (78–195 mitochondria were measured in each experiment). Bars represent SEM. P-values were calculated with the chi-square test. Control vs. Afg3l2: P = 8 × 10−9; Afg3l2 vs. Afg3l2 with NAC: P = 1 × 10−12.
C Quantification of mitochondrial occupancy per axon (n = 4 experiments; 8–10 axons per experiment).
D Quantification of mitochondrial transport types in the axon. Data represent mean ± SEM of four independent experiments. At least 8 axons from each experiment were analyzed.
E, F Average mitochondrial velocity in the anterograde (E) and retrograde (F) direction. Data represent mean ± SEM of four independent experiments. The velocity of mitochondria from 3 axons per experiment was analyzed.
Data information: P-values in (C-E) were determined with Student's t-test.
We then depleted AFG3L2 in cortical neurons grown in medium containing vitamin E. Vitamin E induced slight fragmentation of mitochondria in control neurons, which is reflected in a reduced occupancy (Fig7A and B). Consistently, vitamin E did not rescue mitochondrial morphology and occupancy of AFG3L2-depleted neurons when compared to controls (Fig7A and B). However, a significant improvement in the anterograde transport of mitochondria was observed (Fig7C, Supplementary Table S1). Mitochondrial velocities of anterogradely and retrogradely moving mitochondria were not significantly affected by vitamin E (Fig7D and E). We conclude that ROS, present at physiological levels, play a pivotal role in coupling quality control of mitochondria to their transport.
Figure 7. Mitochondrial transport defect in AFG3L2-depleted neurons is rescued by vitamin E.

A The percentage of mitochondria belonging to a given length bin was averaged from three independent experiments (130–218 mitochondria were measured in each experiment). Bars represent SEM. P-values were calculated with the chi-square test. Control versus control with vitamin E: P = 0.01, control vs. Afg3l2: P = 5 × 10−7.
B Quantification of mitochondrial occupancy per axon (n = 3 experiments; at least 8 axons per experiment).
C Quantification of mitochondrial transport types in the axon. Data represent mean ± SEM of three independent experiments. At least 8 axons from each experiment were analyzed.
D, E Average mitochondrial velocity in the anterograde (D) and retrograde (E) direction. Data represent mean ± SEM of three independent experiments. The velocity of mitochondria from 3 axons per experiment was analyzed.
Data information: P-values in (C) and (D) were determined with Student's t-test.
Discussion
Neurodegenerative diseases linked to mutations in components of the m-AAA protease, paraplegin and AFG3L2, are characterized by late-onset axonal degeneration in cortical motor or cerebellar neurons. Our study shows for the first time that the frequency of anterograde transport of mitochondria, but not the speed of movement, is affected in neurons with reduced levels of AFG3L2, suggesting that defective mitochondrial trafficking contributes to the pathogenic mechanism in the human diseases. Mitochondrial transport defects in Afg3l2-deficient neurons are restricted to mitochondria moving from the cell body toward the growth cone. A similar phenotype has been previously described in cortical neurons isolated from mice lacking spastin, a microtubule-severing protein involved in HSP (Kasher et al, 2009), in motor neurons derived from SODG93A transgenic mice, a model of amyotrophic lateral sclerosis (De Vos et al, 2007), and in amyloid β-treated neurons (Reddy et al, 2012). Even mild impairment of anterograde transport in patients carrying heterozygous mutations in AFG3L2 could lead to progressive depletion of mitochondria from axons causing late-onset neurodegeneration. It is tempting to speculate that impaired anterograde transport of mitochondria underlies the developmental failure of large myelinated axons in absence of AFG3L2 in the mouse.
Surprisingly, our data uncouple mitochondrial trafficking defects from OMA1 activation and mitochondrial fragmentation. Stress-activated OMA1-mediated processing of OPA1 is not required for mitochondrial transport defects in the absence of AFG3L2. Furthermore, reduced OPA1 levels do not affect mitochondrial transport, despite leading to fragmentation, in agreement with previous studies (Misko et al, 2010). Our data therefore suggest that alteration of mitochondrial transport and increased processing of OPA1 in absence of AFG3L2 are independent events. Notably, we observed a strict correlation between the degree of mitochondrial fragmentation and the reduction in mitochondrial occupancy, suggesting increased mitochondrial turnover likely by a parkin-independent mechanism.
How are Afg3l2-deficient mitochondria hindered from being transported toward the growth cone? We identify tau as a regulator of transport of dysfunctional Afg3l2-deficient mitochondria. The primary function of the microtubule-associated protein tau is to bind and stabilize microtubules (Wang & Liu, 2008). Tau was found to largely affect access of the anterograde motor kinesins to microtubules (Dixit et al, 2008). Consistently, tau overexpression inhibits kinesin-dependent transport of cargos, including mitochondria (Ebneth et al, 1998). Loss of tau function in the mouse still allows normal mitochondrial transport (Yuan et al, 2008), whereas tau depletion is beneficial in certain pathological conditions; for example, it improves axonal transport defects of mitochondria in neurons treated with Aβ peptides (Vossel et al, 2010). Similarly, we show that reducing tau levels can partially rescue the mitochondrial transport defects associated with Afg3l2 downregulation.
Tau hyperphosphorylation is an important downstream event to the lack of AFG3L2 in cortical neurons in vivo. During embryonic and early postnatal periods, tau is hyperphosphorylated at several sites, including Thr181, Ser199, Ser202, and Thr205. Notably, the same residues are also hyperphosphorylated in degenerating neurons of Alzheimer's disease patients (Wang & Liu, 2008). Upon phosphorylation, tau detaches from microtubules and can potentially improve motility of cargos, such as mitochondria. Consistently, degeneration caused by missorting of tau into neurons can be rescued by increasing its phosphorylation via the tau kinase MARK2 (Thies & Mandelkow, 2007). However, prolonged tau hyperphosphorylation promotes self-aggregation into cytoplasmic inclusions and leads to neurodegeneration (Ballatore et al, 2007). Hyperphosphorylated tau at residues recognized by the AT8 antibody was recently found to inhibit mitochondrial transport in cortical neuronal axons by affecting spacing of microtubules (Shahpasand et al, 2012). In conclusion, tau hyperphosphorylation might be beneficial at early stages in the pathogenic process, but it is conceivable that sustained tau hyperphosphorylation impairs anterograde transport of mitochondria in Afg3l2-deficient neurons. In agreement with such a scenario, neuronal microtubules appear severely disrupted in absence of AFG3L2. Notably, interfering with axonal transport by mutating kinesin-1, or by downregulating miro or milton in Drosophila, was sufficient to activate kinase pathways responsible for tau hyperphosphorylation (Falzone et al, 2009, 2010; Iijima-Ando et al, 2012), suggesting that loss of axonal mitochondria enhances tau pathology.
Accumulation of fibrillary tau inclusions (neurofibrillary tangles) in the central nervous system has been implicated in onset and progression of aging-associated disorders that manifest clinically with progressive cognitive and/or motor impairment (Ballatore et al, 2007). However, the causative link between tau hyperphosphorylation and upstream events in tauopathies is still unclear. Here, we demonstrate that mitochondrial dysfunction can directly trigger tau hyperphosphorylation upon genetic ablation of a mitochondrial protease. Similarly, we found abnormal tau hyperphosphorylation and deposition of tau filaments in Phb2-deficient degenerating neurons (Merkwirth et al, 2012). Prohibitins form large protein scaffolds in the inner mitochondrial membrane that interact with the m-AAA protease (Merkwirth & Langer, 2009). Together, our data provide genetic evidence for a causal relationship between mitochondrial dysfunction and tau hyperphosphorylation, as suggested by other studies (Hoglinger et al, 2005; Escobar-Khondiker et al, 2007; Melov et al, 2007; Ohsawa et al, 2008).
In both Phb2 and Afg3l2 mouse models, we observed activation of MAPKs (Merkwirth et al, 2012). MAPKs have been shown to phosphorylate tau at several residues both in vitro and in vivo (Zhu et al, 2002) and are activated by several cell stresses, including oxidative stress (Son et al, 2013). Consistently, we find that antioxidants efficiently reduce levels of phospho-tau in primary neurons. Increased levels of oxidized proteins were detected in the cerebellum of Afg3l2 heterozygous mice (Maltecca et al, 2009), while it is conceivable that neurons die before oxidative damage occurs in the brain of Afg3l2 knockout mice. Failure to detect a significant increase of ROS levels in Afg3l2-deficient neurons may be due to the limited sensitivity of available fluorescent dyes (Murphy et al, 2011). However, in support of the hypothesis that ROS play a signaling role in regulating mitochondrial transport, we found that treatment with antioxidants, such as NAC and vitamin E, can rescue the anterograde transport defects of Afg3l2-deficient mitochondria. Interestingly, NAC regulates both the frequency of transport and the velocity of mitochondria in wild-type neurons, stressing the hypothesis that modulating the physiological levels of ROS can affect mitochondrial transport. NAC has a more pronounced effect than vitamin E on mitochondrial transport in Afg3l2-deficient neurons. NAC was previously shown to rescue the mitochondrial transport defects observed in dopaminergic neurons treated with MPP+, a toxin that inhibits mitochondrial complex I activity (Kim-Han et al, 2011). Neurons treated with NAC display a reduction of the levels of actin and β-tubulin. Thus, the beneficial effect of NAC may be also exerted by modulation of redox modifications of cytoskeletal components. In fact, actin has been previously shown to sense oxidative load and to be affected by NAC (Dalle-Donne et al, 2001; Farah & Amberg, 2007). An intriguing hypothesis is that ROS-activated signaling cascades can affect mitochondrial transport not only by impinging on tau phosphorylation, but also by targeting motor proteins, or mitochondrial adaptors. Future experiments will be required to investigate this possibility in depth.
In conclusion, we propose a pathogenic model, where deficiency of AFG3L2 leads on one side to mitochondrial fragmentation, via OMA1-mediated processing of OPA1, and on the other side to increased production of ROS (Fig8). Increased ROS may result from respiratory incompetence due to defective mitochondrial translation (Almajan et al, 2012). ROS may trigger activation of stress-induced kinases, which phosphorylate tau in an attempt to reduce its attachment to the microtubules, thus promoting mitochondrial transport. However, with time, insoluble hyperphosphorylated tau aggregates, disrupts microtubules, and affects mitochondrial anterograde transport. The axonal transport defect may further contribute to sustain pathological tau hyperphosphorylation (Fig8). Mitochondrial transport defects ultimately lead to late-onset mitochondrial depletion from axons and neurodegeneration. Our data may be of relevance for several neurodegenerative conditions characterized by mitochondrial dysfunction and tau accumulation and open up the possibility that treatment with antioxidants may be beneficial in models of Afg3l2 deficiency.
Figure 8. Schematic representation of the pathogenic cascade in neurodegenerative diseases due to AFG3L2 deficiency.
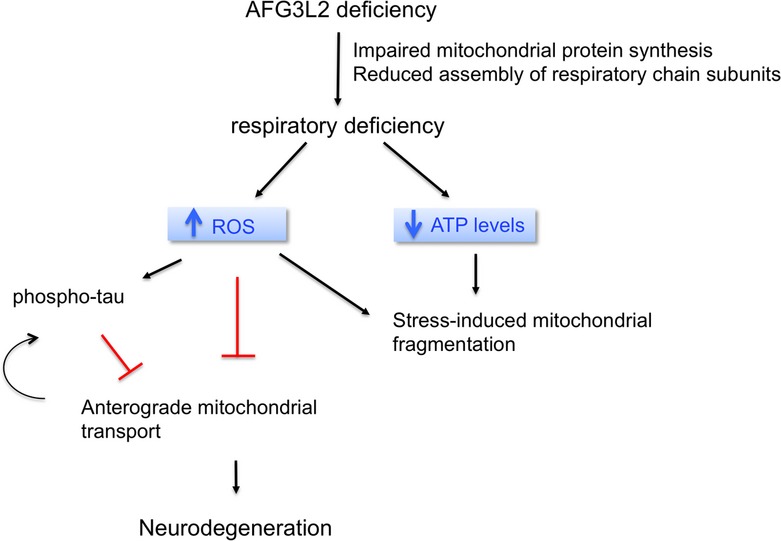
Upon AFG3L2 deficiency, mitochondria become dysfunctional due to impaired mitochondrial protein synthesis and reduced assembly of respiratory chain subunits. The resulting respiratory impairment causes, on one side, stress-induced fragmentation of mitochondria via OMA1 activation and, on the other side, increased ROS production. ROS trigger a pathogenic cascade leading to hyperphosphorylation of tau, which in turn can affect mitochondrial anterograde transport. ROS may also hinder transport via some other yet uncharacterized mechanism. Defects in anterograde transport of mitochondria lead to depletion of mitochondria in axons and neurodegeneration.
Material and Methods
Animal experiments
All animal procedures were conducted in accordance with European (EU directive 86/609/EEC), national (TierSchG), and institutional guidelines and were approved by local authorities (Landesamt für Natur, Umwelt, und Verbraucherschutz Nordrhein-Westfalen) under the license 87–51.04.2010.A219. Afg3l2Emv66/Emv66 mice were obtained by breeding heterozygous Afg3l2+/Emv66 (Maltecca et al, 2008) in a mixed FVB-C57BL/6 background. Conditional Afg3l2 floxed mice (Almajan et al, 2012) were mated with CaMKIIa cre (Minichiello et al, 1999) to achieve Afg3l2 forebrain neuron-specific knockouts. Mice carrying the Oma1 gene exon 3 flanked by loxP sites were generated by gene targeting in C57BL/6 background. Oma1 knockout mice were produced by crossing homozygous Oma1 floxed mice with a transgenic line expressing the Cre recombinase under the β-actin promoter.
Tissue preparation
Mice were deeply anesthetized and perfused transcardially with 4% paraformaldehyde (PFA) in PBS. Tissues were dissected and postfixed in 4% PFA for immunohistochemistry and in 2% glutaraldehyde in 0.12 M phosphate buffer for electron microscopy. For biochemical analyses, tissues were immediately frozen in dry ice and stored at −80°C until use.
RNA in situ hybridization
PCR products obtained by amplification of mouse brain cDNA were used as templates to transcribe either sense or antisense digoxygenin-labeled riboprobes using the DIG RNA labeling kit (Roche). Oligonucleotides used to generate the PCR products contained the SP6 promoter. Sequences are available upon request. RNA in situ hybridization was performed on vibratome sections, as previously described (Tiveron et al, 1996).
Histology and Immunohistochemistry
Postfixed brains were embedded in 6% agar, and 30-μm sections were cut using a vibratome (Leica). Free-floating sections were stained with 0.25% thionine solution. Immunohistochemistry with AT8 antibody (Thermo Scientific) was performed with M.O.M. Immunodetection kit (Vector Laboratories), according to the manufacturer's protocol. Sections were mounted using Eukitt medium (Fluka). For immunofluorescence, sections were permeabilized and blocked in 0.4% Triton X-100 and 10% goat serum in TBS and incubated with SMI 31, SMI 52, and anti-MBP antibodies (Covance) diluted in 5% goat serum in TBS. Following incubation with anti-mouse Alexa Fluor 488 (Life technologies), sections were mounted using FluorSave Reagent (Calbiochem). Images were acquired using an Axio-Imager M2 microscope equipped with Apotome 2 (Zeiss).
Western blot analysis
Tissues were homogenized in RIPA buffer containing 150 mM NaCl, 50 mM Tris–HCl pH 7.4, 5 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, and protease inhibitor cocktail (Roche) on ice. After high-speed centrifugation, the supernatant was harvested, and protein concentration was determined using Bradford assay (Bio-Rad). 20–50 μg proteins were separated by 12% SDS–PAGE and blotted onto PVDF membrane (Millipore). The membrane was blocked in 5% milk containing 0.1% Tween-20 in TBS for 1 h at room temperature and probed with primary antibodies overnight at 4°C. We used AT8, AT270 (detecting Tau phosphorylation site Thr181), anti-phospho-Tau-Ser199, anti-phospho-Tau-Ser396 antibodies from Pierce, anti-Tau1, anti-ERK1/2, anti-GAPDH from Millipore, anti-PKA C-α, anti-phospho-PKA C (Thr197), anti-phospho-p44/42MAPK (ERK1/2; Thr202/Tyr204) from Cell Signaling. Following incubation with HRP-conjugated secondary antibodies, signal was detected using ECL (GE Healthcare).
Electron microscopy
After fixation in 2% glutaraldehyde in 0.12 M phosphate buffer, tissues were treated with 1% osmium tetroxide and embedded in Epon (Fluka). About 70-nm ultrathin sections were cut, collected onto 200 mesh copper grids (Electron Microscopy Sciences), and stained with uranyl acetate (Plano GMBH) and lead citrate (Electron Microscopy Sciences). Images were captured by a transmission electron microscope (CM10, Phillips) equipped with Orius SC200W camera.
Primary neuron culture and transfection
Embryos were harvested from wild-type CD1 mice or Oma1 knockout mice at E18. Cortices were collected after removing the meninges, cut into small pieces, washed in Earle's balanced salt solution (Gibco), dissociated with glass Pasteur pipette, and centrifuged at 100 g for 5 min in neurobasal medium (Gibco). The supernatant was removed, and fresh medium was added to resuspend the neurons and filtered through a 100-μm filter. 125,000 neurons were transfected using a micro-electroporation system (Microporator MP-100 apparatus, Digital Bio) and then plated on glass-bottom dishes (MatTek corporation) coated with poly-D-lysine hydrobromide (0.1 mg/ml, Sigma-Aldrich). The neurons were grown in neurobasal medium supplemented with B-27 (Gibco) and glutamine at 37°C with 5% CO2. In each experiment, 0.5 μg of mito-mCherry was co-transfected together with 100 nM of the respective siRNA.
Stealth-small interfering RNAs (siRNAs) were synthesized by Invitrogen with the following sequences:
Afg3l2: 5′-CCUGCCUCCGUACGCUCUAUCAAUA-3′
Mapt: 5′-CAGUCGAAGAUUGGCUCCUUGGAUA-3′.
The medium GC stealth-negative control (Invitrogen) was used for all experiments.
NAC (Sigma-Aldrich) and vitamin E (Sigma-Aldrich) were supplemented to the complete medium at concentrations of 1 mM and 200 μM, respectively. Vitamin E was replenished every 24 h.
Live-cell imaging
Mitochondria in the axons were imaged using a Perkin-Elmer Ultra View spinning disc confocal microscope, equipped with a CCD camera (Hamamatsu, C9100-50), at 60× magnification (oil-immersion objective, N.A = 1.49, illumination wavelength = 561 nm). Video recordings were acquired at 1,000 × 1,000 pixel resolution every 10 seconds for a period of 10 min (61 image stacks/movie). The stage enclosing the dishes was set to 37°C and 5% CO2. Imaging of the axons was performed 72 h after plating the neurons, and they were identified as processes arising from the soma twice or thrice longer than other processes. Axons selected for analyses had a minimum length of 200 μm. Images were acquired at least 50 μm distal to the soma.
Kymograph generation
Mitochondrial movements along an axon were visualized using kymographs, generated with a custom written script (Matlab R2011b incl. image processing toolbox, Mathworks). Initially, an optimal axon centerline between two manually set endpoints was automatically obtained, based on a maximum projection of all image stacks (imaged mitochondria). Then, kymographs were obtained by generating a maximum projection for each image stack (time point) leading to orthogonal projection of maximum intensities in a neighborhood of approximately 1 μm onto the axon centerline.
Assessment of mitochondrial length, occupancy, and axonal transport
Mitochondrial length was quantified from the first image of the video recordings, by drawing a line along the major axis or along the diameter of circular mitochondria. This analysis was performed in Volocity 6.1 (PerkinElmer). Mitochondrial occupancy was calculated as the total length of all mitochondria in an axon divided by axonal length. For quantification of mitochondrial transport, we classified mitochondrial movement over time as anterograde, retrograde, or oscillatory. Mitochondria were defined as oscillatory if they changed direction to move a distance greater than 5 μm, while they were considered stationary if they moved less than 5 μm during the video recordings. In case of a fission event, daughter mitochondria were considered as separate. In case of a fusion event, mitochondrion after fusion was considered as separate from the parent mitochondria. Classification of mitochondrial movements was manually performed from the video recordings obtained in Volocity 6.1 (PerkinElmer) as the position of soma was noted while image acquisition. Mitochondrial velocity was calculated for anterograde and retrograde movements in all the timeframes where a mitochondrion was moving. After establishing a cut-off value of 0.1 μm/s, the average velocity was calculated for each individual mitochondrion.
Statistical analysis
Mitochondrial length was measured from at least three independent experiments. Data were binned, averaged across experiments, and statistical evaluation was performed using the chi-square test. Unpaired two-tailed Student's t-test was used for statistical comparison of mitochondrial occupancy, transport, and average velocity. Data are presented as mean ± standard error (SEM). In all cases, at least three independent experiments were performed, and 8–10 axons analyzed. The P-values were calculated using Graphpad Prism software (version 6.02) and are indicated when lower than 0.05.
Acknowledgments
The authors wish to thank Jens Brüning, Thomas Wunderlich, and Markus Schubert for generous sharing of antibodies. We are also grateful to Elisa Motori and Nils-Göran Larsson for sharing reagents and for helpful discussion. We thank Peter Frommolt for discussion on statistical evaluation of data. We thank the CECAD Imaging Facility and Christian Jüngst for their support. This work was supported by grants of the Deutsche Forschungsgemeinschaft to E.I.R. (RU1653/1-1), and T.L. (LA918/8-1), a long-term fellowship of the Alexander-von-Humboldt foundation to M.J.B, and a fellowship of the NRW International Graduate School in Development Health and Disease to A.K.K.
Author contributions
AKK performed imaging experiments, with the help of ACS; NK developed the code required for generating kymographs; SW, SM, AK, PM, and DH performed experiments on the mouse models; MJB produced Oma1 knockout mice; TL and EIR conceived the study and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information for this article is available online: http://emboj.embopress.org
References
- Almajan ER, Richter R, Paeger L, Martinelli P, Barth E, Decker T, Larsson NG, Kloppenburg P, Langer T, Rugarli EI. AFG3L2 supports mitochondrial protein synthesis and Purkinje cell survival. J Clin Invest. 2012;122:4048–4058. doi: 10.1172/JCI64604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atorino L, Silvestri L, Koppen M, Cassina L, Ballabio A, Marconi R, Langer T, Casari G. Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. J Cell Biol. 2003;163:777–787. doi: 10.1083/jcb.200304112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MJ, Lampe PA, Stojanovski D, Korwitz A, Anand R, Tatsuta T, Langer T. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 2014;33:578–593. doi: 10.1002/embj.201386474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Bonn F, Tatsuta T, Petrungaro C, Riemer J, Langer T. Presequence-dependent folding ensures MrpL32 processing by the m-AAA protease in mitochondria. EMBO J. 2011;30:2545–2556. doi: 10.1038/emboj.2011.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, De Michele G, Filla A, Cocozza S, Marconi R, Dürr A, Fontaine B, Ballabio A. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–983. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- Chu CT, Levinthal DJ, Kulich SM, Chalovich EM, DeFranco DB. Oxidative neuronal injury. The dark side of ERK1/2. Eur J Biochem. 2004;271:2060–2066. doi: 10.1111/j.1432-1033.2004.04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Milzani A, Di Simplicio P, Colombo R. The actin cytoskeleton response to oxidants: from small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radical Biol Med. 2001;31:1624–1632. doi: 10.1016/s0891-5849(01)00749-3. [DOI] [PubMed] [Google Scholar]
- De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Brownlees J, Ackerley S, Shaw PJ, McLoughlin DM, Shaw CE, Leigh PN, Miller CC, Grierson AJ. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet. 2007;16:2720–2728. doi: 10.1093/hmg/ddm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bella D, Lazzaro F, Brusco A, Plumari M, Battaglia G, Pastore A, Finardi A, Cagnoli C, Tempia F, Frontali M, Veneziano L, Sacco T, Boda E, Brussino A, Bonn F, Castellotti B, Baratta S, Mariotti C, Gellera C, Fracasso V, et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet. 2010;42:313–321. doi: 10.1038/ng.544. [DOI] [PubMed] [Google Scholar]
- Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvezin-Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, Chomyn A, Bauer MF, Attardi G, Larsson NG, Neupert W, Reichert AS. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem. 2006;281:37972–37979. doi: 10.1074/jbc.M606059200. [DOI] [PubMed] [Google Scholar]
- Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J Cell Biol. 1998;143:777–794. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, Martinou JC, Westermann B, Rugarli EI, Langer T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol. 2009;187:1023–1036. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar-Khondiker M, Hollerhage M, Muriel MP, Champy P, Bach A, Depienne C, Respondek G, Yamada ES, Lannuzel A, Yagi T, Hirsch EC, Oertel WH, Jacob R, Michel PP, Ruberg M, Hoglinger GU. Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons. J Neurosci. 2007;27:7827–7837. doi: 10.1523/JNEUROSCI.1644-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzone TL, Gunawardena S, McCleary D, Reis GF, Goldstein LS. Kinesin-1 transport reductions enhance human tau hyperphosphorylation, aggregation and neurodegeneration in animal models of tauopathies. Hum Mol Genet. 2010;19:4399–4408. doi: 10.1093/hmg/ddq363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzone TL, Stokin GB, Lillo C, Rodrigues EM, Westerman EL, Williams DS, Goldstein LS. Axonal stress kinase activation and tau misbehavior induced by kinesin-1 transport defects. J Neurosci. 2009;29:5758–5767. doi: 10.1523/JNEUROSCI.0780-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah ME, Amberg DC. Conserved actin cysteine residues are oxidative stress sensors that can regulate cell death in yeast. Mol Biol Cell. 2007;18:1359–1365. doi: 10.1091/mbc.E06-08-0718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes F, Tatsuta T, Langer T. Mitochondrial AAA proteases–towards a molecular understanding of membrane-bound proteolytic machines. Biochim Biophys Acta. 2012;1823:49–55. doi: 10.1016/j.bbamcr.2011.09.015. [DOI] [PubMed] [Google Scholar]
- Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol. 2007;178:757–764. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol. 2009;187:959–966. doi: 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoglinger GU, Lannuzel A, Khondiker ME, Michel PP, Duyckaerts C, Feger J, Champy P, Prigent A, Medja F, Lombes A, Oertel WH, Ruberg M, Hirsch EC. The mitochondrial complex I inhibitor rotenone triggers a cerebral tauopathy. J Neurochem. 2005;95:930–939. doi: 10.1111/j.1471-4159.2005.03493.x. [DOI] [PubMed] [Google Scholar]
- Hornig-Do HT, Tatsuta T, Buckermann A, Bust M, Kollberg G, Rotig A, Hellmich M, Nijtmans L, Wiesner RJ. Nonsense mutations in the COX1 subunit impair the stability of respiratory chain complexes rather than their assembly. EMBO J. 2012;31:1293–1307. doi: 10.1038/emboj.2011.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima-Ando K, Sekiya M, Maruko-Otake A, Ohtake Y, Suzuki E, Lu B, Iijima KM. Loss of axonal mitochondria promotes tau-mediated neurodegeneration and Alzheimer's disease-related tau phosphorylation via PAR-1. PLoS Genet. 2012;8:e1002918. doi: 10.1371/journal.pgen.1002918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasher PR, De Vos KJ, Wharton SB, Manser C, Bennett EJ, Bingley M, Wood JD, Milner R, McDermott CJ, Miller CC, Shaw PJ, Grierson AJ. Direct evidence for axonal transport defects in a novel mouse model of mutant spastin-induced hereditary spastic paraplegia (HSP) and human HSP patients. J Neurochem. 2009;110:34–44. doi: 10.1111/j.1471-4159.2009.06104.x. [DOI] [PubMed] [Google Scholar]
- Kelly GS. Clinical applications of N-acetylcysteine. Altern Med Rev. 1998;3:114–127. [PubMed] [Google Scholar]
- Kim-Han JS, Antenor-Dorsey JA, O'Malley KL. The parkinsonian mimetic, MPP+, specifically impairs mitochondrial transport in dopamine axons. J. Neurosci. 2011;31:7212–7221. doi: 10.1523/JNEUROSCI.0711-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppen M, Metodiev MD, Casari G, Rugarli EI, Langer T. Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia. Mol Cell Biol. 2007;27:758–767. doi: 10.1128/MCB.01470-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltecca F, Aghaie A, Schroeder DG, Cassina L, Taylor BA, Phillips SJ, Malaguti M, Previtali S, Guenet JL, Quattrini A, Cox GA, Casari G. The mitochondrial protease AFG3L2 is essential for axonal development. J Neurosci. 2008;28:2827–2836. doi: 10.1523/JNEUROSCI.4677-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltecca F, Magnoni R, Cerri F, Cox GA, Quattrini A, Casari G. Haploinsufficiency of AFG3L2, the gene responsible for spinocerebellar ataxia type 28, causes mitochondria-mediated Purkinje cell dark degeneration. J Neurosci. 2009;29:9244–9254. doi: 10.1523/JNEUROSCI.1532-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin ML, Yardin C, Terro F. Tau protein kinases: involvement in Alzheimer's disease. Ageing Res Rev. 2013;12:289–309. doi: 10.1016/j.arr.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Melov S, Adlard PA, Morten K, Johnson F, Golden TR, Hinerfeld D, Schilling B, Mavros C, Masters CL, Volitakis I, Li QX, Laughton K, Hubbard A, Cherny RA, Gibson B, Bush AI. Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS ONE. 2007;2:e536. doi: 10.1371/journal.pone.0000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C, Langer T. Prohibitin function within mitochondria: essential roles for cell proliferation and cristae morphogenesis. Biochim Biophys Acta. 2009;1793:27–32. doi: 10.1016/j.bbamcr.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Merkwirth C, Martinelli P, Korwitz A, Morbin M, Bronneke HS, Jordan SD, Rugarli EI, Langer T. Loss of prohibitin membrane scaffolds impairs mitochondrial architecture and leads to tau hyperphosphorylation and neurodegeneration. PLoS Genet. 2012;8:e1003021. doi: 10.1371/journal.pgen.1003021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kuhn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci. 2010;30:4232–4240. doi: 10.1523/JNEUROSCI.6248-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP, Holmgren A, Larsson NG, Halliwell B, Chang CJ, Kalyanaraman B, Rhee SG, Thornalley PJ, Partridge L, Gems D, Nystrom T, Belousov V, Schumacker PT, Winterbourn CC. Unraveling the biological roles of reactive oxygen species. Cell Metab. 2011;13:361–366. doi: 10.1016/j.cmet.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niki E, Traber MG. A history of vitamin E. Ann Nutr Metab. 2012;61:207–212. doi: 10.1159/000343106. [DOI] [PubMed] [Google Scholar]
- Nolden M, Ehses S, Koppen M, Bernacchia A, Rugarli EI, Langer T. The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell. 2005;123:277–289. doi: 10.1016/j.cell.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Ohsawa I, Nishimaki K, Murakami Y, Suzuki Y, Ishikawa M, Ohta S. Age-dependent neurodegeneration accompanying memory loss in transgenic mice defective in mitochondrial aldehyde dehydrogenase 2 activity. J Neurosci. 2008;28:6239–6249. doi: 10.1523/JNEUROSCI.4956-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RB, Nunomura A, Lee HG, Casadesus G, Perry G, Smith MA, Zhu X. Signal transduction cascades associated with oxidative stress in Alzheimer's disease. J Alzheimer's Dis. 2007;11:143–152. doi: 10.3233/jad-2007-11202. [DOI] [PubMed] [Google Scholar]
- Pierson TM, Adams D, Bonn F, Martinelli P, Cherukuri PF, Teer JK, Hansen NF, Cruz P Mullikin For The Nisc Comparative Sequencing Program JC. Blakesley RW, Golas G, Kwan J, Sandler A, Fuentes FajardoK, Markello T, Tifft C, Blackstone C, Rugarli EI, Langer T, Gahl WA, Toro C. Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases. PLoS Genet. 2011;7:e1002325. doi: 10.1371/journal.pgen.1002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Tripathi R, Troung Q, Tirumala K, Reddy TP, Anekonda V, Shirendeb UP, Calkins MJ, Reddy AP, Mao P, Manczak M. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer's disease: implications to mitochondria-targeted antioxidant therapeutics. Biochim Biophys Acta. 2012;1822:639–649. doi: 10.1016/j.bbadis.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugarli EI, Langer T. Mitochondrial quality control: a matter of life and death for neurons. EMBO J. 2012;31:1336–1349. doi: 10.1038/emboj.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, Przedborski S. Mitochondria: the next (neurode) generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahpasand K, Uemura I, Saito T, Asano T, Hata K, Shibata K, Toyoshima Y, Hasegawa M, Hisanaga S. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer's disease. J Neurosci. 2012;32:2430–2441. doi: 10.1523/JNEUROSCI.5927-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son Y, Kim S, Chung HT, Pae HO. Reactive oxygen species in the activation of MAP kinases. Methods Enzymol. 2013;528:27–48. doi: 10.1016/B978-0-12-405881-1.00002-1. [DOI] [PubMed] [Google Scholar]
- Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thies E, Mandelkow EM. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci. 2007;27:2896–2907. doi: 10.1523/JNEUROSCI.4674-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiveron MC, Hirsch MR, Brunet JF. The expression pattern of the transcription factor Phox2 delineates synaptic pathways of the autonomic nervous system. J Neurosci. 1996;16:7649–7660. doi: 10.1523/JNEUROSCI.16-23-07649.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JZ, Liu F. Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog Neurobiol. 2008;85:148–175. doi: 10.1016/j.pneurobio.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Kumar A, Peterhoff C, Duff K, Nixon RA. Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J Neurosci. 2008;28:1682–1687. doi: 10.1523/JNEUROSCI.5242-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Lee HG, Raina AK, Perry G, Smith MA. The role of mitogen-activated protein kinase pathways in Alzheimer's disease. Neurosignals. 2002;11:270–281. doi: 10.1159/000067426. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.