

Abstract
Endocrine-resistant breast cancer is a major clinical obstacle. The use of 17β-estradiol (E2) has re-emerged as a potential treatment option following exhaustive use of tamoxifen (TAM) or aromatase inhibitors although side effects have hindered its clinical usage. Protein kinase C alpha (PKCα) expression was shown to be a predictor of disease outcome for patients receiving endocrine therapy and may predict a positive response to an estrogenic treatment. Here, we have investigated the use of novel benzothiophene selective estrogen mimics (SEMs) as an alternative to E2 for the treatment of TAM-resistant breast cancer. Following in vitro characterization of SEMs, a panel of clinically relevant PKCα-expressing, TAM-resistant models were used to investigate the antitumor effects of these compounds. SEM treatment resulted in growth inhibition and apoptosis of TAM-resistant cell lines in vitro. In vivo SEM treatment induced tumor regression of TAM-resistant T47D:A18/PKCα and T47D:A18-TAM1 tumor models. T47D:A18/PKCα tumor regression was accompanied by translocation of ERα to extranuclear sites, possibly defining a mechanism through which these SEMs initiate tumor regression. SEM treatment did not stimulate growth of E2-dependent T47D:A18/neo tumors. Additionally, unlike E2 or TAM, treatment with SEMs did not stimulate uterine weight gain. These findings suggest the further development of SEMs as a feasible therapeutic strategy for the treatment of endocrine-resistant breast cancer without the side effects associated with E2.
Keywords: PKCα, endocrine resistance, Selective Estrogen Receptor Modulators
Introduction
The selective estrogen receptor modulator (SERM) tamoxifen (TAM) is the most widely prescribed endocrine therapy for the treatment and prevention of breast cancer in premenopausal women, while aromatase inhibitors are the drug of choice for post-menopausal women. De novo or acquired resistance to these endocrine therapies limits their clinical effectiveness leading to disease progression. As such, there is a clinical need for therapeutic alternatives for women who no longer respond to conventional endocrine therapies.
Protein kinase C alpha (PKCα) belongs to a family of serine/threonine protein kinases (1, 2). PKCα expression in breast cancer is associated with TAM-resistance, poor patient survival and breast cancer aggressiveness (3–5). To further substantiate these clinical observations we reported that ectopic overexpression of PKCα in the T47D:A18 breast cancer cell line resulted in a hormone-independent, TAM-resistant phenotype (6). Interestingly, these TAM-resistant T47D:A18/PKCα tumors are growth inhibited by 17β-estradiol (E2) in vivo (7). Yao and colleagues describe an MCF-7 tumor model in which long-term exposure (5 years) to TAM led to an E2-inhibited phenotype (8) and elevated PKCα expression (7). Together these studies provide important therapeutic implications, suggesting that PKCα expression may predict both resistance to conventional endocrine therapies as well as a predicted response to E2 or estrogen-like compounds.
Before the introduction of TAM, breast cancer patients were treated with high-dose E2 or diethystilbesterol (DES). Although, similar response rates were observed (9, 10), TAM treatment became the mainstay due to a lower incidence of side effects such as nausea, emesis and edema. Since treatment with E2, DES and TAM are now all associated with side effects including increased risk of thromboembolic disorders and unwanted agonist-driven uterine growth, we sought an alternative treatment strategy that would have therapeutic efficacy in the TAM-resistant setting. We have previously reported that TAM-resistant T47D:A18/PKCα tumors regress upon treatment with both E2 and the benzothiophene SERM raloxifene (RAL), although the effects of RAL did not persist after treatment withdrawal (11). RAL has a favorable antiestrogenic profile in the uterus and has proven safety over 15 years of clinical use in postmenopausal osteoporosis and breast cancer chemoprevention.
In this study, we tested the in vivo effects of two novel benzothiophene SEMs, BTC [2-(4-hydroxyphenyl)benzo[b]thiophen-6-ol] and TTC-352 [3-(4-fluorophenyl)-2-(4-hydroxyphenoxy)benzo[b]thiophen-6-ol] that in contrast to RAL, acted as estrogen agonists in T47D:A18 and MCF-7 cells as reflected by increased cell proliferation and ERE-luciferase reporter activity. Both of these SEMs induced regression of TAM-resistant, hormone-independent T47D:A18/PKCα and T47D:A18-TAM1 xenograft tumors in vivo, but remarkably, neither compound was able to support the growth of hormone-dependent, TAM-sensitive T47D:A18/neo tumors. Of particular importance is that neither SEM caused any significant increase in the uterine weights of treated mice. These data suggest that patients exhibiting endocrine resistance may respond to benzothiophene SEMs using PKCα as a predictive biomarker.
Materials and Methods
Reagents
DMSO, E2 and TAM were obtained from Sigma-Aldrich (St. Louis, MO USA). RAL (Evista®, Eli Lilly and Company, Indianapolis, IN USA) was purchased from the University of Illinois at Chicago Hospital Pharmacy. Cell culture reagents were obtained from Life Technologies (Carlsbad, CA USA). Tissue culture plasticware was purchased from Becton-Dickinson (Franklin Lakes, NJ USA). The following antibodies were used: rabbit polyclonal PKCα (C-20, Santa Cruz Biotechnology, Dallas, TX USA), rabbit polyclonal ERα (SP1, Lab Vision, Thermo Scientific, Kalamazoo, MI USA), mouse monoclonal β-actin (Sigma, St. Louis, MO USA), anti-rabbit Alexa Fluor 488 (Life Technologies, Carlsbad, CA USA) and anti-mouse Cy3 (Jackson Immunoresearch Laboratories, West Grove, PA).
Cell culture conditions
Stable transfectant cell lines T47D:A18/neo and T47D:A18/PKCα were produced and maintained as previously described (6) in RPMI1640 (phenol red) supplemented with 10% fetal bovine serum (FBS) containing G418 (500 μg/ml). T47D cells were initially obtained from ATCC in 1996 and stored during early passage. MCF-7 cells were originally obtained from the Michigan Cancer Foundation (Detroit, MI, USA) in 1992 and stored during early passage. MCF-7:5C cells were maintained E2-depleted media as previously described (12). The T47D:A18-TAM1 cell line was created by maintaining T47D:A18 breast cancer cells long-term (12 months) in 1 uM of 4-hydroxytamoxifen (4-OHT) in E2-depleted media. Single cell clones were derived using the limiting dilution method. All TAM-resistant cell lines retain estrogen receptor alpha (ERα) expression at varying levels compared to their TAM-sensitive counterparts (Supplemental Figure 1). Prior to treatment cell lines were cultured in E2-depleted media for 3 days. Cell lines were routinely tested for mycoplasma contamination (MycoAlert™ Mycoplasma Detection Kit, Lonza Ltd., Rockland, ME, USA). All cell lines were authenticated in April 2014 using short tandem repeat (STR) and ATCC analysis (Promega Corporation Core Genomics Facility, UIC).
Synthesis and oral bioavailability of BTC
The synthesis of BTC and TTC-352 has been described (13). Dansyl derivatization of BTC was employed to increase limits of detection and quantitation for LC-MS/MS analysis of plasma samples (Supplemental Figures 2 and 3). Working solutions of BTC and internal standard (3-Br-BTC) were prepared by serial dilution of 1 mg/mL acetonitrile stocks. Calibration standards were prepared by spiking BTC or 3-Br-BTC (20 ng/mL) into blank mouse plasma to give a final concentration range of 5–100 ng/mL (Supplemental Figure 4). After addition of cold acetonitrile, samples were kept at 4 °C for 2 h, centrifuged at 10,000 rpm for 15 min, and supernatants were concentrated under N2 stream. Resulting residues were reconstituted in 0.1 mL of 100 mM sodium bicarbonate buffer (pH=10.5) and derivatized by addition of 0.1 mL dansyl chloride (2 mg/mL in acetone) followed by incubation at 60°C for 5 min. After removal of solvent, residues were reconstituted in 0.25 mL acetonitrile/water (1:1, v/v) and analyzed by LC-MS/MS.
BTC was administered in ethanol using a vehicle of propylene glycol/carboxymethylcellulose (10 mg/kg p.o) to ovariectomized 4–6 week old athymic mice (Harlan-Sprague-Dawley) (n=3). Blood samples were collected in EDTA tubes at 20 min, 2 h, and 6 h after treatment. Plasma was separated from whole blood by centrifugation at 4 °C. Prior to analysis, plasma was spiked with internal standard and extracted 3x with cold acetonitrile. Recovery of analyte was measured by spiking known amounts of BTC into blank plasma samples. Work up of plasma samples were identical to that described above for standard curve determination.
LC-MS/MS analysis was performed using an API 3000 (Applied Biosystems) triple quadrupole mass spectrometer equipped with Agilent 1200 HPLC (Agilent Technologies, Santa Clara, CA USA). Multiple reaction monitoring (MRM) for the dissociations of m/z 709→171 and m/z 789→171 (loss of 5-dimethylaminonaphthalene) were optimized to measure dansyl-BTC and dansylBr-BTC, respectively (Supplemental Figure 5). Separation was performed using a Hypersil BDS C18 (2.1 mm × 30 mm; 3 μm) column (Thermo Quest Corporation, MA) at a flow rate of 0.3 mL/min. The elution solvent consisted of water with 10% MeOH and 0.3% formic acid (A) and MeCN with 0.3% formic acid (B). The mobile phase was initially held at 10% B for 5 min, increased to 60% B over 1.5 min, and then increased to 90% B over 15 min, with dansyl-BTC and dansylBr-BTC eluting at 17.8 and 19.7 min, respectively (Supplemental Figure 5).
DNA growth assay
T47D:A18/neo, T47D:A18/PKCα and T47D:A18-TAM1 cells were maintained in E2-depleted media 3 days before plating in 24-well plates (15,000 cells/well). Medium containing compound was added the following day and total DNA was determined by incubating cells with Hoechst 33342 cell permeable dye for 1 hour and reading fluorescence at excitation 355 nm/emission 460 nm on a Perkin Elmer Victor3 V plate reader (Waltham, MA USA). Treatment medium was changed every 2–3 days.
Proliferation assay
Following 3 days of growth in E2-depleted media, 2 × 105 cells were seeded into T25 tissue culture flasks. The following day (day 1) treatment medium was added. Cells were counted on Day 9 and medium was changed every 3 days.
Western Blot
Whole cell extracts of cultured cells were prepared in lysis buffer (200 mM Tris, 1% Triton X-100, 5 mM EDTA) with protease and phosphatase inhibitor cocktails (1:50, both from Sigma-Aldrich) after scraping from the culture plates. Protein concentration was measured using the Bradford method (Bio-Rad, Hercules, 26 CA). Proteins were separated under denaturing conditions, blotted onto nitrocellulose membrane (Bio-Rad) using a wet transfer system (Bio-Rad). Images of blots were acquired on a Bio-Rad ChemiDoc System following incubation with SuperSignal West Dura luminol solution (Thermo Fisher Scientific). Protein bands were quantified using densitometry measured in Adobe Photoshop CS4 (San Jose, CA) and normalized to β-actin.
Transient transfection and luciferase assays
Cells were transiently transfected by electroporation with 5 μg ERE-tk-Luc plasmid containing the luciferase reporter gene controlled by a triplet vitellogenin consensus ERE (14)and 1 μg pCMVβ-galactosidase (β-gal) expressing plasmid. After 24 hours the cells were treated and incubated overnight at 37°C. Cells were lysed and luciferase activity and β-gal signals were read by a Monolight 3010 luminometer (Becton Dickinson, Franklin Lakes, NJ USA).
Colony formation assay in Matrigel™ Matrix
Matrigel™ (Becton Dickinson, Franklin Lakes, NJ USA) was thawed overnight at 4°C. Twelve-well plates were coated with 200 μL Matrigel™/well and incubated at 37°C for 30 min. Cells were suspended at 5 × 103 in 400 μL of phenol red-free RPMI 1640 and spread onto pre-gelled Matrigel™ followed by the addition of 360 μL of treatment media containing 40 μL Matrigel™. Plates were incubated at 37°C for 10 days; medium containing 10% Matrigel™ was replaced to the top of the Matrigel™ every 3 days. Colonies were stained with crystal violet on day 10 and each well was counted by light microscopy (20X).
Annexin V analysis of apoptosis
The AlexaFluor 488 Annexin V/Dead Cell Apoptosis kit (Invitrogen, Carlsbad, CA USA) was used to quantify cell death by flow cytometry according to the manufacturer’s instructions. In brief, MCF-7:5C cells were treated with control and test compounds for 6 days with treatment renewal on day 3. Cells (1 × 105) were suspended in 1X Annexin V binding buffer and were stained with AlexaFluor 488 Annexin V and Propidium Iodide (PI). The cells were then analyzed by FACS using a Gallios Flow Cytometer (Beckman Coulter).
Animal experiments
T47D:A18/PKCα, T47D:A18/neo andT47D:A18-TAM1 tumors were established as previously described (7). E2 was administered via silastic capsules (1.0 cm) implanted subcutaneously between the scapulae, producing a mean serum E2 level of 379.5 pg/mL (11, 15). BTC and TTC-352 were administered p.o. at a dose of 1.5 mg/animal daily for 2 weeks as previously described for other SERMs (7). RAL was administered p.o. at a dose 1.5 mg/animal daily for 2 weeks. Tumor cross-sectional area was determined weekly using Vernier calipers and calculated using the formula: length/2 × width/2 × π. Mean tumor area was plotted against time in weeks to monitor tumor growth. The mice were sacrificed by CO2 inhalation and cervical dislocation, and tumors and uteri were excised, cleaned of connective tissue, and immediately weighed. The Animal Care and Use Committee of the University of Illinois at Chicago approved all of the procedures involving animals.
Tumor immunofluorescence and confocal microscopy
Tumor sections (4 μm) were cut from paraffin blocks and prepared for IF staining by deparaffinization and rehydration as previously described (11). Briefly, antigen retrieval was performed by incubating slides in Tris-EDTA (pH = 9.0) buffer. Slides were blocked with antibody diluent (DAKO, Carpinteria, CA USA) followed by primary antibodies at 1:100 in antibody diluent for 1 h, then incubated with secondary antibodies at 1:100 in antibody diluent for 45 min followed by DAPI (4′, 6-diamidino-2-phenylindole [1 μg/mL], DAKO, Carpinteria, CA USA). Confocal analysis was performed with a Zeiss LSM 510 microscope (Carl Zeiss, Incorporated, North America, Thornwood, NY USA).
Statistical analyses
Statistics were run using GraphPad Prism Version 5.0. Statistical analyses used were one-way ANOVA followed by Tukey’s post-testor Student’s t-test where appropriate.
Results
PKCα expression correlates with sensitivity to E2
PKCα-expression in clinical specimens predicted resistance to TAM (4). The ectopic overexpression of PKCα in T47D cells (T47D:A18/PKCα) led to a TAM-resistant, E2-inhibited phenotype in vivo (7), suggesting that PKCα may also predict a positive response to estrogenic therapeutic intervention. To derive an independent model of TAM resistance, T47D:A18 cells were cultured long-term in the presence of 1 μM 4-hydroxytamoxifen (4-OHT) and several TAM-resistant clones were identified including T47D:A18-TAM1. When evaluated in vitro, T47D:A18-TAM1 cell growth was independent of E2 in addition to exhibiting TAM-resistance (Figure 1A). In vivo T47D:A18-TAM1 xenografts were found to act similar to T47D:A18/PKCα xenografts, growing hormone-independently, TAM-resistant, and regress when treated with E2 (Figure 1B). Similar to the T47D:A18/PKCα cell line, T47D:A18-TAM1 cells display increased PKCα expression compared to the TAM-sensitive parental T47D:A18 cell line (Figure 1C and 1D, Lanes 3 and 4).
Figure 1. PKCα-overexpression in breast cancer cells correlates with sensitivity to E2-induced growth inhibition.

A) Effect of E2 and 4-OHT on T47D:A18-TAM1 proliferation in vitro. DNA assays were performed as described in Materials and Methods. Graph shows mean ± SEM and is representative of three independent experiments. B) Effect of TAM and E2 on T47D:A18-TAM1 xenograft growth. 2 mice/group were bilaterally injected with T47D:A18-TAM1 cells and either left untreated or were given oral TAM (1.5 mg/day). Dashed line represents the start of E2 treatment. C) Western blot analysis of PKCα expression in E2-stimulated (T47D:A18/neo, T47D:A18, MCF-7:WS8) and E2-inhibited (T47D:A18/PKCα, T47D:A18-TAM1, and MCF-7:5C) cell lines. D) Densitometric quantification of three western blots from three independent cell lysates. Student’s t-test was used to compare each E2-inhibited cell line to the E2-stimulated counterpart. *, P ≤ 0.05.
The TAM-resistant MCF-7:5C cell cloneis growth inhibited by E2 in vivo and in contrast to T47D:A18/PKCα and T47D:A18-TAM1 cells, growth is also inhibited by E2 in 2D culture (12). MCF-7:5C cells display increased expression of PKCα compared to TAM-sensitive MCF-7:WS8 cells (Figure 1C and 1D, Lanes 5 and 6). Since all three TAM-resistant cell clones display elevated PKCα expression compared to the parental lines, and since PKCα knockdown in T47D:A18/PKCα cells led to a partial reversal of the E2-inhibited phenotype in vivo (16), these results suggest that PKCα may play a role in E2-induced growth inhibition and/or be a predictive biomarker for E2-induced growth inhibition.
SEMs are estrogenic and antiproliferative in endocrine-resistant, PKCα-expressing breast cancer cell lines in vitro
We have previously reported that the E2 growth-inhibitory phenotype observed with T47D:A18/PKCα tumors can be partially recapitulated when these cells are cultured in 3D Matrigel™ as characterized by inhibition of colony formation (7, 17). In an effort to find a potential alternative to E2, and to expand on positive data obtained in vivo with RAL (11), two benzothiophene SEMs, BTC and TTC-352 (Figure 2A and 2B), were selected from a library of compounds and screened by DNA growth assay in 2D.
Figure 2. Effect of BTC and TTC-352 on proliferation of T47D:A18/neo and T47D:A18/PKCα cells in vitro.

Structures of BTC (A) and TTC-352 (B). Effect of BTC and TTC-352 treatment on the growth T47D:A18/neo cells (C and D respectively), T47D:A18/PKCα cells (E and F respectively) and T47D:A18-TAM1 cells (G and H respectively). DNA assays were performed as described in Materials and Methods. Graphs show mean ± SEM and are representative of three independent experiments.
To determine whether BTC and TTC-352 act as estrogen agonists we treated cells in 2D culture and measured DNA content as an index of proliferation. The higher concentrations of BTC and TTC-352 (100 nM) stimulated proliferation of T47D:A18/neo cells comparable to E2 (1 nM)(Figure 2C and 2D). Both T47D:A18/PKCα and T47D:A18-TAM1 cells proliferated equally in the presence of E2, BTC and TTC-352 (1–100 nM)(Figures 2E–H).
To determine if BTC and TTC-352 mirror the antiproliferative effect of E2 on T47D:A18/PKCα cells in 3D culture, colony formation in Matrigel™ was examined. BTC and TTC-352 treatment resulted in increased T47D:A18/neo colony formation (Figure 3A), while significantly preventing T47D:A18/PKCα and T47D:A18-TAM1 colony formation in 3D (Figure 3B and 3C).
Figure 3. BTC and TTC-352 inhibit T47D:A18/PKCα and T47D:A18-TAM1 colony formation in 3D Matrigel™ and induce apoptosis in MCF-7:5C cells in vitro.

Colonies were established as described in Materials and Methods and treated for 10 days (Control [0.1% DMSO], E2 1 nM, BTC 100 nM, TTC-352 100 nM). A) T47D:A18/neo. B) T47D:A18/PKCα. C) T47D:A18-TAM1. Proliferation assay was performed following 9 days of treatment. D) MCF-7:WS8. E) MCF-7:5C. F) Apoptosis was assessed in MCF-7:5C cells following 6 days of treatment as described in Materials and Methods. *, P ≤ 0.05 versus DMSO. **, P ≤ 0.01 versus DMSO. ***, P ≤ 0.001 versus DMSO. Graph shows mean ± SEM of 3 independent experiments.
To confirm the inhibitory activity of SEMs in a third in vitro model of E2-induced growth inhibition we determined the effects in TAM-resistant MCF-7:5C cells: MCF-7:5C cells are growth inhibited by E2 in 2D culture (12). Parental hormone-dependent MCF-7:WS8 cells were growth stimulated when subjected to BTC and TTC-352 treatment (Figure 3D); however, BTC and TTC-352 (100 nM) significantly inhibited the growth of MCF-7:5C cells over 9 days (Figure 3E). Additionally, apoptosis significantly increased in these cells6 days post-treatment with E2, BTC and TTC-352 (Figure 3F).
To confirm the estrogenic activity of BTC and TTC-352 we examined transcriptional activation of ERα using an estrogen response element (ERE)-luciferase reporter construct (14). In T47D:A18/neo, T47D:A18/PKCα and T47D:A18-TAM1 cells BTC and TTC-352 treatment resulted in an increase in ERα transcriptional activity that was concentration dependent (Figure 4A, 4B and 4C). BTC acted as a full agonist with respect to ERE-luciferase induction in all cell lines, whereas TTC-352 appears to act as a partial ER agonist yielding a significant but partial induction at 100 nM concentration. Taken together, our in vitro findings suggest that SEMs exhibit estrogenic activity and mimic the inhibitory activity of E2 in MCF-7:5C cells in 2D culture and in T47D:A18/PKCα and T47D:A18-TAM1 cells in 3D culture.
Figure 4. BTC and TTC-352 induce ERα transcriptional activity in T47D:A18/neo, T47D:A18/PKCα and T47D:A18-TAM1 cell lines.
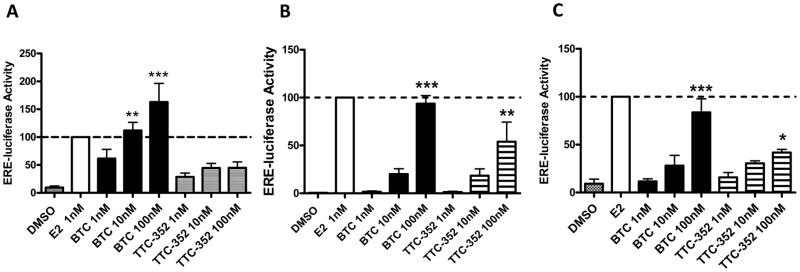
A) T47D:A18/neo, B) T47D:A18/PKCα and C) T47D:A18-TAM1 cell lines. Data is expressed normalized to E2 (100%).*, P ≤ 0.05 versus DMSO. **, P ≤ 0.01 versus DMSO. ***, P ≤ 0.001 versus DMSO. Graph shows mean ± SEM of 3 independent experiments.
Bioavailability of BTC and benzothiophene SERMs
In humans, the absolute bioavailability of orally administered RAL is reported as 2%, with oral clearance of 44 l/kg/h (18, 19). Desmethylarzoxifene (DMA) is a more potent estrogen antagonist in the breast and maintains estrogen agonist actions in bone tissues, however, it also has poor bioavailability (20). Arzoxifene was designed as a DMA prodrug to overcome the problems associated with low bioavailability (21–24). As part of a comparative study of biological activity of DMA, arzoxifene and F-DMA in juvenile female rats, metabolism was assessed by quantification of remaining drug in plasma after 3 days of drug administration: DMA was not observed above detection limits, whereas arzoxifene and F-DMA were easily quantifiable (25–27). BTC is the benzothiophene core of DMA and RAL, whereas TTC-352 bears structural similarity with F-DMA. Consequently, we were concerned that the bioavailability of BTC would be too low for study in vivo (24, 25, 27, 28). Therefore, plasma levels of BTC were measured after oral administration to ovariectomized athymic nude mice. Since BTC ionizes poorly by electrospray ionization ESI-MS, a chemical derivatization method was developed using tandem mass spectroscopic MRM analysis. Drug was detected in plasma at a peak concentration of 40 ng/mL at 30 min, which fell to 10 ng/mL after 6 h (Supplemental Figure 6). Given the observation of BTC well above detection limits in mouse plasma, it was decided to compare the effects of BTC and TTC-352 with RAL and E2 in the T47D:A18/PKCα xenograft model using oral delivery.
SEMs cause regression of hormone-independent, TAM-resistant xenografts
We recently reported that RAL treatment results insignificant regression of TAM-resistant T47D:A18/PKCα tumors. However, RAL did not yield complete tumor regression and importantly was without activity in 3D cultures (11). In contrast to RAL, the SEMs, BTC and TTC-352, are ER agonists in 2D cultures (Figure 2) and are able to inhibit both T47D:A18/PKCα and T47D:A18-TAM1 colony formation in 3D (Figures 3B and 3C). To determine if these compounds could induce T47D:A18/PKCα tumor regression, T47D:A18/PKCα cells were injected into 40 athymic mice and tumors were allowed to grow in the absence of any treatment for seven weeks reaching a mean tumor size of 0.5 cm2 (100%). At that time, the mice were randomized to four treatment groups; continued no treatment (control arm, 9 mice), E2 capsule (9 mice), oral RAL 1.5 mg/day (9 mice), oral BTC 1.5 mg/day (9 mice), or oral TTC-352 1.5 mg/day (4 mice). Following two weeks, all treatments significantly reduced tumor volume compared to non-treated controls (P< 0.05) (Figure 5A): BTC (~88%)and TTC-352(~70%) elicited a decrease in tumor volume that exceeded the regression elicited by RAL (~50%). Most notably, regression induced by E2, BTC, and TTC-352 was sustained for at least four weeks after drug withdrawal, in contrast to RAL (Figure 5A).
Figure 5. BTC and TTC-352 inhibit T47D:A18/PKCα and T47D:A18-TAM1 xenograft tumors.

A) BTC and TTC-352 treatment result in regression of T47D:A18/PKCα tumors. Graph shows percentage of tumor regression (100% ~0.5 cm2). Dotted line indicates when treatment was ended. Arrow designates where tumors from (B) were obtained. *, P ≤ 0.001 versus control. Graph shows mean ± SEM. B) ERα localization in T47D:A18/PKCα tumors by immunofluorescence staining. Total magnification: 630X.C) T47D:A18-TAM1 tumors regress when treated with BTC and TTC-352. Dotted lines represents start of treatment.
We previously reported that regression of T47D:A18/PKCα tumors, induced by both RAL and E2, is accompanied by the exit of ERα from the nucleus suggesting the possible mechanistic involvement of extranuclear ERα (11, 17). We show here that tumor regression following treatment with BTC and TTC-352 is also accompanied by exit of ERα from the nucleus (Figure 5B). These results suggest that the mechanism of regression by benzothiophene SEMs is similar to that of E2 and may involve extranuclear ERα.
To test the effects of E2 and SEMs on T47D:A18-TAM1 xenograft tumors, tumors were established in absence of treatment and at 10 weeks, 11 athymic nude mice were randomized into four treatment groups: untreated control arm (2 mice), E2 capsule (3 mice), oral BTC 1.5 mg/day (3 mice), or oral TTC-352 1.5 mg/day (3 mice). Following 2 weeks of daily BTC and TTC-352 treatment, we observed significant T47D:A18-TAM1 tumor regression (Figure 5C).
Therefore the SEMs BTC and TTC-352 which are weak agonists in vitro, show similar efficacy to E2 causing significant tumor regression in two TAM-resistant breast cancer xenografts.
SEMs do not support growth of hormone-dependent xenografts
The ER-positive, hormone-dependent T47D:A18/neo breast cancer cell line requires E2 for growth in vitro and in vivo (6, 7). Since BTC and TTC-352 support the growth of T47D:A18/neo cells in 2D (Figure 1A and 1B) and 3D cultures (Figure 3A), we next asked whether these SEMs would sustain the growth of T47D:A18/neo tumors in vivo. T47D:A18/neo cells were bilaterally injected into 20 athymic mice and divided into six treatment groups; 3 non-treated control, 3 E2 capsule, 3 oral TAM 1.5 mg/day, 3 oral RAL 1.5 mg/day, 4 oral BTC 1.5 mg/day, or 4 oral TTC-352 1.5 mg/day. Following seven weeks of treatment, mice treated with E2, as expected, harbored T47D:A18/neo tumors that reached an average size of ~0.35 cm2 (100%), whereas no significant tumor growth was observed in the other treatment groups (Figure 6A). Thus, the growth of T47D:A18/neo cells seen at higher concentrations of BTC and TTC-352 in vitro (Figure 1E and 1F) was not recapitulated in vivo. Importantly, the dose of SEMs capable of causing robust regression of T47D:A18/PKCα and T47D:A18-TAM1 tumors did not stimulate T47D:A18/neo tumor growth. Additionally, no significant weight loss was observed over the seven-week treatment period (Figure 6B). Therefore SEMs are capable of causing regression of endocrine-resistant breast cancer xenografts, and importantly they are unable to support the growth of hormone-dependent xenografts in vivo. This result is significant as it suggests that SEMs will not stimulate the growth of any endocrine-sensitive breast cancer cells that may be present in heterogeneous tumors.
Figure 6. BTC and TTC-352 have no effect on T47D:A18/neo tumor growth, body weight or uterine weights of ovariectomized mice.

A) BTC and TTC-352 do not result in growth of T47D:A18/neo tumors. B) Body weights of treated mice from (A). C) Uterine weights from mice in (A). Weights are reported as uterine weight (mg)/body weight (g).***, P ≤ 0.001 versus control. Graphs show mean ± SEM.
SEMs are without uterotrophic actions in athymic mice
E2 has a proliferative effect on the endometrium resulting in an increase in uterine weight. TAM has an estrogenic effect on endometrial growth, which leads to an increased risk of developing endometrial cancer (29). In ovariectomized rats at a minimally effective dose, RAL did not increase uterine weight in contrast to E2 and TAM, and at doses up to 10 mg/kg/day did not increase luminal epithelial cell thickness (30–33). Mindful of the in vitro estrogen agonist actions of BTC and TTC-352 described above, we sought to compare the effects on uterine weight of BTC, TTC-352, RAL, TAM, and E2. Following 7 weeks of treatment the uteri from the ovariectomized mice used in the T47D:A18/neo xenograft study (Figure 6A) were excised and weighed. Interestingly, BTC and TTC-352 caused no significant increase in the uterine weights of the treated mice (Figure 6C). The SEM-treated mice were indistinguishable from those that were treated with RAL, which is known to be without uterotrophic actions. In contrast, and as expected, TAM and E2 caused significant increases in uterine weight.
Discussion
Before the introduction of TAM, the ER agonists, E2 and DES, were recognized to have clinical utility in the treatment of breast cancer (9, 34). Discovery of alternative ER agonists that might mimic the anticancer effects of E2 and DES, without the unacceptable side effects, is a reasonable and novel strategy. To identify such “selective estrogen mimics” (SEMs), we chose to take advantage of the knowledge that the growth of several ER-positive, but endocrine-resistant, breast cancer cell lines are inhibited by E2. These TAM-resistant cell lines overexpress PKCα, the role of which in the antiproliferative actions of E2 is not yet defined. Using three such cell lines (T47D:A18/PKCα, T47D:A18-TAM1, MCF-7:5C), two novel SEMs were identified.
BTC and TTC-352 showed estrogenic activity in vitro in 2D cultures with respect to proliferation and ERE-mediated transcription, although: i) both SEMs were less potent than E2; and ii) TTC-352 displayed partial agonist properties. In 2D cultures of TAM-resistant MCF-7:5C cells, both SEMs and E2 inhibited growth and induced apoptosis (Figure 3E and 3F, respectively). BTC and TTC-352 mimicked E2 and inhibited TAM-resistant T47D:A18/PKCα and T47D:A18-TAM1 colony formation (Figure 3B and 3C). Regression of T47D:A18/PKCα tumors by BTC and TTC-352 was accompanied by exit of ERα from the nucleus to extranuclear sites, again mimicking the actions of E2 (Figure 5B), and hinting at a role for extranuclear ERα in BTC and TTC-352 induced tumor regression. Withdrawal of SEM treatment did not lead to relapse and regrowth. E2 and SEM treatment also induced regression of established T47D:A18-TAM1 tumors in a xenograft model (Figure 5C). These xenograft models which represent both exogenous and endogenous PKCα expression, are similarly inhibited by SEMs.
We have observed that translocation of ERα from the nucleus to cytoplasm is a common feature of treatments that cause regression of T47D:A18/PKCα tumors, but not those that are ineffective, i.e. TAM (11). The similarity with the diarylthiohydantoin antiandrogens (e.g. enzalutamide, ARN509, RD162) that cause a similar translocation of the androgen receptor (AR) in prostate cancer cells is of interest, in particular because this feature is seen as a clinical advantage over older antiandrogens (35, 36). Enzalutamideis currently approved for treatment of castration-resistant prostate cancer. We have previously reported an increased physical interaction of ERα and caveolin-1 (cav-1) in E2-induced tumor regression (11), suggesting that cav-1 may be responsible for the transport of ERα to extranuclear sites following E2 or SEM treatment. Cav-1 serves as a scaffold protein recruiting signaling molecules including ERα to the plasma membrane to form a signalosome complex. Contrary to the growth promoting effects and activation of kinase cascades generally associated with extranuclear ERα and E2 treatment, we have previously reported a downregulation and inactivation of Akt following E2 treatment in tumors overexpressing PKCα (17). We hypothesize that the overexpression of PKCα in the presence of E2 or SEMs may lead to a modified signalosome complex in the cytoplasm, altering the canonical effects of E2 thus leading to apoptosis. We are currently overexpressing kinase dead PKCα mutants in T47D cells, as it is likely that the kinase activity of PKCα contributes to the increased sensitivity of estrogenic compounds in endocrine resistant breast cancer cells.
Use of TAM in breast cancer therapy is currently recommended for at least 5 years. Although only used following TAM treatment failure, there is a potential risk with the use of E2 or SEMs in promoting tumorigenesis of endocrine-dependent neoplasms. Although E2 induced growth of hormone-dependent, TAM-sensitive, parental T47D:A18/neo tumors in vivo, neither BTC nor TTC-352 supported T47D:A18/neo tumor growth (Figure 6A). Furthermore, in contrast to E2 and TAM, neither BTC nor TTC-352 treatment resulted in an increase in the uterine weight of mice (Figure 6C) indicating that BTC and TTC-352 act in vivo as SEMs with selective E2-like activity in endocrine-resistant mammary tumors. It is fascinating that structurally related compounds, variously showing classical ERα antagonist activity (RAL), or classical agonist activity (BTC, TTC-352) should elicit the same tumor regressing actions in T47D:A18/PKCα and T47D:A18-TAM1 xenografts, although the failure of RAL-induced regression to persist after drug withdrawal is noted. That the estrogen agonists, BTC and TTC-352, did not stimulate growth of estrogen-sensitive T47D:A18/neo xenografts or uterine tissues is most simply rationalized by the relatively low potency of these agonists, again indicating involvement of a pathway that is not simply classically mediated by ERα in T47D:A18/PKCα and T47D:A18-TAM1 xenografts.
Resistance to endocrine therapies is a major obstacle encountered in the clinical setting. Currently there is a lack of effective therapeutic options for women who no longer respond to conventional, endocrinetherapy. The combination of the aromatase inhibitor exemestane with the mTOR inhibitor everolimus improved progression-free survival for endocrine-resistant breast cancer patients, however an increased toxicity profile was observed in patients who took the combined treatment (37). There is a clinical need to provide endocrine-therapy resistant patients with effective and safe alternatives. Our findings and those of others suggest that PKCα expression is a predictive marker of disease outcome for patients on endocrine therapy (3–5). Further PKCα expression may predict a positive response to E2 or an E2-like compound(7). E2 has clinical efficacy (9, 34, 38, 39), but due to unfavorable side effects it is no longer used for treatment.
Recently, the use of E2 or an E2-like compound has re-emerged as a possible treatment strategy for patients exhibiting endocrine therapy resistant breast cancers(40, 41). Clinical trials have demonstrated the efficacy of E2 in this setting (38, 39). In fact, a long-term follow up study indicated a survival advantage for patients treated with the synthetic estrogen DES compared to patients treated with TAM (42). The basis for the clinical use of estrogens is supported by a number of preclinical laboratory models (7, 8, 12, 43–48). The ability to predict a patient’s response to therapy prior to treatment would be a very attractive clinical option. Patients presenting with tumors overexpressing PKCα would likely benefit from an E2-like therapy, of which there are currently few options.
In the present study, we sought to identify possible alternative therapeutic options for TAM-resistant breast cancers. As the presence of PKCα dictated an enhanced estrogenic response to BTC and TTC-352 as well as a tumor regressing phenotype, these compounds may have potential clinical value in the endocrine–resistant setting. Our findings support the use of SEMs in patients that no longer respond to conventional endocrine therapies and whose tumors overexpress PKCα. Importantly treatment with BTC and TTC-352 had minimal effects on proliferation within the uteri of mice in vivo suggesting that the estrogenic effects of these agents are specific to the breast. Both BTC and TTC-352 are potential alternatives to E2 treatment and represent chemical probes and lead compounds for further optimization towards new treatment options in the management of endocrine resistant breast cancer.
Supplementary Material
Acknowledgments
We would like to acknowledge the Histopathology Core and the Confocal Microscopy Facility of the Research Resources Center at the University of Illinois at Chicago for providing services and expertise.
Grant Support
These studies were supported by NCI/NIH RO1 CA122914 (to D.A. Tonetti) and R01 102590 to (G.R.J. Thatcher).
Footnotes
The authors declare they have no competing interests.
References
- 1.Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA, et al. Protein kinase C isozymes and the regulation of diverse cell responses. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2000;279:L429–L38. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- 2.Mackay HJ, Twelves CJ. Protein kinase C: a target for anticancer drugs? Endocrine-Related Cancer. 2003;10:389–96. doi: 10.1677/erc.0.0100389. [DOI] [PubMed] [Google Scholar]
- 3.Assender JW, Gee JMW, Lewis I, Ellis IO, Robertson JFR, Nicholson RI. Protein kinase C isoform expression as a predictor of disease outcome on endocrine therapy in breast cancer. Journal of Clinical Pathology. 2007;60:1216–21. doi: 10.1136/jcp.2006.041616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tonetti DA, Morrow M, Kidwai N, Gupta A, Badve S. Elevated protein kinase C alpha expression may be predictive of tamoxifen treatment failure. Br J Cancer. 2003;88:1400–2. doi: 10.1038/sj.bjc.6600923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lonne G, Cornmark L, Zahirovic I, Landberg G, Jirstrom K, Larsson C. PKCalpha expression is a marker for breast cancer aggressiveness. Molecular Cancer. 2010;9:76. doi: 10.1186/1476-4598-9-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tonetti DA, Chisamore MJ, Grdina W, Schurz H, Jordan VC. Stable transfection of protein kinase C alpha cDNA in hormone-dependent breast cancer cell lines. Br J Cancer. 2000;83:782–91. doi: 10.1054/bjoc.2000.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chisamore MJ, Ahmed Y, Bentrem DJ, Jordan VC, Tonetti DA. Novel antitumor effect of estradiol in athymic ice injected with a T47D breast cancer cell line overexpressing protein kinase C alpha. Clinical Cancer Research. 2001;7:3156–65. [PubMed] [Google Scholar]
- 8.Yao K, Lee E-S, Bentrem DJ, England G, Schafer JIM, ORegan RM, et al. Antitumor action of physiological estradiol on tamoxifen-stimulated breast tumors grown in athymic mice. Clinical Cancer Research. 2000;6:2028–36. [PubMed] [Google Scholar]
- 9.Ingle JN, Ahmann DL, Green SJ, Edmonson JH, Bisel HF, Kvols LK, et al. Randomized clinical trial of diethylstilbestrol versus tamoxifen in postmenopausal women with advanced breast cancer. New England Journal of Medicine. 1981;304:16–21. doi: 10.1056/NEJM198101013040104. [DOI] [PubMed] [Google Scholar]
- 10.Cole M, Jones C, Todd I. A new anti-oestrogenic agent in late breast cancer: an early clinical appraisal of ICI46474. British Journal of Cancer. 1971;25:270. doi: 10.1038/bjc.1971.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perez White B, Molloy M, Zhao H, Zhang Y, Tonetti D. Extranuclear ERalpha is associated with regression of T47D PKCalpha-overexpressing, tamoxifen-resistant breast cancer. Molecular Cancer. 2013;12:34. doi: 10.1186/1476-4598-12-34. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Lewis JS, Osipo C, Meeke K, Jordan VC. Estrogen-induced apoptosis in a breast cancer model resistant to long-term estrogen withdrawal. The Journal of Steroid Biochemistry and Molecular Biology. 2005;94:131–41. doi: 10.1016/j.jsbmb.2004.12.032. [DOI] [PubMed] [Google Scholar]
- 13.Abdelhamid R, Luo J, Vandevrede L, Kundu I, Michalsen B, Litosh VA, et al. Benzothiophene selective estrogen receptor modulators provide neuroprotection by a novel GPR30-dependent mechanism. ACS chemical neuroscience. 2011;2:256–68. doi: 10.1021/cn100106a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catherino WH, Jordan VC. Increasing the number of tandem estrogen response elements increases the estrogenic activity of a tamoxifen analogue. Cancer Letters. 1995;92:39–47. doi: 10.1016/0304-3835(95)03755-l. [DOI] [PubMed] [Google Scholar]
- 15.O’Regan RM, Cisneros A, MacGregor JL, Muenzner HD, Assikis VJ, Piette M, et al. Effects of the Antiestrogens Tamoxifen, Toremifene, and ICI 182,780 on Endometrial Cancer Growth. J Natl Cancer Inst. 1998;90:1552–8. doi: 10.1093/jnci/90.20.1552. [DOI] [PubMed] [Google Scholar]
- 16.Lin X, Yu Y, Zhao H, Zhang Y, Manela J, Tonetti DA. Overexpression of PKCα is required to impart estradiol inhibition and tamoxifen-resistance in a T47D human breast cancer tumor model. Carcinogenesis. 2006;27:1538–46. doi: 10.1093/carcin/bgl002. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, Zhao H, Asztalos S, Chisamore M, Sitabkhan Y, Tonetti DA. Estradiol-induced regression in T47D:A18/PKCα tumors requires the estrogen receptor and interaction with the extracellular matrix. Molecular Cancer Research. 2009;7:498–510. doi: 10.1158/1541-7786.MCR-08-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Snyder KR, Sparano N, Malinowski JM. Raloxifene hydrochloride. Am J Health Syst Pharm. 2000;57:1669–75. quiz 76–8. [PubMed] [Google Scholar]
- 19.Hochner-Celnikier D. Pharmacokinetics of raloxifene and its clinical application. Eur J Obstet Gynecol Reprod Biol. 1999;85:23–9. doi: 10.1016/s0301-2115(98)00278-4. [DOI] [PubMed] [Google Scholar]
- 20.Palkowitz AD, Glasebrook AL, Thrasher KJ, Hauser KL, Short LL, Phillips DL, et al. Discovery and synthesis of [6-hydroxy-3-[4-[2-(1-piperidinyl)ethoxy]phenoxy]-2-(4-hydroxyphenyl)]benzo[b]thiophene: a novel, highly potent, selective estrogen receptor modulator. J Med Chem. 1997;40:1407–16. doi: 10.1021/jm970167b. [DOI] [PubMed] [Google Scholar]
- 21.Suh N, Glasebrook AL, Palkowitz AD, Bryant HU, Burris LL, Starling JJ, et al. Arzoxifene, a new selective estrogen receptor modulator for chemoprevention of experimental breast cancer. Cancer Res. 2001;61:8412–5. [PubMed] [Google Scholar]
- 22.Burke TW, Walker CL. Arzoxifene as therapy for endometrial cancer. Gynecol Oncol. 2003;90:S40–6. doi: 10.1016/s0090-8258(03)00343-3. [DOI] [PubMed] [Google Scholar]
- 23.Buzdar A, O’Shaughnessy JA, Booser DJ, Pippen JE, Jr, Jones SE, Munster PN, et al. Phase II, randomized, double-blind study of two dose levels of arzoxifene in patients with locally advanced or metastatic breast cancer. J Clin Oncol. 2003;21:1007–14. doi: 10.1200/JCO.2003.06.108. [DOI] [PubMed] [Google Scholar]
- 24.Liu H, Liu J, van Breemen RB, Thatcher GRJ, Bolton JL. Bioactivation of the selective estrogen receptor modulator desmethylated arzoxifene to quinoids: 4′-fluoro substitution prevents quinoid formation. Chem Res Toxicol. 2005;18:162–73. doi: 10.1021/tx049776u. [DOI] [PubMed] [Google Scholar]
- 25.Yu B, Dietz BM, Dunlap T, Kastrati I, Lantvit DD, Overk CR, et al. Structural modulation of reactivity/activity in design of improved benzothiophene selective estrogen receptor modulators: induction of chemopreventive mechanisms. Mol Cancer Ther. 2007;6:2418–28. doi: 10.1158/1535-7163.MCT-07-0268. [DOI] [PubMed] [Google Scholar]
- 26.Overk CR, Peng KW, Asghodom RT, Kastrati I, Lantvit DD, Qin Z, et al. Structure-activity relationships for a family of benzothiophene selective estrogen receptor modulators including raloxifene and arzoxifene. Chem Med Chem. 2007;2:1520–6. doi: 10.1002/cmdc.200700104. [DOI] [PubMed] [Google Scholar]
- 27.Qin Z, Kastrati I, Ashgodom RT, Lantvit DD, Overk CR, Choi Y, et al. Structural modulation of oxidative metabolism in design of improved benzothiophene selective estrogen receptor modulators. Drug Metab Dispos. 2009;37:161–9. doi: 10.1124/dmd.108.023408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu H, Bolton JL, Thatcher GRJ. Chemical modification modulates estrogenic activity, oxidative reactivity, and metabolic stability in 4′ F-DMA, a new benzothiophene selective estrogen receptor modulator. Chem Res Toxicol. 2006;19:779–87. doi: 10.1021/tx050326r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fisher B, Costantino JP, Redmond CK, Fisher ER, Wickerham DL, Cronin WM, et al. Endometrial cancer in tamoxifen-treated breast cancer patients: findings from the National Surgical Adjuvant Breast and Bowel Project (NSABP) B-14. J Natl Cancer Inst. 1994;86:527–37. doi: 10.1093/jnci/86.7.527. [DOI] [PubMed] [Google Scholar]
- 30.Grese TA, Sluka JP, Bryant HU, Cullinan GJ, Glasebrook AL, Jones CD, et al. Molecular determinants of tissue selectivity in estrogen receptor modulators. Proc Natl Acad Sci U S A. 1997;94:14105–10. doi: 10.1073/pnas.94.25.14105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grese TA, Cho S, Finley DR, Godfrey AG, Jones CD, Lugar CW, 3rd, et al. Structure-activity relationships of selective estrogen receptor modulators: modifications to the 2-arylbenzothiophene core of raloxifene. J Med Chem. 1997;40:146–67. doi: 10.1021/jm9606352. [DOI] [PubMed] [Google Scholar]
- 32.Black LJ, Sato M, Rowley ER, Magee DE, Bekele A, Williams DC, et al. Raloxifene (LY139481 HCI) prevents bone loss and reduces serum cholesterol without causing uterine hypertrophy in ovariectomized rats. J Clin Invest. 1994;93:63–9. doi: 10.1172/JCI116985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuchs-Young R, Glasebrook AL, Short LL, Draper MW, Rippy MK, Cole HW, et al. Raloxifene is a tissue-selective agonist/antagonist that functions through the estrogen receptor. Ann N Y Acad Sci. 1995;761:355–60. doi: 10.1111/j.1749-6632.1995.tb31392.x. [DOI] [PubMed] [Google Scholar]
- 34.Kennedy BJ. Massive estrogen administration in premenopausal women with metastatic breast cancer. Cancer. 1962;15:641–8. doi: 10.1002/1097-0142(196205/06)15:3<641::aid-cncr2820150330>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 35.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–90. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A, et al. ARN-509: A novel antiandrogen for prostate cancer treatment. Cancer Research. 2012;72:1494–503. doi: 10.1158/0008-5472.CAN-11-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T, et al. Everolimus in Postmenopausal Hormone-Receptor-Positive Advanced Breast Cancer. New England Journal of Medicine. 2012;366:520–9. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ellis MJ, Gao F, Dehdashti F, Jeffe DB, Marcom PK, Carey LA, et al. Lower-dose vs high-dose oral estradiol therapy of hormone receptor-positive, aromatase inhibitor-resistant advanced breast cancer. JAMA: The Journal of the American Medical Association. 2009;302:774–80. doi: 10.1001/jama.2009.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lønning PE, Taylor PD, Anker G, Iddon J, Wie L, Jørgensen L-M, et al. High-dose estrogen treatment in postmenopausal breast cancer patients heavily exposed to endocrine therapy. Breast Cancer Res Treat. 2001;67:111–6. doi: 10.1023/a:1010619225209. [DOI] [PubMed] [Google Scholar]
- 40.Ingle J. Estrogen as therapy for breast cancer. Breast Cancer Res. 2002;4:133– 6. doi: 10.1186/bcr436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jordan VC, Obiorah I, Fan P, Kim HR, Ariazi E, Cunliffe H, et al. The St. Gallen Prize Lecture 2011: Evolution of long-term adjuvant anti-hormone therapy: consequences and opportunities. The Breast. 2011;20(Supplement 3):S1–S11. doi: 10.1016/S0960-9776(11)70287-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peethambaram PP, Ingle JN, Suman VJ, Hartmann LC, Loprinzi CL. Randomized trial of diethylstilbestrol vs. tamoxifen in postmenopausal women with metastatic breast cancer. An updated analysis. Breast Cancer Res Treat. 1999;54:117–22. doi: 10.1023/a:1006185805079. [DOI] [PubMed] [Google Scholar]
- 43.Osipo C, Gajdos C, Cheng D, Jordan VC. Reversal of tamoxifen resistant breast cancer by low dose estrogen therapy. The Journal of Steroid Biochemistry and Molecular Biology. 2005;93:249–56. doi: 10.1016/j.jsbmb.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 44.Osipo C, Gajdos C, Liu H, Chen B, Jordan VC. Paradoxical action of fulvestrant in estradiol-induced regression of tamoxifen-stimulated breast cancer. J Natl Cancer Inst. 2003;95:1597–608. doi: 10.1093/jnci/djg079. [DOI] [PubMed] [Google Scholar]
- 45.Liu H, Lee E-S, Gajdos C, Pearce ST, Chen B, Osipo C, et al. Apoptotic action of 17β-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo. J Natl Cancer Inst. 2003;95:1586–97. doi: 10.1093/jnci/djg080. [DOI] [PubMed] [Google Scholar]
- 46.Santen RJ, Song RX, Zhang Z, Yue W, Kumar R. Adaptive hypersensitivity to estrogen. Clinical Cancer Research. 2004;10:337s–45s. doi: 10.1158/1078-0432.ccr-031207. [DOI] [PubMed] [Google Scholar]
- 47.Song RX-D, Mor G, Naftolin F, McPherson RA, Song J, Zhang Z, et al. Effect of long-term estrogen deprivation on apoptotic responses of breast cancer cells to 17beta-estradiol. J Natl Cancer Inst. 2001;93:1714–23. doi: 10.1093/jnci/93.22.1714. [DOI] [PubMed] [Google Scholar]
- 48.Shim W-S, Conaway M, Masamura S, Yue W, Wang J-P, Kumar R, et al. Estradiol hypersensitivity and mitogen-activated protein kinase expression in long-term estrogen deprived human breast cancer cells in vivo. Endocrinology. 2000;141:396–405. doi: 10.1210/endo.141.1.7270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.