

Summary
Papillary thyroid carcinoma (PTC) is the most common type of thyroid cancer. Here, we describe the genomic landscape of 496 PTCs. We observed a low frequency of somatic alterations (relative to other carcinomas) and extended the set of known PTC driver alterations to include EIF1AX, PPM1D and CHEK2 and diverse gene fusions. These discoveries reduced the fraction of PTC cases with unknown oncogenic driver from 25% to 3.5%. Combined analyses of genomic variants, gene expression, and methylation demonstrated that different driver groups lead to different pathologies with distinct signaling and differentiation characteristics. Similarly, we identified distinct molecular subgroups of BRAF-mutant tumors and multidimensional analyses highlighted a potential involvement of oncomiRs in less-differentiated subgroups. Our results propose a reclassification of thyroid cancers into molecular subtypes that better reflect their underlying signaling and differentiation properties, which has the potential to improve their pathological classification and better inform the management of the disease.
Introduction
The incidence of thyroid cancer has increased 3-fold over the past 30 years (Chen et al., 2009) and the prevalence of different histologies and genetic profiles has changed over time (Jung et al., 2014). All thyroid cancers, except medullary carcinoma, are derived from follicular cells that comprise the simple unicellular epithelium of normal thyroid. Eighty percent of all thyroid cancers are papillary thyroid carcinomas (PTCs), named for their papillary histological architecture. In addition, PTCs encompass several subtypes, including the follicular variant (FV), characterized by a predominantly follicular growth pattern. PTCs are usually curable with 5-year survival of over 95% (Hay et al., 2002); however, occasionally they dedifferentiate into more aggressive and lethal thyroid cancers. Current treatment involves surgery, thyroid hormone and radioactive iodine (RAI) therapy (which exploits thyroid follicular cells' avidity for iodine).
Previous genetic studies report a high frequency (70%) of activating somatic alterations of genes encoding effectors in the mitogen-activated protein kinase (MAPK) signaling pathway, including point mutations of BRAF and the RAS genes (Cohen et al., 2003; Kimura et al., 2003; Lemoine et al., 1988; Suarez et al., 1988), as well as fusions involving the RET (Grieco et al., 1990) and NTRK1 tyrosine kinases (Pierotti et al., 1995). These mutations are almost always mutually exclusive (Soares et al., 2003), suggesting similar or redundant downstream effects. The various MAPK pathway alterations are strongly associated with distinct clinicopathological characteristics (Adeniran et al., 2006), and gene expression (Giordano et al., 2005) and DNA methylation profiles (Ellis et al., 2014). Mutations in members of the phosphoinositide 3-kinase (PI3K) pathway, such as PTEN, PIK3CA and AKT1, have also been reported at low frequencies (Xing, 2013).
We present The Cancer Genome Atlas (TCGA) project results from a comprehensive multiplatform analysis of 496 PTCs, the largest cohort studied to date. Clinically aggressive thyroid cancers (poorly and undifferentiated carcinomas) were excluded to maximally develop the compendium of tumor-initiating alterations. While excluding histological types of aggressive tumors limited some aspects of the study, the homogeneous PTC cohort allowed robust correlative analyses of multidimensional molecular data. The relatively quiet PTC genome allowed us to assess the signaling and differentiation consequences of the common drivers. Furthermore, the cohort allowed us to define integrated molecular subtypes that correspond to histology, signaling, differentiation state and risk assessment. We put forth that our results will lead to improved clinicopathologic classification and management of patients.
Results
Samples, Clinical Data and Analytical Approach
Tumor samples and matched germline DNA from blood or normal thyroid from 496 patients included 324 (69.4%) classical-type (CT), 99 (21.2%) follicular-variant (FV), 35 (7.5%) tall cell variant (TCV), 9 (2.0%) uncommon PTC variants and 29 without histological annotation, primarily from non-irradiated patients (Table S1A). We estimated risk of tumor recurrence based on the 2009 American Thyroid Association guidelines (American Thyroid Association Guidelines Taskforce on Thyroid et al., 2009), and assessed mortality risk using MACIS scores (Hay et al., 1993) (see Supplement). We generated comprehensive and high-quality molecular data at TCGA genome sequencing and characterization centers with one proteomic and six genomic platforms (Table S1B), and analyzed the data at multiple genomic data analysis centers (Table S1C). Although 496 primary tumors were studied, the number of informative cases varied across platforms mostly for technical reasons and 390 tumors were analyzed on all major platforms (SNP arrays, exomes, RNA-seq, miRNA-seq and DNA methylation).
Our analysis strategy consisted of three parts, each based on integrating several molecular datasets. First, we identified somatic mutations that included single nucleotide variants, small insertions and deletions, gene fusions and copy number alterations in order to characterize the genomic landscape of PTC and identify driver events in cases without any previously known driver (i.e., the so-called ‘dark matter’ of the PTC genome). Next, we focused on the consequences of the driver mutations. We developed a gene expression signature of samples with these mutations and characterized tumors based on this signature. We then determined the differential signaling consequences of BRAFV600E and RAS mutations, the most common pathogenic mutations in the cohort, using protein and mRNA expression data. Finally, we used the molecular data to derive molecular classifications of PTC and integrated these classifications with data related to genotype, signaling, differentiation and risk.
Somatic Single Nucleotide Variants and Insertions and Deletions
Whole exome DNA sequencing of 402 (of 496) tumor/normal pairs (average depth-of-coverage; 97.0× for tumors, 94.9× for normals) showed a low somatic mutation density (0.41 non-synonymous mutations per Mb, on average) (Figures 1A and S1A; Table S3) relative to other cancers (Lawrence et al., 2014; Lawrence et al., 2013). Mutation density correlated with age (Pearson correlation p=5.2×10-18, Figure S1B), risk of recurrence (Kruskal-Wallis test p=3.4×10-4), and MACIS score (Pearson correlation p=4×10-15, Figure S1C). To compensate for the confounding effect of age-at-diagnosis, we regressed the effect of age from mutation density and found that the association of risk with age-corrected mutation density remained (Kruskal-Wallis test p=9.7×10-3; MACIS scores did not, p=0.19, Figure S1C). This association was maintained for the CT cohort (p=0.0044), but not for other variants (Figure S1D). The correlation between mutation density and age suggests age should be used as a continuous variable in risk stratification (Bischoff et al., 2013)), instead of a threshold of 45 years used in many staging systems.
Figure 1. Landscape of Genomic Alterations in 402 Papillary Thyroid Carcinomas.

(A) Mutation density (mutations/Mb) across the cohort. (B) Tumor purity, patient age, gender, history of radiation exposure, risk of recurrence, MACIS score, histological type, and BRS score. (C) Number and frequency of recurrent mutations in genes (left) ranked by MutSig significance (right), gene-sample matrix of mutations (middle) with TERT promoter mutations (bottom). (D) Number and frequency of fusion events (left), gene-sample matrix of fusions across the cohort (middle). (E) Number and frequency of SCNAs (left), chromosome-sample matrix of SCNAs across the cohort (middle) with focal deletions in BRAF and PTEN (bottom), GISTIC2 significance (right). (F) Driving variant types across the cohort. Samples were sorted by driving variant type with dark matter on the left. See also Figures S1, S2, S3, S4 and Tables S1, S2, S3, S4A, and S5A,B,E.
Mutation densities were not associated with other variables, like genotype or radiation exposure (Mann-Whitney p=0.579 and p=0.173, respectively; Figure S1E). TCVs had the highest mutation density (Figure S1F), consistent with their known aggressive behavior. Five BRAFV600E-mutant tumors with aggressive histologic features had higher mutation densities (Figures S1E,G).
Ten tumors with the highest mutation densities (> 1/Mb) were enriched for mutations associated with the APOBEC process (Roberts et al., 2013) (Mann-Whitney p=4×10-5), similar to bladder cancer (Cancer Genome Atlas Research, 2014).
The relatively large number of patients in this study and the low background mutation density provides the statistical power to detect significantly mutated genes (SMGs) in as low as 3% of cases (>90% power for 90% of genes) (Lawrence et al., 2014). MutSig (Lawrence et al., 2013) detected seven SMGs (q<0.1) (Figure 1C and Table S2) with four of the seven in <3% of patients. SMGs included the MAPK-related genes, BRAF, NRAS, HRAS and KRAS, which were virtually mutually exclusive (Figure 1C, Fisher's exact test p=1.1×10-5, MEMo (Ciriello et al., 2012) corrected p<0.01, Table S4A) in 300/402 (74.6%) patients. The 248 (61.7%) BRAF mutations were mostly V600E substitution (Figures 1C and S2A and Table S3C). Somatic single nucleotide variants (SSNVs) were identified in 52 patients (12.9%) within codons 12 and 61 of RAS genes (Figures 1C and S2B and Table S3D). We observed strong associations between BRAF and RAS mutation status and histology, with BRAFV600E characterizing CT and TCV and RAS mutations characterizing FV (Figures 1B,C and Table S3E). These observations confirm the critical role of MAPK pathway alterations in PTC and illustrate that having more than one mutation confers no clonal advantage.
MutSig also identified EIF1AX (eukaryotic translation initiation factor 1A, X-linked) as significantly mutated (q=5.3×10-8; Figure 1C and Table S3B). EIF1AX encodes a protein that mediates transfer of Met-tRNAf to 40S ribosomal subunits to form the 40S preinitiation complex for protein translation. Six (1.5%) mutations (Figure S2C) were identified in tumors lacking other known driver mutations (Fisher's exact test of EIF1AX vs. RAS/BRAF, p=0.013) with one exception: one case with 3 driver mutations in KRAS, BRAF and EIF1AX; here the KRAS mutation was clonal (cancer cell fraction estimated at 100% with 95% CI [56-100%]), while EIF1AX and BRAF were subclonal at 76% [38-100%] and 53% [22-94%] cancer cell fractions, respectively (see Supplement). The near-mutual exclusivity of EIF1AX alterations with MAPK pathway mutations (Figure 1C), together with recurrent mutations in other tumors (Forbes et al., 2011; Martin et al., 2013), suggests that EIF1AX is a novel cancer gene in PTC.
The two remaining SMGs (PPM1D, CHEK2, Figures S2D,E) encode interacting proteins related to DNA repair (Oliva-Trastoy et al., 2007) and occurred concomitant with MAPK-pathway driver mutations (Figure 1C). Germline PPM1D mutations are associated with breast and ovarian cancer predisposition and may impair p53 function (Kleiblova et al., 2013). Although not statistically significant, there were 8 additional DNA repair-related mutations in 26 (6.5%) tumors, all mutually exclusive (Figure S3A, MEMo corrected p=0.24). Tumors carrying these mutations had a significantly higher median mutation density (Table S5A, Mann Whitney p=0.022), although not after age-adjustment (p=0.111), and were enriched with high-risk patients (Fisher's exact test p=0.018). These observations, together with our finding of a mutated network of FANCA-associated genes (see Integrated Analysis of Somatic Alterations), suggest that acquisition of a defect in DNA repair represents a mechanism for development of aggressive PTC.
We also observed alterations in other cancer genes, pathways and functional groups (chromatin remodeling, PI3K, WNT and tumor suppressor genes). Although not statistically significant, they are known to play a role in thyroid cancer pathogenesis and progression (Xing, 2013). We identified 93 mutations within 57 epigenetic regulatory genes in 80/402 (20.0%) tumors, 9 of which possessed more than one mutation (Figure S3D). Mutations in MLL (1.7%), ARID1B (1.0%) and MLL3 (1%) were most frequent (Figures 1C and S3D). In the PI3K and PPARγ pathways, we observed 20 nearly mutually exclusive mutations in PTEN, AKT1/2, PAX8/PPARG (Figure S3E), representing 4.5% (18/402) of cases. Mutations of five WNT pathway-related genes were found in 6/402 (1.5%) tumors (Figure S3B), a lower frequency than reported for aggressive tumor types (Xing, 2013). Mutations of tumor suppressor genes (TP53, RB1, NF1/2, MEN1, PTEN) were identified in 15/402 (3.7%) tumors (Figure S3C). In addition, two genes that may be tumor suppressors were near significance: (i) ZFHX3, a zinc finger homeobox transcription factor (Minamiya et al., 2012), 7/402 (1.7%) tumors (q=0.79); and (ii) BDP1, which may regulate AKT signaling (Woiwode et al., 2008), in 5/402 (1.2%) tumors (q=0.58). Finally, mutations in thyroid-related genes were infrequent: 11/402 (2.7%) mutations in thyroglobulin, 2/402 (0.5%) mutations in thyroid-stimulating hormone receptor (TSHR). No mutations in thyroid hormone receptor genes (TRHA and TRHB) were found.
We identified TERT promoter mutations in 36 (9.4%) of 384 informative tumors, with 27 (7.0%) C228T, 1 (0.3%) C228A and 8 (2.1%) C250T substitutions. These mutations were present in PTCs of all histological types. TERT promoter mutations had modest association with mutation drivers (Fisher's exact test p=0.029) and arm-level somatic copy number alterations (p=0.023), but not BRAF mutations or gene fusions. They showed strong associations with older age, MACIS scores (Kruskal-Wallis test p=2.6×10-9, and p=1.3×10-11, Figure 2B, respectively), and high risk of recurrence (Fisher's exact test p=7×10-8, Figure 2A), within the entire cohort, and these associations remained within the BRAFV600E tumors. Finally, TERT promoter mutations occurred in less-differentiated PTCs (lower TDS values, see Signaling and Differentiation) (Kruskal-Wallis test p=4.2×10-5, Figure2C). These associations are consistent with published results (Melo et al., 2014; Xing et al., 2014) and suggest that molecular diagnostic assessment of TERT promoter mutations may be used to identify high risk patients.
Figure 2. TERT Promoter Mutations and Clonality Assessment of Driver Mutations.

(A-C) Association of TERT promoter mutations with (A) risk of recurrence, (B) MACIS score, and (C) thyroid differentiation score (TDS). See also Table S2.
(D) Mutation cancer cell fraction distribution. The majority of all mutations, including driver mutations BRAF, NRAS, HRAS, KRAS, and EIF1AX, have a calculated cancer cell fraction close to 1.0, indicating their presence all tumor cells.
A recent report suggested that BRAFV600E mutations in PTC are often present only in a small subset of the cancer cells (Guerra et al., 2012), information relevant to therapeutic application of BRAF-inhibitors. To address this issue, we used the ABSOLUTE package (Carter et al., 2012) to estimate the cancer cell fraction (CCF) of mutations in BRAF, NRAS, HRAS, KRAS and EIF1AX (see Supplement). In our study, all of these driver mutations were present in the majority of tumor cells (Figure 2D), i.e. they are largely clonal. Our results instead confirm other studies showing homogeneous cancer cell immunohistochemical staining with BRAFV600E-specific antibodies (Ghossein et al., 2013).
We performed targeted sequencing validation experiments on a subset of 318 tumors at 333 mutated sites. Two hundred sixty-four of 265 (99.6%) of mutations in driver genes were confirmed. We also carried out targeted validation on a random set of 54 mutations, of which 49 (91%) were confirmed, for an overall validation rate of 96%.
Gene Fusions
Chromosomal rearrangements and translocations contribute to PTC pathogenesis (Xing, 2013). We identified both known and novel fusions, including new partners of previously described fusions, in 74 (15.3%) of 484 informative cases, based on multiple platforms (Figure 3). Fusions were mutually exclusive with each other and with BRAF, RAS and EIF1AX mutations (Fisher's exact test p=4.9×10-43; Figures 1C,D and Figure S3F). Fusion-positive tumors were associated with younger age of diagnosis (Wilcoxon ranksum test p=0.005), but not with risk of recurrence (Fisher's exact test p=0.55) or age-corrected mutation density (ranksum test p=0.341, Table S5A).
Figure 3. Candidate “Driver” Gene Fusions in Papillary Thyroid Carcinoma.
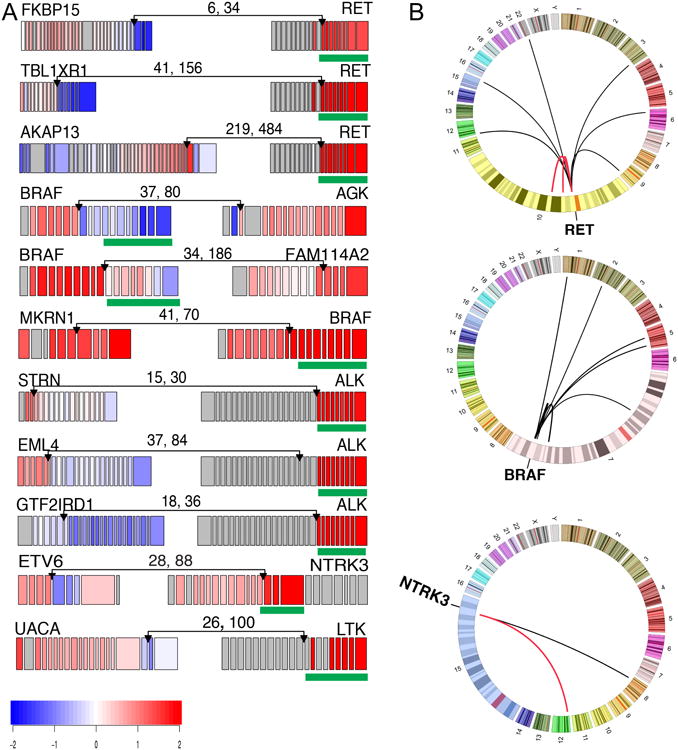
(A) RNA expression fusion plots for representative novel candidate genes involving RET, BRAF, ALK, NTRK3 and LTK fusions. Each gene in the fusion plot is drawn 5′ to 3′, exon specific relative expression data is represented with low (blue) and high expression (red), and the kinase domain is mapped with a green box. The pairs of numbers across the links indicate the number of split reads and paired-end supporting reads from RNA-seq. (B) Circos plots (http://circos.ca) of RET, BRAF and NTRK3 fusions. Red links represent recurrent fusions, black non-recurrent. See also Figures S3F and Table S5B.
RET fusions were most frequent (33/484, 6.8%) (Table S5B and Figure S3F); however, less frequent than previously reported in sporadic or radiation-associated PTC (Ricarte-Filho et al., 2013). We identified four novel unique RET fusions that preserved the kinase domain (Figure 3A and Table S5B).
Fusions involving BRAF have been identified in PTC (Ciampi et al., 2005) and other cancers. In melanoma these events define a molecular subclass with distinct response to MEK inhibition (Hutchinson et al., 2013). In our cohort, we identified 13/484 (2.7%) BRAF fusions with diverse gene partners (Figure 3A and Table S5B); three tumors exhibited the SND1/BRAF gene fusion seen in a gastric cancer cell line (Lee et al., 2012). Some fusions supported BRAF signaling with expression and conservation of its kinase domain (MKRN1/BRAF), while others suggested an alternative activating mechanism. Six of nine BRAF fusions were validated by independent PCR experiments, while PCR evidence for the other three was inconclusive. These diverse fusions, together with the BRAF point mutations and indels described above, illustrate the various possible mechanisms of activating BRAF and highlight its oncogenic importance in PTC.
We identified PAX8/PPARG fusions in 4/484 (0.8%) tumors. Originally found in follicular carcinomas (Kroll et al., 2000), PAX8/PPARG translocations have been reported with low frequency in PTC, especially FV. ETV6/NTRK3 and RBPMS/NTRK3 fusions were uncovered in 6/484 (1.2%) tumors. These fusions are more prevalent in radiation-induced thyroid cancers but have lower prevalence in sporadic PTC (Ricarte-Filho et al., 2013). THADA fusions were identified in 6/484 (1.2%) tumors. Fusions involving ALK presented in 4/484 (0.8%) tumors, including EML4/ALK (Figure 3A), which is observed in lung adenocarcinomas and rare thyroid cancers (Kelly et al., 2014), suggesting opportunities for targeted inhibition of ALK. In addition, two cases had FGFR2 fusions and two cases had non-recurrent fusions of MET and LTK (Table S4B).
Somatic Copy Number Alterations
Somatic copy number alterations (SCNAs) were identified in 135 (27.2%) of 495 informative tumors. These 135 cases were significantly enriched in cases with no driver mutation or fusion (Fisher's exact test p=4.4×10-4; Figures 1C,D,E), suggesting that SCNAs may also drive PTC. Arm-level alterations occurred significantly more frequently in FV than in CT subtypes (Figure S4A) (FDR<0.1, p<0.008), providing evidence for a close relationship between FV and follicular neoplasms (in which SCNAs are more common (Wreesmann et al., 2004a)).
Unsupervised clustering of chromosomal arm-level alterations defined 4 distinct classes (Figure S4B). The largest class (72.9%) lacked significant gains or losses (SCNA-quiet), reflecting the highly differentiated nature of PTC; this group was not enriched for any particular genotype or histologic type. A second class (9.9%) was characterized by an isolated loss of 22q (SCNA-22q-del), a region that includes NF2 and CHEK2 and reported to be lost with significant frequency in PTC (Kjellman et al., 2001). Seventy tumors had 22q loss and 5 tumors possessed CHEK2 mutations, with 4 cases containing both mutations (p=0.0035). The SCNA-22q-del cohort contained few TCV and was enriched for FV (p<0.05) (Figure S4C). This result suggests that loss of CHEK2 and/or the NF2 tumor suppressor may be important in PTC, particularly in the FV subtype. A third class (14.8%) was characterized by a few SCNA events and gain of 1q (SCNA-low-1q-amp), was enriched for TCV (p<0.0001) and BRAF mutations (p<0.05) (Figure S4C), and was associated with significantly higher MACIS scores (p <0.0001) (Figure S4D), risk profiles (Figure S4E), and tumor stage (Figure S4F), consistent with reports of 1q gains in aggressive PTC (Wreesmann et al., 2004b). The final and smallest class (2.4%) was defined by a higher frequency of focal gains and losses (SCNA-high).
We found few significantly recurring focal alterations using GISTIC2 (Mermel et al., 2011). In 5/13 tumors with BRAF fusions, we detected by SNP-array the resulting focal alterations at 7q34. Five tumors with 10q23.31 deletions lacked PTEN expression (Figure S4G). We also found single case amplifications containing oncogenic driver genes (e.g. FGFR3). Together, these results, in the context of SSNVs and fusions, suggest that SCNAs may represent both tumor-initiating and progression-related events in PTC.
Integrated Analyses of Somatic Alterations
Next we sought to identify additional genes that may harbor driver mutations (point mutations, fusions or copy-number changes) that did not meet significance, by searching within protein-protein interaction networks for subnetworks enriched with frequently mutated genes. To this end, we applied the HotNet2 algorithm (Leiserson et al., submitted) and identified 17 significantly mutated subnetworks (p<0.004, Table S5C and Figure S5A). As expected, the largest subnetwork (16 genes) included four known members of the MAPK signaling pathway (BRAF and three RAS genes) and 12 additional genes (Figure S5B). Some of these additional genes (e.g. RAP1GAP) displayed mutations that were mutually exclusive with BRAF (SSNVs, indels and fusions) and RAS, and may represent additional MAPK drivers. Other mutated genes in this subnetwork (e.g. PIK3CA) overlapped with BRAFV600E and may alter the biology of these cancers by activating PI3K signaling, a hypothesis that requires validation.
Other HotNet2 subnetworks significantly overlapped with known pathways (Table S5C and Figures S5C-E). Identification of the ECM-receptor interactions pathway is consistent with the role of ECM microenvironment in PTC (Nucera et al., 2011). The finding of a FANCA-associated protein complex subnetwork provides additional evidence for DNA repair playing a role in PTC.
We used network-based stratification (NBS) (Hofree et al., 2013) to discover three somatic mutation-based PTC subtypes (NBS1-3, Figures S6A-C). As expected, a strong association with histologic subtype was found, with a significant association between NBS1 and FV histology (Figure S6B(b), Fisher's exact test p<2×10-16). The subtypes were also significantly associated with lymph node status, extrathyroidal extension, stage and risk of recurrence (Figure S6B(a,c-e), Fisher's exact test p=4.4×10-4, 3.1×10-7, 3.8×10-3, 8.2×10-6, respectively), as well as other molecular characteristics (Figure S6C). In order to identify somatic events that characterize each subnetwork, we applied the HotNet algorithm to each NBS subtype (Figure S6D and Table S5D). Subtype NBS1 was associated with perturbations in RAS, PTEN, PPARG and TSHR. Subtype NBS2 was associated with alterations in RET and related genes such as NTRK3. Subtype NBS3 appears to be predominantly BRAFV600E-associated. These results confirm three broad classes of PTC determined by the common drivers.
We also looked for the presence of viral pathogens in PTC using two independent methods to assess the RNA-seq data: PathSeq (Kostic et al., 2011) and BioBloom Tools (Chu et al., 2014). We identified two tumors with hepatitis B virus (HBV) and one tumor with human papillomavirus 45 (HPV45) at relative frequencies exceeding 0.1 viral reads per million human reads (RPM) for PathSeq and 0.2 RPM for BBT (see Supplement and Tables S4G-I), indicating that viral pathogens are unlikely significant contributors to PTC pathogenesis.
‘Dark Matter’ Summary
Starting with 402 cases with informative exome DNA sequence data, we examined tumors that lacked apparent driver mutations, so-called ‘dark matter’ samples, for the presence of novel potential driver alterations. SSNVs involving drivers accounted for 299 (73.6%) cases. Mutually exclusive fusions increased the number of cases with drivers to 358 (89.0%). Three mutually exclusive focal deletions (2 PTEN and 1 BRAF) brought the number of cases to 361 (89.8%). Mutually exclusive arm-level SCNAs were present in 27 additional cases, which were mostly FV. Although we cannot pinpoint the driving genes, if we assume that some of these SCNAs indeed act as drivers, the total number of cases with apparent drivers increased to 96.5%, leaving 14 (3.5%) as ‘dark matter’ cases. By reviewing events in these cases, we observed additional potential drivers such as APC, ATM, NF1, and SPOP, mutations of chromatin remodeling genes (e.g. MLL) and potential gene fusions (Table S5E). Including these events and considering arm-level SCNAs as drivers, we have identified putative cancer drivers in 397/402 PTCs (98.8%).
Signaling and Differentiation
PTC is a MAPK-driven cancer that has two mutually exclusive drivers with distinct signaling consequences: BRAFV600E and mutated RAS. Tumors driven by BRAFV600E do not respond to the negative feedback from ERK to RAF (since it signals as a monomer), resulting in high MAPK-signaling (Pratilas et al., 2009). Conversely, tumors driven by RAS and RTK fusions signal via RAF dimers that respond to ERK feedback, resulting in lower MAPK-signaling. This differential signaling results in profound phenotypic differences. For example, expression of genes responsible for iodine uptake and metabolism are greatly reduced in BRAFV600E tumors, in contrast to the “RAF-dimer” tumors in which expression of these genes is largely preserved (Durante et al., 2007). These observations, together with the relatively low number of other genomic alterations, allow for a clear view of the signaling and transcriptional outputs of these two primary drivers. The distinct profile of expression of genes involved in thyroid hormone biosynthesis that we observed between BRAFV600E and RAS-driven tumors is recapitulated closely in mouse PTC models induced by knock-in mutations of BrafV600E or HrasG12V (Charles et al., 2011; Franco et al., 2011), suggesting that these arise as a consequence of the constitutive activation of these drivers.
To explore these relationships across our cohort, we developed a BRAFV600E-RAS score (BRS) to quantify the extent to which the gene expression profile of a given tumor resembles either the BRAFV600E- or RAS-mutant profiles. Using 391 samples with both exome and RNA sequencing data, we compared BRAFV600E-mutated and RAS-mutated tumors to derive a 71-gene signature. Correlations with this signature were used to derive a continuous measure (-1 to +1) with BRAFV600E-like (BVL) PTCs being negative and RAS-like (RL) PTCs positive (see Supplement). As expected, this signature showed strong separation of the BRAFV600E- and RAS-mutant tumors (Figures S7A,B).
We then used the BRS as a reference continuous scale from most BVL to most RL to interrogate the signaling consequences of the other, less common, mutations (Figures 4A,B). All BRAF mutations other than BRAFV600E exhibited RL behavior, including one BRAFK601E, a splice-site mutation and three indels. This is consistent with previous observations that BRAFK601E occurs in FV tumors that are mostly RL-PTCs (Park et al., 2013). All of the BRAF fusions were BVL. Four of the six EIF1AX mutations were RL, one neutral and one weakly BVL. All of the PAX8/PPARG fusions were RL, consistent with their prevalence in follicular-patterned tumors. Nearly all of the RET fusions were weakly BVL and the NTRK1/3 and ALK fusions were largely neutral.
Figure 4. The BRAFV600E-RAS Score.
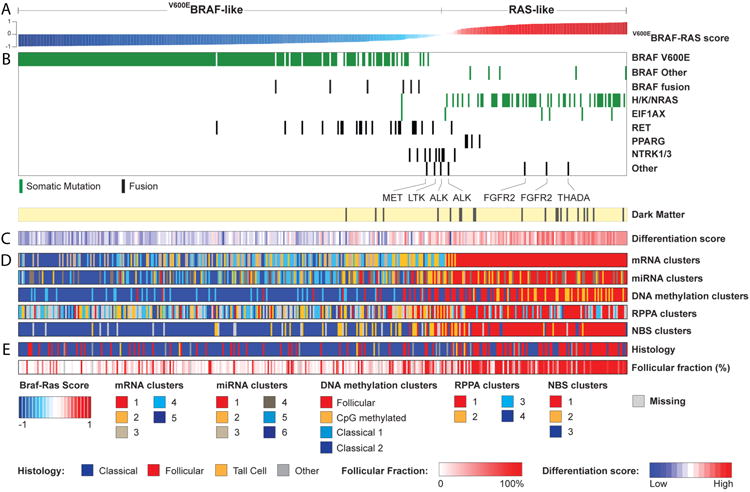
(A) Thyroid samples (n=391) were ranked by BRAFV600E-RAS score (BRS), with BRAFV600E-like and RAS-like samples having negative (-1 to 0) and positive scores (0 to 1), respectively. The BRAFV600E-RAS score is strongly associated with: (B) driver mutation status; (C) thyroid differentiation score (TDS); (D) single data-type clusters and (E) histology and follicular fraction. The RAS-like samples (normalized score > 0, in red on the top bar) consistently emerged as a distinct subgroup characterized by a higher TDS. See also Figures S6 and S7A, B and Tables S2 and S4B.
Next, we focused on thyroid differentiation, which plays a central role in thyroid cancer. We summarized the expression levels of 16 thyroid metabolism and function genes (Table S5F), which were highly correlated across our cohort, and produced a single metric, designated the Thyroid Differentiation Score (TDS). The TDS and BRS measures were highly correlated across all tumors (Spearman = 0.78, P=3.1×10-80), despite being derived from different gene sets. This correlation was mainly driven by RL-PTCs having relatively high TDS values. The BRAFV600E PTC cohort, considered a homogeneous group in numerous studies, showed a wide range of TDS values (Figure 5), and maintained the TDS and BRS correlation, albeit to a lesser degree (Spearman = 0.38, p=3.0×10-9, Figures 4,5 and S7C-E). To gain insights regarding the observed TDS variation, we identified other genes whose expression levels correlated with TDS across all tumors and within the BRAFV600E cohort. We discovered that the TDS was associated with global expression changes, significantly correlated and anti-correlated with thousands of genes in both cohorts (Figures S7F,G). We obtained similar observations using previously reported DNA microarray data (Giordano et al., 2005) (Figure S7H). Next, to test whether differences in the TDS within the BRAFV600E-mutant cohort were associated with subtle architectural changes, we histologically graded the tumors (Figure S7I) and showed that TDS was indeed correlated with grade (Kruskal-Wallis test p=4×10-6, Figure S7J). TDS also correlated with risk (p=2×10-5) and MACIS (Spearman correlation p=1.3×10-6) but only weakly with tumor purity (p=1.5×10-3) (Figure S7J), indicating that the observed differences in TDS and global expression levels were not strongly influenced by variations in levels of tumor stroma or lymphocyte infiltration. These results support the validity of the TDS and BRS and illustrate that, while independently derived, these measures reflect similar biological properties that are profoundly reflected by gene expression in PTC.
Figure 5. Role of Thyroid Differentiation in Papillary Thyroid Carcinomas.

Thyroid Differentiation Score (TDS) across the cohort with tumors sorted by driver mutation and TDS. Below TDS are the BRAFV600E-RAS score (BRS), ERK signature, histological type, MACIS score, risk of recurrence, driver mutations, gene expression data for nine thyroid genes used to derive the TDS (TG, TPO, SLC26A4 (pendrin), SLC5A5 (Na/I symporter), SLC5A8 (apical iodide transporter), DIO1, DIO2, DUOX1, DUOX2), four selected mRNAs correlated to TDS, and three selected miRs correlated to TDS. Featured mRNA (except for 16 thyroid genes) and miRNA genes were selected based on Spearman correlation to TDS in the BRAFV600E cohort (*) and the full cohort (**) (see Supplement). See also Figures S7C-J and Table S5F.
Although the TDS was correlated with many genes, among the most correlated were several genes with cancer relevance (TFF3, KIT, PVRL4 and FHL1, q=8.02×10-30, 5.62×10-29, 4.08×10-13 and 7.52×10-115, respectively, Figures 5 and S7F-G). Among miRs with cancer relevance, miR-21, miR-146b and miR-204 were highly correlated with the TDS (Figure 5 and Figures S7F-G), with miR-21 being the most negatively correlated miR in both the entire and BRAFV600E cohorts. miR-21 and miR-146b are oncogenic miRs in several tumor types (Di Leva et al., 2014) and miR-204 is down-regulated in several tumors types and may be a tumor suppressor (Imam et al., 2012). We subsequently identified variable expression of these miRs in distinct miR clusters with different TDS and BRS values (see Molecular Classification).
Our data demonstrate significant gene expression variation across the BRAFV600E cohort, which may account for the range of differentiation observed and may explain the uncertainty regarding the prognostic and predictive power of BRAFV600E mutation (Xing et al., 2013). Dedifferentiation likely plays a role in dampening responses to RAI therapy and is consistent with observations that RAI-refractory metastases are enriched for BRAFV600E mutants (Sabra et al., 2013). Although other factors are likely involved, our results support the view that potent constitutive activation of the MAPK transcriptional output by oncogenic BRAFV600E downregulates the expression of genes involved in iodine metabolism (Chakravarty et al., 2011; Franco et al., 2011). Of note, the loss of differentiation within the BRAFV600E cohort is likely smaller than that observed in histologically aggressive thyroid cancers.
An integrated view of our TDS analysis (Figure 5) summarizes how RL-PTCs result in highly differentiated tumors enriched for follicular histology with distinct gene expression and DNA methylation patterns. Conversely, BVL-PTCs result in predominantly less differentiated tumors enriched for classical and tall cell histology, with distinct gene expression and DNA methylation patterns. Further assessment of BRS and TDS might be useful in the setting of a clinical trial and even pathology practice using immunohistochemistry.
To better understand the downstream signaling effects of the main driver events (BRAFV600E, RAS), we examined mRNA expression, protein and phosphoprotein levels of various signaling pathways. We used a 52-gene signature derived by inhibiting MEK in a BRAFV600E melanoma cell line (Pratilas et al., 2009) to assess the ERK (and MAPK) activation level. BRS was highly correlated to ERK activation level (i.e. ERK score, see Supplement); BVL-PTCs showed over-activation of the pathway (Figure S8A), and increased expression of DUSP genes (Figure 6). Note that, although the two scores are highly correlated, they were derived independently and have no genes in common. In wild-type cells, ERK induces a negative feedback on effectors upstream in the pathway, resulting in impairment of RAF dimerization. Because BRAFV600E signals as a monomer, it is insensitive to feedback leading to ERK over-activation (Poulikakos et al., 2010).
Figure 6. Downstream Signaling of BVL and RL PTCs.
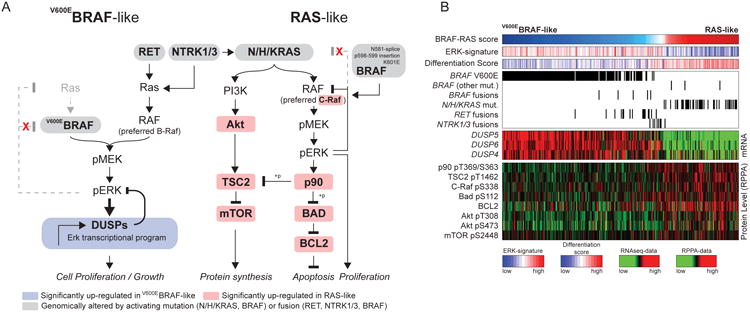
(A) MAPK and PI3K pathways are differentially activated in the BVL and RL PTCs.
(B) BRAFV600E-mutated cases show robust activation of MAPK signaling resulting in higher output of the ERK transcriptional program, represented in particular by DUSP (DUSP4, 5 and 6) mRNAs. This may be due to insensitivity of BRAFV600E to ERK inhibitory feedback. By contrast, RAS-like tumors activated both MAPK and PI3K/AKT signaling, as shown by higher pAKT levels in these tumors. The mechanism by which RAS-like tumors activated MAPK signaling was distinct from that of BRAFV600E tumors, as they had higher CRAF phosphorylation, consistent with engagement of RAF dimers. Paradoxically, RL-PTCs had higher phosphorylation of the ERK substrate p90RSK, which was associated with mTOR activation, likely through phosphorylation and consequent inhibition of TSC2. RL-PTCs also showed activation of an anti-apoptotic program, characterized by S112-BAD phosphorylation (a target of P90RSK) and BCL2 over-expression. See also Figures S8 and Tables S4B,F.
RET fusions consistently had BVL phenotype, high MAPK activity and were associated with low pS338-CRAF and pS299-ARAF (Figure S8B), consistent with preferential signaling via BRAF homodimers (Mitsutake et al., 2006). RL-PTCs had concurrent activation of PI3K/AKT and MAPK signaling, the latter mostly through c-RAF phosphorylation (Figure 6). Despite having lower MAPK activity than BVL-PTCs, RL-PTCs showed significantly higher phosphorylation of p90RSK, a direct ERK substrate. Activation of p90RSK was associated with robust inhibition of TSC2, a distinguishing hallmark of the two functional classes, and likely to induce mTOR. Elevated p90RSK in the RL-PTCs was also associated with phosphorylation of its substrate BAD, and with concurrent BCL2 over-expression, leading to anti-apoptotic signaling (Figure 6 and Table S4B). Finally, we used TieDIE (Paull et al., 2013) to assess differential pathway activation between BVL-PTC and RL-PTC (see Supplement). This approach identified the small GTPase RHEB, a known regulator of mTOR activity (Groenewoud and Zwartkruis, 2013), as a contributing factor to the differences observed between BVL-PTC and RL-PTC (Figure S8C, Table S4F and Data File S1). These findings confirm many of the known signaling changes induced by BRAFV600E and RAS mutations in PTC, provide a framework for MAPK downstream activity in tumors with other driver alterations, and importantly shed new light on the role of p90RSK as a crucial crossroad for MAPK, mTOR and BCL2 signaling in RAS-driven tumors.
Molecular Classification
The comprehensive and multiplatform molecular data and large sample size in this study provide an opportunity to derive and refine the classification of PTC into molecular subtypes and associate them with clinically relevant parameters. To this end, we leveraged the BRS and TDS measures to inform the relationships between tumor cluster, histology, genotype, signaling and differentiation.
Applying unsupervised clustering methods to four genomic datasets yielded a different number of subtypes for each dataset: five for mRNA expression, six for miR expression, four for DNA methylation and four for protein expression (Figures S9A-D). All clustering results were consistent with two meta-clusters that separated the BRAFV600E-driven tumors (BVL-PTCs) from ones with RAS mutations (RL-PTCs), recapitulating the BRS-partitioning and association with histological subtypes (Figures 4 and S9A). We used Stratome× (Streit et al., 2014) to visually highlight these relationships (see THCA publication page); in particular, the significant distinction between BVL-PTCs and RL-PTCs that is evident in each of the molecular datasets (Table S5G). We also applied SuperCluster (see Supplement) to the four genomic datasets, which supported the overarching separation of BVL-PTCs and RL-PTCs (Figure S9E).
Next, we focused on the internal structure reported by different data types for these two meta-clusters. The RL-PTC group was associated with a single cluster in all datasets except DNA methylation (described below). Overall, this group was characterized by FV histology, relatively low risk of recurrence, distinct mRNA expression profiles with lower expression of immune response genes, higher expression of miR-182-5p and miR-183-5p, low levels of fibronectin, VHL and CHK2 proteins and high expression of claudin-7, TIGAR and BRCA2 proteins (Figures 7, S9B and S9D). RL tumors were highly differentiated and associated with younger patients. The DNA methylation data partitioned these tumors into two clusters: the larger, termed Meth-follicular showed few methylation changes compared to normal thyroid, while the other, termed the Meth-CpG Island cluster, was characterized by hypermethylation of a large number of CpG sites in islands and shores (Figure S9C). The significance of this distinction is unclear, although the Meth-CpG Island cluster tended to have high tumor purity with less lymphocyte infiltration and stromal cells (Figure S9C(d)).
Figure 7. Unsupervised clusters for miRNA-seq data.
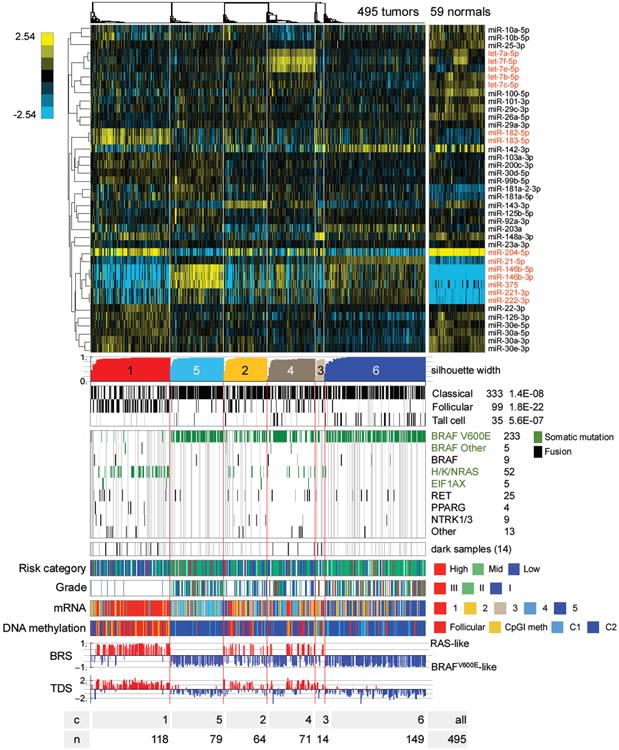
Heatmap showing discriminatory miRs (5p or 3p mature strands) with the largest 6% of metagene matrix scores (see Supplement), as well as miR-204-5p, 221-3p and 222-3p, which were highlighted in correlations to BRS and TDS scores (see Figure S10D). The scalebar shows log2 normalized (reads-per-million, RPM), median-centered miR abundance. miR names in red are discussed in the text. Gray vertical lines in the clinical information tracks mark samples without clinical data, and in the mutation tracks gray lines identify samples without sequence data. See also Figures S9, S10 and Tables S4C,D,E, 5G, 6.
The different datasets partitioned the BVL-PTC group into different numbers of subtypes (from 2 based on DNA methylation data to 5 based on miR data), which did not overlap with each other such as to form a fully consistent lower level partitioning of BVL-PTCs (Figures 4 and S9). Regardless of which dataset was used, the clusters were significantly different based on parameters like proportions of driver mutations and gene fusions, mutational densities, histological and risk profiles, age, BRS and TDS values (Table S5G). The most striking relationship between subtypes was that mRNA-cluster 5 was nearly fully embedded within miR-cluster 6 (86/106 mRNA-cluster 5 tumors were part of the 144 tumors in miR-cluster 6; Fisher's exact test p=1.3×10-36). This mRNA cluster, which we termed tall cell-like, contained most of the TCV tumors (74%), had the highest frequency of BRAFV600E mutations (78%), and the lowest BRS and TDS values (i.e. the strongest BVL phenotype and least differentiated). Since the tall cell-like cluster was associated with more advanced stage (see THCA publication page) and higher risk (Table S5G), this may be clinically relevant. We again used Stratome× to highlight the relationships between mRNA and miR clusters with recurrence risk and histology (see THCA publication page). The tall cell-like cluster was also identified by SuperCluster (Figure S9E).
Integrated miR Analysis
Given the increased role of miRs in determining cancer phenotype, we further examined the association between miR expression, molecular subtypes and clinical parameters. In addition to miR-182 and miR-183 in the RL-PTC group, several other cancer-relevant miRs were relatively abundant in other miR clusters (Figures 7 and S9B). These included oncomiRs (miR-21 and miR-146b) and tumor suppressor miRs (let-7 family, miR-204, and miR-375). OncomiRs miR-221 and miR-222 were reported to play a role in PTC aggressiveness (Mardente et al., 2012) and, in our data, were associated with less differentiated tumors (Figures 7 and S10D(b)).
We focused integrative analysis on miR-21, miR-146b, and miR-204 because they were epigenetically regulated, correlated with BRS and TDS, and/or differentially expressed between PTCs and normal thyroid, as well as between clusters derived from miRNA-seq data (Figures S9B and S10A,B,F). miR-21 expression correlated with highly variable DNA methylation (Figure S10C), defined the tall cell-like mRNA and DNA methylation Meth-classical-1 clusters, and was highly correlated with low BRS and TDS values (Figures 7 and S10D). miR-21 is a regulator of several cancer-related genes (Di Leva et al., 2014). In our data, using Regulome Explorer's pairwise associations, its expression was anti-correlated with expression of cancer-promoting genes and regulators of apoptosis (e.g. PDCD4) (Figure S10E). PDCD4 is a miR-21 target gene reported to function as a tumor suppressor in diverse tumors (Zhu et al., 2008). These observations raise the possibility that increased miR-21 expression via epigenetic dysregulation may contribute to the clinically aggressive nature of this BVL-PTC sub-cluster and may partly explain the aggressive nature of TCV.
miR-146b expression exhibited similar patterns of differential expression and correlations to DNA methylation, BRS, and TDS (Figures 7 and S10C-D), and likely influenced expression of, for example, IRAK1, KIT and TRAF6 (Figure S10E). These results are consistent with observations that miR-146b is associated with risk of recurrence and promotes cell migration and invasion (Chou et al., 2013).
miR-204 expression, while less influenced by DNA methylation, was preferentially lost in miR clusters 5 and 6, the two BVL-PTC sub-clusters with the lowest BRS and TDS values (Figures 7 and S9B). This is consistent with data from other tumors, i.e. miR-204 functions as a tumor suppressor and high levels suppress cell migration, invasion and EMT (Qiu et al., 2013). These results suggest that loss of miR-204 may also contribute to aggressive PTCs with BRAFV600E mutations. Collectively, our results are consistent with prior studies and suggest that miRs may regulate fundamental aspects of the PTC phenotype, i.e. signaling, differentiation, invasion and metastasis, by fine-tuning gene expression.
Discussion
This study illustrates the dominant role and mutually exclusive nature of driving somatic genetic alterations, be they SSNVs, indels, or fusions, in the MAPK and PI3K pathways in PTC. The relative low overall density of somatic mutations may be the biological basis for the indolent clinical behavior of PTC. We discovered new driver mutations in PTC, either entirely novel in this cancer (EIF1AX) or novel alterations of known drivers (RET, BRAF and ALK fusions). As a result of these discoveries, the ‘dark matter’ of the PTC genome has been reduced substantially from ∼25% to less than 4%, which should have profound consequences for preoperative cancer diagnosis in thyroid nodules. Molecular testing of mutation hotspots, rearrangements and gene expression using fine-needle aspiration specimens has become an effective diagnostic tool to more precisely select patients for thyroid surgery (Alexander et al., 2012; Nikiforov et al., 2011), thereby reducing the number of thyroidectomies done for benign nodules and tumors (Nikiforov et al., 2013), and determining the extent of initial thyroid surgery (i.e. lobectomy vs. total thyroidectomy) (Yip et al., 2014). Through these advances, molecular diagnostics has improved the care of patients with thyroid nodules and cancer. Our expansion of the PTC somatic genetic landscape has the potential to even further enhance the care of these patients. This study also offers conclusive evidence that mutated BRAF and other driving mutations are clonal events present in the majority of cells within tumors and identified novel fusion partners of oncogenes (e.g. RET, BRAF, ALK), expanding the biological basis for targeted therapy.
Beyond the driver mutations, we discovered individual genes (CHEK2, ATM, TERT) and sets of functionally related genes (chromatin-remodeling) with alterations or expression patterns (miR-21 and miR-146b) that define clinically-relevant subclasses and may contribute to loss of differentiation and tumor progression. Specifically, increased expression of miR-21 was associated with a known aggressive form of PTC (tall cell variant) and may be a critical event in its pathogenesis. Similarly, TERT promoter mutations identified a subset of aggressive, less differentiated PTCs, consistent with recent reports (Melo et al., 2014; Xing et al., 2014). Our study also indicates that BRAFV600E PTC represents a diverse group of tumors, consisting of at least four molecular subtypes, with variable degrees of thyroid differentiation. Collectively, our results suggest that BRAFV600E PTC should not be considered a homogeneous group in clinical studies and that future studies should include molecular components designed to capture the breadth of genetic diversity among PTCs.
We demonstrate striking signaling differences in RAS- and BRAFV600E-driven PTCs. In particular, BVL-PTCs signal preferentially through MAPK while RL-PTCs signal through both MAPK and PI3K. The relative simplicity of the PTC genome, with dominant mutually exclusive driving events, together with the large cohort and comprehensive data analyzed in this study enabled us to clearly dissect these signaling differences.
Our overarching conclusion is that RL-PTCs and BVL-PTCs are fundamentally different in their genomic, epigenomic and proteomic profiles. This is consistent with their known histological differences and the published literature. However, the breadth and depth of our integrative findings have wide implications for basic pathobiology, tumor classification schemes, traditional and targeted therapies. This view is supported by recent data suggesting differential response to a MEK inhibitor related to thyroid cancer genotype (Ho et al., 2013). We feel, based on the strength of our multidimensional genomic findings, that a pathologic reclassification of follicular-patterned thyroid lesions is justified. There was a time when follicular-patterned PTCs (i.e. RL-PTCs) were classified as follicular carcinomas. Perhaps the time has come to revise the classification of thyroid cancer to reunite the FV of PTC with follicular carcinomas. Moreover, a refined classification scheme that more accurately reflects the genotypic and phenotypic differences between and within RL and BVL PTCs would lead to more precise surgical and medical therapy, especially as thyroid cancer therapy enters the realm of precision medicine.
Methods summary
Tumor and normal thyroid samples were obtained from patients with approval from local Institutional Review Boards. DNA, RNA and protein were purified and distributed throughout the TCGA network. In total, 496 primary tumors and 8 metastatic tumors with associated clinicopathologic data were assayed on at least one molecular profiling platform. Platforms included exome and whole genome DNA sequencing, RNA sequencing, miRNA sequencing, SNP arrays, DNA methylation arrays, and reverse phase protein arrays. Integrated multi-platform analyses were performed. The data and analysis results can be explored through the Broad Institute GDAC portal (http://dx.doi.org/10.7908/C17P8WZG) and FireBrowse portal (http://firebrowse.org/?cohort=THCA), Memorial Sloan Kettering Cancer Center cBioPortal (http://www.cbioportal.org/public-portal/study.do?cancer_study_id=thca_tcga), TieDIE (http://sysbiowiki.soe.ucsc.edu/tiedie), MBatch batch effects assessor (http://bioinformatics.mdanderson.org/tcgambatch/), Regulome Explorer (http://explorer.cancerregulome.org/) and Next-Generation Clustered Heat Maps (http://bioinformatics.mdanderson.org/TCGA/NGCHMPortal/). See also the Supplement and the THCA publication page (https://tcga-data.nci.nih.gov/docs/publications/thca_2014/).
Supplementary Material
Acknowledgments
We are grateful to all the patients and families who contributed to this study, to Chris Gunter for editing and Margi Sheth and Jiashan (Julia) Zhang for project management. Thomas Giordano thanks his colleagues who covered his clinical duties. Supported by the following grants from the United States National Institutes of Health: 5U24CA143799, 5U24CA143835, 5U24CA143840, 5U24CA143843, 5U24CA143845, 5U24CA143848, 5U24CA143858, 5U24CA143866, 5U24CA143867, 5U24CA143882, 5U24CA143883, 5U24CA144025, U54HG003067, U54HG003079, and U54HG003273, P30CA16672.
Footnotes
Competing financial interests: The authors declare no competing financial interests.
Data access: The primary and processed data used to generate the analyses presented here can be downloaded by registered users from The Cancer Genome Atlas at https://tcga-data.nci.nih.gov/tcga/tcgaDownload.jsp. The primary sequence files are deposited in CGHub (https://cghub.ucsc.edu/) and all other molecular, clinical and pathological data are deposited at the Data Coordinating Center (DCC) for public access (http://cancergenome.nih.gov/, https://cghub.ucsc.edu/ and https://tcga-data.nci.nih.gov/docs/publications/thca_2014/) and the TCGA Data Portal (https://tcga-data.nci.nih.gov/tcga/).
Consortia: The members of The Cancer Genome Atlas Research Network for this project are Nishant Agrawal, Rehan Akbani, B. Arman Aksoy, Adrian Ally, Harindra Arachchi, Sylvia L. Asa, J. Todd Auman, Miruna Balasundaram, Saianand Balu, Stephen B. Baylin, Madhusmita Behera, Brady Bernard, Rameen Beroukhim, Justin A. Bishop, Aaron D. Black, Tom Bodenheimer, Lori Boice, Moiz S. Bootwalla, Jay Bowen, Reanne Bowlby, Christopher A. Bristow, Robin Brookens, Denise Brooks, Robert Bryant, Elizabeth Buda, Yaron S.N. Butterfield, Tobias Carling, Rebecca Carlsen, Scott L. Carter, Sally E. Carty, Timothy A. Chan, Amy Y. Chen, Andrew D. Cherniack, Dorothy Cheung, Lynda Chin, Juok Cho, Andy Chu, Eric Chuah, Kristian Cibulskis, Giovanni Ciriello, Amanda Clarke, Gary L. Clayman, Leslie Cope, John Copland, Kyle Covington, Ludmila Danilova, Tanja Davidsen, John A. Demchok, Daniel DiCara, Noreen Dhalla, Rajiv Dhir, Sheliann S. Dookran, Gideon Dresdner, Jonathan Eldridge, Greg Eley, Adel K. El-Naggar, Stephanie Eng, James A. Fagin, Timothy Fennell, Robert L. Ferris, Sheila Fisher, Scott Frazer, Jessica Frick, Stacey B. Gabriel, Ian Ganly, Jianjiong Gao, Levi A. Garraway, Julie M. Gastier-Foster, Gad Getz, Nils Gehlenborg, Ronald Ghossein, Richard A. Gibbs, Thomas J. Giordano, Karen Gomez-Hernandez, Jonna Grimsby, Benjamin Gross, Ranabir Guin, Angela Hadjipanayis, Hollie A. Harper, D. Neil Hayes, David I. Heiman, James G. Herman, Katherine A. Hoadley, Matan Hofree, Robert A. Holt, Alan P. Hoyle, Franklin W. Huang, Mei Huang, Carolyn M. Hutter, Trey Ideker, Lisa Iype, Anders Jacobsen, Stuart R. Jefferys, Corbin D. Jones, Steven J.M. Jones, Katayoon Kasaian, Electron Kebebew, Fadlo R. Khuri, Jaegil Kim, Roger Kramer, Richard Kreisberg, Raju Kucherlapati, David J. Kwiatkowski, Marc Ladanyi, Phillip H. Lai, Peter W. Laird, Eric Lander, Michael S. Lawrence, Darlene Lee, Eunjung Lee, Semin Lee, William Lee, Kristen M. Leraas, Tara M. Lichtenberg, Lee Lichtenstein, Pei Lin, Shiyun Ling, Jinze Liu, Wenbin Liu, Yingchun Liu, Virginia A. LiVolsi, Yiling Lu, Yussanne Ma, Harshad S. Mahadeshwar, Marco A. Marra, Michael Mayo, David G. McFadden, Shaowu Meng, Matthew Meyerson, Piotr A. Mieczkowski, Michael Miller, Gordon Mills, Richard A. Moore, Lisle E. Mose, Andrew J. Mungall, Bradley A. Murray, Yuri E. Nikiforov, Michael S. Noble, Akinyemi I. Ojesina, Taofeek K. Owonikoko, Bradley A. Ozenberger, Angeliki Pantazi, Michael Parfenov, Peter J. Park, Joel S. Parker, Evan O. Paull, Chandra Sekhar Pedamallu, Charles M. Perou, Jan F. Prins, Alexei Protopopov, Suresh S. Ramalingam, Nilsa C. Ramirez, Ricardo Ramirez, Benjamin J. Raphael, W. Kimryn Rathmell, Xiaojia Ren, Sheila M. Reynolds, Esther Rheinbay, Matthew D. Ringel, Michael Rivera, Jeffrey Roach, A. Gordon Robertson, Mara W. Rosenberg, Matthew Rosenthall, Sara Sadeghi, Gordon Saksena, Chris Sander, Netty Santoso, Jacqueline E. Schein, Nikolaus Schultz, Steven E. Schumacher, Raja R. Seethala, Jonathan Seidman, Yasin Senbabaoglu, Sahil Seth, Samantha Sharpe, Kenna R. Mills Shaw, John P. Shen, Ronglai Shen, Steven Sherman, Margi Sheth, Yan Shi, Ilya Shmulevich, Gabriel L. Sica, Janae V. Simons, Payal Sipahimalani, Robert C. Smallridge, Heidi J. Sofia, Matthew G. Soloway, Xingzhi Song, Carrie Sougnez, Chip Stewart, Petar Stojanov, Joshua M. Stuart, Barbara Tabak, Angela Tam, Donghui Tan, Jiabin Tang, Roy Tarnuzzer, Barry S. Taylor, Nina Thiessen, Leigh Thorne, Vésteinn Thorsson, R. Michael Tuttle, Christopher B. Umbricht, David J. Van Den Berg, Fabio Vandin, Umadevi Veluvolu, Roel G.W. Verhaak, Michelle Vinco, Doug Voet, Vonn Walter, Zhining Wang, Scot Waring, Paul M. Weinberger, John N. Weinstein, Daniel J. Weisenberger, David Wheeler, Matthew D. Wilkerson, Jocelyn Wilson, Michelle Williams, Daniel A. Winer, Lisa Wise, Junyuan Wu, Liu Xi, Andrew W. Xu, Liming Yang, Lixing Yang, Travis I. Zack, Martha A. Zeiger, Dong Zeng, Jean Claude Zenklusen, Ni Zhao, Hailei Zhang, Jianhua Zhang, Jiashan (Julia) Zhang, Wei Zhang, Erik Zmuda, and Lihua Zou.
Author Contributions: Project leaders: G.G and T.J.G. Analysis coordinator: C.S. Data coordinator J.C. Manuscript coordinator T.J.G. Project coordinators: M.S. and J.Z. Clinical expertise and data analysis: S.L.A., J.B., J.A.F., I.G., T.J.G., E.K., L.I., K.L., T.M.L., D.G.M., A.P., M.D.R., R.C.S., C.B.U., Y.E.N. and M.A.Z. Supplemental pathology review: S.L.A., T.J.G., R.G., J.A.B. and Y.E.N. DNA sequence and copy number analysis: A.D.C., J.C., G.G., K.K., J.K., D.J.K., L.L., B.A.M., E.R., M.R., G.S., C.S. and C.S. TERT promoter sequencing and analysis: C.S., C.S., J.G., F.W.H., L.A.G., S.D.D., Y.E.N., A.H., R.K., and G.G. Gene fusions: A.H., K.A.H., R.K., C.S., and T.J.G. Viral sequence detection: A.I.O., M.M., C.S.P., S.S. and Y.M. DNA methylation analysis: L.C. and L.D. mRNA analysis: K.A.H., C.S., V.W., N.Z. and A.H. miRNA analysis: L.I. and A.G.R. Pathway/Network/Integrated analysis: G.C., J.A.F., N.G., M.H., L.I., E.O.P., B.J.R., M.D.R., A.G.R., J.M.S., N.S., Y.S. and F.V. Dark matter analysis: J.C., C.S. and T.J.G. Thyroid differentiation analysis: J.K., L.D., L.I., A.G.R., C.S., G.G. and T.J.G. RPPA analysis: R.A., W.L., Y.L. and G.B.M. Supercluster: R.A. Batch effect analysis: R.A., S.L. and J.N.W. Manuscript review: M.M. and R.K.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adeniran AJ, Zhu Z, Gandhi M, Steward DL, Fidler JP, Giordano TJ, Biddinger PW, Nikiforov YE. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. The American journal of surgical pathology. 2006;30:216–222. doi: 10.1097/01.pas.0000176432.73455.1b. [DOI] [PubMed] [Google Scholar]
- Alexander EK, Kennedy GC, Baloch ZW, Cibas ES, Chudova D, Diggans J, Friedman L, Kloos RT, LiVolsi VA, Mandel SJ, et al. Preoperative diagnosis of benign thyroid nodules with indeterminate cytology. The New England journal of medicine. 2012;367:705–715. doi: 10.1056/NEJMoa1203208. [DOI] [PubMed] [Google Scholar]
- American Thyroid Association Guidelines Taskforce on Thyroid, N.,Differentiated Thyroid, C. Cooper DS, Doherty GM, Haugen BR, Kloos RT, Lee SL, Mandel SJ, Mazzaferri EL, McIver B, et al. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid : official journal of the American Thyroid Association. 2009;19:1167–1214. doi: 10.1089/thy.2009.0110. [DOI] [PubMed] [Google Scholar]
- Bischoff LA, Curry J, Ahmed I, Pribitkin E, Miller JL. Is above age 45 appropriate for upstaging well-differentiated papillary thyroid cancer? Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2013;19:995–997. doi: 10.4158/EP13029.OR. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nature biotechnology. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty D, Santos E, Ryder M, Knauf JA, Liao XH, West BL, Bollag G, Kolesnick R, Thin TH, Rosen N, et al. Small-molecule MAPK inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional BRAF activation. The Journal of clinical investigation. 2011;121:4700–4711. doi: 10.1172/JCI46382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles RP, Iezza G, Amendola E, Dankort D, McMahon M. Mutationally activated BRAF(V600E) elicits papillary thyroid cancer in the adult mouse. Cancer research. 2011;71:3863–3871. doi: 10.1158/0008-5472.CAN-10-4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AY, Jemal A, Ward EM. Increasing incidence of differentiated thyroid cancer in the United States, 1988-2005. Cancer. 2009;115:3801–3807. doi: 10.1002/cncr.24416. [DOI] [PubMed] [Google Scholar]
- Chou CK, Yang KD, Chou FF, Huang CC, Lan YW, Lee YF, Kang HY, Liu RT. Prognostic implications of miR-146b expression and its functional role in papillary thyroid carcinoma. The Journal of clinical endocrinology and metabolism. 2013;98:E196–205. doi: 10.1210/jc.2012-2666. [DOI] [PubMed] [Google Scholar]
- Chu J, Sadeghi S, Raymond A, Jackman SD, Nip KM, Mar R, Mohamadi H, Butterfield YS, Robertson AG, Birol I. BioBloom tools: fast, accurate and memory-efficient host species sequence screening using bloom filters. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciampi R, Knauf JA, Kerler R, Gandhi M, Zhu Z, Nikiforova MN, Rabes HM, Fagin JA, Nikiforov YE. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. The Journal of clinical investigation. 2005;115:94–101. doi: 10.1172/JCI23237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello G, Cerami E, Sander C, Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome research. 2012;22:398–406. doi: 10.1101/gr.125567.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen Y, Xing M, Mambo E, Guo Z, Wu G, Trink B, Beller U, Westra WH, Ladenson PW, Sidransky D. BRAF mutation in papillary thyroid carcinoma. Journal of the National Cancer Institute. 2003;95:625–627. doi: 10.1093/jnci/95.8.625. [DOI] [PubMed] [Google Scholar]
- Di Leva G, Garofalo M, Croce CM. MicroRNAs in cancer. Annual review of pathology. 2014;9:287–314. doi: 10.1146/annurev-pathol-012513-104715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durante C, Puxeddu E, Ferretti E, Morisi R, Moretti S, Bruno R, Barbi F, Avenia N, Scipioni A, Verrienti A, et al. BRAF mutations in papillary thyroid carcinomas inhibit genes involved in iodine metabolism. The Journal of clinical endocrinology and metabolism. 2007;92:2840–2843. doi: 10.1210/jc.2006-2707. [DOI] [PubMed] [Google Scholar]
- Ellis RJ, Wang Y, Stevenson HS, Boufraqech M, Patel D, Nilubol N, Davis S, Edelman DC, Merino MJ, He M, et al. Genome-wide methylation patterns in papillary thyroid cancer are distinct based on histological subtype and tumor genotype. The Journal of clinical endocrinology and metabolism. 2014;99:E329–337. doi: 10.1210/jc.2013-2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic acids research. 2011;39:D945–950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco AT, Malaguarnera R, Refetoff S, Liao XH, Lundsmith E, Kimura S, Pritchard C, Marais R, Davies TF, Weinstein LS, et al. Thyrotrophin receptor signaling dependence of Braf-induced thyroid tumor initiation in mice. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:1615–1620. doi: 10.1073/pnas.1015557108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghossein RA, Katabi N, Fagin JA. Immunohistochemical detection of mutated BRAF V600E supports the clonal origin of BRAF-induced thyroid cancers along the spectrum of disease progression. The Journal of clinical endocrinology and metabolism. 2013;98:E1414–1421. doi: 10.1210/jc.2013-1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano TJ, Kuick R, Thomas DG, Misek DE, Vinco M, Sanders D, Zhu Z, Ciampi R, Roh M, Shedden K, et al. Molecular classification of papillary thyroid carcinoma: distinct BRAF, RAS, and RET/PTC mutation-specific gene expression profiles discovered by DNA microarray analysis. Oncogene. 2005;24:6646–6656. doi: 10.1038/sj.onc.1208822. [DOI] [PubMed] [Google Scholar]
- Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, Pierotti MA, Della Porta G, Fusco A, Vecchio G. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell. 1990;60:557–563. doi: 10.1016/0092-8674(90)90659-3. [DOI] [PubMed] [Google Scholar]
- Groenewoud MJ, Zwartkruis FJ. Rheb and mammalian target of rapamycin in mitochondrial homoeostasis. Open biology. 2013;3:130185. doi: 10.1098/rsob.130185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra A, Sapio MR, Marotta V, Campanile E, Rossi S, Forno I, Fugazzola L, Budillon A, Moccia T, Fenzi G, et al. The primary occurrence of BRAF(V600E) is a rare clonal event in papillary thyroid carcinoma. The Journal of clinical endocrinology and metabolism. 2012;97:517–524. doi: 10.1210/jc.2011-0618. [DOI] [PubMed] [Google Scholar]
- Hay ID, Bergstralh EJ, Goellner JR, Ebersold JR, Grant CS. Predicting outcome in papillary thyroid carcinoma: development of a reliable prognostic scoring system in a cohort of 1779 patients surgically treated at one institution during 1940 through 1989. Surgery. 1993;114:1050–1057. discussion 1057-1058. [PubMed] [Google Scholar]
- Hay ID, Thompson GB, Grant CS, Bergstralh EJ, Dvorak CE, Gorman CA, Maurer MS, McIver B, Mullan BP, Oberg AL, et al. Papillary thyroid carcinoma managed at the Mayo Clinic during six decades (1940-1999): temporal trends in initial therapy and long-term outcome in 2444 consecutively treated patients. World journal of surgery. 2002;26:879–885. doi: 10.1007/s00268-002-6612-1. [DOI] [PubMed] [Google Scholar]
- Ho AL, Grewal RK, Leboeuf R, Sherman EJ, Pfister DG, Deandreis D, Pentlow KS, Zanzonico PB, Haque S, Gavane S, et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. The New England journal of medicine. 2013;368:623–632. doi: 10.1056/NEJMoa1209288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofree M, Shen JP, Carter H, Gross A, Ideker T. Network-based stratification of tumor mutations. Nature methods. 2013;10:1108–1115. doi: 10.1038/nmeth.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson KE, Lipson D, Stephens PJ, Otto G, Lehmann BD, Lyle PL, Vnencak-Jones CL, Ross JS, Pietenpol JA, Sosman JA, et al. BRAF fusions define a distinct molecular subset of melanomas with potential sensitivity to MEK inhibition. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:6696–6702. doi: 10.1158/1078-0432.CCR-13-1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imam JS, Plyler JR, Bansal H, Prajapati S, Bansal S, Rebeles J, Chen HI, Chang YF, Panneerdoss S, Zoghi B, et al. Genomic loss of tumor suppressor miRNA-204 promotes cancer cell migration and invasion by activating AKT/mTOR/Rac1 signaling and actin reorganization. PloS one. 2012;7:e52397. doi: 10.1371/journal.pone.0052397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CK, Little MP, Lubin JH, Brenner AV, Wells SA, Jr, Sigurdson AJ, Nikiforov YE. The increase in thyroid cancer incidence during the last four decades is accompanied by a high frequency of BRAF mutations and a sharp increase in RAS mutations. The Journal of clinical endocrinology and metabolism. 2014;99:E276–285. doi: 10.1210/jc.2013-2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly LM, Barila G, Liu P, Evdokimova VN, Trivedi S, Panebianco F, Gandhi M, Carty SE, Hodak SP, Luo J, et al. Identification of the transforming STRN-ALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:4233–4238. doi: 10.1073/pnas.1321937111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer research. 2003;63:1454–1457. [PubMed] [Google Scholar]
- Kjellman P, Lagercrantz S, Hoog A, Wallin G, Larsson C, Zedenius J. Gain of 1q and loss of 9q21.3-q32 are associated with a less favorable prognosis in papillary thyroid carcinoma. Genes, chromosomes & cancer. 2001;32:43–49. doi: 10.1002/gcc.1165. [DOI] [PubMed] [Google Scholar]
- Kleiblova P, Shaltiel IA, Benada J, Sevcik J, Pechackova S, Pohlreich P, Voest EE, Dundr P, Bartek J, Kleibl Z, et al. Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. The Journal of cell biology. 2013;201:511–521. doi: 10.1083/jcb.201210031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic AD, Ojesina AI, Pedamallu CS, Jung J, Verhaak RG, Getz G, Meyerson M. PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nature biotechnology. 2011;29:393–396. doi: 10.1038/nbt.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroll TG, Sarraf P, Pecciarini L, Chen CJ, Mueller E, Spiegelman BM, Fletcher JA. PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma [corrected] Science. 2000;289:1357–1360. doi: 10.1126/science.289.5483.1357. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NV, Lira ME, Pavlicek A, Ye J, Buckman D, Bagrodia S, Srinivasa SP, Zhao Y, Aparicio S, Rejto PA, et al. A novel SND1-BRAF fusion confers resistance to c-Met inhibitor PF-04217903 in GTL16 cells through [corrected] MAPK activation. PloS one. 2012;7:e39653. doi: 10.1371/journal.pone.0039653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine NR, Mayall ES, Wyllie FS, Farr CJ, Hughes D, Padua RA, Thurston V, Williams ED, Wynford-Thomas D. Activated ras oncogenes in human thyroid cancers. Cancer research. 1988;48:4459–4463. [PubMed] [Google Scholar]
- Mardente S, Mari E, Consorti F, Di Gioia C, Negri R, Etna M, Zicari A, Antonaci A. HMGB1 induces the overexpression of miR-222 and miR-221 and increases growth and motility in papillary thyroid cancer cells. Oncology reports. 2012;28:2285–2289. doi: 10.3892/or.2012.2058. [DOI] [PubMed] [Google Scholar]
- Martin M, Masshofer L, Temming P, Rahmann S, Metz C, Bornfeld N, van de Nes J, Klein-Hitpass L, Hinnebusch AG, Horsthemke B, et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nature genetics. 2013;45:933–936. doi: 10.1038/ng.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo M, da Rocha AG, Vinagre J, Batista R, Peixoto J, Tavares C, Celestino R, Almeida A, Salgado C, Eloy C, et al. TERT promoter mutations are a major indicator of poor outcome in differentiated thyroid carcinomas. The Journal of clinical endocrinology and metabolism. 2014;99:E754–765. doi: 10.1210/jc.2013-3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome biology. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamiya Y, Saito H, Ito M, Imai K, Konno H, Takahashi N, Motoyama S, Ogawa J. Suppression of Zinc Finger Homeobox 3 expression in tumor cells decreases the survival rate among non-small cell lung cancer patients. Cancer biomarkers : section A of Disease markers. 2012;11:139–146. doi: 10.3233/CBM-2012-00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsutake N, Miyagishi M, Mitsutake S, Akeno N, Mesa C, Jr, Knauf JA, Zhang L, Taira K, Fagin JA. BRAF mediates RET/PTC-induced mitogen-activated protein kinase activation in thyroid cells: functional support for requirement of the RET/PTC-RAS-BRAF pathway in papillary thyroid carcinogenesis. Endocrinology. 2006;147:1014–1019. doi: 10.1210/en.2005-0280. [DOI] [PubMed] [Google Scholar]
- Nikiforov YE, Ohori NP, Hodak SP, Carty SE, LeBeau SO, Ferris RL, Yip L, Seethala RR, Tublin ME, Stang MT, et al. Impact of mutational testing on the diagnosis and management of patients with cytologically indeterminate thyroid nodules: a prospective analysis of 1056 FNA samples. The Journal of clinical endocrinology and metabolism. 2011;96:3390–3397. doi: 10.1210/jc.2011-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforov YE, Yip L, Nikiforova MN. New strategies in diagnosing cancer in thyroid nodules: impact of molecular markers. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:2283–2288. doi: 10.1158/1078-0432.CCR-12-1253. [DOI] [PubMed] [Google Scholar]
- Nucera C, Lawler J, Parangi S. BRAF(V600E) and microenvironment in thyroid cancer: a functional link to drive cancer progression. Cancer research. 2011;71:2417–2422. doi: 10.1158/0008-5472.CAN-10-3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva-Trastoy M, Berthonaud V, Chevalier A, Ducrot C, Marsolier-Kergoat MC, Mann C, Leteurtre F. The Wip1 phosphatase (PPM1D) antagonizes activation of the Chk2 tumour suppressor kinase. Oncogene. 2007;26:1449–1458. doi: 10.1038/sj.onc.1209927. [DOI] [PubMed] [Google Scholar]
- Park JY, Kim WY, Hwang TS, Lee SS, Kim H, Han HS, Lim SD, Kim WS, Yoo YB, Park KS. BRAF and RAS mutations in follicular variants of papillary thyroid carcinoma. Endocrine pathology. 2013;24:69–76. doi: 10.1007/s12022-013-9244-0. [DOI] [PubMed] [Google Scholar]
- Paull EO, Carlin DE, Niepel M, Sorger PK, Haussler D, Stuart JM. Discovering causal pathways linking genomic events to transcriptional states using Tied Diffusion Through Interacting Events (TieDIE) Bioinformatics. 2013;29:2757–2764. doi: 10.1093/bioinformatics/btt471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierotti MA, Bongarzone I, Borrello MG, Mariani C, Miranda C, Sozzi G, Greco A. Rearrangements of TRK proto-oncogene in papillary thyroid carcinomas. Journal of endocrinological investigation. 1995;18:130–133. doi: 10.1007/BF03349721. [DOI] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, Rosen N. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:4519–4524. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu YH, Wei YP, Shen NJ, Wang ZC, Kan T, Yu WL, Yi B, Zhang YJ. miR-204 inhibits epithelial to mesenchymal transition by targeting slug in intrahepatic cholangiocarcinoma cells. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2013;32:1331–1341. doi: 10.1159/000354531. [DOI] [PubMed] [Google Scholar]
- Ricarte-Filho JC, Li S, Garcia-Rendueles ME, Montero-Conde C, Voza F, Knauf JA, Heguy A, Viale A, Bogdanova T, Thomas GA, et al. Identification of kinase fusion oncogenes in post-Chernobyl radiation-induced thyroid cancers. The Journal of clinical investigation. 2013;123:4935–4944. doi: 10.1172/JCI69766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nature genetics. 2013;45:970–976. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabra MM, Dominguez JM, Grewal RK, Larson SM, Ghossein RA, Tuttle RM, Fagin JA. Clinical outcomes and molecular profile of differentiated thyroid cancers with radioiodine-avid distant metastases. The Journal of clinical endocrinology and metabolism. 2013;98:E829–836. doi: 10.1210/jc.2012-3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares P, Trovisco V, Rocha AS, Lima J, Castro P, Preto A, Maximo V, Botelho T, Seruca R, Sobrinho-Simoes M. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene. 2003;22:4578–4580. doi: 10.1038/sj.onc.1206706. [DOI] [PubMed] [Google Scholar]
- Streit M, Lex A, Gratzl S, Partl C, Schmalstieg D, Pfister H, Park PJ, Gehlenborg N. Guided visual exploration of genomic stratifications in cancer. Nature methods. 2014;11:884–885. doi: 10.1038/nmeth.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez HG, Du Villard JA, Caillou B, Schlumberger M, Tubiana M, Parmentier C, Monier R. Detection of activated ras oncogenes in human thyroid carcinomas. Oncogene. 1988;2:403–406. [PubMed] [Google Scholar]
- Woiwode A, Johnson SA, Zhong S, Zhang C, Roeder RG, Teichmann M, Johnson DL. PTEN represses RNA polymerase III-dependent transcription by targeting the TFIIIB complex. Molecular and cellular biology. 2008;28:4204–4214. doi: 10.1128/MCB.01912-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wreesmann VB, Ghossein RA, Hezel M, Banerjee D, Shaha AR, Tuttle RM, Shah JP, Rao PH, Singh B. Follicular variant of papillary thyroid carcinoma: genome-wide appraisal of a controversial entity. Genes, chromosomes & cancer. 2004a;40:355–364. doi: 10.1002/gcc.20049. [DOI] [PubMed] [Google Scholar]
- Wreesmann VB, Sieczka EM, Socci ND, Hezel M, Belbin TJ, Childs G, Patel SG, Patel KN, Tallini G, Prystowsky M, et al. Genome-wide profiling of papillary thyroid cancer identifies MUC1 as an independent prognostic marker. Cancer research. 2004b;64:3780–3789. doi: 10.1158/0008-5472.CAN-03-1460. [DOI] [PubMed] [Google Scholar]
- Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nature reviews Cancer. 2013;13:184–199. doi: 10.1038/nrc3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing M, Alzahrani AS, Carson KA, Viola D, Elisei R, Bendlova B, Yip L, Mian C, Vianello F, Tuttle RM, et al. Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. JAMA : the journal of the American Medical Association. 2013;309:1493–1501. doi: 10.1001/jama.2013.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing M, Liu R, Liu X, Murugan AK, Zhu G, Zeiger MA, Pai S, Bishop J. BRAF V600E and TERT Promoter Mutations Cooperatively Identify the Most Aggressive Papillary Thyroid Cancer With Highest Recurrence. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014;32:2718–2726. doi: 10.1200/JCO.2014.55.5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip L, Wharry LI, Armstrong MJ, Silbermann A, McCoy KL, Stang MT, Ohori NP, LeBeau SO, Coyne C, Nikiforova MN, et al. A clinical algorithm for fine-needle aspiration molecular testing effectively guides the appropriate extent of initial thyroidectomy. Annals of surgery. 2014;260:163–168. doi: 10.1097/SLA.0000000000000215. [DOI] [PubMed] [Google Scholar]
- Zhu S, Wu H, Wu F, Nie D, Sheng S, Mo YY. MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell research. 2008;18:350–359. doi: 10.1038/cr.2008.24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.