

Abstract
The Grb2-associated binding protein 1 (GAB1) integrates signals from different signaling pathways and is over-expressed in many cancers, therefore representing a new therapeutic target. In the present study, we aim to target the pleckstrin homology (PH) domain of GAB1 for cancer treatment. Using homology models we derived, high-throughput virtual screening of five million compounds resulted in five hits which exhibited strong binding affinities to GAB1 PH domain. Our prediction of ligand binding affinities is also in agreement with the experimental K D values. Furthermore, molecular dynamics studies showed that GAB1 PH domain underwent large conformational changes upon ligand binding. Moreover, these hits inhibited the phosphorylation of GAB1 and demonstrated potent, tumor-specific cytotoxicity against MDA-MB-231 and T47D breast cancer cell lines. This effort represents the discovery of first-in-class GAB1 PH domain inhibitors with potential for targeted breast cancer therapy and provides novel insights into structure-based approaches to targeting this protein.
Author Summary
In this paper, we described the identification and evaluation of a set of first-in-class potent inhibitors targeting a new cancer target, Grb2-associated binder-1 (GAB1), which integrates signals from different signaling pathways, and is frequently over-expressed in cancer cells. To achieve our goals, we have employed intensive computational modeling to understand the structure of the GAB1 pleckstrin homology (PH) domain and screened five million compounds. Upon biological evaluation, we found that several inhibitors that induced large conformational changes of the target structure exhibited strong selective binding to GAB1 PH domain. Particularly, these inhibitors demonstrated potent and tumor-specific cytotoxicity in breast cancer cells. This represents a groundbreaking discovery in targeting GAB1 signaling which may be used for cancer therapy, especially for triple negative breast cancer patients.
Introduction
Overexpression of Grb2-associated binding protein 1 (GAB1) has been observed in several human cancers, such as breast and lung cancers [1]–[4]. This protein is a substrate of several growth factors and interleukin receptors, and it is involved in the integration of different signal transductions [1]–[4]. Particularly, GAB1 mediates the activation of mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI-3K) cascades [5], [6]. It belongs to a family of scaffolding proteins closely related to the insulin receptor substrates (e.g., IRS1) [2]. It contains an N-terminal pleckstrin homology (PH) domain binding to phosphatidylinositol-(3,4,5)-triphosphate (PtdIns(3,4,5)P3), tyrosine phosphorylation sites for the Src homology 2 (SH2) binding and a proline-rich domain interacting with Src homology 3 (SH3) domain [6]. PH domains can be subdivided into four groups based on their selective binding to phosphoinositides [7], and GAB1 PH domain belongs to Group 1 which exhibits the strongest binding to PtdIns(3,4,5)P3, but weak affinity and specificity to PtdIns(3,4)P2 or PtdIns(4,5)P2 [4], [8]. Additionally, the phosphorylation of GAB1 on Y627 depends on the intracellular translocation from cytosol to membrane by binding to PtdIns(3,4,5)P3 via its PH domain [9]. Therefore, inhibition of GAB1 PH domain functions may prevent the recruitment of GAB1 to the membrane and suppress cancer cell (e.g., breast cancer) proliferation and metastasis [10]. Herein, we attempt to identify novel small molecule inhibitors selectively targeting the PH domain of GAB1 and suggest that these small molecules exhibit high therapeutic potency for cancer treatment.
Unfortunately, no three-dimensional (3D) structure is available to date for GAB1 PH domain or any PH domain in complex with drug-like small molecules. Challenges remain for accurate structural prediction due to its low sequence identity (<30%) to other PH domains with known structures [11]. However, the core β-sandwich fold among PH domains is conserved [11], making it possible to construct a reliable homology model structure of GAB1 PH domain. Here, based on the position-site specific matrixes (PSSM) obtained from all non-redundant PH domain structures, we performed fold recognition and homology modeling, followed by intensive structural refinement. The resulted model was then applied to high-throughput virtual screening of our unique collection of over five million drug and lead-like compounds with our in-house drug discovery workflow ( Fig. 1 ) [12]. Upon biological evaluation, five out of the initially tested 20 hits exhibited positive activities to form direct binding to GAB1 PH domain, inhibit GAB1 Y627 phosphorylation and suppress breast cancer cell proliferation with low micromolar IC50. As is known, triple negative breast cancers are more aggressive with poor prognosis and difficult to treat clinically [13], but our inhibitors showed high potency against these malicious cells. Therefore, this study validates the effectiveness of our in silico platform for drug discovery, and demonstrates that targeting the PH domain of GAB1 provides a promising and novel therapeutic strategy for cancer treatment.
Figure 1. Structure-based drug discovery workflow.

All of the available PH domain 3D structures in the PDB were used to build our PSSM scoring functions which were employed to construct the homology model of GAB1 PH domain. The derived structure was used to perform high-throughput virtual screening of five million drug/lead-like compounds. Through a funnel-like process 20 hits were selected for experimental testing and five were confirmed as consistently active in all of the assays.
Results
Fold recognition and sequence alignment
PH domains are unique due to their conserved secondary structures and 3D folds, all with seven β-sheets and a C-terminal helix. However, the pairwise sequence identities among different PH domains are usually below 30%, and the loop regions are hypervariable in length and amino acid sequence [11]. Herein, we collected all available 34 non-redundant crystal structures of PH domains from Protein Data Bank (PDB) [14] and performed secondary structure-based sequence alignment using STRAP [15]. From the sequence alignment, we generated PSSMs for β1, β2, β3, β6, β7 and α1 (presented as sequence logos in S1 Fig.) to guide secondary structure prediction of new PH domain (e.g., GAB1). As no reliable PSSMs for β4 and β5 were generated due to low sequence similarity, we used PSIPRED server [16] to predict these two β-sheets.
S1 Fig. shows the sequence logos derived from the collected 34 PH domains, in which the size of residue indicates the relative frequency of that residue at the corresponding position. As expected, we found that most conserved residues are in the hydrophobic cores of PH domains. The residues responsible for phosphoinositide binding are generally located at β1[7], β2[2], β2[5], β3[4], β3[+1] and β7 [1] (the number in the brackets indicates the residue position at the secondary structure element). Predominantly, they are basic residues such as lysine and arginine. We combined these observations with PSSM and PSIPRED to predict the secondary structure of GAB1 PH domain, and found the predicted structure preserves a typical β-sandwich fold where C8-K14, W26-L33, V44-Y48, R58-D61, Q66-G71, I84-N88, and R92-V97 form the respective seven β-sheets, while E101-I114 forms the C-terminal α-helix ( Fig. 2 ). However, the GAB1 PH domain is unique with: 1) a long β1,2 loop landmarked by the conserved K14 and W26, similar to myosin X (PDB ID: 3TFM [17]); 2) a long β2,3 loop, similar to IRS1 (PDB ID: 1QQG [18]); 3) a long β5,6 loop, similar to TAPP1 (PDB ID: 1EAZ [19]); 4) the highest sequence identity of active-site residues (except for β1,2 loop region) to DAPP1 (PDB ID: 1FAO [20]) (shadowed residues in Fig. 2 ). Therefore, we have chosen the above four proteins as the templates for the following-up homology modeling studies.
Figure 2. Sequence alignment of the PH domains.

IRS1 (PDB ID: 1QQG), TAPP1 (PDB ID: 1EAZ), Myosin X (PDB ID: 3TFM) and DAPP1 (PDB ID: 1FAO). The secondary structure of the generated GAB1 homology model and the crystal structures of the templates are illustrated, using α for α-helix, β for β-sheet. Except for the highly variable β1,2 loop regions, the active site residues directly interacting with PtdIns(3,4,5)P3 are highlighted with shadows.
Homology modeling and structural optimization with molecular dynamics
We constructed 1,000 homology models of GAB1 PH domain in complex with inositol-tetrakisphosphate (IP4) based on the X-ray crystal structures of four aforementioned templates. After loop refinement and molecular dynamics (MD) simulation, we selected one reliable model in which IP4 binds stably to GAB1 PH domain with a minor fluctuation of phosphates (RMSF<1.1 Å), shown in S2 Fig. The simulation of this model reached the equilibrium after 5 ns, as judged by the RMSD of all of the backbone atoms (C, CA and N) (S2 Fig.). Large fluctuations of the Cα atoms were only observed in the β1,2, β2,3 and β5,6 loops (S2 Fig.). The quality of the lowest-energy model was assessed by QMEAN [21], ProSA [22] and PROCHECK [23]. The Ramachandran plot showed reasonable backbone dihedral angles: 92.2% of the residues were in the most favored regions, and eight residues in the additional or generously allowed regions. Both the ProSA Z-score (−4.04) and QMEAN Z-score (−0.13) of final model were within the range as typically seen for the native proteins of the similar size (S3 Fig.). In addition, the DOPE per-residue profile demonstrated a significant decrease in the DOPE scores at the β2,3 loop, β4,5 loop, β5, β5,6 loop and β6 for the refined structure compared with the initial homology model (S4 Fig., and homology model coordinate file is available at http://imdlab.org/supporting/PLOSCompBio).
As illustrated by Fig. 3A , the 3D model of GAB1 PH domain maintained the conserved β-sandwich folding. Similar to other Group 1 PH domains (e.g., Grp1 [20] and Btk [24]), the phosphoinositide-binding site of GAB1 was surrounded by the β1,2, β3,4 and β6,7 loops. The 2-hydroxyl group of IP4 oriented towards the β1,2 loop, and the 3,4,5-phosphates intensively interacted with the aforementioned basic residues in the β1, β2, β4 and β7. Particularly, K19 and R23 in the β1,2 loop formed hydrogen bonds with 5-P and 1-P, respectively ( Fig. 3B ). This explains why GAB1 PH domain specifically binds to PtdIns(3,4,5)P3 but not PtdIns(3,4)P2 or PtdIns(4,5)P2. Strikingly, the sequence motif NKKEFE in the β5,6 loop folded into an additional α-helix, as we termed α′. This additional α-helix also occurs in phospholipase Cδ PH domain (PDB ID: 1MAI [25]), and it interacts with W26, F79 and Y95 in the β1,2 loop via hydrogen bonding networks and hydrophobic interactions ( Fig. 3C ). This α′-helix was likely to stabilize the IP4-bound conformation of β1,2 loop, as W26A or W26C mutation impairs the PtdIns(3,4,5)P3 binding [8]. Furthermore, the motif SPP in the β1,2 loop formed intensive vdW interactions with the β7 and inositol scaffold ( Fig. 3C ). Finally, GAB1 PH domain had an extra hydrophobic region (later defined as Region II) due to the smaller side chains of those hydrophobic residues around β6,7 loop compared to IRS1. All these specific structural features intrinsically offered possibility of designing selective inhibitors against GAB1 over other PH domains, as further discussed in the ligand-induced conformational changes section.
Figure 3. The 3D model of GAB1 PH domain.

A. The overall structure of IP4-bound GAB1 PH domain. The protein secondary structure is shown in ribbons and IP4 is shown in sticks. B. A close view of the IP4 binding site. The critical residues for IP4 binding are labeled, and IP4 is in black stick with position labeled. The hydrogen bonds are illustrated with dashes. C. Interactions between α′-helix and β5,6 loop. The residues mediating the interactions are highlighted with sticks and hydrogen bonds are illustrated with dashes.
In silico hit identification
To identify novel inhibitors of GAB1 PH domain, we performed structure-based virtual screening using our MD-refined structural model. Additionally, a protein-based pharmacophore filter was derived using GRID method to select those high-throughput virtual screening hits of which the docked poses matched the pharmacophores [26], [27]. Residues K14, R23, K27, R29, R58 and R92 were identified as the residues that favorably interact with hydrogen bond acceptors, whereas Y47, F94 and I60 were specified as the preferential areas for hydrophobic moieties (S5 Fig.). The residues responsible for PtdIns(3,4,5)P3 binding were predicted to be K14, K27, R29, Y47, K49, R58 and R92, consistent to the mutagenesis studies [6], [8]. These critical residues were employed to define the protein pharmacophores to select docking poses of those 10,000 top ranked hits (only based on docking scores ranging 43.47–101.39) from the virtual screening of over five million compounds of our in-house collection. The resulted 2,783 hits were subjected to cluster analysis based on their chemical diversity (Tanimoto coefficient <0.65), and we obtained 268 clusters and selected the best-scored hits from each cluster ( Fig. 1 ). Upon visualizing their molecular interactions with the GAB1 PH domain, we chose 20 hits as listed in Table 1 and S1 Table.
Table 1. Biochemical and biological activities of hits.
| KD or Ki (µM) | % pGAB1 inhibition | IC50 (µM) | ||||
| ID | GAB1 | IRS1 | AKT1 | MDA-MB-231 | T47D | |
| GAB-001 | 9.38±1.60* | NB | ND | 55 | 40.3±1.3 | 23.7±2.7 |
| GAB-002 | 2.1±0.1 | 16.4±4.3 | ND | 32 | 142.7±11.6 | 137.6±6.9 |
| GAB-003 | 2.7±0.8 | 3.3±1.5 | ND | 14 | 85.2±7.2 | 13.9±0.6 |
| GAB-004 | 2.2±1.1 | NB | ND | 81 | 66.6±4.6 | 19.4±2.9 |
| GAB-005 | NB | NB | ND | 6 | IA | IA |
| GAB-006 | NB | NB | ND | 16 | ND | 100.8±1.8 |
| GAB-007 | 42.3±8.9 | 1.6±0.2 | ND | 53 | 4.6±0.7 | 20.1±4.3 |
| GAB-008 | NB | 16.2±1.7 | ND | 8 | IA | 43.2±3.2 |
| GAB-009 | NB | NB | ND | 38 | 162.9±6.0 | 97.6±6.1 |
| GAB-010 | NB | 0.12±0.02 | ND | 92 | 66.0±4.4 | 23.8±4.2 |
| GAB-011 | NB | NB | ND | 24 | IA | IA |
| GAB-012 | NB | NB | 4.58±1.72 | 43 | IA | 119.2±3.6 |
| GAB-013 | NB | NB | 6.27±1.16 | 27 | IA | IA |
| GAB-014 | NB | NB | NB | 42 | 69.9±0.1** | 177.5±3.9 |
| GAB-015 | 13.1±1.5* | 0.05 | 5.0±0.4* | 77 | 71.7±1.6 | 125.4±5.4 |
| GAB-016 | 0.9±0.1 | 0.15 | 4.3±0.1* | 96 | 79.0±1.1 | 45.9±1.6 |
| GAB-017 | 0.68±0.03 | 0.05 | 18.9±1.2* | 82 | 40.6±0.8 | 41.4±2.7 |
| GAB-018 | 36.6±5.0* | NB | 6.0±1.0* | 71 | 85.3±1.5 | 185.4±2.5 |
| GAB-019 | ND | ND | ND | 91 | ND | 39.4±6.2 |
| GAB-020 | 1.33±4.5 | ND | 2.4±0.6* | 87 | ND | 68.2±5.5 |
| DPIEL | 2.8 | ND | 5.04±0.48 | 72 | ND | 30.6±4.3 |
| IP4 | ND | ND | 3.08±0.49 | ND | ND | ND |
*Ki measurement.
**MDA-MB-468 cell line used.
ND: Not Determined. NB: No Binding. IA: Inactive.
Biological evaluation of identified hits
To validate our in silico identified hits, we performed three types of experimental assays to evaluate their bioactivities: direct binding to GAB1 PH domain, inhibition of Y627 phosphorylation of GAB1, and cytotoxicity IC50 in triple negative MDA-MB-231 and T47D human breast cancer cells. Our experiments revealed that 10 out of 20 hits demonstrated submicromolar to micromolar binding affinity (<50 µM) to GAB1 PH domain measured by surface plasmon resonance (SPR). Among them, GAB-001, GAB-004, GAB-007, GAB-016 and GAB-017 demonstrated promising bioactivity in the subsequent in vitro assays ( Table 1 , Fig. 4 and Fig. 5 ).
Figure 4. Inhibition of Y627 phosphorylation in GAB1.

No. 1-20 are GAB-001 to GAB-020, and No. 21 is DPIEL.
Figure 5. Cell proliferation assay for T47D and MDA-MB-231 breast cancer cell line and MCF-10A breast epithelial cell line.

GAB-001 exhibited selective binding to GAB1 (K i = 9.4±1.6 µM) (S6 Fig.), but not to IRS1 PH domain. In addition, it inhibited Y627 phosphorylation and killed breast cancer cells at IC50 = 23.7±2.7 µM (for T47D). GAB-004 achieved similar binding selectivity as GAB-001, but had a stronger inhibition of pY627 (81%) and a lower IC50 (19.4±2.9 µM). Interestingly GAB-007 demonstrated weak binding (K D = 42.3±8.9 µM) and mild pY627 inhibition (53%), but it showed high cytotoxicity in both MDA-MB-231 (IC50 = 4.6±0.7 µM) and T47D (IC50 = 20.1±4.3 µM) cell lines ( Fig. 5 ), probably due to other off-target mechanisms. GAB-016 and GAB-017 are N-(1,3,4-thiadiazol-2-yl)benzenesulfonamide derivatives which were previously synthesized through our search for AKT PH domain inhibitors [28]. They demonstrated nanomolar binding affinity for GAB1 PH domain (S6 Fig.), and were 5-fold and 28-fold more selective to the GAB1 than AKT, respectively. Consistent to their high binding affinities, GAB-016 and GAB-017 also inhibited over 80% of Y627 phosphorylation.
GAB1 targeted tumor-specific cytotoxicity
All the aforementioned active inhibitors showed potent cytotoxicity to cancer cell lines (T47D and MDA-MB-231). More excitingly the cytotoxicity is specific to cancer cells as the inhibitors exhibit little inhibition in the non-cancer MCF-10A breast cell line ( Fig. 5 ). Expectedly, as GAB1 and IRS1 pathways are intertwined [29], some inhibitors could suppress IRS1 phosphorylation (S7 Fig.). In addition, some compounds that selectively bind AKT PH domain (e.g., GAB-012, GAB-013 and GAB-018) did not effectively kill MDA-MB-231 or T47D breast cancer cell lines at 50 µM ( Table 1 ).
MD simulation of protein-ligand complexes and binding free energy calculation
To further investigate the structural mechanisms of our inhibitors to interact with the GAB1 PH domain, we performed MD simulations of the protein-inhibitor complexes (listed in Table 2 ). As expected, the active compounds (GAB-001, GAB-004, GAB-007, GAB-016 and GAB-017) demonstrated stable bindings to GAB1 PH domain in three independent simulations (RMSD<2.5 Å), whereas GAB-002 and GAB-003 dissociated with the protein after around 25 ns (S8 Fig.). In addition, MD simulations showed that GAB-007, GAB-010 and GAB-016 could form stable binding to IRS1 PH domain (S8 Fig.), consistent to the SPR results in Table 1 .
Table 2. Computation of the absolute binding free energy using PMF-based routine.
| GAB1 | |||||
| Component | GAB-001 | GAB-004 | GAB-007 | GAB-016 | GAB-017 |
| ΔGc,site (kcal/mol) | 1.14 | 0.98 | 0.92 | 1.42 | 1.31 |
| ΔGc,bulk (kcal/mol) | 1.27 | 1.00 | 1.09 | 1.52 | 1.65 |
| ΔGo,bulk (kcal/mol) | 1.04 | 0.9 | 0.85 | 0.98 | 0.67 |
| S* (Å2) | 6.27×103 | 8.74×103 | 9.94×103 | 7.00×103 | 7.91×103 |
| I* (Å) | 8.52×105 | 3.76×105 | 1.53×104 | 1.32×107 | 3.43×106 |
| ΔGbind (kcal/mol) | −7.76 | −7.72 | −5.79 | −9.55 | −8.89 |
| Exp. ΔGbind (kcal/mol) | −6.91 | −7.78 | −6.01 | −8.31 | −8.47 |
To add another layer of validation of the binding modes predicted by MD simulations, we calculated the absolute binding free energies of our inhibitors to GAB1/IRS1 PH domain using an in-house potential of mean force (PMF) method, which aims to circumvent the insufficient sampling issue by introducing hypothetical intermediate states representing the association pathway of ligand from the unbound “bulk” regions to the ligand-binding “site” (S9 Fig.). The principle of this approach has been described elsewhere [30], [31]. Here, we implemented this method using ff99SB force field [32]. Briefly, the umbrella sampling and weighted histogram analysis were used as the primary tools to derive two sets of PMF: ligand conformational PMF w(ξ) and protein-ligand separation PMF w(r). The details of mathematical calculations were available in S1 Method, and the w(ξ) and w(r) plots for eight protein-ligand complexes were available in S10 Fig. and S11 Fig. As indicated by Fig. 6 and Table 2 , the predicted absolute binding free energies via PMF method were in a good agreement with the experimental values (RMSE = 0.64 kcal/mol, R2 = 0.85). One may notice that these predictions encompassed two different PH domain targets (GAB1 and IRS1) and a variety of ligand chemotypes. The good correlation between experimental binding free energies and predicted free energies implied that the predicted inhibitor binding modes by MD simulations were accurate.
Figure 6. The correlation between the predicted binding free energies (PMF method) and the experimental ones.

The grey line indicates the calculated correlation between predicted and experimental binding free energies, and the black line indicates the ideal correlation. The number beside each point is the corresponding inhibitor ID.
Ligand-induced conformational changes of PH domain
We have generated eight reliable PH domain-inhibitor complex models from MD simulations (listed in Table 2 ) which have been validated by the PMF absolute binding free energy calculations as described in the previous section. When comparing the bound and unbound protein structures, we observed for the first time the ligand-induced conformational changes in three regions around the phoshpoinositide-binding pocket (termed as Region I, Region II and Region III) for both GAB1 and IRS1 PH domains.
The Region I is comprised of the conserved K14GAB1/K21IRS1 (β1[7]), K27GAB1/K21IRS1 (β2[2]), Y47GAB1/Y46IRS1 (β3[4]) and F94GAB1/F93IRS1 (β7[3]) ( Fig. 7A–E ). The MD simulations showed significant conformational changes in Region I (RMSD>2 Å) for both GAB1 and IRS1, as illustrated by the RMSD analysis (red plots in Fig. 7A ). The side chain rearrangement of these residues, especially K14GAB1/K21IRS1 and Y47GAB1/Y46IRS1, created a pocket which favorably binds an aromatic moiety connecting with a H-bond acceptor group (Movie S1, S2, S3 available at http://imdlab.org/supporting/PLOSCompBio). This moiety could form cation-π and hydrophobic interactions with the surrounding K14GAB1/K21IRS1 and F94GAB1/F93IRS1, respectively ( Fig. 7B–E ). All inhibitors we identified in this study contain such a pharmacophore (phenylthiazole in GAB-004, phenylisoxazole in GAB-010, S-phenyl carbothioate in GAB-007, benzenesulfone in GAB-001, GAB-016 and GAB-017) (S12 Fig.). We had mentioned that β1[7], β2[2] and β3[4] were the PIP3-binding residues, thus Region I conformational changes were attributable to the activities of the inhibitors. Generally, the conformational changes of Region I residues in GAB1 were more substantial than IRS1, except F94GAB1 ( Fig. 7A ). In comparison, GAB-010 could induce an alternative conformation of F93IRS1 ( Fig. 7E ), which also occurred in ArhGAP9 crystal structure (PDB ID: 2P0D [33]). The function of conformational change of F93IRS1 is likely to further open the pocket to accommodate larger moiety such as phenylisoxazole (GAB-010), as other IRS1 Region I residues were less flexible.
Figure 7. Inhibitor-induced conformational changes for PH domain structures.

A. Heavy atom RMSDs for the residues with conformational changes. RMSD of each residue is calculated from the snapshots taken from three independent MD simulations and compared with the unbound control structure. W107GAB1 and W106IRS1 (in black) are used as the controls for no conformational change. Red: Region I residues; Blue: Region II residues; Magenta: Region III residues. “..” for p<0.1; “*” for p<0.05; “**” for p<0.01. B–E. The critical residues in the three regions are shown in sticks. Pink: the protein in unbound state; Green: the protein in inhibitor-bound state. The inhibitors are depicted with gray sticks and surfaces. The arrows illustrate large conformational changes. B. GAB1 PH domain in complex with GAB-001; C. GAB1 PH domain in complex with GAB-017; D. IRS1 PH domain in complex with GAB-007; E. IRS1 PH domain in complex with GAB-010.
The Region II is formed by β4, β6,7 loop and the first several amino acids of β7 ( Fig. 7A–E ). The key residues are R58GAB1/R62IRS1 (β4[2]) and R92GAB1/E91IRS1 (β7[1]). Compared with Region I residues, more significant conformational changes were observed in the Region II residues in the GAB1 PH domain (RMSD>2.5 Å) (blue plots in Fig. 7A ). These conformational changes created a new pocket which binds aliphatic (GAB-016 and GAB-017) and aromatic moieties (e.g., chlorobenzothiophene in GAB-001 and furan in GAB-004). Remarkably, we found that the bulky aromatic moieties (GAB-001 and GAB-004) generally induced more movement of GAB1 Region II residues than the aliphatic moieties (GAB-016 and GAB-017) (blue plots in Fig. 7A ). We also observed that significant conformational changes only occurred in GAB1, but not in IRS1 PH domain (blue plots in Fig. 7A ), probably because the electrostatic attraction between R62IRS1 and E91IRS1 significantly restrained the fluctuation of these two residues (Movie S1 available at http://imdlab.org/supporting/PLOSCompBio), while the electrostatic repelling between R58GAB1 and R92GAB1 made these two residues more flexible (Movie S3). These findings imply that the flexibility of Region II residues of PH domain may correlate the size of binding group.
The Region III is located on the solvent-accessible side of the β7, especially I92GAB1 or H92IRS1 (β7[3]) ( Fig. 7A–E ). When GAB-010 binds IRS1 PH domain, the benzimidazole moiety induced a significant side chain movement of H92 (RMSD = 3.39 Å) as compared with unbound form (magenta plots on the right in Fig. 7A and Movie S3 available at http://imdlab.org/supporting/PLOSCompBio). In contrast, this region in GAB1 PH domain did not exhibit significant conformational changes when binding any inhibitor (magenta plots on the left in Fig. 7A ). Upon comparison of GAB1 and IRS1 PH domain sequences, we speculated that the accessibility of Region III was affected by the length of β1,2 loop: GAB1 PH domain has a longer β1,2 loop than IRS1 ( Fig. 2 ), and the residues P16 and P17 forms intensive vdW interactions with β7 ( Fig. 2C ), which would in turn block the access of inhibitors to Region III. This explains the selective binding of GAB-010 to IRS1, but not GAB1 PH domain.
Discussion
GAB1 is a critical protein in cellular signaling, and its PH domain has been suggested as an attractive target for various cancer treatments [34], [35]. However, the absence of its 3D structure makes it challenging for structure-based drug discovery. We herein present a rigorously designed workflow for inhibitor identification by integrating various techniques ranging from structural bioinformatics, homology modeling, ligand-steered refinement, molecular dynamics, and virtual screening, followed by experiment evaluation with biochemical/biophysical and cellular assays. With our integrated protocol, we have successfully identified several selective inhibitors targeting the GAB1 PH domain and they are selective to breast cancer cells. This discovery offers us a great starting point to target this critical protein for cancer treatment, particularly for the triple negative breast cancer.
Our results also showed that the triple-negative breast cancer cell line, MDA-MB-231, was more resistant to GAB1 inhibitors than ER-positive breast cancer cell line, T47D ( Table 1 ). It has been reported that MDA-MB-231, but not T47D, has mutations on GAB1 downstream proteins, such as KRas and BRaf mutations [36]. Since KRas and BRaf mutations are known to reduce the dependency on the upstream activators, such as EGFR [37], it was not surprising that MDA-MB-231 was more resistance to GAB1 inhibitors. Strikingly, we observed a concomitant inhibition of pGAB1 and pIRS1 by either GAB1-specific or IRS1-specific inhibitors (S7 Fig.). This could be due to the crosstalk between c-Met and α6β4 integrin pathway [29], which couples the phosphorylation of GAB1 and IRS1 upon HGF stimulation. These observations may bring us new insights of combined PH domain-targeted cancer therapeutic strategies. Further mechanistic studies are ongoing to investigate these hypotheses.
While it is exciting to see that we have identified selective inhibitors of the GAB1 PH domain using our unique computation-experimentation integrated platform, we have to admit that some of the other hits also bind to multiple PH domains (e.g., IRS1 and AKT1), as demonstrated by Table 1 . For example, GAB-001 and GAB-004 are selectively inhibitor GAB1, but GAB-016 and GAB-017 are pan inhibitors against GAB1, IRS1 and AKT1. More follow-up experiments also showed that GAB-016 targets GAB2 PH domain as well. This is not surprising because PH domain is defined by their common β-sandwich structure. In addition, GAB1 and GAB2 PH domains are highly homologous (76% sequence identity), and IRS1 is one of the templates used in our homology modeling to build the 3D structure of GAB1 PH domain. Of note, all GAB1-selective or IRS1-selective inhibitors showed much better IC50 against T47D and MDA-MB-231 breast cancer cell lines than the non-tumorigenic MCF-10A cell line ( Fig. 5 ). More intriguingly, we also observed that AKT1-selective inhibitors (e.g., GAB-012 and GAB-013) were toxic to MCF-10A at 100 µM, but not for T47D and MDA-MB-231 at the same concentration (data not shown). This may imply that targeting GAB1 or IRS1, but not AKT1, might be a better targeted strategy for breast cancer treatment.
Although PH domains have been intensively studied as cancer target for drug discovery, to date there are no available protein structures in complex with any drug-like small molecules. As mentioned, this has significantly limited the structure-based drug discovery efforts. In the present work, we utilized several inhibitors to investigate the dynamics of GAB1 PH domain and evaluate their selectivity in potential cancer cell inhibition. Interestingly, we found that the apo-structure of the PH domain protein could undergo large conformational changes in three regions to accommodate different inhibitors. The side-chain conformations of the residues in Region I determines the binding of either multiple electronegative groups (e.g., the multiple phosphates in IP4) or an aromatic moiety conjugated with a group containing H-bond acceptors (e.g., benzenesulfone), as shown in Fig. 7 . The accessibility of Region II and Region III depend on several critical amino acids on β4 and β7 and the length of β1,2 loop, respectively. The selectivity of PH domain inhibitors may be designed based on our modeling of the protein structures. For instance, GAB-010 is highly selective to IRS1 but no binding to GAB1 or AKT1, largely due to the short β1,2 loop. The knowledge that GAB1 PH domain undergoes conformational change upon ligand binding provides novel insights of guiding the future structure-based drug design efforts, and of course more experimental validation will increase our understanding of GAB1 structure and functions.
Materials and Methods
Chemical dataset
A collection of five million drug and lead-like compounds which were curated from various sources (e.g., PubChem [38] and MayBridge) was used for virtual screening. LigPrep [39] was employed for ligand preparation, including the removal of salts, assignment of appropriate protonation, tautomerization and ring conformations, and generation of 3D structures by energy minimization with OPLS2001 force field [40]. Additionally, an internal collection of 167 previously synthesized inhibitors targeting AKT PH domain were included for virtual screening.
PSSM and sequence logo representation
A total of 65 high-resolution crystal structures of PH domains were obtained from PDB, and we curated 34 non-redundant proteins. Their PDB IDs are listed in S2 Table. They were used for secondary structure-based sequence alignment with STRAP [15]. We extracted the multiple sequence alignments for β1, β2, β3, β6, β7 and α1 secondary structural fragments. The individual alignment will be used as input to PSI-BLAST [41] which could generate a PSSM for each individual fragment as shown in S3 Table. These PSSMs can be represented by WebLogo for more intuitive visualization and understanding (S1 Fig.). These figures were generated using the WebLogo server [42].
3D structure prediction of GAB1 PH domain in complex with IP4
The sequence of GAB1 PH domain was retrieved from UniProt database (accession number Q13480) [43]. The secondary structure was predicted by PSSM combined with PSIPRED [16] and aligned to the templates (myosin X (PDB ID: 3TFM [17]), IRS1 (PDB ID: 1QQG [18]), TAPP1 (PDB ID: 1EAZ [19]) and DAPP1 (PDB ID: 1FAO [20]) for homology modeling. To improve the quality of homology modeling, we manually corrected the multiple sequence alignment generated by ClustalX to ensure each secondary structure elements (e.g., α-helix and β-sheets) were properly aligned. GAB1 PH domain homology models were built using MODELLER 9v10 [44]. As the active site residues of DAPP1 have the highest homology to those of GAB1 PH domain, the coordinates of the IP4 co-crystallized with DAPP1 was used as the initial structure. We generated initial 1,000 homology models. Since the lysine-rich loop β1,2 is important for phosphoinositide binding, especially for Group 1 PH domain [7], [45], the β1,2 loop of top ten initial models (evaluated by DOPE score) were subjected to ligand-steered refinement using built-in function of MODELLER. We selected five models out of the 100 generated loop models based on the overall DOPE scores [46], Ramachandran plot, and the consistencies to IP4 binding site features [24], [25], [47]–[51] and the reported mutagenesis studies [6], [8]. These five GAB1-IP4 complex models were refined by MD simulations using AMBER10 available at Texas Advanced Computing Center. All MD simulations were performed in triplicates with different initial velocities. The MD simulations were performed using ff99SB force field [32] in TIP3P explicit solvent with particle mesh Ewald (PME), periodic boundary conditions and SHAKE. The topology and charges of the ligand were generated by Antechamber with AM1-BCC charges [52]. The system is solvated and neutralized in the cuboid box in which the closest distance between any atom originally in solute and the edge of the box is 12 Å. The system was equilibrated for 100 ps, and the production MD simulations were run in NPT ensemble for 20 ns, with the time step = 2 fs. The snapshots were taken every 1 ps. The root mean square deviation (RMSD) relative to the first frame and the root mean square fluctuation (RMSF) relative to the average structure were analyzed with cpptraj implemented in AmberTools12 [52]. The average structures were minimized, and the model quality was evaluated by QMEAN [21], ProSA [22] and PROCHECK [23]. A reasonable protein model should have both ProSA and QMEAN Z-scores within the range for the native proteins of similar size, as illustrated by S3 Fig.
Virtual screening
GOLD 5.1 [53] was employed for virtual screening on our high performance computing cluster using the GAB1-IP4 complex model derived above. Molecular docking was performed with flexible side chains of the residues involved in IP4 binding, and the conformation with the best score of each compound was ranked based on their ChemPLP scores. Protein pharmacophore modeling was performed using GRID v22c [54]. Briefly, the GRID calculations were performed with a grid box enclosing the target with 1 Å beyond each dimension. During the calculations, the GRID directive Move was set (MOVE = 1) to allow the flexibility of the side chains. The molecular interaction fields (MIFs) [55] were computed to determine the interaction between the receptor atoms and three different probes: the hydrophobic (DRY), the amide nitrogen (N1, H bond donor), and the carbonyl oxygen (O, H bond acceptor). Via visual inspection of the local minima of the GRID energy maps, the favorable binding sites of these three probes were used to define the features of a pharmacophore query. The derived pharmacophores were used to evaluate the binding poses of the initially selected 10,000 hits out of the five million compounds. If the docked hit poses fit the pharmacophore, they would be selected and subjected to clustering analysis based on the MACCS fingerprints and Tanimoto coefficient. The best scored compound from each cluster was chosen and the binding poses of these hits were individually inspected based on molecular visualization.
3D structure refinement of GAB1/IRS1 PH domain in complex with inhibitors
In order to evaluate the selectivity of our inhibitors, we optimized the also PH domain (IRS1 or GAB1) -inhibitor complex structures using MD simulations. The starting conformation for MD simulation is the binding mode which obtained the best score in molecular docking. The MD simulations were performed in triplicates for 50 ns using the parameters described in “3D structure prediction of GAB1 PH domain in complex with IP4” section. We also generated the trajectory of GAB1-GAB-001 complex (Movie S1), GAB1-GAB-017 (Movie S2) and IRS1-GAB-010 (Movie S3). Each trajectory contained 1,000 snapshots which were taken every 50 ps. The ligands and the critical residues were in sticks, whereas the backbones of PH domain proteins were in ribbons. Starting from the docking conformation, these MD trajectories vividly demonstrated the conformational changes of the PH domain proteins upon ligand binding. The movies were available at http://imdlab.org/supporting/PLOSCompBio.
PMF-based binding free energy calculation
The routine of PMF-based computation of protein-ligand absolute binding free energy has been previously described [30], [31]. Briefly, the average structure of protein-ligand complex obtained from three independent 50 ns MD simulations was subject to energy minimization to remove clashes. The resulted structure was considered as the reference frame to define the position and orientation constraints. The PMF as a function of mass-weighted RMSD (ξ) relative to the reference ligand or the protein-ligand distance (r) was sampled by umbrella sampling and weighted-histogram analysis method (WHAM). The full description of this method is available in S1 Method. The experimental binding free energies were derived from experimental K
D (or K
i) using the equation 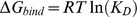 or
or 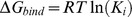 .
.
Surface plasmon resonance (SPR) spectroscopy binding assays
The DNA sequences of human GAB1 and IRS1 PH domain (IRS1 is for selectivity evaluation) were cloned into pGEX-4T1 inducible bacterial expression plasmid (GeneStorm, Invitrogen, Carlsbad, CA) transformed into BL21 (DE3) E. Coli. Expression and purification of the recombinant proteins were performed as previously described [51]. Binding assays were performed using a Biacore 2000 instrument with the Biacore Control Software v3.2 and BIAevaluation v4.1 analysis software (Biacore, Piscataway, NJ) as previously described [51]. Briefly, the PH domain GST-fusion proteins were immobilized on a CM5 Sensorchip (Biacore BR-1000-12) using Biacore's Amine Coupling Kit (Biacore BR-1000-50) to a level of 10,000 Response units (RUs). Small molecule analytes at concentrations ranging from one tenth to ten times the predicted K D were injected at a high flow rate (30 µL/min). Dimethylsulfoxide (DMSO) concentrations in all samples and running buffer were 1% (v/v) or less. For the competitive binding assays and K i determination, PtdIns(3,4,5)P3-biotin labeled liposomes (Echelon Biosciences, Salt Lake City, UT) and SA chips were used with increasing concentrations of the compound tested. We did triplicate SPR assays for each concentration.
Cellular proliferation assay
Two human breast cancer cell lines and one normal breast cell line were used for this study: T47D ductal breast epithelial tumor cell line, MDA-MB-231 epithelial tumor cell line and MCF-10A non-tumorigenic epithelial cell line (American Type Culture Collection, Rockville, MD). T47D and MDA-MB-231 cells were maintained in bulk culture in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 4.5 g/L glucose, 100 U/mL penicillin and 100 mg/mL streptomycin in a 5% CO2 atmosphere. MCF-10A cells were maintained in MEGM with other conditions same as the cancer cell lines. Cells were passaged using 0.25% trypsin and 0.02% EDTA. Cells were confirmed to be mycoplasma free by testing them with an ELISA kit (Roche-Boehringer Mannheim, Indianapolis, IN). Our hit compounds were freshly prepared in DMSO at a stock concentration of 10 mM. For the evaluation of cellular proliferation, a standard 96-well micro-cytotoxicity assay was performed as described in reference [51]. Briefly, the assay was set up by plating cells at 5,000–10,000 cells per well (depending on cell doubling time) for a growth period of 4 days. The identified hits were added directly to the media, dissolved in DMSO at various concentrations ranging from 1 to 200 µM. The endpoint was spectrophotometric determination of the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide. All assays were performed in triplicates.
Inhibition of GAB1 and IRS1 phosphorylation
For all biological assays, hit compounds were added at 20 µM concentration directly into the culture media of the cells for 4 hr following a 16 hr incubation of T47D cells without FBS. Cells were stimulated with HGF for 20 min at 50 ng/ml. Following this treatment, cells were lysed as previously published [51] and equal amounts of total cell lysate were loaded on a pSer312-IRS-1/Total IRS-1 Meso Scale Discovery plate as described by the manufacturer. The plate was read using a Sector Imager 2400A instrument (Meso Scale Discovery protein profiling system, Gaithersburg, MD). For the measurement of GAB1 phosphorylation, T47D cells were treated as for the phosphorylation of IRS1 evaluation. Cell lysates were run on a 7% SDS-PAGE and membrane were probed with specific anti-phospho-Tyr627 GAB1 (Cell signaling). Each experiment was performed at least three times.
Supporting Information
Sequence logos for PH domain. Position-specific logos for the secondary structure elements of PH domain (generated by Weblogo server). β4 and β5 are not included because of the high variation within these two β-sheets. The size of residue indicates the relative frequency of the residue at the corresponding position.
(PDF)
Backbone RMSD and per-residue backbone RMSF for GAB1-IP4 complex. The simulation lasted for 20 ns. The mass-weighted average RMSFs were calculated for each residue based on backbone atoms. Atomic RMSFs of four phosphates in IP4 were shown in the last table.
(PDF)
Quality of GAB1 PH domain model. This model was subjected to energy minimization prior to quality assessment. (A) QMEAN; (B) ProSA; (C); Ramachandran plot and summary generated by PROCHECK.
(PDF)
Comparison of DOPE score profiles. IRS1 (PDB ID: 1QQG), GAB1 homology model and the lowest energy GAB1 model with MD simulation refinement were compared. The MD refinement model was subject to 1000 steps energy minimization with ff99SB force field to remove transient structural defects. The secondary structure is annotated at the bottom.
(PDF)
Isovolume of GAB1 PH domain generated by GRID. The area favoring H-bond acceptor (−6.0 kcal/mol) was displayed in magenta surface, whereas area favoring hydrophobic (−2.0 kcal/mol) probes was displayed in green surface. The protein backbone is illustrated with cyan lines, and the ligand (IP4) is in blue sticks. The crucial residues are labeled around isovolume surface.
(PDF)
SPR of GAB-001, GAB-016 binding to GAB1 PH domain.
(PDF)
Inhibition of S312 phosphorylation of IRS1. The percentage of inhibition values are calculated from three separated experiments.
(PDF)
Backbone RMSD of GAB1PH/IRS1PH – inhibitor complex.
(PDF)
Schematic representation of frame of reference used to define the position and orientation restraints. To reduce the degrees of freedom, P1, P2, P3 and L1, L2, L3 were employed to represent the coordinates of the protein and the ligand, respectively, as described in S1 Method .
(PDF)
Ligand conformational PMF as a function of mass-weighted RMSD (ξ). The ligand in the bulk is in black lines, whereas the ligand in the active site is in read lines. (A–E). PMFs derived from GAB1-inhibitor complexes. (F–H). PMFs derived from IRS1-inhibitor complexes.
(PDF)
Separation PMF as a function of L1-P1 distance (r). (A–E). PMFs derived from GAB1-inhibitor complexes. (F–H). PMFs derived from IRS1-inhibitor complexes.
(PDF)
Pharmacophore of the inhibitors. Green: hydrophobic or aromatic region. Blue: the projection of H-bond acceptor.
(PDF)
Docking scores for 20 htis. The hits which are consistently active in three assays are labeled with bold IDs.
(PDF)
The crystal structures employed to generate PSSM.
(PDF)
PSSM profile for PH domain (β1, β2, β3, β6, β7 and α1).
(PDF)
Technical details for computational modeling.
(PDF)
Acknowledgments
Special thanks to OpenEye Scientific Software and ChemAxon for their free academic licenses. We also thank the HPC resources from Texas Advanced Computing Center (TACC) and the University of Texas M.D. Anderson Cancer Center.
Funding Statement
This work was partially supported by the NIH 5R01CA138702-03 and the University Cancer Foundation via the Institutional Research Grant program at the University of Texas M.D. Anderson Cancer Center. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Gu H, Neel BG (2003) The Gab in signal transduction. Trends in Cell Biology 13: 122–130. [DOI] [PubMed] [Google Scholar]
- 2. Holgado-Madruga M, Emlet DR, Moscatello DK, Godwin AK, Wong AJ (1996) A Grb2-associated docking protein in EGF- and insulin-receptor signalling. Nature 379: 560–564. [DOI] [PubMed] [Google Scholar]
- 3. Gillgrass A, Cardiff RD, Sharan N, Kannan S, Muller WJ (2003) Epidermal growth factor receptor-dependent activation of Gab1 is involved in ErbB-2-mediated mammary tumor progression. Oncogene 22: 9151–9155. [DOI] [PubMed] [Google Scholar]
- 4. Wohrle FU, Daly RJ, Brummer T (2009) Function, regulation and pathological roles of the Gab/DOS docking proteins. Cell Commun Signal 7: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nishida K, Hirano T (2003) The role of Gab family scaffolding adapter proteins in the signal transduction of cytokine and growth factor receptors. Cancer Science 94: 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Holgado-Madruga M, Moscatello DK, Emlet DR, Dieterich R, Wong AJ (1997) Grb2-associated binder-1 mediates phosphatidylinositol 3-kinase activation and the promotion of cell survival by nerve growth factor. Proceedings of the National Academy of Sciences of the United States of America 94: 12419–12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hurley JH, Misra S (2000) Signaling and subcellular targeting by membrane-binding domains. Annu Rev Biophys Biomol Struct 29: 49–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maroun CR, Moscatello DK, Naujokas MA, Holgado-Madruga M, Wong AJ, et al. (1999) A conserved inositol phospholipid binding site within the pleckstrin homology domain of the Gab1 docking protein is required for epithelial morphogenesis. Journal of Biological Chemistry 274: 31719–31726. [DOI] [PubMed] [Google Scholar]
- 9. Eulenfeld R, Schaper F (2009) A new mechanism for the regulation of Gab1 recruitment to the plasma membrane. Journal of Cell Science 122: 55–64. [DOI] [PubMed] [Google Scholar]
- 10. Ren Y, Wu J (2003) Simultaneous suppression of Erk and Akt/PKB activation by a Gab1 pleckstrin homology (PH) domain decoy. Anticancer Res 23: 3231–3236. [PubMed] [Google Scholar]
- 11. Lemmon MA, Ferguson KM, Abrams CS (2002) Pleckstrin homology domains and the cytoskeleton. FEBS Lett 513: 71–76. [DOI] [PubMed] [Google Scholar]
- 12. Chan CH, Morrow JK, Li CF, Gao Y, Jin G, et al. (2013) Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 154: 556–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Foulkes WD, Smith IE, Reis-Filho JS (2010) Triple-negative breast cancer. N Engl J Med 363: 1938–1948. [DOI] [PubMed] [Google Scholar]
- 14. Bernstein FC, Koetzle TF, Williams GJ, Meyer EF Jr, Brice MD, et al. (1977) The Protein Data Bank: a computer-based archival file for macromolecular structures. J Mol Biol 112: 535–542. [DOI] [PubMed] [Google Scholar]
- 15. Gille C, Frommel C (2001) STRAP: editor for STRuctural Alignments of Proteins. Bioinformatics 17: 377–378. [DOI] [PubMed] [Google Scholar]
- 16. McGuffin LJ, Bryson K, Jones DT (2000) The PSIPRED protein structure prediction server. Bioinformatics 16: 404–405. [DOI] [PubMed] [Google Scholar]
- 17. Lu Q, Yu J, Yan J, Wei Z, Zhang M (2011) Structural basis of the myosin X PH1(N)-PH2-PH1(C) tandem as a specific and acute cellular PI(3,4,5)P(3) sensor. Mol Biol Cell 22: 4268–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dhe-Paganon S, Ottinger EA, Nolte RT, Eck MJ, Shoelson SE (1999) Crystal structure of the pleckstrin homology-phosphotyrosine binding (PH-PTB) targeting region of insulin receptor substrate 1. Proceedings of the National Academy of Sciences of the United States of America 96: 8378–8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thomas CC, Dowler S, Deak M, Alessi DR, van Aalten DM (2001) Crystal structure of the phosphatidylinositol 3,4-bisphosphate-binding pleckstrin homology (PH) domain of tandem PH-domain-containing protein 1 (TAPP1): molecular basis of lipid specificity. Biochem J 358: 287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ferguson KM, Kavran JM, Sankaran VG, Fournier E, Isakoff SJ, et al. (2000) Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Mol Cell 6: 373–384. [DOI] [PubMed] [Google Scholar]
- 21. Benkert P, Kunzli M, Schwede T (2009) QMEAN server for protein model quality estimation. Nucleic Acids Res 37: W510–W514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35: W407–W410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26: 283–291. [Google Scholar]
- 24. Baraldi E, Carugo KD, Hyvonen M, Lo Surdo P, Riley AM, et al. (1999) Structure of the PH domain from Bruton's tyrosine kinase in complex with inositol 1,3,4,5-tetrakisphosphate. Structure (London) 7: 449–460. [DOI] [PubMed] [Google Scholar]
- 25. Ferguson KM, Lemmon MA, Schlessinger J, Sigler PB (1995) Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell 83: 1037–1046. [DOI] [PubMed] [Google Scholar]
- 26. Du-Cuny L, Chen L, Zhang S (2011) A critical assessment of combined ligand- and structure-based approaches to HERG channel blocker modeling. J Chem Inf Model 51: 2948–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morrow JK, Du-Cuny L, Chen L, Meuillet EJ, Mash EA, et al. (2011) Recent development of anticancer therapeutics targeting Akt. Recent Pat Anticancer Drug Discov 6: 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ahad AM, Song ZH, Du-Cuny L, Moses SA, Zhou LL, et al. (2011) Development of sulfonamide AKT PH domain inhibitors. Bioorganic & Medicinal Chemistry 19: 2046–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shaw LM (2001) Identification of insulin receptor substrate 1 (IRS-1) and IRS-2 as signaling intermediates in the alpha6beta4 integrin-dependent activation of phosphoinositide 3-OH kinase and promotion of invasion. Mol Cell Biol 21: 5082–5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lau AY, Roux B (2011) The hidden energetics of ligand binding and activation in a glutamate receptor. Nature Structural & Molecular Biology 18: 283–U262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Woo HJ, Roux B (2005) Calculation of absolute protein-ligand binding free energy from computer simulations. Proceedings of the National Academy of Sciences of the United States of America 102: 6825–6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Onufriev A, Bashford D, Case DA (2004) Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 55: 383–394. [DOI] [PubMed] [Google Scholar]
- 33. Ceccarelli DF, Blasutig IM, Goudreault M, Li Z, Ruston J, et al. (2007) Non-canonical interaction of phosphoinositides with pleckstrin homology domains of Tiam1 and ArhGAP9. J Biol Chem 282: 13864–13874. [DOI] [PubMed] [Google Scholar]
- 34. Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, et al. (2007) The genomic landscapes of human breast and colorectal cancers. Science 318: 1108–1113. [DOI] [PubMed] [Google Scholar]
- 35. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, et al. (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316: 1039–1043. [DOI] [PubMed] [Google Scholar]
- 36. Hollestelle A, Elstrodt F, Nagel JH, Kallemeijn WW, Schutte M (2007) Phosphatidylinositol-3-OH kinase or RAS pathway mutations in human breast cancer cell lines. Mol Cancer Res 5: 195–201. [DOI] [PubMed] [Google Scholar]
- 37. Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, et al. (2012) Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst 104: 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bolton EE, Wang Y, Thiessen PA, Bryant SH (2008) Chapter 12 PubChem: Integrated Platform of Small Molecules and Biological Activities. In: Ralph AW, David, editors. Annual Reports in Computational Chemistry: Elsevier. pp. 217–241.
- 39.(2010) Suite 2010: LigPrep. 2.4 ed. New York, NY: Schrodinger, LLC.
- 40. Jorgensen WL, Maxwell DS, TiradoRives J (1996) Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. Journal of the American Chemical Society 118: 11225–11236. [Google Scholar]
- 41. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schneider TD, Stephens RM (1990) Sequence logos: a new way to display consensus sequences. Nucleic Acids Res 18: 6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Uniprot C (2012) Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Research 40: D71–D75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sali A, Blundell TL (1993) Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol 234: 779–815. [DOI] [PubMed] [Google Scholar]
- 45. DiNitto JP, Lambright DG (2006) Membrane and juxtamembrane targeting by PH and PTB domains. Biochim Biophys Acta, Mol Cell Biol Lipids 1761: 850–867. [DOI] [PubMed] [Google Scholar]
- 46. Shen MY, Sali A (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci 15: 2507–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Du-Cuny L, Song Z, Moses S, Powis G, Mash EA, et al. (2009) Computational modeling of novel inhibitors targeting the Akt pleckstrin homology domain. Bioorg Med Chem 17: 6983–6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lietzke SE, Bose S, Cronin T, Klarlund J, Chawla A, et al. (2000) Structural basis of 3-phosphoinositide recognition by pleckstrin homology domains. Mol Cell 6: 385–394. [DOI] [PubMed] [Google Scholar]
- 49. Mahadevan D, Powis G, Mash EA, George B, Gokhale VM, et al. (2008) Discovery of a novel class of AKT pleckstrin homology domain inhibitors. Mol Cancer Ther 7: 2621–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Meuillet EJ, Mahadevan D, Vankayalapati H, Berggren M, Williams R, et al. (2003) Specific inhibition of the Akt1 pleckstrin homology domain by D-3-deoxy-phosphatidyl-myo-inositol analogues. Mol Cancer Ther 2: 389–399. [PubMed] [Google Scholar]
- 51. Moses S, Ali MA, Zuohe S, Du-Cuny L, Zhou LL, et al. (2009) In Vitro and In Vivo Activity of a Novel Small Molecule Inhibitor Targeting the Pleckstrin Homology Domain of Protein Kinase B/AKT. Cancer Research 69: 5073–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, et al. (2012) AMBER 12.
- 53.(2007) GOLD 5.1. 5.1 ed. Cambridge, UK: CCDC.
- 54.(2004) GRID v22c. Perugia, Italy: Molecular Discovery Ltd.
- 55. Baroni M, Cruciani G, Sciabola S, Perruccio F, Mason JS (2007) A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for Ligands and Proteins (FLAP): theory and application. J Chem Inf Model 47: 279–294. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequence logos for PH domain. Position-specific logos for the secondary structure elements of PH domain (generated by Weblogo server). β4 and β5 are not included because of the high variation within these two β-sheets. The size of residue indicates the relative frequency of the residue at the corresponding position.
(PDF)
Backbone RMSD and per-residue backbone RMSF for GAB1-IP4 complex. The simulation lasted for 20 ns. The mass-weighted average RMSFs were calculated for each residue based on backbone atoms. Atomic RMSFs of four phosphates in IP4 were shown in the last table.
(PDF)
Quality of GAB1 PH domain model. This model was subjected to energy minimization prior to quality assessment. (A) QMEAN; (B) ProSA; (C); Ramachandran plot and summary generated by PROCHECK.
(PDF)
Comparison of DOPE score profiles. IRS1 (PDB ID: 1QQG), GAB1 homology model and the lowest energy GAB1 model with MD simulation refinement were compared. The MD refinement model was subject to 1000 steps energy minimization with ff99SB force field to remove transient structural defects. The secondary structure is annotated at the bottom.
(PDF)
Isovolume of GAB1 PH domain generated by GRID. The area favoring H-bond acceptor (−6.0 kcal/mol) was displayed in magenta surface, whereas area favoring hydrophobic (−2.0 kcal/mol) probes was displayed in green surface. The protein backbone is illustrated with cyan lines, and the ligand (IP4) is in blue sticks. The crucial residues are labeled around isovolume surface.
(PDF)
SPR of GAB-001, GAB-016 binding to GAB1 PH domain.
(PDF)
Inhibition of S312 phosphorylation of IRS1. The percentage of inhibition values are calculated from three separated experiments.
(PDF)
Backbone RMSD of GAB1PH/IRS1PH – inhibitor complex.
(PDF)
Schematic representation of frame of reference used to define the position and orientation restraints. To reduce the degrees of freedom, P1, P2, P3 and L1, L2, L3 were employed to represent the coordinates of the protein and the ligand, respectively, as described in S1 Method .
(PDF)
Ligand conformational PMF as a function of mass-weighted RMSD (ξ). The ligand in the bulk is in black lines, whereas the ligand in the active site is in read lines. (A–E). PMFs derived from GAB1-inhibitor complexes. (F–H). PMFs derived from IRS1-inhibitor complexes.
(PDF)
Separation PMF as a function of L1-P1 distance (r). (A–E). PMFs derived from GAB1-inhibitor complexes. (F–H). PMFs derived from IRS1-inhibitor complexes.
(PDF)
Pharmacophore of the inhibitors. Green: hydrophobic or aromatic region. Blue: the projection of H-bond acceptor.
(PDF)
Docking scores for 20 htis. The hits which are consistently active in three assays are labeled with bold IDs.
(PDF)
The crystal structures employed to generate PSSM.
(PDF)
PSSM profile for PH domain (β1, β2, β3, β6, β7 and α1).
(PDF)
Technical details for computational modeling.
(PDF)