

Abstract
Background
A high-throughput, sensitive, specific, mass spectrometry-based method for quantitating estrone (E1), estradiol (E2), and testosterone (T) in postmenopausal human serum has been developed for clinical research. The method consumes 100 ul human serum for each measurement (triplicates consume 300 ul) and does not require derivatization. We adapted a commercially available 96-well plate for sample preparation, extraction, and introduction into the mass spectrometer on a single platform.
Methods
Steroid extraction from serum samples and mass spectrometer operational parameters were optimized for analysis of estradiol and subsequently applied to other analytes. In addition to determining the limit of detection (LOD) and limit of quantitation (LOQ) from standard curves, a serum LOQ (sLOQ) was determined by addition of known steroid quantities to serum samples. Mass spectrometric method quantitative data were compared to results using a state-of-the-art ELISA (enzyme-linked immunosorbent assay) using stored serum samples from menopausal women.
Results
The LOD, LOQ, sLOQ was (0.1 pg, 0.3 pg, 1 pg/ml) for estrone, (0.3 pg, 1 pg, 3 pg/ml) for estradiol, and (0.3 pg, 1 pg, 30pg/ml) for testosterone, respectively. Mass spectrometry accurately determined concentrations of E2 that could not be quantified by immunochemical methods. E1 concentrations measured by mass spectrometry were in all cases significantly lower than the ELISA measurements, suggesting immunoreactive contaminants in serum may interfere with ELISA. The testosterone measurements broadly agreed with each other in that both techniques could differentiate between low, medium and high serum levels.
Conclusions
We have developed and validated a scalable, sensitive assay for trace quantitation of E1, E2 and T in human serum samples in a single assay using sample preparation method and stable isotope dilution mass spectrometry.
Keywords: estradiol, estrone, estriol, testosterone, progesterone, tandem mass spectrometry
Introduction
The estrogens (estrone E1, estradiol E2, and estriol E3), testosterone (T), and progesterone (P4) are steroid hormones with numerous, characterized functions in adults, where the steroid concentrations are relatively abundant and can be routinely measured [1] . The biology of steroids at lower concentrations is less understood, primarily because the methods to quantitate steroids in low abundance are insufficiently accurate, specific, sensitive, or reproducible [2, 3] . Peri- or postmenopausal women and geriatric men constitute two growing populations whose quality of life can likely be improved by assessment of levels of the estrogens, testosterone and perhaps progesterone, below current reliable detection limits [4]. The menopausal transition can negatively impact women’s lives both psychologically (irritability, anxiety, insomnia, mood imbalance, and depression [4, 5] and physically (hot flashes, metabolic syndrome and osteoporosis [4, 6] . Elderly men also experience a decline in their physical stature and cognitive function, which are thought to be caused by an age-related decrease in testosterone production [7] . Similar issues related to accurate sex steroid measurement complicate pediatric diagnosis and therapy for abnormalities of pubertal maturation, because the cardinal sex steroids are often at or below currently available detection limits in these individuals [2].
For hormone measurement at relatively low circulating concentrations, traditional immunoassays such as ELISAs (enzyme-linked immunosorbent assay) suffer from nonspecific antibody interactions, inconsistent reproducibility, inadequate sensitivity, and require separate assays for each compound of interest [8]. Alternately, a stable isotope dilution mass spectrometry based method directly analyzes the steroid of interest for unambiguous identification and quantitation of multiple hormones in a single sample. The stable isotope standard normalizes for sample loss during preparative steps, indicates an elution time to compensate for any chromatographic drift and can highlight the presence of isobaric contaminants with identical mass transitions by comparing the ratio of several mass transitions [9]. Furthermore, this method is compatible with time-course studies with frequent, small volume, blood sampling and with studies that address the low steroid concentration biology.
We sought to develop a mass spectrometry-based method to quantitate the sex steroids E1, E2, E3, P4 and T primarily for postmenopausal clinical studies and secondarily for elderly men and children. The elderly populations have low concentrations of the above mentioned sex steroids while the sample volume drawn from children is small. A volume and sensitivity constraint of 100 µl and 1 pg/ml for each steroid was established as a goal for method development. We chose a non-derivatized approach because derivatization introduces an additional source of variation, increases sample preparation time and cost, and requires multiple chemical derivatives if the compounds of interest have different functional groups. Moreover, it has been documented that derivatization conditions can hydrolyze some estrogens resulting in erroneous measurements [8, 10]. We also sought to implement a reproducible sample preparation method amenable to the considerable number of samples utilized in clinical studies.
While there are a number of excellent mass spectrometry based assays, none were sufficient to overcome the constraints listed above. A sampling of the literature for current E2 mass spectrometry based quantitative methods reveals current methods are quite sensitive but require chemical derivatization [11, 12] , larger sample volume requirement [13–15], or sample preparation methods not amenable to the number of samples in clinical studies.
We present herein the development of a readily scalable sample preparation technique along with a sensitive and reproducible stable isotope dilution tandem mass spectrometry based method to quantitate E2, E1, E3, and T in postmenopausal serum without the use of derivatization reagents. In addition to the four steroids listed above, we also analyzed P4 but excluded it from our final assay because of an unidentified, isobaric contaminant with identical mass transitions to those used for quantitating P4. Our assay consumes little serum (100 µl per measurement) and utilizes a commercially available 96-well plate.
Materials and Methods
Reagents and Solvents
Estrone, 17-β-estradiol, estriol, progesterone, testosterone, methanol, acetonitrile, isopropyl alcohol, and sodium hydroxide were of the highest grade commercially available from Sigma-Aldrich (St. Louis, MO). The following stable isotopes were purchased from Cambridge Isotope Laboratories (Andover, MA): [D4]-estradiol (2,4,16,16, 95–97 atom% [D4]), [D4]-estriol (2,4,16,17, 98 atom% [D4]), [D4]-estrone (2,4,16,16, 97 atom% [D4]), and [D9]-progesterone (2,2,4,6,6,17A,21,21,21, 98 atom% [D9]). [13C3]-Testosterone (2, 3, 4 99 atom% [13C3]) was purchased from Cerilliant (Round Rock, TX). Stable isotope structures listed in Supplementary Table 1.
LC-MS/MS
Two Shimadzu UFLCXR 50326 LC-20AD pumps (Kyoto, Japan) and a Leap Technologies PAL HTC-xt Sample Handler (Carrboro, North Carolina) were coupled to an AB Sciex Triple Quad QTRAP 5500 ESI-LC-MS/MS mass spectrometer (Framingham, MA) for all experiments. The mobile phase consisted of (A) water and (B) methanol at a flow rate of 200 µl/min; 10% NH4OH in acetonitrile was added post-column via 25 ml syringe pump at a flow-rate of 6 µl/min. Post column addition of the ammonium hydroxide ionizing solution (pH 13) was used to preserve the column’s functionality per manufacturer’s recommendations for pH range (pH 1.5–8.5 under gradient conditions). LC parameters were as follows: 0–1 min 60% B, 1.10–5 min 70–75% B, 5.10–10 min 85% B, 10.10–14 min 95% B, 14.10–17.10 min 60% B. The stationary phase employed was a C18 column (Phenomenex Kinetex 2.6µ C18 100Å pore size, 100×3.0mm) protected by a C18 guard cartridge (Phenomenex).
Initial MS experiments were used to optimize solvent and instrument conditions for abundant precursor ions. MS/MS experiments were used to determine the mass transitions used for quantitative MRM (multiple reaction monitoring) experiments; we utilized both ESI+ (testosterone and progesterone) and ESI− (estradiol, estrone and estriol) (Table 1). Polarity switching allowed for the detection of both positive and negative ions in the same analytical experiment. The variables that contributed to the greatest gains in sensitivity were nebulizer gas flow rate (35 psi), turbo drying gas temperature and flow rate for solvent evaporation (400 °C, 45 psi), and collision energy (25 and −55 V). Other instrument parameters that were optimized included: electrospray ion source (4500 V and −4500 V), declustering potential (40 and −100 V), entrance potential (10 and −10 V), collision gas (medium), collision cell exit potential (13 and - 18 V), quadrupole resolution (Q1 high, Q3 high), and settling time (50.0 milliseconds).
Table 1.
Mass Spectrometry Method Characteristics
| Analyte | Mass Transitions (Quantifier/Qualifier) m/z |
LC tR (min)c |
Ext. Rec. (%)d |
LOD (pg)e |
LOQ (pg)e |
Within Assay Var. (%)f |
Bet. Assay Var. (%)f |
Serum LOQ (sLOQ) (pg/ml)g,i |
CoV at sLOQ (%) |
|---|---|---|---|---|---|---|---|---|---|
| E3a | 287.2 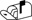 171.1/145.1 171.1/145.1 |
4.7 | 96 | 1.0 | 1.0 | 6.5 | 3.0 | 30.0 | 5.9 |
| [D4]-E3 | 291.2 173.1/147.1 |
||||||||
| E1a | 269.2 145.1/183.1 |
7.1 | 86 | 0.1 | 0.3 | 6.9 | 4.5 | 1.0 | 4.3 |
| [D4]-E1 | 273.2 147.1/187.1 |
||||||||
| E2a | 271.2 183.1/145.1 |
7.3 | 96 | 0.3 | 1.0 | 8.2 | 3.0 | 3.0 | 3.4 |
| [D4]-E2 | 275.2 187.1/147.1 |
||||||||
| Tb | 289.2 97.1/109.1 |
7.6 | 82 | 0.3 | 1.0 | 7.2 | 3.5 | 30.0 | 8.5 |
| [13C3]-T | 292.2 100.1/112.1 |
||||||||
| P4b | 315.2 97.1/109.1 |
9.0 | 50 | 0.3 | 1.0 | 7.3 | 2.9 | N/Ah | N/Ah |
| [D9]-P4 | 324.2 100.1/113.1 |
||||||||
Estrone, estradiol, and estriol are analyzed as negative ions.
Testosterone and progesterone are analyzed as positive ions.
Liquid chromatography retention time.
Extraction recovery.
Limit of Detection (LOD) and Quantitation (LOQ) calculated from standard curve (methanol solvent).
Calculated from standard curves (within assay variability n = 5, between assay variability n = 3).
LOQ and variability calculated from addition of standards to unaltered serum samples (n = 3).
Cannot determine due to chemical interference (see Figure 1)
To convert E3 (pg/ml) to pM, multiply by 3.47, E2: 3.67, E1: 3.72, T: 3.47, P4: 3.18.
Standards Preparation
The standard curve calibrants were carefully prepared by initially weighing the steroids and dissolving them in MeOH. The concentration of these standards were double checked by utilizing the unique extinction coefficients of each steroid; we utilized a spectrophotometer (HP 8452A Diode Array Spectrophotometer, Palo Alto, CA) and the extinction coefficients of E2 (280 nm ε 103.28), E1 (280 nm ε 103.49), T (239 nm ε 104.23), P (240 nm ε 104.25) [16] and E3 (280 nm ε 103.4) [17] . The standard curves all have a R2 greater than 0.999 (Supplementary Figure 3). Standards were prepared and then stored at −20°C.
To determine the purity of each stable isotope standard, atom percent excess MS experiments were performed and resulted in no detectable [D0] or [12C] peaks for any of the analytes (data not shown). To account for the potential isobaric interference of estrone isotopes (m/z 269) with estradiol (m/z 271), we utilized the combination of liquid-chromatography retention time (7.1 min for E1, 7.3 min for E2) and mass transition ratio (m/z 271→183 and m/z 271→145 are nearly identical in area) to monitor for interference of estrone with estradiol (Supplementary Figure 2). We did not detect any discernible difference in the mass transition ratio of an estradiol standard and estradiol in serum (Figure 1).
Figure 1.

Data comparison highlighting the mass spectrometric method’s criteria. A quantifiable compound (E2) and a compound of interest (P4) with a confounding co-eluting compound are used as examples to demonstrate the method’s ability to accurately quantitate steroids of interest. All spectra are from LC-MS/MS MRM of E2 and P4. A Representative spectra illustrating the two mass transitions for qualifying and quantitating E2 and B the stable isotope [D4]-E2. C Endogenous E2 in postmenopausal serum at a higher concentration (83 pg/ml) and D at a lower concentration (9 pg/ml). E Representative MRM spectra of mass transitions used to qualify and quantitate P4 and F [D9]-P4. G Representative spectra for the P4 transitions m/z 315→97/109 in serum samples. Notice the multiple mass transitions that co-elute with P4 at 9 minutes. The ratio of the two mass transitions was drastically different than the standards in E and resulted in exclusion of P4 because it could not be differentiated from the co-eluting compound(s).
Sample Preparation and Extraction
Blood samples were collected from human subjects, stored at 4 °C overnight, and centrifuged at 4 °C (22,000 RCF). Serum was pipetted into cryo-tubes and stored at −80 °C. All of the postmenopausal samples used in this report were taken from women aged 45–57 in good general health, with a BMI of less than 35 kg/m2, who were nonsmokers not taking any exogenous hormones. All had had at least 6 months and no more than 3 years of amenorrhea, consistent with menopause. They were being screened for enrollment into the Kronos Early Estrogen Prevention Study (KEEPS) [18] and, at the time of screening, donated an extra tube of blood for future research. This use of the blood specimens was considered exempt research and was registered with the University of Colorado’s IRB (COMIRB).
Human serum (100 µl), labeled standards (100 pg in 25 µl methanol) and 5% isopropyl alcohol in water (175 µl) were pipetted into wells of a 2 ml, 96 square-well plate (Biotage, Uppsala, Sweden), covered with a 96-square silicone sealing mat (Phenomenex Torrance, CA), vortexed, placed into a sonication bath with a lead weight on top such that the plate sits on the bottom of the bath with water up to approximately half the height of the plate, and then sonicated for 10 min. Preconditioned samples from each well were transferred into a 96-well SLE+ plate (Supported Liquid Extraction, Biotage Isolute SLE+ 400 µl) and allowed to absorb into the matrix for approximately 5 minutes after a pulse of vacuum to help penetrate the top frit. The steroids were eluted with 4×400 µl aliquots of dichloromethane under gravity; following the last addition, a vacuum manifold (Biotage VacMaster 96) was used for the final elution. The combined eluent was dried down in the 96-well collection plate, resuspended in 100 µl of 40% methanol in water, and injected (90 µl) directly from the 96-well plate for mass spectrometry analysis.
The extraction efficiency was calculated from the recovery of stable isotope standards before and after the extraction process (Table 1). Our extraction method was constrained by several important operational parameters: each measurement could not employ more than 100 µl starting serum, applied no derivatization, and the method had to be amenable to high-throughput conditions. The variables that significantly enhanced our extraction recovery of estradiol were the use of a supported-liquid-extraction (SLE+, Biotage) step over a liquid-liquid based approach, the use of a relatively high concentration of internal standard that also served as a carrier standard, the use of isopropyl alcohol instead of methanol or ethanol during sample incubation, the use of sonication to increase mixing of the unlabeled and labeled standards in serum, and the use of multiple dichloromethane solvent elutions instead of methyl tert-butyl ether, 1:1 ethyl acetate:isooctane, or ethyl acetate. The final extraction method resulted in sample preparation, extraction, and introduction into the mass spectrometer all in a 96-well plate format.
Limit of Detection (LOD), Limit of Quantitation (LOQ), and Variability Determination
The LOD for each analyte was determined by measuring the signal to noise (s/n) ratio of our standards; a s/n of 3 was the minimally accepted value. The limit of quantitation for each analyte was determined two ways: as a statistical measure from repeated measurements of the standard curve with less than 20% variability [19], and as a practical determination spiking known amounts of analytes into serum samples. An increase in signal corresponding to the added amount of analyte was used to determine this serum limit of quantitation (sLOQ).
Values for the coefficient of variability were calculated for standards (E3, E1, E2, T, and P4) at the values used for determination of standard curves (0.1, 0.3, 1.0, 3.0, 10.0, 30.0 and 100 pg/100 µl) with 5 replicate samples prepared for each concentration. The ‘within assay variability’ was determined as the average of the coefficients of variability at concentrations with measured values above the limit of quantitation in this analysis (CoV < 20 %) [19]. These measurements were repeated on three different dates and the ‘between assay variability’ was determined by averaging measured values for the compounds studied at each concentration prepared. Coefficients of variability were determined at each concentration, and these were averaged within the range of values above the limit of quantitation.
ELISA
ELISA assays were performed using Siemens and LDN Company (Nordhorn, Germany) reagents, including standards, on a Siemens Advia Centaur (Erlangen, Germany) per manufactures’ instructions. The LOQ for the assays are 12–3000 pg/ml (E2, Siemens), 100–15,000 pg/ml (T, Siemens) and 15–2000 pg/ml (E1, LDN). The inter-assay and intra-assay coefficients of variation for the ELISA method routinely employed in clinical studies were as follows: E2 (10.6%, 3.7%), E1 (11.7%, 6.4%), and T (6.2%, 1.6%).
Results and Discussion
We demonstrate the successful development of a high-throughput sample preparation method coupled to a sensitive mass spectrometry based analytical method. The limit of detection (LOD) for each compound was determined from the same unlabeled standards used in our standard curve with a minimum signal-to-noise ratio of 3 on the quantifier mass transition (Table 1). The limit of quantitation (LOQ) was determined by statistical measures based on quantities from the standard curve, and by the addition of unlabeled standards to postmenopausal serum samples with subsequent extraction and quantitation. In this more realistic determination, the LOQ was determined from detection of the lowest standard added to serum greater than the standard deviation (data summarized in Table 1). It should be noted that the negative ion analysis for E1 and E2 had much less chemical/electronic noise than the positive ion based analysis of T during serum-based LOQ determination experiments. The LOQ based on statistics from the standard curves for all three is similar (0.3 pg E1, 1.0 pg E2, 1.0 pg T) when standards are used but differ significantly when serum is employed for LOQ determination (1 pg/ml E1, 3 pg/ml E2, 30 pg/ml T) . This highlights the importance of our sLOQ determination instead of the more commonly utilized charcoal stripped serum [11, 13] ; matrix can be variable from batch to batch, dramatically affecting the data acquired, whereas solvent is much more consistent.
The specificity of the mass spectrometry assay derives from directly measuring the analyte of interest. The isolated precursor ion is collisionally dissociated into diagnostic product ions in a tandem mass spectrometry experiment. Usually the two most abundant product ions are used for qualifying and quantifying a compound; sometimes a less abundant product ion is utilized if the biological matrix has a competing mass transition. The ratio of these two mass transitions must remain the same between a standard and biological matrix; a difference indicates the presence of another compound with similar physical properties. The precursor and product ions of the three steroids quantitated in this assay have been well characterized [20–26], Supplementary Figure 1). The product ions for progesterone have not been rigorously pursued but are presumed to be similar to the structures suggested for testosterone’s product ions m/z 97 and m/z 109. It should also be noted that the mass transitions utilized in the mass spectrometry method were also verified in serum to determine the presence of a competing compound and select an alternate mass transition if necessary. We determined our LOD and LOQ in serum samples by a standard addition approach to more accurately reflect serum samples collected for clinical research.
Our method is compatible with measuring estriol and progesterone; however, we encountered issues with each. The postmenopausal serum samples had no detectable E3 present, consistent with E3 being abundant only during pregnancy [27]. Our method detected an abundant, serum-specific, unidentified compound with the same elution time (9.0 minutes) and selected ion transitions chosen for progesterone (m/z 315.2→97.1 and 109.1). The contaminant was identified because the ratio of the mass transition abundances observed from the serum samples significantly differed from the mass transitions of the progesterone standard (Figure 1). Thus, accurate and specific quantitation of progesterone was not possible. Although we did not further characterize the competing compound, we were able to make the decision to not quantify endogenous progesterone because the specific criteria established for this assay were not achieved. This is an important observation because there are numerous endogenous (e.g. other isobaric steroids and metabolites) and exogenous small molecules (e.g. prescription drugs and environmental endocrine disruptors) that could interfere with any assay, whether it is an immunochemical or mass spectrometry based assay.
Our extraction optimization centered on estradiol and results in high recoveries for the estrogens and testosterone (96% E2 and E2, 86% E1, 82% T) but was far less efficient with progesterone (50% P4) (Table 1). Our method was optimized for post-menopausal serum samples and, of note, did not use charcoal stripped serum; this was done to account for endogenous contaminants that would not be present in a charcoal stripped serum standard. We chose a commercially available, single platform, 96-well plate format for sample preparation, extraction, and instrument injection because it is amenable to automation facilitating future studies of larger population sizes and/or frequent blood sampling.
We compared our mass spectrometry based method to our in-house state-of-the-art ELISA assay to demonstrate the validity and capability of our method. We quantitated E1, E2, and T from previously collected postmenopausal serum samples in quintuplicate (MS) and duplicate (ELISA) (Table 2). We observed several key differences in quantitation between the MS and ELISA methods; this discrepancy agrees with previous reports [28]. The mass spectrometric method was able to detect E2 in all six serum samples but could only quantitate in three samples above the sLOQ (3 pg/ml). ELISA could only quantitate E2 in the most abundant sample.
Table 2.
Comparison of Mass Spectrometry and ELISA with Postmenopausal Serum Samples
| Serum Sample |
E2 (pg/mlc) | E1 (pg/mlc) | T (pg/mlc) | |||
|---|---|---|---|---|---|---|
| MSa | ELISAb | MSa | ELISAb | MSa | ELISAb | |
| 1 | 82.5 ± 2.1 | 67 | 58.6 ± 2.6 | 73 | 422.5 ± 14 | 431 |
| 2 | <3 | <11.8 | 7.9 ± 0.4 | 49 | 143.9 ± 8.0 | 100 |
| 3 | <3 | <11.8 | 8.8 ± 0.6 | 67 | 132.7 ± 5.2 | 130 |
| 4 | <3 | ND | 9.8 ± 0.6 | 48 | 137.3 ± 7.5 | 218 |
| 5 | 4.1 ± 0.6 | <11.8 | 6.9 ± 0.3 | 29 | 62.4 ± 2.8 | 109 |
| 6 | 7.5 ± 0.7 | <11.8 | 45.0 ± 1.3 | 67 | 63.3 ± 2.8 | 18 |
Concentration as determined by mass spectrometry, n = 5.
Average concentration as determined by ELISA, n = 2.
To convert E2 (pg/ml) to pM, multiply by 3.67, E1: 3.72, T: 3.47.
MS and ELISA based methods were able to quantitate E1 in all six samples, however, there was significant discrepancy in the reported values in that ELISA measurements for Samples 2–5 were at least four times higher than the mass spectrometry values reported.
Some inconsistencies between the MS and ELISA assay were observed for testosterone measurements. Both assays detected a relatively high level of testosterone in the first sample, 423 vs 431 pg/ml. The MS method indicated a similar concentration in serum samples #2–4 (143 pg/ml, 133 pg/ml, 137 pg/ml) whereas the ELISA method demonstrated much more variation (100 pg/ml, 130 pg/ml, 218 pg/ml). Serum samples 5 and 6 demonstrated significant discrepancies between the two methods (62 and 63 pg/ml with MS compared to 18 and 109 pg/ml with ELISA).
The mass spectrometry measurements between serum samples were similar in concentration for each steroid; estradiol and testosterone measurements from serum sample #1 were higher than the #2–6 but this was also reflected in the ELISA measurements. In these cases, we believe the mass spectrometry data to be more accurate because MS directly measures the analyte of interest and both the quantifier and qualifier transition must be detected in a specific ratio at the correct HPLC retention times. These simultaneous criteria ensure specific quantitation of an analyte. None of the postmenopausal women whose serum was assayed were taking exogenous hormones.
A sampling of current methods reveals a shortcoming in one or more of the requirements we set forth. Tai et al. [11] developed a NIST (National Institutes of Standards) recognized method that is very sensitive (0.6 pg), however, it requires derivatization with dansyl chloride and 3–5 ml of serum. Fiers et al. [13] also developed a sensitive assay (1 pg/ml) without derivatization but this method consumes 500 µl serum. Ray et al. [20] developed a sensitive assay to measure free Estradiol but it requires 250 µl serum per measurement, a 22 hour dialysis step and utilizes dansyl chloride derivatization.
The serum samples utilized reflected a small sampling of the postmenopausal population. A larger number of samples would have provided greater evidence of the repeatability of our methods and its suitability for high-throughput assessments. The small number of serum samples nevertheless demonstrates the feasibility of our method. The mass spectrometric portion of the method is sensitive enough to quantitate E2, E1, and T and generate meaningful clinical data [2]. The sample preparation method can accommodate up to 96 samples at a time, yielding a preparation method conducive to clinical studies.
The mass spectrometry method proposed was developed centered on the sensitivity and extraction recovery of estradiol from postmenopausal serum as being paramount to the other steroids; the low concentrations of estradiol in postmenopausal serum samples necessitated this approach. The result was a sensitive, reproducible, specific estradiol assay, which is compatible with estrone, testosterone, and estriol. The exclusion of progesterone, due to the mass transition ratio discrepancy between the standard and serum sample highlights the stringent requirements set forth to significantly reduce inaccurate measurements in our mass spectrometry assay. The use of a 96-well plate greatly expedited sample preparation compared to other methods considered. The scalable sample preparation method and sensitive, specific, reproducible mass spectrometry analysis are ideally suited for clinical research studies requiring quantification of E2, E1 and T in postmenopausal women, aging men and prepubertal juveniles.
Supplementary Material
Highlights.
-
-
We present a high-throughput sample preparation method for serum steroids.
-
-
We present a sensitive mass spectrometric method for quantitating steroids.
-
-
LOD, LOQ, sLOQ: E1 (0.1 pg, 0.3 pg, 1 pg/ml), E2 (0.3 pg, 1 pg, 3 pg/ml), T (0.3 pg, 1 pg, 30 pg/ml)).
-
-
Comparison between ELISA and LC-MS/MS with postmenopausal samples.
-
-
LC-MS/MS able to accurately and reproducibly quantitate samples ELISA cannot.
Acknowledgment
This work was supported in part by grants from the National Institutes of Health HD058155 (NS), RR027216 (RCM) and Lipid Maps Collaborative Grant GM069338 (RCM), and Colorado Clinical and Translational Sciences Institute RR025780.
Abbreviations, in order cited
- E1
estrone
- E2
estradiol
- T
testosterone
- µl
microliter
- LOD
limit of detection
- LOQ
limit of quantitation
- sLOQ
serum limit of quantitation
- ELISA
enzyme-linked immunosorbent assay
- pg
picogram
- ml
milliliter
- E3
estriol
- P4
progesterone (pregn-4-ene-3,20-dione)
- LC
liquid chromatography
- MS/MS
tandem mass spectrometry
- MS
mass spectrometry
- MRM
multiple reaction monitoring
- ε
extinction coefficient
- m/z
mass to charge ratio
- RCF
relative centrifugal force
- s/n
signal to noise ratio
- CoV
coefficient of variation
- NIST
National Institutes of Standards
- HPLC
high performance liquid chromatography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure summary (see Disclosure of Potential Conflict of Interest form for instructions): KMW, CAJ, JAH, JC, APB, RCM and NS have nothing to declare. SB is currently employed with Biotage.
References
- 1.Kushnir MM, Rockwood AL, Roberts WL, Yue B, Bergquist J, Meikle aW. Liquid chromatography tandem mass spectrometry for analysis of steroids in clinical laboratories. Clin. Biochem. 2011;44:77–88. doi: 10.1016/j.clinbiochem.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 2.Rosner W, Hankinson SE, Sluss PM, Vesper HW, Wierman ME. Challenges to the measurement of estradiol: an endocrine society position statement. J. Clin. Endocrinol. Metab. 2013;98:1376–1387. doi: 10.1210/jc.2012-3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosner W, Auchus RJ, Azziz R, Sluss PM, Raff H. Position statement: Utility, limitations, and pitfalls in measuring testosterone: an Endocrine Society position statement. J. Clin. Endocrinol. Metab. 2007;92:405–413. doi: 10.1210/jc.2006-1864. [DOI] [PubMed] [Google Scholar]
- 4.Weismiller DG. Menopause. Prim. Care. 2009;36:199–226. doi: 10.1016/j.pop.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 5.Bromberger JT, Kravitz HM. Mood and Menopause: Findings from the Study of Women’s Health Across the Nation (SWAN) over ten years. Obstet. Gynecol. Clin. North Am. 2012;38:609–625. doi: 10.1016/j.ogc.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Polotsky HN, Polotsky AJ. Metabolic implications of menopause. Semin. Reprod. Med. 2010;28:426–434. doi: 10.1055/s-0030-1262902. [DOI] [PubMed] [Google Scholar]
- 7.Maggio M, Dall’Aglio E, Lauretani F, Cattabiani C, Ceresini G, Caffarra P, Valenti G, Volpi R, Vignali A, Schiavi GCG. The hormonal pathway to cognitive impairment in older men. J. Nutr. Health Aging. 2012;16:40–54. doi: 10.1007/s12603-012-0002-7. [DOI] [PubMed] [Google Scholar]
- 8.Soldin SJ, Soldin OP. Steroid hormone analysis by tandem mass spectrometry. Clin. Chem. 2009;55:1061–1066. doi: 10.1373/clinchem.2007.100008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wooding KM, Auchus RJ. Mass spectrometry theory and application to adrenal diseases. Mol. Cell. Endocrinol. 2013;371:201–207. doi: 10.1016/j.mce.2012.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo T, Gu J, Soldin OP, Singh RJ, Soldin SJ. Rapid measurement of estrogens and their metabolites in human serum by liquid chromatography-tandem mass spectrometry without derivatization. Clin. Biochem. 2008;41:736–741. doi: 10.1016/j.clinbiochem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tai SS, Welch MJ. Development and Evaluation of a Reference Measurement Procedure for the Determination of Estradiol-17B in Human Serum Using Isotope-Dilution Liquid Chromatography-Tandem Mass Spectrometry. Anal. Chem. 2005;77:6359–6363. doi: 10.1021/ac050837i. [DOI] [PubMed] [Google Scholar]
- 12.Kushnir MM, Rockwood AL, Bergquist J, Varshavsky M, Roberts WL, Yue B, Bunker AM, Meikle, a W. High-sensitivity tandem mass spectrometry assay for serum estrone and estradiol. Am. J. Clin. Pathol. 2008;129:530–539. doi: 10.1309/LC03BHQ5XJPJYEKG. [DOI] [PubMed] [Google Scholar]
- 13.Fiers T, Casetta B, Bernaert B, Vandersypt E, Debock M, Kaufman J-M. Development of a highly sensitive method for the quantification of estrone and estradiol in serum by liquid chromatography tandem mass spectrometry without derivatization. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2012;893–894:57–62. doi: 10.1016/j.jchromb.2012.02.034. [DOI] [PubMed] [Google Scholar]
- 14.Harwood DT, Handelsman DJ. Development and validation of a sensitive liquid chromatography-tandem mass spectrometry assay to simultaneously measure androgens and estrogens in serum without derivatization. Clin. Chim. Acta. 2009;409:78–84. doi: 10.1016/j.cca.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 15.Pauwels S, Antonio L, Jans I, Lintermans A, Neven P, Claessens F, Decallonne B, Billen J, Vanderschueren D, Vermeersch P. Sensitive routine liquid chromatography-tandem mass spectrometry method for serum estradiol and estrone without derivatization. Anal. Bioanal. Chem. 2013;405:8569–8577. doi: 10.1007/s00216-013-7259-5. [DOI] [PubMed] [Google Scholar]
- 16.Rex MC, Dawson Daphne C, Elliott William H, Elliott KMJ. Data for Biochemical Research Third. Oxford: Clarendon Press; 1986. [Google Scholar]
- 17.Slaunwhite R, Engel L, Olmsted C. Extinction and of estrogens. J. Biol. Chem. 1951:627–631. [PubMed] [Google Scholar]
- 18.Miller VM, Black DM, Brinton Ea, Budoff MJ, Cedars MI, Hodis HN, Lobo Ra, Manson JE, Merriam GR, Naftolin F, Santoro N, Taylor HS, Harman SM. Using basic science to design a clinical trial: baseline characteristics of women enrolled in the Kronos Early Estrogen Prevention Study (KEEPS) J. Cardiovasc. Transl. Res. 2009;2:228–239. doi: 10.1007/s12265-009-9104-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shah VP, Midha KK, Dighe S, McGilveray IJ, Skelly JP, Yacobi A, Layloff T, Viswanathan CT, Cook CE, McDowall RD. Analytical methods validation: bioavailability, bioequivalence and pharmacokinetic studies. Conference report. Eur. J Drug Metab. Pharmacokinet. 1992;16:249–255. doi: 10.1007/BF03189968. [DOI] [PubMed] [Google Scholar]
- 20.Ray Ja, Kushnir MM, Bunker A, Rockwood AL, Meikle aW. Direct measurement of free estradiol in human serum by equilibrium dialysis-liquid chromatography-tandem mass spectrometry and reference intervals of free estradiol in women. Clin. Chim. Acta. 2012;413:1008–1014. doi: 10.1016/j.cca.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 21.Bourcier S, Poisson C. Elucidation of the decomposition pathways of protonated and deprotonated estrone ions: application to the identification of photolysis products. Rapid Commun. Mass Spectrom. 2010;24:2999–3010. doi: 10.1002/rcm.4722. [DOI] [PubMed] [Google Scholar]
- 22.Wooding KM, Barkley RM, Hankin JA, Johnson CA, Bradford AP, Santoro N, Murphy RC. Mechanism of Formation of the Major Estradiol Product Ions Following Collisional Activation of the Molecular Anion in a Tandem Quadrupole Mass Spectrometer. J. Am. Soc. Mass Spectrom. 2013:1451–1455. doi: 10.1007/s13361-013-0705-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Magi E, Scapolla C, Di Carro M, Liscio C. Determination of endocrine-disrupting compounds in drinking waters by fast liquid chromatography-tandem mass spectrometry. J. Mass Spectrom. 2010;45:1003–1011. doi: 10.1002/jms.1781. [DOI] [PubMed] [Google Scholar]
- 24.Gentili A, Perret D, Marchese S, Mastropasqua R, Curini RDC. Analysis of Free Estrogens and their Conjugates in Sewage and River Waters by Solid-Phase Extraction then Liquid Chromatography- Electrospray-Tandem Mass Spectrometry. Chromatographia. 2002;56:25–32. [Google Scholar]
- 25.Thevis M, Beuck S, Höppner S, Thomas A, Held J, Schäfer M, Oomens J, Schänzer W. Structure elucidation of the diagnostic product ion at m/z 97 derived from androst-4-en-3-one-based steroids by ESI-CID and IRMPD spectroscopy. J. Am. Soc. Mass Spectrom. 2012;23:537–546. doi: 10.1007/s13361-011-0308-4. [DOI] [PubMed] [Google Scholar]
- 26.Williams TM, Kind aJ, Houghton E, Hill DW. Electrospray collision-induced dissociation of testosterone and testosterone hydroxy analogs. J. Mass Spectrom. 1999;34:206–216. doi: 10.1002/(SICI)1096-9888(199903)34:3<206::AID-JMS785>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 27.Gruber CJ, Tschugguel W, Schneeberger C, Huber JC. Production and Actions of Estrogens. N. Engl. J. Med. 2002;346:340–352. doi: 10.1056/NEJMra000471. [DOI] [PubMed] [Google Scholar]
- 28.Ohlsson C, Nilsson ME, Tivesten A, Ryberg H, Mellström D, Karlsson MK, Ljunggren Ö, Labrie F, Orwoll ES, Lee DM, Pye SR, O’Neill TW, Finn JD, Adams JE, Ward KA, Boonen S, Bartfai G, Casanueva FF, Forti G, Giwercman A, Han TS, Huhtaniemi IT, Kula K, Lean MEJ, Pendleton N, Punab M, Vanderschueren D, Wu FCW, Vandenput L. Comparisons of immunoassay and mass spectrometry measurements of serum estradiol levels and their influence on clinical association studies in men. J. Clin. Endocrinol. Metab. 2013;98:E1097–E1102. doi: 10.1210/jc.2012-3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.