

Abstract
Brentuximab vedotin (BV) is an antibody-drug conjugate that specifically delivers the potent cytotoxic drug MMAE to CD30-positive cells. BV is FDA-approved for treatment of relapsed/refractory Hodgkin lymphoma (HL) and anaplastic large cell lymphoma (ALCL); however, many patients do not achieve complete remission and develop BV resistant disease. We selected for BV-resistant HL (L428) and ALCL (Karpas-299) cell lines using either constant (ALCL) or pulsatile (HL) exposure to BV. We confirmed drug resistance by MTS assay, and analyzed CD30 expression in resistant cells by flow cytometry, qRT-PCR, and Western blotting. We also measured drug exporter expression, MMAE resistance, and intracellular MMAE concentrations in BV-resistant cells. Additionally, tissue biopsy samples from 10 HL and 5 ALCL patients who had relapsed or progressed after BV treatment were analyzed by immunohistocytochemistry for CD30 expression. The resistant ALCL cell line, but not the HL cell line, demonstrated downregulated CD30 expression compared to the parental cell line. In contrast, the HL cell line, but not the ALCL cell line, exhibited MMAE resistance and increased expression of the MDR1 drug exporter compared to the parental line. For both HL and ALCL, samples from patients relapsed/resistant on BV persistently expressed CD30 by immunohistocytochemistry. One HL patient sample expressed MDR1 by immunohistocytochemistry. Although loss of CD30 expression is a possible mode of BV resistance in ALCL in vitro models, this has not been confirmed in patients. MMAE resistance and MDR1 expression are possible modes of BV resistance for HL both in vitro and in patients.
Introduction
About 9,200 cases of Hodgkin lymphoma (HL) and 2,000 cases of anaplastic large cell lymphoma (ALCL) are diagnosed in the US annually (1). Although induction chemotherapy has a high response rate, 30% of HL and 40–65% of ALCL patients will experience relapse (2, 3). Roughly half of these patients can be salvaged with high dose chemotherapy followed by autologous stem cell transplantation (ASCT) (4, 5). For the 50% of patients who relapse after ASCT, options are limited. HL is characterized by the presence of Reed-Sternberg cells, which comprise only a minority of cells in the tumor mass and express CD30 surface antigen (6). Alternatively, ALCL is comprised of CD30-expressing lymphoma cells in the majority of the tumor mass. Brentuximab vedotin (BV) is a novel therapeutic in the class of antibody-drug conjugates (ADC) that consists of three components: the cAC10 chimeric IgG1 antibody specific for CD30, the microtubule-disrupting agent monomethyl auristatin E (MMAE), and a protease-cleavable linker that covalently attaches MMAE to cAC10 (7). The entire ADC is internalized upon binding to cell surface CD30 and lysosomal enzymes digest the protease cleavable linker, releasing MMAE, which disrupts the microtubule network and causes cell cycle arrest and apoptosis.
In a pivotal phase II trial for relapsed/refractory HL, BV demonstrated an overall response rate (ORR) of 75% and a complete response (CR) rate of 34% (8). In a phase II trial in patients with relapsed/refractory ALCL, BV demonstrated an ORR of 86% and CR rate of 57% (9). Patients who achieve CR may have durable remissions; however, those achieving only partial responses (PR) have relatively short response durations, with medians of 3.5 months in HL and 2.5 months in ALCL (8, 9). All patients who do not attain CR eventually develop progressive disease despite active treatment with BV. Given that BV is the only therapy approved by the FDA for relapsed/refractory HL in the last 20 years (10), and one of two approved therapies for ALCL, it is imperative that we understand its resistance mechanisms.
Currently, it is unknown whether BV-resistant tumors escape through alterations in surface expression of CD30 (resistance to antibody moiety), by development of resistance to the antimicrotubule agent MMAE, or by expression of one or more transporters that export MMAE out of the cell. To explore possible BV resistance mechanisms, we have selected cell lines for BV resistance and also have analyzed tumor samples from patients who progressed on BV therapy.
Materials and Methods
Cell culture
The L428 (HL) and Karpas-299 (ALCL) cell lines were purchased from the Leibniz Institute DSMZ German Collection of Microorganisms and Cell Cultures, which authenticates cell lines using short tandem repeat (STR) DNA typing. Cells were passaged in the laboratory for fewer than 6 months following purchase and original authentication. Cells were grown in RPMI-1640 (Cellgro Inc.) supplemented with 10% heat inactivated fetal bovine serum (FBS), 2mM glutamine, 100 μg/ml streptomycin and 100 units/ml penicillin. All cell lines were cultured at 37°C in a humidified, 5% CO2 atmosphere.
Selection of BV-resistant cell lines
BV was obtained from City of Hope Pharmacy. Selection of BV-resistant cell lines used two different approaches. For the constant exposure approach, cells were incubated at sub-IC50 concentrations of BV and monitored for changes in cell number over 1 month. BV concentration was then increased incrementally up to the IC50 concentration as long as the cell numbers increased from prior passage. For the pulsatile approach, cells were incubated at supra-IC50 concentration until proliferation halted (as determined by twice a week cell sampling and counting from the selection culture), and then cells were rescued with BV-free media. When consistent proliferation was seen, BV was added back at the same supra-IC50 concentration. Selection was deemed successful when consistent proliferation was seen even in supra-IC50 concentrations of BV.
RNA extraction and quantitative real time PCR
Total RNA was extracted using Trizol (Invitrogen), following the manufacturer’s protocol. Residual DNA was digested using the DNA-free kit following the manufacturer’s instructions (Ambion). cDNA was obtained by reverse transcribing 10 μg of RNA with SuperScript III reverse transcriptase and random primers according to manufacturer’s instructions (Invitrogen). Expression of human mRNAs encoding CD30 (CD30-F: 5′-CCAGGATCAAGTCACTCATCTCA-3′, CD30-R: 5′-AAGTCCCTGGGCAAAGTAAAG-3′), MRD1 (MRD1-F: 5′-GCTCCTGACTATGCCAAAGCC-3′, MRD1-R,5′-CTTCACCTCCAGGCTCAGTCCC-3′), GAPDH (GAPDH-F: 5′-CGCTCTCTGCTCCTCCTGTT-3′, GAPDH-R: 5′-CCATGGTGTCTGAGCGATGT-3′), TBP (TBP-F: 5′-TGCACAGGAGCCAAGAGTGAA-3′, TBP-R: 5′-CACATCACAGCTCCCCACCA-3′) was determined by quantitative real-time PCR (CFX96, BIO-RAD) using 2X iQ SYBR® Green Mastermix (BIO-RAD) according to manufacturer’s protocol. GAPDH expression was used as an internal control for CD30 expression and TBP expression was used as an internal control for MDR1. Primers were ordered from Integrated DNA Technologies.
Protein extraction and immunoblotting
Cell lysates were collected in Trizol. Protein concentrations were determined with the BCA Protein Assay (Pierce) according to manufacturer’s instructions. 10 μg protein was loaded onto a 7.5% SDS-PAGE gel and proteins were transferred to Hybond-LFP membrane (Amersham), followed by primary and secondary antibody incubation. The following antibodies were used: rabbit monoclonal anti-CD30 (Abcam), rabbit monoclonal anti-MDR1 (Cell Signaling), rabbit monoclonal anti-GAPDH (Cell Signaling) and goat anti-rabbit-horse radish peroxidase conjugated secondary antibody (Abcam). Secondary antibody was detected using an ECL Plus kit (Thermo Fisher Scientific). Signals were detected using BIO-RAD Molecular Imager ChemiDoc ™ XRS+.
MTS proliferation assay
Cells were seeded in 96-well plates at 5,000 cells (L-428) and 10,000 cells (karpas-299) per well. Cells were incubated with increasing amounts of BV (0.1 nM to 100μM for Karpas-299; 1 μg/ml to 2400 μg/ml for L428), in triplicate. Cell viability was measured after 72 hours using the CellTiter 96R Aqueous Non-Radioactive Cell Proliferation Assay (Promega) according to manufacturer’s instructions. IC50 value is the concentration of drug which produced a 50% reduction in viability compared with 0 drug control and was calculated from the dose-response curves.
Flow cytometry
2 x 105 cells were incubated with 0.5ng/μl of PE-conjugated monoclonal antibodies at room temperature in the dark for 20 min. Anti CD30 antibody was purchased from Beckton Dickinson and used at concentrations titrated for optimal staining. Flow cytometry was performed on a Beckman Coulter CyAn ADP 9color Cytometer. Lymphocytes or abnormal cell populations were gated on CD45/side scatter (SSC) or forward scatter (FSC) dot plot.
Immunohistochemistry
Ten HL patient samples and 5 ALCL patient samples were obtained at the time of relapse or progressive disease and analyzed by IHC. Samples were obtained from leftover tissue on COH IRB approved protocol. For CD30 immunostaining, we used the monoclonal mouse anti-human CD30, clone Ber-H2. For MDR1 immunostaining, we used the monoclonal mouse anti-human MDR1, clone PG-M1. Both immunostaining was visualized with DAKO Envision/HRP kit (Dakocytomation). Immunohistochemical staining was performed on 5-μm thick paraffin embedded tissue. Tissue sections were deparaffinized in xylene followed by 100%–70% alcohol. Samples were then quenched in 3% hydrogen peroxide and pretreated to promote antigen retrieval with High pH. Slides were incubated in primary antibody at 1/30 dilution for 20 minutes at RT. After rinsing in Dako Wash, slides were incubated in EnVision FLEX/HRP (DAKO K-8000) for 20 minutes. Slides were further washed in Dako buffer and then incubated with diaminobenzidine tetrahydrochloride (DAB) from Dako, counterstained with hematoxylin, and mounted.
Iintracellular MMAE accumulation
Karpas-299 and Karpas-299R cells were treated with 16 ng/ml of BV for 0, 2, 6, 24, and 48 hours. L428 and L428-R cells were treated with 20 μg/ml of BV for 0, 2, 6, 24, and 48 hours. MMAE concentration in cells was measured by LC-MS/MS according to a modification of a previously published method (11). Briefly, following addition of an internal standard, cell membranes were disrupted by sonication and proteins were precipitated using methanol. MMAE concentrations in the extracted cell pellets were then analyzed by gradient reversed phase HPLC separation and tandem mass spectrometric detection. The Waters Quattro Premier XE mass spectrometer (Milford, MA, USA) was operated in positive electrospray ionization mode and quantitation was performed using multiple reaction monitoring (MRM). MassLynx version 4.1 software was used for data acquisition and processing. Under optimized assay conditions, the lower limit of quantitation was 0.01 ng/106 cells or 0.17 pg on column. Inter- and intra-day precision and accuracy of the method was within ± 10% of target values.
Statistical Considerations
Cell proliferation/growth over time and across different concentrations are represented as means with 95% confidence intervals (error bars). For measurements over time, a repeated measures ANOVA model was also used to examine possible time and group interactions. For dose-response measurements, a four parameter log-logistic model was fitted to the curves and IC50 values were estimated accordingly. Statistical analyses were performed by two-tailed unpaired t test unless otherwise noted. An effect was considered statistically significant when the corresponding p-value was less than 0.05. All statistical analysis was carried out using both R software and Excel.
Results
BV resistant HL and ALCL cell line selection
MTS assays determined the IC50 of parental cell lines to be: L428 (HL) 27 μg/ml ± 4.8 μg/ml and Karpas-299 (ALCL) 29 ng/ml ± 21 ng/ml (Table 1). BV-resistant cell models were selected using two different approaches. For the constant exposure approach, L428 and Karpas-299 cell lines were incubated at sub-IC50 concentrations of BV (25 μg/ml for L428 and 10 ng/ml for Karpas-299) and cell numbers were monitored over the course of 3 months in culture. The BV concentration was increased when consistent cell proliferation at the initial selecting concentration was achieved. This approach was successful for selecting Karpas-299 resistant cells as they were able to grow in concentrations of BV as high as 20 ng/ml, but we were unable to obtain resistant L428 cells by means of constant exposure to drug. We therefore used a pulsatile approach, in which L428 cells were incubated in a supra-IC50 concentration of BV (50 μg/ml) and cell numbers were assessed twice weekly until no further proliferation was seen. Cells were then rescued with BV-free media until proliferation was again observed (increase in cell number for three consecutive weeks), at which point 50 μg/ml BV was added back to the cells. This process was continued until consistent proliferation in 50 μg/ml of BV was achieved.
Table 1.
| L428-P | L428-R | Karpas-P | Karpas-R | |
|---|---|---|---|---|
| IC50 to BV | 27 ± 4.8 μg/ml | 236 ± 22 μg/ml | 29 ± 21 ng/ml | 19 ± 1.9 μg/ml |
| Relative resistance | 1 | 8.7 | 1 | 655 |
| % cells CD30+ | 98% | 97% | 96% | 59% |
| Median intensity CD30 | 280 ± 10 | 265 ± 5 | 592 ± 51 | 78 ± 17 |
We confirmed BV resistance in both cell lines using cell proliferation assays and MTS assays (Figure 1). In cell proliferation assays, resistant cell lines were able to proliferate in the presence of BV at concentrations above their respective parental line IC50s (Figure 1A, 1B). At the same concentrations, the parental cell lines quickly died. In MTS assays, resistant cell lines demonstrated IC50s shifted to higher BV concentrations: L428-R (236 μg/ml ± 22 μg/ml, 8.7-fold relative resistance), Karpas-R (19 μg/ml ± 1.9 μg/ml, 655-fold relative resistance) (Figure 1C, 1D, Table 1).
Figure 1. BV-resistant ALCL and HL in vitro cell models.

Proliferation experiments were performed in duplicate wells and averaged over 2 separate experiments, and a repeated measures ANOVA model is applied to show that time, group, and their interactions are all very significant (Panel A and B). Viable cells counts were performed by hemocytometer with methylene blue. Panel A: L428 and L428-R were seeded at 100,000 cells per well and incubated with BV at 80 μg/ml. L428-R cells are able to proliferate at this concentration whereas L428 can not. (p < 0.0001). Panel B: Karpas-299 and Karpas-R were seeded at 40,000 cells per well and incubated with BV at 30 ng/ml. Karpas-R cells are able to prliferate at this concentration whereas Karpas-299 can not (p < 0.0003). MTS assays were performed in triplicate wells and averaged over 3 separate experiments, and cells were seeded in 96-well plates at 5,000 cells (L428) and 10,000 cells (Karpas-299) per well. Panel C: A four-parameter log-logistic model was fitted to assess inhibitory effect of L428-P and L428-R, respectively. The estimated IC50 (standard error)s are 27.46 (4.76) and 236.08 (22.15) respectively. Panel D: A four-parameter log-logistic model was fitted to assess inhibitory effect of Karpas-P and Karpas-R, respectively. The estimated IC50 (standard error)s are 0.0029 (0.021) and 19.45 (1.87) respectively.
CD30+ cells are resistant to BV
Flow cytometry showed down-regulation of surface CD30 expression for Karpas-R (Figure 2A-D, Table 1) but not for L428-R (Table 1 and data not shown). For Karpas-R, there was a 38% reduction in the percentage of CD30+ cells and a 79% reduction in median intensity, relative to Karpas-P cells. There was no reduction in the percentage of CD30+ cells or in median intensity of CD30 staining in L428-R cells compared with L428-P cells (Table 1).
Figure 2. BV-resistance ALCL and HL in vitro cell models.

Panel A–D. Flow cytometry showing surface CD30 expression in Karpas P, Karpas-R, and Karpas-R CD30+ and Karpas-R CD30− cells after cell sorting. Panel E shows CD30 mRNA expression by qRT-PCR.(qRT-PCR performed with triplicate wells and repeated twice). A two-sample t-test showed that there are significant expression differences for comparisons Karpas-P vd Karpas-R (P-value = 0.003) and Karpas-R CD30+ vs Karpas-R CD30− (P-value = 0.002). Panel F: MTS assay were performed with triplicate wells and averaged over 3 experiments. A four-parameter log-logistic model was fitted to assess inhibitory effect of Karpas-P, Karpas-R, Karpas-R CD30+, and Karpas-R CD30-, respectively. The estimated IC50 (standard error)s are 0.0058 (0.0071), 14.18 (1.46), 10.82 (0.89), and 16.99 (2.06) respectively.
The sustained CD30 expression in L428-R cells led us to ask if the resistance phenotype for Karpas-R cells was wholly or partially due to loss of the CD30 target or if resistance was independent of CD30 expression. To address this question, we sorted Karpas-R cells into CD30+ and CD30− sub-populations and examined the respective CD30 levels and BV resistance profiles of these sorted sub-populations. The CD30− sub-population had lower surface CD30 protein (Fig. 2A-D) and mRNA (Fig. 2E), compared to the CD30+ sub-population and to parental Karpas-P cells. In the experiment shown here, the sorted CD30+ sub-population had 79% CD30+ cells with a median intensity of 98, whereas the CD30− sub-population had 19% CD30+ cells at a median intensity of 36 after one week of culture in the absence of BV. The unsorted Karpas-R population was 59% CD30+ with a median intensity of 74 under these same conditions, whereas the parental Karpas-P cells were essentially 100% CD30+ with a median intensity of 556. MTS assays showed that the unsorted Karpas-R population, the sorted CD30+ sub-population, and the sorted CD30− sub-population were all equally resistant to BV (Figure 2F). This experiment was performed in triplicate wells and repeated three times with essentially the same results.
BV resistance phenotype is not permanent in Karpas-R cells
We also asked whether down-regulation of CD30 and resistance were stable phenotypes in the Karpas-R cell line. Karpas-R cells were cultured in BV-free growth medium for 26 weeks and then re-assayed for CD30 expression by flow cytometry and for BV resistance by MTS. After 26 weeks without drug selection, 100% of the Karpas-R cells were CD30+ and the median intensity was 340, suggesting a partial reversion of the CD30 down-regulation phenotype (Supplemental Figure 1). MTS assay showed that these cells were 11-fold more resistant to BV, a partial reversion of the phenotype, but still with substantial resistance despite regained CD30 expression (Supplemental Figure 2).
CD30 is not down-regulated in primary HL and ALCL lymphomas
Given the transient reduction of CD30 expression that we observed in Karpas-R cells (but not in L428-R cells), we wanted to determine CD30 status in patients with acquired BV resistance. We previously reported that CD30 continued to be expressed in two HL patients with BV resistance (12) and we now report the analysis of samples from 15 additional patients (10 HL, 5 ALCL) who initially achieved response to BV and later became resistant while on therapy or developed relapsed disease while off therapy. We obtained tissue samples from their HL or ALCL tumors before treatment and also at the time of their relapsed or progressive disease. Regardless of the histology, or whether relapse occurred during or after treatment with BV, all primary lymphoma samples showed persistent CD30 expression by IHC. Figure 3 shows representative CD30 staining on three patients with ALCL; our HL data have been reported previously (12). Thus, it does not appear that treatment with BV commonly leads to loss of CD30 in either HL or ALCL tumors that have progressed or relapsed.
Figure 3. CD30 staining in tissue from ALCL patients resistant to BV.
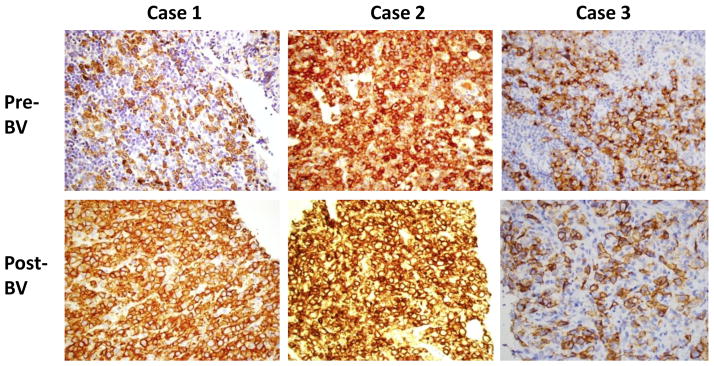
Please see methods section on CD30 immunohistochemical staining. Biopsy post BV was done at the time of disease relapse while off BV or disease progression while on BV. CD 30 expression is retained in both scenarios.
MMAE resistance as a mechanism of BV resistance
To investigate other possible mechanisms of resistance, we first asked whether BV resistance might stem from resistance to the anti-microtubule agent MMAE, the cytotoxic component of BV. We performed MTS assays to determine the IC50 of MMAE in parental L428-P and Karpas-P cells and their resistant counterparts. L428-R cells were about 39-fold more resistant to MMAE, compared to L428-P cells (Figure 4A), whereas they exhibited only a 8.7-fold resistance to BV (Figure 1C). In contrast, the MMAE IC50 of Karpas-R cells was similar to the IC50 of parental Karpas-P cells (Figure 4B). These results again suggest different mechanisms of resistance to BV in the two cell types, with resistance to the cytotoxic agent itself possibly contributing to the resistance phenotype in L428-R but not in Karpas-R cells.
Figure 4. MMAE resistance and intracellular accumulation.

Panel A shows MTS assay performed on L428P and L428 R cells using MMAE. L428 R is resistant to MMAE as compared to L428 P (MTS assay performed in triplicate wells and repeated three times). A four-parameter log-logistic model was fitted to assess inhibitory effect of L428-P and L428-R, respectively. The estimated IC50 (standard error)s are 0.63 (0.07) and 24.66 (4.87) respectively. Panel B shows MTS assay performed on Karpas-P and Karpas-R cells using MMAE. Karpas R is equally sensitive to MMAE as compared to Karpas-P (MTS assay performed in triplicate wells and repeated three times). A four-parameter log-logistic model was fitted to assess inhibitory effect of Karpas-P and Karpas-R, respectively. The estimated IC50 (standard error)s are 0.28 (0.05) and 0.52 (0.05) respectively. Panels C–E shows intracellular MMAE concentration from 0–48 hours of exposure with BV (experiments done in duplicate wells and repeated twice). Data are expressed as mean curves with 95% confidence interval and a repeated measures ANOVA model applied to show that th time, group, and their interactions are significant. In Panel C, L428P had much more intracellular MMAE as compared to L428 R (p < 0.0001). Panel D shows Karpas R and P had same intracellular concentration of MMAE when incubated with low concentration of BV. Panel E shows Karpas-P had more intracellular concentration of MMAE as compared to Karpas-R when incubated with higher concentration of BV (p ≤ 0.0001).
Reduced drug accumulation as a possible mechanism of BV resistance
One possible mechanism of BV (and MMAE) resistance is through altered intracellular accumulation of MMAE after the drug is internalized. To determine if this were the case, we measured intracellular MMAE concentrations in L428-P and L428-R cells incubated with 20 μg/ml of BV (near the IC50 of the parental line) at multiple time points over the course of 48 hours. L428-R cells consistently showed a decreased amount of intracellular MMAE as compared to L428-P cells, with 6.7 fold (±3.4 fold) more MMAE accumulated in L428-P cells by the 48 hour time point (Figure 4C).
We performed the same experiment with Karpas-P and Karpas-R cells using 16 ng/ml of BV (near the IC50 of parental line), but found no differences in intracellular MMAE accumulation at any time point (Figure 4D). Interestingly, when we incubated Karpas-P and Karpas-R cells with 20 μg/ml of BV (near the IC50 of Karpas-R cells), we observed a decreased amount of intracellular MMAE in Karpas-R cells at each time point, with 5.2 fold (± 2.1 fold) higher amounts in Karpas-P than in Karpas-R cells by 48 hours (Figure 4E).
As an additional test for altered drug accumulation activity, we loaded both the L428-P and L428-R cells with rhodamine-123 dye and incubated them on ice or at 37°C for two hours. Rhodamine fluorescence was quantified by flow cytometry. L428-R cells had 10-fold lower fluorescence than L428-P cells after two days (Supplemental Figure 3). To determine whether the lower drug accumulation in L428-R cells could be due to altered expression of one or more drug transporters, we used quantitative RT-PCR to measure mRNA levels for MDR1, MRP1 and MRP3, in both sets of parental and resistant cell lines. We found an overexpression of MDR1 mRNA in L428-R cells relative to L428-P cells (Figure 5A). We did not find overexpression of MRP1 or MRP3 mRNA in L428-R cells. Protein levels of P-glycoprotein, the product of the MDR1 gene, were similarly elevated in L428-R cells (Figure 5B). This phenotype might also be unstable, since L428-R cells grown in the absence of BV for 5 months had decreased MDR1 mRNA and P-glygoprotein expression relative to L428-R cells continuously exposed to BV (Figure 5). We then used verapamil to inhibit P-glycoprotein function and performed MTS assay to determine if inhibition of P-glycoprotein would restore BV sensitivity. We found that we could lower the BV IC50 of L428-R from 296 μg/ml to 78 μg/ml (Figure 5C). Unlike L428-R cells, Karpas-R cells did not demonstrate overexpression of MDR1/P-glycoprotein, as measured by qRT-PCR or Western analysis, and neither cell line had elevated expression of MRP1 or MRP3.
Figure 5. MDR1/P-glycoprotein Drug Exporter Expressed in L428 BV-resistant model.

Panel A shows MDR1 mRNA expression in L428-P, L428-R, and L428-R off drug for 5 months. qRT-PCR for MDR1 mRNA expression performed in triplicate wells and repeated three times. A two-sample t-test showed that there is significant expression level difference for comparisons L 428-P vs L 428-R (p-value<0.0001), L 428-P vs L 428-R Off (p-value<0.001), and L 428-R Off vs L 428-R (p-value = 0.003). Panel B shows Western blot for P-glycoprotein (PgP, protein product of MDR1 mRNA) in L428-P, L428-R, and L428-R off drug for 5 months. L428-R had more MDR1 mRNA and PgP as compared to L428-R off drug or L428-P. Panel C shows MTS assays performed in triplicate wells and averaged over 3 separate experiments. Cells were seeded in 96-well plates at 5,000 cells per well. A four-parameter log-logistic model was fitted to assess inhibitory effect of L428-R with and without verapamil, respectively. The estimated IC50s (±standard error) are 76 (± 23) and 297 (±12), respectively.
We also examined P-glycoprotein, MRP1, and MRP3 expression by immunohistochemistry in our HL tumor samples. Out of four paired pre- and post-treatment HL samples, we detected one positive P-glycoprotein signal (Supplemental Figure 4), one positive MRP1 signal, and two positive MRP3 signals at the time of relapse. All of these patients were initially sensitive to BV treatment, but became resistant at the time of disease relapse, after BV therapy.
Discussion
One of the major goals of this study was to investigate the role of CD30 in BV resistance in HL and ALCL. We performed an in vitro selection for BV-resistant HL and ALCL cell lines and determined their CD30 expression by flow cytometry, qRT-PCR, and Western analysis. We found that the resistant HL cell line, L428-R, did not have altered CD30 levels, whereas the resistant ALCL cell line, Karpas-R, did show down-regulated CD30 levels. We did not see complete loss of expression in Karpas-R cells, however. Moreover, by sorting the Karpas-R cells into CD30+ and CD30− sub-populations, we demonstrated that the CD30+ cells were still resistant to BV despite their significant CD30 expression. It appears that the percentage of CD30+ cells in the population does not correlate with the degree of BV resistance.
Although loss of CD30 is not necessary for resistance to BV in ALCL, the level of expression could be important. As demonstrated by flow cytometry, parental Karpas-P cells had a high median intensity of CD30 expression, whereas the unsorted Karpas-R population had an approximately 8-fold lower median intensity. The lower median intensity of CD30 staining in the resistant population could reflect an altered dynamic of CD30 internalization and recycling to the cell surface, but further study is required to determine if this is the case. The intensity of CD30 expression could also be an indirect measure of the number of CD30 molecules present on the cell surface at the time of fixation. Regardless of the mechanism, it would not be surprising for an altered steady-state level of receptor on the cell surface to affect BV sensitivity, since BV must bind CD30 to enter the cell.
The intracellular MMAE accumulation study was consistent with lower cell-surface CD30 affecting drug uptake. When incubated in a low concentration of BV, Karpas-R cells appeared to have sufficient CD30 expression to allow efficient drug entry. In a higher concentration of BV, however, Karpas-R had 5.2-fold lower intracellular MMAE accumulation than Karpas-P cells. The difference in intracellular MMAE accumulation between Karpas-P and Karpas-R at the high BV concentration, and lack of difference at the low BV concentration, could suggest a saturation effect of CD30. Limited BV cellular entry due to lower cell-surface CD30 might explain a 3-fold reduction in MMAE sensitivity, but it does not match the 1100-fold reduction in BV IC50 that we measured for Karpas-R cells. Indeed, resistance in our cell lines was not correlated with either the percentage of CD30+ cells or the median intensity of cell-surface CD30 signal in a given cell population, suggesting that the resistance phenotype must involve another mechanism that is not related to CD30 accessibility.
We also found that CD30 down-regulation in Karpas-R cells was not permanent. The resistant Karpas-R population was able to re-express CD30 and regain partial sensitivity to BV after a prolonged period of growth in the absence of drug. This phenomenon has significant clinical correlations. Although IHC analysis of primary ALCL specimens did not show CD30 down-regulation, the majority of our patient samples were obtained from patients who had not been exposed to BV for weeks or months. Given that CD30 can be re-expressed in cell lines during a period without drug exposure in cell lines, it is perhaps not surprising that our patient samples were all CD30 positive as well. It remains to be determined whether CD30 down-regulation occurs in ALCL patients during or immediately after BV therapy, as in our cell line model, and likewise whether patients with regained or persistent CD30 positivity after a prolonged period off of therapy are nevertheless resistant to BV.
There are other targets on the surface of HL tumors, such as CD70 or CD25 that can be used as a way to efficiently deliver potent cytotoxic molecules such as MMAE or related agents (13–15). SGN-75 which has been tested in renal cell carcinoma and B cell lymphomas, can deliver a derivative of MMAF (monomethyl auristatin F) directly to CD 70 expressing cells (16). Another drug, SGN-CD70A, which is currently undergoing phase I testing, can deliver a pyrrolobenzodiazepine (PBD) dimer directly to CD 70 expressing cells (17). It is possible that both of these drugs could be utilized in HL patients who are resistant to BV.
In terms of HL, our MTS assays showed that L428-R cells were resistant to unconjugated MMAE and that surface expression of CD30 was not altered relative to L428-P cells. We also discovered that L428-R cells overexpressed MDR1. Although this is the first report of MDR1 overexpression in an HL cell line resistant to an ADC, MDR1 and other drug exporters have been implicated in drug resistance for many other tumor types (18–21). This overexpression was confirmed by qRT-PCR and Western analysis. Drug exporter activity was confirmed by a rhodamine efflux assay and inhibition of P-glycoprotein function was able to reverse the resistance phenotype. We also found that 3 of 4 HL paired patient samples stained positive for one of the class of drug transporters at the time of relapse and those patients were resistant to BV treatment. This suggests that drug transporters can play a role in BV drug resistance in HL. Although MMAE can be actively pumped out of the cell by P-glycoprotein or other transporters, there are other cytotoxic agents that can be linked to antibody drug conjugates that are not substrates for transport, and these could be utilized as CD30 immunoconjugates in the future.
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Institutes of Health (NIH) National Cancer Institute (NCI) K12 CA01727 (PI: J. Mortimer) awarded to R. Chen, the NCI Lymphoma SPORE (CA107399) with Development Research Program award to R. Chen, NCI CA33572 (PI: S. Rosen) with Team Leadership Supplement to R. Chen, NCI CA062505 (PI: E. Newman), NCI CA186717 (PI: R. Morgan) and the Tim Nesvig Lymphoma Research Fund (award to R. Chen).
We would like to thank the Flow Cytometry Core and the Analytical Pharmacology Core facilities of the City of Hope Comprehensive Cancer Center grant NCI CA33572. These studies were also supported in part by, NCI CCITLA, U01 CA062505, UM1 CA186717, the City of Hope Lymphoma SPORE CA107399 and the Tim Nesvig Lymphoma Research Foundation. RC is an NCI K12 Calabresi Career Development Scholar.
Footnotes
Disclosures: Dr. Chen has received research funding for clinical trials and speaker honoraria from Seattle Genetics, Inc. The other authors have no potential financial conflicts to disclose.
References
- 1.American Cancer Society. Cancer Facts and Figures. 2014 Available from: http://www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2014/index.
- 2.Quddus F, Armitage JO. Salvage therapy for Hodgkin’s lymphoma. Cancer J. 2009;15:161–3. doi: 10.1097/PPO.0b013e3181a1438a. [DOI] [PubMed] [Google Scholar]
- 3.Savage KJ, Harris NL, Vose JM, Ullrich F, Jaffe ES, Connors JM, et al. ALK anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International Peripheral T-Cell Lymphoma Project. Blood. 2008;111:5496–504. doi: 10.1182/blood-2008-01-134270. [DOI] [PubMed] [Google Scholar]
- 4.Moskowitz CH, Nimer SD, Zelenetz AD, Trippett T, Hedrick EE, Filippa DA, et al. A 2-step comprehensive high-dose chemoradiotherapy second-line program for relapsed and refractory Hodgkin disease: analysis by intent to treat and development of a prognostic model. Blood. 2001;97:616–23. doi: 10.1182/blood.v97.3.616. [DOI] [PubMed] [Google Scholar]
- 5.Kuruvilla J, Nagy T, Pintilie M, Tsang R, Keating A, Crump M. Similar response rates and superior early progression-free survival with gemcitabine, dexamethasone, and cisplatin salvage therapy compared with carmustine, etoposide, cytarabine, and melphalan salvage therapy prior to autologous stem cell transplantation for recurrent or refractory Hodgkin lymphoma. Cancer. 2006;106:353–60. doi: 10.1002/cncr.21587. [DOI] [PubMed] [Google Scholar]
- 6.Pileri SA, Ascani S, Leoncini L, Sabattini E, Zinzani PL, Piccaluga PP, et al. Hodgkin’s lymphoma: the pathologist’s viewpoint. J Clin Pathol. 2002;55:162–76. doi: 10.1136/jcp.55.3.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Francisco JA, Cerveny CG, Meyer DL, Mixan BJ, Klussman K, Chace DF, et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood. 2003;102:1458–65. doi: 10.1182/blood-2003-01-0039. [DOI] [PubMed] [Google Scholar]
- 8.Younes A, Gopal AK, Smith SE, Ansell SM, Rosenblatt JD, Savage KJ, et al. Results of a Pivotal Phase II Study of Brentuximab Vedotin for Patients With Relapsed or Refractory Hodgkin’s Lymphoma. J Clin Oncol. 2012;30:2183–9. doi: 10.1200/JCO.2011.38.0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pro B, Advani R, Brice P, Bartlett NL, Rosenblatt JD, Illidge T, et al. Brentuximab Vedotin (SGN-35) in Patients With Relapsed or Refractory Systemic Anaplastic Large-Cell Lymphoma: Results of a Phase II Study. J Clin Oncol. 2012;30:2190–6. doi: 10.1200/JCO.2011.38.0402. [DOI] [PubMed] [Google Scholar]
- 10.de Claro RA, McGinn K, Kwitkowski V, Bullock J, Khandelwal A, Habtemariam B, et al. U.S. Food and Drug Administration approval summary: brentuximab vedotin for the treatment of relapsed Hodgkin lymphoma or relapsed systemic anaplastic large-cell lymphoma. Clin Cancer Res. 2012;18:5845–9. doi: 10.1158/1078-0432.CCR-12-1803. [DOI] [PubMed] [Google Scholar]
- 11.Boswell CA, Mundo EE, Zhang C, Bumbaca D, Valle NR, Kozak KR, et al. Impact of drug conjugation on pharmacokinetics and tissue distribution of anti-STEAP1 antibody-drug conjugates in rats. Bioconjug Chem. 2011;22:1994–2004. doi: 10.1021/bc200212a. [DOI] [PubMed] [Google Scholar]
- 12.Nathwani N, Krishnan AY, Huang Q, Kim Y, Karanes C, Smith EP, et al. Persistence of CD30 expression in Hodgkin lymphoma following brentuximab vedotin (SGN-35) treatment failure. Leuk Lymphoma. 2012;53:2051–3. doi: 10.3109/10428194.2012.666543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaffer DR, Savoldo B, Yi Z, Chow KK, Kakarla S, Spencer DM, et al. T cells redirected against CD70 for the immunotherapy of CD70-positive malignancies. Blood. 2011;117:4304–14. doi: 10.1182/blood-2010-04-278218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Malley DP, Chizhevsky V, Grimm KE, Hii A, Weiss LM. Utility of BCL2, PD1, and CD25 immunohistochemical expression in the diagnosis of T-cell lymphomas. Appl Immunohistochem Mol Morphol. 2014;22:99–104. doi: 10.1097/PAI.0b013e31828ef1f7. [DOI] [PubMed] [Google Scholar]
- 15.Berkowitz JL, Janik JE, Stewart DM, Jaffe ES, Stetler-Stevenson M, Shih JH, et al. Safety, efficacy, and pharmacokinetics/pharmacodynamics of daclizumab (anti-CD25) in patients with adult T-cell leukemia/lymphoma. Clin Immunol. 2014;155:176–87. doi: 10.1016/j.clim.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tannir NM, Forero-Torres A, Ramchandren R, Pal SK, Ansell SM, Infante JR, et al. Phase I dose-escalation study of SGN-75 in patients with CD70-positive relapsed/refractory non-Hodgkin lymphoma or metastatic renal cell carcinoma. Invest New Drugs. 2014;32:1246–57. doi: 10.1007/s10637-014-0151-0. [DOI] [PubMed] [Google Scholar]
- 17.Seattle Genetics I. NCT02216890: Safety Study of SGN-CD70A in Cancer Patients. 2014 Available from: https://clinicaltrials.gov/ct2/show/NCT02216890.
- 18.O’Brien C, Cavet G, Pandita A, Hu X, Haydu L, Mohan S, et al. Functional genomics identifies ABCC3 as a mediator of taxane resistance in HER2-amplified breast cancer. Cancer Res. 2008;68:5380–9. doi: 10.1158/0008-5472.CAN-08-0234. [DOI] [PubMed] [Google Scholar]
- 19.Greaves W, Xiao L, Sanchez-Espiridion B, Kunkalla K, Dave KS, Liang CS, et al. Detection of ABCC1 expression in classical Hodgkin lymphoma is associated with increased risk of treatment failure using standard chemotherapy protocols. Journal of hematology & oncology. 2012;5:47. doi: 10.1186/1756-8722-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marchetti S, Pluim D, Beijnen JH, Mazzanti R, van Tellingen O, Schellens JH. Effect of the drug transporters ABCB1, ABCC2, and ABCG2 on the disposition and brain accumulation of the taxane analog BMS-275,183. Invest New Drugs. 2014;32:1083–95. doi: 10.1007/s10637-014-0143-0. [DOI] [PubMed] [Google Scholar]
- 21.Du Y, Su T, Zhao L, Tan X, Chang W, Zhang H, et al. Associations of polymorphisms in DNA repair genes and MDR1 gene with chemotherapy response and survival of non-small cell lung cancer. PloS one. 2014;9:e99843. doi: 10.1371/journal.pone.0099843. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.