

ABSTRACT
Although it is established that oxygen availability regulates cellular metabolism and growth, little is known regarding how intracellular pathogens use host factors to grow at physiological oxygen levels. Therefore, large-scale human small interfering RNA screening was performed to identify host genes important for growth of the intracellular protozoan parasite Toxoplasma gondii at tissue oxygen tensions. Among the genes identified by this screen, we focused on the hexokinase 2 (HK2) gene because its expression is regulated by hypoxia-inducible transcription factor 1 (HIF-1), which is important for Toxoplasma growth. Toxoplasma increases host HK2 transcript and protein levels in a HIF-1-dependent manner. In addition, parasite growth at 3% oxygen is restored in HIF-1-deficient cells transfected with HK2 expression plasmids. Both HIF-1 activation and HK2 expression were accompanied by increases in host glycolytic flux, suggesting that enhanced HK2 expression in parasite-infected cells is functionally significant. Parasite dependence on host HK2 and HIF-1 expression is not restricted to transformed cell lines, as both are required for parasite growth in nontransformed C2C12 myoblasts and HK2 is upregulated in vivo following infection. While HK2 is normally associated with the cytoplasmic face of the outer mitochondrial membrane at physiological O2 levels, HK2 relocalizes to the host cytoplasm following infection, a process that is required for parasite growth at 3% oxygen. Taken together, our findings show that HIF-1-dependent expression and relocalization of HK2 represent a novel mechanism by which Toxoplasma establishes its replicative niche at tissue oxygen tensions.
IMPORTANCE
Little is known regarding how the host cell contributes to the survival of the intracellular parasite Toxoplasma gondii at oxygen levels that mimic those found in tissues. Our previous work showed that Toxoplasma activates the expression of an oxygen-regulated transcription factor that is required for growth. Here, we report that Toxoplasma regulates the abundance and activity of a key host metabolic enzyme, hexokinase 2, by activating HIF-1 and by promoting dissociation of hexokinase 2 from the mitochondrial membrane. Collectively, our data reveal HIF-1/hexokinase 2 as a novel target for an intracellular pathogen that acts by reprograming the host cell’s metabolism to create an environment conducive for parasite replication at physiological oxygen levels.
INTRODUCTION
Cells adapt to changes in O2 levels in part by inducing large-scale transcriptional changes to alter metabolic and growth pathways (1). The hypoxia-inducible factor (HIF) family of transcription factors are the key transcriptional regulators that mediate these changes (1). HIFs are heterodimeric transcription factors composed of α (HIF-1α, -2α, or -3α) and β (HIF-1β) subunits. While both subunits are constitutively expressed, under normoxic conditions, HIFα is rapidly targeted to the proteasome after it has been prolyl hydroxylated by a family of O2-dependent prolyl hydroxylases (PHD1, 2, and 3) (2, 3). During hypoxia, HIFα is stabilized and translocates to the nucleus, where it binds HIF-1β and activates gene expression.
Hexokinases catalyze the first rate-limiting glycolytic reaction: conversion of glucose to glucose-6-phosphate (G6P). Hexokinase 1 (HK1) is the most abundant and ubiquitously expressed isoform, whereas HK2 is a HIF-1 target gene important in shifting a cell from oxidative phosphorylation to glycolytic metabolism (4, 5). This shift occurs when cells are exposed to hypoxia (<1.5% O2) or when they rely on glycolysis (e.g., transformed cells or activated macrophages) under oxygen-replete conditions (5–7).
Both HK isoforms localize to the cytoplasm and outer mitochondrial membrane (OMM). Two properties of HK2 mediate OMM association—a 15-amino-acid N-terminal polypeptide that inserts into the OMM and binding to a mitochondrial porin termed the voltage-dependent anion channel (VDAC) (8, 9). HK1/2 association with VDAC promotes catabolic glucose metabolism and protects cells from apoptosis by limiting cytochrome c release from the mitochondria (10, 11). In addition, OMM-bound HK1/2 cannot use cytoplasmic ATP as a cosubstrate but rather uses intramitochondrial ATP as it diffuses through VDAC, preventing excess lactate accumulation (12).
Most intracellular pathogens grow in tissues whose O2 levels are lower than the atmospheric level (21% O2) found in tissue culture-based experiments. Toxoplasma gondii, the causative agent of toxoplasmosis, grows and causes disease in tissues with diverse O2 tensions (13). Following ingestion, the parasite infects and replicates within the intestine, a hypoxic environment, and then disseminates to peripheral tissues (14). Host factors that allow Toxoplasma and other pathogens to replicate at decreased O2 levels are poorly understood. Previous microarray studies revealed that several HIF-1-regulated transcripts are upregulated following infection, including the transferrin receptor and glycolytic-pathway genes such as HK2 (15). Consistent with these data, Toxoplasma activates HIF-1 and parasite growth in murine embryonic fibroblasts with HIF-1α knockout mutations (HIF-1αKO MEFs) is decreased by >70% at 21% O2 and by >95% at a physiological O2 level (3%) (16). The goal of this study was to identify host cell factors important for Toxoplasma growth at both normoxic and physiological O2 levels and determine which HIF-1 target gene(s) is (are) important in Toxoplasma-infected cells.
RESULTS
Large-scale siRNA screening identifies host genes important for parasite growth at normoxic and physiological oxygen levels.
A large-scale human small interfering RNA (siRNA) screen was performed with HeLa cells to identify host genes important for Toxoplasma growth at 21% (normoxic) and/or 3% (physiological) O2. siRNA libraries, plated in 96-well plates, were used in which each well contained three siRNAs targeting different regions of a transcript (Fig. 1A). Each plate targeted 88 different genes, and the remaining wells were used to generate a standard curve or contained siRNAs targeting Polo-like kinase 1 (PLK1) or HIF-1α as controls. PLK1 assessed transfection efficiency because its depletion kills cells, resulting in decreased parasite growth (17). HIF-1α was used because its loss leads to decreased parasite growth at 3% O2 (Fig. 1D) (16). β-Galactosidase (βgal)–green fluorescent protein (GFP)-expressing (RH-βgal/GFP) parasites were added 48 h later to siRNA-transfected host cells and grown at either 21 or 3% O2 for 72 h, and then the parasites were enumerated. Control assays showed a transfection efficiency of >95% and that the siRNAs reduced target gene expression for at least 96 h (see Fig. S1A and B in the supplemental material). Similarly, we found that of a small siRNA library targeting 11 human caspases, nine caspase siRNAs reduced target gene expression by at least 90% without impacting parasite growth (see Fig. S1C and D). Caspase-14 could not be assessed because it is not expressed in HeLa cells (18), and PCR primers to assess caspase-5 expression could not be developed (data not shown).
FIG 1 .

HK2 is important for parasite growth at 3% oxygen. (A) siRNA screen overview. (B) Z scores of siRNA screening data at 21% (x axis) and 3% (y axis) O2. Dotted lines are Z scores of less than −2 or >2. siRNAs that reduce host cell viability are highlighted in red. (C) HK2 Western blot of lysates from mock- or parasite-infected negative-control (Neg Ctrl) or HK2 siRNA-transfected HeLa cells grown at 21% oxygen. Parasite growth was measured in HIF-1α (D) or HK2 (E) siRNA-transfected cells. Shown are averages and standard deviations from three assays. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (Student’s t test).
Primary screening targeting 9,102 human genes revealed that 316 siRNAs reduced parasite growth and 71 improved growth at 21% O2 while 293 reduced growth and 109 improved growth at 3% O2 (Fig. 1B; see Table S1 in the supplemental material). Using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) host cell viability assay, we eliminated 168 of these from further consideration because they significantly reduced viability (Z scores of less than −1.67) of uninfected host cells at 21% O2. Secondary screening with different siRNAs confirmed the inhibitory effect of 28 (13.6%) of the 206 genes (see secondary-screening tab in Table S1). Pathway analysis using the COGNOSCENTE software program (http://vanburenlab.tamhsc.edu/) revealed no apparent interactions between the proteins encoded by these genes.
Hexokinase 2 is a HIF-1-regulated gene important for Toxoplasma growth at physiologically relevant oxygen concentrations.
Host genes were hypothesized to be HIF-1 targets important for parasite growth if they are known HIF-1 target genes whose siRNAs limited parasite growth more at 3% O2 than at 21% O2. Only HK2 fulfilled this criterion, and quantitative real-time PCR (qRT-PCR) and Western blotting verified HK2 siRNA specificity (Fig. 1C; see Fig. S1E in the supplemental material). A detailed time course revealed O2-dependent parasite growth phenotypes due to HK2 and HIF-1α depletion (Fig. 1D and E). While HK2 siRNAs decrease the viability of some cells exposed to hypoxia (<1.5% O2) (5), they had no apparent effect at 3% O2 (see Fig. S1E).
The effects of Toxoplasma on HK2 mRNA abundance in wild-type HIF-1α (HIF-1αWT) and HIF-1αKO MEFs were compared at 18 hpi with RH strain parasites. Infection increased HK2 mRNA abundance in a HIF-1-dependent manner at both O2 tensions (Fig. 2A and B). HIF-1α was also required at 3% O2 to increase levels of other glycolytic-pathway transcripts (ALDOA, ALDOB, glyceraldehyde 3-phosphate dehydrogenase [GAPDH], PGAM1, and PKM2) (see Fig. S2 in the supplemental material). Infection increased HK2 protein at 21% O2 in HIF-1αWT but not HIF-1αKO MEFs. Relative to uninfected HIF-1αWT MEFs at 21%, HK2 protein abundance at 3% O2 was similarly elevated in mock- and parasite-infected HIF-1αWT MEFs but not in HIF-1αKO MEFs suggesting that to 3% O2 HK2 was maximally expressed (Fig. 2C and D). HK2 antibody specificity was demonstrated by an inability of the antibody to detect a protein in purified parasite lysates (Fig. 1C and 2C). Together with our finding that glycolytic transcripts are increased in human fibroblasts infected with Toxoplasma type II ME49 tachyzoites (15), these data indicate that HIF-1 regulates HK2 expression in Toxoplasma-infected cells.
FIG 2 .

HK2 is regulated by HIF-1 in Toxoplasma-infected cells. HK2 transcript levels were measured in mock- or parasite-infected HIF-1αWT or HIF-1αKO MEFs grown at 21% (A) or 3% (B) O2. (C) Lysates from mock- or parasite-infected HIF-1αWT or HIF-1αKO MEFs (18 hpi) were blotted for HK2, SAG1, and β-actin. The value below each HK2 band represents the relative amount of HK2 in each sample normalized to the HK2 abundance in uninfected HIF-1αWT MEFs at 21% oxygen. (D) Quantification of four independent Western blot assays. Shown are average values and standard deviations from four assays. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001 (one-way ANOVA).
We next tested whether HK2 expression complements parasite growth in HIF-1αKO MEFs by infecting HIF-1αWT or HIF-1αKO MEFs transfected with empty-vector (EV), HK2, or enhanced-GFP (eGFP; an irrelevant protein control) expression plasmids and assessing parasite growth 72 h later. HK2, but not eGFP, significantly increased parasite growth at both O2 levels only in HIF-1αKO MEFs (Fig. 3B). Similar results were noted when GFP fluorescence was used to detect RH-βgal/GFP parasites growing in EV- or HK2-transfected HIF-1αWT or HIF-1αKO MEFs (see Fig. S3 in the supplemental material). Next, parasite growth was compared between host cells transfected with either an HK2 or an HK1 expression vector (the proteins were expressed at similar levels) (Fig. 3A). Parasite growth increased in HK1-transfected HIF-1αKO MEFs at 21% O2 but not at 3% O2 (Fig. 3C).
FIG 3 .
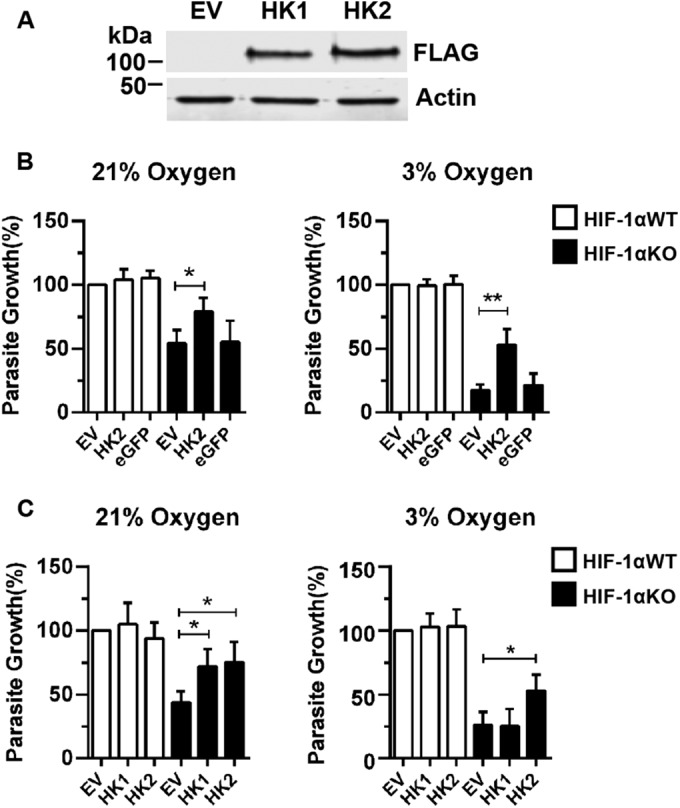
HK2 expression in HIF-1αKO MEFs restores Toxoplasma growth. (A) Representative Western blots of FLAG-tagged HK1 and HK2 expression in HIF-1αWT MEFs. (B) Parasite growth was measured 72 hpi in HIF-1αWT or HIF-1αKO MEFs transfected with EV, HK2, or eGFP expression constructs. (C) Parasite growth measured 72 hpi in HIF-1αWT or HIF-1αKO MEFs that were mock transfected or transfected with an HK2 or HK1 expression construct. Shown are averages and standard deviations of three experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (Student’s t test).
Infection increases host glycolysis in a HIF-1- and HK2-dependent manner.
Upregulation of HK2 should lead to increased G6P consumption by glycolysis and other G6P-regulated metabolic pathways. Therefore, levels of the glycolytic by-product lactate were measured in tissue culture supernatants to determine whether host glycolytic flux is elevated following infection. At both 21% and 3% O2, extracellular lactate abundance significantly increased in supernatants from infected HIF-1αWT MEFs (18 h postinfection [hpi]), which is consistent with earlier work (19). In contrast, lactate did not increase after HIF-1αKO MEFs were infected (Fig. 4A), even though parasite growth in HIF-1αKO MEFs at that time point was unaffected at either O2 tension (see Fig. S4B in the supplemental material). Next, we found that lactate levels were not increased at either O2 tension at 18 hpi in HK2 siRNA-transfected host cells (Fig. 4B), a time at which the siRNAs did not affect parasite growth at 21% O2 and only minimally at 3% O2 (see Fig. S4C).
FIG 4 .
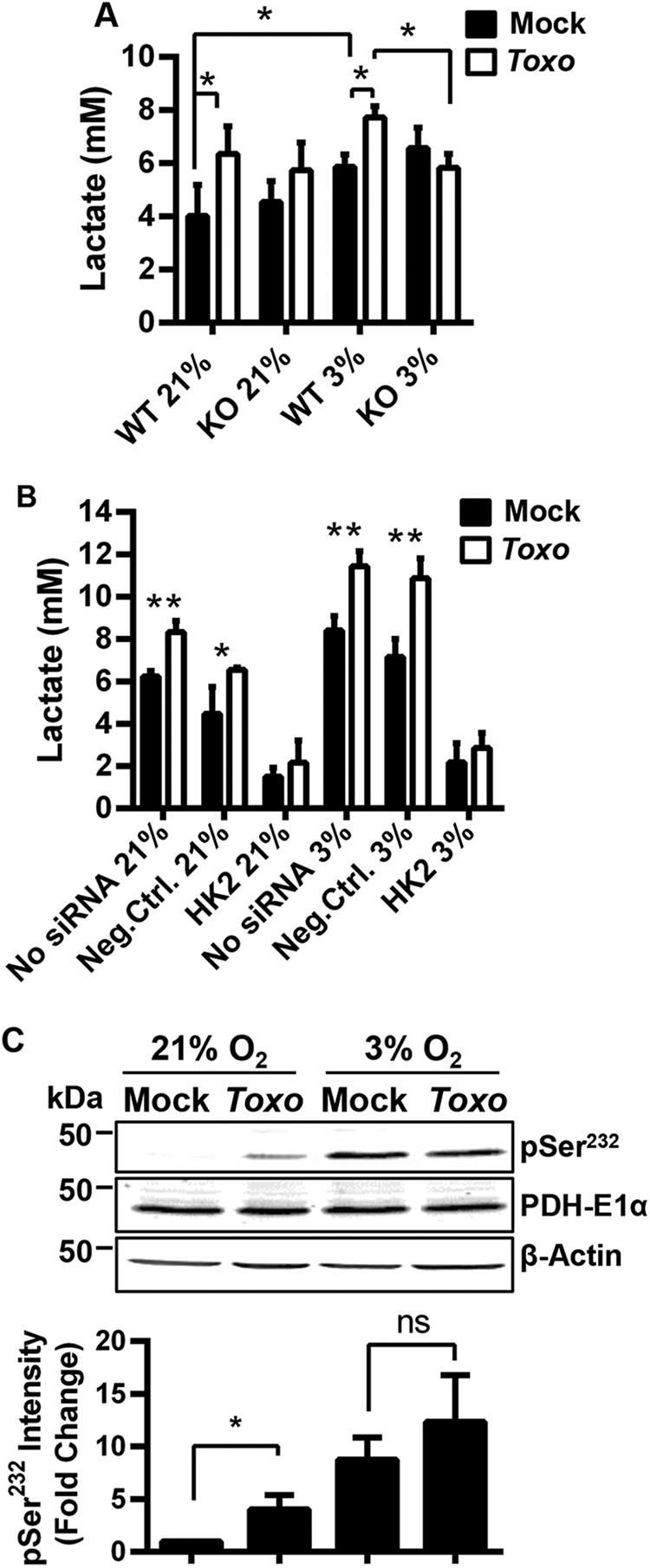
HK2 is functionally expressed in Toxoplasma-infected cells. (A and B) Extracellular lactate was measured in supernatants harvested at 18 hpi from HIF-1αWT and HIF-1αKO (A) or HeLa cells (B) that were mock transfected or transfected with negative-control or HK2 siRNAs. (C) Lysates from mock- or parasite-infected HIF-1αWT MEFs grown at 21 or 3% O2 for 18 h and blotted for total PDH E1α, phospho-PDH E1pSer232, or β-actin. The graph represents averages and standard deviations from three assays. ns, not significant; *, P < 0.05; **, P < 0.01 (one-way ANOVA [A and C] or Student’s t test [E]).
Monocarboxylic acid transporter 4 (MCT4) is a lactate efflux pump and HIF-1 target gene that is upregulated by infection (see Fig. S2) (15, 20). We tested whether decreased extracellular lactate levels from infected HIF-1αKO MEFs was due to decreased lactate export. Thus, intracellular lactate levels were measured in mock- and parasite-infected HIF-1αWT and HIF-1αKO cell extracts. No significant differences were observed, indicating that decreased lactate abundance in HIF-1αKO cell supernatants is not due to insufficient lactate export (see Fig. S4).
These data suggest that infection-induced increases in lactate are HIF-1- and HK2-dependent. However, some lactate measured in the culture supernatants could be parasite derived. While this cannot be resolved biochemically because of an inability to discriminate between host- and parasite-derived lactate, several lines of evidence indicate that even if parasite-derived lactate was being released, it was not significantly impacting our measurements. First, the time point at which the supernatants were collected was before parasite replication significantly decreased in either HIF-1αKO or HK2 siRNA-transfected cells (see Fig. S4B and C). Second, parasites grown in the HIF-1αKO MEFs at that time point take up larger amounts of [14C]glucose, which may be due to the increased intracellular glucose pools that accumulate when HK2 expression is decreased (not shown). We hypothesized that if infection increased host cell aerobic glycolysis and lactate production via elevated HK2, then the tricarboxylic acid (TCA) cycle would be reduced. One way to regulate metabolic flux through the TCA cycle is to limit the conversion of pyruvate to acetyl coenzyme A by inhibiting the pyruvate dehydrogenase (PDH) complex (21). Phosphorylation of the PDH E1α subunit on serine residues 232, 293, and/or 300 by PDH kinases inhibits PDH activity (21). Lysates from mock- or parasite-infected cells (18 hpi) were blotted with anti-PDH E1α or -phospho-PDH E1αSer232 antibodies, neither of which cross-react with the parasite (not shown). Infection increased PDH E1αS232 phosphorylation at 21% O2, while at 3% O2, its phosphorylation was similarly elevated in infected and uninfected cells (Fig. 4C), suggesting that PDH E1α is maximally phosphorylated at 3% O2. Together, these data indicate that Toxoplasma infection leads to a HIF-1-dependent increase in HK2 activity.
Toxoplasma growth requires HIF-1α and HK2 expression in nontransformed cells.
We next tested whether the dependence of Toxoplasma on host HIF-1α/HK2 was a phenotype associated with parasites growing in transformed HeLa and MEF cells (22) that rely on aerobic glycolysis even under O2-replete conditions. C2C12 cells are nontransformed immortalized myoblasts previously used to study Toxoplasma growth and differentiation (23, 24). HIF-1α and HK2 siRNAs efficiently reduced target gene expression without affecting C2C12 cell viability (Fig. 5A and B). Parasite growth in HIF-1α siRNA-transfected C2C12 cells was significantly reduced at 72 hpi at 3% but not 21% O2 (Fig. 5C). Toxoplasma growth was also reduced at 72 hpi at 21% O2 in HK2 siRNA-transfected C2C12 cells. But unlike in HIF-1α siRNA-transfected cells, parasite growth was decreased ~50% at 21% O2 in HK2 siRNA-transfected C2C12 cells (Fig. 5C). Thus, the requirement for HIF-1α and HK2 to support parasite growth is not restricted to transformed cells, such as HeLa cells and MEFs.
FIG 5 .
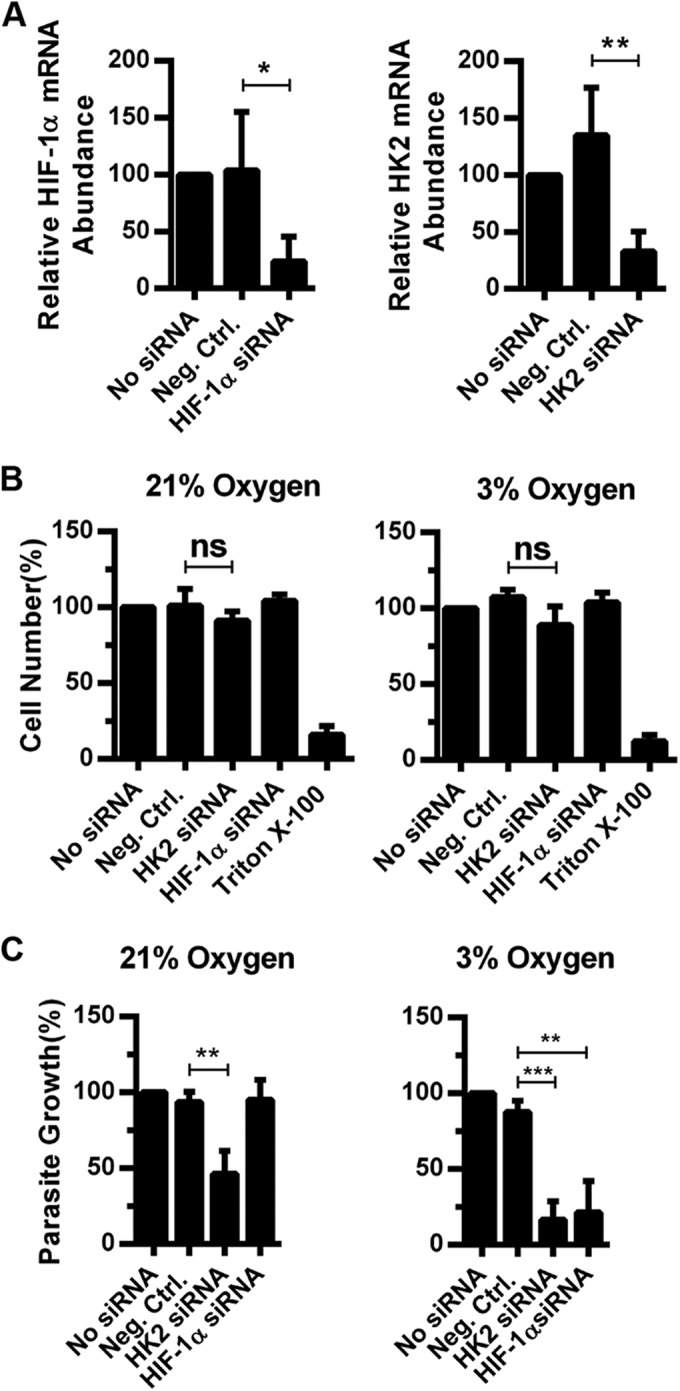
Parasite growth in C2C12 murine myoblasts is HIF-1α and HK2 dependent at physiological O2 levels. C2C12 cells were transfected with HIF-1α, HK2, or negative-control (Neg.Ctrl.) siRNAs, and 72 h later, target gene expression (A) or relative cell numbers (B) were measured. (C) siRNA-transfected C2C12 cells were infected and parasite growth was determined 72 h later. Shown are the averages and standard deviations of three assays. ns, not significant; *, P < 0.05; **, P < 0.01 (one-way ANOVA).
In vivo HK2 protein levels increase in Toxoplasma-infected mice.
In vivo HK2 expression was examined by intraperitoneally infecting mice with 105 RH-βgal/GFP parasites for 7 days. Protein lysates prepared from lungs, livers, and spleens were Western blotted to assess HK2 protein levels since these tissues contain tachyzoites at that time point (25). HK2 protein levels were significantly increased in livers and spleens but not in lungs (Fig. 6A).
FIG 6 .
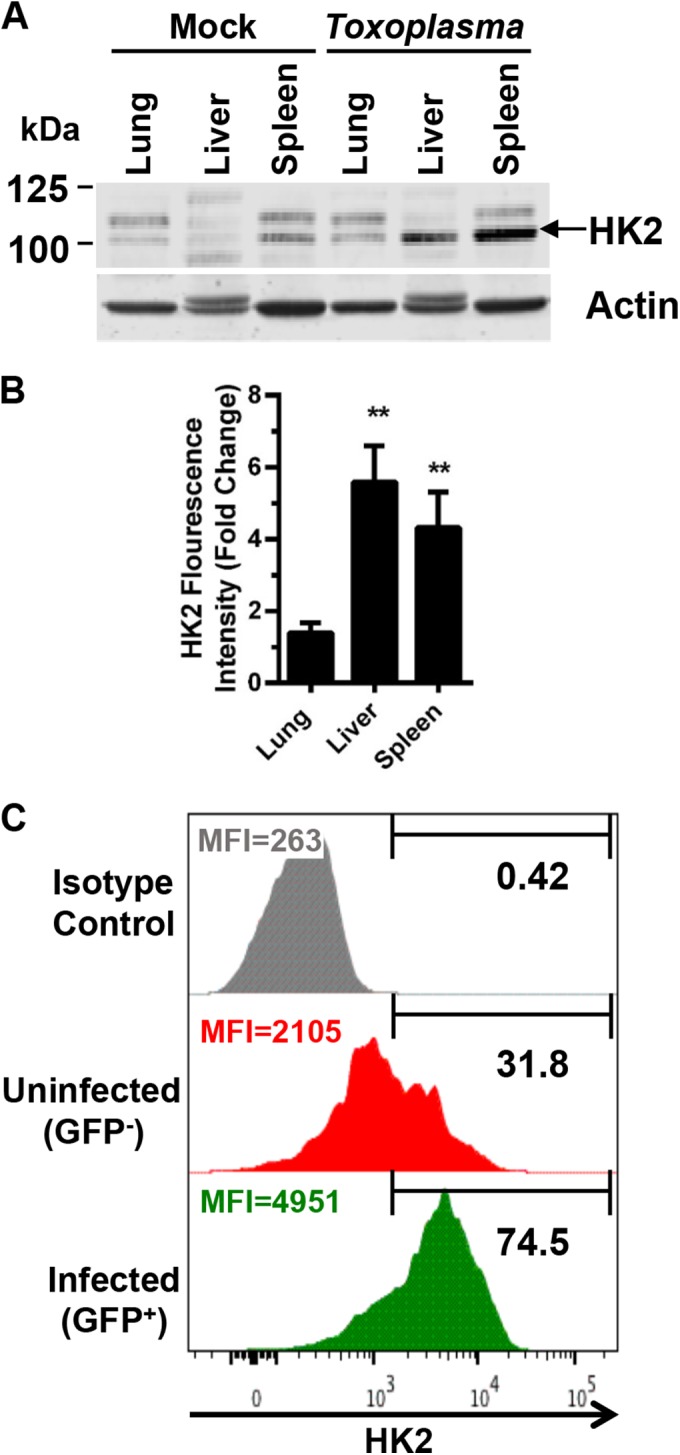
Toxoplasma increases HK2 expression in vivo. (A) Lungs, livers, and spleens were collected from mice 5 days postinfection and Western blotted to detect HK2 (HK2 band denoted by arrow). (B) Quantification of HK2 from three assays. Shown are averages and standard deviations. **, P < 0.01 (Student’s t test). (C) HK2 expression in infected (GFP+) and uninfected (GFP−) monocytes harvested from peritoneal cavities of intraperitoneally infected mice. The HK2hi expression gate was set empirically on the basis of fluorescence-activated cell sorter plots. Shown is a representative sample of five infected mice. MFI, mean fluorescence intensity.
To directly assess whether infected host cells increase HK2 expression, we intraperitoneally infected mice with 105 RH-βgal/GFP parasites, harvested the peritoneal exudate, and used flow cytometry to compare HK2 expression in parasite-infected (GFP+) and uninfected (GFP−) inflammatory monocytes, which are innate immune cells in which RH strain parasites proliferate (26). HK2 expression was significantly increased in the infected monocytes (Fig. 6C). It was unknown why approximately 31% of the GFP− monocytes were HK2hi. One possibility is that these cells were infected with Toxoplasma but killed the parasite.
Toxoplasma requires HK2 dissociation from the OMM for growth at 3% oxygen.
HK2 contains highly conserved N- and C-terminal catalytic domains and glucose recognition by both domains is dependent on aspartic acid residues D209 and D657 (in the N- and C-terminal domains, respectively) (Fig. 7A) (27). A third domain (yellow M in Fig. 7A) at the N terminus of HK2 inserts into the OMM to facilitate VDAC binding (8) (Fig. 7A; see Fig. S5 in the supplemental material). To test whether Toxoplasma growth requires HK2 catalytic activity and/or mitochondrial membrane association, HIF-1αWT or HIF-1αKO MEFs were transfected with wild-type, catalytically inactive (MuHK2), or OMM binding-deficient (TrHK2) HK2 constructs, all of which were expressed at similar levels (Fig. 7B). While parasite growth did not increase in any of the transfected HIF-1αWT MEFs or in MuHK2-transfected HIF-1αKO MEFs, parasite growth was similarly increased at 21% and 3% O2 in HIF-1αKO MEFs transfected with either the wild-type HK2 or TrHK2 mutant construct (Fig. 7C).
FIG 7 .

Toxoplasma promotes mitochondrial dissociation of HK2, but not HK1, at 3% O2. (A, B) Schematic of HK2 expression constructs (A) and Western blot assays confirming expression (B). (C) Parasite growth was measured 72 hpi in HIF-1αWT or HIF-1αKO MEFs transfected with the HK2 constructs indicated. (D and E) Mitochondrial (lanes M) and cytoplasmic (lanes C) fractions from uninfected or infected HIF-1αWT MEFs (18 hpi) were blotted to detect HK2 (D) or HK1 (E) and succinate dehydrogenase complex subunit A (SDHA), as a mitochondrial marker. Graphs show averages and standard deviations of relative HK2 or HK1 abundance in each fraction from three experiments. *, P < 0.05; **, P < 0.01 (one-way ANOVA). WCL, whole-cell lysate.
Since TrHK2 complemented parasite growth, we analyzed endogenous HK2 protein localization by Western blotting cytoplasmic and mitochondrial fractions from mock- or parasite-infected HIF-1αWT MEFs. In both uninfected and infected cells at 21% O2, ~75% of the HK2 was present in the cytoplasmic fraction (Fig. 7D). At 3% O2, ~83% of HK2 was mitochondrially associated in mock-infected cells, which is consistent with other work (28). Following infection, however, HK2 dissociated from the OMM (Fig. 7D). Similar results were noted by fluorescence microscopy (see Fig. S6A in the supplemental material). HK2 mitochondrial dissociation was specific since HK1 localization were unaffected by parasite infection (Fig. 7E). Moreover, HIF-1α was not required for HK2 mitochondrial dissociation (see Fig. S6B), indicating that HK2 expression and dissociation from the OMM are mutually exclusive.
To test whether Toxoplasma requires HK2 cytoplasmic localization to grow at 3% O2, we took advantage of the fact that HK1 remains associated with the OMM following infection and assayed parasite growth in cells expressing a truncated HK1 (TrHK1) mutant (Fig. 8A) that cannot bind to the OMM. TrHK1 restored parasite growth at 21% O2 similar to full-length HK1 (Fig. 8C); the proteins were expressed at similar levels (Fig. 8B). However, at 3% O2 only TrHK1 complemented parasite growth in HIF-1αKO MEFs. Together, these data indicate that at physiological O2 levels, Toxoplasma growth is dependent on its ability to promote HK2 dissociation from the OMM.
FIG 8 .
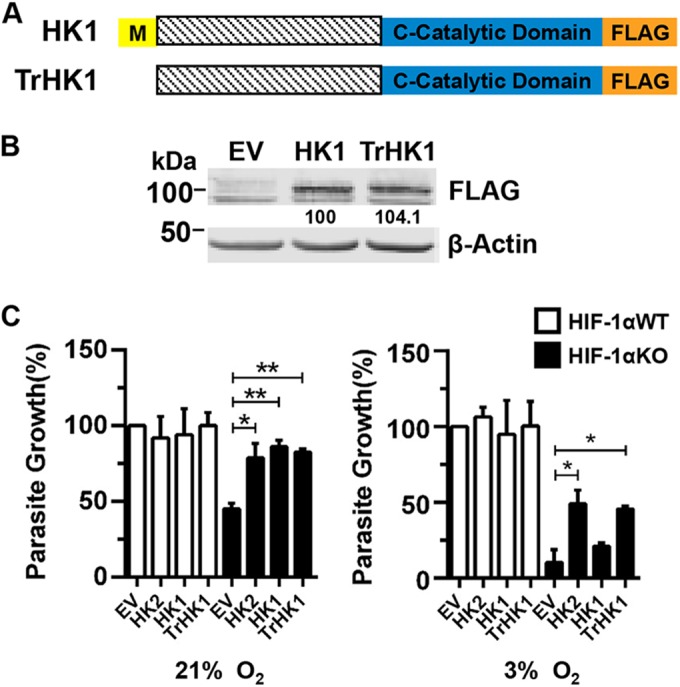
OMM binding-deficient HK1 complements parasite growth in HIF-1αKO MEFs at 3% O2. (A, B) Schematic of HK1 expression constructs (A) and Western blot assays confirming expression levels in HIF-1αWT MEFs (B). (C) Parasite growth was measured 72 hpi in HIF-1αWT or HIF-1αKO MEFs transfected with the constructs indicated. *, P < 0.05; **, P < 0.01 (one-way ANOVA).
DISCUSSION
A forward genetic screen was used to identify the HIF-1-regulated gene(s) needed for parasite growth at 3% O2. Of the genes identified, HK2 was the only known HIF-1 target important for parasite growth. We further showed that (i) HK2 expression in Toxoplasma-infected cells is HIF-1 dependent, (ii) parasite growth is restored in HK2-transfected HIF-1αKO MEFs, (iii) HIF-1/HK2 expression alters host metabolism, (iv) HK2 is upregulated in vivo and in nontransformed cells, and (v) Toxoplasma promotes and requires HK2 cytoplasmic localization at 3% O2.
siRNA screening is a powerful tool to define host cell processes used by pathogens to establish their replicative niches. While most genes from the screen were not suspected of functioning in Toxoplasma growth, their identification may reveal novel cellular processes that support parasite growth. For example, UBE2D2 functions in mitophagy, suggesting that Toxoplasma uses this specific autophagic process (29). Validating the screen was difficult since few host genes were known to be important for Toxoplasma growth. But EGLN1 (PHD2) siRNAs increased growth at 3% O2, which was expected since inhibition of PHD2 stabilizes and activates HIF-1 (30). We found that a significant number of hits from the primary screen were false positives, which was likely due to off-target effects of the siRNAs (31). For example, ACVR1C (ALK7), a member of the ALK4,5,7 receptor family, was a primary hit that was intriguing since ALK4,5,7 signaling mediates Toxoplasma activation of HIF-1 (30). Yet, the secondary screen did not confirm ACVR1C as a result of an off-target effect since ACVR1C mRNA was undetectable in HeLa cells (not shown). False negatives are another limitation of siRNA screens and are more challenging to identify. For example, HIF-1α siRNAs in the siRNA library did not reduce parasite growth, although control HIF-1α siRNAs purchased from a different company were included in each screening plate since they effectively decreased HIF-1α mRNA abundance and Toxoplasma growth at 3% O2.
The gene for HK2 and other glycolytic transcripts are HIF-1 targets that allow cells to shift from aerobic to anaerobic metabolism during hypoxic exposure. But we noted that while HIF-1 was important for HK2 expression, other transcription factors are likely involved in HK2 regulation. These could include c-myc and p53, which are activated by Toxoplasma and regulate HK2 levels (32–35). We also found that HK2 mRNA and protein levels were not coupled, implying that posttranslational mechanisms are also involved to fine tune HK2 protein abundance following infection.
At 3% O2, infection induced HK2 dissociation from the OMM to the cytosol. How HK2-OMM localization is regulated is largely unknown. AKT kinase, which promotes HK2 mitochondrial association by phosphorylating VDAC (36), is likely not involved since Toxoplasma activates host AKT (37). GSK-3β signaling promotes HK2 dissociation from the OMM (38), but Toxoplasma is not known to activate GSK-3β. OMM cholesterol content (39) and G6P levels (40) also affect HK-OMM association, and future work will define how Toxoplasma alters HK2 localization.
Full-length HK1 complemented parasite growth at 21% O2 but not at 3% O2. We speculate that at 21% O2, a threshold amount of endogenous HK1/2 is cytoplasmically localized and that transgenic HK1 expression yields enough additional cytoplasmic HK activity in the knockout cells to support parasite growth. But at 3% O2, HK1 does not dissociate from the OMM following infection. Similarly, differences in parasite growth between cell lines (e.g., HIF-1α siRNAs [Fig. 1D] versus HIF-1αKO MEFs [Fig. 3] at 21% O2) may be due, in part, to differences between cells in cytoplasmic HK expression.
It is unknown what role HK2 has during infection. One possibility is that it mediates ATP production to meet the increased metabolic demands that infection places on a host cell, especially at decreased O2 levels when mitochondria are less active. The ability of TrHK2 to complement parasite growth supports this hypothesis, since cytoplasmic HK2 uses cytoplasmic ATP as a substrate, whereas OMM-associated HK2 uses intramitochondrial ATP (41). This may indicate a requirement for increased host glycolysis that we showed is HIF-1α and HK2 regulated in parasite-infected cells and is supported by our finding that infection upregulates glycolytic transcript abundance, as well as phosphorylation of PDH E1α. Alternatively, HK2 may fuel the pentose phosphate pathway that supplies the cell with ribose 5-phosphate and NADPH. Metabolomic studies are needed to resolve these models, but these studies are complicated by the difficulty of specifically isolating host metabolites in a manner suitable for downstream analysis.
HK2 expression may impact Toxoplasma growth in other ways. First, loss of HK2 may increase intracellular glucose levels that could form advanced glycation end products such as carboxyethyl lysine (CEL) (42). But CEL was undetectable in infected HK2-deficient cells (see Fig. S7 in the supplemental material). Alternatively, Toxoplasma may scavenge a G6P-derived metabolite. But, this is doubtful since Toxoplasma scavenges glucose at 21% O2 and likely at 3% O2 (43). In addition, nutrient starvation triggers Toxoplasma development into bradyzoites. But, type II ME49 parasites (used because RH strain does not form bradyzoites) do not form bradyzoites in HK2 siRNA-transfected host cells (see Fig. S8 in the supplemental material). These data are consistent with our finding that bradyzoites do not form in HIF-1αKO MEFs, as well as other work showing that lactate restricts bradyzoite development (16, 19). Finally, Toxoplasma, like Theileria annulata, may activate HIF-1 and induce aerobic glycolysis in order to decrease oxygen radical production. But, this model conflicts with the finding that reactive oxygen species levels are reduced when HK2 is OMM associated (44, 45).
Aerobic glycolysis is a well-established feature of tumorigenic cells, as well as other types of cells. For example, macrophages and T cells must switch to glycolysis-fueled metabolism in order to function properly (46–48). This is consistent with our finding that HK2 is elevated in Toxoplasma-infected monocytes. However, macrophage activation of HIF-1 and aerobic glycolysis are features of M1 macrophages (7), whereas the RH strain parasites used here elicit M2 macrophages (49, 50). This discrepancy may be resolved by recent work showing that macrophages from Toxoplasma-infected mice display both M1 and M2 phenotypes (51). Thus, we hypothesize that parasite growth in monocytes requires activated HIF-1/HK2 to promote replication while the immunological features of the host cell are dictated by the parasite strain.
In summary, we report that Toxoplasma regulates HK2 through two distinct mechanisms. First, Toxoplasma upregulates HK2 by activating HIF-1. Second, a critical level of cytoplasmic HK2 is needed to support parasite replication, and at 21% O2 this is likely achieved solely by increased HK2 expression. But, at decreased O2 levels Toxoplasma triggers HK2 release from the OMM. We speculate that HK2 dissociation is important because mitochondrial ATP production at physiological O2 levels is reduced, leading to HK2 dissociation from the OMM in order for the kinase to use cytoplasmic ATP as a cosubstrate. Our future work will test this hypothesis, as well as determine which HK2-dependent pathways are required for Toxoplasma. However, HK2 could not fully complement growth in HIF-1αKO MEFs, suggesting that other genes/pathways are needed for parasite growth.
MATERIALS AND METHODS
Ethics.
Animal protocols (MIC12093Y) were approved by the SUNY at Buffalo IACUC and carried out in accordance with the Public Health Service Policy on the Humane Care and Use of Laboratory Animals and AAALAC accreditation guidelines.
Cells and parasites.
All cells and parasites were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), glutamine, and sodium pyruvate, unless otherwise noted (16). GFP-histone H2-B HeLa cells were provided by Geoff Wahl, and HIF-1αWT and HIF-1αKO MEFs were from Randy Johnson. All other cells (human foreskin fibroblasts, HeLa cells, and C2C12 cells) were from the ATCC. Host cells and parasites were regularly tested for mycoplasma with the Mycoplasma Detection kit (Lonza; Basel, Switzerland) and found to be negative. Unless noted otherwise, all experiments used the RH-βgal/GFP strain provided by Gustavo Arrizabalaga.
Plasmids.
All HK expression constructs were purchased from Addgene (Cambridge, MA). Inserts were digested out of pGFP-N3 with NheI and KpnI and ligated into the C-terminal FLAG tag-containing vector pFLAG-CMV5.1 with T4 DNA ligase (Invitrogen). All constructs were verified by sequencing.
siRNA screening and siRNA transfections.
A siRNA library targeting 9,102 human genes with three siRNAs per gene (for a list of the genes, see Table S1 in the supplemental material) was purchased from Ambion (Austin, TX). siRNA sets were pooled and aliquoted into individual wells of 96-well plates. Eight thousand HeLa cells were added to each well and reverse transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA). After 24 h, wells were rinsed twice with medium and incubated 24 h longer; the cells were then infected with 4,000 parasites/well and grown for 72 h at either 21% or 3% O2 in an InVivo300 Hypoxia Workstation (Baker Ruskinn; Sanford, ME). Medium was removed, and β-galactosidase activity was measured by adding 20 µM chlorophenol red-β-d-galactopyranoside (CPRG) in 100 µl of Z buffer (52). Parasite numbers were determined by linear regression analysis from a standard curve prepared in each plate and normalized to the average number of parasites in all wells of a plate. Each siRNA was tested at least three times, and averages were calculated. Z scores [(x − mean)/standard deviation] were calculated by using the mean and standard deviation of the entire library. Z scores of less than or equal to −2 or ≥2 indicated significant decreases or increases, respectively, in parasites growth. The MTT assay assessed HeLa cell viability at 96 h posttransfection.
Murine C2C12 myoblasts were seeded into 96-well plates and 24 h later transfected with RNAiMAX (Invitrogen). After 6 h, the siRNAs were removed and the cells were grown for 48 h and then infected with parasites at 21% or 3% O2 for 72 h. Cell viability was determined with the CellTiter-Blue Assay (Promega, Madison, WI).
Western blotting.
Unless stated otherwise, host cells were infected with type I RH strain parasites at a multiplicity of infection (MOI) of 4 (4:1 ratio of parasites to host cells). Whole-cell lysates were prepared in ice-cold radioimmunoprecipitation assay (RIPA) buffer and Western blotted as previously described (30). Mitochondrial and cytoplasmic fractions were prepared from 107 cells/sample by Dounce homogenization using the mitochondrial isolation kit (Thermo Scientific, Waltham, MA). For the antibodies used, see Table S2 in the supplemental material. Western blots were imaged with an Odyssey CLx infrared scanner (LI-COR, Lincoln, NE), and fluorescence intensity was quantified with Image Studio 3.1.
qRT-PCR.
qRT-PCR was performed as previously described (53). Briefly, DNase-treated total RNA was reverse transcribed with random hexamers. cDNA (25 ng) was mixed with gene-specific primers (see Table S2 in the supplemental material) and SYBR green PCR mix (Applied Biosystems). Data were collected and analyzed with an ABI 7500 Fast RT-PCR machine (Applied Biosystems). Changes were calculated by the 2−CtΔΔ method.
HK2 complementation assays.
HIF-1αWT and HIF-1αKO MEFs were transfected as previously described (30) and then infected with RH-βgal/GFP parasites. Parasite growth was determined 72 h later with CPRG. Relative parasite growth was calculated by using a standard curve generated with HIF-1αWT MEFs.
Lactate assays.
Medium collected from mock- or parasite-infected HIF-1αWT and HIF-1αKO MEFs grown for 18 h in phenol red-free DMEM lacking sodium pyruvate with 1% FBS was centrifuged at 16,000 × g at 4°C to remove cell debris. Samples were diluted, and lactate was measured with the lactate assay kit (Eton Bioscience, San Diego, CA). Intracellular lactate levels were measured in cells lysed with 95% ethanol.
Fluorescence microscopy.
Coverslips in 24-well plates were fixed with 3% paraformaldehyde in phosphate-buffered saline, permeabilized, and stained as follows. Mitochondria were detected by incubation with 250 mM Mitotracker CMXRos (Invitrogen) at 37°C for 30′ before fixation. Bradyzoites were induced by high-pH medium and detected by Dolichos biflorus agglutinin (DBA) staining as previously described (16). Rabbit anti-SAG1 antibody (from John Boothroyd) was used to detect tachyzoites. Coverslips were mounted in 4′,6-diamidino-2-phenylindole (DAPI)-containing Vectashield (Vector Labs, Burlingame, CA) and imaged by fluorescence microscopy.
Murine infections.
C57BL/6 mice were mock injected or intraperitoneally injected with 105 RH parasites, and their lungs, livers, and spleens were collected 7 days postinfection. Lysates were prepared by sonicating 100 mg of tissue in 500 µl of lysis buffer (RIPA buffer plus protease inhibitors). HK2 expression in peritoneal exudate Ly6C+ inflammatory monocytes (54) was assessed in cells that were harvested 5 days after intraperitoneal infection of C57BL/6 mice with 105 RH-βgal/GFP parasites. HK2 expression was compared in the infected (GFP+) and uninfected (GFP−) cells. Data were analyzed with FlowJo software.
Statistical analysis.
When appropriate, one-way ANOVA with Tukey’s post hoc test or Student’s t test was performed with GraphPad Prism (GraphPad, La Jolla, CA).
SUPPLEMENTAL MATERIAL
Optimization of siRNA-mediated gene knockdown in Toxoplasma-infected cells. (A) GFP-histone H2B-HeLa cells were transfected with GAPDH or GFP siRNAs, and GFP expression was imaged 96 h later. (B) GAPDH transcript abundance was measured by qRT-PCR in HeLa cells transfected with negative control or GAPDH siRNAs. (C) HeLa cells were transfected with the indicated siRNAs, and target gene expression was measured 72 h later. (D) Parasite growth was measured 72 h after infecting HeLa cells that were transfected with the indicated siRNAs. (E) HK2 transcript levels were measured by qRT-PCR 96 h after HeLa cells were transfected with individual HK2 siRNAs or with pooled siRNAs. (F) HeLa cell numbers were measured 72 h after transfection with negative-control, HK2, or HIF-1α siRNAs. As a control, cells were lysed with 0.25% Triton X-100. Shown are averages and standard deviations of three assays. Download
Glycolytic enzyme mRNA abundance in Toxoplasma-infected HIF-1αWT or HIF-1αKO MEFs. Total RNA was collected from mock- or parasite-infected HIF-1αWT or KO MEFs and used to assess the indicated transcript levels. Shown are averages and standard deviations from three assays. *, P < 0.05; **, P < 0.01 (Student’s t test). Download
GFP fluorescence intensity parallels parasite growth in HK2-transfected HIF-1αKO MEFs. Shown are representative images of wells from the experiment in Fig. 3B showing GFP expression from RH-βgal/GFP parasites in each sample. Download
Infection does not increase intracellular lactate levels in HIF-1αKO MEFs. Intracellular lactate levels were measured 18 h after HIF-1αWT or HIF-1αKO MEFs were mock- or parasite-infected. Shown are averages and standard deviations from three assays. (B and C) Number of parasites per vacuole in HIF-1αWT and HIF-1αKO MEFs (B) or HeLa cells transfected with negative control or HK2 siRNAs (C) were determined 18 hpi. Shown are averages and standard deviations from three experiments. Download
The HK2 mitochondrial binding domain-deficient mutant protein (TrHK2) localizes to the cytoplasm. Representative images of WT HK2, MuHK2, and TrHK2 subcellular localization in HIF-1αWT MEFs. Download
HIF-1α is not required for HK2 translocation at 3% O2. (A) Cells were transfected with an HK2-GFP-tagged construct and then mock infected or infected with red fluorescent protein-expressing strain RH parasites for 18 h. The cells were fixed, mounted on slides with DAPI-containing mounting medium, and analyzed by fluorescence microscopy. Note the altered HK2-GFP staining at 3% O2 between the uninfected and infected cells. (B) Mitochondrial and cytoplasmic fractions prepared from uninfected or infected HIF-1αKO MEFs (18 hpi) were blotted to detect HK2 and SDHA as a mitochondrial marker. Graphs show average values and standard deviations from three independent experiments. Download
CEL does not accumulate in Toxoplasma-infected, HK2-depleted cells. HeLa cells transfected with either negative-control or HK2 siRNA were infected at 21% or 3% O2 (MOI of 0.1) for 72 h and treated with 200 µM methylglyoxal (MG) for the last 24 h. Cells were stained with SAG1 to detect parasites or with an anti-CEL antibody. Shown are representative images from three independent experiments. Download
Reduced HK2 expression does not induce bradyzoite differentiation. Negative-control- or HK2 siRNA-transfected HeLa cells on coverslips were infected with ME49 tachyzoites (MOI of 0.1) and grown at 21% or 3% O2 in the absence or presence of SB505124. After 72 h, the cells were fixed and labeled with anti-SAG1 (tachyzoite-specific surface protein) and DBA (a bradyzoite cyst wall-specific lectin). Fifty vacuoles were counted on each coverslip. Shown are average values and standard deviations from three assays. **, P < 0.01; Student’s t test. Download
(Primary Sceen Tab) siRNA screening data for the 9,102 genes that were screened. (Secondary Screen Tab) Included are parasite and host cell growth data for the 218 genes that were screened. Note that #NUM! indicates that the number was incalculable.
Names and sequences of the qRT-PCR primers used in this study.
ACKNOWLEDGMENTS
We thank Karen Sweeney for writing the custom Excel data analysis program and Jim Ajioka and members of the Blader laboratory for helpful discussions.
This work was supported by grants from the NIH (AI069986 and AI087485) and the American Cancer Society (MBC-114461) to I.J.B.
Footnotes
Citation Menendez MT, Teygong C, Wade K, Florimond C, Blader IJ. 2015. siRNA screening identifies the host hexokinase 2 (HK2) gene as an important hypoxia-inducible transcription factor 1 (HIF-1) target gene in Toxoplasma gondii-infected cells. mBio 6(3):e00462-15. doi:10.1128/mBio.00462-15.
REFERENCES
- 1.Ratcliffe PJ. 2013. Oxygen sensing and hypoxia signalling pathways in animals: the implications of physiology for cancer. J Physiol 591:2027–2042. doi: 10.1113/jphysiol.2013.251470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bruick RK, McKnight SL. 2001. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 3.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr. 2001. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 4.Mathupala SP, Rempel A, Pedersen PL. 2001. Glucose catabolism in cancer cells: identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J Biol Chem 276:43407–43412. doi: 10.1074/jbc.M108181200. [DOI] [PubMed] [Google Scholar]
- 5.Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, Hawkins C, Guha A. 2011. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med 208:313–326. doi: 10.1084/jem.20101470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koppenol WH, Bounds PL, Dang CV. 2011. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer 11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 7.Galván-Peña S, O’Neill LA. 2014. Metabolic reprograming in macrophage polarization. Front Immunol 5:420. doi: 10.3389/fimmu.2014.00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie GC, Wilson JE. 1988. Rat brain hexokinase: the hydrophobic N-terminus of the mitochondrially bound enzyme is inserted in the lipid bilayer. Arch Biochem Biophys 267:803–810. doi: 10.1016/0003-9861(88)90090-2. [DOI] [PubMed] [Google Scholar]
- 9.Abu-Hamad S, Zaid H, Israelson A, Nahon E, Shoshan-Barmatz V. 2008. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J Biol Chem 283:13482–13490. doi: 10.1074/jbc.M708216200. [DOI] [PubMed] [Google Scholar]
- 10.Arzoine L, Zilberberg N, Ben-Romano R, Shoshan-Barmatz V. 2009. Voltage-dependent anion channel 1-based peptides interact with hexokinase to prevent its anti-apoptotic activity. J Biol Chem 284:3946–3955. doi: 10.1074/jbc.M803614200. [DOI] [PubMed] [Google Scholar]
- 11.John S, Weiss JN, Ribalet B. 2011. Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS One 6:e17674. doi: 10.1371/journal.pone.0017674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.BeltrandelRio H, Wilson JE. 1992. Coordinated regulation of cerebral glycolytic and oxidative metabolism, mediated by mitochondrially bound hexokinase dependent on intramitochondrially generated ATP. Arch Biochem Biophys 296:667–677. doi: 10.1016/0003-9861(92)90625-7. [DOI] [PubMed] [Google Scholar]
- 13.Kim K, Weiss LM. 2004. Toxoplasma gondii: the model apicomplexan. Int J Parasitol 34:423–432. doi: 10.1016/j.ijpara.2003.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Munoz M, Liesenfeld O, Heimesaat MM. 2011. Immunology of Toxoplasma gondii. Immunol Rev 240:269–285. doi: 10.1111/j.1600-065X.2010.00992.x. [DOI] [PubMed] [Google Scholar]
- 15.Blader IJ, Manger ID, Boothroyd JC. 2001. Microarray analysis reveals previously unknown changes in Toxoplasma gondii-infected human cells. J Biol Chem 276:24223–24231. doi: 10.1074/jbc.M100951200. [DOI] [PubMed] [Google Scholar]
- 16.Spear W, Chan D, Coppens I, Johnson RS, Giaccia A, Blader IJ. 2006. The host cell transcription factor hypoxia-inducible factor 1 is required for Toxoplasma gondii growth and survival at physiological oxygen levels. Cell Microbiol 8:339–352. doi: 10.1111/j.1462-5822.2005.00628.x. [DOI] [PubMed] [Google Scholar]
- 17.Gaji RY, Huynh MH, Carruthers VB. 2013. A novel high throughput invasion screen identifies host actin regulators required for efficient cell entry by Toxoplasma gondii. PLoS One 8:e64693. doi: 10.1371/journal.pone.0064693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krajewska M, Kim H, Shin E, Kennedy S, Duffy MJ, Wong YF, Marr D, Mikolajczyk J, Shabaik A, Meinhold-Heerlein I, Huang X, Banares S, Hedayat H, Reed JC, Krajewski S. 2005. Tumor-associated alterations in caspase-14 expression in epithelial malignancies. Clin Cancer Res 11:5462–5471. doi: 10.1158/1078-0432.CCR-04-2527. [DOI] [PubMed] [Google Scholar]
- 19.Weilhammer DR, Iavarone AT, Villegas EN, Brooks GA, Sinai AP, Sha WC. 2012. Host metabolism regulates growth and differentiation of Toxoplasma gondii. Int J Parasitol 42:947–959. doi: 10.1016/j.ijpara.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ullah MS, Davies AJ, Halestrap AP. 2006. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem 281:9030–9037. doi: 10.1074/jbc.M511397200. [DOI] [PubMed] [Google Scholar]
- 21.Patel MS, Korotchkina LG. 2006. Regulation of the pyruvate dehydrogenase complex. Biochem Soc Trans 34:217–222. doi: 10.1042/BST20060217. [DOI] [PubMed] [Google Scholar]
- 22.Ryan HE, Lo J, Johnson RS. 1998. HIF-1alpha is required for solid tumor formation and embryonic vascularization. EMBO J 17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swierzy IJ, Lüder CG. 2015. Withdrawal of skeletal muscle cells from cell cycle progression triggers differentiation of Toxoplasma gondii towards the bradyzoite stage. Cell Microbiol 17:2–17. doi: 10.1111/cmi.12342. [DOI] [PubMed] [Google Scholar]
- 24.Yaffe D, Saxel O. 1977. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 270:725–727. doi: 10.1038/270725a0. [DOI] [PubMed] [Google Scholar]
- 25.Hitziger N, Dellacasa I, Albiger B, Barragan A. 2005. Dissemination of Toxoplasma gondii to immunoprivileged organs and role of toll/interleukin-1 receptor signalling for host resistance assessed by in vivo bioluminescence imaging. Cell Microbiol 7:837–848. doi: 10.1111/j.1462-5822.2005.00517.x. [DOI] [PubMed] [Google Scholar]
- 26.Fentress SJ, Behnke MS, Dunay IR, Mashayekhi M, Rommereim LM, Fox BA, Bzik DJ, Taylor GA, Turk BE, Lichti CF, Townsend RR, Qiu W, Hui R, Beatty WL, Sibley LD. 2010. Phosphorylation of immunity-related GTPases by a Toxoplasma gondii-secreted kinase promotes macrophage survival and virulence. Cell Host Microbe 8:484–495. doi: 10.1016/j.chom.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ardehali H, Yano Y, Printz RL, Koch S, Whitesell RR, May JM, Granner DK. 1996. Functional organization of mammalian hexokinase II: retention of catalytic and regulatory functions in both the NH- and COOH-terminal halves. J Biol Chem 271:1849–1852. [DOI] [PubMed] [Google Scholar]
- 28.Cheung EC, Ludwig RL, Vousden KH. 2012. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc Natl Acad Sci U S A 109:20491–20496. doi: 10.1073/pnas.1206530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geisler S, Vollmer S, Golombek S, Kahle PJ. 2014. The ubiquitin-conjugating enzymes UBE2N, UBE2L3 and UBE2D2/3 are essential for Parkin-dependent mitophagy. J Cell Sci 127:3280–3293. doi: 10.1242/jcs.146035. [DOI] [PubMed] [Google Scholar]
- 30.Wiley M, Sweeney KR, Chan DA, Brown KM, McMurtrey C, Howard EW, Giaccia AJ, Blader IJ. 2010. Toxoplasma gondii activates hypoxia inducible factor by stabilizing the HIF-1alpha subunit via type I activin like receptor kinase receptor signaling. J Biol Chem 285:26852–26860. doi: 10.1074/jbc.M110.147785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS. 2003. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol 21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- 32.Bougdour A, Durandau E, Brenier-Pinchart MP, Ortet P, Barakat M, Kieffer S, Curt-Varesano A, Curt-Bertini RL, Bastien O, Coute Y, Pelloux H, Hakimi MA. 2013. Host cell subversion by Toxoplasma GRA16, an exported dense granule protein that targets the host cell nucleus and alters gene expression. Cell Host Microbe 13:489–500. doi: 10.1016/j.chom.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 33.Mathupala SP, Heese C, Pedersen PL. 1997. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J Biol Chem 272:22776–22780. doi: 10.1074/jbc.272.36.22776. [DOI] [PubMed] [Google Scholar]
- 34.Franco M, Shastri AJ, Boothroyd JC. 2014. Infection by Toxoplasma gondii specifically induces host c-Myc and the genes this pivotal transcription factor regulates. Eukaryot Cell 13:483–493. doi: 10.1128/EC.00316-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. 2007. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol 27:7381–7393. doi: 10.1128/MCB.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. 2001. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev 15:1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim L, Denkers EY. 2006. Toxoplasma gondii triggers Gi-dependent PI 3-kinase signaling required for inhibition of host cell apoptosis. J Cell Sci 119:2119–2126. doi: 10.1242/jcs.02934. [DOI] [PubMed] [Google Scholar]
- 38.Pastorino JG, Hoek JB, Shulga N. 2005. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res 65:10545–10554. doi: 10.1158/0008-5472.CAN-05-1925. [DOI] [PubMed] [Google Scholar]
- 39.Baggetto LG, Clottes E, Vial C. 1992. Low mitochondrial proton leak due to high membrane cholesterol content and cytosolic creatine kinase as two features of the deviant bioenergetics of Ehrlich and AS30-D tumor cells. Cancer Res 52:4935–4941. [PubMed] [Google Scholar]
- 40.Pastorino JG, Shulga N, Hoek JB. 2002. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem 277:7610–7618. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- 41.Cesar Mde C, Wilson JE. 1998. Further studies on the coupling of mitochondrially bound hexokinase to intramitochondrially compartmented ATP, generated by oxidative phosphorylation. Arch Biochem Biophys 350:109–117. doi: 10.1006/abbi.1997.0497. [DOI] [PubMed] [Google Scholar]
- 42.Ramasamy R, Vannucci SJ, Yan SS, Herold K, Yan SF, Schmidt AM. 2005. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 15:16R–28R. doi: 10.1093/glycob/cwi053. [DOI] [PubMed] [Google Scholar]
- 43.Blume M, Rodriguez-Contreras D, Landfear S, Fleige T, Soldati-Favre D, Lucius R, Gupta N. 2009. Host-derived glucose and its transporter in the obligate intracellular pathogen Toxoplasma gondii are dispensable by glutaminolysis. Proc Natl Acad Sci U S A 106:12998–13003. doi: 10.1073/pnas.0903831106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Metheni M, Echebli N, Chaussepied M, Ransy C, Chéreau C, Jensen K, Glass E, Batteux F, Bouillaud F, Langsley G. 2014. The level of H(2)O(2) type oxidative stress regulates virulence of Theileria-transformed leukocytes. Cell Microbiol 16:269–279. doi: 10.1111/cmi.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.da-Silva WS, Gómez-Puyou A, de Gómez-Puyou MT, Moreno-Sanchez R, De Felice FG, de Meis L, Oliveira MF, Galina A. 2004. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J Biol Chem 279:39846–39855. doi: 10.1074/jbc.M403835200. [DOI] [PubMed] [Google Scholar]
- 46.Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley C, Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley CA, Ramalho-Santos J, Van Houten B, Schatten G. 2011. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS One 6:e20914. doi: 10.1371/journal.pone.0020914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, van den Bosch MW, Quinn SR, Domingo-Fernandez R, Johnston DG, Jiang JK, Israelsen WJ, Keane J, Thomas C, Clish C, Vanden Heiden M, Xavier RJ, O’Neill LA. 2015. Pyruvate kinase M2 regulates HIF-1alpha activity and IL-1beta induction and is a critical determinant of the Warburg effect in LPS-Activated macrophages. Cell Metab 21:65–80. doi: 10.1016/j.cmet.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, Weber JD, Pearce EJ, Jones RG, Pearce EL. 2013. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jensen KD, Wang Y, Wojno ED, Shastri AJ, Hu K, Cornel L, Boedec E, Ong YC, Chien YH, Hunter CA, Boothroyd JC, Saeij JP. 2011. Toxoplasma polymorphic effectors determine macrophage polarization and intestinal inflammation. Cell Host Microbe 9:472–483. doi: 10.1016/j.chom.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Butcher BA, Fox BA, Rommereim LM, Kim SG, Maurer KJ, Yarovinsky F, Herbert DR, Bzik DJ, Denkers EY. 2011. Toxoplasma gondii rhoptry kinase ROP16 Activates STAT3 and STAT6 resulting in cytokine inhibition and arginase-1-dependent growth control. PLoS Pathog 7:e1002236. doi: 10.1371/journal.ppat.1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patil V, Zhao Y, Shah S, Fox BA, Rommereim LM, Bzik DJ, Yap GS. 2014. Co-existence of classical and alternative activation programs in macrophages responding to Toxoplasma gondii. Int J Parasitol 44:161–164. doi: 10.1016/j.ijpara.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eustice DC, Feldman PA, Colberg-Poley AM, Buckery RM, Neubauer RH. 1991. A sensitive method for the detection of beta-galactosidase in transfected mammalian cells. Biotechniques 11:739–740, 742. [PubMed] [Google Scholar]
- 53.Phelps ED, Sweeney KR, Blader IJ. 2008. Toxoplasma gondii rhoptry discharge correlates with activation of the EGR2 host cell transcription factor. Infect Immun 76:4703–4712. doi: 10.1128/IAI.01447-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Charles E, Joshi S, Ash JD, Fox BA, Farris AD, Bzik DJ, Lang ML, Blader IJ. 2010. CD4 T-cell suppression by cells from Toxoplasma gondii-infected retinas is mediated by surface protein PD-L1. Infect Immun 78:3484–3492. doi: 10.1128/IAI.00117-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Optimization of siRNA-mediated gene knockdown in Toxoplasma-infected cells. (A) GFP-histone H2B-HeLa cells were transfected with GAPDH or GFP siRNAs, and GFP expression was imaged 96 h later. (B) GAPDH transcript abundance was measured by qRT-PCR in HeLa cells transfected with negative control or GAPDH siRNAs. (C) HeLa cells were transfected with the indicated siRNAs, and target gene expression was measured 72 h later. (D) Parasite growth was measured 72 h after infecting HeLa cells that were transfected with the indicated siRNAs. (E) HK2 transcript levels were measured by qRT-PCR 96 h after HeLa cells were transfected with individual HK2 siRNAs or with pooled siRNAs. (F) HeLa cell numbers were measured 72 h after transfection with negative-control, HK2, or HIF-1α siRNAs. As a control, cells were lysed with 0.25% Triton X-100. Shown are averages and standard deviations of three assays. Download
Glycolytic enzyme mRNA abundance in Toxoplasma-infected HIF-1αWT or HIF-1αKO MEFs. Total RNA was collected from mock- or parasite-infected HIF-1αWT or KO MEFs and used to assess the indicated transcript levels. Shown are averages and standard deviations from three assays. *, P < 0.05; **, P < 0.01 (Student’s t test). Download
GFP fluorescence intensity parallels parasite growth in HK2-transfected HIF-1αKO MEFs. Shown are representative images of wells from the experiment in Fig. 3B showing GFP expression from RH-βgal/GFP parasites in each sample. Download
Infection does not increase intracellular lactate levels in HIF-1αKO MEFs. Intracellular lactate levels were measured 18 h after HIF-1αWT or HIF-1αKO MEFs were mock- or parasite-infected. Shown are averages and standard deviations from three assays. (B and C) Number of parasites per vacuole in HIF-1αWT and HIF-1αKO MEFs (B) or HeLa cells transfected with negative control or HK2 siRNAs (C) were determined 18 hpi. Shown are averages and standard deviations from three experiments. Download
The HK2 mitochondrial binding domain-deficient mutant protein (TrHK2) localizes to the cytoplasm. Representative images of WT HK2, MuHK2, and TrHK2 subcellular localization in HIF-1αWT MEFs. Download
HIF-1α is not required for HK2 translocation at 3% O2. (A) Cells were transfected with an HK2-GFP-tagged construct and then mock infected or infected with red fluorescent protein-expressing strain RH parasites for 18 h. The cells were fixed, mounted on slides with DAPI-containing mounting medium, and analyzed by fluorescence microscopy. Note the altered HK2-GFP staining at 3% O2 between the uninfected and infected cells. (B) Mitochondrial and cytoplasmic fractions prepared from uninfected or infected HIF-1αKO MEFs (18 hpi) were blotted to detect HK2 and SDHA as a mitochondrial marker. Graphs show average values and standard deviations from three independent experiments. Download
CEL does not accumulate in Toxoplasma-infected, HK2-depleted cells. HeLa cells transfected with either negative-control or HK2 siRNA were infected at 21% or 3% O2 (MOI of 0.1) for 72 h and treated with 200 µM methylglyoxal (MG) for the last 24 h. Cells were stained with SAG1 to detect parasites or with an anti-CEL antibody. Shown are representative images from three independent experiments. Download
Reduced HK2 expression does not induce bradyzoite differentiation. Negative-control- or HK2 siRNA-transfected HeLa cells on coverslips were infected with ME49 tachyzoites (MOI of 0.1) and grown at 21% or 3% O2 in the absence or presence of SB505124. After 72 h, the cells were fixed and labeled with anti-SAG1 (tachyzoite-specific surface protein) and DBA (a bradyzoite cyst wall-specific lectin). Fifty vacuoles were counted on each coverslip. Shown are average values and standard deviations from three assays. **, P < 0.01; Student’s t test. Download
(Primary Sceen Tab) siRNA screening data for the 9,102 genes that were screened. (Secondary Screen Tab) Included are parasite and host cell growth data for the 218 genes that were screened. Note that #NUM! indicates that the number was incalculable.
Names and sequences of the qRT-PCR primers used in this study.