

Abstract
Cells are constantly confronted with endogenous and exogenous factors that affect their genomes. Eons of evolution have allowed the cellular mechanisms responsible for preserving the genome to adjust for achieving contradictory objectives: to maintain the genome unchanged and to acquire mutations that allow adaptation to environmental changes. One evolutionary mechanism that has been refined for survival is genetic variation. In this review, we describe the mechanisms responsible for two biological processes: genome maintenance and mutation tolerance involved in generations of genetic variations in mitotic cells of both Saccharomyces cerevisiae and Schizosaccharomyces pombe. These processes encompass mechanisms that ensure the fidelity of replication, DNA lesion sensing and DNA damage response pathways, as well as mechanisms that ensure precision in chromosome segregation during cell division. We discuss various factors that may influence genome stability, such as cellular ploidy, the phase of the cell cycle, transcriptional activity of a particular region of DNA, the proficiency of DNA quality control systems, the metabolic stage of the cell and its respiratory potential, and finally potential exposure to endogenous or environmental stress.
Keywords: fidelity of replication, dNTP pool, homologous recombination, transcription-associated genome instability, aneuploidy, mitochondrial genome maintenance
The stability of budding and fission yeast genomes is influenced by two contradictory factors: (1) the need to be fully functional, which is ensured through the replication fidelity pathways of nuclear and mitochondrial genomes through sensing and repairing DNA damage, through precise chromosome segregation during cell division; and (2) the need to acquire changes for adaptation to environmental challenges.
Graphical Abstract Figure.
The stability of budding and fission yeast genomes is influenced by two contradictory factors: (1) the need to be fully functional, which is ensured through the replication fidelity pathways of nuclear and mitochondrial genomes through sensing and repairing DNA damage, through precise chromosome segregation during cell division; and (2) the need to acquire changes for adaptation to environmental challenges.
INTRODUCTION
The efficiency of the processes that maintain stable DNA is important for cell viability. However, due to the unpredictability of their rapidly changing environment, cells that are unable to adapt might not persist. With respect to evolution, populations must secure a margin of genome variability that allows for the adjustment to new environmental conditions. This state is achieved through mutations. Mutations contribute to genome variations that provide the population with a reservoir of variability that enables it to survive in a changing environment; in this manner, mutations certainly drive evolution. Some mutations appear spontaneously as a consequence of replication errors, error-prone repair of DNA lesions provoked by endogenous factors (e.g. metabolites or reactive oxygen species) and unequal segregation of chromosomes during mitosis (even in cells not exposed to any genotoxic stress conditions). The types of emerging mutations and their rate depend on the availability and efficiency of DNA quality control systems, cell age and ploidy, cell-cycle phase and the metabolic stage of the cell. Types of mutations also rely on specific features of the DNA sequence itself. For example, repetitive DNA sequences, such as mononucleotide tracts, are difficult templates for DNA polymerases, causing frequent sliding (Fortune et al. 2005). Moreover, trinucleotide repeats are susceptible to the formation of DNA secondary structures and are likely to undergo expansions or contractions (Razidlo and Lahue 2008). In diploid cells in which the second copy of the genome sequence is present, loss of heterozygosity (LOH) occurs at a high rate, predominantly through allelic crossover but also via gene conversion and chromosome loss (Ohnishi et al. 2004). The frequency of rearrangements between two homologous loci in yeast genomes strongly depends on their chromosomal localization; therefore, nuclear architecture also influences genome maintenance (Agmon et al. 2013).
Both simple unicellular organisms and metazoan cells have similar mechanisms for genome maintenance; the same is true for the mechanisms that cause mutations. Because these mechanisms are evolutionarily conserved and their efficiency translates into similar error rates in the cells of all mesophilic organisms (Drake et al. 1998; Drake 2009), the rules governing the preservation of the intact genome established for yeast should be easily applicable to other cell types. Because yeast cells have served as the model for genome stability research for years, the wealth of knowledge in this field has been gathered for this organism. In this review, we present information collected in this field, not only for the budding yeast Saccharomyces cerevisiae that has served as the most frequent model organism but also for the fission yeast Schizosaccharomyces pombe. We hope that these data might be useful for work on all types of cells.
REPLICATION ERRORS
Replication is one of the main sources of spontaneous mutations despite the evolution of mechanisms to avoid mutations and maintain unchanged DNA sequences. Spontaneous mutations result from imperfections in any of three processes: (1) accurate nucleotide selection by DNA polymerases; (2) proofreading 3′→5′ exonuclease activity of replicative DNA polymerases; and (3) correction by DNA repair systems of the mistakes made by all DNA polymerases and filling the gaps left after replication.
Nobody is perfect: the DNA polymerases–enzymes imperfecta
Eukaryotic cells usually possess more than 10 DNA polymerases. For example, S. cerevisiae contains 11 (polymerases: α, β, γ, δ, ε σ, φ, θ, ζ, η and Rev1) (Saccharomyces Genome Database. http://www.yeastgenome.org/, 12 November 2014 date last accessed), Sc. pombe contains 12 (all homologs of S. cerevisiae polymerases plus Pol κ) (PomBase database. http://www.pombase.org/, 12 November 2014 date last accessed) and human cells contain up to 18 (polymerases: α, β, γ, δ, ε ι, κ, λ, μ, ν, σ, φ, θ, ζ, η, REV1, PRIMPOL and DNTT) (GeneCards. http://www.genecards.org/, 12 November 2014 date last accessed) (Table 1). These DNA polymerases belong to several polymerase families including A, B, X and Y. The role they play in cells is determined by their fidelity and processivity (Table 1). The enzymes that are the most precise in DNA synthesis belong to the B and A families of polymerases and are involved in replication. The less accurate enzymes belong mostly to the Y and X families of polymerases and are involved in DNA repair (e.g. in translesion synthesis, TLS). Because the functional mechanisms and roles of DNA polymerases in various processes were extensively studied in yeast S. cerevisiae cells, we will focus on data obtained from this model organism.
Table 1.
DNA polymerases and their functions in budding and fission yeast.
| Family | DNA polymerase | Fidelity of replication | Saccharomyces cerevisiae gene | Schizosaccharomyces pombe gene | Molecular functiona | Cellular processesa | Interactions important for DNA synthesis |
|---|---|---|---|---|---|---|---|
| B | Replicative | 10−4–10−5 | POL1 | pol1 | DNA polymerase | Replication initiation; DSB repair | Pol32, Ctf4 |
| polymerases | POL12 | spb70 | primase | Telomere capping | |||
| Pol α | PRI1 | spp1 | |||||
| PRI2 | spp2 | ||||||
| B | Pol ε | 10−6–10−7 | POL2 | pol2 | DNA polymerase 3′→5′ exonuclease | Leading-strand synthesis, telomere silencing, MMR, NER, DSB repair via NHEJ, mitotic sister chromatid cohesion | Mrc1 |
| DPB2 | dpb2 | Replisome assembly | |||||
| DPB3 | dpb3 | DNA binding | |||||
| DPB4 | dpb4 | ||||||
| B | Pol δ | 10−5–10−7 | POL3 | cdc6 | DNA polymerase, 3′→5′ exonuclease | Lagging-strand synthesis, telomere silencing, MMR, BER, NER, PRR | |
| POL31c | cdc1 | ||||||
| POL32c | cdc27 | PCNA, Pol1 | |||||
| cdm1 | |||||||
| TLS polymerases | |||||||
| B | Pol ζ | 10−4–10−5 | REV3 | rev3 | DNA polymerase | TLS, PRR | Rev1, Rad30, Pol3 |
| REV7 | rev7 | The cell-cycle control | |||||
| POL31c | |||||||
| POL32c | PCNA, Pol1 | ||||||
| Y | Rev1p | REV1 | rev1 | DNA polymerase, dCTP transferase, | TLS, PRR, DSB repair | PCNA, Pol ζ | |
| Y | Pol η | 10−1–10−2 | RAD30 | eso1 | DNA polymerase | TLS, PRR, DSB repair | |
| Y | Pol κ | dinB | TLSb | ||||
| The other DNA polymerases | |||||||
| A | Pol γ | 10−5–10−6 | MIP1 | pog1 | DNA polymerase, 3′→5′ exonuclease | mtDNA replication | |
| X | Pol β | 10−3–10−4 | POL4 | pol4 | DNA polymerase, DNA-directed RNA polymerase, dRP lyaseb | DSB repair via NHEJ, BER, gap filling | |
| B | Pol φ | POL5 | pol5 | DNA polymerase, rDNA binding | rRNA transcription | ||
| B | Pol σ | PAP2/TRF4 | cid14 | Poly(A) polymerase, dRP lyase | Nuclear poly(A)-dependent RNA catabolism BER | ||
| B | TRF5 | Poly(A) polymerase | BER |
All information listed in this Table 2 is available at (Saccharomyces Genome Database. http://www.yeastgenome.org/; PomBase. http://www.pombase.org/on-line-database) and the reader is referred to these sources, and the references therein for further details. Additional data have been published in (Kunkel et al. 1989; Shcherbakova et al. 2003; Fortune et al. 2005; Zhong et al. 2006; McCulloch et al. 2007; McCulloch and Kunkel 2008; Burgers 2009).
aGene Ontology annotations for S. cerevisiae protein. Orthologs in other fungi mostly play similar role in the cell. In some cases more information is available for gene product from other fungi than for its S. cerevisiae ortholog.
bGene Ontology annotations for Sc. pombe.
cPol31p and Pol32p are shared subunits for two DNA polymerases Pol δ and Pol ζ.
Budding yeast possesses three replicative DNA polymerases: polymerase alpha-primase, Pol α (Lucchini et al. 1987; Brooke and Dumas 1991; Brooke et al. 1991); polymerase delta, Pol δ (Chang 1977); and polymerase epsilon, Pol ε (Morrison et al. 1990). Pol α makes one mistake per 104–105 bases replicated (Kunkel et al. 1989), whereas Pol ε (operating on the leading strand) and Pol δ (operating on the lagging strand) (Karthikeyan et al. 2000; Larrea et al. 2010) exhibit error frequencies of 10−5–10−7 due to their proofreading 3′→5′ exonuclease activity (Shcherbakova et al. 2003; Fortune et al. 2005; McCulloch and Kunkel 2008; Burgers 2009). Polymerase gamma (Pol γ) has a similar error rate (10−5–10−6) and is responsible for replication of the mitochondrial genome (McCulloch and Kunkel 2008). The fallibility of DNA synthesis increases when the polymerase does not have proofreading activity. Under these conditions, the error frequency rises to 10−3–10−4, as was shown for Pol ζ and Pol β, or even to 10−1–10−2, as was calculated for Pol η (Zhong et al. 2006; McCulloch et al. 2007). These polymerases perform poorly on undamaged templates, and their contribution to spontaneous mutagenesis is limited due to their low processivity and strictly regulated access to the DNA template during normal undisturbed DNA synthesis. Instead, they have a unique ability to synthesize DNA from a damaged template, often allowing a proper reconstitution of the sequence in the nascent DNA strand (McCulloch et al. 2004b; Gibbs et al. 2005). These DNA polymerases are known by several different names in the literature. Sometimes they are called alternative polymerases because they are alternatives to replicative polymerases, while other times they are called TLS polymerases because they synthesize across lesions in DNA templates. They have even been called specialized polymerases because they are able to insert the right nucleotide across from the damaged one. Each alternative polymerase has its own cognate substrate; we will describe this phenomenon in more detail in a later section. Only Pol ζ differs from this scheme because it synthesizes longer DNA stretches. Due to its low fidelity, it is responsible for the majority of mutations (Stone et al. 2009; Northam et al. 2010; Stone, Lujan, Kunkel 2012). In fact, Pol ζ is the most mutagenic among all DNA polymerases found in yeast.
DNA synthesis errors can be introduced during two stages of the process: during selection of a correct dNTP for synthesis and at the extension step. dNTP selection occurs mainly by Watson–Crick base pairing and discrimination against each of the possible mismatched bases prior to covalent incorporation (Echols and Goodman 1991). Misincorporation might take place as a result of the balance between synthesis and excision that compromises the rate of DNA extension, both of which can be influenced by template sequence, abundance of dNTP pools and their bias (Bebenek and Kunkel 1990, 2000). Thus, insertion of an improper dNTP during replication may lead to the creation of a mismatch, as well as the occurrence of a frameshift mutation, according to the following factors: (1) base to base hydrogen bonding between nucleotides in the template and nascent strands; (2) base pair geometry and substrate-induced conformational changes by the DNA polymerase; (3) strength of contact between the polymerase and the DNA minor groove at and upstream of the active site of the polymerase, which influences nucleotide selectivity; and (4) the efficiency of 3′→5′ exonucleolytic proofreading and strand misalignment (Kunkel and Bebenek 2000) (Fig. 1). DNA polymerases are influenced differently by the factors listed above; therefore, each polymerase generates its unique pattern of errors by which it can be recognized. Each polymerase also has its own function in the cell, such as acting on the leading or lagging strand, gap filling and synthesizing mitochondrial DNA (mtDNA). The same factors influence both replicative and non-replicative DNA polymerases, with the exception that the latter group lacks 3′→5′ exonuclease activity. Additionally, Rev1 does not use canonical Watson–Crick base pairing during synthesis. Probably for this reason, this enzyme has the lowest DNA fidelity among all known yeast DNA polymerases (Nair et al. 2005; Prakash, Johnson and Prakash 2005; Waters et al. 2009). On the opposite side of the DNA synthesis fidelity scale are the major replicative polymerases Pol δ and Pol ε. Pol δ was shown to have high selectivity for the correct nucleotide based on kinetic studies of the insertion of individual dNTPs opposite a known nucleotide in a DNA template. This ability together with its proofreading 3′→5′ exonuclease activity establishes Pol δ as the most accurate polymerase in yeast. Nonetheless, Pol δ often generates single nucleotide deletions in homopolymeric runs and wastefully proofreads these types of mismatches with a much higher error rate (approximately three single nucleotide deletions per 104 nucleotides polymerized). Additionally, Pol δ may also slide over templates containing direct repeats spaced by three or more base pairs, resulting in deletions of the nucleotides between these direct repeats. Pol δ strand slippage on repetitive sequences during replication is believed to be the major source of insertions and deletions in yeast genomes (Fortune et al. 2005). As mentioned above, DNA polymerases can be distinguished by the pattern of mistakes they make. Although both Pol δ and Pol ε incorporate nucleotides with high fidelity, Pol δ has a higher probability of sliding on repetitive sequences. In contrast, the high fidelity of Pol ε results from the high effectiveness of its 3′→5′ exonuclease proofreading activity rather than from base selection correctness; this finding is based on the in vitro spectrum of errors (mutation spectra) observed for a proofreading-deficient form of Pol ε that showed a unique error signature with a high proportion of transversions resulting from T-T, T-C and C-T mispairs (Shcherbakova et al. 2003). In turn, the disruption of S. cerevisiae Pol γ exonuclease activity increased the mtDNA deletion rate 160-fold, indicating that exonuclease activity is crucial for avoiding deletions during mtDNA replication (Stumpf and Copeland 2013). This result also suggested a possible source of mtDNA deletions of the progeroid phenotype in exonuclease-deficient DNA polymerase γ in mice (Stumpf and Copeland 2013). Pol γ proofreading 3′→5′ exonuclease activity minimizes the frequency of point mutations and prevents deletions, thereby contributing to the stabilization of mtDNA in yeast cells (Vanderstraeten et al. 1998). However, apart from its proofreading activity, the exonuclease domain of Pol γ contributes to the coordination of its polymerase and exonuclease functions. This was documented by the analysis of phenotypes of mip1 (Pol γ) alleles, in which mutations were localized to the DNA-binding channel of the exonuclease domain in close vicinity to the polymerase domain. In these mutants, the imbalance between DNA synthesis and degradation caused poor mtDNA replication (Szczepanowska and Foury 2010). However, increased mutagenesis was also detected in strains encoding mutant mip1 variants that were unable to maintain mtDNA, although they were not affected by polymerase fidelity or exonuclease proofreading activity. Increased mutagenesis was in this case caused by slowing down the replication fork, thereby predisposing the template DNA to irreparable damage that was bypassed with a poor fidelity (Stumpf and Copeland 2014).
Figure 1.
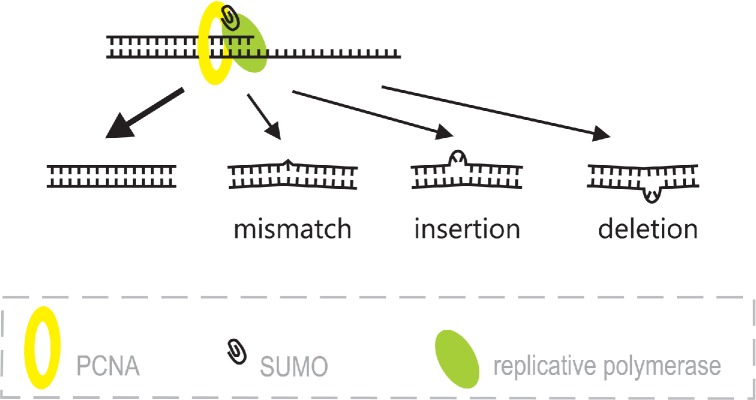
Various effects of DNA synthesis on undamaged template. DNA polymerase is most often accurate; however, from time to time it makes mistakes, such as mismatches and frameshifts (insertions or deletions), which cause DNA distortions.
During normal replication, three DNA polymerases (Pol α, Pol ε and Pol δ) work together at the replication fork to duplicate the DNA. The replication fork polymerases are programed to replicate opposite DNA strands; Pol ε synthesizes the leading strand, while primases Pol α and Pol δ polymerize the Okazaki fragments on the lagging strand (Karthikeyan et al. 2000; Larrea et al. 2010). This spatial arrangement causes replication fork asymmetry, which is observed in both budding and fission yeast and influences DNA synthesis fidelity (Kunkel 2011; Miyabe, Kunkel and Carr 2011). Nonetheless, the replicative polymerases might operate at the very same cytological foci (Hiraga et al. 2005), correcting one another's mistakes (Morrison and Sugino 1994; Pavlov et al. 2004; Burgers 2009).
Yeast replicative DNA polymerases work in complexes composed of several subunits that require proper assembly (Table 1). Formation of the holoenzyme depends on the Mms19 (Met18) protein belonging to the cytosolic iron–sulfur protein assembly machinery and encompasses not only assembly of the active enzyme from several subunits (which most likely requires chaperone assistance) but also installation of the necessary cofactors (Stehling et al. 2012). Mms19 is involved in assembly of the [4Fe-4S] cluster in the CysB motif of several DNA polymerases, including Pol α, Pol ε Pol δ and Pol ζ. This prosthetic group is needed for enzyme stabilization. Installation of another cofactor (the Zn2+ ion in the CysA motif of Pol δ) is required for its PCNA-mediated processivity (Netz et al. 2011). The major subunit in the DNA polymerase holoenzyme is responsible for DNA polymerization and optionally for its 3′→5′ exonuclease proofreading activity. In S. cerevisiae cells, the accessory proteins contribute to the activity of the enzyme and influence its fidelity and processivity. The accessory subunits play an additional role in maintaining contact between the holoenzyme and other cellular components via various interactions. These interactions permit both access to the DNA template and the transmission of important cellular signals to the polymerase, allowing for a proper response. Thus, the accessory subunits may modulate polymerase activity. For example, the interaction between Pol32 (one of the non-catalytic subunit of Pol δ) and Pol30 determines Pol δ processivity. The homotrimer of Pol30 forms a circular structure called PCNA (proliferating cell nuclear antigen) that serves as the DNA polymerase processivity factor. The PCNA works as a sliding clamp encircling the DNA strand and tethering the polymerase to the template, thereby preventing its dissociation (Fukuda et al. 1995; Johansson, Garg and Burgers 2004). Pol32 interacts with the Pol1 subunit of Pol α. This interaction enables cooperation between Pol α and Pol δ that is critical for lagging-strand synthesis (Johansson, Garg and Burgers 2004). The accessory subunits of both replicases Pol δ and Pol ε permit their interaction with different proteins engaged in the DNA repair pathways. They compete for the same interactors (e.g. PCNA) with specialized polymerases, influencing their access to the DNA template. The Pol31 and Pol32 subunits of Pol δ and the Dpb2, Dpb3 and Dpb4 subunits of Pol ε contribute to the fidelity of replication by influencing dNTP selection and/or the proofreading activity of catalytic subunits of the respective holoenzyme and by stabilizing its interaction with DNA (Giot et al. 1997; Huang et al. 2002; Aksenova et al. 2010). Accordingly, mutant versions of the DPB2 gene significantly influence spontaneous mutagenesis (Jaszczur et al. 2009). The polymerase subunits also have an impact on regulation of the replication process. Disturbed interactions between Pol ε subunits and the GINS complex result in increased mutagenesis (Grabowska et al. 2014). The Pol1 interaction with chromatin-binding protein Ctf4 associated with the GINS complex couples the heterohexameric Mcm2-7 helicase to Pol α. This interaction is important for the formation of the correct replisome progression complex on every Okazaki fragment during lagging-strand synthesis (Gambus et al. 2009). In contrast, the Pol2 Pol ε catalytic subunit binds to Mrc1 when this S-phase checkpoint protein is associated with the Mcm2-7 helicase. This binding permits matching of the polymerization with the unwinding rate on the leading DNA strand of the replication fork during normal replication (Lou et al. 2008). Another possible source of replication errors is continued DNA synthesis when the DNA is subjected to damage. To avoid this scenario during DNA damage stress, the checkpoint protein Mrc1 is phosphorylated in a Mec1-dependent manner. Then, phosphorylated Mrc1 interacts with Tof1 to form a pausing complex that is required for Pol ε stabilization at stalled replication forks (Osborn and Elledge 2003). Thus, transient interactions of replicative polymerases with the Ctf4 and Mrc1 proteins protect cells against chronic activation of the DNA damage checkpoint during chromosome replication and permit the finishing of DNA synthesis and subsequent finalization of the cell cycle. Replication blocks can also be overcome by activating the checkpoint response to regulate template switching or origin firing, fork restart and cell-cycle progression, all of which will engage DNA repair (Friedel, Pike and Gasser 2009). Presumably, almost all components of the replication fork machinery contribute to replication accuracy. Indeed, improper functioning of proteins engaged in initiation, elongation or replication control manifest as a mutator phenotype. Among other abnormalities, genome instability phenotypes are present in strains defective in origin recognition complex, various replication factor C (RFC) complexes, and Mcm2-7 helicase complex, and these phenotypes have been reported for cdc45, csm3, ctf18, dpb11, pol30 and sld4 mutant cells (Stone et al. 2008; Alabrudzinska, Skoneczny and Skoneczna 2011; Li and Tye 2011; Cheng et al. 2012). Significantly, mutations in genes encoding proteins functioning in the replication fork that frequently cause replication stalling often lead to gross chromosomal rearrangements (GCR), whereas mutations arising as a consequence of polymerase defects are mainly base substitutions and frameshifts.
The availability of DNA synthesis precursor dNTPs affects the quality of the nascent DNA
DNA synthesis requires a constant supply of dNTPs to proceed. The size of the dNTP pools and their bias (i.e. relative concentrations) must be tightly regulated to ensure optimized DNA metabolism. dNTP pool concentrations oscillate during the cell cycle, reaching a maximum during transition through the G1/S boundary. This feature helps cells set up for the next round of replication and allows progression of the cell cycle (Koç et al. 2003; Chabes and Stillman 2007). dNTP pools also rise in response to DNA damage and stalling of the replication fork, facilitating replication through DNA lesions (Chabes et al. 2003; Koç et al. 2004; Chabes and Stillman 2007; Lis et al. 2008; Sabouri et al. 2008;Davidson et al. 2012; Poli et al. 2012). However, according to data obtained for various eukaryotic cells, this bypass is often mutagenic (Echols and Goodman 1991; Lis et al. 2008; Sabouri et al. 2008). As was shown for human cells, low dNTP concentrations favor high-fidelity replication, whereas high dNTP concentrations stimulate polymerization reactions and inhibit the proofreading activity of replicases, thereby enhancing the dNTP misincorporation rate (Kunkel, Silber and Loeb 1982; Echols and Goodman 1991). However, the amount of dNTPs is not the only factor that contributes to this phenomenon. As was shown for yeast cells, increased levels of misinsertions, strand misalignments and mismatch extensions at the expense of proofreading can result from an imbalance in the dNTP pools (Kumar et al. 2011). Interestingly, changes in the dNTP pool size or bias can induce not only point mutations but also DNA breaks, which can lead to GCR (Ouspenski, Elledge and Brinkley 1999; Chabes et al. 2003; Fasullo et al. 2010; Davidson et al. 2012).
The maintenance of proper dNTP levels is essential for cellular viability and genome stability, therefore, the level and activity of the ribonucleotide reductase complex (RNR), the enzyme responsible for the rate-limiting step in dNTP synthesis, is one of the most tightly regulated in the cell (Fig. 2) (Sanvisens, de Llanos and Puig 2013). The RNR complex is composed of small and large subunits: R2 and R1, respectively. In S. cerevisiae, the small subunits (R2) are encoded by the RNR2 and RNR4 genes, and the large subunits (R1) are encoded by the RNR1 and RNR3 genes (Elledge and Davis 1987, 1990). At the transcriptional level, RNR genes are regulated by the cell cycle and DNA damage response networks. Cell-cycle-regulated expression of RNR genes in S. cerevisiae depends on the MBF (Mbp1-Swi6) transcription factor and leads to gradual accumulation of their transcripts during the G1 phase of the cell cycle. This accumulation reaches a maximum during late G1, just before the G1/S transition (Koç et al. 2003; Harris et al. 2013). MBF-dependent transcription of RNR is repressed by the unphosphorylated form of Stb1, which binds the Swi6 subunit of the complex. During the G1 phase, Stb1 undergoes Sin3-mediated phosphorylation by the Cln/cyclin-dependent kinase CDK, which turns on transcription of the RNR genes (Ho et al. 1999). During late G1, Stb1 is released from the RNR promoter and gradually replaced by accumulating Nrm1. The transcriptional corepressor Nrm1 stably associates with RNR promoters via MBF to repress transcription upon exit from the G1 phase (De Bruin, Kalashnikova and Wittenberg 2008; Travesa et al. 2012). DNA damage-induced transcription of RNR genes is controlled by checkpoint kinase Rad53 (homolog of Sc. pombe cds1 and human CHK2). In response to DNA damage or a block in replication, the Rad53 kinase is phosphorylated in a Mec1-, Tel1- or trans-autophosphorylation-dependent fashion. This phosphorylation causes activation of Rad53. One target of Rad53 is Nrm1, which does not bind to MBF target promoters when phosphorylated (Travesa et al. 2012). Another Rad53 target is Dun1 kinase, which hyperphosphorylates Crt1, a transcriptional repressor of DNA damage-regulated genes, including RNR2,3,4, leading to the induction of Crt1-dependent genes (Huang, Zhou and Elledge 1998). Activated Rad53 is also responsible for Ixr1-dependent induction of RNR1, Dun1-dependent induction of RNR2,3,4 and the subsequent upregulation of dNTP levels (Koc and Merrill 2007; Tsaponina et al. 2011), followed by modulation of the replication fork speed and delayed entry into mitosis under replication stress (Poli et al. 2012). The constitutively high level of dNTPs transiently arrests cells in late G1. Normally, dATP feedback inhibition is sufficient to couple dNTP production to utilization. The low dNTP pools found during G1 prevents firing of the DNA replication origins. High dNTP levels during G1 result in the activation of defective pre-replicative complexes based on the slow assembly of Cdc45 (a component of the preinitiation complex) onto chromatin. Even a moderate increase in dNTPs due to RNR overproduction results in synthetic sickness, with orc2-1 and orc5-1 mutants in genes encoding origin recognition complex subunits. Moreover, high levels of dNTP pools not only negatively affect the activation of late origins of replication but also inhibit the DNA damage checkpoint (Chabes and Stillman 2007). The exposure to RNR inhibitor hydroxyurea reduces dNTP pools, triggering cell-cycle arrest in S phase. Hence, the timing of Rad53 deactivation is important. Dephosphorylation of Rad53 allows DNA synthesis to resume and is managed by type 2A-like protein phosphatase activity of the Pph3-Psy2 complex. It was shown that Rad53 independently regulates restart at the replication forks and firing of late origins and that regulation of these processes is mediated by specific Rad53 phosphatases (O'Neill et al. 2007). In addition, rad53 mutants treated with hydroxyurea accumulate unusual DNA structures at the replication forks, resulting in genome rearrangements. This finding indicates that Rad53 prevents the collapse of the fork and avoids genome destabilization when replication pauses (Lopes et al. 2001). Notably, impaired function of other DNA damage or replication block checkpoint proteins, such as Mec1, Tel1, Rad9, Ctf18 or Mrc1, also lead to elevated genome instability (Lustig and Petes 1986; Yuen et al. 2007; Lou et al. 2008; Razidlo and Lahue 2008; Alabrudzinska, Skoneczny and Skoneczna 2011).
Figure 2.

The RNR regulation in S. cerevisiae. The transcription of RNR genes in the cell cycle is MBF dependent, and occurs in G1 reaching the maximum near G1/S transition. When Stb1 is unphosphorylated, it binds the Swi6 subunit of the MBF complex causing repression of RNR genes. In early G1 phase, Stb1 is phosphorylated by CDK which turns on RNR transcription. In late G1, Nrm1 promotes the MBF-dependent repression of RNR genes. In response to DNA damage in the S phase, the Mec1/Rad53/Dun1 kinase cascade activates RNR transcription. Hyperphosphorylation of Crt1 repressor turns on the expression of RNR2,3,4 genes. Moreover, phosphorylation by Rad53 of Nrm1 causes its release from RNR promoters; and phosphorylation of Ixr1 activates the transcription of RNR1 gene. Additionally, the Mec1/Rad53/Dun1 pathway activates RNR activity by promoting the phosphorylation and degradation of the RNR inhibitor, Sml1 and enhancing the relocalization of small RNR subunits to the cytoplasm via modifications of Wtm1 (nuclear anchor protein for RNR small subunits) and Dif1 (importin). Protein names in white refer to transcription factors, in black to proteins with other functions.
An optimal dNTP pool supply is also ensured by the post-transcriptional regulation of RNR complex biogenesis and activity (Ouspenski, Elledge and Brinkley 1999; Chabes et al. 2003; Fasullo et al. 2010; Kumar et al. 2010, 2011; Davidson et al. 2012). In response to DNA damage, RNR activity inhibitor Sml1 (Spd1 in Sc. pombe) is phosphorylated and degraded in a Mec1/Rad53/Dun1-dependent manner; thus, the production of dNTPs necessary for DNA repair and replication effectively increases (Zhao and Rothstein 2002; Nestoras et al. 2010). In Sc. pombe, Spd1 impedes RNR holoenzyme formation by sequestering the R2 subunit in the nucleus far from the cytoplasmic R1 subunit (Fleck et al. 2013). Additionally, RNR activity is regulated by the compartmentalization of its subunits. It was shown that the small subunits of S. cerevisiae RNR (Rnr2 and Rnr4) are imported to the nucleus in a Dif1-dependent manner and then anchored by Wtm1 (Lee and Elledge 2006). Similarly to Sml1 and Spd1, Dif1 is phosphorylated and degraded following DNA damage (Wu and Huang 2008), permitting the redistribution of RNR subunits to the cytoplasm, where they can assemble into an active complex (Yao et al. 2003). Proper assembly of RNR also requires a number of cofactors, including an atypical iron prosthetic group called diferric-tyrosyl radical cofactor [FeIII2-Y•], monothiol glutaredoxin heme protein complex Grx3-Grx4 and iron–sulfur assembly complex Dre2-Tah18 (Zhang et al. 2008; Netz et al. 2010; Stehling et al. 2012; Zhang 2014). The active holoenzyme also requires thioredoxin and glutaredoxin as electron donors during DNA precursor synthesis; thus, dNTP production can also be influenced by cytoplasmic thioredoxins (Trx1 and Trx2), glutathione reductase (Glr1) and gamma glutamylcysteine synthetase (Gsh1). In fact, trx1Δ trx2Δ cells are unable to accumulate dNTPs and display elevated levels of glutathione reductase (Koc et al. 2006). It has also been shown that threonine metabolism is employed to buffer deficiencies in RNR by enabling a compensatory increase in de novo purine biosynthesis that provides additional rate-limiting substrates for dNTP production (Hartman 2007).
The next level of RNR regulation involves allosteric control of its enzymatic activity, permitting an adequate and balanced supply of all four dNTPs and fast adaptation to perturbation of cellular dNTP pools. The R2 subunit of RNR generates a tyrosyl radical that is continuously shuttled to a cysteine residue in the active site of the R1 subunit during catalysis (Kolberg et al. 2004). In addition to the active site, the R1 subunit also contains two separate allosteric sites (the S site and A site) to control activity and substrate specificity (Jordan and Reichard 1998; Eklund et al. 2001). The allosteric sites bind specific NTPs and dNTPs as effectors. Through binding ATP, dATP, dTTP or dGTP, the S site controls the specificity for each of the four substrates. Through binding ATP and dATP, the A site adjusts enzyme activity. Binding of ATP to the A site stimulates enzyme activity, while binding of dATP inhibits it (Cooperman and Kashlan 2003; Zhang, Liu and Huang 2014).
Interestingly, the interaction between Spd1 (an allosteric regulator of RNR) and DNA-associated PCNA has recently been reported in Sc. pombe. This interaction is required for Spd1 degradation following ubiquitination by ubiquitin ligase CRL4Cdt2 (Salguero et al. 2012). This finding places RNR directly at the replication fork. Moreover, it has been demonstrated that a mutation in the R1 subunit of RNR (cdc22-D57N) that alleviates allosteric feedback inhibition resulted in highly elevated dNTP pools that were further increased by deletion of the SPD1 gene. The Δspd1 cdc22-D57N double mutant showed increased mutation rates and was more sensitive to damaging agents, causing double-strand breaks (DSBs) stress compared to single mutants. Thus, Spd1 can protect the genome when dNTP pools are high. In contrast, overexpression of Spd1 generates replication stress and provokes genome instability (Fleck et al. 2013).
Correction systems of replication errors
Cleaning up after replication-mismatch repair and ribonucleotide excision repair
When the proofreading activity of replicases fails to remove the deoxynucleotides misinserted during DNA synthesis, the last chance to correct the mistake is employment of the mismatch repair (MMR) system. The functioning of the MMR system was nicely reviewed by Li (2008), Spampinato et al. (2009) and Kolodner and Marsischky (1999). The first step in this pathway depends on specialized protein complexes Msh2-Msh3 or Msh2-Msh6 that recognize structural abnormalities in the DNA helix. The structural anomalies may be caused by mispaired bases or the insertion/deletion of one or more nucleotides. To properly correct the spatial anomaly detected in the DNA, the nascent and template DNA strands have to be distinguished. In yeast, the nascent strand is discriminated based on the discontinuity associated with DNA replication. Consistent with this model, nuclease Exo1 was found to preferentially repair errors made by Pol α on the lagging strand (Liberti, Larrea and Kunkel 2013). It has been postulated that MMR initiates at Okazaki fragment termini in the lagging strand and at nicks generated in the leading strand by the mismatch-activated Mlh1-Pms2 endonuclease. Because cells lacking RNase H2 display a partial MMR defect combined with an increase in mutagenesis, it was recently proposed that a single ribonucleotide in the vicinity of a mismatch incised by RNase H2 can also act as an initiation site for MMR. Therefore, ribonucleotides misincorporated during DNA replication may serve as physiological markers of the nascent DNA strand (Ghodgaonkar et al. 2013). Following recognition of the template and nascent strands, they are marked via asymmetric binding of the Mlh1-Pms1, Mlh1-Mlh2 or Mlh1-Mlh3 complexes. Then, the fragment of nascent DNA containing the distortion can be removed by flap endonucleases, mainly Exo1, but also Rad27 (Fen1) or Sgs1-Top3 (Kao et al. 2002; Tran et al. 2002; Fricke and Brill 2003). The recent studies established that also Mlh1-Pms1 endonuclease is required for MMR in Exo1-independent MMR subpathway (Smith et al. 2013; Goellner et al. 2014). Excision of the DNA fragment containing the replication error creates an ssDNA gap, which not only provides Pol δ with an opportunity to fill it using the original DNA as template but also contributes to checkpoint activation that is needed to maintain genome integrity because ssDNA is believed to be the checkpoint activation signal (Mojas, Lopes and Jiricny 2007; Reha-Krantz et al. 2011). The final step of MMR is ligation. Other components of this pathway include the ssDNA-binding complex RPA (Rfa1-Rfa2-Rfa3), the PCNA-loading clamp (RFC complex) and PCNA. PCNA serves various DNA repair proteins including those involved in MMR as a handy platform facilitating access to DNA (Clark et al. 2000; Shell, Putnam and Kolodner 2007; Stone et al. 2008).
Bowen et al. (2013) performed a series of amazing experiments reconstituting MMR reactions using purified S. cerevisiae proteins engaged in MMR and a set of DNA substrates containing diverse defects. It was found that a mixture of Msh2-Msh6, Exo1, RPA, RFC-Δ1N, PCNA and Pol δ was sufficient to repair substrates containing various mispairs, frameshifts and a 3′- or 5′-strand break with a range of efficiencies. Furthermore, Bowen et al. showed that the Msh2-Msh3 complex could substitute for the Msh2-Msh6 complex; however, analysis of heterodimers showed a different specificity towards repair of the various mispairs and the addition of the Mlh1-Pms1 complex had no effect on MMR. The Msh2-Msh6 complexes and all three Mlh1-heterocomplexes (Mlh1-Pms1, Mlh1-Mlh3 and Mlh1-Mlh2) also function in the resolution of recombination intermediates and bind with high affinity to a cross-shaped four-stranded structure called a Holliday junction (HJ) that contributes to reciprocal meiotic recombination (Marsischky et al. 1999; Argueso et al. 2003; Rogacheva et al. 2014).
As mentioned above, the MMR system can effectively correct not only mispaired bases but also various frameshifts that are particularly difficult for polymerases to correct using their proofreading activities (Romanova and Crouse 2013). Frameshifts appear most often when the polymerase reaches a polynucleotide tract, which is a hotspot for mutagenesis during DNA synthesis. These tracts result from spontaneous, replication-associated strand slippage (Kunkel 1990) or one of two possible template misalignment mechanisms (primer-template misalignment or dNTP-stabilized misalignment). The insertion of an incorrect nucleotide creates a mispaired primer terminus that is difficult for the DNA polymerase to extend. Subsequent primer-template misalignment can restore proper base pairing, thereby promoting efficient primer extension by relocation of the terminus. This results in a +1 frameshift if the misinserted nucleotide is complementary to the next base of the template strand or a −1 frameshift if it is complementary to the previous base (Bebenek and Kunkel 1990). In the dNTP-stabilized misalignment mechanism, a dNTP substrate is paired correctly, although not with the next available template base. If the next base does not match, the mismatched base is paired with a downstream base on a ‘looped out’ template strand instead. This mechanism generates a −1 frameshift due to the skipping of nucleotides on the template strand by the polymerase (Efrati et al. 1997; Kobayashi et al. 2002). Recently, it was also suggested that the flanking sequence of mononucleotide tracts can play a role in initiating primer-template misalignment; additionally, non-homologous end-joining pathway (NHEJ) defects and MMR system deficiencies can generate frameshifts in a replication-independent manner. Moreover, it was shown that NHEJ is uniquely required for the de novo creation of tandem duplications from non-iterated sequences (Lehner et al. 2012).
The results obtained recently for both S. cerevisiae and Sc. pombe cells suggest an additional class of replicative errors. These errors arise from ribonucleotide monophosphate (rNMP) incorporation into nascent DNA (Watt et al. 2011). These are very common because they happen on average once per 2 kb of newly synthesized DNA; their high frequency is thought to be due to high cellular levels of ribonucleotide triphosphates, although their incorporation is mostly limited by the selectivity of DNA replicases. However, if these misinsertions persist after the DNA synthesis step, they cannot be corrected by MMR; instead, they require the special enzyme RNase H2 for repair. RNase H2 is normally used for the removal of RNA primers from Okazaki fragments, but this enzyme is also responsible for incision of the ribonucleotide, which is the initiation step of ribonucleotide excision repair (RER). During the RER pathway, the incision step is followed by excision by the flap endonuclease Fen1 (or with lower efficiency by Exo1). Next, strand displacement synthesis performed by DNA Pol δ or Pol ε and the process is finally completed by DNA ligase I (Miyabe, Kunkel and Carr 2011; Sparks et al. 2012). The RER pathway also employs PCNA, which is loaded onto DNA by the RFC clamp loader complex. Due to the interaction with PCNA, all successive components of the pathway gain access to the DNA (Sparks et al. 2012). In RNase H2-deficient strains, rNMP incorporated into the leading strand can be removed by processing outside the context of replication in a process that requires Top1 and repetitive sequences and gives rise to misaligned intermediates, resulting in short deletions (Clark et al. 2011; Kunkel 2011; Miyabe, Kunkel and Carr 2011). Another way to overcome rNMP incorporation into DNA in RNase H2-deficient strains is to use post-replication repair (PRR) pathways. In these strains, PCNA is constantly mono- or polyubiquitinated, so PRR is constitutively activated, and misincorporated ribonucleotides can be efficiently bypassed by Pol ζ or omitted in a Mms2-dependent template switch manner (Lazzaro et al. 2012).
Recent studies have demonstrated the importance of yet another protein for the proper functioning of the RER pathway. DNA ligases can generate DNA damage through abortive ligation that produces chemically adducted, toxic 5′-adenylated DNA lesions, (e.g. adenylated 5′ ends containing ribose), which is characteristic of RNase H2 incision. Fortunately, both Sc. pombe and S. cerevisiae also harbor aprataxin (Aptx and Hnt3, respectively), an enzyme with 5′-adenylated RNA–DNA deadenylase activity. Aprataxin efficiently repairs 5′-adenylated RNA–DNA and acts in the RNA–DNA damage response to promote cellular survival and prevent S-phase checkpoint activation in budding yeast undergoing RER (Tumbale et al. 2014).
PRR: a specialized pathway to repair replication stall-borne problems
The processes described so far are designed to make replication error free. They generally perform flawlessly while all components remain functional. However, when any of them fails, mutations arise. Another error-prone situation is the presence of DNA lesions in the template. DNA lesions appear frequently in response to endogenous or exogenous stresses, but as long as they are recognized and corrected by appropriate repair pathways, they do not cause any problems. DNA lesions become mutagenic if they persist until the next DNA replication round. Unrepaired damage in the template strand causes difficulties during DNA synthesis. The type of lesion emerging in DNA determines selection of the appropriate response pathway. Possible responses include (1) TLS performed either by the replicative polymerase or specialized DNA polymerase(s), which is facilitated by an increase in the dNTP pool; and (2) the DNA damage avoidance pathway, in which lesions may be passed by the DNA template change. However, in some cases, the lesions block replication completely and cause a permanent stall in the replication fork, followed by cell-cycle arrest and, eventually, death. The repair pathway responsible for overcoming DNA lesion problems during replication is the PRR pathway. Recently, a controversy arose regarding the actual time window during which PRR occurs. The previous belief was that PRR operates during replication. Now, the scientific community has split into two groups. Some believe that PRR functions during replication, while others favor the view that PRR operates on single-stranded gaps left behind the replication fork or formed during the repair of lesions arising at the G2/M phase (Daigaku, Davies and Ulrich 2010; Karras and Jentsch 2010; Putnam, Hayes and Kolodner 2010). Nevertheless, PRR is the cellular pathway designed to cope with replication of the problematic template.
Two PRR subpathways exist in S. cerevisiae: one that is error prone and one that is error free. It has been shown that PCNA trimer stability is required for TLS by Pol δ and by Pol η (Dieckman and Washington 2013). Moreover, PCNA functions as a molecular switch between the error-prone and error-free PRR subpathways. A change in the operating pathway occurs in response to a DNA damage signal followed by checkpoint activation and is realized by alternative covalent modifications of PCNA (Hoege et al. 2002; Watts 2006; Andersen, Xu and Xiao 2008; Zhang et al. 2011) and the different affinity of DNA polymerases for its modified forms (Garg et al. 2005; Parker et al. 2007; Acharya et al. 2011). Access to the perturbed replication fork is thought to be regulated by competition between polymerases, leading to the conclusion that it can also be influenced by a change in the relative abundance of polymerases in the nucleus. In turn, the relative abundance of polymerases in the nucleus is regulated by their varied expression levels during the cell cycle [e.g. expression of the replicases peaks at the G1/S phase (Verma et al. 1991), while the expression of Pol ζ peaks at G2/M (Waters and Walker 2006)], by their transport and assembly (Netz et al. 2011), and finally by their stability (e.g. Pol η and Rev1 levels are regulated via ubiquitin-dependent degradation; Podlaska et al. 2003; Skoneczna et al. 2007; Wiltrout and Walker 2011). The fluctuations in polymerase delivery to the replication fork may influence the probability of their interaction with PCNA because various polymerases have similar affinities for the same modified form of PCNA, as do Pol η, Pol ζ and Rev1 in budding yeast. It was reported that in the cells of higher eukaryotes, DNA polymerases are also subject to modifications that control their PCNA-binding abilities or even exclude them from the chromatin (Lehmann 2011). Recently, the first report was published, concerning ubiquitin-dependent degradation of yeast Pol3 (catalytic subunit of Pol δ), which excludes it from the replication fork after DNA damage, allowing access of Rev1 (Daraba et al. 2014). This finding is in line with transient stabilization of Pol η observed after UV irradiation (Skoneczna et al. 2007). Together, these data suggest that polymerase switch (specialized polymerase in place of replicative one in case of DNA damage) can be the ubiquitin-mediated feature.
During undisturbed replication, PCNA is sumoylated by the Ubc9–Siz1 complex (Stelter and Ulrich 2003; Huang et al. 2007), which permits interaction with replicative Pols δ and ε and fast replisome progression at the replication forks. Detection of bulky lesions on the DNA template impedes fork progression and induces Rad6–Rad18-mediated ubiquitination of PCNA, therefore promoting Ubi–PCNA-dependent translesion DNA synthesis (TLS); further polyubiquitination of Ubi–PCNA performed by the Ubc13-Mms2-Rad5 ubiquitin ligase complex in turn favors the damage avoidance pathway (Fig. 3). When synthesis over a lesion is finished, Ubp10 deubiquitinates PCNA, allowing replisome remodeling to switch back to the replicase and the efficient resumption of replication (Gallego-Sánchez et al. 2012). Desumoylation of PCNA is performed by the SUMO proteases Ulp1 that removes SUMO and Ulp2 that removes poly-SUMO chains (Stelter and Ulrich 2003; Huang et al. 2007; Drag and Salvesen 2008).
Figure 3.

RFC/sliding clamp-dependent regulation of various DNA damage response pathways. Various RFC complexes affect PCNA and 9–1–1 complex in different ways. Each RFC complex has distinct ability to load/unload of sliding clamps and their modified forms and/or to stimulate particular modification (ubiquitination or sumoylation) of sliding clamps. The sliding clamp type and its modification channels DNA damage response to particular DNA repair pathway. Therefore, RFC complexes control cellular response to DNA damage adjusting it to particular kind of stress. Green arrows refer to stimulating effect of RFC complex on loading the particular form of sliding clamp onto DNA, and stimulating effect of one RFC complex on another one (Elg1 contributes to Ctf18 recruitment to chromatin which influences the PCNA positioning on DNA). Red lines refer to sliding clamp unload stimulation. The presence of additional RFC complex on the picture denotes the possible existence of other, yet to be discovered, RFC complexes.
Information on PRR in Sc. pombe is limited. It is known that Sc. pombe and S. cerevisiae share homologous sets of PRR genes, but they are used in different ways. The major distinctions of Sc. pombe cells in the control of lesion tolerance pathways are the existence of pathways that involve various TLS polymerases, the TLS requirement for enzymes needed for mono- and polyubiquitination of PCNA and, surprisingly, the mostly error-free bypass of various DNA lesions regardless of the polymerase or polymerases synthesizing through them (Coulon et al. 2010). Although Siz1 and Ubc9 homologs exist in Sc. pombe, PCNA sumoylation has not been documented. The data obtained for cells carrying mutations in various DNA polymerase genes showed that even though all four Sc. pombe TLS polymerases operate on DNA templates containing lesions (i.e. the substrate that is recognized by the receptors as a DNA damage signal), only cells lacking Pol κ or Pol η (but nor Pol ζ or Rev1) activated the checkpoint after UV irradiation. Interestingly, the enhanced checkpoint response in cells lacking Pol κ and Pol η was not due to stalled replication forks but was instead caused by post-replication DNA gaps with unrepaired UV lesions in the template that acted as both substrates for TLS polymerases and as signals for checkpoint activation (Callegari et al. 2010). Previous results demonstrated that checkpoint activation in fission yeast contributes to Pol κ function in the DNA damage-tolerant pathway differently than in budding yeast. The Rad17 checkpoint protein provides both activation of dinB (encoding Pol κ) expression and supports its physical interaction with the checkpoint-clamp components Hus1 and Rad1 (Kai and Wang 2003). Thus, it could be expected that TLS is a part of the checkpoint response.
Pol η is the only polymerase for which transcriptional activation of the encoding gene following DNA damage has been demonstrated in budding yeast. After UV induction, RAD30 transcript levels increase about 2-fold (Pavlov, Nguyen and Kunkel 2001). However, because the overexpression of Rad30 (Pol η) and the Pol ζ subunits Rev3 or Rev7 increase the mutation rate (Rajpal, Wu and Wang 2000; Pavlov, Nguyen and Kunkel 2001), they should be maintained at low levels under non-mutagenic conditions. Indeed, all budding yeast TLS polymerases (Pol η, Pol ζ and Rev1) need to be removed from the replication fork as quickly as possible after their task is performed; otherwise, they will frequently generate mistakes on the undamaged template due to their poor fidelity (Zhong et al. 2006; McCulloch et al. 2007). In some cases, they can even produce complex mutations (e.g. tandem base pair substitutions and clusters of multiple, closely spaced mutations) as was shown recently for Pol ζ (Stone, Lujan, Kunkel 2012). For this reason, TLS polymerases are actively removed from the cell when they are no longer required. Rev1 and Pol η are short-lived proteins in vivo, with half-lives of approximately 20 min, at least for their tagged versions (Skoneczna et al. 2007; Wiltrout and Walker 2011). The Rev1 protein is additionally regulated during the cell cycle, and is most unstable during G1 phase (Wiltrout and Walker 2011). Because at least two TLS polymerases are degraded in a ubiquitin-dependent manner, it is not surprising that the mutator effect was also observed in yeast strains with reduced proteasomal activities (Podlaska et al. 2003; McIntyre et al. 2006). Furthermore, Pol η was shown to be transiently stabilized after UV irradiation (Skoneczna et al. 2007), when its cognate substrates (e.g. T-T dimers) emerge in the DNA. Pol η specializes in the bypass of T-T dimers and inserts appropriate nucleotides (i.e. AA) relatively accurately opposite these lesions (Washington et al. 2000; Gibbs et al. 2005).
Sumoylated PCNA-, Srs2- and replicative polymerase-dependent DNA synthesis on damaged templates
The PCNA of S. cerevisiae has two SUMO acceptor sites: a major one at Lys164 (that may also be ubiquitinated) that is crucial for DNA damage tolerance and a minor one at Lys127 that does not appear to play a role in DNA damage tolerance activity but is required for the establishment of sister chromatid cohesion during S phase (Hoege et al. 2002; Stelter and Ulrich 2003; Moldovan, Pfander and Jentsch 2006). Because PCNA forms homotrimeric complexes, there are three Lys164 residues available for potential modification. Sumoylation of one Lys164 facilitates modification of its neighboring Lys164 in the complex, but the ligand for subsequent modification may not necessarily be the same as the primary ligand. Therefore, various modifications can exist for one complex, including poly-SUMO chains (Windecker and Ulrich 2008). In addition to PCNA monoubiquitination, Siz1-mediated PCNA sumoylation is required for TLS stimulation. Epistatic analysis of relationships between SIZ1, MMS2 and RAD5 with respect to UV sensitivity demonstrated that in the absence of PCNA sumoylation, Mms2-Ubc13 and Rad5 independently influence TLS (Halas et al. 2011).
The fraction of sumoylated PCNA increases during S phase, as well as during extensive DNA damage. Sumoylated PCNA is able to bind replicative polymerases, an event that permits synthesis over the lesion. Sumoylated PCNA also has high affinity for Srs2 (Kolesar et al. 2012), whose binding prevents homologous recombination (HR) (Papouli et al. 2005; Pfander et al. 2005). Srs2 is a 3′→5′ DNA helicase (Rong and Klein 1993) that unwinds hairpin intermediates that can be formed by triplet repeats in the DNA and promotes fork reversal in these repetitive sequences, thereby preventing their instability and fragility and consequently contributing to protection against DNA expansions and contractions (Dhar and Lahue 2008; Kerrest et al. 2009). Srs2 also possesses translocase activity (Antony et al. 2009) and disrupts Rad51 nucleoprotein filaments (Krejci et al. 2003; Veaute et al. 2003), thereby eliminating the checkpoint activation signal, initiating checkpoint recovery (Hishida et al. 2010; Yeung and Durocher 2011) and preventing recombination (Broomfield and Xiao 2002). The Rad51-translocating activity of the Srs2 helicase is stimulated by interaction with sumoylated PCNA (Pfander et al. 2005). Recently, an additional mechanism was elucidated, whereby sumoylated PCNA and Srs2 block the synthesis-dependent extension of a recombination intermediates, limiting their resolution associated with a crossing over and thus preventing spontaneous genome rearrangements (see also the section on recombination). This newly reported activity of Srs2 requires the SUMO interaction motif at its C-terminus but is independent of its Rad51 translocase activity. It has been shown that binding between Srs2 and sumoylated PCNA results in Pol δ and Pol η dissociation from the repair foci. These results suggest that Siz1-dependent sumoylation of PCNA limits the length of DNA synthesis during template switch or HR and attenuates reciprocal DNA-strand exchange to maintain genome stability (Burkovics et al. 2013).
While the binding of Srs2 to sumoylated PCNA inhibits the recombination pathway, the binding of replicative polymerases stimulates replication over the lesion (Fig. 4). This bypass is facilitated by expanding dNTP pools in response to DNA damage checkpoint activation (Lis et al. 2008; Sabouri et al. 2008). Inhibition of Rnr4- and Pol δ-dependent EMS-induced mutations by hydroxyurea (Lis et al. 2008) and suppression of the high mutation rate observed in pol3–01 mutants by dun1 deletion (Datta et al. 2000) consistently demonstrate that S-phase checkpoint-mediated upregulation of dNTP levels stimulates Pol δ-mediated TLS. Yeast Pol α is also able to insert nucleotides opposite DNA lesions at high dNTP concentrations (Niimi et al. 2004). Moreover, Pol ε can replicate over 7,8-dihydro-8-oxoguanine even when dNTPs are at normal cellular concentrations (Sabouri et al. 2008), and the insertion of nucleotides by Pol δ opposite the same lesion can be efficiently extended by TLS Pol ζ, as was shown by Haracska, Prakash and Prakash (2003). Because Pol δ and Pol ζ share the accessory subunits Pol31 and Pol32, one could assume that the polymerase switch should be smooth. However, Siebler et al. recently showed that the switch was not as easy as expected. This study demonstrated an additional role for Pol32 in TLS beyond its role as a Pol ζ subunit. This newly reported role is related to Pol δ because mutants lacking the C-terminal part of Rev3, responsible for Pol ζ catalytic activity and for interaction with the accessory subunits, are partially proficient in Pol32-dependent UV-induced mutagenesis (Siebler et al. 2014). The polymerase switch scenario was also demonstrated for other polymerases. When Pol η manages to overcome T-T dimers that blocked DNA replication by DNA Pol δ or Pol ε another polymerase switch occurs that allows normal replication to resume (McCulloch et al. 2004a). Thus, DNA synthesis on templates containing lesions usually requires the cooperation of two polymerases.
Figure 4.

DNA synthesis on damaged template. The localization of lesion in the DNA template influences the choice of DNA damage repair/tolerance pathway. The damage of already replicated DNA strand is subject to DNA repair by pathway dedicated to deal with particular lesion, such as BER, NER, HR, etc (left panel of the figure). When DNA lesion happens in ssDNA, e.g. in the replication fork (the middle part of the figure) or in ssDNA gaps left after replication (right panel of the figure), the DNA synthesis process has to be applied. However, replication over the lesion is not an easy task, and typically requires switching of polymerases. Various TLS polymerases are able to overcome the lesion that blocked DNA replication; switching back to DNA replicative polymerase resumes normal replication (one TLS polymerase scenario, A). Sometimes, the polymerase that is able to add the nucleotide opposite the lesion is unable to elongate synthesis. Therefore, resumption of synthesis requires the cooperation of three DNA polymerases (B). To fill the gap one more, RPA-dependent step is needed: the recruitment of a sliding clamp to the 3′-OH end of the DNA gap. Then the repair can proceed as described above.
Daraba et al. recently demonstrated that this polymerase switch may be achieved by degradation. They showed that upon DNA damage, Def1 promotes the ubiquitination of the Pol13 catalytic subunit of replicative polymerase δ. This ubiquitination is followed by proteasomal degradation. In contrast, the Pol31 and Pol32 subunits of polymerase δ are not affected and are able to form a complex with Rev1; thus, this TLS polymerase may perform DNA lesion bypasses at stalled replication forks (Daraba et al. 2014).
Ubiquitinated PCNA- and specialized polymerase-dependent TLS
Following DNA damage, PCNA is monoubiquitinated at Lys164 by the Rad6–Rad18 complex (Hoege et al. 2002). This modification enables PCNA to interact with TLS polymerases and provides access to the damaged template (Fig. 4). TLS is performed in various arrangements by polymerases operating independently (e.g. Pol η and Pol ζ) or in tandem (Pol η then Pol ζ or Pol ζ with Rev1). The final result of synthesis depends on the polymerases engaged in the bypass as well as on the specific lesion (Haracska, Prakash and Prakash 2000, 2003; Gibbs et al. 2005; Northam et al. 2006; Parker et al. 2007; Stone, Lujan, Kunkel 2012). Notably, some TLS polymerases bypass specific DNA lesions with perfect reproduction of the initial content of the template, displaying high effectiveness towards the cognate substrate [e.g. Pol η inserts C opposite 7,8-dihydro-8-oxo-guanine (Haracska, Prakash and Prakash 2000) and A-A opposite a T-T dimer (Gibbs et al. 2005)]. Other polymerases synthesize sequences that differ from the original sequence. Moreover, the sequence context of the lesion influences the relative likelihood that specific nucleotides will be used for DNA synthesis by different TLS polymerases, as was shown by Chan, Resnick and Gordenin (2013).
Polyubiquitinated PCNA- and Rad5-dependent damage avoidance pathway
Following DNA damage, Mms2 and Ubc13 proteins are redistributed from the cytoplasm to the nucleus, where they associate with Rad5. The Rad5-Mms2-Ubc13 complex is responsible for ubiquitination of the PCNA Lys164 residue (Ulrich and Jentsch 2000). Ubiquitins in the chain are linked via Lys63; however, it is not known if the polyubiquitin chain is built sequentially or transferred en block on PCNA. Polyubiquitinated PCNA stimulates the error-free PRR damage avoidance pathway (Branzei, Seki and Enomoto 2004; Branzei, Vanoli, Foiani 2008) (Fig. 3). Using this pathway, the lesion in the DNA template can be omitted as follows: (1) by template switch or sister chromatid junctions (SCJ) using the HR proteins Rad51, Rad52 and Rad54, as well as the Sgs1–Top3 complex, permitting resolution of cross-structures generated during repair (Gangavarapu et al. 2006; Branzei and Foiani 2007; Ball et al. 2009); or (2) by Rad5-dependent fork regression (a ‘chicken foot’ DNA structure), followed by nascent-strand annealing and DNA synthesis (Blastyák et al. 2007). Both branches of this pathway are error free.
The 9–1–1/RPA-dependent pathway involved in gap filling and telomere maintenance
As previously mentioned, PCNA serves as a platform for various proteins operating at the replication fork. The role of PCNA modification in the tunneling of cellular responses to DNA damage was also described. However, having one type of sliding clamp per cell is not sufficient (Fig. 3). In yeast, another PCNA-like sliding clamp exists that is designed especially for interaction with various proteins activated after DNA damage and engaged in DNA repair and cell-cycle checkpoints. This 9–1–1 complex (the Ddc1-Rad17-Mec3 complex, a homolog of human Rad9-Rad1-Hus1 complex) is a heterotrimeric checkpoint clamp consisting of the Ddc1, Rad17 and Mec3 subunits in S. cerevisiae and the Rad9, Rad1 and Hus1 subunits in Sc. pombe. The 9–1–1 complex is an early response factor to DNA damage that activates checkpoints (Majka and Burgers 2003). It can be loaded onto DNA using two different configurations: at the 5′ end of ssDNA coated with RPA or at both the 5′ and 3′ junctions of naked DNA at gaps left on the lagging strand after replication (Majka et al. 2006a). This complex subsequently stimulates Mec1 kinase and/or checkpoint sensor Dpb11, depending on the cell-cycle phase (Majka, Niedziela-Majka and Burgers 2006b; Navadgi-Patil and Burgers 2009; Puddu et al. 2011).
The 9–1–1 complex promotes both Dna2-Sgs1 and Exo1-dependent resection in response to uncapped telomeres (Ngo et al. 2014). The support of resection by the 9–1–1 complex is strongly inhibited by the DNA damage-dependent checkpoint protein Rad9. However, the 9–1–1 complex together with the Exo1 nuclease also participates in the error-free branch of the DNA damage tolerance pathway where it promotes template switching in a manner that is distinct from its canonical checkpoint functions and uncoupled from the replication fork. Recent work by Karras et al. (2013) revealed cooperation in the error-free pathway between the two related clamps PCNA and 9–1–1. The 9–1–1 complex also plays a major role in DNA repair by interacting with and stimulating activity of specialized but not replicative DNA polymerases involved in TLS by filling the gaps left behind the replication fork (Kai and Wang 2003; Cardone, Brendel and Henriques 2008). Additionally, it was shown that the Mcm10 protein that was proposed to play a role in replication fork restart and DNA repair interacts with the Mec3 subunit of the 9–1–1 clamp in response to nucleotide shortages or UV-induced replication stress (Alver et al. 2014). The 9–1–1 checkpoint clamp is also involved in preventing the deleterious effects of dUTP-related apurinic/apyrimidinic sites (called also abasic or AP sites) that may cause replication-related genetic instability (Collura, Kemp and Boiteux 2012). Another role for the PCNA-like sliding clamp is based on its interaction with the Rad51 and Dmc1 proteins, which stimulate efficient repair of meiotic DSBs by facilitating proper assembly of the meiotic recombination complex (Shinohara et al. 2003).
Similar to PCNA, the 9–1–1 complex is ubiquitinated; however, published data disagree on whether this modification is dependent on Rad6 and if Lys197 or Lys164 of Rad17 is the major acceptor site for ubiquitin. It is also not clear if ubiquitination of the 9–1–1 complex participates in its known functions in DNA repair stimulation and cell-cycle control or merely labels it for proteasomal degradation (Fu et al. 2008; Davies, Neiss and Ulrich 2010). Nevertheless, an interaction between Rad18, the E3 ubiquitin ligase and subunits of the 9–1–1 complex was detected (Fu et al. 2008). Moreover, Fu et al. showed that the Rad6–Rad18 complex involved in PRR ubiquitination is required for increased transcription of a large number of yeast genes in response to DNA damage. They also suggested a crucial role for the 9–1–1 complex in DNA repair and checkpoint control under DNA damage stress, possibly through the recruitment or maintenance of the 9–1–1 clamp at lesion sites.
Surprisingly, a link between the 9–1–1 clamp and nucleosome assembly was recently found. The rad17Δ deletion is synthetic lethal with the deficiency of the histone chaperone Rtt106. Moreover, multiple genetic interactions between the 9–1–1 clamp and DNA replication-coupled nucleosome assembly factors, including Rtt106, CAF-1 and lysine residues of H3-H4 were found. Furthermore, rad17Δ cells showed (1) defects in the deposition of newly synthesized histones H3–H4 onto replicated DNA; (2) a reduction in the Asf1–H3 interaction due to an increased association of Asf1 with checkpoint kinase Rad53; and (3) an increase in the interaction between histones H3–H4 with histone chaperone CAF-1 or Rtt106. All of these results suggest a role for 9–1–1-dependent regulation in DNA replication-coupled nucleosome assembly via histone chaperone interactions (Burgess, Han and Zhang 2014).
Crosstalk between RFC complexes adapts cellular responses to various types of DNA damage
The PCNA and 9–1–1 clamps play a crucial role during DNA replication and repair, serving as platforms capable of enabling access to the DNA by various proteins. These clamps are engaged in unwinding DNA, sensing cellular stresses and deciding which repair pathway will be employed under particular conditions. However, the clamps themselves are also subject to regulation. There are several complexes in the cell that control the interaction of PCNA and 9–1–1 with DNA (Fig. 3). The first complex that was shown to possess the ability to load and unload PCNA onto DNA was RFC. In eukaryotic cells, four independent sliding clamp loaders exist (Majka and Burgers 2004). All interact with Mps3, a protein attached to the nuclear envelope, suggesting that these clamp loaders are recruited to the nuclear envelope, e.g. in response to DNA damage (Haas et al. 2012). RFCs are heteropentamers consisting of four smaller Rfc2–5 subunits (Yao et al. 2006) and one large subunit that is unique to each complex. Thus, Rfc1 from the classical RFC complex may be substituted for Ctf18, Elg1 or Rad24, creating alternative RFC complexes (Green et al. 2000; Mayer et al. 2001; Kanellis, Agyei and Durocher 2003). The RFCs work as chromatin-associated factors that load and unload sliding clamps and promote DNA replication and repair; however, each of them has a special predisposition to operate under particular circumstances. The canonical RFC is responsible for PCNA loading and unloading on/off DNA during undisturbed replication (Bowman, O'Donnell and Kuriyan 2004), but it is also required for repair synthesis of large looped heteroduplexes in S. cerevisiae (Corrette-Bennett et al. 2004). Ctf18-RFC and Elg1-RFC can influence PCNA positioning on DNA. Ctf18-RFC unloads PCNA from DNA (Bylund and Burgers 2005) and promotes sister chromatid cohesion, while Elg1–RFC plays an opposing role in this process (Hanna et al. 2001; Maradeo and Skibbens 2009, 2010). Furthermore, Elg1 contributes to Ctf18 recruitment to chromatin. Elg1 and Ctf18 with the aid of Ctf4 may coordinate the relative movement of the replication fork with respect to the cohesin ring (Parnas et al. 2009).
The Elg1–RFC complex interacts effectively with the sumoylated form of PCNA and mediates a decrease in the level of sumoylated PCNA bound to chromatin (Parnas et al. 2010). It is still unclear whether Elg1–RFC actively unloads sumoylated PCNA from chromatin or recruits the desumoylating enzyme Ulp1 to chromatin to reduce the level of sumoylated PCNA. As was noted in the previous section, PCNA sumoylation prevents recombination in the replication fork. Notably, the elg1Δ mutant displays a hyperrecombination phenotype (Ogiwara et al. 2007) and is synthetic lethal with deletions in genes involved in the HR pathway (Aroya and Kupiec 2005). In turn, Ctf18-RFC is involved in the initiation step of replication (Ma et al. 2010) and replication checkpoint activation (Kubota et al. 2011). Ctf18–RFC is essential for Mrc1-dependent activation of Rad53 and for the maintenance of paused replication forks, most likely by bridging Mrc1 and primed ssDNA fragments that serve as a signal of paused DNA synthesis (Crabbé et al. 2010). The Ctf18–RFC loading clamp facilitates the bypass of triplet repeats during replication (Gellon et al. 2011) to maintain the telomeres (Gao et al. 2014) and is also involved in preserving proper genome copy number in the cell during division (Alabrudzinska, Skoneczny and Skoneczna 2011).
The 9–1–1-complex is placed on DNA by the dedicated clamp loader Rad24–RFC in a reaction directed by RPA (Majka et al. 2006a). Interestingly, the Rad24–RFC DNA damage checkpoint clamp loader also unloads PCNA clamps from DNA. Therefore, Rad24–RFC may clear PCNA from DNA to facilitate the shutdown of replication when faced with DNA damage (Yao et al. 2006); however, it is less effective at performing this task than is canonical RFC (Thompson et al. 2012). Rad24–RFC functions as a checkpoint protein that is crucial for DNA processing, recombination partner choice and cell survival after various stresses and the subsequent DNA repair (Aylon and Kupiec 2003; Aylon et al. 2003).
Cells lacking one of the alternative RFC complexes exhibit a strong genome instability phenotype (Aroya and Kupiec 2005; Banerjee, Sikdar and Myung 2007). The alternative RFCs channel cellular responses to DNA-borne stress into two major repair pathways: first, employing PCNA to enable replication resumption, and second, employing the cohesion complex to preserve the integrity of chromosomes. Moreover, alternative RFCs function in a cell-cycle-dependent fashion, adjusting the method of repair to the cell-cycle phase and possibly to cell ploidy (Delacôte and Lopez 2008; Li and Tye 2011). However, the molecular basis through which RFCs exert these diverse effects remains to be uncovered.
RECOMBINATION AND GENOMIC STABILITY
In general terms, DNA recombination can be defined as a DNA transaction pathway in which a strand exchange occurs between different DNA molecules. HR pathway uses a homologous strand as a template for the repair of a damaged strand. The homologous strand might be a sister chromatid, a homologous chromosome or an ectopically located sequence. Non-HR ligates DNA molecules without the step of a homology search for DNA replication priming. Conserved recombination pathways have evolved as a system for reactivating blocked replication forks and for repairing DSBs and interstrand cross-links, lesions that would preclude replication fork progression.
Regulation of the choice between HR and NHEJ
DSBs can arise spontaneously due to endogenous and exogenous factors, such as replication through a nick, metabolism-derived reactive oxygen species (ROS), topoisomerase failures and ionizing radiation. These lesions, however, can also occur during programed gene rearrangements and other recombination events, such as mating-type switching in budding yeast or during meiosis (reviewed in Symington and Gautier 2011). The DSB repair has been studied extensively in the budding yeast. These lesions are processed through two competing pathways, depending on the cell type, the stage in the cell cycle and the structure of the DNA ends: HR and NHEJ. HR is initiated when the DSB end is resected by nucleases and helicases to generate 3′-ssDNA overhangs. In budding yeast, the binding of the Mre11-Rad50-Xrs2 (MRX) complex and the Sae2 protein to DNA initiates 5′→3′-end resection, which is further processed by Exo1 and Dna2–Sgs1 nucleases (reviewed in Mimitou and Symington 2011). The resulting long ssDNA bound to RPA activates the Mec1 (Sc. pombe rad3 and human ATR) DNA damage checkpoint, which coordinates DNA repair reactions with cell-cycle progression (Zou and Elledge 2003). Thereafter, the various HR processes differ in the processing of intermediates, resulting in different error-free and mutagenic repair pathway outcomes. These HR subpathways are further described below in the section ‘HR sub-pathways’. In the NHEJ pathway, DSB ends are blocked from 5′-end resection by bound Ku proteins (Yku70 and Yku80), which form a heterodimer. The Ku heterodimer promotes the direct ligation of the DSB ends by the specialized ligase complex Dnl4–Lif1, requiring ‘clean’ DNA ends, i.e. 3′-OH and 5′-phosphate groups, for ligation. If the DSB ends are not clean, then additional processing by nucleases and a dedicated polymerase (Pol β) are needed for ligation to occur, and during this process, small deletions and insertions might be introduced at the junction site. Therefore, this pathway is considered error prone. Indeed, many lines of evidence suggest that in budding yeast increased mutagenesis can be dependent on NHEJ (Halas et al. 2009; Lehner et al. 2012; Shor, Fox and Broach 2013). HR and NHEJ are separate pathways that are differently regulated (see below), but the MRX complex is shared by both pathways, because, in addition to its important role in HR, MRX also functions as a DNA end-bridging factor in NHEJ (Chen et al. 2001). Nevertheless, Ku proteins and Dnl4 inhibit DNA end resection (Zhang et al. 2007), and, in turn, DNA end resection inhibits NHEJ ligation, reflecting the end requirements of Dnl4–Lif1-mediated ligation. Consistently, defects of sae2 null cells in DSB processing after exposure to ionizing radiation are suppressed by inactivation of YKU70, and this suppression requires both nucleases mediating the extensive end resection, Exo1 and Sgs1–Dna2 (Mimitou and Symington 2010). As previously mentioned, these two pathways are also differently regulated in budding yeast cells according to cell type and cell-cycle stage. In haploid cells, both HR and NHEJ pathways are efficient, whereas in diploid cells NHEJ is inhibited due to the mating-type control-mediated repression of the NEJ1 (LIF2) gene encoding the critical NHEJ regulator (Frank-Vaillant and Marcand 2001; Kegel, Sjöstrand and Aström 2001). The choice of a DSB repair pathway is also dependent on the phase of the cell cycle. In both haploid and diploid cell types, DSB repair through HR primarily occurs during the S and G2 phases when DNA has replicated and one sister chromatid is available as a template to repair the other sister chromatid. This control is mediated through cyclin-dependent Cdk1 kinase, which phosphorylates many DNA repair proteins in the cell (reviewed in Enserink and Kolodner 2010). However, the most critical step to initiate HR involves overcoming the Ku-mediated inhibition of end resection (reviewed in Symington and Gautier 2011). This is accomplished through two Cdk1-dependent mechanisms: one pathway that is associated with the regulation of Ku/Dnl4-Lif1 affinity for DNA ends (Zhang et al. 2009) but has not yet been characterized, and another pathway involving the activation of Sae2 through phosphorylation at Ser267 (Huertas et al. 2008). Interestingly, the CDK-dependent regulation of CtIP, the human ortholog of Sae2, is evolutionarily conserved (reviewed in Symington and Gautier 2011). The requirement for the early end resection HR factors, Mre11 and Sae2, depends on the structure of the DNA ends in budding yeast. These factors are not required for the resection of clean DSB ends, but are necessary for the resection of dirty or complex ends, induced through ionizing radiation, DSB ends adopting secondary structures or DSBs with a protein covalently bound to the 3′ end, e.g. the binding of topoisomerase to a DNA end after treatment with camptothecin (reviewed in Symington and Gautier 2011). The end resection step during DSB repair has also been studied in fission yeast, and the mechanism is essentially conserved between the two yeast species, although there are some interesting differences (Langerak et al. 2011). Similar to the HR in S. cerevisiae, in fission yeast cells defects in the initiation of end resection are suppressed in the absence of Ku proteins.
HR subpathways
The RPA-coated ssDNA generated during the extensive end resection mediated through HR nucleases is processed via several HR subpathways, depending on the homologous DNA availability (reviewed in San Filippo, Sung and Klein 2008). These subpathways include (1) the double HJ pathway, also known as the DSBs repair pathway, (2) synthesis-dependent strand annealing (SDSA), (3) break-induced replication (BIR), (4) single-strand annealing (SSA) and (5) the error-free lesion bypass pathway. In these pathways, except SSA, RPA covering ssDNA is replaced by the Rad51 recombinase in a reaction mediated by Rad52 (Sung 1997). The Rad51 protein combined with ssDNA forms a nucleoprotein filament that catalyzes homologous pairing and strand invasion into a homologous duplex (reviewed in San Filippo, Sung and Klein 2008). The invading strand is used by a DNA polymerase for priming DNA synthesis to extend the ensuing displacement loop (D-loop) with the assistance of the translocases Rad54 or Rdh54 (reviewed in San Filippo, Sung and Klein 2008; Symington, Rothstein and Lisby 2014).
In the double HJ pathway, the strand displaced by the extending D-loop anneals to the second ssDNA overhang on the other side of the DSB (second-end capture) and primes DNA synthesis to fill in the gap. Ligation generates double HJs (crossed-strand structures). The resolution of double HJs by structure-selective endonucleases (resolvases) in different orientations leads to either crossover or non-crossover of flanking markers. HJ intermediates are also dissolved by branch migration, which is mediated by the Sgs1-Top3-Rmi1 (STR) complex (reviewed in Ashton and Hickson 2010), giving rise to only non-crossover products. SDSA differs from the double HJ pathway in that it produces a less extensive D-loop that is dismantled before the second-end capture occurs. Because there is no HJ in SDSA, the products are non-crossover products. In mitotic cells, there are several mechanisms to restrict crossover recombination. One mechanism involves HJ dissolution by the above-mentioned STR complex. In S. cerevisiae, there are two other mechanisms involving two other helicases that promote SDSA, thereby suppressing crossover recombination. First, the Srs2 helicase binds Rad51 and disrupts Rad51 presynaptic filaments (Krejci et al. 2003; Veaute et al. 2003). As mentioned in section ‘Postreplication repair: a specialized pathway to repair replication stall-borne problems’, Srs2 binding to SUMOylated PCNA also limits the access of DNA polymerases to recombination intermediates in a mechanism independent from the Srs2 interaction with Rad51 (Burkovics et al. 2013). Second, the Mph1 helicase dissociates Rad51-dependent D-loops with high efficiency (Prakash et al. 2009).
In both the double HJ and SDSA pathways, DNA replication utilizes only leading-strand synthesis machinery to extend both the first and second ends (see below). Single-ended DSBs, e.g. at telomeres or broken replication forks, are resected similarly to two-ended DSBs, and the resulting 3′-ssDNA overhangs invade a homologous duplex and prime DNA synthesis only from one side. BIR continues to the end of the invaded duplex and engages the machinery of both leading- and lagging-strand DNA synthesis (Lydeard et al. 2010). BIR leads to the non-reciprocal exchange of a part of a donor chromosome to a recipient chromosome (Kraus, Leung and Haber 2001); however, this pathway is also highly mutagenic for other reasons, as described below. Sometimes a DSB is flanked by direct repeats. Following end resection, exposed complementary 3′-ssDNA overhangs anneal, forming a duplex in the new strand configuration, with the concomitant deletion of the intervening sequence. This is a SSA pathway, which is mechanistically different than the other HR pathways, because it does not require Rad51 for homologous strand pairing. However, the strand annealing reaction is not spontaneous (it is inhibited by RPA bound to ssDNA); it is mediated by an additional Rad52 protein function that is specific for ssDNA annealing (reviewed in Symington, Rothstein and Lisby 2014). This pathway also requires the activity of the Rad1–Rad10 endonuclease (Fishman-Lobell and Haber 1992), which cuts branched DNA structures at the transition between dsDNA and ssDNA to remove ssDNA flaps that fail to anneal (reviewed in Lyndaker and Alani 2009). The SSA pathway is efficient, even for end resection and annealing between short (5–25 nt) direct repeats flanking a DSB. This ‘small’ variant has been described as a microhomology-mediated end joining (MMEJ) pathway in Ku-deficient cells (Ma et al. 2003). However, the MMEJ pathway is not well characterized and its genetic requirements remain controversial (see also Symington and Gautier 2011). In S. cerevisiae, this ‘small’ SSA variant might have a modified requirement for annealing (reviewed in Decottignies 2013) because instead of Rad52, Rad59 is necessary for the annealing of short homologous sequences (Sugawara, Ira and Haber 2000). An SSA-like pathway has also been studied in fission yeast as a microhomology-mediated recombination pathway repressed by Ku proteins (Decottignies 2007). Indeed, repetitive elements are numerous in yeast genomes, consequently SSA could mediate large-scale rearrangements that cause deletions of sequences located between the repeats.
The last HR subpathway for error-free lesion bypass is one of the two main mechanisms of DNA damage tolerance (the other mechanism is TLS, which occurs either in an error-free or error-prone manner as mentioned in the section ‘Postreplication repair: a specialized pathway to repair replication stall-borne problems’) in response to stalled replication forks. Various endogenous and exogenous factors might cause replication fork stalling (reviewed in Aguilera and Gómez-González 2008; Branzei and Foiani 2010), including secondary DNA structures often adopted by repetitive sequences, highly transcribed DNA, tightly bound proteins forming replication fork barriers and a diverse array of template lesions. As mentioned in section ‘Postreplication repair: a specialized pathway to repair replication stall-borne problems’, a recombination-dependent mechanism mediates transient template switching, which uses the sequence of the newly synthesized sister chromatid to bypass the lesion, providing adequate time for damage removal and facilitating both gap filling and the restart of replication forks stalled by replication stress. Template switching is associated with the formation of SCJ, in the proximity of replication forks, that are cruciform pseudo-double HJ structures (Liberi et al. 2005; reviewed in Vanoli et al. 2010). These recombinant intermediates accumulate in a Rad51/Rad52-dependent manner in MMS-treated cells lacking the Sgs1 helicase. The pathway utilizes a subset of recombination proteins (Rad51, Rad55, Rad57, Exo1 and Rad59) and both replicative polymerases, Pol ε and Pol δ (Vanoli et al. 2010). Recently, template switch intermediates have been characterized using the 2D gel electrophoresis and electron microscopy (Giannattasio et al. 2014). The choice between different bypass mechanisms is regulated by ubiquitin and SUMO modifications of PCNA at sites of stalled replication forks (reviewed in Moldovan, Pfander and Jentsch 2007) and mentioned in the previous sections). In S. cerevisiae, the template switch mechanism depends on the Rad18 ubiquitin ligase- and Rad5-mediated polyubiquitylation of PCNA, whereas Ubc9-dependent SUMOylation of PCNA and Mms21-dependent SUMOylation of Smc5-Smc6 and Sgs1-Top3 complexes are required to remove the cruciform recombinant intermediates (Liberi et al. 2005; Branzei et al. 2006; Branzei, Vanoli, Foiani 2008; Sollier et al. 2009). Thus, this process is strictly controlled, highlighting its importance for the maintenance of genome stability in conditions of replication stress. Interestingly, González-Prieto et al. (2013) have found evidence for the Rad52-mediated recruitment of Rad51 to chromatin during the unperturbed replication. Upon exposure to an alkylating agent, the recruited Rad51 facilitates DNA synthesis on a damaged template in a DSB repair-independent process, most probably by promoting gap filling of single-stranded lesions that accumulate during replication of alkylated DNA. Future research will determine how the Rad52-dependent recruitment of Rad51 to unperturbed replication forks is linked to the above-mentioned lesion bypass mechanisms. As an aside, in higher eukaryotic cells, the Rad51 ortholog and other HR factors are indispensable for DNA replication, and the inactivation of genes encoding these proteins is lethal, whereas equivalent recombination proteins are dispensable for viability in both yeast species (reviewed in San Filippo, Sung and Klein 2008; Errico and Costanzo 2010).
Mutations dependent on recombination
Although HR is typically an error-free process, error-free recombination products are only produced during allelic-strand exchanges between sister chromatids, and as mentioned above, recombination between sister chromatids in the S and G2 phases of the cell cycle is favored by the cell-cycle mechanisms that regulate recombination. Aside from recombination between sister chromatids, there are many sources of genetic alterations in the final recombination products.
First, Rad51-mediated strand invasion of homologous sequences containing few heterologies can occur, thus generating a heteroduplex DNA molecule bearing mismatches typically processed by the MMR pathway (Chen and Jinks-Robertson 1998). The MMR-mediated repair of these mismatches results in gene conversion, defined as the unidirectional (non-reciprocal) transfer of information from one DNA duplex to another. If mismatches are not repaired, then the mismatched strands will segregate during the next round of replication, resulting in a sectored colony, or when there are too many mismatches in the heteroduplex DNA intermediate, the MMR-mediated anti-recombination pathway blocks the formation of recombinant products (heteroduplex rejection). Thus, the final recombinants could include gene conversion changes introduced by the MMR of heteroduplex DNA molecules initiating recombination.
Second, the two HR pathways, double HJ and SDSA, share a common intermediate, the D-loop, in which only donor (displaced) strands are copied (Ira, Satory and Haber 2006). The modified replication forks in the D-loop use only the leading-strand synthesis machinery to extend the first and second ends. Importantly, D-loop extension does not require the replicative helicase complex comprising the heterohexameric Mcm2–7 (MCM) helicase proteins and Cdc45 (and presumably GINS proteins) (Wang et al. 2004). However, a recent study shows that the proper functioning of GINS in the replisome is necessary for the fidelity of DNA replication in budding yeast (Grabowska et al. 2014). Accordingly, several lines of evidence suggest that the replication forks in D-loops, which form during HR-mediated DSB repair, replicate DNA with significantly decreased fidelity (reviewed in Malkova and Haber 2012). In the region surrounding an induced DSB, mutations arise at a rate 1000-fold higher than the spontaneous mutation rate. Although the majority of these mutations are base substitutions, other types of mutations can occur through multiple microhomology-mediated template switches between various, even highly diverged, sequences during replication, indicating that D-loop replication is less effective than normal replication. Consistently, template switch mutations are eliminated in strains with a proofreading-defective Pol δ because defective polymerases are less likely to dissociate from the template than the wild-type enzyme. Thus, mutations also result from errors in the replicative polymerases Pol δ and Pol ε. In addition, the mutations detected in the region of the DSB repair where both strands have been newly synthesized are not dependent on either TLS polymerases, Pol ζ and Pol η, or Pol32, despite the dependence on Pol δ, or the MMR system (Hicks, Kim and Haber 2010). During DSB repair, DNA replication also occurs at regions outside the D-loop, where extensively resected ssDNA is filled in to complete the repair. This replication is also mutagenic, generating an increased proportion of frameshifts rather than base pair substitutions. The base pair substitutions are associated with the Pol ζ-mediated fill-in replication of ssDNA, and the frameshift mutagenesis is halved in rev3 null strains. Therefore, multiple polymerases are responsible for the mutagenesis occurring during fill-in replication. In addition, MMR defects do not change either the rate or the spectrum of the mutations (Hicks, Kim and Haber 2010). It is not clear why the MMR system is active on the heteroduplexes formed during strand invasion in HR pathways, but becomes inactive once the DNA replication associated with HR is initiated. Nevertheless, MMR is temporally coupled to replication in the S phase, whereas heteroduplex rejection is largely cell cycle independent (Hombauer et al. 2011). It is possible that the replication associated with recombination lacks a cell-cycle-dependent factor for the specific activation of MMR in the S phase. Interestingly, Rattray et al. (2002) showed that in the absence of Rad57, the DSB-induced recombination of inverted repeats significantly increases mutagenesis associated with repair events. Rad57 forms a heterodimer with Rad55, which mediates Rad51-dependent homologous DNA pairing and strand exchange, as an alternative to the Rad52 mediator function (Sung 1997) mentioned in the section ‘HR subpathways’. Therefore, it is conceivable that after some template switching events during DNA replication associated with DSB repair, the polymerase resumes replication on the correct template after a reiterated engagement of the extending strand with Rad51, suggesting that in budding yeast cells, the function of Rad51 contributes to the fidelity of DNA replication, as mentioned in the section ‘HR subpathways’ in relation to the recombination-dependent error-free lesion bypass.
The fidelity of DNA replication associated with DSB repair has also been studied in the context of concomitant exposure to MMS or UV and a short oligonucleotide template for repair (Yang et al. 2008). In this system, extensive end resection typically proceeds after DSB induction, but only a small region in the immediate vicinity of the DSB is available for D-loop replication, resulting in the persistence of ssDNA next to the DSB that has to be filled in. Not surprisingly, treatment with MMS or UV leads to a further 10-fold increase in mutagenesis, but in addition, in the region that has been rendered single-stranded prior to treatment, mutations occur in widely separated clusters of multiple mutations. The UV-induced mutations also show a strand-specific bias. Thus, lesions in exposed ssDNA are hypermutagenic. As mentioned above, although the active replication forks in BIR require most of the S-phase replication proteins (Lydeard et al. 2010), BIR is highly inaccurate and, in contrast to two-sided DSB events, the lack of replication fidelity extends over the entire path of the replication fork (Deem et al. 2011). The rate of frameshift mutagenesis increases 2500-fold during BIR compared to mutagenesis during normal replication, although MMR and proofreading activities are proficient. The frameshift mutations suggest that replication becomes unstable due to template-switching errors. Interestingly, the replication fork generated during BIR is unusual. The replication proceeds in a Pif1-dependent migrating D-loop structure that is extended by Pol δ (Saini et al. 2013; Wilson et al. 2013). Similar to the results mentioned above, BIR intermediates are a strong source of clustered mutagenesis induced after MMS treatment (Sakofsky et al. 2014). Furthermore, in conditions of replication stress or premature mitosis due to checkpoint deficiency, BIR leads to a surge of GCR events. The results of research on BIR mechanisms are particularly significant because these experiments model the fates of collapsed forks or eroded telomeres in the cell, suggesting that failed replication can lead to genome rearrangements and clustered mutagenesis. Studies in fission yeast have shown similar results on BIR. Iraqui et al. (2012) reported that a single collapsed fork leads to mutations and large-scale genomic rearrangements (deletions and translocations) due to erroneous DNA synthesis during the recovery of replication forks.
The third source of genetic modifications during the HJ pathway, but not the SDSA pathway, is the resolution step, in which resolvases catalyze the cleavage of HJ intermediates to generate either non-crossover or crossover (reciprocal exchange of the flanking sequences) products. The double HJ, leading to crossover in the final recombination products, does not have genetic consequences when the HJ intermediates have linked sister chromatids in the same allelic positions. However, under other circumstances, the resolution may result in genetic rearrangements, e.g. LOH in mitotic diploid cells when the HJ intermediates have linked homologous chromosomes. There are three mitotic resolvases in S. cerevisiae (reviewed in Sarbajna and West 2014): Yen1 (the homolog of the human GEN1 protein), Mus81–Mms4 and Slx1–Slx4 (however, the report by Muñoz-Galván et al. 2012 questioned the nuclease activity of Slx1 in budding yeast). These enzymes differ in the range of branched molecules processed and the products of the catalyzed HJ cleavage. The results of several studies on the regulation of S. cerevisiae resolvases have indicated a number of mechanisms by which cells control the actions of these structure-selective nucleases during mitosis and meiosis via cell-cycle-dependent phosphorylation. For example, there is strong evidence for cell-cycle regulation of joint molecule resolution during recombination-based template switching events between sister chromatids (error-free lesion bypass mentioned in sections ‘Postreplication repair: a specialized pathway to repair replication stall-borne problems’ and ‘HR subpathways’). The pivotal element in this control circuit is the conserved Dpb11–Slx4 complex (in Sc. pombe, Rad4/Cut5 and Slx4, respectively) of scaffold proteins that control the joint molecule resolution between sister chromatids mediated by the Mus81–Mms4 endonuclease. Cell-cycle-dependent phosphorylation of Slx4 by Cdk1 promotes the Dpb11–Slx4 interaction. In mitosis, at the appropriate time to resolve joint molecules before cell division, phosphorylation of Mms4 by Polo-like kinase Cdc5 (in Sc. pombe Plo1) stimulates the association of Mus81–Mms4 with the Dpb11–Slx4 complex, thereby promoting joint molecule resolution (Gritenaite et al. 2014). To close the regulatory circuit, the DNA checkpoint inhibits the interaction of Mus81–Mms4 with Dpb11–Slx4 and consequently prevents resolution (Gritenaite et al. 2014). Another line of recent research concentrates on the regulation of HJ resolution in meiosis, because crossovers are essential for the establishment of cohesin-mediated interactions between homologs and the ultimate segregation of these molecules (Matos et al. 2011; Matos, Blanco, West 2013; reviewed in Sarbajna and West 2014). Furthermore, there is a meiosis-specific pathway for crossover production that is presumably dependent on the recently described heterodimeric Mlh1–Mlh3 endonuclease (Ranjha, Anand and Cejka 2014; Rogacheva et al. 2014; reviewed in Kohl and Sekelsky 2013). The metabolism of HJs in the fission yeast is notably different from that of the budding yeast, since Sc. pombe cells lack a clear Yen1/GEN1 homolog and depend on the Mus81–Eme1 (the fission yeast orthologs of Mus81 and Mms4) resolvase for HJ processing in mitosis and meiosis. For example, Mus81–Eme1 is upregulated by DNA damage through Rad3 (human ATR; S. cerevisiae Mec1)- and Cdc2/Cdk1-dependent phosphorylation of Eme1 (Dehé et al. 2013), whereas, in S. cerevisiae, cell-cycle-dependent phosphoactivation of Mus81–Mms4 is inhibited by DNA-damaging agents, because upon DNA damage the activated Mec1 checkpoint pathway delays expression/activation of cell-cycle kinases and cell-cycle progression. However, HJ resolution by endonucleases is not promoted in unchallenged mitotic cells. In S. cerevisiae during mitosis, HJs are rather processed by non-crossover dissolution mediated by the STR complex, as mentioned in the section ‘HR subpathways’. The STR complex suppresses crossover formation (Gangloff, Soustelle and Fabre 2000; Ira et al. 2003) and the accumulation of recombinant intermediates at damaged replication forks (Liberi et al. 2005). Biochemical studies, initially for the human ortholog BLM complex and subsequently for the S. cerevisiae complex, have shown that indeed the BLM/Sgs1 complex dissolves double HJ intermediates into products that are exclusively non-crossover (Wu and Hickson 2003; Raynard, Bussen and Sung 2006; Cejka et al. 2010). Surprisingly, the conserved Sgs1 helicase (the Sc. pombe ortholog Rqh1) has multiple functions linked to recombination-related DNA transactions. In mitotic cells, in addition to being a component of the crossover-suppressing STR complex, the helicase functions also as a prorecombination protein in end resection, as described in the section ‘HR subpathways’.
Recombination is a double-edged sword: it facilitates DNA replication and protects genome stability, but it can also lead to genomic rearrangements. In fission yeast, recombination promotes cell viability under replication stress at the expense of genetic stability (Lambert et al. 2005). Therefore, recombination must be tightly controlled through multiple mechanisms, such as those listed above, and coordinated with the cell-cycle checkpoint response (reviewed in Labib and De Piccoli 2011). Last but not least, structural maintenance of chromosomes (SMC) complexes, cohesin, condensin and Smc5–Smc6 are indispensable for chromosome organization, DNA transactions and cell viability (reviewed in Jeppsson et al. 2014). The SMC complexes have already been implicated in the regulation of DSB repair by HR (reviewed in Symington, Rothstein and Lisby 2014), whereas the Smc5–Smc6 complex in particular has been shown to be required for preventing the accumulation of inappropriate recombination structures arising during mitosis under replicative stress (Sollier et al. 2009; Choi et al. 2010) and during meiosis (Copsey et al. 2013; Lilienthal, Kanno and Sjögren 2013; Xaver et al. 2013). Future studies will focus on understanding how these factors link recombination and the maintenance of chromosome structure.
THE INFLUENCE OF PLOIDY ON GENOME STABILITY
One important aspect of the optimization of genome maintenance processes is the number of copies of genetic material found in various organisms or during various growth stages of a given organism. Having more than one copy of the genome helps to create a ‘reservoir of variability’ that is beneficial for adaptation. Second copy of the genome is also crucial for a HR, the usually accurate DNA repair mechanism. Therefore, it is not surprising that most eukaryotes are diploid or polyploid for the most of their lives. However, having multiple copies of the genome also has a downside. Sometimes, especially under conditions that cause DSBs, it allows for genome rearrangements that can result in excessive genome shuffling with disastrous rather than beneficial effects on the survival. In haploid cells, the presence of a second copy of the genome that enables HR is limited to the S and G2 phases of the cell cycle when DNA is duplicated. During G1, rearrangements may persist in the cell only if they do not perturb essential genes. Thus, sensitivity to DSBs is greater in haploid cells than in diploid cells. The distinction in mutation frequencies between haploid and diploid cells becomes comprehensible when the disparity in their DNA rearrangement survival rates and the differences in the availability of homologous sequences in their genomes are considered. In diploid S. cerevisiae cells, the frequency of spontaneous mutagenesis in forward mutation assays at CAN1/can1Δ and URA3/ura3Δ heterozygous markers is two orders of magnitude higher than in respective loci in haploid cells (approximately 10−4 versus 10−6, respectively) (Hiraoka et al. 2000; Ohnishi et al. 2004; Alabrudzinska, Skoneczny and Skoneczna 2011). The frequency of point mutations, such as base substitutions or frameshifts, remains similar in both cell types. The higher mutagenesis level in diploid cells is due to recombination events, including gene conversion, crossover or chromosome loss, as revealed by mutation spectra analysis (Hiraoka et al. 2000; Ohnishi et al. 2004). These differences may suggest that genome maintenance mechanisms specific for various ploidy levels exist. However, this part of the genome stability story needs further experimental support.
One of the first results showing differences in budding yeast cellular responses to DNA damage with respect to ploidy were published by Li and Tye (2011). The authors showed that the mcm4Chaos3 mutation leading to an MCM helicase defect caused replication stress in both haploid and diploid cells. However, only diploid mutants exhibited a G2/M transition delay, displayed signs of severe genetic instability and subsequently, reduced viability. These unforeseen outcomes are due to differences in repair pathways choice; while haploid cells use the Rad6-dependent pathways that resumes stalled replication forks, diploid cells use the DSB repair pathways that depend on the Rad52 and MRX complex (Li and Tye 2011). A study of the nature and frequency of chromosomal rearrangements at a marker gene placed in the telomeric region showed a decrease in chromosome rearrangement frequency and an increase in the complexity of the rearrangements occurring at the target gene with the increase in ploidy level. The presence of short DNA tandem repeat sequences seems to be a key requirement for deletion and reciprocal translocation processes to occur in diploids (Tourrette et al. 2007). A genomic screen for sensitivity to the Top2 inhibitor doxorubicin revealed a substantial difference between haploid and diploid cells in response to this agent. Experiments in which cells from homozygous diploid and haploid yeast gene deletion clone collections were exposed to doxorubicin revealed much higher levels of damage and cell death or severely reduced growth fitness (up to 5-fold) of diploid mutant clones compared to their respective haploid mutant clones. In addition, the toxic effect of doxorubicin was observed only in diploid cells of strains lacking such genes as bem1Δ, ctf4Δ, ctk1Δ, hfi1Δ, nup133Δ or tho2Δ. These mutant strains displayed severe G1/S phase cell-cycle progression defects following doxorubicin treatment, and some of the mutant strains were either significantly enhanced (ctk1Δ and hfi1Δ) or deficient (tho2Δ) in recombination (Westmoreland et al. 2009). As mentioned in the section ‘Regulation of the choice between HR and NHEJ’, the efficiency of NHEJ is significantly reduced in diploid cells due to mating-type locus-dependent regulation of NHEJ activity. Because the choice of one DSB repair pathway over the other may secure or endanger stable genome maintenance, the time window for NHEJ activity is limited to the G1 phase even in haploids. In contrast, HR, in which the sister chromatid is used to efficiently repair DSBs, occurs during the post-replication stage. Delacôte and Lopez (2008) suggested that the association between the cell-cycle checkpoint and the appropriate DNA repair pathway determines the maintenance of genome stability. In the deletion strains that are specifically sensitive to doxorubicin as diploid homozygous strains, this sensitivity is clearly correlated with defects in G1/S phase cell-cycle progression. Doxorubicin-induced DSBs produced in the G1 phase are thus not repaired by NHEJ because the DNA checkpoint does not function properly to compensate for the decreased efficiency of NHEJ in diploids and, consequently, the DSBs can progress through S phase and be processed by HR in the late S/G2 phase. However, in this case, HR cannot use the sister chromatid, which is also broken at the same locus, but must use ectopic homologous sequences dispersed in the genome, leading to genetic instability.
DIVISION ABNORMALITIES
Whatever the reason for DNA content change, it always arises in the progeny cells as a consequence of cell division imperfections, resulting in another enormous reservoir of genetic variability. Factors such as unrepaired DNA damage, faulty mitotic spindle organization, functioning or positioning, defective septum organization or perturbations in mitotic checkpoints may affect the final DNA content of the cells. Abnormal cell division drives two possible anomaly types: aneuploidization and ploidy shift.
Chromosome instability in yeast
Having two copies of the genome, diploid cells are oversensitive to factors causing rearrangements. For example, DSBs stress can lead to changes in the DNA sequence, including aneuploidization (i.e. the gain or loss of individual chromosomes). The latter phenotype can also result from dysfunction of genes involved in DSB recognition, signaling or repair. Recent discoveries reveal further causative agents that may lead to aneuploidy in S. cerevisiae, such as hydroquinone-induced delay at G2/M transition checkpoint, which is normally activated by the Hog1–Swe1 pathway (Shiga et al. 2010).
Chromosome instability (CIN) is a widely observed phenotype in yeast and other fungi, including different pathogenic species (Hughes et al. 2000; Infante et al. 2003; Morrow and Fraser 2013; Harrison et al. 2014). Interestingly, various species differently tolerate aneuploidy, e.g. Sc. pombe are primarily euploid (with a species-specific number of chromosomes), because aneuploid cells of this organism display low viability (Molnar and Sipiczki 1993). The tolerance of CIN is influenced by two factors: (1) the character of the change, i.e. chromosome gain or loss events, and (2) the chromosome type, i.e. which chromosome is affected. In S. cerevisiae, monosomy (loss of one chromosome from diploid genome) can be tolerated, particularly in the case of chromosome III (Haber 1974). In standard laboratory conditions, the spontaneous rate of individual chromosome loss in S. cerevisiae varies between 10−4 and 10−6 events per cell division (Hartwell and Smith 1985; Klein 2001; Kumaran, Yang and Leu 2013). The same rate of chromosome loss has been observed in Sc. pombe (Bodi, Gysler-Junker and Kohli 1991). Notably, there is no strong correlation between the frequency of chromosome loss and the size of the chromosome. The largest yeast chromosome, XII, is lost at a frequency similar to chromosome III, one of the smallest chromosomes of S. cerevisiae. Both chromosomes show a much higher loss frequency compared with other chromosomes of similar sizes (Kumaran, Yang and Leu 2013). The frequency of chromosome XV gain under normal conditions is approximately 10−6–10−7 events per cell division. After antifungal drug treatment, the frequency increases by approximately one to two orders of magnitude (Howlett and Schiestl 2000).
Diverse stresses can induce CIN. Proteotoxic stress caused by transient Hsp90 inhibition or heat shock markedly increased CIN producing cell populations with various karyotypes. This elevated CIN is most likely linked to the chaperone role of Hsp90 in the kinetochore complex assembly (Stemmann et al. 2002). Cells exposed to Hsp90 inhibitor exhibit changes in chromosome XV, resulting in a multi-drug-resistant phenotype and disturbed chromosome stoichiometries. These results strongly suggest that aneuploidy is a form of stress-induced mutation capable of fueling rapid phenotypic evolution and drug resistance and may be related to a Hsp90-dependent adaptive mechanism under stress conditions (Chen et al. 2012). However, Yona et al. showed that chromosomal duplications are only the first ‘quick fix’ reaction in response to acquired stress. They suggested that aneuploidy is only a transient stage involving a short-lived intermediate that facilitates further adaptation and gives cells time to develop more refined and sustainable solutions (Yona et al. 2012). This may explain why aneuploidy is so widespread among yeast strains (Hughes et al. 2000).
The concept of the selective advantage of chromosome gain or loss under various stresses was systematically analyzed in S. cerevisiae by two groups (Pavelka et al. 2010; Sheltzer et al. 2011). Experimental data showed numerous examples of aneuploidy in yeast cells, but regardless of the origin of this biological phenomenon, the possession of more chromosomes did not necessary benefit the yeast. The addition of an extra copy of a single chromosome initially decreased cellular fitness. Cells displayed increased chromosome loss, elevated mitotic recombination and defective DNA damage repair, causing aneuploidy-induced genomic instability that could facilitate the development of subsequent genetic alterations and drive genome instability (Sheltzer et al. 2011). However, quantitative growth assays revealed that some aneuploid strains with distinct karyotypes and genome contents up to 3n showed significantly improved growth compared with isogenic euploid strains when cultured in parallel and subjected to various suboptimal growth conditions or the presence of a panel of chemotherapeutic or antifungal drugs. Aneuploidy directly affects the cell proteome and generates significant phenotypic variations that could help the cell adapt to changing conditions. Thus, aneuploidy can drive evolution (Pavelka et al. 2010). It was also demonstrated for several independent translocation events that despite their common origin from the integration of the same linear DNA construct, each translocation mutant strain had a different phenotype, sporulation rate, level of gene expression and cell morphology. These phenotypic variations are the result of applying various methods to cope with an initial translocation event in individual yeast cells (Rossi, Noel and Bruschi 2010).
In parallel with these findings, the hypothesis was proposed for the fungi Candida albicans claiming aneuploidy as a form of large-scale mutation able to confer adaptive phenotypes under diverse stress conditions (Zacchi et al. 2010). Candida albicans strains lacking two of the four histone H4 variants showed a decrease in histone H4 dosage followed by a severe growth defect, unstable colony morphology and the production of fast-growing morphologically stable suppressors. These suppressors displayed an increased histone H4 gene copy number due to partial or whole chromosomal trisomies. In budding yeast, hypoacetylated histone H4 is present at centromere regions associated with the Cse4 protein. Inhibition of histone deacetylases in cse4 or hhf1–20 kinetochore mutants is lethal. An increase in the acetylation of H4 in a sir2 (histone deacetylase) mutant or in cells overexpressing acetylase Sas2 led to increased rates of chromosome loss and to synthetic dosage lethality in kinetochore mutants. Therefore, the hypoacetylation of H4K16 at centromeres plays an important role in accurate chromosome segregation (Choy et al. 2011). In contrast, a malfunction in sister chromatid cohesion led to chromosome gain (Covo et al. 2014).
Other mechanisms, in addition to exogenous stress, could also lead to aneuploidization. Alterations in chromosome segregation can happen due to failed control of DNA quality (replication checkpoint) or cell division (mitotic checkpoint), improper spindle organization or orientation and defects in attachment of chromosomes to spindle filaments or in system correcting misattachments (spindle assembly checkpoint), as well as deficiencies in the mechanisms of chromosome segregation (anaphase-promoting complex checkpoint), cytokinesis or septation (Suijkerbuijk and Kops 2008; McCulley and Petes 2010; Thompson, Bakhoum and Compton 2010; Silva et al. 2011; Stirling, Crisp et al. 2012). The proteins engaged in the complex process of mitosis frequently influence the final DNA content in daughter cells. Abnormal DNA content imposed by defects in mitosis can be repaired, provided that the causative agent is eliminated. Otherwise, a dividing aneuploid cell undergoes mitotic arrest triggered by unbalanced DNA content and generates a damage signal (prolonged spindle assembly checkpoint activation) for its elimination by mitotic catastrophe (Vitale et al. 2011). If cell death is not executed at this point, various scenarios may occur, including senescence followed by cell death, continuation of aberrant divisions leading to aneuploidization or cell death (depending on whether daughter cells receive all essential genes), mitotic slippage or cytokinesis failure leading to polyploidization. For example, in S. cerevisiae failure to establish the cohesion complex in ctf18Δ or eco1 mutants causes aneuploidization (Spencer et al. 1990; Kouprina et al. 1993; Unal, Heidinger-Pauli and Koshland 2007). However, inability to dissolve the cohesion complex triggers polyploidization (Bermúdez-López et al. 2010). Lack of the ESP1 gene encoding separase or an excess of Pds1 encoding securin aborts sister chromatid separation and also causes polyploidization (Lu and Cross 2009). Mitotic exit network components directly control cytokinesis by targeting the Inn1, Cyk3 and Chs2 proteins to the bud neck. Dysfunction of any of these proteins results in aneuploidization (Meitinger et al. 2010). Another process that contributes to proper chromosome segregation involves sensing chromosome attachment to the mitotic spindle (reviewed in Biggins 2013). When chromosomes are attached improperly, a signal of no tension is sensed to activate the spindle assembly checkpoint (SAC). Components of this checkpoint include the Mad1, Mad2, Mad3, Bub1, Bub3 and Mps1 proteins (Hoyt, Totis and Roberts 1991; Li and Murray 1991). The SAC mediates cell-cycle arrest by inhibiting the anaphase-promoting complex (APC) via inhibition of Cdc20 (Li et al. 1997; Kim et al. 1998). This prevents the APC from promoting the ubiquitylation of cyclin B and securin, two key substrates required for mitotic progression (Peters 2006). The checkpoint signal that prevents cells with defective spindles from initiating chromosome segregation is generated at the kinetochore. Although all SAC proteins except Mad3 localize to the kinetochore (Gillett, Espelin and Sorger 2004), the time of binding and the roles of the specific proteins differ. Mad1 and Mad2 are specifically recruited to unattached kinetochores (Gillett, Espelin and Sorger 2004). Although Mad1 is stably bound to unattached kinetochores, Mad2 exists in two different pools. The Mad2 in a ‘closed’ conformation (Mad2-C) remains stably bound to Mad1 and serves as a receptor for soluble Mad2 in an ‘open’ conformation (Mad2-O) (Luo et al. 2002; De Antoni et al. 2005). Mad2-O cycles onto the kinetochore, where it binds to Mad2-C, promoting the conversion of Mad2-O to Mad2-C. In the ‘closed’ conformation, the Mad2 protein exposes the dimerization surface, which is also required downstream of kinetochores to mount a checkpoint response. Interestingly, downstream of kinetochores, the dimerization surface does not mediate Mad2 dimerization but interaction with Mad3 (Mariani et al. 2012). Because Mad1 and Cdc20 have similar Mad2-binding motifs, Mad2-C is able to bind to Cdc20 as well (Luo et al. 2002; Mapelli et al. 2007). Thus, the kinetochore Mad1-bound pool of Mad2-C promotes the formation of a soluble Mad3-Mad2-Cdc20 complex (stabilized by the Mad3–Mad2 interaction), subsequently promoting APC inhibition. The Bub1 and Bub3 checkpoint proteins always localize to kinetochores during mitosis, but similar to Mad1 and Mad2, they are also required for regulation of kinetochore biorientation (Warren et al. 2002; Lee and Spencer 2004). Due to this additional function, mutation of these genes results in the strongest segregation defects (Warren et al. 2002; Kawashima et al. 2010; Storchova et al. 2011). A mutation in the SGO1 gene encoding the spindle checkpoint component involved in sensing the presence of that tension leads to chromosome loss as well (Indjeian, Stern and Murray 2005; Fernius and Hardwick 2007). The iron-responsive transcription factor Aft1 associates with the kinetochore complex through Iml3 and, similar to Iml3, is required for the increased association of cohesin with pericentric chromatin, which in turn is required to resist microtubule tension. Thus, aft1Δ cells display chromosome segregation defects (Hamza and Baetz 2012). Another protein involved in activation of the SAC in response to a defect in mitosis is histone H3. A histone H3 mutation impairs the ability of yeast cells to activate the checkpoint in a tensionless crisis, leading to missegregation and aneuploidy. This defect results from an attenuated H3–Sgo1 interaction that is essential for pericentric recruitment of Sgo1. Histone H3 seems to be a key factor in transmitting tension status to the SAC (Luo et al. 2010).
Ploidy shifts in yeast
Studies have shown that ploidy reduction towards diploidy occurs after several hundred generations in both triploid and tetraploid lines of S. cerevisiae (Gerstein et al. 2006; Gerstein, McBride, Otto 2008). However, after a sufficient number of generations (approximately 1800 generations), haploid strains also convert into diploid strains (Gerstein et al. 2006). These results suggest that the diploid state is favorable for standard laboratory S. cerevisiae strains maintained under typical conditions. However, ploidy change occurs quite often in diploid cells.
Similar to aneuploidy, ploidy changes in yeast cells can be provoked by several mechanisms: (1) defects in condensation of not fully replicated or unrepaired damaged DNA that persist till mitosis; (2) defects in interaction between DNA, kinetochore and spindle microtubules; (3) perturbations in mitotic spindle organization, positioning, orientation or microtubule dynamics; (4) disturbances in septum organization; (5) malfunction of control pathways of chromosome division and (6) various exogenous factors, such as gamma irradiation, certain anticancer drugs or heat shock, that can affect the aforementioned factors. Two experimental approaches revealed this intriguing phenomenon, which is exclusive to diploid cells. Saccharomyces cerevisiae diploid cells constantly exposed to rearrangement stress as a consequence of deletion of RAD52 (engaged in HR) or CTF18 (regulating PCNA and cohesion complex access to DNA) led to GCR, which can reduce the genome to the haploid level. This process occurs sequentially in rad52/rad52 cells, resulting in the loss of the chromosomes one by one during successive divisions (Song and Petes 2012), whereas ctf18/ctf18 cells reduce their genome in a single step (Alabrudzinska, Skoneczny and Skoneczna 2011). Both haploidization types result in the limitation of rearrangement events. Due to the shortage of homologous sequences in the haploid genome, GCR frequency is low in haploid compared to diploid cells (10−9 versus 10−4, respectively) (Chen and Kolodner 1999; Hiraoka et al. 2000; Ohnishi et al. 2004). It is still not clear if the tendency to reduce the ploidy level observed in diploid strains bearing rad52 or ctf18 mutations is incidental or a way to avoid further genome destabilization. However, haploid strains that appeared in the process of ploidy reduction have much lower mutation rates than initial diploids; moreover, they are fertile and more vital, so they likely dominate the cell population and have better chances for survival (Alabrudzinska, Skoneczny and Skoneczna 2011). Although it should be noticed that the frequency of haploidization is not high, approximately 90% of cells in the initial diploid population drift in the opposite direction and increase their DNA content to the aneuploid or polyploid level. These cells show elevated mutation rates, decreased fitness and escalating division problems, leading to permanent cell-cycle arrest and finally to cell death (Alabrudzinska, Skoneczny and Skoneczna 2011).
Ploidy changes have also been reported for other yeast mutants, but those cases involved the conversion of haploids into diploids. These changes accompany deletions of a number of genes. Haploid cells devoid of the ZDS1 gene (which encodes a factor involved in mitotic exit) will become diploid cells when exposed to Ca2+ ions (Miyakawa and Mizunuma 2007). Mutations in any of three adjacent residues (L97, Y98 or G99) near the C-terminus of histone H4 led to polyploidy as well (Yu et al. 2011). The deletion of the genes encoding components of the RSC chromatin remodeling complex required for G1/S transition (SFH1 or RSC3) resulted in a ploidy shift that could be rescued by CLB5 deletion (S-phase cyclin) or by transient depletion of the replication origin licensing factor Cdc6 (Campsteijn, Wijnands-Collin and Logie 2007). Increased ploidy is also observed in ipl1Δ and bem2Δ strains (Chan and Botstein 1993). Ipl1 is an Aurora kinase in the chromosomal passenger complex required for spindle pool body cohesion, regulation of chromosome segregation, spindle checkpoint and cytokinesis. The Rho GTPase-activating protein Bem2 is involved in the control of cytoskeleton organization and is required for bud emergence (Chan and Botstein 1993). These data highlight the role of regulatory systems in the control of cellular ploidy.
Schizosaccharomyces pombe is a valuable model for studying the mechanisms governing the accuracy of chromosome segregation. Because fission yeast cells divide equally, much like the cells of higher eukaryotes, these organisms are often used as models for studying chromosome segregation. However, both S. cerevisiae and Sc. pombe undergo closed mitosis, whereas the majority of higher eukaryotes perform open mitosis, during which the nuclear envelope is disassembled. The release of the nuclear content enables spindle microtubules to access kinetochores (Kanoh 2013). Nuclear envelope breakdown does not occur during closed mitosis. This feature of yeast cells changes the nature of the chromosome segregation process. In Sc. pombe, telomeres are tethered to the nuclear envelope in interphase. Chromosome tethering is required for various nuclear processes, such as transcription, transport and DNA repair. But chromosome segregation in mitosis requires efficient chromosome movements to assure equal division. Cdc2-dependent phosphorylation of the telomere-binding protein Rap1 during mitosis promotes the transient dissociation of telomeres from the nuclear envelope, which is required for accurate chromosome segregation (Fujita et al. 2012). The tethering of chromosomes ends has an additional function: it protects telomeres from illegitimate end-to-end fusions that would otherwise lead to dicentric chromosome formation. Dicentric chromosomes trigger breakage–fusion–bridge cycles and subsequent genome instability (Almeida and Godinho Ferreira 2013). The creation of chromosome fusions in telomerase mutant strains depends on the MMEJ SSA repair pathway and requires MRN/Ctp1. In strains lacking the telomere regulator Taz1/TRF2, end-joining reactions occur via NHEJ (Almeida and Godinho Ferreira 2013).
Studies have shown that the putative Ras1 effector in Sc. pombe, Scd1, alters microtubule dynamics within the spindle to affect spindle assembly and chromosome capture. Scd1 physically associates with Moe1, a factor that contributes to the inherent instability of microtubules and is required for proper spindle function. Defects in both Ras1 and Scd1 lead to genome instability (Segal and Clarke 2001). Recent studies have implicated two other pathways that contribute to the regulation of accurate chromosome segregation: Chk1/Wat1-dependent regulation of the mitotic spindle microtubule structure (Verma et al. 2014) and histone variant H2A.Z-dependent regulation of cohesin dynamics. Indeed, it has been shown that H2A.Z and the SMC complex ensure genome integrity through accurate chromosome segregation (Tapia-Alveal et al. 2014).
Mechanisms of ploidy change in yeast
Ploidy shifts occur frequently in yeast cells. One can assume that most of these events happen when cells evade death initiated by improper division. Cells that are arrested at the mitotic checkpoint are expected to die if they unable to restore proper mitosis. Ongoing aberrant division must result in abnormal DNA content. Division arrest may be initiated by severe DNA damage, spindle perturbation or even heat shock and should not be revoked until the problem is solved. If the signal recognizing the presence of anomalies does not disappear and division arrest persists, the cell normally is marked for removal by mitotic catastrophe similar to the process in mammalian cells (Vitale et al. 2011). However, when cell-cycle control fails or there is a mutation predisposing the cell to genomic instability, the cell may avoid execution and the aberrant division may be completed with various consequences, such as the gain or loss of single chromosomes, haploidization or entering into sequential steps of illegitimate polyploidization and depolyploidization (Fig. 5).
Figure 5.

The DNA content disturbances mechanisms in yeast. When cells arrested at the mitotic checkpoint are unable to restore proper mitosis, they supposedly die due to mitotic catastrophe. If cells evade death at this point, the ploidy shifts may occur. Aberrant division results in various consequences, such as the gain or loss of single chromosomes, haploidization or entering into sequential steps of illegitimate polyploidization and depolyploidization. Genome duplication may occur by ‘mitotic slippage’ or by ‘cytokinesis failure’ leading to binucleated or polyploidal cell, respectively. Polyploidal cells frequently undergo aneuploidization. Ploidy reduction may occur by multipolar spindle formation and subsequent loss of chromosome set in one step, by successive loss of chromosomes leading to haploid state, or by selective elimination of the parental chromosomes from the cell via nucleoautophagy.
What mechanisms could lead to a ploidy shift? In general, it is believed that genome duplication may occur by two independent mechanisms: ‘mitotic slippage’ or ‘cytokinesis failure’. Aneuploidization can be provoked by DNA discontinuities, by errors in chromosome attachment (e.g. monotelic, syntelic or merotelic chromosomes) (Fig. 6), or by non-synchronous and delayed spindle movement.
Figure 6.
The types of chromosome attachment errors. Properly attached chromosomes (amphitelic attachment), and different types of attachment errors: monotelic (only one chromosome from a pair is attached), syntelic (both chromosomes from pair are attached to one side of division spindle) and merotelic chromosomes (chromosomes are attached unequally).
However, in other fungi, the ploidy shift also results from the successive loss of chromosomes through different stages of aneuploidy, ending with haploid state. This mechanism has been described for different species of imperfect fungi after treatment with benomyl (Stahl and Esser 1992; Samsonova et al. 1996) and γ-radiation (Mortimer, Contopoulou and Schild 1981), and after protoplasts fusion (Fournier et al. 1977; Büttner et al. 1990) or in early zygote progeny (Kurischko 1986). The polyploidy and subsequent ploidy reduction via multipolar spindle formation has been observed in the tetraploid cells of C. albicans (Suzuki et al. 1986). Indeed, other unexpected ploidy changes have also been described in the ‘obligate diploid’ C. albicans. This fungi can become haploid through a chromosome loss mechanism, and the haploids of different mating types efficiently mate to regenerate the diploid form. Homozygous diploids arise spontaneously through autodiploidization. Haploids and autodiploids show a reduction in fitness, while heterozygous diploids restore original fitness. Therefore, homozygous diploid cells transiently appear in mixed populations (Hickman et al. 2013). It has also been shown that after treatment with antifungal drugs, e.g. fluconazole, C. albicans strains frequently form aneuploid cells that are drug resistant. Fluconazole induces abnormal cell-cycle progression, leading to binucleate cells, as mother and daughter cells fail to separate after chromosome segregation. After the next growth cycle, these cells form an unusual cell type called a ‘trimera’ yielding four daughter cells. Two of these cells undergo mitotic collapse to form a tetraploid cell with extra spindle components. The subsequent aberrant/unequal chromosome segregation causes aneuploidization of the progeny (Harrison et al. 2014).
Haploidization can occur following at least three scenarios: (1) loss of the full sets of chromosomes at once (e.g. by multipolar spindle formation) (Alabrudzinska, Skoneczny and Skoneczna 2011); (2) successive loss of chromosomes through various stages of aneuploidy, ending with a haploid state (Song and Petes 2012); or (3) selective elimination of the parental chromosomes from the cell via a nucleoautophagy-related mechanism (Fig. 5). The latter possibility has not yet been documented in yeast; however, it cannot be excluded at this point. A similar mechanism comprising micronuclei formation, heterochromatinization and fragmentation of micronucleated chromatin in the final step during haploidization in a process resembling apoptosis (named programed DNA elimination) was observed in other eukaryotic cells (Zelesco and Graves 1983; Gernand et al. 2005). In mammalian cells, the formation of micronuclei containing aggregated, DSB-fragmented DNA, kinetochores and centromeres is widely used to score genomic instability, genotoxic exposure or replication stress (Norppa and Falck 2003; Huang et al. 2011; Xu et al. 2011). Determining whether nucleoautophagy that occurs in yeast cells (Krick et al. 2008; Robert et al. 2011) may work as a similar mechanism requires further study. Gerstein et al. provided additional evidence supporting the first haploidization mechanism described above. In their experiments, polyploid S. cerevisiae cells were cultured for hundreds of generations, resulting in ploidy reduction towards diploidy. Remarkably, the chromosome loss was not random. Instead, nearly complete sets of chromosomes were lost at once, with some additional chromosome missegregation events. The authors proposed a mitotic mechanism for the elimination of an entire set of chromosomes in S. cerevisiae, which would reduce the ploidy level (Gerstein et al. 2006; Gerstein, McBride, Otto 2008).
TRANSCRIPTION CAN AFFECT GENOME STABILITY
Although most mutagenic changes are introduced into DNA during its replication or due to erroneous DNA repair, the importance of transcription as a cause of genetic instability is increasingly being recognized. It is hardly surprising that transcription can affect genome stability because RNA synthesis involves significant temporary changes in DNA strand conformation and, especially when passing through highly expressed genes, transcription machinery may often come into conflict with other protein complexes operating on the DNA, primarily the replication complex. Remarkably, approximately 40 human genes are between 1 and 2.3 million base pairs in length (Scherer 2008). Because the rate of transcription in eukaryotes is 18–42 nt per second (Pérez-Ortín, Alepuz and Moreno 2007), in such extreme cases the gene transcription can last for more than 10 h and span several cell division cycles (Tennyson, Klamut and Worton 1995). Thus, encounters between the transcription and replication machineries are inevitable. For this reason, those loci experience frequent mutagenic events (Helmrich, Ballarino and Tora 2011). Intensively transcribed DNA stretches are also prone to mutagenesis inflicted by transcription in S. cerevisiae cells.
The first indications that transcription could destabilize the genome came from early studies on bacteria demonstrating with the reversion assay that increased transcription from a certain region was accompanied by elevated mutagenesis of that region (Herman and Dworkin 1971). Later, this phenomenon was found and studied in both prokaryotic and eukaryotic models with a substantial contribution from S. cerevisiae. Its densely packed, mostly intronless genome makes studies of this phenomenon especially convenient.
As mentioned above, the RNA polymerase complex (RNAP) may collide with the replication fork. This is most likely to occur when the complexes move in opposite directions (head-on collisions); however, in bacteria in which replication is more than 10 times faster than transcription (approximately 600 nt per second), collisions can also occur when they move codirectionally. Transcription involves the separation of DNA strands; thus, the non-transcribed strand (NTS) is exposed to potential attack by mutagenic agents. During transcription, newly synthesized RNA may form a heteroduplex with the transcribed strand (TS), whereas the NTS remains single stranded and may form alternative secondary structures. Moreover, the movement of RNAP along the DNA strand distorts its double helix uniformity. All of these transcription-associated incidents make the transcribed DNA regions prone to mutagenic changes introduced through various mechanisms that have been described in previous sections of this review. These mechanisms are collectively classified as transcription-associated genome instability (TAGIN) and can be divided into two broad categories: transcription-associated mutagenesis (TAM) and transcription-associated recombination (TAR).
Sources and mechanisms of TAGIN
During transcription, DNA strands are separated by the RNAP complex, creating the transcription bubble. This enforces positive supercoiling in front of the complex and negative supercoiling behind it (Liu and Wang 1987). Although this torsional stress is constantly relaxed by topoisomerases (Wang 2002), the journey of RNAP along the transcribed genome region is accompanied by local distortion of the native DNA helix. Nascent RNA molecule forms hybrid 9 nucleotides in length with the TS, whereas the complementary NTS remains single stranded (Gnatt et al. 2001). Whether this short stretch of ssDNA, confined within the RNAP particle, is susceptible to DNA damaging agents is debatable (Martinez-Rucobo et al. 2011). However, the distorted DNA helix (and especially the negatively supercoiled zone that follows RNAP) is susceptible to reshaping into unusual non-B DNA structures, such as stem-loops, triplexes or G-quadruplexes (reviewed in Belotserkovskii, Mirkin and Hanawalt 2013) and ssDNA stretches (Liu and Wang 1987). These structures are known to be preferentially attacked by endogenous or exogenous damaging agents (Wright et al. 2003).
In S. cerevisiae, actively transcribed genes are affixed in the proximity of the nuclear pores (Casolari et al. 2004; Cabal et al. 2006), which may also prevent those regions of the DNA helix from unrestricted rotation. Thus, this phenomenon might represent yet another transcription-related source of torsional stress, resulting in negative supercoil accumulation with all its associated consequences (Fig. 7A).
Figure 7.
Encounters between RNA polymerase and the replication fork. During transcription, the RNAP causes both positive and negative supercoiling of the DNA strand exerting a torsional stress on it. This stress aggravates when RNA polymerase gets into head-on collision with the moving replication fork. However, in healthy cells these conflicts are resolved by the action of topoisomerases. Newly synthesized RNA strand is able to invade the negatively supercoiled DNA strand; however, under normal circumstances it is drawn off by the RNA maturation and export machineries. When any of these mechanisms malfunctions an R-loop may develop with non-transcribed strand exposed to potential attack of mutagenic agents.
One of the structures that may form in the trail of the RNAP moving along the DNA is an R-loop (Fig. 7B). This structure consists of a stretch of ssDNA of the NTS and a hybrid between the newly synthesized RNA and TS. The formation of an R-loop is favored by the negative DNA supercoils that facilitate invasion of the DNA helix by the RNA strand. RNA:DNA heteroduplexes are thermodynamically more stable than the respective DNA strands within the double helix (Roberts and Crothers 1992), especially when the NTS strand is G-rich (Ginno et al. 2013); thus, one might wonder how the cell is able to dissociate them once they form. Yet under normal conditions, there are various processes that are synchronized with transcription that can counteract R-loop formation. In prokaryotes, transcription is coupled to translation (Gowrishankar and Harinarayanan 2004), which is able to pull RNA off the transcribed operon. In eukaryotes, RNA is post-transcriptionally processed and exported from the nucleus (Li and Manley 2006). Incidentally, although R-loop formation is considered to be a threat to genome stability (see below), it has also been shown to play a positive regulatory role in transcription in human cells (Ginno et al. 2013).
The consequences of collisions between transcription and replication may be even more serious. Distinct from bacteria, in eukaryotes, transcription and replication are usually temporally separated within the cell cycle, although some genes, including those encoding rRNA, tRNA and histones, are transcribed during S phase. Replication and transcription seem to be also spatially separated to some extent (Wei et al. 1998). Nevertheless, collisions are unavoidable. Intuitively, head-on conflicts would seem to inflict more damage than codirectional ones. Transcription and replication travelling in the opposite direction force the buildup of positive supercoils between them, which may impede replication fork movement. Indeed, replication fork pauses have been observed in yeast at sites of head-on encounters between replication and transcription of tRNA genes (Deshpande and Newlon 1996). These collisions can lead to replication fork arrest or reversal, resulting in formation of potentially harmful structure called ‘chicken-foot’, as was shown for Escherichia coli (Postow et al. 2001). This type of structure has been demonstrated in S. cerevisiae when the replication block leads to activation of the PRR damage avoidance pathway via fork regression (Blastyák et al. 2007).
Transcription-associated genome instability
Transcription as a source of point mutations and small insertions or deletions
The first suggestions of the occurrence of TAM in S. cerevisiae appeared in the early 1990s. It was shown that the induction of gene expression could increase the mutation rate of that gene by as much as 150-fold (Korogodin et al. 1991). This finding was later supported by results obtained with pGAL- and pTET-inducible promoter systems by Datta and Jinks-Robertson (1995). More recently, it was shown that the mutagenesis rate is directly proportional to the level of transcription (Kim et al. 2007). By cloning the reversion system in either orientation, the authors demonstrated that the overall mutation rate was similarly increased regardless of the direction of replication fork movement, but an influence on the mutation spectra was observed. An impact of the transcription level on the mutation spectra was also demonstrated in another study. While low transcription levels resulted mostly in base substitutions, high transcription levels predominately led to short insertion–deletion mutations (Lippert et al. 2004), with two nucleotide deletions comprising 21% of all mutations. Therefore, the authors deemed 2-nt deletions as a TAM signature.
The TAM phenomenon was mostly studied on individual loci, sometimes artificially generated that may be prone to increased instability due to the heterologous context or might in some other way be sequence specific. Therefore, one would wonder if this phenomenon manifests across the whole genome. This question was addressed in a study analyzing the whole genomic sequence and transcriptome data (Park, Qian and Zhang 2012). The authors compared genome sequences of closely related S. cerevisiae and S. paradoxus as well as human and macaque sequences selecting only differences present in transcribed yet selectively neutral intron sequences. Then the authors compared the frequency of mutational changes within those sequences with their transcription levels and were able to demonstrate simultaneously for numerous loci that positive correlation between transcription and mutagenesis does exist.
A characteristic feature of TAM is its asymmetry. It was demonstrated in E. coli by transcription-dependent preferential deamination of cytosines on the NTS (Beletskii and Bhagwat 1996). These data were more recently substantiated by a demonstration of strand asymmetry in the incidence of spontaneous oxidative and alkylating lesions (Klapacz and Bhagwat 2005; Fix, Canugovi and Bhagwat 2008). In a whole genomic comparison study of two related yeast species, all types of mutations were found in correlation with increased transcription. However, the most frequent base substitutions were C→T, A→G, G→T and A→T (Park, Qian and Zhang 2012), suggesting that TAM is a result of cytosine or adenine deamination and the consequence of oxidative damage because 8-oxo-guanine (the major oxidative lesion) can mispair with adenine (Cheng et al. 1992). Another type of lesion that seems to be associated with a high level of transcription is the generation of AP sites (Morey, Greene and Jinks-Robertson 2000). Such sites are repaired by preferential incorporation of dUTP, leading to mutation and allowing detection of the lesions (Kim and Jinks-Robertson 2009). However, the mechanisms of transcription-associated base removal from the DNA strand are currently unknown. Thus, it seems clear that the predominant source of TAM is DNA damage.
Transcription-associated recombination
Transcription can also invoke higher order changes in the genome. It was demonstrated in S. cerevisiae that TAR may be the consequence of the activity of all three RNAPs (Keil and Roeder 1984; Pratt-Hyatt et al. 2006). Conflicts between transcription and replication are considered the main source of TAR. Head-on conflicts stimulate TAR more than codirectional conflicts (Prado and Aguilera 2005) (Fig. 7); however, a genome-wide study revealed correlations of both types of conflict with high levels of transcription in yeast (Azvolinsky et al. 2009). These conflicts can lead to replication fork arrest (Bermejo, Lai and Foiani 2012), thereby increasing the probability of replication fork collapse and causing recombinogenic DSBs and ssDNA gaps (Aguilera and Gómez-González 2008; Branzei and Foiani 2010). Likewise, recombination may be mediated by R-loops (González-Aguilera et al. 2008; Stirling 2012), since in yeast, deficiency of the THO complex that binds to the nascent mRNA during transcription elongation (Rondón, Jimeno and Aguilera 2010) shows a strong hyperrecombination phenotype (Piruat and Aguilera 1998). However, the exact mechanism(s) of recombination originating from R-loop formation or transcription–replication conflicts is unknown.
One of the regions of potential intense conflicts between replication and transcription is the rDNA region of the genome. In S. cerevisiae, this region consists of more than 100 identical tandem repeats located on chromosome XII (James et al. 2009). Each repeat encodes 35S rRNA and on the opposite strand, 5S rRNA. The direction of transcription of both of rRNAs is outward from the ARS element that lies between both sequences. This configuration prevents head-on collision between replication and transcription within a single rDNA unit but would not prevent conflicts with transcription running from the adjacent unit. However, each repeat is separated by a short sequence called the replication fork barrier. The binding of Fob1 protein to the barrier blocks the progression of the replication fork in one direction, into 35S rDNA sequence of the neighboring rDNA unit (Kobayashi 2003; Mohanty and Bastia 2004).
Multiple copies of rDNA make this region exceptionally susceptible to lesions that can instigate recombination and detrimental diversification of the number of repeats within the cell population. Nevertheless, maintaining the optimal number of rDNA copies also depends on recombination (Kobayashi 2011). A study demonstrated that the transcription of rDNA is necessary to accomplish this maintenance (Kobayashi et al. 1998). In this case, tightly controlled TAR is beneficial to the S. cerevisiae cell.
TAM as an adaptive mechanism
Contrary to mutations introduced during replication, those imposed by transcription can occur in non-dividing cells under starvation conditions. Notably, starvation-induced expression of specific genes leads to increased mutagenesis within their sequences and provides potential benefits to cells under strong selection pressure (reviewed in Wright 2004). In fact, the first paper reporting data indicating the existence of TAM in budding yeast (Korogodin et al. 1991), mentioned on the beginning of this section, describes exactly this type of experimental setup. It is likely that TAM is common in starved or resting cells because in a genome-wide study of stationary-phase cells, a correlation was found between the level of transcription and DNA turnover (probably reflecting the repair reactions) at various loci (de Morgan et al. 2010).
Transcription-coupled DNA repair
In addition to mutagenesis, one of the mechanisms of DNA repair is coupled to RNA synthesis, which increases the complexity of connections between transcription and genome stability even further. Various DNA lesions, including UV-induced photoproducts and bulky adducts present on the TS, create a physical barrier to movement of the RNAP along the DNA strand. The stalled complex recruits nucleotide–excision repair (NER) machinery (Hanawalt and Spivak 2008). Recently, it was demonstrated in yeast that AP sites on the TS are repaired by the NER pathway, indicating that non-bulky lesions also trigger TCR (Kim and Jinks-Robertson 2010). Therefore, transcription-coupled DNA repair (sometimes called TC-NER) preferentially repairs the transcribed DNA strand. In yeast, the Rad26 protein is required for efficient TC-NER (van Gool et al. 1994).
MITOCHONDRIA AND GENOME STABILITY
Mitochondria are double-membrane-bound organelles that are found in all eukaryotic cells. These organelles are thought to have evolved through endosymbiosis of α-proteobacteria-like cells that colonized a primordial eukaryotic cell for mutual benefit (Gray 1999). Mitochondria play an important role in the transformation of the energy stored in the bonds of carbohydrate molecules into the ATP synthesis via oxidative phosphorylation during respiration. Oxidative phosphorylation is accomplished in the inner mitochondrial membrane through the sophisticated assembly of four respiratory multisubunit protein complexes (although both budding and fission yeast lack complex I) of the electron transport chain and the fifth respiratory complex of ATP synthase (Saraste 1999). During evolution, mitochondria lost most of the ancestral proteobacterial genome, reflecting gene transfer to the nucleus and the loss of redundant information. However, these organelles preserved a residual genome encoding a few subunits of the respiratory complex, together with ribosomal RNAs and tRNAs necessary for mitochondrial genome expression. Budding and fission yeast share some features of their mitochondrial biology (see Table 2), but, as with many other aspects of Sc. pombe cell biology (Rhind et al. 2011), fission yeast mitochondria are more similar to mammalian mitochondria (Schäfer 2003). Specifically, the mitochondrial genome of Sc. pombe cells is compact compared to S. cerevisiae mtDNA (19 kb versus 75 kb), with few transcriptional initiation sites, short intergenic regions and the tRNA punctuation mode for mitochondrial messenger maturation, as in higher eukaryotic cells. Surprisingly, despite differences in genome organization, the sets of mtDNA genes in these two yeast species are remarkably convergent in spite of 500 million years of evolution after the two lineages diverged (Dashko et al. 2014). The most significant difference between the two yeast species is dependence on wild-type mtDNA, and consequently, dependence on respiration. Budding yeast cells are petite positive, i.e. able to survive without the wild-type mtDNA (so called rho+ mitochondrial genome) and the ability to respire. These mutants form smaller colonies than respiring cells on a glucose-limiting medium. In contrast, fission yeast cells are petite negative (Bulder 1964). The petite-positivity of budding yeast cells has greatly facilitated studies on the mechanisms responsible for the maintenance of mtDNA in this species. Cytoplasmic petite mutations occur spontaneously in budding yeast at a frequency of 1% per generation (Ephrussi 1953). There are several varieties of cytoplasmic petites (reviewed in Dujon 1981; Contamine and Picard 2000). (1) The most frequent varieties are those that possess inactive mtDNA products of mitochondrial genome rearrangements, including partial deletions and amplifications of remaining fragments (rho− mutants). (2) The mitochondria of another class of petites are entirely devoid of mtDNA (rho0 mutants). These yeasts are thought to arise spontaneously due to a failure to correctly segregate mtDNA with the mitochondria during their transmission to the incipient bud. (3) Further, there are cytoplasmic petites that have lost the ability to respire due to a point mutation or mutations in only one or two of the mitochondrial genes necessary for respiration (mit− mutants). In addition to respiratory deficiencies, the rho− and rho0 mutants display pleiotropic phenotypes associated with their inability to synthesize mitochondrial proteins. In contrast, mit− mutants are proficient in mitochondrial gene expression, although these yeasts are also unable to respire. It is not easy to distinguish between rho−/rho0 and mit− mutants upon first sight; therefore, to estimate point mutagenesis in yeast mtDNA, researchers test yeast cultures to determine the frequency of mutants that are respiratory proficient, but resistant to inhibitors that target specific mitochondrial processes. Often, the resistance phenotypes are associated with point mutations in mtDNA. Several mitochondrial inhibitors have been used in numerous studies for the isolation of mitochondrial point mutations, but erythromycin is the most specific for S. cerevisiae mtDNA (Baruffini, Ferrero and Foury 2010). Resistance to erythromycin is usually conferred through mutations at several positions in the mitochondrial 21S rRNA gene (Sor and Fukuhara 1982; Vanderstraeten et al. 1998). Notably, mitochondrial point mutations (whether mit− or mitochondrial drug resistance mutations) occur rarely compared with rho− deletions (see above). The frequency of spontaneous and induced mitochondrial point mutations is several orders of magnitude lower than the incidence of petite mutants (Foury and Vanderstraeten 1992; O'Rourke et al. 2002). This difference suggests that point mutations and rho− deletions in mtDNA result from distinct mechanisms. In contrast to S. cerevisiae, fission yeast cannot survive the loss or even a large deletion of mtDNA. The metabolism of these yeasts strictly depends on respiration, even when grown on glucose (Flores et al. 2000). Nevertheless, several genetic modifications were shown to convert fission yeast into petite-positive organisms: (1) nuclear mutations ptp1–1 or ptp2–1 (Haffter and Fox 1992), until recently unidentified, (2) expression of S. cerevisiae YME1 gene, encoding a mitochondrial protease associated with the inner membrane (Kominsky and Thorsness 2000) and (3) an mtDNA mutator allele of the mitochondrial gene rps3/urf A, encoding a putative mitoribosomal protein (Seitz-Mayr and Wolf 1982; reviewed in Schäfer 2003). Mutations suppressing the mtDNA-loss lethality of another petite-negative yeast species Kluyveromyces lactis have been found to be alleles of genes encoding subunits of the mitochondrial F1-ATPase (reviewed in Clark-Walker 2003). These mutations increase the affinity of F1-ATPase for ATP in the hydrolysis reaction (Clark-Walker 2003). Many lines of evidence indicate that the robust ATP hydrolysis is critical for the viability of cells lacking the functional mtDNA, because the mitochondrial inner membrane potential, ΔΨ, in petite cells is maintained, in lieu of the respiratory chain activity, by hydrolysis of ATP imported from cytosol (Giraud and Velours 1997; Clark-Walker 2003). Consequently, in petite cells, ATP is imported from cytosol into mitochondria by the reversal of the ATP/ADP translocator that generates in the process an electrogenic potential sufficient to maintain the mitochondrial protein import. The mitochondrial protein import, in turn, is essential for cell viability (reviewed in Chacinska et al. 2009), because the mitochondria are essential organelles even in cells of petite-positive species that do not require respiration (the essential function of mitochondria is described in the section ‘Mitochondrial influence on the nuclear genome’). Thus, it is likely that similar mutations to those from K. lactis conferring petite-positivity can be isolated in the fission yeast. However, although the feature of petite-positivity in S. cerevisiae has significantly contributed to the progress of studies on mtDNA stability, the potential advantage of using petite-positive strains of the fission yeast for studying the mechanisms of mtDNA maintenance remains unknown. Thus, most data on the stability of mtDNA in yeast come from studies on budding yeast, and, therefore, these results are reviewed below, with occasional forays into the Sc. pombe mitochondria if such data are available.
Table 2.
Comparison of budding and fission yeast cells in respect to their energy-yielding metabolisms and mitochondrial genomes in reference to the human mtDNA (based on Kühl 2010 and Flores et al. 2000).
| Feature | Saccharomyces cerevisiae | Schizosaccharomyces pombe |
|---|---|---|
| Metabolism | ||
| Ability to ferment glucose and produce ethanol. | Yes | Yes |
| Ability to use ethanol as a sole carbon source. | Yes | No |
| Mitochondrial genome | ||
| Petite-positivity | Yes | No |
| Introns in mitochondrial genesa | Yes | Yes |
| rRNA coded in mtDNA | 15S, 21S | rns, rnl |
| RNA component of RNase P coded in mtDNAb | 9S | rnp |
| Ribosomal subunit genec | VAR1 | rps3 |
| Major ORFs | 8 | 8 |
| Complex Id | 0 | 0 |
| Complex III | COB (CYTb) | cytb |
| Complex IV | COX1, -2, -3 | cox1, -2, -3 |
| Complex Ve | ATP6, -8, -9 | atp6, -8, -9 |
aIn human mtDNA, there are no introns interrupting mitochondrial genes.
bThere is no RNase P RNA coded in human mtDNA.
cThere is no gene for a ribosomal protein in human mtDNA.
dIn human mtDNA, there are seven genes coding for subunits of complex I.
eIn human cells, subunit 9 of complex V is encoded by a nuclear gene.
Nucleoids
Mitochondrial DNA in eukaryotic cells is packaged into compact nucleoprotein structures called nucleoids (Chen and Butow 2005; Kucej and Butow 2007). Nucleoids form dynamic complexes, and the number of nucleoids, mtDNA molecules/nucleoids and nucleoid protein content varies depending on growth conditions. An aerobically grown budding yeast cell might contain 40–60 nucleoids, whereas under conditions of limited aeration, the number of nucleoids is less than 10 per cell. The number of mtDNA molecules per nucleoid is inversely related to the number of nucleoids: the more nucleoids per cell, the less mtDNA molecules per nucleoid. The protein components of mitochondrial nucleoids have been identified in various organisms and are vastly divergent for organisms from different lineages (Kucej and Butow 2007). Surprisingly, in addition to the obvious proteins implicated in DNA transactions (e.g. replication and transcription) and DNA packaging, and in addition to the chaperonins and proteases involved in regulating other proteins, nucleoid components include coopted metabolic enzymes that, in a few instances, have been shown to be bifunctional (see below). This group of nucleoid proteins is the most lineage specific (Kucej and Butow 2007). Nevertheless, the main packaging proteins in nucleoids, HMG (high-mobility group) proteins, are conserved in eukaryotic cells. Abf2 is the HMG protein in S. cerevisiae mitochondria, whereas the mitochondrial transcription factor A (TFAM) is the HMG protein in human mitochondria.
In contrast to TFAM, the S. cerevisiae nucleoid protein does not play a specific role in regulating transcription in yeast. Instead, Abf2 binds mtDNA with some preference for GC-rich sequences (GC clusters in AT-rich sequences are dominant features of S. cerevisiae mtDNA) and determines the compaction and structure of mitochondrial nucleoids (Newman et al. 1996). Cells lacking the ABF2 gene lose rho+ mtDNA when grown on glucose-rich medium (Diffley and Stillman 1991), but these cell maintain rho+ genomes on media with either non-fermentable or non-repressing substrates as sole carbon sources (Chen et al. 2005), or under conditions of general amino acid control pathway induction in cells grown without the amino acids isoleucine, leucine and valine (Zelenaya-Troitskaya, Perlman and Butow 1995). In the former case, it was shown that another mitochondrial nucleoid protein, Aco1, whose expression is repressed in glucose-rich media, partially replaces Abf2 in the nucleoid. In the latter case, another component of the mitochondrial nucleoid replaces the Abf2 mtDNA-packaging function, the Ilv5 protein. Ilv5 is the acetohydroxy acid reductoisomerase that catalyzes one of the reactions in the branched chain amino acid biosynthesis pathway. The Aco1 protein is a bifunctional iron–sulfur cluster (ISC)-containing enzyme, which functions as both a mitochondrial aconitase in the tricarboxylic cycle and a nucleoid protein for the maintenance of rho+ genomes (Chen et al. 2005). These two functions are accomplished by two separate domains of the protein. Ilv5 binds to dsDNA independently of Abf2, and a deficiency of this protein destabilizes rho+ mtDNA (Macierzanka et al. 2008). However, when Aco1 replaces the Abf2 mtDNA-packaging function in nucleoids, mtDNA is less protected and becomes sensitive to nuclease attack (Newman et al. 1996) and oxidative damage (O'Rourke et al. 2002). It has also been shown that, under these conditions, microsatellite repeats and longer direct repeats in mtDNA are destabilized, although point mutations are detected at wild-type levels (Sia et al. 2008). These results indicate that the lack of Abf2 in mitochondrial nucleoids increases polymerase slippage events during mtDNA replication, and this defect might lead to mitochondrial genome rearrangements. Therefore, this mechanism most likely reflects the rho+ instability observed in cells lacking Abf2. However, there is also evidence that Abf2 is a positive factor in some HR pathways. In a study using 2D gel electrophoresis, Abf2 promoted HJ intermediates in rho+ mtDNA in vivo but not recombination intermediates detected in the mtDNA of rho− petite strains (MacAlpine, Perlman and Butow 1998). Thus, Abf2 not only helps to prevent genome rearrangements during replication, but it may also be a positive factor of a mitochondrial recombination process that helps to stabilize rho+ genomes. Interestingly, the report by Taylor et al. (2005) has shown that moderate overexpression of ABF2 increases the mtDNA copy number. Abf2 overproduction also increases the level of recombination intermediates (MacAlpine, Perlman and Butow 1998), suggesting that elevated Abf2-dependent recombination stimulates mtDNA replication.
Unfortunately, mitochondrial nucleoids from fission yeast cells have not yet been studied. Moreover, there are many predicted HMG proteins in the Sc. pombe genome (PomBase, http://www.pombase.org/), but none of these proteins have been identified as an obvious ortholog of Abf2 or TFAM, and none have been found to have a clear cut mitochondrial targeting sequence (based on MitoProt prediction; MitoProt, http://ihg.gsf.de/ihg/mitoprot.html/, date last accessed 12 November 2014). Therefore, the protein content in the mitochondrial nucleoids of fission yeast requires further study, which will likely reveal more instances of ‘evolutionary tinkering with nucleoids’ (Kucej and Butow 2007).
Replication of mtDNA
Saccharomyces cerevisiae cells harbor the mitochondrial DNA polymerase, Mip1 (Pol γ), which mediates mtDNA replication. The MIP1 gene deletion leads to the loss of mtDNA (rho0 strains; Foury 1989; Merz and Westermann 2009), indicating that this polymerase is a major, if not unique (see below), replicative polymerase in the mitochondria of budding yeasts. Similar results were obtained for the Sc. pombe pog1 gene, encoding the fission yeast ortholog of Mip1 (Chu et al. 2007). Mip1 functions as a single catalytic subunit enzyme (Lucas et al. 2004; Viikov, Väljamäe and Sedman 2011), in contrast to the mitochondrial polymerase from mammalian cells, which requires an accessory subunit (reviewed in Kaguni 2004). The mitochondrial DNA polymerase from the fission yeast, Pog1, is likely also a single subunit polymerase. The catalytic subunits of polymerase γ are phylogenetically related to the prokaryotic PolA family and well conserved in eukaryotic cells (Kaguni 2004), suggesting that Pol γ was an early invention in the evolution of mitochondrial endosymbiosis. In vitro studies have shown that Mip1 is a highly processive DNA polymerase with strong strand-displacement activity (Viikov, Väljamäe and Sedman 2011). Strand-displacement activity is important for the long-patch base-excision repair (BER) pathway, as described below, underscoring that Pol γ is responsible not only for mtDNA duplication, but also plays a role in various mitochondrial DNA repair pathways that require polymerase activity. Pol γ has three main activities: a polymerase activity, a 3′→5′ exonuclease proofreading activity and a 5′-dRP lyase activity (reviewed in Kaguni 2004). The high fidelity of mtDNA replication (Table 1) is largely determined by the N-terminal domain, which exhibits 3′→5′ exonuclease activity for the excision of misincorporated bases before polymerase extension. A mutation in this domain results in the substitution of a conserved residue, mip1-D347A, leading to a several hundred fold increase in the frequency of point mutations in mtDNA and a several fold increase in the incidence of rho− petite mutants (Foury and Vanderstraeten 1992). Moreover, defects in the Mip1 exonuclease activity combined with a defective mutation in the MSH1 gene, encoding the mitochondrial mutS homolog required in MMR in yeast mitochondria (see below), resulted in a synergistic mitochondrial error catastrophe and the complete loss of rho+ mtDNA (and conversion to rho−) after a few generations (Vanderstraeten et al. 1998). Thus, the exonuclease proofreading activity of Mip1 and the Msh1-dependent MMR pathway are the main, at least partially independent, guardians of genome stability during mtDNA replication in S. cerevisiae. The 5′-dRP lyase activity of the Mip1 polymerase is important for the elimination of intermediates during BER, which is reviewed below.
Interestingly, despite the fact that Mip1 is a major replicative DNA polymerase in S. cerevisiae mitochondria, it is not the only DNA polymerase in this compartment. Two subunits of Pol α, including the largest catalytic subunit, Pol1, and the accessory subunit Pol12, have been detected in yeast mitochondria (Lasserre et al. 2013). It is not known whether the mitochondrial subunits of Pol α are actually involved in the replication of mtDNA. However, several lines of evidence clearly indicate that the TLS polymerases, DNA Pol ζ (Rev3/Rev7) and Rev1 function in yeast mitochondria in pathways that are not equivalent to the nuclear functions of the polymerases. First, Zhang, Chatterjee and Singh (2005) showed that all three polymerases localize to yeast mitochondria, and inactivations of REV3/REV7 and REV1 differently interact with the MIP1-deficient mutation in an in vivo assay of mtDNA frameshift mutagenesis. Second, in contrast to TLS repair in the nucleus (Friedberg, Lehmann and Fuchs 2005), cells lacking Rev3 or Rev7 show increased spontaneous and UV-induced point mutagenesis in mtDNA (Kalifa and Sia 2007). The mutagenesis of mtDNA is also increased in rev1 null cells, but only upon exposure to UV. Thus, the TLS polymerases in yeast mitochondria protect mtDNA from accumulating base substitutions. Considering that the nuclear mutagenesis mediated through Rev3/Rev7 and Rev1 is dependent on the formation of the four-subunit complex between Rev3, Rev7, Pol31 and Pol32 and its interaction with PCNA (Makarova, Stodola and Burgers 2012) and that the formation of this complex in mitochondria is not likely, it is not so unexpected that Pol ζ and Rev1 are not as mutagenic in mitochondria as observed in the nucleus. However, both Pol ζ and Rev1 are responsible for the majority of spontaneous and UV-induced frameshift mutations associated with mitochondrial microsatellite sequences and UV-induced petite formation (Kalifa and Sia 2007). This result clearly indicates that Pol ζ and Rev1 contribute to the generation of mtDNA rearrangements during mtDNA replication, even in unstressed cells. On the other hand, the instability of rho+ mtDNA in certain mip1 mutants is partially suppressed through the overexpression of REV3, but not REV1, whereas increased mitochondrial point mutagenesis in these mutants is suppressed through the overexpression of both REV3 and REV1, but not of each individual gene (Baruffini et al. 2012). These results suggest that Pol ζ and Rev1 function in yeast mitochondria in several, at least partly separate, lesion-dependent and as yet uncharacterized pathways. To complicate the matter even more, Pol η (Rad30) is also localized to yeast mitochondria (Chatterjee, Pabla and Siede 2013). In the mitochondria of budding yeast, Rad30 acts in at least two mtDNA repair pathways: one pathway reduces UV-induced mitochondrial point mutagenesis, together with Pol ζ, and the other pathway counteracts UV-induced rearrangements of mtDNA and petite generation, opposing the activity of Pol ζ (Chatterjee, Pabla and Siede 2013). Orthologs of the S. cerevisiae polymerases described in this section are in Sc. pombe; however, there are no reports about the potential mitochondrial functions of these enzymes.
Initiation of mtDNA replication and transmission of mtDNA during cell division
The initiation of mtDNA replication in S. cerevisiae is a controversial topic. The debate stems from the long-held opinion that the topology of yeast mtDNA should be similar to the topology of the circular mtDNA in mammalian cells and the circular genomes of the endosymbiotic precursors of mitochondria. Moreover, the genetic map of the budding yeast is also circular. The sequence of S. cerevisiae mtDNA contains eight GC (guanine and cytosine)-rich cluster elements that resemble the structures in the heavy-strand replication origin of mammalian mtDNA. These clusters were named ori/rep sequences, and analogous to those detected in mammalian mitochondria, three to four of these elements (Lecrenier and Foury 2000) are thought to be active origins of mtDNA replication initiated via transcription mediated through the mitochondrial RNA polymerase, the Rpo41 protein in S. cerevisiae (Fig. 8A). However, cells lacking the Rpo41 polymerase, although unable to maintain rho+ genomes, maintain rho− genomes, even those containing only repeated ori sequences (Lorimer, Brewer and Fangman 1995). In S. cerevisiae cells, mitochondrial protein synthesis is required for the maintenance of the rho+ mitochondrial genome (Myers, Pape and Tzagoloff 1985). Thus, it remains unclear whether Rpo41 is actually needed for the initiation of mtDNA replication in budding yeast. Furthermore, many lines of evidence in various yeast species (S. cerevisiae and Sc. pombe included) have demonstrated that only a small fraction of mtDNA in these cells is actually circular; a majority of the mtDNA molecules are linear and have heterogeneous sizes due to tandem (concatameric) arrays of genomic units (Maleszka, Skelly and Clark-Walker 1991; Bendich 1996). Maleszka et al. also detected circular molecules with single- or double-stranded tails (lariats) and proposed a rolling-circle replication (RCR) mechanism as a model for yeast mtDNA replication, featuring uncoupled synthesis of the leading and lagging strand. In this model, circular mtDNA molecules arise only through the recombination-mediated looping out of a single genetic unit from the concatameric mtDNA array. 2D gel studies on Sc. pombe mtDNA confirmed this model of mtDNA replication for fission yeast cells as well (Han and Stachow 1994). There is now prevailing genetic evidence from budding yeast that the RCR model is true for mtDNA replication in this species (Fig. 8B). Ling and Shibata (2002) have proposed this model to explain the finding that, in S. cerevisiae, mother cells primarily contain concatameric mtDNA arrays, whereas in buds mtDNA is predominantly in the form of circular monomers. These authors also identified Mhr1 as the protein required for the transmission of nascent concatameric mtDNA into buds, and this protein catalyzes a recombinase reaction involving ssDNA pairing with homologous dsDNA, thereby forming a heteroduplex, the basic intermediate of HR. Surprisingly, Mhr1 is not structurally related to the RecA family of recombinases. In the model by Ling and Shibata, Mhr1-mediated pairing generates a primer for mtDNA RCR, producing a concatameric mtDNA array. This concatameric molecule is subsequently transmitted to buds with a concomitant monomerization of mtDNA. In three follow-up studies, the same authors confirmed this model, showing that (1) Mhr1-dependent RCR initiated at the DSB generated at ori5 is responsible for concatamer formation and the biased inheritance of the rho− genome containing this ori element (Ling, Hori and Shibata 2007); (2) ROS exposure leads to increased mtDNA copy numbers by promoting Mhr1-initiated RCR of mtDNA at ROS-generated DSB ends (Hori et al. 2009); (3) the mitochondrial 5′ exonuclease Din7 participates with Mhr1 in ROS-enhanced mtDNA replication and recombination at ori5 (Ling et al. 2013). Therefore, it can be concluded that Mhr1 mediates, at least partially, recombination-dependent replication (RDR) in yeast mitochondria. However, strains with the mhr1–1 allele, encoding an Mhr1 variant that is inactive in the ssDNA pairing activity in vitro (Ling and Shibata 2002), exhibit a moderate phenotype (i.e. temperature dependent loss of rho+ DNA), whereas the deletion of the MHR1 gene results in the unconditional loss of rho+ genomes (conversion to rho−). Consequently, it is highly likely that another, yet unidentified, Mhr1 activity is indispensable for the maintenance of rho+ mtDNA. In addition, there is recent evidence (Fritsch et al. 2014) supporting the notion that there are more recombinases in S. cerevisiae mitochondria than only Mhr1 (Ling et al. 1995). Interestingly, the Mhr1 function is conserved in fission yeast. There is a clear ortholog protein, and inactivation of the coding gene is lethal for the petite-negative yeast Sc. pombe (PomBase. http://www.pombase.org/). However, in the Mhr1-dependent concatamer formation model, the mechanism of mtDNA partitioning into buds, i.e. how mtDNA monomers are actually looped out from nascent Mhr1-initiated concatamers before being transmitted to buds, has not yet been elucidated. Future studies should identify other nucleoid proteins that are involved in the partitioning process. This will provide a better understanding of yeast mitochondrial genomes. Currently, another protein, conserved in both yeast species, has been implicated in the Mhr1-related RDR pathway. The mitochondrial S. cerevisiae Cce1/Mgt1 structure-selective endonuclease resolves HJs (Kleff, Kemper and Sternglanz 1992). In Sc. pombe, an orthologous nuclease, Ydc2, exhibits the same activity as the budding yeast homolog (Oram, Keeley and Tsaneva 1998). Strains in both yeast species lacking these nucleases display strikingly similar phenotypes with their mtDNA aggregated in an interlinked network joined by unresolved recombination junctions (Lockshon et al. 1995; Doe et al. 2000). A double S. cerevisiae mutant lacking Cce1 and harboring the defective allele mhr1–1 (see above) lacks mtDNA (Ling and Shibata 2002), and thus, these two mutations interact synergistically, suggesting that the two proteins, Cce1 and Mhr1, function in two parallel pathways necessary for mtDNA transmission to progeny cells.
Figure 8.

Current models of initiation of mtDNA replication in S. cerevisiae. (A) Transcription-dependent mtDNA replication. In the model, mtDNA replication is primed by the mitochondrial RNA polymerase Rpo41 at ori/rep sequences and the primer is extended afterwards by the Mip1 polymerase. However, the resulting expanding bubble mtDNA structures (θ structures), that are expected according the model, have not been detected (Maleszka et al. 1991; Bendich 1996). (B) Rolling-circle mtDNA replication mechanism proposed by Maleszka et al. (1991) and later expanded by Ling et al. (2007) and Hori et al. (2009) to explain the increases in mtDNA copy number triggered by ROS. In the model, mtDNA RCR is primed by homologous pairing catalyzed by the Mhr1 recombinase between ssDNA derived from the resection of DSB ends generated by the Ntg1-mediated processing of mtDNA lesions caused by DNA-damaging agents, e.g. ROS, as it has been shown for H2O2-induced Ntg1-dependent DSBs at the ori5 sequence that result in the Mhr1-mediated initiation of mtDNA replication. Two strands of the schematic representation reflect the later stages of RCR, when the lagging-strand synthesis (mediated by the putative mitochondrial Okazaki fragment processing apparatus whose factors are listed in the box) has been initiated on the single-stranded tail produced by RCR which ultimately results in the production of concatameric linear molecules that are the prevalent topological form of mtDNA in yeast cells. In this model, rare circular mtDNA, monomeric or oligomeric, are derived from multimeric molecules through intramolecular recombination events.
Okazaki fragment processing in mitochondria
Information on lagging-strand synthesis in yeast mitochondria is limited. Surprisingly, almost the entire apparatus for Okazaki fragment processing is present in budding yeast mitochondria and in human mitochondria (reviewed in Holt 2009). The Okazaki fragment processing apparatus comprises (1) the largest subunit of Pol α, Pol1, with its accessory subunit Pol12 (Lasserre et al. 2013), (2) the Pif1 helicase (Lahaye et al. 1991; Schulz and Zakian 1994) and (3) the Rad27 endonuclease (Kalifa et al. 2009). The essential Dna2 nuclease/helicase is likely also localized to yeast mitochondria based on genetic evidence (Budd et al. 2006), but direct evidence is lacking. Yeast Pol1 and Dna2 demonstrate that it is exceptionally challenging to study the mitochondrial function of an essential protein with dual nuclear and mitochondrial localization. However, at least for the three classical proteins of the well-characterized Okazaki fragment processing pathway (reviewed in Balakrishnan and Bambara 2011), Pif1, Rad27 and Dna2, the dual nuclear-mitochondrial localization is conserved in evolution, as orthologs of the three proteins display dual localization in human cells (Futami, Shimamoto and Furuichi 2007; Liu et al. 2008; Duxin et al. 2009), indicating that these proteins could function in a fundamental mitochondrial process, likely associated with the lagging-strand synthesis during mtDNA replication. Nevertheless, the proteins do not function in mitochondria exclusively in this process. In this section, one potential mitochondrial Okazaki fragment processing factor, the budding yeast Pif1 helicase and its fission yeast ortholog, Pfh1, are reviewed. Saccharomyces cerevisiae Rad27 and Sc. pombe orthologous Rad2 (Fen1) are reviewed in the section on BER in mitochondria.
The Pif1 helicase in mitochondria
The Pif1 helicase, which has 5′→3′ DNA helicase activity, has been associated with several mtDNA transactions, from replication and recombination (reviewed in Contamine and Picard 2000) to repair (O'Rourke et al. 2002; Doudican et al. 2005; Cheng, Dunaway and Ivessa 2007) in S. cerevisiae yeast mitochondria. This helicase and its paralog, Rrm3, are nuclear non-overlapping regulators of replication fork passage through hard-to-replicate regions, such as rDNA and G4 (G-quadruplex) structures (reviewed in Chung 2014). The ability of Pif1 to unwind G4 structures is likely essential for mtDNA replication because in budding yeast, there is a 10-fold higher concentration of the G4 motifs in mtDNA than in nuclear DNA (Capra et al. 2010). Furthermore, Pif1 has recently been shown to facilitate DNA synthesis in the D-loop during HR (Wilson et al. 2013). Strains lacking Pif1 exhibit high rho+ mtDNA instability; however, depending on genetic background, the final petite pif1Δ phenotype is either rho0 (Merz and Westermann 2009) or rho− (Doudican et al. 2005). In the latter case, a significant portion of pif1Δ cells maintain rho+ mtDNA, but with a marked accumulation of mitochondrial point mutations (O'Rourke et al. 2005), suggesting that the Pif1 helicase is also involved in mtDNA repair pathways. This conclusion is supported by the synergy between PIF1 and SOD2 deletions. The latter gene encodes mitochondrial matrix superoxide dismutase, and loss of this gene leads to increased mtDNA damage and, consequently, increased mtDNA point mutagenesis. However, loss of this gene does not impair the maintenance of rho+ genomes (Doudican et al. 2005; Kaniak et al. 2009). Furthermore, the maintenance of rho+ mtDNA in pif1Δ cells is synergistically destabilized by concomitant inactivation of the NTG1 gene, which encodes a nuclear and mitochondrial N-glycosylase/lyase specific for certain oxidatively modified bases (O'Rourke et al. 2002). Interestingly, this protein also stimulates DSBs (the enzymatic mechanism of this Ntg1 activity has not yet been elucidated) at ori5 for Mhr1-dependent RDR, resulting in concatamer formation and biased inheritance of a rho− ori5-containing genome (Ling, Hori and Shibata 2007). Although the lack of Ntg1 in cells maintaining rho+ mtDNA at wild-type levels does not influence mtDNA stability (e.g. O'Rourke et al. 2002; Doudican et al. 2005; Kaniak et al. 2009), this glycosylase is important for the maintenance of rho+ genomes in cells with destabilized rho+ mtDNA (i.e. pif1Δ cells) and in abf2Δ cells (O'Rourke et al. 2002). These results suggest that Ntg1 might be an element of the recombination-dependent system, regulating mtDNA replication and, consequently, mtDNA copy number. The synergy between the inactivation of either PIF1 or ABF2 and the inactivation of NTG1 suggests that Pif1 and Abf2 are important for rho+ maintenance independently of the Ntg1-mediated pathway, underscoring the complexity of mtDNA transactions in yeast cells.
In Sc. pombe, the unique Pif1 helicase ortholog Pfh1 is essential for the replication of both nuclear and mitochondrial genomes (Pinter, Aubert and Zakian 2008). In fission yeast cells, the helicase from the budding yeast, S. cerevisiae Pif1, and its nuclear paralog, S. cerevisiae Rrm3, failed to replace both essential nuclear and mitochondrial functions of Pfh1. However, Rrm3 partially suppressed the phenotypes of a nuclear Pfh1-depleted strain. These results suggest that the mitochondrial Pif1-like helicases in budding and fission yeast significantly diverged after the evolutionary separation of the two lineages. Nevertheless, it would be interesting to repeat the inactivation of the phf1 gene in a petite-positive Sc. pombe strain to better characterize the Pfh1 functions.
Mitochondrial BER
The exposure of DNA to endogenous threats (e.g. metabolism-derived ROS) and exogenous genotoxic agents produces various modifications to the structure of constituent bases. These lesions, if they do not distort the DNA helix, are repaired through BER (Baute and Depicker 2008; Svilar et al. 2011). Many BER enzymes in budding yeast have dual nuclear-mitochondrial localization (reviewed in Boiteux and Jinks-Robertson 2013). The pathway exists in two basic variants: a short-patch pathway and a long-patch pathway (Fig. 9A).
Figure 9.

Mitochondrial BER pathways in yeast cells. (A) Pathways of mtBER in S. cerevisiae cells. In the box, presumed factors of mt LP BER pathway are listed, with the Dna2 name dimmed, since its presence in mitochondria, though very likely, has not been directly proved yet. (B) BER pathways in Sc. pombe mitochondria. Highlighted in red are names of those proteins, which have been experimentally confirmed to be localized to mitochondria. In the box, presumed factors of mt LP BER pathway are listed.
The short-patch BER pathway (SP BER) employs various enzymes and involves several stages of intermediate processing: (1) recognition and excision of specific bases [in S. cerevisiae mitochondria, Ung1 recognizes and cleaves off uracil (Chatterjee and Singh 2001), Ogg1 excises 7,8-dihydro-8-oxo-2′-deoxyguanosine, 8-oxoG, a common oxidative modification of guanine, opposite cytosine (Singh et al. 2001) and Ntg1 (a homolog of E. coli endonuclease III, mentioned in the section ‘The Pif1 helicase in mitochondria’) excises oxidized pyrimidines (Alseth et al. 1999)]; (2) cleavage of the resulting AP site through either the AP lyase activities of bifunctional Ogg1 or Ntg1 (in contrast to the monofunctional glycosylase Ung1, which is unable to catalyze this reaction), producing phospho-α,β-unsaturated aldehyde (3′-PUA), a DNA polymerase blocking group on the 3′ end of the gap and a phosphate group on the 5′ end, or through the mitochondrial Apn1 AP endonuclease (Vongsamphanh, Fortier and Ramotar 2001), which cuts the phosphodiester backbone of the AP site to generate a strand break with 3′-OH and 5′-terminal deoxyribose-phosphate (5′-dRP) sugar moieties; (3) DNA-end processing to generate DNA 3′ ends that are filled in by a polymerase, and 5′ ends that are subsequently ligated. During the AP lyase reaction, the blocking PUA group is further processed by the 3′-phosphodiesterase activity of Apn1 to generate a 3′-OH end. Upon the AP endonuclease reaction, the 5′-dRP moiety is converted through the 5′-dRP lyase activity of Pol γ (mentioned above) to a 5′-phosphate that can be subsequently ligated; and (4) single-nucleotide gap filling and ligation. The actual role of the Apn1 protein goes beyond the simple SP BER model. Apn1 also functions in DNA repair without the need for a preceding glycosylase. The enzyme nicks DNA on the 5′ side of various oxidatively damaged bases, thereby generating 3′-OH and 5′-phosphate termini (Ischenko and Saparbaev 2002). This nucleotide incision repair (NIR) activity is also exhibited by AP endonucleases from other organisms (reviewed in Daley, Zakaria and Ramotar 2010).
Single mutants lacking the individual SP BER enzymes listed above under normal growth conditions do not display a phenotype associated with mtDNA stability (Apn1: Vongsamphanh, Fortier and Ramotar 2001), or display moderately increased mtDNA point mutagenesis (Apn1: Kaniak et al. 2009) that might differ, however, in the specificity of substitutions fixed in mtDNA depending on some unknown features of a genetic context (Ogg1: Dzierzbicki et al. 2004; Pogorzala, Mookerjee and Sia 2009), or display even moderately decreased mitochondrial point mutagenesis (Ntg1, Apn1: Pogorzala, Mookerjee and Sia 2009; Ntg1: Kaniak et al. 2009). The phenotype of ung1 null mutants was also described as a moderately increased frequency of respiratory-deficient cells with intact mitochondrial genomes; thus, these mutants displayed the properties of mit− mutants (Chatterjee and Singh 2001). Importantly, apn1 null strains exhibit significantly increased mitochondrial point mutagenesis upon exposure to the alkylating agent methyl methanesulfonate, MMS (Vongsamphanh, Fortier and Ramotar 2001), and show delayed kinetics of alkylation-induced damage repair specifically in mtDNA (Acevedo-Torres et al. 2009). Thus, the SP BER enzymes might recognize specific types of environmentally induced mtDNA damage that are rarely observed under normal growth conditions. Consistently, the Ntg1 glycosylase has been implicated, upon treatment of isolated mitochondria with hydrogen peroxide, in the generation of DSBs at ori5 to initiate Mhr1-mediated replication of mitochondrial genome, thereby increasing the mtDNA copy number (Hori et al. 2009) (Fig. 8A).
The long-patch BER pathway (LP BER) assists SP BER enzymes in 5′-blocked intermediate processing. The distinguishing feature of the two BER pathways is the size of the patch being repaired: one nucleotide in the case of SP BER, as described above, and two or more nucleotides in the case of LP BER (reviewed in Fortini and Dogliotti 2007). LP BER takes over the repair when the 5′-blocking group generated in the first steps of BER, typically when 5′-dRP cannot be removed by polymerase, e.g. the end is further oxidized. First, the 5′-blocking group is converted into a 5′-flap through strong strand displacement synthesis mediated by yeast Pol γ (Viikov, Väljamäe and Sedman 2011). Then, a 5′-flap endonuclease removes the short flap, the polymerase fills in the gap of several nucleotides and the ligase completes the repair. The identity of the LP BER flap endonuclease in yeast mitochondria is not clear, but in yeast nuclei, this endonuclease has been identified as Rad27 (Ayyagari et al. 2003), an ortholog of FEN1 in mammalian cells. This nuclease participates in many nuclear pathways. In addition to the cleavage of 5′-DNA flaps created during Okazaki fragment processing and the processing of intermediates during nuclear LP BER, Rad27 plays a role in the prevention of sequence duplications and repeat sequence expansions and in DSB repair through NHEJ (Tishkoff et al. 1997; Freudenreich, Kantrow and Zakian 1998; Schweitzer and Livingston 1998; Wu and Wang 1999; Wu, Wilson and Lieber 1999). Rad27, as mentioned above, is also localized to mitochondria in S. cerevisiae cells (Kalifa et al. 2009). Strains lacking Rad27 exhibit significantly increased mitochondrial point mutagenesis (Kalifa et al. 2009), likely reflecting the decreased mitochondrial LP BER. However, the lack of Rad27 also reduces the instability of dinucleotide tandem (microsatellite) repeats and longer direct repeats in mtDNA (Kalifa et al. 2009). Rearrangements of these sequence elements in yeast mitochondria are presumed to occur primarily through polymerase slippage or template switching events (Sia, Jinks-Robertson and Petes 1997; Phadnis, Sia and Sia 2005). Thus, the absence of Rad27 inhibits replication-mediated rearrangements of mtDNA. Interestingly, strains lacking Rev3, as described in the section ‘Replication of mtDNA’, have phenotypes associated with mtDNA stability that are similar to those of rad27Δ strains: increased mtDNA point mutagenesis, but decreased instability of mitochondrial microsatellite repeats, suggesting that Rad27 and Rev3 may function in the same pathway in yeast mitochondria. It is not clear whether the pathway is a mitochondrial LP BER; however, it is likely that Rev3 assists Mip1 in certain mtDNA repair processes (see the section ‘Replication of mtDNA’).
Surprisingly, there is more information about the mitochondrial LP BER in mammalian cells than in yeast cells. In addition to the human Rad27 ortholog FEN1, the DNA2 and EXOG nucleases have also been implicated in mitochondrial LP BER. In the case of Dna2, the yeast ortholog of DNA2, direct evidence regarding the localization of this protein in yeast mitochondria is needed. The role of EXOG, a paralog of the ENDOG nuclease, in mitochondrial LP BER in human cells has been well established. The nuclease, endowed with a 5′exo-/endonuclease activity, is required for repairing endogenous 5′-blocking single-strand breaks (SSBs) in mtDNA. EXOG depletion results in the accumulation of persistent SSBs in mtDNA, enhances ROS levels and induces apoptosis (Tann et al. 2011). The Nuc1 nuclease, the S. cerevisiae ortholog of mammalian ENDOG and EXOG nucleases (Büttner et al. 2007; Cymerman et al. 2008), is localized to the mitochondria where it functions in HR (Zassenhaus and Denniger 1994). Nuc1 produces recombinogenic DNA ends by introducing single-strand gaps into dsDNA through endonuclease and 5′→3′ exonuclease activities (Dake et al. 1988). Nuc1 has also been implicated in the degradation of damaged mtDNA under conditions of acute oxidative stress accompanied by a shortage of intramitochondrial energy (Dzierzbicki et al. 2012). In response to apoptotic stimuli, mammalian ENDOG translocates to the nucleus, where it participates with other nucleases in the fragmentation of chromosomal DNA (Li, Luo and Wang 2001). The Nuc1 nuclease has also been described to function in a similar pathway in response to apoptotic signals in budding yeast (Büttner et al. 2007). Another nuclease, Din7, a paralog of the Rad27 nuclease, is localized to mitochondria (Fikus et al. 2000). The role of the protein has not been established yet, but its overproduction results in the increase of the mitochondrial HR, elevated petite formation, mitochondrial microsatellite instability and induced point mutagenesis of mtDNA (Koprowski et al. 2003). The Din7 nuclease is also involved in the Mhr1-dependent RCR of mtDNA (Ling et al. 2013), as mentioned in the section ‘Initiation of mtDNA replication and transmission of mtDNA during cell division’. It remains to be established whether Rad27, Din7, Rev3, Nuc1 and Dna2 are indeed involved in the mitochondrial LP BER pathway in budding yeast cells.
The set of SP BER enzymes in Sc. pombe is markedly divergent from the BER enzymes in budding yeast cells (Table 3). There are more mono-functional glycosylases in fission yeast cells, similar to higher eukaryotic cells. Nevertheless, only one bifunctional Nth1 functions in Sc. pombe cells, presumably, providing the major, if not unique, AP lyase activity for processing AP sites (reviewed in Kanamitsu and Ikeda 2010). However, the mitochondrial localization of BER glycosylases in fission yeast cells remains unclear (Fig. 9B). As in S. cerevisiae, there are two AP endonucleases in Sc. pombe: Apn1, the homolog of the bacterial exonuclease III (Xth), and Apn2, the homolog of the bacterial endonuclease IV (Nfo), and these AP endonuclease orthologs evolved differently in the two yeast species (Kanamitsu and Ikeda 2010). Interestingly, Sc. pombe AP endonucleases do not cut AP sites, which are cut by Nth1. Instead, they remove the 3′-blocked ends that remain after Nth1 AP lyase activity. However, another Nfo-like endonuclease in fission yeast cells, Uve1, is localized to both the nucleus and mitochondria. This enzyme functions in an alternative AP endonuclease pathway, as mentioned above for S. cerevisiae Apn1, catalyzing the NIR-mediated removal of a variety of UV and non-UV lesions. Consequently, it is presumed that in fission yeast cells, both dual-localization Apn1 and Uve1 engage in an Nth1-independent NIR, and 5′-blocked DNA ends generated in NIR reactions can be further processed through the LP BER pathway. Interestingly, according to a recent report, most laboratory Sc. pombe strains, but not other independent isolates, carry a defective mutation in the apn1 gene (Laerdahl et al. 2011). Nevertheless, even in the context of the functional Apn1 endonuclease, these authors showed that Nth1 remains responsible for the majority of the AP lyase activity in cell extracts. Considering that the mitochondrial localizations of both Nth1 and Apn2 are unclear, the dual localization of its functional variant, Apn1, might significantly contribute to the AP lyase and NIR activities in fission yeast mitochondria. Future studies are needed to confirm this idea. In Sc. pombe cells, there are also orthologs of all the putative S. cerevisiae mitochondrial LP BER proteins described above. However, there are no reports of mitochondrial functions of these proteins, except for the confirmation of the mitochondrial localization of the fission yeast ortholog of Nuc1, Pnu1 (Oda et al. 2007). In pnu1 null cells, the ectopic production of a truncated Pnu1 lacking the mitochondrial targeting signal sequence, i.e. a fully active form of the nuclease generated during Pnu1 import to the mitochondria, results in cell death due to DNA fragmentation (Oda et al. 2007), suggesting that ENDOG orthologs may indeed be involved in a conserved apoptotic pathway in eukaryotic cells.
Table 3.
Comparison of BER and SSB repair enzymes in Sc. pombe, S. cerevisiae and human cells in respect to their subcellular localization (PomBase. http://www.pombase.org/; Saccharomyces Genome Database. http://www.yeastgenome.org/; Online Mendelian Inheritance in Man. http://www.omim.org/).
| Description of the protein | Saccharomyces cerevisiae protein (subcellular localization) | Schizosaccharomyces pombe protein (subcellular localization) | Human ortholog (subcellular localization) |
|---|---|---|---|
| SP BER pathway | |||
| Uracil/thymine (in G:T) DNA N-glycosylase Thp1 | – | Thp1 (n) | hTDG (n) |
| Uracil DNA N-glycosylase Ung1 | Ung1 (mt/n) | Ung1 (mt?/n) | hUNG (mt/n) |
| DNA-3-methyladenine glycosylase | Mag1 (n) | Mag1 (n) | – |
| DNA-3-methyladenine glycosylase | Mag1 (n) | Mag2 (n) | – |
| Adenine (in 8-oxoG:A) DNA glycosylase | – | Myh1 (n) | hMUTYH (n) |
| Homolog of endonuclease III from E. coli | Ntg1a (mt/n), Ntg2a (n) | Nth1 (mt?/n) | hNTHL1 (mt/n) |
| 8-Oxoguanine glycosylase | Ogg1 (mt/n) | – | hOGG1 (mt/n) |
| Homolog of E.coli exonuclease III [Xth]; AP-endonuclease Apn2 | Apn2 (n) | Apn2 (n) | hAPEX2 (mt/n) |
| Homolog of E. coli endonuclease IV [Nfo]); AP-endonuclease Apn1 | Apn1 (mt/n) | Apn1b (mt/n) | – |
| Endonuclease Uve1 | – | Uve1 (mt/n) | – |
| Homolog of endonuclease VII of E. coli | – | – | NEIL1 (mt/n) |
| Homolog of endonuclease VII of E. coli | – | – | NEIL2 (mt/n) |
| ERCC-8 DNA repair homolog | Rad28 (n) | Ckn1 (mt?/n) | ERCC8 (mt/n) |
| SNF2 family helicase Rhp26 | Rad26 (n) | Rhp26 (mt?/n) | ERCC6 (mt/n) |
| LP BER pathway | |||
| Flap endonuclease; FEN1 ortholog | Rad27 (mt/n) | Rad2 (mt?/n) | FEN1 (mt/n) |
| Mitochondrial endo- and exonuclease | Nuc1 (mt/n) | Pnu1 (m) | ENDOG (mt/n), EXOG (mt) |
| DNA replication endonuclease-helicase | Dna2 (mt?/n) | Dna2 (mt?/n) | DNA2 (mt/n) |
| Exonuclease Exo1 | Exo1 (n) | Exo1 (n) | EXO1 (n) |
| Exonuclease Exo1 | Din7 (mt) | Exo1 (n) | EXO1 (n) |
| SSB repair | |||
| Aprataxin | Hnt3 (n) | Hnt3 (mt?/n) | APTX (mt/n) |
| Tyrosyl-DNA phosphodiesterase Tdp1 | Tdp1 (n) | Tdp1 (n) | TDP1 (mt/n) |
mt—mitochondrial localization; n—nuclear localization.
Dual role for the Msh1 protein in mutation avoidance and rho+ genome stability
The MSH1 gene encodes a homolog of E. coli MutS protein (Reenan and Kolodner 1992), the critical component of the bacterial MMR pathway. In vitro, the Msh1 protein hydrolyzes ATP and recognizes DNA substrates containing mismatches and unpaired nucleotides (Chi and Kolodner 1994). MutS homologs generally act by initiating the repair of base substitution and insertion–deletion mismatches that arise during either replication or recombination (reviewed in Schofield and Hsieh 2003). Strains lacking this gene exhibit a petite phenotype due to rho− deletions, suggesting that Msh1 plays an important role in the maintenance of rho+ mtDNA stability. In addition, Msh1 is required in yeast mitochondria to prevent the accumulation of point mutations in mtDNA as a crucial factor in the proposed, but as yet uncharacterized, pathway of mitochondrial MMR. Strains with partial Msh1 defects, e.g. msh1Δ/MSH1 heterozygous strains, exhibit increased accumulation of point mutations in the mtDNA (Reenan and Kolodner 1992). Thus, the MSH1 gene is haplo insufficient, suggesting that the Msh1 level is fine-tuned for optimal mitochondrial genome maintenance. Accordingly, the overexpression of the MSH1 gene leads to the instability of rho+ genomes and increases the instability of poly(AT), but not poly(GT) repeats, through an unknown mechanism (Koprowski et al. 2002). However, moderate MSH1 overexpression suppresses the mitochondrial mutator phenotype of the ogg1Δ mutant (Dzierzbicki et al. 2004) and the mitochondrial mutator phenotype of the sod2Δ mutant (Kaniak et al. 2009). Additionally, the increased dosage of wild-type MSH1, but not of mutant alleles encoding deficient variants of the protein, results in enhanced allelic mitochondrial HR (Kaniak et al. 2009). On the other hand, the Msh1 activity suppresses direct repeat-mediated deletions, thereby inhibiting genomic rearrangements that may result in defective mtDNA (Mookerjee and Sia 2006). Therefore, Msh1 promotes allelic HR in mitochondria, but inhibits non-allelic direct-repeat-mediated deletion events and GT-repeat frameshift mutations. We propose that the Msh1 protein scans mtDNA duplexes, generated during either replication or recombination-associated strand invasion or strand annealing, for the presence of mismatches, and it directs the duplexes into either HR combined with mismatch correction or heteroduplex rejection and inhibition of the incipient recombination, depending on the quality of the duplex. Thus, on the one hand, Msh1 is a key player in the mitochondrial mutation avoidance system, and on the other hand, it is a chief controller of recombination processes in yeast mitochondria. The two functions of Msh1 are genetically separated (Mookerjee, Lyon and Sia 2005), which should facilitate the discrimination of Msh1 functions. Consistently, Pogorzala, Mookerjee and Sia (2009) also showed that Msh1 plays multiple roles in mitochondrial BER as a part of mitochondrial mutation avoidance, but this function is separate from the Msh1 function required for rho+ genome maintenance. A mitochondrial ortholog of Msh1 has been identified in Sc. pombe cells (Matsuyama et al. 2006); however, its role in fission yeast mitochondria has not been determined.
HR in yeast mitochondria
The pathways of mitochondrial recombination in yeast are not well understood (reviewed in Chen 2013). HR is highly active in S. cerevisiae mitochondria, e.g. markers separated by only 1 kb are practically unlinked (Dujon 1981). As mentioned in the section ‘Initiation of mtDNA replication and transmission of mtDNA during cell division’, the current model of initiation of mtDNA replication posits that HR-dependent processes generate primers for the mitochondrial RCR (Ling and Shibata 2002). The model explains the ROS-induced increase in mtDNA copy numbers through Mhr1-initiated Din7-dependent RCR of mtDNA at ROS-generated DSB ends (Hori et al. 2009; Ling et al. 2013). However, none of the proteins described in the Mhr1-dependent pathway (the DSB-generating Ntg1 glycosylase, the exonuclease Din7 or the Mhr1 recombinase), or in other recombination-related processes in mitochondria, like the Nuc1 nuclease (section ‘Mitochondrial base excision repair’) or the Cce1 resolvase (section ‘Initiation of mtDNA replication and transmission of mtDNA during cell division’), is essential for mitochondrial HR (Ling et al. 1995; Phadnis, Sia and Sia 2005; Fritsch et al. 2014). As mentioned above, genetic evidence suggests that the nuclease-helicase Dna2, the key end resection nuclease in the nuclear HR (reviewed in Mimitou and Symington 2011), functions in the yeast mitochondria and is necessary for the maintenance of rho+ mtDNA (Budd et al. 2006), though direct evidence is missing. The Pif1 helicase has been implicated for some time in the regulation of mitochondrial recombination (reviewed in Contamine and Picard 2000). The finding that during BIR and crossover recombination in the nucleus Pif1 facilitates DNA synthesis by promoting the establishment of a migrating D-loop structure (Wilson et al. 2013) suggests the possibility that Pif1 is a crucial factor mediating RDR of mitochondrial genomes. Consistently, the Pif1 helicase physically interacts with a mitochondrial single-stranded-binding protein, Rim1 (Ramanagoudr-Bhojappa et al. 2013). Nevertheless, there is also evidence that the function of another mitochondrial helicase Hmi1, that is required for the maintenance of rho+ genomes, partially overlaps with that of the Pif1 helicase (reviewed in Chen 2013). Clearly, there are several mitochondrial HR pathways operating in mitochondria that remain to be clarified. In addition, the set of mitochondrial proteins involved in DNA transactions has recently increased by components of the MRX complex and Ku proteins, as described in the following section, showing that the DSB repair pathways in the nucleus and the mitochondria share some central components. Last but not least, a new SSA activity has been uncovered for the Mgm101 protein (Mbantenkhu et al. 2011), as described in the section ‘The Mgm101 protein is required for rho+ 2305 genome stability’, offering the alternative to the Mhr1 recombinase activity.
MRX complex and Ku proteins function in mitochondria
In a recent report, Kalifa et al. (2012) have provided evidence that the factors involved in HR and NHEJ in the nucleus also function in the mitochondria. These authors showed that the inactivation of crucial DSB repair pathways mediated by the MRX complex and Ku proteins synergistically reduces the rate of deletions mediated by direct repeats in yeast mtDNA. The results suggest that these deletions primarily originate from the processing of DSB lesions in the repeated sequences via two independent pathways: mitochondrial MRX and mitochondrial Ku proteins. Surprisingly, the decrease in the direct repeat deletions in mtDNA in a strain lacking both MRX and Ku complexes was associated with a modest but significant increase in spontaneous petite mutants. These results indicate that the mechanisms of direct repeat-mediated deletions and rearrangements responsible for petite formation are mechanistically different. Under certain conditions, both pathways, mitochondrial HR and NHEJ, contribute to the maintenance of the rho+ genome. However, in another study, Dzierzbicki et al. (2012) showed that mutants deficient in the MRX function exhibit increased susceptibility to oxidative stress-induced rearrangements in mtDNA in antimycin A-treated cells. Under these conditions, the inactivation of RAD50 is not synergistic, but epistatic to YKU70 deletion, suggesting that in this case, the function of Rad50 is engaged in a different process than the function detected during the direct deletion-mediated rearrangements observed by Kalifa et al. (2012). In the nucleus, the MRX complex is involved in both HR and NHEJ pathways, therefore the MRX complex likely also participates in a mitochondrial HR pathway that contributes to the maintenance of rho+ genomes under oxidative stress conditions. Consistently, the elevated petite formation in cultures of antimycin A-treated rad50Δ cells correlates with the decreased potential for sustaining mitochondrial allelic HR. Thus, under certain conditions, the mitochondrial MRX-dependent pathway plays a significant role in the maintenance of rho+ mtDNA. Other factors acting in this mtDNA repair pathway remain unknown. Similarly, other components of mitochondrial NHEJ have not yet been identified.
The Mgm101 protein is required for rho+ genome stability
The mitochondrial nucleoid protein Mgm101 is essential for the maintenance of rho+ mtDNA, but not rho−, genomes (Zuo, Clark-Walker and Chen 2002). However, in certain genetic contexts, strains lacking the MGM101 are rho0 (Crider et al. 2012). Mgm101-deficient cells are more sensitive to mtDNA damage induced through UV irradiation and are hypersensitive to mtDNA damage induced through γ rays and hydrogen peroxide treatment (Meeusen et al. 1999). Consequently, Mgm101 performs an essential function in the repair of oxidatively damaged mtDNA and is critical for maintaining the mitochondrial genome. Meeusen and Nunnari (2003) showed that Mgm101 forms part of a two-membrane spanning (TMS) protein structure that traverses both the outer and inner mitochondrial membrane and associates with replicating nucleoids. The Mmm1 outer membrane protein and the Mip1 polymerase are also components of this structure. Interestingly, the Pif1 helicase is also a component of an inner mitochondrial membrane complex (Cheng and Ivessa 2010). It is not clear whether the Mgm101-containing TMS structure is the same as the Pif1 complex.
The report by Mbantenkhu et al. (2011) has shown that Mgm101 shares conserved motifs with the N-terminal ssDNA-annealing domain of the yeast Rad52 protein. Rad52 primarily functions as a mediator of Rad51 recombinase activity, but this protein also has ssDNA-annealing activity nucleus (as mentioned in the section ‘HR subpathways’). Consistently, the authors demonstrated that purified Mgm101 promotes ssDNA-annealing reactions in vitro in the presence of the mitochondrial ssDNA-binding protein Rim1. Moreover, Mgm101-deficient cells exhibit decreased rates of direct repeat-mediated deletions in mtDNA, consistent with Phadnis, Sia and Sia (2005) who proposed that the pathways affecting these rearrangements of mtDNA predominantly lead to non-crossover exchanges. Certainly, new insights about the mitochondrial functions of S. cerevisiae Mgm101, Pif1 and Msh1 will be informative for research on Sc. pombe mitochondria. All of these proteins have orthologs in fission yeast cells and the systems of mtDNA replication and inheritance are comparable between these species. Surprisingly, S. cerevisiae Mgm101 also forms a complex with the FANCM ortholog Mph1 helicase and MutSα (Msh2–Msh6 complex) in the nucleus and participates in the recombination-dependent repair of interstrand cross-link lesions in nuclear DNA (Ward et al. 2012). However, it is unclear whether similar Mgm101-dependent pathways function in yeast mitochondria.
It should be noted that the Mmm1 protein, a component of the above-mentioned TMS structure, has been demonstrated to be also an endoplasmic reticulum (ER) transmembrane protein and a part of the ER-mitochondria encounter structure (ERMES), a multiprotein complex physically tethering the ER with mitochondria (Kornmann et al. 2009). Strains defective in the ERMES function display an abnormal mitochondrial morphology and a deficiency in the maintenance of rho+ mtDNA (Hobbs et al. 2001; Kornmann et al. 2009). It has been suggested that the ERMES contact sites serve to link the segregation of mtDNA with the mitochondrial division (Murley et al. 2013). Studies on the interplay between mitochondrial nucleoids and ERMES proteins as well as other cellular components interacting with them will be important for understanding the pathways of mtDNA inheritance in yeast cells.
MITOCHONDRIAL INFLUENCE ON THE NUCLEAR GENOME
Mitochondria are crucial for diverse array of cellular processes, including energy metabolism, metabolic pathways involving lipids, amino acids, nucleotides and iron, signaling and programed cell death (apoptosis). However, one of the mitochondrial pathways has been recognized in budding yeast as essential for cell viability: the generation and maturation of Fe-S prosthetic groups (ISCs) for both mitochondrial and extramitochondrial Fe-S proteins (Kispal et al. 1999; reviewed in Lill and Kispal 2000; Kaniak-Golik and Skoneczna 2015). ISCs facilitate redox enzyme activities: they are also components of various protein complexes, cytosolic ribosomes included (Kispal et al. 2005), and, in addition, are required for various enzymes, including those engaged in DNA replication (Netz et al. 2012; reviewed in White and Dillingham 2012). It is thus not surprising that functional mitochondria are crucial for maintaining the stability of cellular genetic information. This requirement is not limited to the mitochondrial genome; it also extends to the stability of the nuclear genome (reviewed in Kaniak-Golik and Skoneczna 2015). In yeast cells, mitochondrial dysfunction leads to a nuclear mutator phenotype (measured by the frequency of canavanine-resistant colonies), regardless of whether this dysfunction can be attributed to blocking oxidative phosphorylation with antimycin A at mitochondrial complex III, deletions in mtDNA (rho−) or the complete loss of mtDNA (rho0) (Rasmussen et al. 2003). The mutator phenotype is not caused by increased intracellular ROS levels because although antimycin A treatment elevates ROS levels, ROS levels are decreased in the rho− and rho0 strains. Moreover, nuclear mutations arising in rho0 cells depend on the Rev1 protein and DNA Pol ζ, whereas those resulting from antimycin A treatment do not. In a recent study, Dirick et al. (2014) showed that the genomic instability specific to rho0 diploid cells results from DNA breaks and mitotic recombination, primarily occurs in non-dividing cells, and tends to fluctuate depending on the environmental conditions. Consequently, mitochondrial dysfunction is mutagenic, and multiple pathways are likely responsible for the resulting nuclear mutator phenotype. Similar conclusions can be drawn from other studies showing that ROS elevation due to mitochondrial disorder and subsequent oxidative damage cause the destabilization of the mitochondrial and/or nuclear genome (Malc et al. 2009; Yazgan and Krebs 2012). Other studies have confirmed the notion that the nuclear DNA instability phenotype is not generated by mitochondrial ROS but by defects in other processes (reviewed in Kaniak-Golik and Skoneczna 2015), such as dNTP production (Desler et al. 2007), ISC formation (Veatch et al. 2009), metal ion detoxification/homeostasis (Karthikeyan, Lewis and Resnick 2002) or apoptosis (Gao et al. 2011). When considering mitochondrial dysfunction, it is important to differentiate between a mutation in a single mitochondrial gene encoding a component of the respiratory chain, which is necessary for proper mitochondrial function and rho− or rho0 mutations. All of these mutations affect cellular energy production through oxidative phosphorylation, but the former preserves the mitochondrial genome and the production of other mitochondrially encoded proteins, whereas the latter disrupts the ability to synthesize those proteins (components of the mitochondrial translation apparatus that are required for the process are also encoded by mtDNA, as described in the section ‘Mitochondria and genome stability’). Respiratory-deficient rho+ mutants do not exhibit as robust a nuclear mutator phenotype as that of the rho0 strains (Veatch et al. 2009; Dirick et al. 2014). Therefore, a mere respiratory deficiency is not sufficient for a marked genomic instability. A series of conclusive experiments by Veatch et al. (2009) has shown that the cellular crisis observed in rho0 cells, manifesting as progressively slower growth, cell-cycle arrest and nuclear genome instability, is linked to a defect in ISC biogenesis due to a reduction in the mitochondrial membrane potential ΔΨ. First, the cellular crisis observed upon mtDNA loss is not a consequence of respiratory deficiency, as discussed above. The deletion of three independent nuclear genes required for respiration at various steps of the electron transport chain, namely CAT5 (monooxygenase required for ubiquinone biosynthesis), RIP1 (ubiquinol-cytochrome-c reductase) and COX4 (subunit IV of cytochrome c oxidase), had no effect on the growth rate in the analyzed cells. Second, rho0 cells carrying the ATP1–111 allele, which encodes a hyperactive F1 ATP synthase and generates a higher ΔΨ than in wild-type rho0 cells, did not present any detectable cellular crisis or genome instability. Third, the downregulation of the non-mitochondrial, cytosolic Nar1 protein, which is required for cytosolic and nuclear Fe-S protein maturation, was sufficient to cause increased genomic instability in cells with intact mitochondrial function. These results suggest that a deficiency which is essential for cell viability mitochondrial function, i.e. ISC biogenesis, is associated with the nuclear instability in rho0 strains, because ISCs are catalytic and structural components of many cellular proteins, including those involved in DNA replication and repair. Accordingly, the proteins involved in ISC cluster biogenesis or in the assembly of various Fe-S proteins have been shown to be required to maintain nuclear genome integrity. Mutations in these genes cause a CIN phenotype that is severe for the CIA1 and DRE2 genes (Ben-Aroya et al. 2008) and moderate for the RLI1 gene (Ben-Aroya et al. 2010). The following nuclear proteins contain ISCs: Pri2, a primase involved in lagging-strand DNA synthesis and DNA DSB repair (Klinge et al. 2007); the catalytic subunits of all three replicative DNA polymerases, α, δ and ε and the specialized DNA Pol ζ, that is involved in PRR in yeast (Netz et al. 2012); the glycosylase Ntg2, which is involved in BER (Alseth et al. 1999); various helicases, including Rad3, which are involved in NER (Rudolf et al. 2006); and the essential helicase-nuclease Dna2, which is required for Okazaki fragment processing and recombinational DNA repair (Pokharel and Campbell 2012). Notably, ISC biogenesis, which is dependent on mitochondrial function, is critical for the molecular activities of these proteins. New studies of both budding yeast and fission yeast will identify other interesting connections between mitochondria and the nuclear genome, linking metabolism with cell-cycle progression and arrest and consequent cell proliferation or apoptosis. These connections might eventually help elucidate the role of mitochondrially generated ROS in neurodegenerative diseases, such as Parkinson's and Alzheimer's diseases, and may help to understand the association of mitochondrial disorders with various cancers and other pathologies.
CONCLUDING REMARKS
The long evolutionary history of life has brought mechanisms of genome stability to perfection. Various mechanisms have been employed to maintain constant genome content. In yeast cells, replication is accurate, errors and lesions are effectively removed from DNA, the breaks in DNA are ligated, abnormal DNA structures arising during cell cycle are resolved, cell divisions are causally monitored to assure equal chromosome segregation, and DNA damage and replication blocks are detected and appropriately responded to, all with respect to cell phase, ploidy, metabolic activity and environmental conditions. However, occasionally, the scrupulous protection of the genome can lead directly to death, and only by accepting the risk of mutation can a cell enable itself to survive. This reflects the evolution of mutation tolerance mechanisms in parallel with genome maintenance mechanisms. Optimizing the fidelity of replication and the balance between the efficiency and costs of DNA repair assures success in future generations that must adapt to a changing environment. Thus, genetic variations contribute to survival and evolution.
In this review, we have focused on the mechanisms leading to somatic genome instability in yeast, providing a comprehensive overview of how these pathways are orchestrated in eukaryotic cells and describing how cells pay a high mutagenic price for survival. All processes ensuring genome stability are highly conserved from yeast to humans, so the significant findings in yeast can be extrapolated to vertebrates, greatly facilitating the molecular analysis of these complex regulatory networks.
Acknowledgments
We would like to thank Zygmunt Ciesla, Justyna McIntyre and Iwona Fijalkowska for their critical reading of the manuscript. We are grateful to Kamil Krol for help in figure preparation. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
FUNDING
This work was supported by the Polish National Science Center grant 2011/03/B/NZ2/00293 to AS, and Polish National Science Center grant 2012/07/B/NZ3/02914 to AKG, and Polish National Science Center grant 2011/01/B/NZ3/02904 to MS.
Conflict of interest. None declared.
REFERENCES
- Acevedo-Torres K, Fonseca-Williams S, Ayala-Torres S, et al. Requirement of the Saccharomyces cerevisiae APN1 gene for the repair of mitochondrial DNA alkylation damage. Environ Mol Mutagen. 2009;50:317–27. doi: 10.1002/em.20462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya N, Klassen R, Johnson RE, et al. PCNA binding domains in all three subunits of yeast DNA polymerase δ modulate its function in DNA replication. P Natl Acad Sci USA. 2011;108:17927–32. doi: 10.1073/pnas.1109981108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agmon N, Liefshitz B, Zimmer C, et al. Effect of nuclear architecture on the efficiency of double-strand break repair. Nat Cell Biol. 2013;15:694–9. doi: 10.1038/ncb2745. [DOI] [PubMed] [Google Scholar]
- Aguilera A, Gómez-González B. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet. 2008;9:204–17. doi: 10.1038/nrg2268. [DOI] [PubMed] [Google Scholar]
- Aksenova A, Volkov K, Maceluch J, et al. Mismatch repair-independent increase in spontaneous mutagenesis in yeast lacking non-essential subunits of DNA polymerase ε. PLoS Genet. 2010;6:e1001209. doi: 10.1371/journal.pgen.1001209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabrudzinska M, Skoneczny M, Skoneczna A. Diploid-specific genome stability genes of S. cerevisiae: genomic screen reveals haploidization as an escape from persisting DNA rearrangement stress. PLoS One. 2011;6:e21124. doi: 10.1371/journal.pone.0021124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida H, Godinho Ferreira M. Spontaneous telomere to telomere fusions occur in unperturbed fission yeast cells. Nucleic Acids Res. 2013;41:3056–67. doi: 10.1093/nar/gks1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alseth I, Eide L, Pirovano M, et al. The Saccharomyces cerevisiae homologues of endonuclease III from Escherichia coli, Ntg1 and Ntg2, are both required for efficient repair of spontaneous and induced oxidative DNA damage in yeast. Mol Cell Biol. 1999;19:3779–87. doi: 10.1128/mcb.19.5.3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alver RC, Zhang T, Josephrajan A, et al. The N-terminus of Mcm10 is important for interaction with the 9–1–1 clamp and in resistance to DNA damage. Nucleic Acids Res. 2014;42:8389–404. doi: 10.1093/nar/gku479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen PL, Xu F, Xiao W. Eukaryotic DNA damage tolerance and translesion synthesis through covalent modifications of PCNA. Cell Res. 2008;18:162–73. doi: 10.1038/cr.2007.114. [DOI] [PubMed] [Google Scholar]
- Antony E, Tomko EJ, Xiao Q, et al. Srs2 disassembles Rad51 filaments by a protein–protein interaction triggering ATP turnover and dissociation of Rad51 from DNA. Mol Cell. 2009;35:105–15. doi: 10.1016/j.molcel.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argueso JL, Kijas AW, Sarin S, et al. Systematic mutagenesis of the Saccharomyces cerevisiae MLH1 gene reveals distinct roles for Mlh1p in meiotic crossing over and in vegetative and meiotic mismatch repair. Mol Cell Biol. 2003;23:873–86. doi: 10.1128/MCB.23.3.873-886.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroya SB, Kupiec M. The Elg1 replication factor C-like complex: a novel guardian of genome stability. DNA Repair. 2005;4:409–17. doi: 10.1016/j.dnarep.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Ashton TM, Hickson ID. Yeast as a model system to study RecQ helicase function. DNA Repair. 2010;9:303–14. doi: 10.1016/j.dnarep.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Aylon Y, Kupiec M. The checkpoint protein Rad24 of Saccharomyces cerevisiae is involved in processing double-strand break ends and in recombination partner choice. Mol Cell Biol. 2003;23:6585–96. doi: 10.1128/MCB.23.18.6585-6596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon Y, Liefshitz B, Bitan-Banin G, et al. Molecular dissection of mitotic recombination in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 2003;23:1403–17. doi: 10.1128/MCB.23.4.1403-1417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyagari R, Gomes XV, Gordenin DA, et al. Okazaki fragment maturation in yeast. I. Distribution of functions between FEN1 AND DNA2. J Biol Chem. 2003;278:1618–25. doi: 10.1074/jbc.M209801200. [DOI] [PubMed] [Google Scholar]
- Azvolinsky A, Giresi PG, Lieb JD, et al. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol Cell. 2009;34:722–34. doi: 10.1016/j.molcel.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan L, Bambara RA. Eukaryotic lagging strand DNA replication employs a multi-pathway mechanism that protects genome integrity. J Biol Chem. 2011;286:6865–70. doi: 10.1074/jbc.R110.209502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball LG, Zhang K, Cobb JA, et al. The yeast Shu complex couples error-free post-replication repair to homologous recombination. Mol Microbiol. 2009;73:89–102. doi: 10.1111/j.1365-2958.2009.06748.x. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Sikdar N, Myung K. Suppression of gross chromosomal rearrangements by a new alternative replication factor C complex. Biochem Bioph Res Co. 2007;362:546–9. doi: 10.1016/j.bbrc.2007.07.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruffini E, Ferrero I, Foury F. In vivo analysis of mtDNA replication defects in yeast. Methods. 2010;51:426–36. doi: 10.1016/j.ymeth.2010.02.023. [DOI] [PubMed] [Google Scholar]
- Baruffini E, Serafini F, Ferrero I, et al. Overexpression of DNA polymerase zeta reduces the mitochondrial mutability caused by pathological mutations in DNA polymerase gamma in yeast. PLoS One. 2012;7:e34322. doi: 10.1371/journal.pone.0034322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baute J, Depicker A. Base excision repair and its role in maintaining genome stability. Crit Rev Biochem Mol. 2008;43:239–76. doi: 10.1080/10409230802309905. [DOI] [PubMed] [Google Scholar]
- Bebenek K, Kunkel TA. Frameshift errors initiated by nucleotide misincorporation. P Natl Acad Sci USA. 1990;87:4946–50. doi: 10.1073/pnas.87.13.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebenek K, Kunkel TA. Streisinger revisited: DNA synthesis errors mediated by substrate misalignments. Cold Spring Harb Sym. 2000;65:81–91. doi: 10.1101/sqb.2000.65.81. [DOI] [PubMed] [Google Scholar]
- Beletskii A, Bhagwat AS. Transcription-induced mutations: increase in C to T mutations in the nontranscribed strand during transcription in Escherichia coli. P Natl Acad Sci USA. 1996;93:13919–24. doi: 10.1073/pnas.93.24.13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belotserkovskii BP, Mirkin SM, Hanawalt PC. DNA sequences that interfere with transcription: implications for genome function and stability. Chem Rev. 2013;113:8620–37. doi: 10.1021/cr400078y. [DOI] [PubMed] [Google Scholar]
- Ben-Aroya S, Agmon N, Yuen K, et al. Proteasome nuclear activity affects chromosome stability by controlling the turnover of Mms22, a protein important for DNA repair. PLoS Genet. 2010;6:e1000852. doi: 10.1371/journal.pgen.1000852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Aroya S, Coombes C, Kwok T, et al. Toward a comprehensive temperature-sensitive mutant repository of the essential genes of Saccharomyces cerevisiae. Mol Cell. 2008;30:248–58. doi: 10.1016/j.molcel.2008.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendich AJ. Structural analysis of mitochondrial DNA molecules from fungi and plants using moving pictures and pulsed-field gel electrophoresis. J Mol Biol. 1996;255:564–88. doi: 10.1006/jmbi.1996.0048. [DOI] [PubMed] [Google Scholar]
- Bermejo R, Lai MS, Foiani M. Preventing replication stress to maintain genome stability: resolving conflicts between replication and transcription. Mol Cell. 2012;45:710–8. doi: 10.1016/j.molcel.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Bermúdez-López M, Ceschia A, de Piccoli G, et al. The Smc5/6 complex is required for dissolution of DNA-mediated sister chromatid linkages. Nucleic Acids Res. 2010;38:6502–12. doi: 10.1093/nar/gkq546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins S. The composition, functions, and regulation of the budding yeast kinetochore. Genetics. 2013;194:817–46. doi: 10.1534/genetics.112.145276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blastyák A, Pintér L, Unk I, et al. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol Cell. 2007;28:167–75. doi: 10.1016/j.molcel.2007.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodi Z, Gysler-Junker A, Kohli J. A quantitative assay to measure chromosome stability in Schizosaccharomyces pombe. Mol Gen Genet. 1991;229:77–80. doi: 10.1007/BF00264215. [DOI] [PubMed] [Google Scholar]
- Boiteux S, Jinks-Robertson S. DNA repair mechanisms and the bypass of DNA damage in Saccharomyces cerevisiae. Genetics. 2013;193:1025–64. doi: 10.1534/genetics.112.145219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen N, Smith CE, Srivatsan A, et al. Reconstitution of long and short patch mismatch repair reactions using Saccharomyces cerevisiae proteins. P Natl Acad Sci USA. 2013;110:18472–7. doi: 10.1073/pnas.1318971110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman GD, O'Donnell M, Kuriyan J. Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature. 2004;429:724–30. doi: 10.1038/nature02585. [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M. Template switching: from replication fork repair to genome rearrangements. Cell. 2007;131:1228–30. doi: 10.1016/j.cell.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Bio. 2010;11:208–19. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- Branzei D, Seki M, Enomoto T. Rad18/Rad5/Mms2-mediated polyubiquitination of PCNA is implicated in replication completion during replication stress. Genes Cells. 2004;9:1031–42. doi: 10.1111/j.1365-2443.2004.00787.x. [DOI] [PubMed] [Google Scholar]
- Branzei D, Sollier J, Liberi G, et al. Ubc9- and mms21-mediated sumoylation counteracts recombinogenic events at damaged replication forks. Cell. 2006;127:509–22. doi: 10.1016/j.cell.2006.08.050. [DOI] [PubMed] [Google Scholar]
- Branzei D, Vanoli F, Foiani M. SUMOylation regulates Rad18-mediated template switch. Nature. 2008;456:915–20. doi: 10.1038/nature07587. [DOI] [PubMed] [Google Scholar]
- Brooke RG, Dumas LB. Reconstitution of the Saccharomyces cerevisiae DNA primase-DNA polymerase protein complex in vitro. The 86-kDa subunit facilitates but is not required for complex formation. J Biol Chem. 1991;266:10 093–8. [PubMed] [Google Scholar]
- Brooke RG, Singhal R, Hinkle DC, et al. Purification and characterization of the 180- and 86-kilodalton subunits of the Saccharomyces cerevisiae DNA primase-DNA polymerase protein complex. The 180-kilodalton subunit has both DNA polymerase and 3′-5′-exonuclease activities. J Biol Chem. 1991;266:3005–15. [PubMed] [Google Scholar]
- Broomfield S, Xiao W. Suppression of genetic defects within the RAD6 pathway by srs2 is specific for error-free post-replication repair but not for damage-induced mutagenesis. Nucleic Acids Res. 2002;30:732–9. doi: 10.1093/nar/30.3.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd ME, Reis CC, Smith S, et al. Evidence suggesting that Pif1 helicase functions in DNA replication with the Dna2 helicase/nuclease and DNA polymerase. Mol Cell Biol. 2006;26:2490–500. doi: 10.1128/MCB.26.7.2490-2500.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulder CJ. Lethality of the petite mutation in petite negative yeasts. Anton Leeuw. 1964;30:442–54. doi: 10.1007/BF02046758. [DOI] [PubMed] [Google Scholar]
- Burgers PMJ. Polymerase dynamics at the eukaryotic DNA replication fork. J Biol Chem. 2009;284:4041–5. doi: 10.1074/jbc.R800062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RJ, Han J, Zhang Z. The Ddc1-Mec3-Rad17 sliding clamp regulates histone-histone chaperone interactions and DNA replication-coupled nucleosome assembly in budding yeast. J Biol Chem. 2014;289:10518–29. doi: 10.1074/jbc.M114.552463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkovics P, Sebesta M, Sisakova A, et al. Srs2 mediates PCNA-SUMO-dependent inhibition of DNA repair synthesis. EMBO J. 2013;32:742–55. doi: 10.1038/emboj.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büttner R, Bode R, Samsonova IA, et al. Mapping of the glucoamylase gene of Trichosporon adeninovorans by mitotic haploidization using hybrids from protoplast fusions. J Basic Microbiol. 1990;30:227–31. doi: 10.1002/jobm.3620300402. [DOI] [PubMed] [Google Scholar]
- Büttner S, Eisenberg T, Carmona-Gutierrez D, et al. Endonuclease G regulates budding yeast life and death. Mol Cell. 2007;25:233–46. doi: 10.1016/j.molcel.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Bylund GO, Burgers PMJ. Replication protein A-directed unloading of PCNA by the Ctf18 cohesion establishment complex. Mol Cell Biol. 2005;25:5445–55. doi: 10.1128/MCB.25.13.5445-5455.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabal GG, Genovesio A, Rodriguez-Navarro S, et al. SAGA interacting factors confine sub-diffusion of transcribed genes to the nuclear envelope. Nature. 2006;441:770–73. doi: 10.1038/nature04752. [DOI] [PubMed] [Google Scholar]
- Callegari AJ, Clark E, Pneuman A, et al. Postreplication gaps at UV lesions are signals for checkpoint activation. P Natl Acad Sci USA. 2010;107:8219–24. doi: 10.1073/pnas.1003449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campsteijn C, Wijnands-Collin A-MJ, Logie C. Reverse genetic analysis of the yeast RSC chromatin remodeler reveals a role for RSC3 and SNF5 homolog 1 in ploidy maintenance. PLoS Genet. 2007;3:e92. doi: 10.1371/journal.pgen.0030092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra JA, Paeschke K, Singh M, et al. G-quadruplex DNA sequences are evolutionarily conserved and associated with distinct genomic features in Saccharomyces cerevisiae. PLoS Comput Biol. 2010;6:e1000861. doi: 10.1371/journal.pcbi.1000861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone JM, Brendel M, Henriques JaP. DNA repair by polymerase delta in Saccharomyces cerevisiae is not controlled by the proliferating cell nuclear antigen-like Rad17/Mec3/Ddc1 complex. Genet Mol Res. 2008;7:127–32. doi: 10.4238/vol7-1gmr405. [DOI] [PubMed] [Google Scholar]
- Casolari JM, Brown CR, Komili S, et al. Genome-wide localization of the nuclear transport machinery couples transcriptional status and nuclear organization. Cell. 2004;117:427–39. doi: 10.1016/s0092-8674(04)00448-9. [DOI] [PubMed] [Google Scholar]
- Cejka P, Plank JL, Bachrati CZ, et al. Rmi1 stimulates decatenation of double Holliday junctions during dissolution by Sgs1-Top3. Nat Struct Mol Biol. 2010;17:1377–82. doi: 10.1038/nsmb.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabes A, Georgieva B, Domkin V, et al. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell. 2003;112:391–401. doi: 10.1016/s0092-8674(03)00075-8. [DOI] [PubMed] [Google Scholar]
- Chabes A, Stillman B. Constitutively high dNTP concentration inhibits cell cycle progression and the DNA damage checkpoint in yeast Saccharomyces cerevisiae. P Natl Acad Sci USA. 2007;104:1183–8. doi: 10.1073/pnas.0610585104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacinska A, Koehler CM, Milenkovic D, et al. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–44. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Botstein D. Isolation and characterization of chromosome-gain and increase-in-ploidy mutants in yeast. Genetics. 1993;135:677–91. doi: 10.1093/genetics/135.3.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K, Resnick MA, Gordenin DA. The choice of nucleotide inserted opposite abasic sites formed within chromosomal DNA reveals the polymerase activities participating in translesion DNA synthesis. DNA Repair. 2013;12:878–89. doi: 10.1016/j.dnarep.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LM. DNA polymerases from bakers’ yeast. J Biol Chem. 1977;252:1873–80. [PubMed] [Google Scholar]
- Chatterjee A, Singh KK. Uracil-DNA glycosylase-deficient yeast exhibit a mitochondrial mutator phenotype. Nucleic Acids Res. 2001;29:4935–40. doi: 10.1093/nar/29.24.4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee N, Pabla R, Siede W. Role of polymerase η in mitochondrial mutagenesis of Saccharomyces cerevisiae. Biochem Bioph Res Co. 2013;431:270–3. doi: 10.1016/j.bbrc.2012.12.119. [DOI] [PubMed] [Google Scholar]
- Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23:81–5. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- Chen G, Bradford WD, Seidel CW, et al. Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature. 2012;482:246–50. doi: 10.1038/nature10795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Trujillo K, Ramos W, et al. Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol Cell. 2001;8:1105–15. doi: 10.1016/s1097-2765(01)00388-4. [DOI] [PubMed] [Google Scholar]
- Chen W, Jinks-Robertson S. Mismatch repair proteins regulate heteroduplex formation during mitotic recombination in yeast. Mol Cell Biol. 1998;18:6525–37. doi: 10.1128/mcb.18.11.6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XJ. Mechanism of homologous recombination and implications for aging-related deletions in mitochondrial DNA. Microbiol Mol Biol R. 2013;77:476–96. doi: 10.1128/MMBR.00007-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XJ, Butow RA. The organization and inheritance of the mitochondrial genome: Nature Reviews Genetics. Nat Rev Genet. 2005;6:815–25. doi: 10.1038/nrg1708. [DOI] [PubMed] [Google Scholar]
- Chen XJ, Wang X, Kaufman BA, et al. Aconitase couples metabolic regulation to mitochondrial DNA maintenance. Science. 2005;307:714–7. doi: 10.1126/science.1106391. [DOI] [PubMed] [Google Scholar]
- Cheng E, Vaisica JA, Ou J, et al. Genome rearrangements caused by depletion of essential DNA replication proteins in Saccharomyces cerevisiae. Genetics. 2012;192:147–60. doi: 10.1534/genetics.112.141051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KC, Cahill DS, Kasai H, et al. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G-T and A-C substitutions. J Biol Chem. 1992;267:166–72. [PubMed] [Google Scholar]
- Cheng X, Dunaway S, Ivessa AS. The role of Pif1p, a DNA helicase in Saccharomyces cerevisiae, in maintaining mitochondrial DNA. Mitochondrion. 2007;7:211–22. doi: 10.1016/j.mito.2006.11.023. [DOI] [PubMed] [Google Scholar]
- Cheng X, Ivessa AS. Association of the yeast DNA helicase Pif1p with mitochondrial membranes and mitochondrial DNA. Eur J Cell Biol. 2010;89:742–7. doi: 10.1016/j.ejcb.2010.06.008. [DOI] [PubMed] [Google Scholar]
- Chi NW, Kolodner RD. Purification and characterization of MSH1, a yeast mitochondrial protein that binds to DNA mismatches. J Biol Chem. 1994;269:29984–92. [PubMed] [Google Scholar]
- Choi K, Szakal B, Chen Y-H, et al. The Smc5/6 complex and Esc2 influence multiple replication-associated recombination processes in Saccharomyces cerevisiae. Mol Biol Cell. 2010;21:2306–14. doi: 10.1091/mbc.E10-01-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy JS, Acuña R, Au W-C, et al. A role for histone H4K16 hypoacetylation in Saccharomyces cerevisiae kinetochore function. Genetics. 2011;189:11–21. doi: 10.1534/genetics.111.130781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Z, Li J, Eshaghi M, et al. Adaptive expression responses in the Pol-γ null strain of S. pombe depleted of mitochondrial genome. BMC Genomics. 2007;8:323. doi: 10.1186/1471-2164-8-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W-H. To peep into Pif1 helicase: multifaceted all the way from genome stability to repair-associated DNA synthesis. J Microbiol. 2014;52:89–98. doi: 10.1007/s12275-014-3524-3. [DOI] [PubMed] [Google Scholar]
- Clark AB, Lujan SA, Kissling GE, et al. Mismatch repair-independent tandem repeat sequence instability resulting from ribonucleotide incorporation by DNA polymerase ε. DNA Repair. 2011;10:476–82. doi: 10.1016/j.dnarep.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AB, Valle F, Drotschmann K, et al. Functional interaction of proliferating cell nuclear antigen with MSH2-MSH6 and MSH2-MSH3 complexes. J Biol Chem. 2000;275:36498–501. doi: 10.1074/jbc.C000513200. [DOI] [PubMed] [Google Scholar]
- Clark-Walker GD. Kinetic properties of F1-ATPase influence the ability of yeasts to grow in anoxia or absence of mtDNA. Mitochondrion. 2003;2:257–65. doi: 10.1016/S1567-7249(02)00107-1. [DOI] [PubMed] [Google Scholar]
- Collura A, Kemp PAVD, Boiteux S. Abasic sites linked to dUTP incorporation in DNA are a major cause of spontaneous mutations in absence of base excision repair and Rad17-Mec3-Ddc1 (9–1–1) DNA damage checkpoint clamp in Saccharomyces cerevisiae. DNA Repair. 2012;11:294–303. doi: 10.1016/j.dnarep.2011.12.004. [DOI] [PubMed] [Google Scholar]
- Contamine V, Picard M. Maintenance and integrity of the mitochondrial genome: a plethora of nuclear genes in the budding yeast. Microbiol Mol Biol R. 2000;64:281–15. doi: 10.1128/mmbr.64.2.281-315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooperman BS, Kashlan OB. A comprehensive model for the allosteric regulation of Class Ia ribonucleotide reductases. Adv Enzyme Regul. 2003;43:167–82. doi: 10.1016/s0065-2571(02)00035-3. [DOI] [PubMed] [Google Scholar]
- Copsey A, Tang S, Jordan PW, et al. Smc5/6 coordinates formation and resolution of joint molecules with chromosome morphology to ensure meiotic divisions. PLoS Genet. 2013;9:e1004071. doi: 10.1371/journal.pgen.1004071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrette-Bennett SE, Borgeson C, Sommer D, et al. DNA polymerase delta, RFC and PCNA are required for repair synthesis of large looped heteroduplexes in Saccharomyces cerevisiae. Nucleic Acids Res. 2004;32:6268–75. doi: 10.1093/nar/gkh965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon S, Ramasubramanyan S, Alies C, et al. Rad8Rad5/Mms2-Ubc13 ubiquitin ligase complex controls translesion synthesis in fission yeast. EMBO J. 2010;29:2048–58. doi: 10.1038/emboj.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covo S, Puccia CM, Argueso JL, et al. The sister chromatid cohesion pathway suppresses multiple chromosome gain and chromosome amplification. Genetics. 2014;196:373–84. doi: 10.1534/genetics.113.159202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbé L, Thomas A, Pantesco V, et al. Analysis of replication profiles reveals key role of RFC-Ctf18 in yeast replication stress response. Nat Struct Mol Biol. 2010;17:1391–7. doi: 10.1038/nsmb.1932. [DOI] [PubMed] [Google Scholar]
- Crider DG, García-Rodríguez LJ, Srivastava P, et al. Rad53 is essential for a mitochondrial DNA inheritance checkpoint regulating G1 to S progression. J Cell Biol. 2012;198:793–8. doi: 10.1083/jcb.201205193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cymerman IA, Chung I, Beckmann BM, et al. EXOG, a novel paralog of Endonuclease G in higher eukaryotes. Nucleic Acids Res. 2008;36:1369–79. doi: 10.1093/nar/gkm1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigaku Y, Davies AA, Ulrich HD. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature. 2010;465:951–5. doi: 10.1038/nature09097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dake E, Hofmann TJ, McIntire S, et al. Purification and properties of the major nuclease from mitochondria of Saccharomyces cerevisiae. J Biol Chem. 1988;263:7691–702. [PubMed] [Google Scholar]
- Daley JM, Zakaria C, Ramotar D. The endonuclease IV family of apurinic/apyrimidinic endonucleases. Mutat Res. 2010;705:217–27. doi: 10.1016/j.mrrev.2010.07.003. [DOI] [PubMed] [Google Scholar]
- Daraba A, Gali VK, Halmai M, et al. Def1 promotes the degradation of Pol3 for polymerase exchange to occur during DNA-damage-induced mutagenesis in Saccharomyces cerevisiae. PLoS Biol. 2014;12:e1001771. doi: 10.1371/journal.pbio.1001771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashko S, Zhou N, Compagno C, et al. Why, when, and how did yeast evolve alcoholic fermentation? FEMS Yeast Res. 2014;14:826–32. doi: 10.1111/1567-1364.12161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta A, Jinks-Robertson S. Association of increased spontaneous mutation rates with high levels of transcription in yeast. Science. 1995;268:1616–9. doi: 10.1126/science.7777859. [DOI] [PubMed] [Google Scholar]
- Datta A, Schmeits JL, Amin NS, et al. Checkpoint-dependent activation of mutagenic repair in Saccharomyces cerevisiae pol3-01 mutants. Mol Cell. 2000;6:593–603. doi: 10.1016/s1097-2765(00)00058-7. [DOI] [PubMed] [Google Scholar]
- Davidson MB, Katou Y, Keszthelyi A, et al. Endogenous DNA replication stress results in expansion of dNTP pools and a mutator phenotype. EMBO J. 2012;31:895–907. doi: 10.1038/emboj.2011.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AA, Neiss A, Ulrich HD. Ubiquitylation of the 9–1–1 checkpoint clamp is independent of rad6-rad18 and DNA damage. Cell. 2010;141:1080–7. doi: 10.1016/j.cell.2010.04.039. [DOI] [PubMed] [Google Scholar]
- De Antoni A, Pearson CG, Cimini D, et al. The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr Biol. 2005;15:214–25. doi: 10.1016/j.cub.2005.01.038. [DOI] [PubMed] [Google Scholar]
- De Bruin RAM, Kalashnikova TI, Wittenberg C. Stb1 collaborates with other regulators to modulate the G1-specific transcriptional circuit. Mol Cell Biol. 2008;28:6919–28. doi: 10.1128/MCB.00211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Morgan A, Brodsky L, Ronin Y, et al. Genome-wide analysis of DNA turnover and gene expression in stationary-phase Saccharomyces cerevisiae. Microbiology. 2010;156:1758–71. doi: 10.1099/mic.0.035519-0. [DOI] [PubMed] [Google Scholar]
- Decottignies A. Microhomology-mediated end joining in fission yeast is repressed by pku70 and relies on genes involved in homologous recombination. Genetics. 2007;176:1403–15. doi: 10.1534/genetics.107.071621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decottignies A. Alternative end-joining mechanisms: a historical perspective. Front Genet. 2013;4:48. doi: 10.3389/fgene.2013.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deem A, Keszthelyi A, Blackgrove T, et al. Break-induced replication is highly inaccurate. PLoS Biol. 2011;9:e1000594. doi: 10.1371/journal.pbio.1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehé P-M, Coulon S, Scaglione S, et al. Regulation of Mus81-Eme1 Holliday junction resolvase in response to DNA damage. Nat Struct Mol Biol. 2013;20:598–603. doi: 10.1038/nsmb.2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacôte F, Lopez BS. Importance of the cell cycle phase for the choice of the appropriate DSB repair pathway, for genome stability maintenance: the trans-S double-strand break repair model. Cell Cycle. 2008;7:33–8. doi: 10.4161/cc.7.1.5149. [DOI] [PubMed] [Google Scholar]
- Deshpande AM, Newlon CS. DNA replication fork pause sites dependent on transcription. Science. 1996;272:1030–3. doi: 10.1126/science.272.5264.1030. [DOI] [PubMed] [Google Scholar]
- Desler C, Munch-Petersen B, Stevnsner T, et al. Mitochondria as determinant of nucleotide pools and chromosomal stability. Mutat Res. 2007;625:112–24. doi: 10.1016/j.mrfmmm.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Dhar A, Lahue RS. Rapid unwinding of triplet repeat hairpins by Srs2 helicase of Saccharomyces cerevisiae. Nucleic Acids Res. 2008;36:3366–73. doi: 10.1093/nar/gkn225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieckman LM, Washington MT. PCNA trimer instability inhibits translesion synthesis by DNA polymerase η and by DNA polymerase δ. DNA Repair. 2013;12:367–76. doi: 10.1016/j.dnarep.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diffley JF, Stillman B. A close relative of the nuclear, chromosomal high-mobility group protein HMG1 in yeast mitochondria. P Natl Acad Sci USA. 1991;88:7864–8. doi: 10.1073/pnas.88.17.7864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirick L, Bendris W, Loubiere V, et al. Metabolic and environmental conditions determine nuclear genomic instability in budding yeast lacking mitochondrial DNA. G3. 2014;4:411–23. doi: 10.1534/g3.113.010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe C, Osman F, Dixon J, et al. The Holliday junction resolvase SpCCE1 prevents mitochondrial DNA aggregation in Schizosaccharomyces pombe. Mol Gen Genet. 2000;263:889–97. doi: 10.1007/s004380000256. [DOI] [PubMed] [Google Scholar]
- Doudican NA, Song B, Shadel GS, et al. Oxidative DNA damage causes mitochondrial genomic instability in Saccharomyces cerevisiae. Mol Cell Biol. 2005;25:5196–204. doi: 10.1128/MCB.25.12.5196-5204.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drag M, Salvesen GS. DeSUMOylating enzymes–SENPs. IUBMB Life. 2008;60:734–42. doi: 10.1002/iub.113. [DOI] [PubMed] [Google Scholar]
- Drake JW. Avoiding dangerous missense: thermophiles display especially low mutation rates. PLoS Genet. 2009;5:e1000520. doi: 10.1371/journal.pgen.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW, Charlesworth B, Charlesworth D, et al. Rates of spontaneous mutation. Genetics. 1998;148:1667–86. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon B. Mitochondrial genetics and functions. In: Strathern JN, Jones EW, Broach JR, editors. Molecular Biology of the Yeast Saccharomyces: Life Cycle and Inheritance. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1981. pp. 505–635. [Google Scholar]
- Duxin JP, Dao B, Martinsson P, et al. Human Dna2 is a nuclear and mitochondrial DNA maintenance protein. Mol Cell Biol. 2009;29:4274–82. doi: 10.1128/MCB.01834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzierzbicki P, Kaniak-Golik A, Malc E, et al. The generation of oxidative stress-induced rearrangements in Saccharomyces cerevisiae mtDNA is dependent on the Nuc1 (EndoG/ExoG) nuclease and is enhanced by inactivation of the MRX complex. Mutat Res. 2012;740:21–33. doi: 10.1016/j.mrfmmm.2012.12.004. [DOI] [PubMed] [Google Scholar]
- Dzierzbicki P, Koprowski P, Fikus MU, et al. Repair of oxidative damage in mitochondrial DNA of Saccharomyces cerevisiae: involvement of the MSH1-dependent pathway. DNA Repair. 2004;3:403–11. doi: 10.1016/j.dnarep.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Echols H, Goodman MF. Fidelity Mechanisms in DNA Replication. Annu Rev Biochem. 1991;60:477–511. doi: 10.1146/annurev.bi.60.070191.002401. [DOI] [PubMed] [Google Scholar]
- Efrati E, Tocco G, Eritja R, et al. Abasic translesion synthesis by DNA polymerase beta violates the ‘A-rule’. Novel types of nucleotide incorporation by human DNA polymerase beta at an abasic lesion in different sequence contexts. J Biol Chem. 1997;272:2559–69. doi: 10.1074/jbc.272.4.2559. [DOI] [PubMed] [Google Scholar]
- Eklund H, Uhlin U, Färnegårdh M, et al. Structure and function of the radical enzyme ribonucleotide reductase. Prog Biophys Mol Bio. 2001;77:177–268. doi: 10.1016/s0079-6107(01)00014-1. [DOI] [PubMed] [Google Scholar]
- Elledge SJ, Davis RW. Identification and isolation of the gene encoding the small subunit of ribonucleotide reductase from Saccharomyces cerevisiae: DNA damage-inducible gene required for mitotic viability. Mol Cell Biol. 1987;7:2783–93. doi: 10.1128/mcb.7.8.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge SJ, Davis RW. Two genes differentially regulated in the cell cycle and by DNA-damaging agents encode alternative regulatory subunits of ribonucleotide reductase. Gene Dev. 1990;4:740–51. doi: 10.1101/gad.4.5.740. [DOI] [PubMed] [Google Scholar]
- Enserink JM, Kolodner RD. An overview of Cdk1-controlled targets and processes. Cell Div. 2010;5:11. doi: 10.1186/1747-1028-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ephrussi B. Nucleocytoplasmic Relations in Microorganisms. Oxford: Clarendon Press; 1953. [Google Scholar]
- Errico A, Costanzo V. Differences in the DNA replication of unicellular eukaryotes and metazoans: known unknowns. EMBO Rep. 2010;11:270–8. doi: 10.1038/embor.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasullo M, Tsaponina O, Sun M, et al. Elevated dNTP levels suppress hyper-recombination in Saccharomyces cerevisiae S-phase checkpoint mutants. Nucleic Acids Res. 2010;38:1195–203. doi: 10.1093/nar/gkp1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernius J, Hardwick KG. Bub1 kinase targets Sgo1 to ensure efficient chromosome biorientation in budding yeast mitosis. PLoS Genet. 2007;3:e213. doi: 10.1371/journal.pgen.0030213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fikus MU, Mieczkowski PA, Koprowski P, et al. The product of the DNA damage-inducible gene of Saccharomyces cerevisiae, DIN7, specifically functions in mitochondria. Genetics. 2000;154:73–81. doi: 10.1093/genetics/154.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman-Lobell J, Haber JE. Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science. 1992;258:480–4. doi: 10.1126/science.1411547. [DOI] [PubMed] [Google Scholar]
- Fix D, Canugovi C, Bhagwat AS. Transcription increases methylmethane sulfonate-induced mutations in alkB strains of Escherichia coli. DNA Repair. 2008;7:1289–97. doi: 10.1016/j.dnarep.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleck O, Vejrup-Hansen R, Watson A, et al. Spd1 accumulation causes genome instability independently of ribonucleotide reductase activity but functions to protect the genome when deoxynucleotide pools are elevated. J Cell Sci. 2013;126:4985–94. doi: 10.1242/jcs.132837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores C-L, Rodriguez C, Petit T, et al. Carbohydrate and energy-yielding metabolism in non-conventional yeast. FEMS Microbiol Rev. 2000;24:507–29. doi: 10.1111/j.1574-6976.2000.tb00553.x. [DOI] [PubMed] [Google Scholar]
- Fortini P, Dogliotti E. Base damage and single-strand break repair: mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair. 2007;6:398–409. doi: 10.1016/j.dnarep.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Fortune JM, Pavlov YI, Welch CM, et al. Saccharomyces cerevisiae DNA polymerase delta: high fidelity for base substitutions but lower fidelity for single- and multi-base deletions. J Biol Chem. 2005;280:29980–7. doi: 10.1074/jbc.M505236200. [DOI] [PubMed] [Google Scholar]
- Fournier P, Provost A, Bourguignon C, et al. Recombination after protoplast fusion in the yeast Candida tropicalis. Arch Microbiol. 1977;115:143–9. doi: 10.1007/BF00406367. [DOI] [PubMed] [Google Scholar]
- Foury F. Cloning and sequencing of the nuclear gene MIP1 encoding the catalytic subunit of the yeast mitochondrial DNA polymerase. J Biol Chem. 1989;264:20552–60. [PubMed] [Google Scholar]
- Foury F, Vanderstraeten S. Yeast mitochondrial DNA mutators with deficient proofreading exonucleolytic activity. EMBO J. 1992;11:2717. doi: 10.1002/j.1460-2075.1992.tb05337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank-Vaillant M, Marcand S. NHEJ regulation by mating type is exercised through a novel protein, Lif2p, essential to the ligase IV pathway. Gene Dev. 2001;15:3005–12. doi: 10.1101/gad.206801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenreich CH, Kantrow SM, Zakian VA. Expansion and length-dependent fragility of CTG repeats in yeast. Science. 1998;279:853–6. doi: 10.1126/science.279.5352.853. [DOI] [PubMed] [Google Scholar]
- Fricke WM, Brill SJ. Slx1-Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1-Top3. Gene Dev. 2003;17:1768–78. doi: 10.1101/gad.1105203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC, Lehmann AR, Fuchs RPP. Trading places: how do DNA polymerases switch during translesion DNA synthesis? Mol Cell. 2005;18:499–505. doi: 10.1016/j.molcel.2005.03.032. [DOI] [PubMed] [Google Scholar]
- Friedel AM, Pike BL, Gasser SM. ATR/Mec1: coordinating fork stability and repair. Curr Opin Cell Biol. 2009;21:237–44. doi: 10.1016/j.ceb.2009.01.017. [DOI] [PubMed] [Google Scholar]
- Fritsch ES, Chabbert CD, Klaus B, et al. A genome-wide map of mitochondrial DNA recombination in yeast. Genetics. 2014;198:755–71. doi: 10.1534/genetics.114.166637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Zhu Y, Zhang K, et al. Rad6-Rad18 mediates a eukaryotic SOS response by ubiquitinating the 9–1–1 checkpoint clamp. Cell. 2008;133:601–11. doi: 10.1016/j.cell.2008.02.050. [DOI] [PubMed] [Google Scholar]
- Fujita I, Nishihara Y, Tanaka M, et al. Telomere-nuclear envelope dissociation promoted by Rap1 phosphorylation ensures faithful chromosome segregation. Curr Biol. 2012;22:1932–7. doi: 10.1016/j.cub.2012.08.019. [DOI] [PubMed] [Google Scholar]
- Fukuda K, Morioka H, Imajou S, et al. Structure-function relationship of the eukaryotic DNA replication factor, proliferating cell nuclear antigen. J Biol Chem. 1995;270:22527–34. doi: 10.1074/jbc.270.38.22527. [DOI] [PubMed] [Google Scholar]
- Futami K, Shimamoto A, Furuichi Y. Mitochondrial and nuclear localization of human Pif1 helicase. Biol Pharm Bull. 2007;30:1685–92. doi: 10.1248/bpb.30.1685. [DOI] [PubMed] [Google Scholar]
- Gallego-Sánchez A, Andrés S, Conde F, et al. Reversal of PCNA ubiquitylation by Ubp10 in Saccharomyces cerevisiae. PLoS Genet. 2012;8:e1002826. doi: 10.1371/journal.pgen.1002826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambus A, van Deursen F, Polychronopoulos D, et al. A key role for Ctf4 in coupling the MCM2–7 helicase to DNA polymerase alpha within the eukaryotic replisome. EMBO J. 2009;28:2992–3004. doi: 10.1038/emboj.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangavarapu V, Haracska L, Unk I, et al. Mms2-Ubc13-dependent and -independent roles of Rad5 ubiquitin ligase in postreplication repair and translesion DNA synthesis in Saccharomyces cerevisiae. Mol Cell Biol. 2006;26:7783–90. doi: 10.1128/MCB.01260-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, Soustelle C, Fabre F. Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat Genet. 2000;25:192–4. doi: 10.1038/76055. [DOI] [PubMed] [Google Scholar]
- Gao H, Moss DL, Parke C, et al. The Ctf18RFC clamp loader is essential for telomere stability in telomerase-negative and mre11 mutant alleles. PloS One. 2014;9:e88633. doi: 10.1371/journal.pone.0088633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q, Ren Q, Liou L-C, et al. Mitochondrial DNA protects against salt stress-induced cytochrome c-mediated apoptosis in yeast. FEBS Lett. 2011;585:2507–12. doi: 10.1016/j.febslet.2011.06.034. [DOI] [PubMed] [Google Scholar]
- Garg P, Stith CM, Majka J, et al. Proliferating cell nuclear antigen promotes translesion synthesis by DNA polymerase zeta. J Biol Chem. 2005;280:23446–50. doi: 10.1074/jbc.C500173200. [DOI] [PubMed] [Google Scholar]
- Gellon L, Razidlo DF, Gleeson O, et al. New functions of Ctf18-RFC in preserving genome stability outside its role in sister chromatid cohesion. PLoS Genet. 2011;7:e1001298. doi: 10.1371/journal.pgen.1001298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gernand D, Rutten T, Varshney A, et al. Uniparental chromosome elimination at mitosis and interphase in wheat and pearl millet crosses involves micronucleus formation, progressive heterochromatinization, and DNA fragmentation. Plant Cell. 2005;17:2431–8. doi: 10.1105/tpc.105.034249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein AC, Chun H-JE, Grant A, et al. Genomic convergence toward diploidy in Saccharomyces cerevisiae. PLoS Genet. 2006;2:e145. doi: 10.1371/journal.pgen.0020145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein AC, McBride RM, Otto SP. Ploidy reduction in Saccharomyces cerevisiae. Biol Lett. 2008;4:91–4. doi: 10.1098/rsbl.2007.0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodgaonkar MM, Lazzaro F, Olivera-Pimentel M, et al. Ribonucleotides misincorporated into DNA act as strand-discrimination signals in eukaryotic mismatch repair. Mol Cell. 2013;50:323–32. doi: 10.1016/j.molcel.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio M, Zwicky K, Follonier C, et al. Visualization of recombination-mediated damage bypass by template switching. Nat Struct Mol Biol. 2014;21:884–92. doi: 10.1038/nsmb.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs PEM, McDonald J, Woodgate R, et al. The relative roles in vivo of Saccharomyces cerevisiae Pol eta, Pol zeta, Rev1 protein and Pol32 in the bypass and mutation induction of an abasic site, T-T (6-4) photoadduct and T-T cis-syn cyclobutane dimer. Genetics. 2005;169:575–82. doi: 10.1534/genetics.104.034611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillett ES, Espelin CW, Sorger PK. Spindle checkpoint proteins and chromosome-microtubule attachment in budding yeast. J Cell Biol. 2004;164:535–46. doi: 10.1083/jcb.200308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginno PA, Lim YW, Lott PL, et al. GC skew at the 5′ and 3′ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013;23:1590–600. doi: 10.1101/gr.158436.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giot L, Chanet R, Simon M, et al. Involvement of the yeast DNA polymerase delta in DNA repair in vivo. Genetics. 1997;146:1239–51. doi: 10.1093/genetics/146.4.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud MF, Velours J. The absence of the mitochondrial ATP synthase delta subunit promotes a slow growth phenotype of rho- yeast cells by a lack of assembly of the catalytic sector F1. Eur J Biochem. 1997;245:813–8. doi: 10.1111/j.1432-1033.1997.00813.x. [DOI] [PubMed] [Google Scholar]
- Gnatt AL, Cramer P, Fu J, et al. Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 A resolution. Science. 2001;292:1876–82. doi: 10.1126/science.1059495. [DOI] [PubMed] [Google Scholar]
- Goellner EM, Smith CE, Campbell CS, et al. PCNA and Msh2-Msh6 activate an Mlh1-Pms1 endonuclease pathway required for Exo1-independent mismatch repair. Mol Cell. 2014;55:291–304. doi: 10.1016/j.molcel.2014.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Aguilera C, Tous C, Gómez-González B, et al. The THP1-SAC3-SUS1-CDC31 complex works in transcription elongation-mRNA export preventing RNA-mediated genome instability. Mol Biol Cell. 2008;19:4310–8. doi: 10.1091/mbc.E08-04-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Prieto R, Muñoz-Cabello AM, Cabello-Lobato MJ, et al. Rad51 replication fork recruitment is required for DNA damage tolerance. EMBO J. 2013;32:1307–21. doi: 10.1038/emboj.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar J, Harinarayanan R. Why is transcription coupled to translation in bacteria? Mol Microbiol. 2004;54:598–603. doi: 10.1111/j.1365-2958.2004.04289.x. [DOI] [PubMed] [Google Scholar]
- Grabowska E, Wronska U, Denkiewicz M, et al. Proper functioning of the GINS complex is important for the fidelity of DNA replication in yeast. Mol Microbiol. 2014;92:659–80. doi: 10.1111/mmi.12580. [DOI] [PubMed] [Google Scholar]
- Gray MW. Mitochondrial evolution. Science. 1999;283:1476–81. doi: 10.1126/science.283.5407.1476. [DOI] [PubMed] [Google Scholar]
- Green CM, Erdjument-Bromage H, Tempst P, et al. A novel Rad24 checkpoint protein complex closely related to replication factor C. Curr Biol. 2000;10:39–42. doi: 10.1016/s0960-9822(99)00263-8. [DOI] [PubMed] [Google Scholar]
- Gritenaite D, Princz LN, Szakal B, et al. A cell cycle-regulated Slx4-Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Gene Dev. 2014;28:1604–19. doi: 10.1101/gad.240515.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas J, Lemoncelli A, Morozov C, et al. Physical links between the nuclear envelope protein Mps3, three alternate replication factor C complexes, and a variant histone in Saccharomyces cerevisiae. DNA Cell Biol. 2012;31:917–24. doi: 10.1089/dna.2011.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber JE. Bisexual mating behavior in a diploid of Saccharomyces cerevisiae: evidence for genetically controlled non-random chromosome loss during vegetative growth. Genetics. 1974;78:843–58. doi: 10.1093/genetics/78.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffter P, Fox TD. Nuclear mutations in the petite-negative yeast Schizosaccharomyces pombe allow growth of cells lacking mitochondrial DNA. Genetics. 1992;131:255–60. doi: 10.1093/genetics/131.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halas A, Baranowska H, Podlaska A, et al. Evaluation of the roles of Pol zeta and NHEJ in starvation-associated spontaneous mutagenesis in the yeast Saccharomyces cerevisiae. Curr Genet. 2009;55:245–51. doi: 10.1007/s00294-009-0239-9. [DOI] [PubMed] [Google Scholar]
- Halas A, Podlaska A, Derkacz J, et al. The roles of PCNA SUMOylation, Mms2-Ubc13 and Rad5 in translesion DNA synthesis in Saccharomyces cerevisiae. Mol Microbiol. 2011;80:786–97. doi: 10.1111/j.1365-2958.2011.07610.x. [DOI] [PubMed] [Google Scholar]
- Hamza A, Baetz K. Iron-responsive transcription factor Aft1 interacts with kinetochore protein Iml3 and promotes pericentromeric cohesin. J Biol Chem. 2012;287:4139–47. doi: 10.1074/jbc.M111.319319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Stachow C. Analysis of Schizosaccharomyces pombe mitochondrial DNA replication by two dimensional gel electrophoresis. Chromosoma. 1994;103:162–70. doi: 10.1007/BF00368008. [DOI] [PubMed] [Google Scholar]
- Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Bio. 2008;9:958–70. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- Hanna JS, Kroll ES, Lundblad V, et al. Saccharomyces cerevisiae CTF18 and CTF4 are required for sister chromatid cohesion. Mol Cell Biol. 2001;21:3144–58. doi: 10.1128/MCB.21.9.3144-3158.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haracska L, Prakash S, Prakash L. Replication past O(6)-methylguanine by yeast and human DNA polymerase eta. Mol Cell Biol. 2000;20:8001–7. doi: 10.1128/mcb.20.21.8001-8007.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haracska L, Prakash S, Prakash L. Yeast DNA polymerase zeta is an efficient extender of primer ends opposite from 7,8-dihydro-8-Oxoguanine and O6-methylguanine. Mol Cell Biol. 2003;23:1453–9. doi: 10.1128/MCB.23.4.1453-1459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MR, Lee D, Farmer S, et al. Binding specificity of the G1/S transcriptional regulators in budding yeast. PloS One. 2013;8:e61059. doi: 10.1371/journal.pone.0061059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison BD, Hashemi J, Bibi M, et al. A tetraploid intermediate precedes aneuploid formation in yeasts exposed to fluconazole. PLoS Biol. 2014;12:e1001815. doi: 10.1371/journal.pbio.1001815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman JL. Buffering of deoxyribonucleotide pool homeostasis by threonine metabolism. P Natl Acad Sci USA. 2007;104:11700–5. doi: 10.1073/pnas.0705212104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Smith D. Altered fidelity of mitotic chromosome transmission in cell cycle mutants of S. cerevisiae. Genetics. 1985;110:381–95. doi: 10.1093/genetics/110.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmrich A, Ballarino M, Tora L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell. 2011;44:966–77. doi: 10.1016/j.molcel.2011.10.013. [DOI] [PubMed] [Google Scholar]
- Herman RK, Dworkin NB. Effect of gene induction on the rate of mutagenesis by ICR-191 in Escherichia coli. J Bacteriol. 1971;106:543–50. doi: 10.1128/jb.106.2.543-550.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman MA, Zeng G, Forche A, et al. The ‘obligate diploid’ Candida albicans forms mating-competent haploids. Nature. 2013;494:55–9. doi: 10.1038/nature11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks WM, Kim M, Haber JE. Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science. 2010;329:82–5. doi: 10.1126/science.1191125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraga S-I, Hagihara-Hayashi A, Ohya T, et al. DNA polymerases alpha, delta, and epsilon localize and function together at replication forks in Saccharomyces cerevisiae. Genes Cells. 2005;10:297–309. doi: 10.1111/j.1365-2443.2005.00843.x. [DOI] [PubMed] [Google Scholar]
- Hiraoka M, Watanabe K, Umezu K, et al. Spontaneous loss of heterozygosity in diploid Saccharomyces cerevisiae cells. Genetics. 2000;156:1531–48. doi: 10.1093/genetics/156.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishida T, Hirade Y, Haruta N, et al. Srs2 plays a critical role in reversible G2 arrest upon chronic and low doses of UV irradiation via two distinct homologous recombination-dependent mechanisms in postreplication repair-deficient cells. Mol Cell Biol. 2010;30:4840–50. doi: 10.1128/MCB.00453-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y, Costanzo M, Moore L, et al. Regulation of transcription at the Saccharomyces cerevisiae start transition by Stb1, a Swi6-binding protein. Mol Cell Biol. 1999;19:5267–78. doi: 10.1128/mcb.19.8.5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs AE, Srinivasan M, McCaffery JM, et al. Mmm1p, a mitochondrial outer membrane protein, is connected to mitochondrial DNA (mtDNA) nucleoids and required for mtDNA stability. J Cell Biol. 2001;152:401–10. doi: 10.1083/jcb.152.2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoege C, Pfander B, Moldovan G-L, et al. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–41. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- Holt IJ. Mitochondrial DNA replication and repair: all a flap. Trends Biochem Sci. 2009;34:358–65. doi: 10.1016/j.tibs.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Hombauer H, Srivatsan A, Putnam CD, et al. Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science. 2011;334:1713–6. doi: 10.1126/science.1210770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori A, Yoshida M, Shibata T, et al. Reactive oxygen species regulate DNA copy number in isolated yeast mitochondria by triggering recombination-mediated replication. Nucleic Acids Res. 2009;37:749–61. doi: 10.1093/nar/gkn993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett NG, Schiestl RH. Simultaneous measurement of the frequencies of intrachromosomal recombination and chromosome gain using the yeast DEL assay. Mutat Res. 2000;454:53–62. doi: 10.1016/s0027-5107(00)00097-x. [DOI] [PubMed] [Google Scholar]
- Hoyt MA, Totis L, Roberts BT. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 1991;66:507–17. doi: 10.1016/0092-8674(81)90014-3. [DOI] [PubMed] [Google Scholar]
- Huang M, Zhou Z, Elledge SJ. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell. 1998;94:595–605. doi: 10.1016/s0092-8674(00)81601-3. [DOI] [PubMed] [Google Scholar]
- Huang M-E, Rio A-G, Galibert M-D, et al. Pol32, a subunit of Saccharomyces cerevisiae DNA polymerase delta, suppresses genomic deletions and is involved in the mutagenic bypass pathway. Genetics. 2002;160:1409–22. doi: 10.1093/genetics/160.4.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R-Y, Kowalski D, Minderman H, et al. Small ubiquitin-related modifier pathway is a major determinant of doxorubicin cytotoxicity in Saccharomyces cerevisiae. Cancer Res. 2007;67:765–72. doi: 10.1158/0008-5472.CAN-06-2839. [DOI] [PubMed] [Google Scholar]
- Huang Y, Hou H, Yi Q, et al. The fate of micronucleated cells post X-irradiation detected by live cell imaging. DNA Repair. 2011;10:629–38. doi: 10.1016/j.dnarep.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Huertas P, Cortés-Ledesma F, Sartori AA, et al. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature. 2008;455:689–92. doi: 10.1038/nature07215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TR, Roberts CJ, Dai H, et al. Widespread aneuploidy revealed by DNA microarray expression profiling. Nat Genet. 2000;25:333–7. doi: 10.1038/77116. [DOI] [PubMed] [Google Scholar]
- Indjeian VB, Stern BM, Murray AW. The centromeric protein Sgo1 is required to sense lack of tension on mitotic chromosomes. Science. 2005;307:130–3. doi: 10.1126/science.1101366. [DOI] [PubMed] [Google Scholar]
- Infante JJ, Dombek KM, Rebordinos L, et al. Genome-wide amplifications caused by chromosomal rearrangements play a major role in the adaptive evolution of natural yeast. Genetics. 2003;165:1745–59. doi: 10.1093/genetics/165.4.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Malkova A, Liberi G, et al. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell. 2003;115:401–11. doi: 10.1016/s0092-8674(03)00886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Satory D, Haber JE. Conservative inheritance of newly synthesized DNA in double-strand break-induced gene conversion. Mol Cell Biol. 2006;26:9424–9. doi: 10.1128/MCB.01654-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iraqui I, Chekkal Y, Jmari N, et al. Recovery of arrested replication forks by homologous recombination is error-prone. PLoS Genet. 2012;8:e1002976. doi: 10.1371/journal.pgen.1002976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischenko AA, Saparbaev MK. Alternative nucleotide incision repair pathway for oxidative DNA damage. Nature. 2002;415:183–7. doi: 10.1038/415183a. [DOI] [PubMed] [Google Scholar]
- James SA, O'Kelly MJT, Carter DM, et al. Repetitive sequence variation and dynamics in the ribosomal DNA array of Saccharomyces cerevisiae as revealed by whole-genome resequencing. Genome Res. 2009;19:626–35. doi: 10.1101/gr.084517.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaszczur M, Rudzka J, Kraszewska J, et al. Defective interaction between Pol2p and Dpb2p, subunits of DNA polymerase epsilon, contributes to a mutator phenotype in Saccharomyces cerevisiae. Mutat Res. 2009;669:27–35. doi: 10.1016/j.mrfmmm.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppsson K, Kanno T, Shirahige K, et al. The maintenance of chromosome structure: positioning and functioning of SMC complexes. Nat Rev Mol Cell Bio. 2014;15:601–14. doi: 10.1038/nrm3857. [DOI] [PubMed] [Google Scholar]
- Johansson E, Garg P, Burgers PMJ. The Pol32 subunit of DNA polymerase delta contains separable domains for processive replication and proliferating cell nuclear antigen (PCNA) binding. J Biol Chem. 2004;279:1907–15. doi: 10.1074/jbc.M310362200. [DOI] [PubMed] [Google Scholar]
- Jordan A, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 1998;67:71–98. doi: 10.1146/annurev.biochem.67.1.71. [DOI] [PubMed] [Google Scholar]
- Kaguni LS. DNA polymerase gamma, the mitochondrial replicase. Annu Rev Biochem. 2004;73:293–320. doi: 10.1146/annurev.biochem.72.121801.161455. [DOI] [PubMed] [Google Scholar]
- Kai M, Wang TS-F. Checkpoint activation regulates mutagenic translesion synthesis. Gene Dev. 2003;17:64–76. doi: 10.1101/gad.1043203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalifa L, Beutner G, Phadnis N, et al. Evidence for a role of FEN1 in maintaining mitochondrial DNA integrity. DNA Repair. 2009;8:1242–9. doi: 10.1016/j.dnarep.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalifa L, Quintana DF, Schiraldi LK, et al. Mitochondrial genome maintenance: roles for nuclear nonhomologous end-joining proteins in Saccharomyces cerevisiae. Genetics. 2012;190:951–64. doi: 10.1534/genetics.111.138214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalifa L, Sia EA. Analysis of Rev1p and Pol ζ in mitochondrial mutagenesis suggests an alternative pathway of damage tolerance. DNA Repair. 2007;6:1732–9. doi: 10.1016/j.dnarep.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamitsu K, Ikeda S. Early steps in the DNA base excision repair pathway of a fission yeast Schizosaccharomyces pombe. J Nucleic Acids. 2010;2010:1–9. doi: 10.4061/2010/450926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellis P, Agyei R, Durocher D. Elg1 forms an alternative PCNA-interacting RFC complex required to maintain genome stability. Curr Biol. 2003;13:1583–95. doi: 10.1016/s0960-9822(03)00578-5. [DOI] [PubMed] [Google Scholar]
- Kaniak A, Dzierzbicki P, Rogowska AT, et al. Msh1p counteracts oxidative lesion-induced instability of mtDNA and stimulates mitochondrial recombination in Saccharomyces cerevisiae. DNA Repair. 2009;8:318–29. doi: 10.1016/j.dnarep.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Kaniak-Golik A, Skoneczna A. Mitochondria-nucleus network for genome stability. Free Radical Bio Med. 2015;82:73–104. doi: 10.1016/j.freeradbiomed.2015.01.013. [DOI] [PubMed] [Google Scholar]
- Kanoh J. Release of chromosomes from the nuclear envelope: a universal mechanism for eukaryotic mitosis? Nucleus. 2013;4:100–4. doi: 10.4161/nucl.23984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao HI, Henricksen LA, Liu Y, et al. Cleavage specificity of Saccharomyces cerevisiae flap endonuclease 1 suggests a double-flap structure as the cellular substrate. J Biol Chem. 2002;277:14379–89. doi: 10.1074/jbc.M110662200. [DOI] [PubMed] [Google Scholar]
- Karras GI, Fumasoni M, Sienski G, et al. Noncanonical role of the 9–1–1 clamp in the error-free DNA damage tolerance pathway. Mol Cell. 2013;49:536–46. doi: 10.1016/j.molcel.2012.11.016. [DOI] [PubMed] [Google Scholar]
- Karras GI, Jentsch S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell. 2010;141:255–67. doi: 10.1016/j.cell.2010.02.028. [DOI] [PubMed] [Google Scholar]
- Karthikeyan G, Lewis LK, Resnick MA. The mitochondrial protein frataxin prevents nuclear damage. Hum Mol Genet. 2002;11:1351–62. doi: 10.1093/hmg/11.11.1351. [DOI] [PubMed] [Google Scholar]
- Karthikeyan R, Vonarx EJ, Straffon AF, et al. Evidence from mutational specificity studies that yeast DNA polymerases delta and epsilon replicate different DNA strands at an intracellular replication fork. J Mol Biol. 2000;299:405–19. doi: 10.1006/jmbi.2000.3744. [DOI] [PubMed] [Google Scholar]
- Kawashima SA, Yamagishi Y, Honda T, et al. Phosphorylation of H2A by Bub1 prevents chromosomal instability through localizing shugoshin. Science. 2010;327:172–7. doi: 10.1126/science.1180189. [DOI] [PubMed] [Google Scholar]
- Kegel A, Sjöstrand JO, Aström SU. Nej1p, a cell type-specific regulator of nonhomologous end joining in yeast. Curr Biol. 2001;11:1611–7. doi: 10.1016/s0960-9822(01)00488-2. [DOI] [PubMed] [Google Scholar]
- Keil RL, Roeder GS. Cis-acting, recombination-stimulating activity in a fragment of the ribosomal DNA of S. cerevisiae. Cell. 1984;39:377–86. doi: 10.1016/0092-8674(84)90016-3. [DOI] [PubMed] [Google Scholar]
- Kerrest A, Anand RP, Sundararajan R, et al. SRS2 and SGS1 prevent chromosomal breaks and stabilize triplet repeats by restraining recombination. Nat Struct Mol Bio. 2009;16:159–67. doi: 10.1038/nsmb.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Abdulovic AL, Gealy R, et al. Transcription-associated mutagenesis in yeast is directly proportional to the level of gene expression and influenced by the direction of DNA replication. DNA Repair. 2007;6:1285–96. doi: 10.1016/j.dnarep.2007.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Jinks-Robertson S. dUTP incorporation into genomic DNA is linked to transcription in yeast. Nature. 2009;459:1150–3. doi: 10.1038/nature08033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Jinks-Robertson S. Abasic sites in the transcribed strand of yeast DNA are removed by transcription-coupled nucleotide excision repair. Mol Cell Biol. 2010;30:3206–15. doi: 10.1128/MCB.00308-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Lin DP, Matsumoto S, et al. Fission yeast Slp1: an effector of the Mad2-dependent spindle checkpoint. Science. 1998;279:1045–7. doi: 10.1126/science.279.5353.1045. [DOI] [PubMed] [Google Scholar]
- Kispal G, Csere P, Prohl C, et al. The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J. 1999;18:3981–9. doi: 10.1093/emboj/18.14.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kispal G, Sipos K, Lange H, et al. Biogenesis of cytosolic ribosomes requires the essential iron-sulphur protein Rli1p and mitochondria. EMBO J. 2005;24:589–98. doi: 10.1038/sj.emboj.7600541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapacz J, Bhagwat AS. Transcription promotes guanine to thymine mutations in the non-transcribed strand of an Escherichia coli gene. DNA Repair. 2005;4:806–13. doi: 10.1016/j.dnarep.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Kleff S, Kemper B, Sternglanz R. Identification and characterization of yeast mutants and the gene for a cruciform cutting endonuclease. EMBO J. 1992;11:699. doi: 10.1002/j.1460-2075.1992.tb05102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein HL. Spontaneous chromosome loss in Saccharomyces cerevisiae is suppressed by DNA damage checkpoint functions. Genetics. 2001;159:1501–9. doi: 10.1093/genetics/159.4.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge S, Hirst J, Maman JD, et al. An iron-sulfur domain of the eukaryotic primase is essential for RNA primer synthesis. Nat Struct Mol Bio. 2007;14:875–7. doi: 10.1038/nsmb1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Valentine MR, Pham P, et al. Fidelity of Escherichia coli DNA polymerase IV. Preferential generation of small deletion mutations by dNTP-stabilized misalignment. J Biol Chem. 2002;277:34198–207. doi: 10.1074/jbc.M204826200. [DOI] [PubMed] [Google Scholar]
- Kobayashi T. The replication fork barrier site forms a unique structure with Fob1p and inhibits the replication fork. Mol Cell Biol. 2003;23:9178–88. doi: 10.1128/MCB.23.24.9178-9188.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T. Regulation of ribosomal RNA gene copy number and its role in modulating genome integrity and evolutionary adaptability in yeast. Cell Mol Life Sci. 2011;68:1395–403. doi: 10.1007/s00018-010-0613-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Heck DJ, Nomura M, et al. Expansion and contraction of ribosomal DNA repeats in Saccharomyces cerevisiae: requirement of replication fork blocking (Fob1) protein and the role of RNA polymerase I. Gene Dev. 1998;12:3821–30. doi: 10.1101/gad.12.24.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koc A, Mathews CK, Wheeler LJ, et al. Thioredoxin is required for deoxyribonucleotide pool maintenance during S phase. J Biol Chem. 2006;281:15058–63. doi: 10.1074/jbc.M601968200. [DOI] [PubMed] [Google Scholar]
- Koc A, Merrill GF. Checkpoint deficient rad53-11 yeast cannot accumulate dNTPs in response to DNA damage. Biochem Bioph Res Co. 2007;353:527–30. doi: 10.1016/j.bbrc.2006.12.049. [DOI] [PubMed] [Google Scholar]
- Koç A, Wheeler LJ, Mathews CK, et al. Replication-independent MCB gene induction and deoxyribonucleotide accumulation at G1/S in Saccharomyces cerevisiae. J Biol Chem. 2003;278:9345–52. doi: 10.1074/jbc.m213013200. [DOI] [PubMed] [Google Scholar]
- Koç A, Wheeler LJ, Mathews CK, et al. Hydroxyurea arrests DNA replication by a mechanism that preserves basal dNTP pools. J Biol Chem. 2004;279:223–30. doi: 10.1074/jbc.M303952200. [DOI] [PubMed] [Google Scholar]
- Kohl KP, Sekelsky J. Meiotic and mitotic recombination in meiosis. Genetics. 2013;194:327–34. doi: 10.1534/genetics.113.150581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolberg M, Strand KR, Graff P, et al. Structure, function, and mechanism of ribonucleotide reductases. Biochim Biophys Acta. 2004;1699:1–34. doi: 10.1016/j.bbapap.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Kolesar P, Sarangi P, Altmannova V, et al. Dual roles of the SUMO-interacting motif in the regulation of Srs2 sumoylation. Nucleic Acids Res. 2012;40:7831–43. doi: 10.1093/nar/gks484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodner RD, Marsischky GT. Eukaryotic DNA mismatch repair. Curr Opin Genet Dev. 1999;9:89–96. doi: 10.1016/s0959-437x(99)80013-6. [DOI] [PubMed] [Google Scholar]
- Kominsky DJ, Thorsness PE. Expression of the Saccharomyces cerevisiae gene YME1 in the petite-negative yeast Schizosaccharomyces pombe converts it to petite-positive. Genetics. 2000;154:147–54. doi: 10.1093/genetics/154.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koprowski P, Fikus MU, Dzierzbicki P, et al. Enhanced expression of the DNA damage-inducible gene DIN7 results in increased mutagenesis of mitochondrial DNA in Saccharomyces cerevisiae. Mol Genet Genomics. 2003;269:632–9. doi: 10.1007/s00438-003-0873-8. [DOI] [PubMed] [Google Scholar]
- Koprowski P, Fikus MU, Mieczkowski P, et al. A dominant mitochondrial mutator phenotype of Saccharomyces cerevisiae conferred by msh1 alleles altered in the sequence encoding the ATP-binding domain. Mol Genet Genomics. 2002;266:988–94. doi: 10.1007/s00438-001-0621-x. [DOI] [PubMed] [Google Scholar]
- Kornmann B, Currie E, Collins SR, et al. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477–81. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korogodin VI, Korogodina VL, Fajszi C, et al. On the dependence of spontaneous mutation rates on the functional state of genes. Yeast. 1991;7:105–17. doi: 10.1002/yea.320070204. [DOI] [PubMed] [Google Scholar]
- Kouprina N, Tsouladze A, Koryabin M, et al. Identification and genetic mapping of CHL genes controlling mitotic chromosome transmission in yeast. Yeast. 1993;9:11–9. doi: 10.1002/yea.320090103. [DOI] [PubMed] [Google Scholar]
- Kraus E, Leung WY, Haber JE. Break-induced replication: a review and an example in budding yeast. P Natl Acad Sci USA. 2001;98:8255–62. doi: 10.1073/pnas.151008198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejci L, Van Komen S, Li Y, et al. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature. 2003;423:305–9. doi: 10.1038/nature01577. [DOI] [PubMed] [Google Scholar]
- Krick R, Muehe Y, Prick T, et al. Piecemeal microautophagy of the nucleus requires the core macroautophagy genes. Mol Biol Cell. 2008;19:4492–505. doi: 10.1091/mbc.E08-04-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T, Hiraga S, Yamada K, et al. Quantitative proteomic analysis of chromatin reveals that Ctf18 acts in the DNA replication checkpoint. Mol Cell Proteomics. 2011;10:M110.005561. doi: 10.1074/mcp.M110.005561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucej M, Butow RA. Evolutionary tinkering with mitochondrial nucleoids. Trends Cell Biol. 2007;17:586–92. doi: 10.1016/j.tcb.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Kühl I. Mitochondrial Translation and Transformation in the Fission Yeast Schizosaccharomyces pombe as a Model System. Paris: Defended on Universite de Versailles Saint-Quentin-en-Yvelines; 2010. [Google Scholar]
- Kumar D, Abdulovic AL, Viberg J, et al. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res. 2011;39:1360–71. doi: 10.1093/nar/gkq829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D, Viberg J, Nilsson AK, et al. Highly mutagenic and severely imbalanced dNTP pools can escape detection by the S-phase checkpoint. Nucleic Acids Res. 2010;38:3975–83. doi: 10.1093/nar/gkq128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaran R, Yang S-Y, Leu J-Y. Characterization of chromosome stability in diploid, polyploid and hybrid yeast cells. PloS One. 2013;8:e68094. doi: 10.1371/journal.pone.0068094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA. Misalignment-mediated DNA synthesis errors. Biochemistry. 1990;29:8003–11. doi: 10.1021/bi00487a001. [DOI] [PubMed] [Google Scholar]
- Kunkel TA. Balancing eukaryotic replication asymmetry with replication fidelity. Curr Opin Chem Biol. 2011;15:620–6. doi: 10.1016/j.cbpa.2011.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Bebenek K. DNA replication fidelity. Annu Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Hamatake RK, Motto-Fox J, et al. Fidelity of DNA polymerase I and the DNA polymerase I-DNA primase complex from Saccharomyces cerevisiae. Mol Cell Biol. 1989;9:4447–58. doi: 10.1128/mcb.9.10.4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Silber JR, Loeb LA. The mutagenic effect of deoxynucleotide substrate imbalances during DNA synthesis with mammalian DNA polymerases. Mutat Res. 1982;94:413–9. doi: 10.1016/0027-5107(82)90304-9. [DOI] [PubMed] [Google Scholar]
- Kurischko C. Spontaneous haploidization in early zygote progeny and its use for mapping in the yeast Yarrowia lipolytica. Curr Genet. 1986;10:709–11. doi: 10.1007/BF00410920. [DOI] [PubMed] [Google Scholar]
- Labib K, De Piccoli G. Surviving chromosome replication: the many roles of the S-phase checkpoint pathway. Philos T Roy Soc B. 2011;366:3554–61. doi: 10.1098/rstb.2011.0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laerdahl JK, Korvald H, Nilsen L, et al. Schizosaccharomyces pombe encodes a mutated AP endonuclease 1. DNA Repair. 2011;10:296–305. doi: 10.1016/j.dnarep.2010.11.014. [DOI] [PubMed] [Google Scholar]
- Lahaye A, Stahl H, Thines-Sempoux D, et al. PIF1: a DNA helicase in yeast mitochondria. EMBO J. 1991;10:997. doi: 10.1002/j.1460-2075.1991.tb08034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S, Watson A, Sheedy DM, et al. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell. 2005;121:689–702. doi: 10.1016/j.cell.2005.03.022. [DOI] [PubMed] [Google Scholar]
- Langerak P, Mejia-Ramirez E, Limbo O, et al. Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet. 2011;7:e1002271. doi: 10.1371/journal.pgen.1002271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrea AA, Lujan SA, Nick McElhinny SA, et al. Genome-wide model for the normal eukaryotic DNA replication fork. P Natl Acad Sci USA. 2010;107:17674–9. doi: 10.1073/pnas.1010178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasserre J-P, Plissonneau J, Velours C, et al. Biochemical, cellular and molecular identification of DNA polymerase α in yeast mitochondria. Biochimie. 2013;95:759–71. doi: 10.1016/j.biochi.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Lazzaro F, Novarina D, Amara F, et al. RNase H and postreplication repair protect cells from ribonucleotides incorporated in DNA. Mol Cell. 2012;45:99–110. doi: 10.1016/j.molcel.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecrenier N, Foury F. New features of mitochondrial DNA replication system in yeast and man. Gene. 2000;246:37–48. doi: 10.1016/s0378-1119(00)00107-4. [DOI] [PubMed] [Google Scholar]
- Lee MS, Spencer FA. Bipolar orientation of chromosomes in Saccharomyces cerevisiae is monitored by Mad1 and Mad2, but not by Mad3. P Natl Acad Sci USA. 2004;101:10655–60. doi: 10.1073/pnas.0404102101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YD, Elledge SJ. Control of ribonucleotide reductase localization through an anchoring mechanism involving Wtm1. Gene Dev. 2006;20:334–44. doi: 10.1101/gad.1380506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann AR. Ubiquitin-family modifications in the replication of DNA damage. FEBS Lett. 2011;585:2772–9. doi: 10.1016/j.febslet.2011.06.005. [DOI] [PubMed] [Google Scholar]
- Lehner K, Mudrak SV, Minesinger BK, et al. Frameshift mutagenesis: the roles of primer-template misalignment and the nonhomologous end-joining pathway in Saccharomyces cerevisiae. Genetics. 2012;190:501–10. doi: 10.1534/genetics.111.134890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–9. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- Li R, Murray AW. Feedback control of mitosis in budding yeast. Cell. 1991;66:519–31. doi: 10.1016/0092-8674(81)90015-5. [DOI] [PubMed] [Google Scholar]
- Li X, Manley JL. Cotranscriptional processes and their influence on genome stability. Gene Dev. 2006;20:1838–47. doi: 10.1101/gad.1438306. [DOI] [PubMed] [Google Scholar]
- Li XC, Tye BK. Ploidy dictates repair pathway choice under DNA replication stress. Genetics. 2011;187:1031–40. doi: 10.1534/genetics.110.125450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gorbea C, Mahaffey D, et al. MAD2 associates with the cyclosome/anaphase-promoting complex and inhibits its activity. P Natl Acad Sci USA. 1997;94:12431–6. doi: 10.1073/pnas.94.23.12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberi G, Maffioletti G, Lucca C, et al. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Gene Dev. 2005;19:339–50. doi: 10.1101/gad.322605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti SE, Larrea AA, Kunkel TA. Exonuclease 1 preferentially repairs mismatches generated by DNA polymerase α. DNA Repair. 2013;12:92–6. doi: 10.1016/j.dnarep.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilienthal I, Kanno T, Sjögren C. Inhibition of the Smc5/6 complex during meiosis perturbs joint molecule formation and resolution without significantly changing crossover or non-crossover levels. PLoS Genet. 2013;9:e1003898. doi: 10.1371/journal.pgen.1003898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill R, Kispal G. Maturation of cellular Fe-S proteins: an essential function of mitochondria. Trends Biochem Sci. 2000;25:352–6. doi: 10.1016/s0968-0004(00)01589-9. [DOI] [PubMed] [Google Scholar]
- Ling F, Hori A, Shibata T. DNA Recombination-initiation Plays a role in the extremely biased inheritance of yeast [rho−] mitochondrial DNA that contains the replication origin ori5. Mol Cell Biol. 2007;27:1133–45. doi: 10.1128/MCB.00770-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling F, Hori A, Yoshitani A, et al. Din7 and Mhr1 expression levels regulate double-strand-break-induced replication and recombination of mtDNA at ori5 in yeast. Nucleic Acids Res. 2013;41:5799–816. doi: 10.1093/nar/gkt273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling F, Makishima F, Morishima N, et al. A nuclear mutation defective in mitochondrial recombination in yeast. EMBO J. 1995;14:4090. doi: 10.1002/j.1460-2075.1995.tb00081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling F, Shibata T. Recombination-dependent mtDNA partitioning: in vivo role of Mhr1p to promote pairing of homologous DNA. EMBO J. 2002;21:4730–40. doi: 10.1093/emboj/cdf466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippert MJ, Freedman JA, Barber MA, et al. Identification of a distinctive mutation spectrum associated with high levels of transcription in yeast. Mol Cell Biol. 2004;24:4801–9. doi: 10.1128/MCB.24.11.4801-4809.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis ET, O'Neill BM, Gil-Lamaignere C, et al. Identification of pathways controlling DNA damage induced mutation in Saccharomyces cerevisiae. DNA Repair. 2008;7:801–10. doi: 10.1016/j.dnarep.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LF, Wang JC. Supercoiling of the DNA template during transcription. P Natl Acad Sci USA. 1987;84:7024–7. doi: 10.1073/pnas.84.20.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Qian L, Sung J-S, et al. Removal of oxidative DNA damage via FEN1-dependent long-patch base excision repair in human cell mitochondria. Mol Cell Biol. 2008;28:4975–87. doi: 10.1128/MCB.00457-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockshon D, Zweifel SG, Freeman-Cook LL, et al. A role for recombination junctions in the segregation of mitochondrial DNA in yeast. Cell. 1995;81:947–55. doi: 10.1016/0092-8674(95)90014-4. [DOI] [PubMed] [Google Scholar]
- Lopes M, Cotta-Ramusino C, Pellicioli A, et al. The DNA replication checkpoint response stabilizes stalled replication forks. Nature. 2001;412:557–61. doi: 10.1038/35087613. [DOI] [PubMed] [Google Scholar]
- Lorimer HE, Brewer BJ, Fangman WL. A test of the transcription model for biased inheritance of yeast mitochondrial DNA. Mol Cell Biol. 1995;15:4803–9. doi: 10.1128/mcb.15.9.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou H, Komata M, Katou Y, et al. Mrc1 and DNA polymerase ε function together in linking DNA replication and the S phase checkpoint. Mol Cell. 2008;32:106–17. doi: 10.1016/j.molcel.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Cross F. Mitotic exit in the absence of separase activity. Mol Biol Cell. 2009;20:1576–91. doi: 10.1091/mbc.E08-10-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas P, Lasserre J-P, Plissonneau J, et al. Absence of accessory subunit in the DNA polymerase gamma purified from yeast mitochondria. Mitochondrion. 2004;4:13–20. doi: 10.1016/j.mito.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Lucchini G, Francesconi S, Foiani M, et al. Yeast DNA polymerase–DNA primase complex; cloning of PRI 1, a single essential gene related to DNA primase activity. EMBO J. 1987;6:737–42. doi: 10.1002/j.1460-2075.1987.tb04815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Xu X, Hall H, et al. Histone h3 exerts a key function in mitotic checkpoint control. Mol Cell Biol. 2010;30:537–49. doi: 10.1128/MCB.00980-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Tang Z, Rizo J, et al. The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol Cell. 2002;9:59–71. doi: 10.1016/s1097-2765(01)00435-x. [DOI] [PubMed] [Google Scholar]
- Lustig AJ, Petes TD. Identification of yeast mutants with altered telomere structure. P Natl Acad Sci USA. 1986;83:1398–402. doi: 10.1073/pnas.83.5.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydeard JR, Lipkin-Moore Z, Sheu Y-J, et al. Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Gene Dev. 2010;24:1133–44. doi: 10.1101/gad.1922610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyndaker AM, Alani E. A tale of tails: insights into the coordination of 3′ end processing during homologous recombination. BioEssays. 2009;31:315–21. doi: 10.1002/bies.200800195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J-L, Kim EM, Haber JE, et al. Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol Cell Biol. 2003;23:8820–8. doi: 10.1128/MCB.23.23.8820-8828.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Zhai Y, Feng D, et al. Identification of novel factors involved in or regulating initiation of DNA replication by a genome-wide phenotypic screen in Saccharomyces cerevisiae. Cell Cycle. 2010;9:4399–410. doi: 10.4161/cc.9.21.13679. [DOI] [PubMed] [Google Scholar]
- MacAlpine DM, Perlman PS, Butow RA. The high mobility group protein Abf2p influences the level of yeast mitochondrial DNA recombination intermediates in vivo. P Natl Acad Sci USA. 1998;95:6739–43. doi: 10.1073/pnas.95.12.6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulley JL, Petes TD. Chromosome rearrangements and aneuploidy in yeast strains lacking both Tel1p and Mec1p reflect deficiencies in two different mechanisms. P Natl Acad Sci USA. 2010;107:11465–70. doi: 10.1073/pnas.1006281107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch SD, Kokoska RJ, Chilkova O, et al. Enzymatic switching for efficient and accurate translesion DNA replication. Nucleic Acids Res. 2004a;32:4665–75. doi: 10.1093/nar/gkh777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch SD, Kokoska RJ, Masutani C, et al. Preferential cis-syn thymine dimer bypass by DNA polymerase eta occurs with biased fidelity. Nature. 2004b;428:97–100. doi: 10.1038/nature02352. [DOI] [PubMed] [Google Scholar]
- McCulloch SD, Kunkel TA. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008;18:148–61. doi: 10.1038/cr.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch SD, Wood A, Garg P, et al. Effects of accessory proteins on the bypass of a cis-syn thymine-thymine dimer by Saccharomyces cerevisiae DNA polymerase eta. Biochemistry. 2007;46:8888–96. doi: 10.1021/bi700234t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macierzanka M, Plotka M, Pryputniewicz-Drobinska D, et al. Maintenance and stabilization of mtDNA can be facilitated by the DNA-binding activity of Ilv5p. Biochim Biophys Acta. 2008;1783:107–17. doi: 10.1016/j.bbamcr.2007.09.009. [DOI] [PubMed] [Google Scholar]
- McIntyre J, Podlaska A, Skoneczna A, et al. Analysis of the spontaneous mutator phenotype associated with 20S proteasome deficiency in S. cerevisiae. Mutat Res. 2006;593:153–63. doi: 10.1016/j.mrfmmm.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Majka J, Binz SK, Wold MS, et al. Replication protein A directs loading of the DNA damage checkpoint clamp to 5′-DNA junctions. J Biol Chem. 2006a;281:27 855–61. doi: 10.1074/jbc.M605176200. [DOI] [PubMed] [Google Scholar]
- Majka J, Burgers PMJ. Yeast Rad17/Mec3/Ddc1: a sliding clamp for the DNA damage checkpoint. P Natl Acad Sci USA. 2003;100:2249–54. doi: 10.1073/pnas.0437148100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majka J, Burgers PMJ. The PCNA-RFC families of DNA clamps and clamp loaders. Prog Nucleic Acid Re. 2004;78:227–60. doi: 10.1016/S0079-6603(04)78006-X. [DOI] [PubMed] [Google Scholar]
- Majka J, Niedziela-Majka A, Burgers PMJ. The checkpoint clamp activates Mec1 kinase during initiation of the DNA damage checkpoint. Mol Cell. 2006b;24:891–901. doi: 10.1016/j.molcel.2006.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova AV, Stodola JL, Burgers PM. A four-subunit DNA polymerase complex containing Pol accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res. 2012;40:11618–26. doi: 10.1093/nar/gks948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malc E, Dzierzbicki P, Kaniak A, et al. Inactivation of the 20S proteasome maturase, Ump1p, leads to the instability of mtDNA in Saccharomyces cerevisiae. Mutat Res. 2009;669:95–103. doi: 10.1016/j.mrfmmm.2009.05.008. [DOI] [PubMed] [Google Scholar]
- Maleszka R, Skelly PJ, Clark-Walker GD. Rolling circle replication of DNA in yeast mitochondria. EMBO J. 1991;10:3923. doi: 10.1002/j.1460-2075.1991.tb04962.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkova A, Haber JE. Mutations arising during repair of chromosome breaks. Annu Rev Genet. 2012;46:455–73. doi: 10.1146/annurev-genet-110711-155547. [DOI] [PubMed] [Google Scholar]
- Mapelli M, Massimiliano L, Santaguida S, et al. The Mad2 conformational dimer: structure and implications for the spindle assembly checkpoint. Cell. 2007;131:730–43. doi: 10.1016/j.cell.2007.08.049. [DOI] [PubMed] [Google Scholar]
- Maradeo ME, Skibbens RV. The Elg1-RFC clamp-loading complex performs a role in sister chromatid cohesion. PloS One. 2009;4:e4707. doi: 10.1371/journal.pone.0004707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maradeo ME, Skibbens RV. Replication factor C complexes play unique pro- and anti-establishment roles in sister chromatid cohesion. PloS One. 2010;5:e15381. doi: 10.1371/journal.pone.0015381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani L, Chiroli E, Nezi L, et al. Role of the Mad2 dimerization interface in the spindle assembly checkpoint independent of kinetochores. Curr Biol. 2012;22:1900–8. doi: 10.1016/j.cub.2012.08.028. [DOI] [PubMed] [Google Scholar]
- Marsischky GT, Lee S, Griffith J, et al. Saccharomyces cerevisiae MSH2/6 complex interacts with Holliday junctions and facilitates their cleavage by phage resolution enzymes. J Biol Chem. 1999;274:7200–6. doi: 10.1074/jbc.274.11.7200. [DOI] [PubMed] [Google Scholar]
- Martinez-Rucobo FW, Sainsbury S, Cheung ACM, et al. Architecture of the RNA polymerase-Spt4/5 complex and basis of universal transcription processivity. EMBO J. 2011;30:1302–10. doi: 10.1038/emboj.2011.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos J, Blanco MG, Maslen S, et al. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell. 2011;147:158–72. doi: 10.1016/j.cell.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos J, Blanco MG, West SC. Cell-cycle kinases coordinate the resolution of recombination intermediates with chromosome segregation. Cell Rep. 2013;4:76–86. doi: 10.1016/j.celrep.2013.05.039. [DOI] [PubMed] [Google Scholar]
- Matsuyama A, Arai R, Yashiroda Y, et al. ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol. 2006;24:841–7. doi: 10.1038/nbt1222. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Gygi SP, Aebersold R, et al. Identification of RFC(Ctf18p, Ctf8p, Dcc1p): an alternative RFC complex required for sister chromatid cohesion in S. cerevisiae. Mol Cell. 2001;7:959–70. doi: 10.1016/s1097-2765(01)00254-4. [DOI] [PubMed] [Google Scholar]
- Mbantenkhu M, Wang X, Nardozzi JD, et al. Mgm101 is a Rad52-related protein required for mitochondrial DNA recombination. J Biol Chem. 2011;286:42360–70. doi: 10.1074/jbc.M111.307512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeusen S, Nunnari J. Evidence for a two membrane-spanning autonomous mitochondrial DNA replisome. J Cell Biol. 2003;163:503–10. doi: 10.1083/jcb.200304040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeusen S, Tieu Q, Wong E, et al. Mgm101p is a novel component of the mitochondrial nucleoid that binds DNA and is required for the repair of oxidatively damaged mitochondrial DNA. J Cell Biol. 1999;145:291–304. doi: 10.1083/jcb.145.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meitinger F, Petrova B, Lombardi IM, et al. Targeted localization of Inn1, Cyk3 and Chs2 by the mitotic-exit network regulates cytokinesis in budding yeast. J Cell Sci. 2010;123:1851–61. doi: 10.1242/jcs.063891. [DOI] [PubMed] [Google Scholar]
- Merz S, Westermann B. Genome-wide deletion mutant analysis reveals genes required for respiratory growth, mitochondrial genome maintenance and mitochondrial protein synthesis in Saccharomyces cerevisiae. Genome Biol. 2009;10:R95. doi: 10.1186/gb-2009-10-9-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 2010;29:3358–69. doi: 10.1038/emboj.2010.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. DNA end resection–unraveling the tail. DNA Repair. 2011;10:344–8. doi: 10.1016/j.dnarep.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyabe I, Kunkel TA, Carr AM. The major roles of DNA polymerases epsilon and delta at the eukaryotic replication fork are evolutionarily conserved. PLoS Genet. 2011;7:e1002407. doi: 10.1371/journal.pgen.1002407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa T, Mizunuma M. Physiological roles of calcineurin in Saccharomyces cerevisiae with special emphasis on its roles in G2/M cell-cycle regulation. Biosci Biotech Bioch. 2007;71:633–45. doi: 10.1271/bbb.60495. [DOI] [PubMed] [Google Scholar]
- Mohanty BK, Bastia D. Binding of the replication terminator protein Fob1p to the Ter sites of yeast causes polar fork arrest. J Biol Chem. 2004;279:1932–41. doi: 10.1074/jbc.M309078200. [DOI] [PubMed] [Google Scholar]
- Mojas N, Lopes M, Jiricny J. Mismatch repair-dependent processing of methylation damage gives rise to persistent single-stranded gaps in newly replicated DNA. Gene Dev. 2007;21:3342–55. doi: 10.1101/gad.455407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan G-L, Pfander B, Jentsch S. PCNA controls establishment of sister chromatid cohesion during S phase. Mol Cell. 2006;23:723–32. doi: 10.1016/j.molcel.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Moldovan G-L, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–79. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Molnar M, Sipiczki M. Polyploidy in the haplontic yeast Schizosaccharomyces pombe: construction and analysis of strains. Curr Genet. 1993;24:45–52. doi: 10.1007/BF00324664. [DOI] [PubMed] [Google Scholar]
- Mookerjee SA, Lyon HD, Sia EA. Analysis of the functional domains of the mismatch repair homologue Msh1p and its role in mitochondrial genome maintenance. Curr Genet. 2005;47:84–99. doi: 10.1007/s00294-004-0537-1. [DOI] [PubMed] [Google Scholar]
- Mookerjee SA, Sia EA. Overlapping contributions of Msh1p and putative recombination proteins Cce1p, Din7p, and Mhr1p in large-scale recombination and genome sorting events in the mitochondrial genome of Saccharomyces cerevisiae. Mutat Res. 2006;595:91–106. doi: 10.1016/j.mrfmmm.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Morey NJ, Greene CN, Jinks-Robertson S. Genetic analysis of transcription-associated mutation in Saccharomyces cerevisiae. Genetics. 2000;154:109–20. doi: 10.1093/genetics/154.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison A, Araki H, Clark AB, et al. A third essential DNA polymerase in S. cerevisiae. Cell. 1990;62:1143–51. doi: 10.1016/0092-8674(90)90391-q. [DOI] [PubMed] [Google Scholar]
- Morrison A, Sugino A. The 3′–>5′ exonucleases of both DNA polymerases delta and epsilon participate in correcting errors of DNA replication in Saccharomyces cerevisiae. Mol Gen Genet. 1994;242:289–96. doi: 10.1007/BF00280418. [DOI] [PubMed] [Google Scholar]
- Morrow CA, Fraser JA. Ploidy variation as an adaptive mechanism in human pathogenic fungi. Semin Cell Dev Biol. 2013;24:339–46. doi: 10.1016/j.semcdb.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Mortimer RK, Contopoulou R, Schild D. Mitotic chromosome loss in a radiation-sensitive strain of the yeast Saccharomyces cerevisiae. P Natl Acad Sci USA. 1981;78:5778–82. doi: 10.1073/pnas.78.9.5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Galván S, Tous C, Blanco MG, et al. Distinct roles of Mus81, Yen1, Slx1-Slx4, and Rad1 nucleases in the repair of replication-born double-strand breaks by sister chromatid exchange. Mol Cell Biol. 2012;32:1592–603. doi: 10.1128/MCB.00111-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murley A, Lackner LL, Osman C, et al. ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. eLife. 2013;2:e00422. doi: 10.7554/eLife.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers AM, Pape LK, Tzagoloff A. Mitochondrial protein synthesis is required for maintenance of intact mitochondrial genomes in Saccharomyces cerevisiae. EMBO J. 1985;4:2087. doi: 10.1002/j.1460-2075.1985.tb03896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair DT, Johnson RE, Prakash L, et al. Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science. 2005;309:2219–22. doi: 10.1126/science.1116336. [DOI] [PubMed] [Google Scholar]
- Navadgi-Patil VM, Burgers PM. The unstructured C-terminal tail of the 9–1–1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Mol Cell. 2009;36:743–53. doi: 10.1016/j.molcel.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestoras K, Mohammed AH, Schreurs A-S, et al. Regulation of ribonucleotide reductase by Spd1 involves multiple mechanisms. Gene Dev. 2010;24:1145–59. doi: 10.1101/gad.561910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netz DJA, Stith CM, Stümpfig M, et al. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat Chem Biol. 2011;8:125–32. doi: 10.1038/nchembio.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netz DJA, Stith CM, Stümpfig M, et al. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat Chem Biol. 2012;8:125–32. doi: 10.1038/nchembio.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netz DJA, Stümpfig M, Doré C, et al. Tah18 transfers electrons to Dre2 in cytosolic iron-sulfur protein biogenesis. Nat Chem Biol. 2010;6:758–65. doi: 10.1038/nchembio.432. [DOI] [PubMed] [Google Scholar]
- Newman SM, Zelenaya-Troitskaya O, Perlman PS, et al. Analysis of mitochondrial DNA nucleoids in wild-type and a mutant strain of Saccharomyces cerevisiae that lacks the mitochondrial HMG box protein Abf2p. Nucleic Acids Res. 1996;24:386–9. doi: 10.1093/nar/24.2.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo GHP, Balakrishnan L, Dubarry M, et al. The 9–1–1 checkpoint clamp stimulates DNA resection by Dna2-Sgs1 and Exo1. Nucleic Acids Res. 2014;42:10516–28. doi: 10.1093/nar/gku746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimi A, Limsirichaikul S, Yoshida S, et al. Palm mutants in DNA polymerases alpha and eta alter DNA replication fidelity and translesion activity. Mol Cell Biol. 2004;24:2734–46. doi: 10.1128/MCB.24.7.2734-2746.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norppa H, Falck GC-M. What do human micronuclei contain? Mutagenesis. 2003;18:221–33. doi: 10.1093/mutage/18.3.221. [DOI] [PubMed] [Google Scholar]
- Northam MR, Garg P, Baitin DM, et al. A novel function of DNA polymerase zeta regulated by PCNA. EMBO J. 2006;25:4316–25. doi: 10.1038/sj.emboj.7601320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northam MR, Robinson HA, Kochenova OV, et al. Participation of DNA polymerase in replication of undamaged DNA in Saccharomyces cerevisiae. Genetics. 2010;184:27–42. doi: 10.1534/genetics.109.107482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill BM, Szyjka SJ, Lis ET, et al. Pph3-Psy2 is a phosphatase complex required for Rad53 dephosphorylation and replication fork restart during recovery from DNA damage. P Natl Acad Sci USA. 2007;104:9290–5. doi: 10.1073/pnas.0703252104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke TW, Doudican NA, Mackereth MD, et al. Mitochondrial dysfunction due to oxidative mitochondrial DNA damage is reduced through cooperative actions of diverse proteins. Mol Cell Biol. 2002;22:4086–93. doi: 10.1128/MCB.22.12.4086-4093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke TW, Doudican NA, Zhang H, et al. Differential involvement of the related DNA helicases Pif1p and Rrmp in mtDNA point mutagenesis and stability. Gene. 2005;354:86–92. doi: 10.1016/j.gene.2005.03.031. [DOI] [PubMed] [Google Scholar]
- Oda K, Kawasaki N, Fukuyama M, et al. Ectopic expression of mitochondria endonuclease Pnu1p from Schizosaccharomyces pombe induces cell death of the yeast. J Biochem Mol Biol. 2007;40:1095–9. doi: 10.5483/bmbrep.2007.40.6.1095. [DOI] [PubMed] [Google Scholar]
- Ogiwara H, Ui A, Enomoto T, et al. Role of Elg1 protein in double strand break repair. Nucleic Acids Res. 2007;35:353–62. doi: 10.1093/nar/gkl1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi G, Endo K, Doi A, et al. Spontaneous mutagenesis in haploid and diploid Saccharomyces cerevisiae. Biochem Bioph Res Co. 2004;325:928–33. doi: 10.1016/j.bbrc.2004.10.120. [DOI] [PubMed] [Google Scholar]
- Oram M, Keeley A, Tsaneva I. Holliday junction resolvase in Schizosaccharomyces pombe has identical endonuclease activity to the CCE1 homologue YDC2. Nucleic Acids Res. 1998;26:594–601. doi: 10.1093/nar/26.2.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn AJ, Elledge SJ. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Gene Dev. 2003;17:1755–67. doi: 10.1101/gad.1098303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouspenski II, Elledge SJ, Brinkley BR. New yeast genes important for chromosome integrity and segregation identified by dosage effects on genome stability. Nucleic Acids Res. 1999;27:3001–8. doi: 10.1093/nar/27.15.3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papouli E, Chen S, Davies AA, et al. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol Cell. 2005;19:123–33. doi: 10.1016/j.molcel.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Park C, Qian W, Zhang J. Genomic evidence for elevated mutation rates in highly expressed genes. EMBO Rep. 2012;13:1123–9. doi: 10.1038/embor.2012.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JL, Bielen AB, Dikic I, et al. Contributions of ubiquitin- and PCNA-binding domains to the activity of Polymerase eta in Saccharomyces cerevisiae. Nucleic Acids Res. 2007;35:881–9. doi: 10.1093/nar/gkl1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnas O, Zipin-Roitman A, Mazor Y, et al. The ELG1 clamp loader plays a role in sister chromatid cohesion. PloS One. 2009;4:e5497. doi: 10.1371/journal.pone.0005497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnas O, Zipin-Roitman A, Pfander B, et al. Elg1, an alternative subunit of the RFC clamp loader, preferentially interacts with SUMOylated PCNA. EMBO J. 2010;29:2611–22. doi: 10.1038/emboj.2010.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelka N, Rancati G, Zhu J, et al. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature. 2010;468:321–5. doi: 10.1038/nature09529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov YI, Maki S, Maki H, et al. Evidence for interplay among yeast replicative DNA polymerases alpha, delta and epsilon from studies of exonuclease and polymerase active site mutations. BMC Biol. 2004;2:11. doi: 10.1186/1741-7007-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov YI, Nguyen D, Kunkel TA. Mutator effects of overproducing DNA polymerase eta (Rad30) and its catalytically inactive variant in yeast. Mutat Res. 2001;478:129–39. doi: 10.1016/s0027-5107(01)00131-2. [DOI] [PubMed] [Google Scholar]
- Pérez-Ortín JE, Alepuz PM, Moreno J. Genomics and gene transcription kinetics in yeast. Trends Genet. 2007;23:250–7. doi: 10.1016/j.tig.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Bio. 2006;7:644–56. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- Pfander B, Moldovan G-L, Sacher M, et al. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature. 2005;436:428–33. doi: 10.1038/nature03665. [DOI] [PubMed] [Google Scholar]
- Phadnis N, Sia RA, Sia EA. Analysis of repeat-mediated deletions in the mitochondrial genome of Saccharomyces cerevisiae. Genetics. 2005;171:1549–59. doi: 10.1534/genetics.105.047092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinter SF, Aubert SD, Zakian VA. The Schizosaccharomyces pombe Pfh1p DNA helicase is essential for the maintenance of nuclear and mitochondrial DNA. Mol Cell Biol. 2008;28:6594–608. doi: 10.1128/MCB.00191-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piruat JI, Aguilera A. A novel yeast gene, THO2, is involved in RNA pol II transcription and provides new evidence for transcriptional elongation-associated recombination. EMBO J. 1998;17:4859–72. doi: 10.1093/emboj/17.16.4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlaska A, McIntyre J, Skoneczna A, et al. The link between 20S proteasome activity and post-replication DNA repair in Saccharomyces cerevisiae. Mol Microbiol. 2003;49:1321–32. doi: 10.1046/j.1365-2958.2003.03635.x. [DOI] [PubMed] [Google Scholar]
- Pogorzala L, Mookerjee S, Sia EA. Evidence that msh1p plays multiple roles in mitochondrial base excision repair. Genetics. 2009;182:699–709. doi: 10.1534/genetics.109.103796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokharel S, Campbell JL. Cross talk between the nuclease and helicase activities of Dna2: role of an essential iron-sulfur cluster domain. Nucleic Acids Res. 2012;40:7821–30. doi: 10.1093/nar/gks534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli J, Tsaponina O, Crabbé L, et al. dNTP pools determine fork progression and origin usage under replication stress. EMBO J. 2012;31:883–94. doi: 10.1038/emboj.2011.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow L, Ullsperger C, Keller RW, et al. Positive torsional strain causes the formation of a four-way junction at replication forks. J Biol Chem. 2001;276:2790–6. doi: 10.1074/jbc.M006736200. [DOI] [PubMed] [Google Scholar]
- Prado F, Aguilera A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 2005;24:1267–76. doi: 10.1038/sj.emboj.7600602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R, Satory D, Dray E, et al. Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Gene Dev. 2009;23:67–79. doi: 10.1101/gad.1737809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem. 2005;74:317–53. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- Pratt-Hyatt MJ, Kapadia KM, Wilson TE, et al. Increased recombination between active tRNA genes. DNA Cell Biol. 2006;25:359–64. doi: 10.1089/dna.2006.25.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puddu F, Piergiovanni G, Plevani P, et al. Sensing of replication stress and Mec1 activation act through two independent pathways involving the 9–1–1 complex and DNA polymerase ε. PLoS Genet. 2011;7:e1002022. doi: 10.1371/journal.pgen.1002022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Hayes TK, Kolodner RD. Post-replication repair suppresses duplication-mediated genome instability. PLoS Genet. 2010;6:e1000933. doi: 10.1371/journal.pgen.1000933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajpal DK, Wu X, Wang Z. Alteration of ultraviolet-induced mutagenesis in yeast through molecular modulation of the REV3 and REV7 gene expression. Mutat Res. 2000;461:133–43. doi: 10.1016/s0921-8777(00)00047-1. [DOI] [PubMed] [Google Scholar]
- Ramanagoudr-Bhojappa R, Blair LP, Tackett AJ, et al. Physical and functional interaction between yeast Pif1 helicase and Rim1 single-stranded DNA binding protein. Nucleic Acids Res. 2013;41:1029–46. doi: 10.1093/nar/gks1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjha L, Anand R, Cejka P. The Saccharomyces cerevisiae Mlh1-Mlh3 heterodimer is an endonuclease that preferentially binds to Holliday junctions. J Biol Chem. 2014;289:5674–86. doi: 10.1074/jbc.M113.533810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen AK, Chatterjee A, Rasmussen LJ, et al. Mitochondria-mediated nuclear mutator phenotype in Saccharomyces cerevisiae. Nucleic Acids Res. 2003;31:3909–17. doi: 10.1093/nar/gkg446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattray AJ, Shafer BK, McGill CB, et al. The roles of REV3 and RAD57 in double-strand-break-repair-induced mutagenesis of Saccharomyces cerevisiae. Genetics. 2002;162:1063–77. doi: 10.1093/genetics/162.3.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynard S, Bussen W, Sung P. A double Holliday junction dissolvasome comprising BLM, topoisomerase IIIalpha, and BLAP75. J Biol Chem. 2006;281:13861–4. doi: 10.1074/jbc.C600051200. [DOI] [PubMed] [Google Scholar]
- Razidlo DF, Lahue RS. Mrc1, Tof1 and Csm3 inhibit CAG.CTG repeat instability by at least two mechanisms. DNA Repair. 2008;7:633–40. doi: 10.1016/j.dnarep.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reenan RA, Kolodner RD. Isolation and characterization of two Saccharomyces cerevisiae genes encoding homologs of the bacterial HexA and MutS mismatch repair proteins. Genetics. 1992;132:963–73. doi: 10.1093/genetics/132.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reha-Krantz LJ, Siddique MS, Murphy K, et al. Drug-sensitive DNA polymerase δ reveals a role for mismatch repair in checkpoint activation in yeast. Genetics. 2011;189:1211–24. doi: 10.1534/genetics.111.131938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, Chen Z, Yassour M, et al. Comparative functional genomics of the fission yeasts. Science. 2011;332:930–6. doi: 10.1126/science.1203357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert T, Vanoli F, Chiolo I, et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471:74–9. doi: 10.1038/nature09803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RW, Crothers DM. Stability and properties of double and triple helices: dramatic effects of RNA or DNA backbone composition. Science. 1992;258:1463–6. doi: 10.1126/science.1279808. [DOI] [PubMed] [Google Scholar]
- Rogacheva MV, Manhart CM, Chen C, et al. Mlh1-Mlh3, a meiotic crossover and DNA mismatch repair factor, is a Msh2-Msh3-stimulated endonuclease. J Biol Chem. 2014;289:5664–73. doi: 10.1074/jbc.M113.534644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanova NV, Crouse GF. Different roles of eukaryotic MutS and MutL complexes in repair of small insertion and deletion loops in yeast. PLoS Genet. 2013;9:e1003920. doi: 10.1371/journal.pgen.1003920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondón AG, Jimeno S, Aguilera A. The interface between transcription and mRNP export: from THO to THSC/TREX-2. Biochim Biophys Acta. 2010;1799:533–8. doi: 10.1016/j.bbagrm.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Rong L, Klein HL. Purification and characterization of the SRS2 DNA helicase of the yeast Saccharomyces cerevisiae. J Biol Chem. 1993;268:1252–9. [PubMed] [Google Scholar]
- Rossi B, Noel P, Bruschi CV. Different aneuploidies arise from the same bridge-induced chromosomal translocation event in Saccharomyces cerevisiae. Genetics. 2010;186:775–90. doi: 10.1534/genetics.110.120683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolf J, Makrantoni V, Ingledew WJ, et al. The DNA repair helicases XPD and FancJ have essential iron-sulfur domains. Mol Cell. 2006;23:801–8. doi: 10.1016/j.molcel.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Sabouri N, Viberg J, Goyal DK, et al. Evidence for lesion bypass by yeast replicative DNA polymerases during DNA damage. Nucleic Acids Res. 2008;36:5660–7. doi: 10.1093/nar/gkn555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini N, Ramakrishnan S, Elango R, et al. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature. 2013;502:389–92. doi: 10.1038/nature12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakofsky CJ, Roberts SA, Malc E, et al. Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep. 2014;7:1640–8. doi: 10.1016/j.celrep.2014.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salguero I, Guarino E, Shepherd MEA, et al. Ribonucleotide reductase activity is coupled to DNA synthesis via proliferating cell nuclear antigen. Curr Biol. 2012;22:720–6. doi: 10.1016/j.cub.2012.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samsonova IA, Kunze G, Bode R, et al. A set of genetic markers for the chromosomes of the imperfect yeast Arxula adeninivorans. Yeast. 1996;12:1209–17. doi: 10.1002/(sici)1097-0061(19960930)12:12<1209::aid-yea12>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–57. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- Sanvisens N, de Llanos R, Puig S. Function and regulation of yeast ribonucleotide reductase: cell cycle, genotoxic stress, and iron bioavailability. Biomed J. 2013;36:51–8. doi: 10.4103/2319-4170.110398. [DOI] [PubMed] [Google Scholar]
- Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283:1488–93. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- Sarbajna S, West SC. Holliday junction processing enzymes as guardians of genome stability. Trends Biochem Sci. 2014;39:409–19. doi: 10.1016/j.tibs.2014.07.003. [DOI] [PubMed] [Google Scholar]
- Schäfer B. Genetic conservation versus variability in mitochondria: the architecture of the mitochondrial genome in the petite-negative yeast Schizosaccharomyces pombe. Curr Genet. 2003;43:311–26. doi: 10.1007/s00294-003-0404-5. [DOI] [PubMed] [Google Scholar]
- Scherer S. A Short Guide to the Human Genome. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2008. [Google Scholar]
- Schofield MJ, Hsieh P. DNA mismatch repair: molecular mechanisms and biological function. Annu Rev Microbiol. 2003;57:579–608. doi: 10.1146/annurev.micro.57.030502.090847. [DOI] [PubMed] [Google Scholar]
- Schulz VP, Zakian VA. The Saccharomyces PIF1 DNA helicase inhibits telomere elongation and de novo telomere formation. Cell. 1994;76:145–55. doi: 10.1016/0092-8674(94)90179-1. [DOI] [PubMed] [Google Scholar]
- Schweitzer JK, Livingston DM. Expansions of CAG repeat tracts are frequent in a yeast mutant defective in Okazaki fragment maturation. Hum Mol Genet. 1998;7:69–74. doi: 10.1093/hmg/7.1.69. [DOI] [PubMed] [Google Scholar]
- Segal M, Clarke DJ. The Ras pathway and spindle assembly collide? BioEssays. 2001;23:307–10. doi: 10.1002/bies.1044. [DOI] [PubMed] [Google Scholar]
- Seitz-Mayr G, Wolf K. Extrachromosomal mutator inducing point mutations and deletions in mitochondrial genome of fission yeast. P Natl Acad Sci USA. 1982;79:2618–22. doi: 10.1073/pnas.79.8.2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova PV, Pavlov YI, Chilkova O, et al. Unique error signature of the four-subunit yeast DNA polymerase epsilon. J Biol Chem. 2003;278:43770–80. doi: 10.1074/jbc.M306893200. [DOI] [PubMed] [Google Scholar]
- Shell SS, Putnam CD, Kolodner RD. The N terminus of Saccharomyces cerevisiae Msh6 is an unstructured tether to PCNA. Mol Cell. 2007;26:565–78. doi: 10.1016/j.molcel.2007.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Blank HM, Pfau SJ, et al. Aneuploidy drives genomic instability in yeast. Science. 2011;333:1026–30. doi: 10.1126/science.1206412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiga T, Suzuki H, Yamamoto A, et al. Hydroquinone, a benzene metabolite, induces Hog1-dependent stress response signaling and causes aneuploidy in Saccharomyces cerevisiae. J Radiat Res. 2010;51:405–15. doi: 10.1269/jrr.10014. [DOI] [PubMed] [Google Scholar]
- Shinohara M, Sakai K, Ogawa T, et al. The mitotic DNA damage checkpoint proteins Rad17 and Rad24 are required for repair of double-strand breaks during meiosis in yeast. Genetics. 2003;164:855–65. doi: 10.1093/genetics/164.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shor E, Fox CA, Broach JR. The yeast environmental stress response regulates mutagenesis induced by proteotoxic stress. PLoS Genet. 2013;9:e1003680. doi: 10.1371/journal.pgen.1003680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia EA, Jinks-Robertson S, Petes TD. Genetic control of microsatellite stability. Mutat Res Repair. 1997;383:61–70. doi: 10.1016/s0921-8777(96)00046-8. [DOI] [PubMed] [Google Scholar]
- Sia RA, Carrol S, Kalifa L, et al. Loss of the mitochondrial nucleoid protein, Abf2p, destabilizes repetitive DNA in the yeast mitochondrial genome. Genetics. 2008;181:331–4. doi: 10.1534/genetics.108.095786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebler HM, Lada AG, Baranovskiy AG, et al. A novel variant of DNA polymerase ζ, Rev3ΔC, highlights differential regulation of Pol32 as a subunit of polymerase δ versus ζ in Saccharomyces cerevisiae. DNA Repair. 2014;24:138–49. doi: 10.1016/j.dnarep.2014.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva P, Barbosa J, Nascimento AV, et al. Monitoring the fidelity of mitotic chromosome segregation by the spindle assembly checkpoint. Cell Prolif. 2011;44:391–400. doi: 10.1111/j.1365-2184.2011.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh KK, Sigala B, Sikder HA, et al. Inactivation of Saccharomyces cerevisiae OGG1 DNA repair gene leads to an increased frequency of mitochondrial mutants. Nucleic Acids Res. 2001;29:1381–8. doi: 10.1093/nar/29.6.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoneczna A, McIntyre J, Skoneczny M, et al. Polymerase eta is a short-lived, proteasomally degraded protein that is temporarily stabilized following UV irradiation in Saccharomyces cerevisiae. J Mol Biol. 2007;366:1074–86. doi: 10.1016/j.jmb.2006.11.093. [DOI] [PubMed] [Google Scholar]
- Smith CE, Mendillo ML, Bowen N, et al. Dominant mutations in S. cerevisiae PMS1 identify the Mlh1-Pms1 endonuclease active site and an exonuclease 1-independent mismatch repair pathway. PLoS Genet. 2013;9:e1003869. doi: 10.1371/journal.pgen.1003869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollier J, Driscoll R, Castellucci F, et al. The Saccharomyces cerevisiae Esc2 and Smc5–6 proteins promote sister chromatid junction-mediated intra-S repair. Mol Biol Cell. 2009;20:1671–82. doi: 10.1091/mbc.E08-08-0875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Petes TD. Haploidization in Saccharomyces cerevisiae induced by a deficiency in homologous recombination. Genetics. 2012;191:279–84. doi: 10.1534/genetics.111.138180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sor F, Fukuhara H. Identification of two erythromycin resistance mutations in the mitochondrial gene coding for the large ribosomal RNA in yeast. Nucleic Acids Res. 1982;10:6571–7. doi: 10.1093/nar/10.21.6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spampinato CP, Gomez RL, Galles C, et al. From bacteria to plants: a compendium of mismatch repair assays. Mutat Res. 2009;682:110–28. doi: 10.1016/j.mrrev.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Sparks JL, Chon H, Cerritelli SM, et al. RNase H2-initiated ribonucleotide excision repair. Mol Cell. 2012;47:980–6. doi: 10.1016/j.molcel.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer F, Gerring SL, Connelly C, et al. Mitotic chromosome transmission fidelity mutants in Saccharomyces cerevisiae. Genetics. 1990;124:237–49. doi: 10.1093/genetics/124.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl U, Esser K. Genetic Exchange Processes in Lower Eukaryotes. In: Rehm H-J, Reed G, editors. Biotechnology. Vol. 2. Weinheim, Germany: Wiley-VCH Verlag GmbH; 1992. pp. 73–92. [Google Scholar]
- Stehling O, Vashisht AA, Mascarenhas J, et al. MMS19 assembles iron-sulfur proteins required for DNA metabolism and genomic integrity. Science. 2012;337:195–9. doi: 10.1126/science.1219723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelter P, Ulrich HD. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 2003;425:188–91. doi: 10.1038/nature01965. [DOI] [PubMed] [Google Scholar]
- Stemmann O, Neidig A, Köcher T, et al. Hsp90 enables Ctf13p/Skp1p to nucleate the budding yeast kinetochore. P Natl Acad Sci USA. 2002;99:8585–90. doi: 10.1073/pnas.082223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling PC, Chan YA, Minaker SW, et al. R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Gene Dev. 2012;26:163–75. doi: 10.1101/gad.179721.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling PC, Crisp MJ, Basrai MA, et al. Mutability and mutational spectrum of chromosome transmission fidelity genes. Chromosoma. 2012;121:263–75. doi: 10.1007/s00412-011-0356-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JE, Kissling GE, Lujan SA, et al. Low-fidelity DNA synthesis by the L979F mutator derivative of Saccharomyces cerevisiae DNA polymerase. Nucleic Acids Res. 2009;37:3774–87. doi: 10.1093/nar/gkp238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JE, Lujan SA, Kunkel TA. DNA polymerase zeta generates clustered mutations during bypass of endogenous DNA lesions in Saccharomyces cerevisiae. Environ Mol Mutagen. 2012;53:777–86. doi: 10.1002/em.21728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JE, Ozbirn RG, Petes TD, et al. Role of proliferating cell nuclear antigen interactions in the mismatch repair-dependent processing of mitotic and meiotic recombination intermediates in yeast. Genetics. 2008;178:1221–36. doi: 10.1534/genetics.107.085415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storchova Z, Becker JS, Talarek N, et al. Bub1, Sgo1, and Mps1 mediate a distinct pathway for chromosome biorientation in budding yeast. Mol Biol Cell. 2011;22:1473–85. doi: 10.1091/mbc.E10-08-0673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf JD, Copeland WC. The exonuclease activity of the yeast mitochondrial DNA polymerase γ suppresses mitochondrial DNA deletions between short direct repeats in Saccharomyces cerevisiae. Genetics. 2013;194:519–22. doi: 10.1534/genetics.113.150920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf JD, Copeland WC. MMS exposure promotes increased mtDNA mutagenesis in the presence of replication-defective disease-associated DNA polymerase γ variants. PLoS Genet. 2014;10:e1004748. doi: 10.1371/journal.pgen.1004748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Ira G, Haber JE. DNA length dependence of the single-strand annealing pathway and the role of Saccharomyces cerevisiae RAD59 in double-strand break repair. Mol Cell Biol. 2000;20:5300–9. doi: 10.1128/mcb.20.14.5300-5309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suijkerbuijk SJE, Kops GJPL. Preventing aneuploidy: the contribution of mitotic checkpoint proteins. Biochim Biophys Acta. 2008;1786:24–31. doi: 10.1016/j.bbcan.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Sung P. Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein A to promote DNA strand exchange by Rad51 recombinase. Gene Dev. 1997;11:1111–21. doi: 10.1101/gad.11.9.1111. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Kanbe T, Kuroiwa T, et al. Occurrence of ploidy shift in a strain of the imperfect yeast Candida albicans. J Gen Microbiol. 1986;132:443–53. doi: 10.1099/00221287-132-2-443. [DOI] [PubMed] [Google Scholar]
- Svilar D, Goellner EM, Almeida KH, et al. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid Redox Sign. 2011;14:2491–507. doi: 10.1089/ars.2010.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–71. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- Symington LS, Rothstein R, Lisby M. Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics. 2014;198:795–835. doi: 10.1534/genetics.114.166140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepanowska K, Foury F. A cluster of pathogenic mutations in the 3′-5′ exonuclease domain of DNA polymerase gamma defines a novel module coupling DNA synthesis and degradation. Hum Mol Genet. 2010;19:3516–29. doi: 10.1093/hmg/ddq267. [DOI] [PubMed] [Google Scholar]
- Tann AW, Boldogh I, Meiss G, et al. Apoptosis induced by persistent single-strand breaks in mitochondrial genome: critical role of EXOG (5′-EXO/endonuclease) in their repair. J Biol Chem. 2011;286:31975–83. doi: 10.1074/jbc.M110.215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia-Alveal C, Lin S-J, Yeoh A, et al. H2A.Z-dependent regulation of cohesin dynamics on chromosome arms. Mol Cell Biol. 2014;34:2092–104. doi: 10.1128/MCB.00193-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SD, Zhang H, Eaton JS, et al. The conserved Mec1/Rad53 nuclear checkpoint pathway regulates mitochondrial DNA copy number in Saccharomyces cerevisiae. Mol Biol Cell. 2005;16:3010–8. doi: 10.1091/mbc.E05-01-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennyson CN, Klamut HJ, Worton RG. The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nat Genet. 1995;9:184–90. doi: 10.1038/ng0295-184. [DOI] [PubMed] [Google Scholar]
- Thompson JA, Marzahn MR, O'Donnell M, et al. Replication factor C is a more effective proliferating cell nuclear antigen (PCNA) opener than the checkpoint clamp loader, Rad24-RFC. J Biol Chem. 2012;287:2203–9. doi: 10.1074/jbc.C111.318899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol. 2010;20:R285–95. doi: 10.1016/j.cub.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff DX, Filosi N, Gaida GM, et al. A novel mutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinct from DNA mismatch repair. Cell. 1997;88:253–63. doi: 10.1016/s0092-8674(00)81846-2. [DOI] [PubMed] [Google Scholar]
- Tourrette Y, Schacherer J, Fritsch E, et al. Spontaneous deletions and reciprocal translocations in Saccharomyces cerevisiae: influence of ploidy. Mol Microbiol. 2007;64:382–95. doi: 10.1111/j.1365-2958.2007.05660.x. [DOI] [PubMed] [Google Scholar]
- Tran PT, Erdeniz N, Dudley S, et al. Characterization of nuclease-dependent functions of Exo1p in Saccharomyces cerevisiae. DNA Repair. 2002;1:895–912. doi: 10.1016/s1568-7864(02)00114-3. [DOI] [PubMed] [Google Scholar]
- Travesa A, Kuo D, de Bruin RAM, et al. DNA replication stress differentially regulates G1/S genes via Rad53-dependent inactivation of Nrm1. EMBO J. 2012;31:1811–22. doi: 10.1038/emboj.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsaponina O, Barsoum E, Aström SU, et al. Ixr1 is required for the expression of the ribonucleotide reductase Rnr1 and maintenance of dNTP pools. PLoS Genet. 2011;7:e1002061. doi: 10.1371/journal.pgen.1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbale P, Williams JS, Schellenberg MJ, et al. Aprataxin resolves adenylated RNA-DNA junctions to maintain genome integrity. Nature. 2014;506:111–5. doi: 10.1038/nature12824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich HD, Jentsch S. Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair. EMBO J. 2000;19:3388–97. doi: 10.1093/emboj/19.13.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unal E, Heidinger-Pauli JM, Koshland D. DNA double-strand breaks trigger genome-wide sister-chromatid cohesion through Eco1 (Ctf7) Science. 2007;317:245–8. doi: 10.1126/science.1140637. [DOI] [PubMed] [Google Scholar]
- van Gool AJ, Verhage R, Swagemakers SM, et al. RAD26, the functional S. cerevisiae homolog of the Cockayne syndrome B gene ERCC6. EMBO J. 1994;13:5361–9. doi: 10.1002/j.1460-2075.1994.tb06871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderstraeten S, Van den Brûle S, Hu J, et al. The role of 3′-5′ exonucleolytic proofreading and mismatch repair in yeast mitochondrial DNA error avoidance. J Biol Chem. 1998;273:23690–7. doi: 10.1074/jbc.273.37.23690. [DOI] [PubMed] [Google Scholar]
- Vanoli F, Fumasoni M, Szakal B, et al. Replication and recombination factors contributing to recombination-dependent bypass of DNA lesions by template switch. PLoS Genet. 2010;6:e1001205. doi: 10.1371/journal.pgen.1001205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch JR, McMurray MA, Nelson ZW, et al. Mitochondrial dysfunction leads to nuclear genome instability via an iron–sulfur cluster defect. Cell. 2009;137:1247–58. doi: 10.1016/j.cell.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veaute X, Jeusset J, Soustelle C, et al. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature. 2003;423:309–12. doi: 10.1038/nature01585. [DOI] [PubMed] [Google Scholar]
- Verma R, Patapoutian A, Gordon CB, et al. Identification and purification of a factor that binds to the Mlu I cell cycle box of yeast DNA replication genes. P Natl Acad Sci USA. 1991;88:7155–9. doi: 10.1073/pnas.88.16.7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma SK, Ranjan R, Kumar V, et al. Wat1/pop3, a conserved WD repeat containing protein acts synergistically with checkpoint kinase Chk1 to maintain genome ploidy in fission yeast S. pombe. PloS One. 2014;9:e89587. doi: 10.1371/journal.pone.0089587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viikov K, Väljamäe P, Sedman J. Yeast mitochondrial DNA polymerase is a highly processive single-subunit enzyme. Mitochondrion. 2011;11:119–26. doi: 10.1016/j.mito.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Vitale I, Galluzzi L, Castedo M, et al. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Bio. 2011;12:385–92. doi: 10.1038/nrm3115. [DOI] [PubMed] [Google Scholar]
- Vongsamphanh R, Fortier PK, Ramotar D. Pir1p mediates translocation of the yeast Apn1p endonuclease into the mitochondria to maintain genomic stability. Mol Cell Biol. 2001;21:1647–55. doi: 10.1128/MCB.21.5.1647-1655.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Bio. 2002;3:430–40. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- Wang X, Ira G, Tercero JA, et al. Role of DNA replication proteins in double-strand break-induced recombination in Saccharomyces cerevisiae. Mol Cell Biol. 2004;24:6891–9. doi: 10.1128/MCB.24.16.6891-6899.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward TA, Dudášová Z, Sarkar S, et al. Components of a Fanconi-like pathway control Pso2-independent DNA interstrand crosslink repair in yeast. PLoS Genet. 2012;8:e1002884. doi: 10.1371/journal.pgen.1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren CD, Brady DM, Johnston RC, et al. Distinct chromosome segregation roles for spindle checkpoint proteins. Mol Biol Cell. 2002;13:3029–41. doi: 10.1091/mbc.E02-04-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington MT, Johnson RE, Prakash S, et al. Accuracy of thymine-thymine dimer bypass by Saccharomyces cerevisiae DNA polymerase eta. P Natl Acad Sci USA. 2000;97:3094–9. doi: 10.1073/pnas.050491997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters LS, Minesinger BK, Wiltrout ME, et al. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol R. 2009;73:134–54. doi: 10.1128/MMBR.00034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters LS, Walker GC. The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G(2)/M phase rather than S phase. P Natl Acad Sci USA. 2006;103:8971–6. doi: 10.1073/pnas.0510167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt DL, Johansson E, Burgers PM, et al. Replication of ribonucleotide-containing DNA templates by yeast replicative polymerases. DNA Repair. 2011;10:897–902. doi: 10.1016/j.dnarep.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts FZ. Sumoylation of PCNA: wrestling with recombination at stalled replication forks. DNA Repair. 2006;5:399–403. doi: 10.1016/j.dnarep.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Wei X, Samarabandu J, Devdhar RS, et al. Segregation of transcription and replication sites into higher order domains. Science. 1998;281:1502–6. doi: 10.1126/science.281.5382.1502. [DOI] [PubMed] [Google Scholar]
- Westmoreland TJ, Wickramasekara SM, Guo AY, et al. Comparative genome-wide screening identifies a conserved doxorubicin repair network that is diploid specific in Saccharomyces cerevisiae. PloS One. 2009;4:e5830. doi: 10.1371/journal.pone.0005830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MF, Dillingham MS. Iron-sulphur clusters in nucleic acid processing enzymes. Curr Opin Struct Biol. 2012;22:94–100. doi: 10.1016/j.sbi.2011.11.004. [DOI] [PubMed] [Google Scholar]
- Wilson MA, Kwon Y, Xu Y, et al. Pif1 helicase and Polδ promote recombination-coupled DNA synthesis via bubble migration. Nature. 2013;502:393–6. doi: 10.1038/nature12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltrout ME, Walker GC. Proteasomal regulation of the mutagenic translesion DNA polymerase, Saccharomyces cerevisiae Rev1. DNA Repair. 2011;10:169–75. doi: 10.1016/j.dnarep.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windecker H, Ulrich HD. Architecture and assembly of poly-SUMO chains on PCNA in Saccharomyces cerevisiae. J Mol Biol. 2008;376:221–31. doi: 10.1016/j.jmb.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Wright BE. Stress-directed adaptive mutations and evolution. Mol Microbiol. 2004;52:643–50. doi: 10.1111/j.1365-2958.2004.04012.x. [DOI] [PubMed] [Google Scholar]
- Wright BE, Reschke DK, Schmidt KH, et al. Predicting mutation frequencies in stem-loop structures of derepressed genes: implications for evolution. Mol Microbiol. 2003;48:429–41. doi: 10.1046/j.1365-2958.2003.t01-1-03436.x. [DOI] [PubMed] [Google Scholar]
- Wu L, Hickson ID. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–4. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- Wu X, Huang M. Dif1 controls subcellular localization of ribonucleotide reductase by mediating nuclear import of the R2 subunit. Mol Cell Biol. 2008;28:7156–67. doi: 10.1128/MCB.01388-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Wang Z. Relationships between yeast Rad27 and Apn1 in response to apurinic/apyrimidinic (AP) sites in DNA. Nucleic Acids Res. 1999;27:956–62. doi: 10.1093/nar/27.4.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Wilson TE, Lieber MR. A role for FEN-1 in nonhomologous DNA end joining: the order of strand annealing and nucleolytic processing events. P Natl Acad Sci USA. 1999;96:1303–8. doi: 10.1073/pnas.96.4.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xaver M, Huang L, Chen D, et al. Smc5/6-Mms21 prevents and eliminates inappropriate recombination intermediates in meiosis. PLoS Genet. 2013;9:e1004067. doi: 10.1371/journal.pgen.1004067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Sun Z, Liu Z, et al. Replication stress induces micronuclei comprising of aggregated DNA double-strand breaks. PloS One. 2011;6:e18618. doi: 10.1371/journal.pone.0018618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Sterling J, Storici F, et al. Hypermutability of damaged single-strand DNA formed at double-strand breaks and uncapped telomeres in yeast Saccharomyces cerevisiae. PLoS Genet. 2008;4:e1000264. doi: 10.1371/journal.pgen.1000264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao NY, Johnson A, Bowman GD, et al. Mechanism of proliferating cell nuclear antigen clamp opening by replication factor C. J Biol Chem. 2006;281:17528–39. doi: 10.1074/jbc.M601273200. [DOI] [PubMed] [Google Scholar]
- Yao R, Zhang Z, An X, et al. Subcellular localization of yeast ribonucleotide reductase regulated by the DNA replication and damage checkpoint pathways. P Natl Acad Sci USA. 2003;100:6628–33. doi: 10.1073/pnas.1131932100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazgan O, Krebs JE. Mitochondrial and nuclear genomic integrity after oxidative damage in Saccharomyces cerevisiae. Front Biosci. 2012;17:1079–93. doi: 10.2741/3974. [DOI] [PubMed] [Google Scholar]
- Yeung M, Durocher D. Srs2 enables checkpoint recovery by promoting disassembly of DNA damage foci from chromatin. DNA Repair. 2011;10:1213–22. doi: 10.1016/j.dnarep.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Yona AH, Manor YS, Herbst RH, et al. Chromosomal duplication is a transient evolutionary solution to stress. P Natl Acad Sci USA. 2012;109:21010–5. doi: 10.1073/pnas.1211150109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Srinivasan M, Nakanishi S, et al. A conserved patch near the C terminus of histone H4 is required for genome stability in budding yeast. Mol Cell Biol. 2011;31:2311–25. doi: 10.1128/MCB.01432-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen KWY, Warren CD, Chen O, et al. Systematic genome instability screens in yeast and their potential relevance to cancer. P Natl Acad Sci USA. 2007;104:3925–30. doi: 10.1073/pnas.0610642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacchi LF, Selmecki AM, Berman J, et al. Low dosage of histone H4 leads to growth defects and morphological changes in Candida albicans. PloS One. 2010;5:e10629. doi: 10.1371/journal.pone.0010629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zassenhaus HP, Denniger G. Analysis of the role of the NUC1 endo/exonuclease in yeast mitochondrial DNA recombination. Curr Genet. 1994;25:142–9. doi: 10.1007/BF00309540. [DOI] [PubMed] [Google Scholar]
- Zelenaya-Troitskaya O, Perlman PS, Butow RA. An enzyme in yeast mitochondria that catalyzes a step in branched-chain amino acid biosynthesis also functions in mitochondrial DNA stability. EMBO J. 1995;14:3268. doi: 10.1002/j.1460-2075.1995.tb07330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelesco PA, Graves JA. Hybrids between irradiated and unirradiated mammalian cells: survival and chromosome segregation. J Cell Physiol. 1983;116:98–102. doi: 10.1002/jcp.1041160115. [DOI] [PubMed] [Google Scholar]
- Zhang C. Essential functions of iron-requiring proteins in DNA replication, repair and cell cycle control. Protein Cell. 2014;5:750–60. doi: 10.1007/s13238-014-0083-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Liu G, Huang M. Ribonucleotide reductase metallocofactor: assembly, maintenance and inhibition. Front Biol. 2014;9:104–13. doi: 10.1007/s11515-014-1302-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Chatterjee A, Singh KK. Saccharomyces cerevisiae polymerase zeta functions in mitochondria. Genetics. 2005;172:2683–8. doi: 10.1534/genetics.105.051029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Qin Z, Zhang X, et al. Roles of sequential ubiquitination of PCNA in DNA-damage tolerance. FEBS Lett. 2011;585:2786–94. doi: 10.1016/j.febslet.2011.04.044. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Hefferin ML, Chen L, et al. Role of Dnl4–Lif1 in nonhomologous end-joining repair complex assembly and suppression of homologous recombination. Nat Struct Mol Biol. 2007;14:639–46. doi: 10.1038/nsmb1261. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lyver ER, Nakamaru-Ogiso E, et al. Dre2, a conserved eukaryotic Fe/S cluster protein, functions in cytosolic Fe/S protein biogenesis. Mol Cell Biol. 2008;28:5569–82. doi: 10.1128/MCB.00642-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Shim EY, Davis M, et al. Regulation of repair choice: Cdk1 suppresses recruitment of end joining factors at DNA breaks. DNA Repair. 2009;8:1235–41. doi: 10.1016/j.dnarep.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Rothstein R. The Dun1 checkpoint kinase phosphorylates and regulates the ribonucleotide reductase inhibitor Sml1. P Natl Acad Sci USA. 2002;99:3746–51. doi: 10.1073/pnas.062502299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X, Garg P, Stith CM, et al. The fidelity of DNA synthesis by yeast DNA polymerase zeta alone and with accessory proteins. Nucleic Acids Res. 2006;34:4731–42. doi: 10.1093/nar/gkl465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- Zuo XM, Clark-Walker GD, Chen XJ. The mitochondrial nucleoid protein, Mgm101p, of Saccharomyces cerevisiae is involved in the maintenance of rho(+) and ori/rep-devoid petite genomes but is not required for hypersuppressive rho(−) mtDNA. Genetics. 2002;160:1389–400. doi: 10.1093/genetics/160.4.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]