

Abstract
High-grade serous ovarian cancer (HGSC) is the most common and lethal form of epithelial ovarian cancer (EOC). Two distinct tissues have been suggested as the tissue of origin: ovarian surface epithelia (OSE) and fallopian tube epithelia (FTE). We hypothesized that the DNA methylome of HGSC should more closely resemble the methylome of its tissue of origin. To this end, we profiled HGSC (n=10), and patient-matched OSE and FTE (n=5) primary fresh-frozen tissues, and analyzed the DNA methylome using Illumina 450K arrays (n=20) and Agilent Sure Select methyl-seq (n=7). Methylomes were compared using statistical analyses of differentially methylated CpG sites (DMC) and differentially methylated regions (DMR). In addition, methylation was evaluated within a variety of different genomic contexts, including CpG island shores and Homeobox (HOX) genes, due to their roles in tissue specification. Publically-available HGSC methylome data (n=628) were interrogated to provide additional comparisons with FTE and OSE for validation. These analyses revealed that HGSC and FTE methylomes are significantly and consistently more highly conserved than are HGSC and OSE. Pearson correlations and hierarchal clustering of genes, promoters, CpG islands, CpG island shores, and HOX genes all revealed increased relatedness of HGSC and FTE methylomes. Thus, these findings reveal that the landscape of FTE more closely resembles HGSC, the most common and deadly EOC subtype.
Keywords: DNA methylation, DNA methylome, high grade serous ovarian cancer, fallopian tube epithelia, ovarian surface epithelia
Introduction
HGSC, the most common and lethal subtype of EOC, is frequently diagnosed at an advanced stage, where long-term survival is poor (1). Understanding the mechanisms driving initiation and progression of HGSC is critical for the development of new diagnostic and therapeutic approaches. In this context, the tissue and cellular origin of HGSC remains a critical question in the field (2–4). The first widely-accepted model for the origin of HGSC implicated transformation of the ovarian surface epithelia (OSE), possibly by incessant ovulation (5, 6). This hypothesis is supported by several observations, including the existence of benign epithelial cysts (cystadenomas) in the ovary, a precursor lesion for EOC, and epidemiological links between ovulation and ovarian cancer (3, 4). Based on the OSE origin model, experimental model systems for EOC have utilized primary and immortalized mouse and human OSE cells, and transgenic mice created by genetic manipulation of the OSE (7, 8). A more recent hypothesis for the origin of HGSC invokes transformation of fallopian tube fimbriae epithelia (FTE) (9, 10). This model is supported by the identification of precursor lesions in the distal fallopian tube, including “p53 signatures,” lesions characterized by increased p53 protein expression due to TP53 mutations, and serous tubal intraepithelial carcinoma (STIC) lesions, which are characterized by increased proliferation and multiple cell layers. Cells emanating from STIC lesions are hypothesized to spread to the ovary, where they develop into invasive serous carcinoma (1, 11). The FTE model is supported by several observations: i) identification of common FTE but not OSE precursor lesions in BRCA mutation carriers (12), ii) reduced EOC risk in BRCA mutation carriers following bilateral tubal ligation (13), iii) gene expression profiling studies showing similarity of FTE and HGSC (14, 15), and iv) development of HGSC in vivo following the engineering of specific genetic alterations (Tp53, Brca1/2, Pten) in mouse FTE (16).
In the mammalian embryo, extensive cytosine DNA de-methylation erases the bulk of the gamete methylation pattern, which is followed by coordinated re-methylation establishing cell and tissue type-specific DNA methylation in somatic cells (17). Consequently, DNA methylation participates in the establishment of an epigenetic signature that contributes to cell and tissue type-specific chromatin organization and gene expression (18–21). Whole genome bisulfite sequencing analysis of several human tissues in different individuals has recently revealed widespread tissue-specific DNA methylation variation in humans (22). In addition to its normal function in X-chromosome inactivation, genomic imprinting, tissue differentiation, and transposable element silencing, altered DNA methylation makes a major contribution to human diseases, including cancer (23, 24). In cancer, two general DNA methylation defects are common: gene specific promoter hypermethylation and global DNA hypomethylation (24). Despite these oncogenic changes, tumors might be anticipated to retain tissue-specific DNA methylation that reflect their cellular and tissue origin (25, 26).
In the current study, we hypothesized that the DNA methylome provides a means to investigate cell/tissue origin in HGSC. Comparative analysis of the methylome of HGSC, normal FTE, and normal OSE may provide insight into the tissue of origin of HGSC, due to tissue-specific methylation (18, 22, 27, 28). We interrogated the methylomes of HGSC, FTE, and OSE using two complementary methods, and analyzed methylation in several different genomic contexts, to determine the degree of relatedness of the HGSC methylome to FTE and OSE. Our data indicate that the HGSC methylome is consistently and significantly more similar to FTE than OSE. Beyond the implications of this study for understanding the origin of HGSC, DNA methylome profiling may serve as a useful method for cell of origin mapping for other cancers. The stability of DNA methylation, a covalent DNA modification, is an advantageous aspect of this approach in the clinical setting.
Methods
Human tissues
We obtained fresh-frozen primary HGSC (n=10) from patients undergoing surgical resection at Roswell Park Cancer Institute (RPCI), and patient-matched fresh-frozen normal OSE and FTE (n=5) at RPCI, as described previously (29) (Supplementary Table S1). Briefly, OSE and FTE obtained from patients without malignancy were harvested by mechanical scraping and processing of the epithelial layer of resected ovaries and the fimbriae end of fallopian tubes, immediately upon surgical removal. HGSC tissues were estimated by pathology to contain >80% neoplastic cells. All samples were collected using IRB-approved protocols at RPCI, and sample processing has been described previously (29, 30). We isolated genomic DNA using the Puregene Tissue Kit (Qiagen), which includes RNAse treatment. When selecting HGSC samples, we took into account cancer-specific global DNA hypomethylation, in which repetitive DNA elements are significantly hypomethylated genome-wide (29, 31). Specifically, to eliminate potentially confounding effects due to this phenotype, we utilized HGSC showing similar LINE-1 DNA methylation as FTE and OSE, as determined by pyrosequencing (data not shown) (29, 31). However, an independent analysis of HGSC samples displaying global DNA hypomethylation affirmed the conclusions presented here (data not shown).
DNA methylome data
1) New data: We performed Illumina Infinium 450K bead arrays (450K), which assessed methylation at ~470,000 CpG sites, at the RPCI (n=7) and University of Utah (n=13) Genomics Core Facilities (Supplementary Table S1). We performed Agilent SureSelect Methylome bisulfite sequencing (methyl-seq), a targeted solution hybridization method (32), which analyzed methylation at ~4 × 106 CpGs, at the University of Nebraska Medical Center (UNMC) Epigenomics Core (n=7) (Supplementary Table S1). We used either the Zymo Pico Methyl-Seq or Agilent SureSelect Methyl-Seq Kit for library preparations, and Agilent SureSelect baits to pull down the final sequences. We conducted high-throughput sequencing at the UNMC Sequencing Core, using an Illumina HiSeq 2500 Genome Analyzer. Sequencing parameters and results are shown in Supplementary Table S2. We aligned sequence tags to the human genome (hg19) using Bismark, and selected only those CpG with ≥ 10× coverage (33). For clarity, we refer to newly-generated methylome data as “Karpf 450K data” or “Karpf Methyl-seq data.” 2) Public data. We utilized 450K data from FTE (n=7) and primary HGSC (n=78) from Gene Expression Omnibus (GEO) GSE65821 (Supplementary Table S3); these data have been published and are referred to as “Bowtell 450K data” (34). We only used primary tumor samples, to avoid the influence of disease recurrence and drug resistance. We also utilized Illumina Infinium 27K methylation data (27K) from primary HGSC (n=550) using The Cancer Genome Atlas (TCGA) data portal (35); these data are referred to as “TCGA 27K data.” For analysis of TCGA data, we only utilized CpG sites that overlapped Karpf FTE and OSE 450K data (25,779 CpG sites).
DNA methylome data analysis
We used RnBeads (36) to analyze all methylome data (450K, 27K, Methyl-seq), and restricted our analysis to CpG methylation. We analyzed both differentially methylated CpG sites (DMC; ≥ 25% methylation change) and differentially methylated regions (DMR; contiguous regions of any length containing ≥ 3 CpGs and ≥ 25% methylation change). RnBead data included 5kb genomic tiles (n=131,408), genes [transcriptional start site (TSS) to transcription end, n=30,514], promoters (−1500 to +500bp relative to the TSS, n=30,630) and CpG islands (n=26,595). We additionally analyzed CpG island shores (+/−2000bp of CpG island; n=53,190) (37), and enhancers (n= 32,693; Transcribed Enhancer Atlas Database (http://enhancer.binf.ku.dk/index.php). We determined the overlap of CpG sites with different genomic regions using the Bedtools intersect routine (38). We used an FDR-adjusted p-value of <0.05 within RnBeads. We created hierarchal clustering and methylation heat maps using TM4 microarray software Multi Experiment Viewer (MeV), based on a Pearson correlation metric and average linkage (39, 40).
To compare individual HGSC DNA methylomes with FTE and OSE, we determined DMC for each HGSC using the R software package DSS (41), by smoothing the RnBeads normalized percent methylation values for 0.5kb units, and using a moving average algorithm. DMR were defined as regions of any length containing ≥ 3 CpGs and ≥ 25% methylation change. We quantified the number of HGSC DMC and DMR that showed a significant difference as compared to FTE or OSE, using a Wald test p-value of ≤ 0.05.
HOX gene analysis
We downloaded the coordinates of all human HOX genes and pseudogenes (n=333) from the Homeobox database (http://homeodb.zoo.ox.ac.uk/), and aligned CpG sites using genomic locations from −10kb upstream the TSS to the transcript end. We determined the overlap of CpG sites that were significantly different (FDR-adjusted p-value of <0.05) between FTE and OSE with HOX gene genomic regions using the Bedtools intersect routine (38). We created hierarchal clustering and methylation heat maps using TM4 microarray software Multi Experiment Viewer (MeV), based on a Pearson correlation metric and average linkage (39, 40).
Results
Comparison of the HGSC, FTE, and OSE methylomes
The aim of this study was to compare the degree of relatedness of the DNA methylome of primary HGSC to primary FTE and OSE, in order to infer the tissue lineage of HGSC. For this task, we utilized a set of patient-matched primary FTE and OSE tissues from patients without malignancy (n=5). To verify the origin of these samples, we compared their 450K methylomes to recently reported FTE 450K methylome data (n=7) (34). We compared CpG methylation within a variety of genomic contexts, as well as comparisons of total DMC and DMR (see Methods). This analysis verified conservation of the two independent FTE methylome data and illustrated significant divergence, in all genomic contexts, using patient-matched OSE (Supplementary Table S4).
We analyzed and compared HGSC, FTE, and OSE methylomes from Karpf 450K data. Fig. 1 presents a comparison of methylation of all CpG sites (DMC) and all DMR (5kb tiles). In both comparisons, HGSC methylation was strikingly more similar to FTE than OSE. To validate this finding, we calculated Pearson correlation coefficients for the comparisons, using all CpG sites or methylation restricted to different genomic regions. In each instance, HGSC showed greater correlation with FTE, as compared to OSE (Supplementary Fig. S1). We next used principal component analyses (PCA) to test this relationship, and again observed an increased similarity of HGSC with FTE, as compared to OSE (Supplementary Fig. S2).
Figure 1.
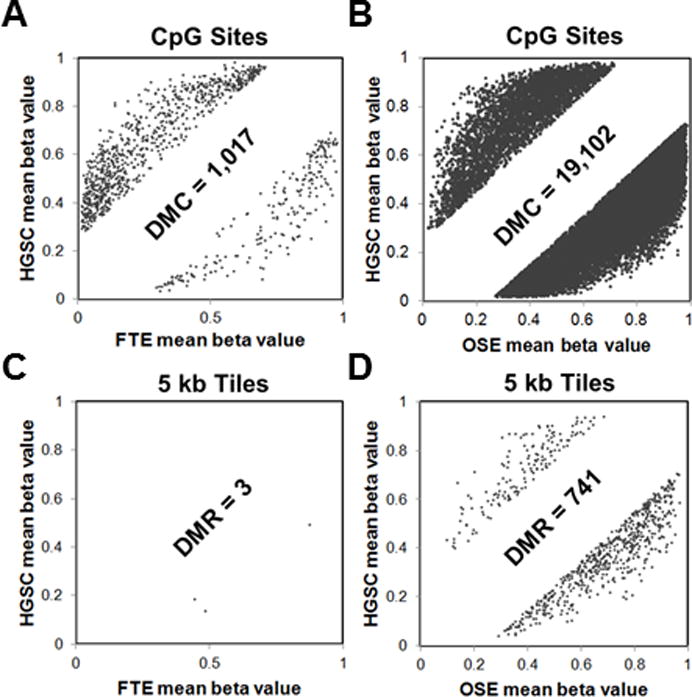
Differential methylation of HGSC vs. patient-matched FTE or OSE, using Karpf 450K data. Each panel shows an x-y plot of the mean methylation beta value of specific CpG sites or 5kb tile regions in HGSC (y-axes) and FTE or OSE (x-axes). Sites or regions without a significant change in methylation are not shown (blank regions in middle of graphs). Sites or regions with increased methylation in HGSC are indicated on the top left of graphs, while sites or regions with increased methylation in FTE or OSE are indicated on the bottom right of graphs. (A) DMC comparison of HGSC (n=10) and FTE (n=5) (DMC = mean CpG methylation beta value difference ≥ 25%; FDR adjusted p-value <0.05). (B) DMC comparison of HGSC (n=10) and OSE (n=5), as described in A. (C) DMR comparison between HGSC (n=10) and FTE (n=5). DMR were 5kb tiles (n=131,408) with a mean beta value difference of ≥ 25% and an FDR-adjusted p-value of <0.05. (D) Comparison of HGSC (n=10) and OSE (n=5), as described in C. The total number of DMC and DMR meeting the differential cutoff in each comparison is indicated on the figure. All analyses were performed using RnBeads (see Methods).
CpG island shores are genomic regions adjacent to CpG islands that display tissue-specific DNA methylation (37), making them relevant for the current study. We identified individual CpG sites within CpG island shores showing differential methylation in FTE and OSE, and used these to perform hierarchical clustering of the three sample groups. Importantly, all HGSC samples clustered more closely to FTE than to OSE (Fig. 2A). We additionally observed that one FTE sample clustered within the OSE samples (Fig. 2A; see also Fig. 2B and Fig. 3). We speculate this is due to a predominance of individual-specific DNA methylation differences in this particular patient.
Figure 2.
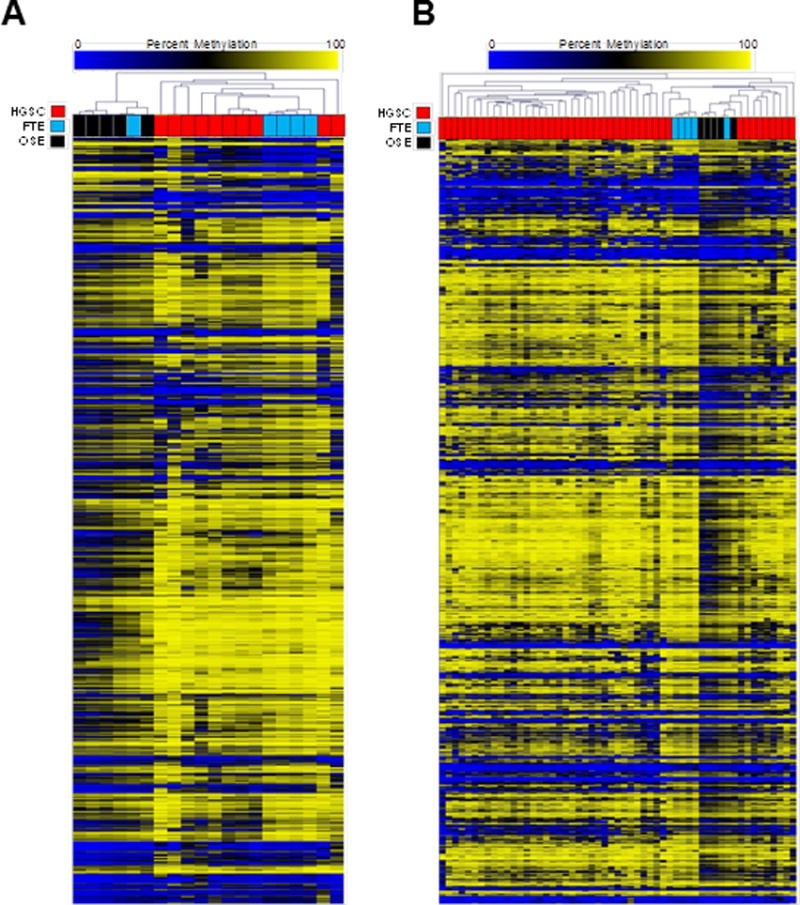
Unsupervised hierarchal clustering of sample groups (HGSC, FTE, OSE) using differentially methylated CpGs associated with CpG island shores. CpGs selected for analysis were: 1) located within CpG island shores, and 2) showed a significant mean beta value difference between FTE and OSE (No beta value cutoff; FDR-adjusted p-value <0.05). (A) Hierarchical cluster dendogram of HGSC (n=10), FTE (n=5), and OSE (n=5) using Karpf 450K data. Data is comprised of 1827 CpG sites. (B) Hierarchical cluster dendogram of HGSC (n=45), FTE (n=5), and OSE (n=5), using Bowtell 450K (HGSC) and Karpf 450K (FTE, OSE) data. Data is comprised of 1753 CpG sites. All analyses were performed using RnBeads (see Methods).
Figure 3.
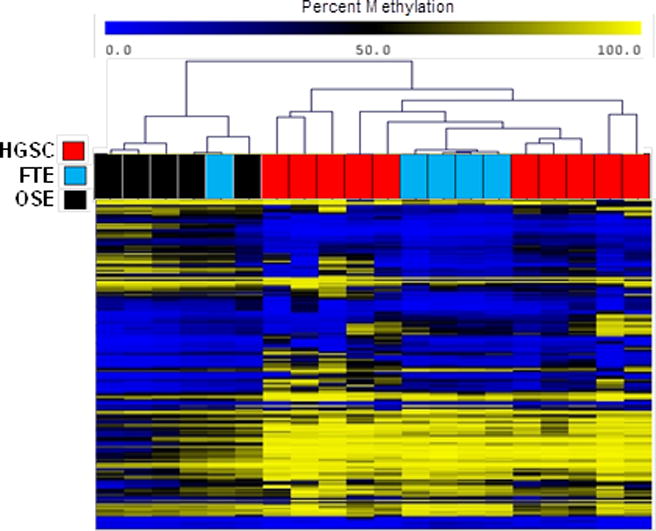
Hierarchal clustering analysis of CpG methylation within HOX genes in HGSC (n=10) and patient-matched FTE and OSE (n=5) (see Methods). The CpGs shown (n=179) had a significant mean beta value methylation difference (FDR-adjusted p-value <0.05, using RnBeads) when compared between patient-matched FTE and OSE.
A summary of all Karpf 450K methylome comparisons is provided in Table 1 (top section). Chi-square testing validated the significantly increased similarity of HGSC and FTE as compared to OSE, in each comparison.
Table 1.
Summary of HGSC vs. patient-matched FTE and OSE DNA methylome comparisons
| Sample Group Comparison | Differentially Methylated CpG sites (DMC)1 | Differentially Methylated Regions (DMR)2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| All Sites | CpG Islands | CpG Island Shores | HOX Genes | Enhancers | 5kb Tiles | CpG Islands | Promoters | Genes | |
| Karpf 450K, HGSC (n=10), Patient-Matched FTE and OSE (n=5) | |||||||||
| HGSC vs. FTE | 1,017 | 388 | 417 | 222 | 29 | 3 | 32 | 0 | 0 |
| HGSC vs. OSE | 19,102 | 3,625 | 6,096 | 1,825 | 530 | 741 | 472 | 223 | 41 |
| Fold Difference3 | 18.8 (****)4 | 9.3 (****) | 14.6 (****) | 8.2 (****) | 18.3 (****) | 247 (****) | 14.8 (****) | 223 (****) | 41 (****) |
| Karpf Methyl-seq, HGSC (n=3), Patient-Matched FTE and OSE (n=2) | |||||||||
| HGSC vs. FTE | 123,586 | 40,298 | 50,322 | 20,850 | 3,019 | 2,649 | 714 | 530 | 325 |
| HGSC vs. OSE | 161,561 | 47,816 | 62,991 | 23,550 | 4,061 | 4,580 | 1,543 | 913 | 510 |
| Fold Difference3 | 1.3 (****) | 1.2 (****) | 1.3 (****) | 1.1 (****) | 1.3 (****) | 1.7 (****) | 2.2 (****) | 1.7 (****) | 1.6 (****) |
| Bowtell 450K, HGSC (n=78), Patient-Matched FTE and OSE (n=5) | |||||||||
| HGSC vs. FTE | 21,646 | 3,130 | 7,209 | 1,732 | 455 | 1,085 | 460 | 379 | 190 |
| HGSC vs. OSE | 39,439 | 5,461 | 12,158 | 2,711 | 991 | 1,879 | 882 | 641 | 302 |
| Fold Difference3 | 1.8 (****) | 1.7 (****) | 1.7 (****) | 1.6 (****) | 2.2 (****) | 1.7 (****) | 1.9 (****) | 1.7 (****) | 1.6 (****) |
| TCGA 27K, HGSC (n=550), Patient-Matched FTE and OSE (n=5) | |||||||||
| HGSC vs. FTE | 1,280 | 134 | 523 | 55 | 9 | N/A | N/A | N/A | N/A |
| HGSC vs. OSE | 1,592 | 180 | 583 | 77 | 8 | N/A | N/A | N/A | N/A |
| Fold Difference3 | 1.2 (****) | 1.3 (**) | 1.1 (ns) | 1.4 (ns) | 0.9 (ns) | N/A | N/A | N/A | N/A |
FDR < 0.05, methylation difference ≥ 25%
FDR < 0.05, mean methylation difference ≥ 25%, ≥ 3 CpGs per region
Increase in HGSC vs. OSE as compared to HGSC vs. FTE
Chi-square p-value:
<0.0001;
<0.001,
<0.01,
<0.05
N/A: Insufficient coverage to conduct DMR measurements
ns: not significant
The methylation patterns of PAX8, mesothelin (MSLN), and Homeobox (HOX) genes are divergent in OSE as compared to HGSC and FTE
We examined the methylation of two genes involved FTE lineage specificity, PAX8 and MSLN (14, 16), hypothesizing that they may show divergence in OSE. As anticipated, PAX8 and MSLN showed relatively similar methylation in HGSC and FTE as compared to OSE (Supplementary Fig. S3). We next examined methylation of HOX genes, as they are involved in development and tissue differentiation, are known to be regulated by DNA methylation, and can show altered methylation and expression in HGSC (42–45). We analyzed methylation of all HOX (and HOX domain-containing) genes (n=333 genes, n=9011 CpGs) using Karpf 450K data. Hierarchical clustering of sample groups using HOX-associated CpGs that showed significant differential methylation between FTE and OSE (n=179) revealed increased similarity of HGSC and FTE (Fig. 3). Specific examples of HOX genes showing divergent methylation patterns in OSE as compared to FTE and HGSC are HOXB1 and HOXB7, which were hypomethylated in OSE as compared to FTE and HGSC (Fig. 4A–B), and HOXD3 and EMX2 (a homeodomain-containing gene), which were hypermethylated in OSE as compared to FTE or HGSC (Fig. 4C–D). We also noted that several HOX genes, including HOXA5, A9, A10, A11, B5, and D11, showed cancer-specific DNA hypermethylation, in agreement with earlier reports (45–47) (Supplementary Figs S4–S5).
Figure 4.
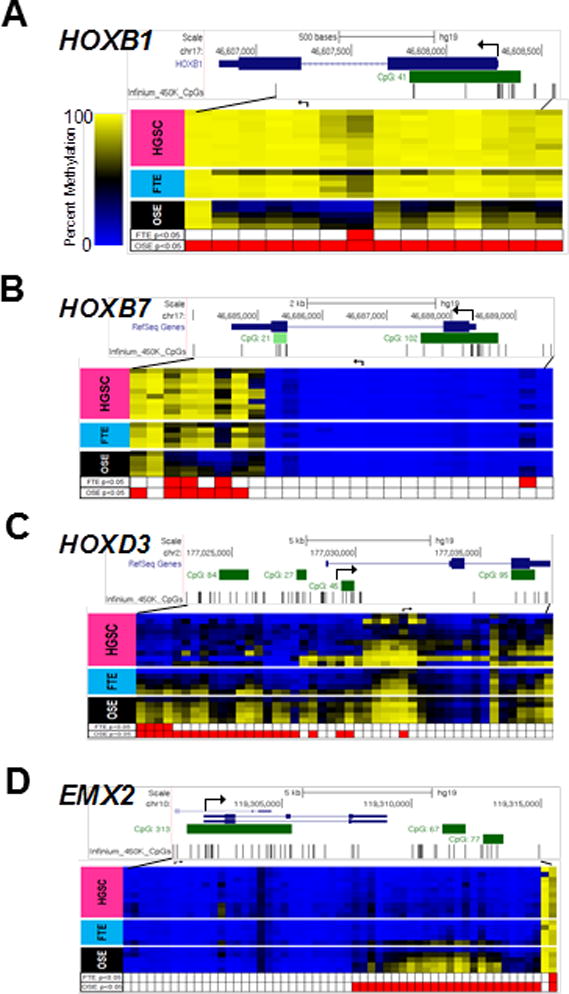
Methylation of specific HOX genes in HGSC, FTE, and OSE. DNA methylation of (A) HOXB1 (B) HOXB7 (C) HOXD3 and (D) EMX2 in HGSC (n=10) and patient-matched FTE (n=5), and OSE (n=5), as determined using Karpf 450K data. Upper: chromosome, gene, and CpG island locations (UCSC genome browser), and map of CpG sites analyzed by 450K. Lower: CpG methylation data for sample groups; red boxes indicate regions that contain differently methylated CpG sites (FDR-adjusted p-value <0.05) between FTE or OSE as compared to HGSC. Broken arrows indicate the TSS.
Methyl-seq confirms the relatedness of the HGSC and FTE methylomes
As a complementary approach to 450K, we utilized methyl-seq to analyze a subgroup of tumor and normal samples, as described in Methods (Supplementary Table S1). Data from 450K and methyl-seq were well-correlated (Supplementary Table S5). The compiled results of methyl-seq are reported in Table 1. As observed with Karpf 450K data, Chi-square testing of methyl-seq revealed significantly increased similarity of HGSC and FTE as compared to HGSC and OSE, in each genomic context.
Independent cohorts of HGSC methylation data validate the relatedness of the HGSC and FTE methylomes
We utilized two published primary HGSC data sets, Bowtell 450K data (n=78) (34) and TCGA 27K data (n=550) (35), and compared these to our patient-matched primary FTE and OSE 450K data. Given the low genomic coverage of Illumina 27K, we confined our analysis of methylation within different genomic contexts to Bowtell 450K data. Pearson correlation and PCA analyses both indicated increased similarity of HGSC to FTE as compared to OSE (Supplementary Figs. S6–S7). Hierarchical clustering of CpG island shore methylation data also revealed increased relatedness of HGSC with FTE as compared to OSE (Fig. 2B). Chi-square analysis revealed significantly greater similarity of HGSC with FTE as compared to OSE, in all genomic contexts (Table 1). Despite the reduced coverage of 27K data, it still illustrated the similarity of HGSC to FTE as compared to OSE (Supplementary Fig. S8; Table 1).
Individual HGSC sample methylome analysis validated the similarity of HGSC and FTE, as compared to OSE
The above data report group-based comparative methylome analyses. To determine the extent to which individual HGSC samples are related to FTE and OSE, we used Karpf 450K and Bowtell 450K data to classify individual samples according to total DMC or total DMR. For total DMC, 76/78 (97%) HGSC showed increased similarity to FTE, while, for DMR, 77/78 (99%) HGSC showed increased similarity to FTE (Fig. 5A–B). These data affirm the conclusions drawn from group-wise analyses.
Figure 5.
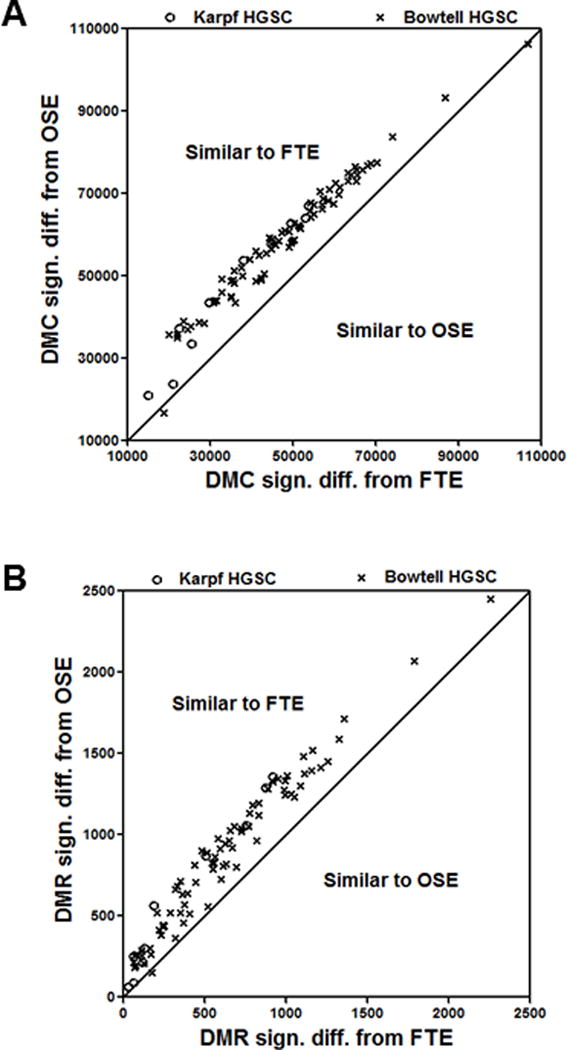
Individual HGSC sample methylation differences as compared to patient-matched FTE or OSE group averages. DNA methylation data from Karpf 450K for HGSC (n=10), FTE (n=5), and OSE (n=5), and Bowtell 450K HGSC data (n=78) were utilized. (A) DMC between Karpf and Bowtell HGSC samples and FTE or OSE. (B) DMR between Karpf and Bowtell HGSC samples and FTE or OSE. In both panels, a Wald test p-value of ≤ 0.05 was used.
Discussion
Using the DNA methylome as a classifier, our data reveal that HGSC more closely resembles FTE than OSE. This relationship was conserved regardless of the HGSC sample population, methylome analysis method, or genomic location, including all CpGs, CpG islands, genes, promoters, and all DMR. In addition, CpG island shores and enhancers maintained this relationship, which is notable based on previous studies indicating the importance of tissue-specific differential methylation in CpG island shores, and the well-established role of enhancers in driving tissue specification (37, 48). Thus, our study provides support for the model that HGSC originates in the fimbriae end of the fallopian tube. We also note that use of the proper normal control is an essential aspect of molecular studies of HGSC, and our data indicate that FTE should be used for this purpose.
Within the FTE, there are two major cell types, secretory and ciliated cells. In addition, FTE harbors a minor population of basal cells, which includes the stem cell niche (3). Secretory cells are proliferative and help regenerate the epithelium during cell turnover, while ciliated cells are terminally differentiated (11). Determination of the DNA methylomes of individual FTE cell types will provide further insight into the cell lineage of HGSC, and how the methylome becomes altered in cancer.
HOX proteins play key roles in cell and tissue identity and HOX genes can be regulated by DNA methylation (42–44). We therefore analyzed HOX genes and observed that overall, HOX methylation in HGSC more closely resembled FTE as compared to OSE. In particular, we show that HOXB1, HOXB7, HOXD3, and EMX2 illustrate a methylation signature that was conserved between HGSC and FTE, but divergent in OSE.
Although overall our data implicate strong conservation between the HGSC and FTE methylomes as compared to OSE, we noted in one comparison (of CpG island shore methylation) that a small subset of HGSC clustered more closely to OSE (Fig. 2B). Therefore, we cannot formally exclude that a sub-population of HGSC has greater similarity to, and therefore potentially arises from, OSE. In addition, we cannot exclude that the robust relative conservation of the HGSC and FTE methylomes observed overall, rather than implicating FTE as the cell/tissue origin, could result from a trans-differentiation event, e.g. Müllerian metaplasia (4). A final limitation of our study is that it utilized clinically-advanced HGSC. Methylome studies of early stage tumors or HGSC precursor lesions will provide important additional insight into the cellular origin of HGSC.
Beyond the relevance of the current study for addressing the tissue of origin of HGSC, our data more generally suggest that DNA methylome analyses may serve as a useful method for mapping cellular and tissue origins in other cancers. The stability of DNA methylation, as both a covalent chemical modification and a stable (replication-coupled) epigenetic mark, is advantageous when considering methylome profiling in the clinical setting. DNA methylation can be measured in biological fluids, fresh-frozen tissue samples, and formalin-fixed paraffin-embedded (FFPE) archival materials (49). Moreover, emerging methylome data implicate DNA methylation in the processes of tissue differentiation and the maintenance of tissue specificity (18, 19, 22, 27, 28). These two traits (biochemical stability and tissue specificity) support the utility and relevance of DNA methylome profiling to investigate the cellular origins of cancer.
Supplementary Material
Implications.
DNA methylome analyses supports the hypothesis that high grade serous ovarian cancer arise from the fallopian tube and that due to its tissue-specificity and biochemical stability, interrogation of the methylome may be a valuable approach to examine cell/tissue lineage in cancer.
Acknowledgments
We thank Ann-Marie Patch and David Bowtell for helpful assistance and data sharing, and Shashikant Lele for support. We thank the University of Buffalo, Center of Excellence in Genomics and Bioinformatics, RPCI Bioinformatics and Genomics Core, University of Utah Genomics Core, UNMC Epigenomics Core, UNMC Bioinformatics Core, and the UNMC DNA Sequencing and Microarray Core (supported by NIGMS 8P20GM103427 and P20GM103471) for assistance. This work was supported by The Otis Glebe Medical Research Foundation (ARK), The Betty J. and Charles D. McKinsey Ovarian Cancer Research Fund (ARK), The Fred & Pamela Buffett Cancer Center (NIH P30 CA036727) (ARK), and NIH T32CA108456 (SNA). KO acknowledges support from NIH P30 CA016056, NIH R01CA158318, NIH P50CA159981, and the Roswell Park Alliance Foundation.
Abbreviations
- DMC
differentially methylated CpG site
- DMR
differentially methylated region
- EOC
epithelial ovarian cancer
- FDR
false discovery rate
- FTE
fallopian tube epithelia
- HGSC
high-grade serous ovarian cancer
- OSE
ovarian surface epithelia
- PCA
principal component analysis
Footnotes
Author Contributions
A.R.K. designed the research, S.N.A. and K.O. provided clinical samples, D.K. and W.Z. performed the research, D.K. analyzed the data, and D.K and A.R.K. wrote the paper.
Competing Financial Interests
The authors declare no competing financial interests.
Genomic data deposit and public access
DNA methylation data (450K, *.idat files, and methyl-seq, *.fastq files) were deposited into the NCBI Gene Expression Omnibus (GEO #).
References
- 1.Bowtell DD, Bohm S, Ahmed AA, Aspuria PJ, Bast RC, Jr, Beral V, et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nature reviews Cancer. 2015;15:668–79. doi: 10.1038/nrc4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silva EG. The Origin of Epithelial Neoplasms of the Ovary: An Alternative View. Advances in anatomic pathology. 2016;23:50–7. doi: 10.1097/PAP.0000000000000098. [DOI] [PubMed] [Google Scholar]
- 3.Ng A, Barker N. Ovary and fimbrial stem cells: biology, niche and cancer origins. Nature reviews Molecular cell biology. 2015;16:625–38. doi: 10.1038/nrm4056. [DOI] [PubMed] [Google Scholar]
- 4.Dubeau L, Drapkin R. Coming into focus: the nonovarian origins of ovarian cancer. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2013;24(Suppl 8):viii28–viii35. doi: 10.1093/annonc/mdt308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Auersperg N, Edelson MI, Mok SC, Johnson SW, Hamilton TC. The biology of ovarian cancer. Seminars in oncology. 1998;25:281–304. [PubMed] [Google Scholar]
- 6.Fathalla MF. Incessant ovulation–a factor in ovarian neoplasia? Lancet. 1971;2:163. doi: 10.1016/s0140-6736(71)92335-x. [DOI] [PubMed] [Google Scholar]
- 7.Connolly DC. Animal models of ovarian cancer. Cancer treatment and research. 2009;149:353–91. doi: 10.1007/978-0-387-98094-2_17. [DOI] [PubMed] [Google Scholar]
- 8.Garson K, Shaw TJ, Clark KV, Yao DS, Vanderhyden BC. Models of ovarian cancer–are we there yet? Molecular and cellular endocrinology. 2005;239:15–26. doi: 10.1016/j.mce.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 9.Kurman RJ, Shih le M. The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. The American journal of surgical pathology. 2010;34:433–43. doi: 10.1097/PAS.0b013e3181cf3d79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crum CP, Drapkin R, Kindelberger D, Medeiros F, Miron A, Lee Y. Lessons from BRCA: the tubal fimbria emerges as an origin for pelvic serous cancer. Clinical medicine & research. 2007;5:35–44. doi: 10.3121/cmr.2007.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karst AM, Drapkin R. Ovarian cancer pathogenesis: a model in evolution. Journal of oncology. 2010;2010:932371. doi: 10.1155/2010/932371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piek JM, van Diest PJ, Zweemer RP, Jansen JW, Poort-Keesom RJ, Menko FH, et al. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. The Journal of pathology. 2001;195:451–6. doi: 10.1002/path.1000. [DOI] [PubMed] [Google Scholar]
- 13.Cibula D, Widschwendter M, Majek O, Dusek L. Tubal ligation and the risk of ovarian cancer: review and meta-analysis. Human reproduction update. 2011;17:55–67. doi: 10.1093/humupd/dmq030. [DOI] [PubMed] [Google Scholar]
- 14.O’Shannessy DJ, Jackson SM, Twine NC, Hoffman BE, Dezso Z, Agoulnik SI, et al. Gene expression analyses support fallopian tube epithelium as the cell of origin of epithelial ovarian cancer. International journal of molecular sciences. 2013;14:13687–703. doi: 10.3390/ijms140713687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marquez RT, Baggerly KA, Patterson AP, Liu J, Broaddus R, Frumovitz M, et al. Patterns of gene expression in different histotypes of epithelial ovarian cancer correlate with those in normal fallopian tube, endometrium, and colon. Clinical cancer research: an official journal of the American Association for Cancer Research. 2005;11:6116–26. doi: 10.1158/1078-0432.CCR-04-2509. [DOI] [PubMed] [Google Scholar]
- 16.Perets R, Wyant GA, Muto KW, Bijron JG, Poole BB, Chin KT, et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer cell. 2013;24:751–65. doi: 10.1016/j.ccr.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seisenberger S, Peat JR, Hore TA, Santos F, Dean W, Reik W. Reprogramming DNA methylation in the mammalian life cycle: building and breaking epigenetic barriers. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2013;368:20110330. doi: 10.1098/rstb.2011.0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lokk K, Modhukur V, Rajashekar B, Martens K, Magi R, Kolde R, et al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome biology. 2014;15:r54. doi: 10.1186/gb-2014-15-4-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wan J, Oliver VF, Wang G, Zhu H, Zack DJ, Merbs SL, et al. Characterization of tissue-specific differential DNA methylation suggests distinct modes of positive and negative gene expression regulation. BMC genomics. 2015;16:49. doi: 10.1186/s12864-015-1271-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oliver VF, Wan J, Agarwal S, Zack DJ, Qian J, Merbs SL. A novel methyl-binding domain protein enrichment method for identifying genome-wide tissue-specific DNA methylation from nanogram DNA samples. Epigenetics & chromatin. 2013;6:17. doi: 10.1186/1756-8935-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang P, Song F, Ghosh S, Morien E, Qin M, Mahmood S, et al. Genome-wide survey reveals dynamic widespread tissue-specific changes in DNA methylation during development. BMC genomics. 2011;12:231. doi: 10.1186/1471-2164-12-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schultz MD, He Y, Whitaker JW, Hariharan M, Mukamel EA, Leung D, et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature. 2015;523:212–6. doi: 10.1038/nature14465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robertson KD. DNA methylation and human disease. Nature reviews Genetics. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 24.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nature genetics. 2009;41:178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sproul D, Kitchen RR, Nestor CE, Dixon JM, Sims AH, Harrison DJ, et al. Tissue of origin determines cancer-associated CpG island promoter hypermethylation patterns. Genome biology. 2012;13:R84. doi: 10.1186/gb-2012-13-10-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ziller MJ, Gu H, Muller F, Donaghey J, Tsai LT, Kohlbacher O, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500:477–81. doi: 10.1038/nature12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–30. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akers SN, Moysich K, Zhang W, Collamat Lai G, Miller A, Lele S, et al. LINE1 and Alu repetitive element DNA methylation in tumors and white blood cells from epithelial ovarian cancer patients. Gynecologic oncology. 2014;132:462–7. doi: 10.1016/j.ygyno.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woloszynska-Read A, James SR, Link PA, Yu J, Odunsi K, Karpf AR. DNA methylation-dependent regulation of BORIS/CTCFL expression in ovarian cancer. Cancer immunity. 2007;7:21. [PMC free article] [PubMed] [Google Scholar]
- 31.Woloszynska-Read A, Zhang W, Yu J, Link PA, Mhawech-Fauceglia P, Collamat G, et al. Coordinated cancer germline antigen promoter and global DNA hypomethylation in ovarian cancer: association with the BORIS/CTCF expression ratio and advanced stage. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:2170–80. doi: 10.1158/1078-0432.CCR-10-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee EJ, Pei L, Srivastava G, Joshi T, Kushwaha G, Choi JH, et al. Targeted bisulfite sequencing by solution hybrid selection and massively parallel sequencing. Nucleic acids research. 2011;39:e127. doi: 10.1093/nar/gkr598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics (Oxford, England) 2011;27:1571–2. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patch A-M, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, et al. Whole–genome characterization of chemoresistant ovarian cancer. Nature. 2015;521:489–94. doi: 10.1038/nature14410. [DOI] [PubMed] [Google Scholar]
- 35.Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Assenov Y, Muller F, Lutsik P, Walter J, Lengauer T, Bock C. Comprehensive analysis of DNA methylation data with RnBeads. Nature methods. 2014;11:1138–40. doi: 10.1038/nmeth.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nature genetics. 2009;41:1350–3. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics (Oxford, England) 2010;26:841–2. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, et al. Methods in Enzymology. Academic Press; 2006. [9] TM4 Microarray Software Suite; pp. 134–93. [DOI] [PubMed] [Google Scholar]
- 40.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:14863–8. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu H, Xu T, Feng H, Chen L, Li B, Yao B, et al. Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic acids research. 2015;43:e141. doi: 10.1093/nar/gkv715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holland PW. Evolution of homeobox genes. Wiley interdisciplinary reviews Developmental biology. 2013;2:31–45. doi: 10.1002/wdev.78. [DOI] [PubMed] [Google Scholar]
- 43.Marzese DM, Scolyer RA, Huynh JL, Huang SK, Hirose H, Chong KK, et al. Epigenome-wide DNA methylation landscape of melanoma progression to brain metastasis reveals aberrations on homeobox D cluster associated with prognosis. Human molecular genetics. 2014;23:226–38. doi: 10.1093/hmg/ddt420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oda M, Yamagiwa A, Yamamoto S, Nakayama T, Tsumura A, Sasaki H, et al. DNA methylation regulates long-range gene silencing of an X-linked homeobox gene cluster in a lineage-specific manner. Genes & development. 2006;20:3382–94. doi: 10.1101/gad.1470906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller KR, Patel JN, Ganapathi MK, Tait DL, Ganapathi RN. Biological role and clinical implications of homeobox genes in serous epithelial ovarian cancer. Gynecologic oncology. 2016 doi: 10.1016/j.ygyno.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 46.Montavon C, Gloss BS, Warton K, Barton CA, Statham AL, Scurry JP, et al. Prognostic and diagnostic significance of DNA methylation patterns in high grade serous ovarian cancer. Gynecologic oncology. 2012;124:582–8. doi: 10.1016/j.ygyno.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 47.Fang F, Munck J, Tang J, Taverna P, Wang Y, Miller DF, et al. The novel, small-molecule DNA methylation inhibitor SGI-110 as an ovarian cancer chemosensitizer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:6504–16. doi: 10.1158/1078-0432.CCR-14-1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ong CT, Corces VG. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nature reviews Genetics. 2011;12:283–93. doi: 10.1038/nrg2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.How Kit A, Nielsen HM, Tost J. DNA methylation based biomarkers: practical considerations and applications. Biochimie. 2012;94:2314–37. doi: 10.1016/j.biochi.2012.07.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.