


Abstract
We introduce RNA2DNAlign, a computational framework for quantitative assessment of allele counts across paired RNA and DNA sequencing datasets. RNA2DNAlign is based on quantitation of the relative abundance of variant and reference read counts, followed by binomial tests for genotype and allelic status at SNV positions between compatible sequences. RNA2DNAlign detects positions with differential allele distribution, suggesting asymmetries due to regulatory/structural events. Based on the type of asymmetry, RNA2DNAlign outlines positions likely to be implicated in RNA editing, allele-specific expression or loss, somatic mutagenesis or loss-of-heterozygosity (the first three also in a tumor-specific setting). We applied RNA2DNAlign on 360 matching normal and tumor exomes and transcriptomes from 90 breast cancer patients from TCGA. Under high-confidence settings, RNA2DNAlign identified 2038 distinct SNV sites associated with one of the aforementioned asymetries, the majority of which have not been linked to functionality before. The performance assessment shows very high specificity and sensitivity, due to the corroboration of signals across multiple matching datasets. RNA2DNAlign is freely available from http://github.com/HorvathLab/NGS as a self-contained binary package for 64-bit Linux systems.
INTRODUCTION
Single nucleotide variations (SNV) are considered a common and important form of genetic variation, and have been linked to various allele-specific features across a spectrum of diseases including cancer (1). Yet functional SNVs can be difficult to identify. One approach to outline potential function-implicated SNVs is to distinguish loci with imbalanced variant and reference allele distribution between RNA and DNA. Such imbalances can indicate regulatory events such as RNA editing, in which a post-transcriptional alteration introduces a new allele (2,3), and variant specific expression or loss (4), where the expression level of the variant allele is elevated or declined due to differential regulation. When both germline and somatic datasets are analyzed, the above events could be assessed in their tissue-specific setting, the most prominent example of which is a tumor versus normal tissue. Furthermore, comparison between the tumor and normal DNA at SNV loci can reveal somatic mutagenesis (5) and somatic allelic loss, including loss of heterozygosity (6), two events of crucial importance for tumorigenesis. All of the above events have been associated with tumorigenesis and other diseases, in addition to normal regulatory mechanisms.
Large-scale identification of allelic imbalances from NGS datasets is challenged by data complexity, the requirement for intense high-precision computation, and the demand for compatibility of the allele quantitation across different types of data (7). We have developed RNA2DNAlign—a robust and efficient computational framework that scans matching RNA and DNA sequencing datasets for SNV loci with differential distribution of the reference and variant allele. To do that, RNA2DNAlign accesses the read counts at every called SNV position and generates a (log-) likelihood ratio score for genotype (DNA) and allelic status (RNA) for all of the matching alignments. Next, the tool screens for deviations of the expected read distribution and association of the position with one of the aforementioned events: RNA editing, variant-specific expression/loss, somatic mutagenesis and loss of heterozygosity. The assessment is based on the relative number of the variant and reference reads between the matching datasets, and relevance to placement in each individual dataset: germline, somatic (normal tissue) or tumor DNA, and normal or tumor RNA.
In the current genomic era when different types of genome-scale data from the same individual are increasingly available, development of applications allowing meaningful integration of the information layers has become imperative. While such integration has been shown to reveal essential disease-implicated mutational profiles at individual scale (8), to our knowledge, there are no existing tools for large-scale quantitative integration of signals between RNA and DNA, or for simultaneous identification of all types of allelic imbalances. Due to corroboration of significant hits on more than one dataset, RNA2DNAlign performs with very high specificity, as we demonstrate through applications on previously analyzed datasets. In this paper, we present and describe the tool, and provide it for public use. We discuss the rationale behind the usage of RNA2DNAlign algorithms and elaborate on the considerations of their settings. We also demonstrate the high efficiency and the large-scale capacity of RNA2DNAlign by analyzing 360 tumor and normal tissue exomes and transcriptomes from 90 breast cancer patients downloaded from the TCGA (9), for which we present and discuss major findings.
MATERIALS AND METHODS
Sample selection and analysis
The 360 exome and transcriptome datasets derived from 90 female breast cancer patients from The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/) were accessed through the Cancer Genomic Hub (https://cghub.ucsc.edu) (9). The samples’ identifiers are listed in Supplementary Table S1A. All the used datasets were generated through paired-end sequencing on an Illumina HiSeq platform. The downloaded datasets were converted to fastq files using Picard tools (http://picard.sourceforge.net) version1.96 and processed through an in-house pipeline. Briefly, we mapped the exome sequencing reads to the human reference genome, build hg19, utilizing Bowtie2 (10) The RNAseq data were aligned to hg19 genome by TopHat2 (11) version 2.0.8, employing default settings and allowing two mismatches. For both DNA and RNA datasets variants were called using the mpileup module of SAMtools (12). The variants were further annotated by SeattleSeq 138 (http://snp.gs.washington.edu/SeattleSeqAnnotation138/). The resulting alignments (.bam) were sorted and indexed, and, together with the variant calls (.vcf) were used as input to RNA2DNAlign. In addition, we tested RNA2DNAlign on pre-aligned indexed datasets (.bam and bam.bai) downloaded directly from TCGA (Bowtie2 and BWA (13) for DNA, and TopHat2 and STAR (14) for RNA, Supplementary Table S1B).
Graphical user interface (GUI) design and implementation
The GUI interface is implemented using wxPython. The GUI can be initiated either from the command line or by double-clicking the program icon, displaying the GUI dialog for setting options. The user selects input file(s) of variant calls (tabular or VCF format) and the indexed, binary read alignment files (BAM format) and chooses a destination folder for the output files. Read alignment file names should indicate the sequencing read-type and sample-type. The default substrings are: ‘GDNA’ for germline or normal exomes or transcriptomes, ‘SDNA’ for somatic or tumor exomes or genomes, ‘NRNA’ for blood or normal tissue transcriptome, and ‘SRNA’ for somatic or tumor tissue transcriptome; these can be changed to desired substrings from the original file names, if needed. Reads and SNV-loci aligned to mitochondrial coordinates are ignored. Variant calls format supports common chromosome labels and must provide chromosome, position, reference, and variant nucleotide. Common chromosome label formats are supported. Importantly, both in-house produced and pre-existing alignments and variant call lists must refer to the same reference genome assembly. The ‘Advanced Options’ selection allows several self-explanatory options for run control, including number of minimum required reads at each locus (10 by default). The requirement for minimum reads is included to remove incomparable genomic regions, for example genes that are not expressed in the tissue from which the RNA is derived. Processing is initiated after input files and parameters are submitted, and the progress of the analysis is shown on the console. The processing speed is 13–25 SNV sites per second (4 read sets, normal/tumor/exome/transcriptome, exonic filter applied), depending on the sequencing depth and the size of the files.
Filtering of exonic variants
The filtering of exonic variants uses a python-based script to consider only SNVs situated within known exon intervals. An exon coordinate reference file (GRCh37/hg19) is included with the package (15). The use of this module is optional and can be skipped for applications that do not require it. User provided intervals may be supplied instead, if desired. Using the exon coordinates filter for applications comparing exomes and transcriptomes is strongly recommended for consideration of region compatibility and processing time.
Tools utilized for statistical and graphical purposes
An in house python script was designed in order to arrange and ascertain chromosome location and variation through 100MB of genome amongst the 90 sample sets. The file was then visualized using CIRCOS version 0.67–7 scatter plots (16). Manhattan Plot was generated using R-Studio Version 0.99.451. The alignments were visually inspected through the Integrative genome viewer (IGV) version 2.3.32 (17).
RESULTS
Overview of RNA2DNAlign
RNA2DNAlign comprises a computational framework for quantitative integration of variant and reference read counts distribution between comparable regions of experimentally derived RNA and DNA sequencing datasets from the same individual. RNA2DNAlign assesses the variant and reference read counts at each SNV position called in any of the matching datasets and assigns a probability for a genotype (for DNA) and allelic status (for RNA). The algorithm then aligns the genotypes and allelic statuses across the matching datasets, and outlines positions with deviations from the expected read distribution (i.e. similar variant fraction across the matching datasets). Based on relative placement of the observed deviations between the datasets, RNA2DNAlign identifies allelic distributions corresponding to the following events: RNA editing (RNAed), variant-specific expression/loss (VSE/VSL), somatic mutagenesis (SOM), and loss of heterozygosity (LOH). If different source datasets derived from the same individual are available (such as normal and tumor tissue) RNA2DNAlign simultaneously screens for tissue-specific subsets of events, such as tumor-specific RNA editing or variant expression/loss (T-RNAed, T-VSE and T-VSL, respectively) (Figure 1). To illustrate the use of RNA2DNAlign, we have selected matching datasets representing a widely accessible combination of normal tissue exome (referred from here on as Nex), normal tissue transcriptome (Ntr), tumor exome (Tex) and tumor transcriptome (Ttr).
Figure 1.

(A) Schematic representation of allelic asymmetries across normal and tumor RNA and DNA datasets, corresponding to eight nucleotide events. An estimation of the range of the variant allele fraction for each of the events is represented through shaded areas. Briefly, loci classified as RNAed have an R>0 exclusively in the RNA sets; for T-RNAed, R > 0 is confined to the tumor RNA sets only. VSE and VSL have 0 < R < 1 in the DNA sets, while R ∼ 1 or 0 in the RNA sets. When the latter applies exclusively to the tumor RNA, the asymmetry matches T-VSE or T-VSL events, respectively. In the case of LOH, the tumor datasets show an R ∼ 0 or 1, and for SOM R = 0 in the normal datasets while R>0 in the tumor sets. (B) IGV visualization of examples of SNVs associated with each of the eight events. The gene and position are shown on the top of each IGV panel. The variant nucleotide is positioned at the middle. The grey lines represent sequencing reads, and the colored letters show differences from the reference sequence. The colored blocks at the top (middle) of each of the four sub-panels depict the quantitative ratio between the variance and reference reads (color-coded); grey at this position indicates a lack of reads bearing the variant nucleotide.
The main framework of RNA2DNAlign is shown on Figure 2A. As an input, RNA2DNAlign requires two types of files derived from matching RNA and DNA: (i) lists of variants in variant call format (.vcf), and, (ii) corresponding aligned reads in binary alignment map (.bam) format. For each combination of four matching datasets of the type normal/tumor/RNA/DNA, RNA2DNAlign generates ten outputs: eight lists of SNVs meeting the definition for the described events, as follows: (a) RNAed, (b) T-RNAed, (c) VSE, (d) T-VSE, (e) VSL, (f) T-VSL), (g) SOM and (h) LOH, accompanied by (i) read count file containing reference and variant read counts for every examined SNV position and (j) summary report on the significant findings.
Figure 2.

(A) RNA2DNAlign workflow. RNA2DNAlign uses generated variant call files (.vcf) and binary alignments (.bam) derived from matching RNA and/or DNA sequencing datasets. Upon filtering of exonic positions (Module 1) the algorithm accesses each alignment file (Module 2) to read the counts and compute the likelihoods for all the possible genotypes (DNA) and allelic statuses (RNA). Module 3 then screens for significant genotypes and allelic statuses and compares them between the matching datasets to outline variants associated with any type of imbalance. (B) Relationship between input and output datasets. To assess for allele distribution matching an events, RNA2DNAlign requires a minimum of two datasets. SOM and LOH can be extracted from normal and tumor exomes; RNAed, VSE and VSL, can be assessed through comparisons between normal exomes and transcriptomes, and T-RNAed, T-VSE and T-VSL can be assessed through alignment of the tumor exomes to the normal and tumor transcriptomes. When all 4 datasets are available, RNA2DNAlign generates all 8 outcomes; otherwise it produces only the possible outcomes.
RNA2DNAlign can also work with less than four matching datasets (Figure 2B). The number of matching datasets (minimum two, maximum four per sample) is based on availability and specificity/sensitivity considerations. As we demonstrate below, using information from all four datasets increases the specificity, due to dual confirmation of the detected allele distribution. On the other hand, using the information from two datasets increases the comparable genomic regions.
To extract SNVs associated with differential allele distribution, RNA2DNAlign employs three consequent modules (see Figure 2A). The first module confines the testing to SNV loci within the genomic regions of interest. Depending on the intended comparisons, the filtering can range from a single position, to exonic regions, or to the entire genome (no filtering applied). In the herein demonstrated application, we have used a filter removing variants positioned outside known exonic intervals, and retaining only exonic variants for further analyses (exonic intervals on hg19 are provided with the package). We applied this filter to keep the tests within regions of cover for both exomes and transcriptomes, thus allowing meaningful comparisons between RNA and DNA when exome datasets are used.
The second step of the algorithm employs the pysam Python module to assess the read counts at every SNV position called in at least one of the datasets, in each of the matching alignments (.bam). A critical contribution from this module is the reference read count for datasets in which the variant is not called (such as exomes for RNA-editing, transcriptomes with monoallelic reference expression, etc.) as this value is typically not output by other tools. The script accesses every SNV position in each of the alignment files, filters aligned reads for length, gaps, mapping quality, and other read and alignment quality metrics, and tallies the remaining reads as having the expected reference or variant nucleotides.
Using the qualifying counts with reference (nR), variant (nV) or other (nO) nucleotides at each locus, this module computes scores to represent the strength of the read-count evidence for homozygous reference, homozygous variant, and heterozygous genotypes in the DNA-sequencing reads, and reference dominant, variant dominant, and bi-allelic expression in the RNA-Seq data. We construct binomial distribution-based probability models for each of the above three typical read-count patterns in DNA- and RNA-Seq reads. We will refer to these scores and models using the genomic terminology, even though we apply them to both DNA- and RNA-Seq reads. The primary role of these simple probability models is not to distinguish all possible allelic ratios, but instead, to reliably reject allelic read-count patterns that show little deviation from expected behavior. Of note, the read counts are provided in a separate output file and can be re-examined or re-analyzed to identify other genomic events of interest.
The binomial models we use assume that the allelic status of each read covering a position is sampled independently with respect to some underlying probability, such as 0.5 for heterozygote loci. Given X ∼ Binomial (0.5, nR + nV) we compute the heterozygous genotype likelihood as pHET = P (X ≥ max (nR,nV)). For homozygous likelihood models, we account for the possible, but unlikely observation of the variant (or reference allele) due to incorrect base calls instead of the alternative allele and use pseudo-counts to compensate for the limited number of observations. Using pseudo-count c = 0.5, we define nR' = nR + c, nV' = nV + c, nO' = nO + 2c, and the probability of the alternative allele from a homozygous genotype as q = nO'/2(nR' + nV' + nO'). Given Y ∼ Binomial (q, nR + nV + nO), we compute the reference homozygous genotype likelihood as pRefHOM = P(Y ≥ nV), and the variant homozygous genotype likelihood as pVarHOM = P(Y ≥ nR). The likelihoods are corrected for multiple testing with respect to the number of variant positions using the Benjamini–Hochberg false discovery rate (FDR) technique (18) and FDR values converted to a score via –10 log10 FDR. We transform the probabilities to FDR so that fixed score thresholds can be applied regardless of the number of variant loci.
The extent of the reference or variant imbalance in the RNA-Seq data is assessed by the RefDOM and VarDOM scores:
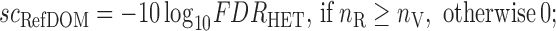 |
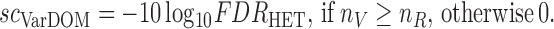 |
A log likelihood ratio style statistic is used to evaluate the competing genotype models, comparing each model's likelihoods against the most likely alternative:
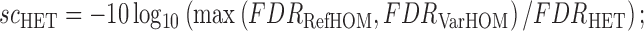 |
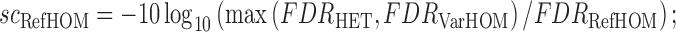 |
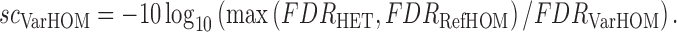 |
In each case the scores can be used as multiple-trial corrected tests for acceptance (large scores) or rejection (small scores) of specific genotype models at each locus at the level of DNA (scHET, scRefHOM, scVarHOM,) or the strength of the imbalance in the reference and variant allele counts (scRefDOM, scVarDOM) at the level of RNA. Accordingly, for each of the matching datasets, the SNVs are classified as heterozygous (HET), homozygous reference (RefHOM), or homozygous variant (VarHOM) for DNA, and bi-allelic (using scHET), reference dominant (via scRefHOM), or variant dominant (via scVarHOM) for RNA. For loci with heterozygous DNA alleles, we can use scRefDOM and scVarDOM to assess the degree of imbalance in RNA-Seq allelic expression.
The third module screens the scores to find loci matching the rules selected for read distribution corresponding to each event in a diploid genome (Table 1). We apply thresholds to the above binomial-distribution-based scores computed for normal and tumor, genomic and transcript reads in a rule-based fashion. These thresholds are empirically hand-tuned based on extensive validation using more than 3000 SNVs with known allelic asymmetries, such as known RNA-editing and somatic mutations, and variants in known imprinted genes (Supplementary Table S2; rules and score thresholds can be readily modified in an accompanying configuration file).
Table 1. Rules used to define events based on distribution of variant and reference read counts in matching RNA and DNA datasets from the same individual.
| RNAed | T-RNAed | VSE | T-VSE | VSL | T-VSL | LOH | SOM | |
|---|---|---|---|---|---|---|---|---|
| gDNA | REF HOM | REF HOM | HET | HET | HET | HET | HET | REF HOM |
| nRNA | BiAl or VAR DOM | REF DOM | VAR DOM | BiAl | REF DOM | BiAl | BiAl or REF DOM or VAR DOM | REF DOM |
| sDNA | REF HOM | REF HOM | HET | HET | HET | HET | REF HOM or VAR HOM | HET or VAR HOM |
| tRNA | BiAl or VAR DOM | BiAl or VAR DOM | VAR DOM | VAR DOM | REF DOM | REF DOM | REF DOM or VAR DOM | HET or VAR DOM |
Implementation and availability
The open-source RNA2DNAlign software is implemented in Python, and is available as a self-contained binary package for 64-bit Linux distributions source from http://github.com/HorvathLab/NGS and as Python source. The pysam package, plus a variety of common third-party python packages including numpy and scipy must be installed to use in Python source form. See the install instructions for more details. The self-contained binary package is appropriate for most Linux users. The software can be configured and executed on the command-line or via an interactive GUI.
RNA2DNAlign analysis of 360 datasets from 90 individuals
To demonstrate the use of the RNA2DNAlign, we applied it on 360 TCGA datasets from 90 female breast cancer patients from whom all of the following four sequencing datasets were available: Nex, Tex, Ntr, and Ttr (see Supplementary Table S1A). The results presented below are the outcome of RNA2DNAlign using high stringency settings (minimum of 10 reads aligned across each examined position in all four datasets), after pre-processing of the raw sequencing reads with an in-house pipeline (10–14).
RNA editing is acknowledged to significantly contribute to the transcriptome diversity in tumors (19–22). Using the information from all 4 datasets from the 90 breast cancer patients, RNA2DNAlign identified a total of 191 distinct exonic SNVs matching the read distribution corresponding to RNAed. The distribution, type and predicted function of the RNA editing events are plotted on Figure 3A. Of the 191 variants, 73 have not been previously reported in the DARNED, DbSNP and/or other RNA editing variation sources (23–29). Of note, the nonsynonymous variant in COG3 identified through integrated genome and transcriptome assessment of a single breast cancer patient (8), was seen in 18 samples of our dataset. The majority of the SNVs corresponded to transitions of either C>T (39%) or A>G (29%). The most frequently seen RNAed SNV was the previously reported RNA editing substitution on chr13:52604880 A>G, leading to a missense change (Q647R) in the gene UTP14C; this variant was called in 62 (69%) of the 90 samples (Figure 3B). Overall, a total of 632 exonic RNA editing events were called across the 90 individuals (Supplementary Table S3). In comparison, RNA2DNAlign called as T-RNAed 95 distinct exonic variants (Figure 3C, Supplementary Table S4), 37 of which were not present in RNA editing variation sources. An example of novel potential tumor-specific RNA-editing G>A at chr3:63981802, in the gene ATXN7 seen in multiple samples (Figure 3D). To ensure that the previously unreported positions sufficiently match the rules established for RNAed we examined each call individually (Supplementary Figure S1).
Figure 3.

(A) Circos plot of the variants matching the read distribution requirements for RNA editing across the 90 studied samples. The size of the peak corresponds to the number of samples (n) in which the variant was called (cut-off 1.5 after log10(n + 1)). Positions commonly assigned as RNAed are seen on chromosomes 3, 13, 16, 17, 22 and X. The outer layer displays a color scheme of the type of nucleotide substitution—C>T (light yellow) and A>G (pink) dominate over the rest of the changes. The central plot shows the substitution type (synonymous—teal, missense—blue), and the inner plot depicts the conservation score for the position (low—light yellow, medium—orange, high—dark orange). (B) IGV visualization of the most commonly called RNAed variant in the dataset (A>G at chr13:52604880 in UTP14C). (C) Circos plot of the variants matching the read distribution for T-RNAed across the 90 studied samples. The plots (from inner-to-outer) follow exactly the legend of (A). (D) IGV visualization of a novel variant called as T-RNAed—G>A at chr3:63981802 in ATXN7.
Variant specific expression/loss. Common regulatory mechanisms that lead to monoallelic expression include imprinting, (30), and cis-acting expression advantage or disadvantage provided by the variant-harboring allele (31). In addition, monoallelic expression can be observed where the transcripts carrying deleterious variants are degraded through surveillance mechanisms such as Nonsense Mediated mRNA Decay (NMD) (32). RNA2DNAlign identified 172 and 215 distinct exonic positions with overexpressed variant (VSE) or the reference (VSL) allele, respectively (Supplementary Tables S5 and S6). While, expectedly, some of the VSE/VSL variants resided in known imprinted genes, most of the genes from those datasets have not previously been associated with monoallelic expression. Examples of monoallelic SNVs include the overexpressed variant rs7334587 in the gene PARP4 (Supplementary Figure S2A), and the depressed variant allele of rs75085951 in PSPC1 (Supplementary Figure S2B). When we assessed the tumor-specific allelic expression, we identified 815 T-VSE and 830 T-VSL variants (Supplementary Tables S7 and S8). Among the redundant across samples observations are the T-VSE rs156697 in GSTO2 and the T-VSL rs144610753 in PER3 (33,34).
Somatic mutations. RNA2DNAlign identified 309 distinct exonic somatic mutations (total of 326) across the 90 breast cancer samples (Figure 4, Supplementary Table S9), 299 of which (99.6%) were present in COSMIC (35).
Figure 4.

Circos plot representation of the somatic variants found by RNA2DNAlign across the 90 studied samples. The grey area shows the distribution and the frequency of all somatic variants. The red band represents the least frequent but significant somatic mutation regions on various regions of the genome (lower frequency limit). The green band signifies the most frequent somatic mutations identified by RNA2DNAlign on different regions of genome. The orange color represents novel somatic variants. The highest frequency of somatic mutations was observed for regions chr19:44625648-55782060 followed by other regions of chromosome 19, 17, 13 and 22, respectively.
Loss of heterozygosity. RNA2DNAlign identified 668 distinct (685 total) SNV sites woth LOH (Figure 5, Supplementary Table S10).
Figure 5.

Summary of significant LOH loci throughout the genome for the 90 samples illustrated on Manhattan plot based on –log10(P) (A) and qqman plot (B). Altering colors dissociate each chromosome.
Performance analysis
To analyze the performance of RNA2DNAlign, we used two approaches in parallel. First, we used simulated datasets to assess the sensitivity of RNA2DNAlign as a function of the call base quality and the sequencing depth. Next, when applicable, we analyzed the intersection between our findings on the 90 breast cancer patients, and well-established databases of variants with the corresponding functionality.
To assess how the RNA2DNAlign sensitivity depends on the variant base quality and sequencing depth, we analyzed the variant detection rate on simulated data, containing artificially introduced SNVs (Figure 6). To generate the simulated data we used partitions of real fastq files, in which we replaced the reference with a variant base, at the same time keeping the original quality score. We used real data fastq files for our simulations in order to supply realistic quality and coverage metrics. Three different fastq partitions were extracted to represent genomic regions with low, medium and high coverage; the tests were run with the requirement for a minimum of 3, 5 and 10 total sequencing reads spanning each SNV position of consideration. As illustrated on Figure 6, for low quality calls, the variant detection rate strongly correlated with the base quality and the depth of sequencing, while for positions with quality scores over 50 the sensitivity approached 100% for all three levels of depth.
Figure 6.

Sensitivity of RNA2DNAlign as a function of base quality and minimum required total number of reads for consideration; performance on simulated datasets. Quality scores were broken into 10-point intervals; between 20 and 40 artificially introduced SNVs for each quality interval were assessed. For each of the eight types of imbalances, the variant read fractions were set according to Table 1; the tests were run with the requirement for a minimum of 3, 5 and 10 total sequencing reads spanning each SNV position of consideration. The sensitivity if RNA2DNAlign was measured as the fraction of positive calls over all introduced SNVs. Overall, similar sensitivity was observed across the different variation types. For quality scores below 50, the detection rate strongly correlated with the base quality and the depth of sequencing, while for positions with higher quality the sensitivity approached 100% for all three levels of depth. On positions covered with more than 10 reads. RNA2DNAlign showed higher than 80% detection rate for quality ranges above 20, and reached over 95% for quality ranges above 30, respectively.
To evaluate the performance of the binomial model at loci with very high coverage, we have explored the properties of scores across all loci. We currently cap the maximum score at 100 (representing FDR-corrected likelihood ratios of 1E-10), in part to avoid unreasonably large scores due to high coverage. First, we observe that most loci receive scores near zero or 100, with relatively few loci with intermediate values (Supplementary Figure S3), indicating that our analysis is not strongly affected by the specific choice of absolute score threshold. Second, if we partition the loci by coverage, we do not observe proportionately more high scores in the high-coverage loci; in fact, we observe the opposite, with more high scores in the low-coverage loci (See Supplementary Figure S3C). These empirical results suggest that false-positives due to the simple binomial based model and very high coverage loci are not common in our results.
Next, because the 90 samples in our study have been previously studied by other groups, genetic variants of different types have been reported in genomic sources. Among the types of variants identified by RNA2DNAlign, somatic mutations are the most extensively curated and documented (35–37); therefore, we selected to analyze the overlap between RNA2DNAlign output on somatic mutations and COSMIC. We extracted the somatic mutations in COSMIC on our dataset of 90 samples, and intersected with SOM lists called by RNA2DNAlign under different minimum read thresholds. As seen in Supplementary Figure S4, the sensitivity was inversely related to the minimum required read number. Next, we examined the somatic mutations listed in COSMIC and not called by RNA2DNAlign. The majority of the variants not called by RNA2DNAlign resided in positions with low or absent RNA-seq reads in the corresponding sample. Because RNA-seq read counts generally reflect expression levels, a major proportion of these missed variants is likely due to low or absent expression of the corresponding transcript. The remaining missed variants were not called by our default pipeline and hence were not considered by RNA2DNAlign.
The RNA2DNAlign output did not show significant deviations using datasets pre-processed through the aligners tested in this study (default settings). Expectedly, tuning the settings towards higher number of called variants (for example, including variants called on unpaired reads) led to an increased number of calls (data not shown).
The overall analysis shows that the major factors that impact the RNA2DNAlign sensitivity are: (i) minimum required read number, (ii) expression of the gene/transcript in the tissue from which RNA was derived and (iii) number and combination of considered datasets. Naturally, the design confines the analyses to (a) loci expressed in the tissue of interest, and (b) regions comparable between the analyzed datasets (exons, in our example). Hence the lists of variants called by RNA2DNAlign are not exhaustive for the entire genome, but rather represent positions co-covered by the studied sequencing libraries, thus allowing estimation of read distribution asymmetries. Therefore, substantially higher number of calls is achieved when using two matching datasets. Since comparisons outside exome-to-transcriptome, such as exome-to-exome, or transcriptome-to-transcriptome, are not restricted to exonic positions only, using two datasets can increase the comparable sequenced regions with informative loci, such as exon flanking areas when comparing exomes, or non-coding RNAs when comparing transcriptomes, thus additionally increasing the number of the identified SNVs. Also, using only two datasets reduces the number of variants ignored due to random sequencing inconsistencies, such as accidental low coverage in some datasets. Of note, running the same matching datasets in different combinations allows aiming both at high specificity (4 matching datasets) and higher detection rate (<4 appropriate matching datasets according to Figure 2B).
It is noteworthy that RNA2DNAlign identified somatic mutations that have not been reported before (Supplementary Figure S5). Our analysis shows that the main proportion of these variants was called exclusively in the tumor transcriptome with our default pipeline (and not in the tumor exome), which is likely the reason why they have not been reported so far. Because RNA2DNAlign assesses each dataset at every position called in at least one of the datasets, it is poised to identify variants challenging for some of the call settings, likely due to a different stringency dynamics of the variant calls between datasets (exome and transcriptome in our example).
DISCUSSION
With the increasing availability of individual omics-scale sequencing data, the bio-medical community will significantly benefit from tools enabling integration of DNA and RNA data, which is expected to reach far beyond linear addition of the separate layers of information. Apart from several recent efforts (38–41), tools integrating DNA and RNA sequencing information at nucleotide resolution on a large scale are missing. RNA2DNAlign is a free software application for large-scale quantitation of variant and reference reads' distribution between DNA and RNA sequencing datasets.
In the herein demonstrated application, RNA2DNAlign identified a total of 2038 high confidence distinct exonic variants (5800 across the 90 breast cancer patients), associated with allelic imbalance of different kinds (Table 2 and Figure 7). Many of the 2038 SNVs were already linked to the respective event, which supported the general confidence of our algorithms. Most, however, have never been reported in the regarded context, and thus comprise novel findings. An example of significant novel outcome is the comprehensive list of variants presenting with monoallelic expression—VSE, T-VSE, VSL and T-VSL—totaling 2133 SNVs, the vast majority of which have not been reported associated with imprinting or other allele-specific expression. It is important to note that while the RNA2DNAlign called SNVs can cause, contribute, or result from the respective event, they may also randomly reside on the highlighted allele, and downstream studies are required to distinguish between driving and passengers variants.
Table 2. Overall characteristics of the frequency and distribution of exonic SNVs identified through RNA2DNAlign to be associated with allelic imbalance reflecting eight regulatory events. The numbers reflect the summary statistics across 90 samples.
| RNAed | T-RNAed | VSE | T-VSE | VSL | T-VSL | LOH | SOM | TOTAL | |
|---|---|---|---|---|---|---|---|---|---|
| Unique | 191 | 94 | 171 | 815 | 215 | 830 | 685 | 309 | 2038 |
| Total | 632 | 103 | 616 | 929 | 585 | 929 | 668 | 329 | 5800 |
| Average | 9 | 2 | 9 | 19 | 9 | 20 | 22 | 4 | 64 |
| Median | 7 | 1 | 6 | 6 | 5 | 6 | 2 | 3 | 22 |
| Range | 1-22 | 0-16 | 1-16 | 0-91 | 0-26 | 0-65 | 0-211 | 0-15 | 6-229 |
Figure 7.

Distribution and frequency of all 8 types of allele imbalances in the 90 studied samples.
Of note, all the above variants are identified using very high stringency settings of RNA2DNAlign, defined through: (i) consideration of four NGS datasets of the type tumor/normal/exome/transcriptome, (ii) minimum requirement of 10 sequencing reads in all four datasets for each examined position and (iii) restriction of the tests to exonic regions only. Relaxing of any of the above requirements substantially increases the number of the calls. For example, setting the minimum required read number to three resulted in 8.6-fold increase in the number of significant calls in the same samples (17 577 distinct significant exonic positions, data not shown). Using the information from all four datasets, expression-specific elements could be found through exome-to-transcriptome comparison, and cancer-related changes could be outlined through comparison between the normal and tumor datasets. Such approach is set to provide higher specificity due to cross-validation of the observation in more than one dataset (i.e. VSE is assigned at positions with variant specific expression in both the normal and the tumor transcriptome, LOH is assigned to positions with monoallelic reads in both tumor exome and tumor transcriptome, etc.). Thus, RNA2DNAlign addresses the well-regarded challenge of high noise of variant calls produced by NGS sequencing.
For applications of RNA2DNAlign that use previously generated alignments and variation calls, it is important to consider that its outputs will depend on the used pipelines and their stringency settings, with a major expected impact towards the false negative variant calls (i.e. calls missed by the variant caller). By default, the RNA2DNAlign design reduces this effect as it inspects the matching datasets for all positions called in any of the datasets. Thus, only SNVs missed in all of the relevant datasets will not be assessed for allele distribution. Of note, in addition to post-aligner-caller applications, RNA2DNAlign could work directly with pre-aligned datasets and lists of positions of interest (for example TCGA alignments, and dbSNP, COSMIC or DARNED variants, or any custom pre-defined list). Such an approach can be used to determine the allelic behavior of positions of interest in the particular datasets. Importantly, in addition to the positions involved in the eight events, RNA2DNAlign outputs the read counts for all the submitted positions, including those not qualifying for any of the 8 events; this output can be used for variety of custom allele-quantifying applications.
Notably, RNA2DNAlign is designed for diploid genomes, and the tumor genomes often present with local or massive ploidy alterations. While the generic definitions used by RNA2DNAlign are expected to be preserved for many ploidy alterations, loci different from diploid need to be treated with conscious. For example, for regions of unknown ploidy, LOH output positions need to be considered as indicative for change from hetero- to mono-allelic status, inclusive but not restricted to changes from di- to monoploid status.
An important challenge addressed by RNA2DNAlign is the limited compatibility between RNA and DNA datasets, more obvious when using exomes. While largely consistent with transcriptomes over exonic regions, exomes present with extra coverage at the exon-flanking areas (usually targeted by the exome capture), and with all the known genes, as compared to only the expressed ones in the tissue from which the RNA was derived. On the other hand, transcriptomes present with a multitude of non-coding expressed sequences that are not targeted by the exome capture. Our design addresses this incompatibility in two ways: (i) the option for filtering, which confines the comparisons within regions targeted of both sequencing approaches, and, (ii) the requirement for minimum of reads at every assessed SNV position, which directs the tests to the expressed genes in the tissue of the RNA-origin. It is noteworthy that RNA2DNAlign is different from approaches optimized towards identifying of allelic imbalances from RNA data alone, which often address biases caused by unavailability of matching DNA (26,42–50). By design, RNA2DNAlign integrates multiple signals from regions co-covered by RNA and DNA sequencing, aiming to identify all possible biologically meaningful allelic asymmetries.
In summary, RNA2DNAlign possesses several important advantages that support novel types of applications. First, the simultaneous assessment of a position in multiple matching datasets supports novel nucleotide-resolution analyses, for example, expression for the DNA-confined events LOH and SOM, normal vs tumor comparisons, etc. Second, this is the first tool to simultaneously produce eight different outputs of SNVs associated with major molecular events, thus allowing multilevel within-sample variant functionality assessments. Third, the read-count output supports numerical operations towards fine quantitation of allelic abundance, including the reference allele-count for positions with no variant, which can prompt various customized downstream analyses. Next, in terms of specificity and sensitivity, the application supports user control both through number of considered datasets, and adjustment of the stringency settings. Related to that, RNA2DNAlign workflow efficiency and high processing speed allows running datasets under multiple settings in parallel, aiming both at specificity and sensitivity in a high-throughput manner. In addition, to screen for allele-specific variants potentially implicated in alternative splicing RNA2DNAlign can be run in parallel with a tool for molecular phasing of variants with an alternative junction/boundary – SNPlice (41), which utilizes the same input files and interface. Finally, the application of RNA2DNAlign does not require computational skills or script writing, and can be run through user-friendly interface by wide range of research- and clinically-oriented users.
Supplementary Material
Acknowledgments
The authors would like to thank Nick Hathaway for his guidance in the use of the qqman Package for data analysis.
Author's contribution: M.M., with assistance of N.A., wrote the computational script. N.E. implemented the read counting and statistical significance framework for genotype models. M.M. performed the raw large data analysis. M.M. and N.A., with the assistance of P.M., M.D., C.D., P.R., E.C., S.B., K.K., M.L., W.W. and K.A. handled further analyses of the large data. M.M. and A.H. prepared the manuscript. A.H. and N.E. conceived the framework and devised the analytical approach. A.H. designed the project and the analytical strategy and supervised the work.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
McCormick Genomic and Proteomic Center (MGPC), GWU; NIH National Center for Advancing Translational Sciences [UL1TR000075]; Clinical and Translational Science Institute at Children's National Medical Center [CTSI-CN to A.H.]; Katzen Innovative Cancer Research Pilot Grant Program [RG0982 to A.H.]. Funding for open access charge: McCormick Genomic and Proteomic Center (MGPC), George Washington University.
Conflict of interest statement. None declared.
REFERENCES
- 1.He Q., He Q., Liu X., Wei Y., Shen S., Hu X., Li Q., Peng X., Wang L., Yu L. Genome-wide prediction of cancer driver genes based on SNP and cancer SNV data. Am. J. Cancer Res. 2014;4:394–410. [PMC free article] [PubMed] [Google Scholar]
- 2.Chan T.H., Lin C.H., Qi L., Fei J., Li Y., Yong K.J., Liu M., Song Y., Chow R.K., Ng V.H., et al. A disrupted RNA editing balance mediated by ADARs (Adenosine DeAminases that act on RNA) in human hepatocellular carcinoma. Gut. 2014;63:832–843. doi: 10.1136/gutjnl-2012-304037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gallo A. RNA editing enters the limelight in cancer. Nat. Med. 2013;19:130–131. doi: 10.1038/nm.3072. [DOI] [PubMed] [Google Scholar]
- 4.Valle L., Serena-Acedo T., Liyanarachchi S., Hampel H., Comeras I., Li Z., Zeng Q., Zhang H.T., Pennison M.J., Sadim M., et al. Germline allele-specific expression of TGFBR1 confers an increased risk of colorectal cancer. Science. 2008;321:1361–1365. doi: 10.1126/science.1159397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watson I.R., Takahashi K., Futreal P.A., Chin L. Emerging patterns of somatic mutations in cancer. Nat. Rev. Genet. 2013;14:703–718. doi: 10.1038/nrg3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiagalingam S., Laken S., Willson J.K., Markowitz S.D., Kinzler K.W., Vogelstein B., Lengauer C. Mechanisms underlying losses of heterozygosity in human colorectal cancers. Proc. Natl. Acad. Sci. U.S.A. 2001;98:2698–2702. doi: 10.1073/pnas.051625398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacArthur D.G., Manolio T.A., Dimmock D.P., Rehm H.L., Shendure J., Abecasis G.R., Adams D.R., Altman R.B., Antonarakis S.E., Ashley E.A., et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508:469–476. doi: 10.1038/nature13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah S.P., Morin R.D., Khattra J., Prentice L., Pugh T., Burleigh A., Delaney A., Gelmon K., Guliany R., Senz J., et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 9.Wilks C., Cline M.S., Weiler E., Diehkans M., Craft B., Martin C., Murphy D., Pierce H., Black J., Nelson D., et al. The Cancer Genomics Hub (CGHub): overcoming cancer through the power of torrential data. Database (Oxford) 2014;2014:bau093. doi: 10.1093/database/bau093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. Genome project data processing S: the sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cunningham F., Amode M.R., Barrell D., Beal K., Billis K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fitzgerald S., et al. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–D669. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thorvaldsdóttir H., Robinson J.T., Mesirov J.P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinformatics. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reiner A., Yekutieli D., Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 2003;19:368–375. doi: 10.1093/bioinformatics/btf877. [DOI] [PubMed] [Google Scholar]
- 19.Li S., Mason C.E. The pivotal regulatory landscape of RNA modifications. Annu. Rev. Genomics Hum. Genet. 2014;15:127–150. doi: 10.1146/annurev-genom-090413-025405. [DOI] [PubMed] [Google Scholar]
- 20.Zipeto M.A., Jiang Q., Melese E., Jamieson C.H. RNA rewriting, recoding, and rewiring in human disease. Trends Mol. Med. 2015;21:549–559. doi: 10.1016/j.molmed.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Paz-Yaacov N., Bazak L., Buchumenski I., Porath H.T., Danan-Gotthold M., Knisbacher B.A., Eisenberg E., Levanon E.Y. Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep. 2015;13:267–276. doi: 10.1016/j.celrep.2015.08.080. [DOI] [PubMed] [Google Scholar]
- 22.Han L., Diao L., Yu S., Xu X., Li J., Zhang R., Yang Y., Werner H.M., Eterovic A.K., Yuan Y., et al. The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell. 2015;28:515–528. doi: 10.1016/j.ccell.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiran A.M., O'Mahony J.J., Sanjeev K., Baranov P.V. Darned in 2013: inclusion of model organisms and linking with Wikipedia. Nucleic Acids Res. 2013;41:D258–D261. doi: 10.1093/nar/gks961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fumagalli D., Gacquer D., Rothe F., Lefort A., Libert F., Brown D., Kheddoumi N., Shlien A., Konopka T., Salgado R., et al. Principles Governing A-to-I RNA Editing in the Breast Cancer Transcriptome. Cell Rep. 2015;13:277–289. doi: 10.1016/j.celrep.2015.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramaswami G., Zhang R., Piskol R., Keegan L.P., Deng P., O'Connell M.A., Li J.B. Identifying RNA editing sites using RNA sequencing data alone. Nat. Methods. 2013;10:128–132. doi: 10.1038/nmeth.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paul M.R., Levitt N.P., Moore D.E., Watson P.M., Wilson R.C., Denlinger C.E., Watson D.K., Anderson P.E. Multivariate models from RNA-Seq SNVs yield candidate molecular targets for biomarker discovery: SNV-DA. BMC Genomics. 2016;17:263. doi: 10.1186/s12864-016-2542-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng Z., Cheng Y., Tan B.C., Kang L., Tian Z., Zhu Y., Zhang W., Liang Y., Hu X., Tan X., et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol. 2012;3:253–260. doi: 10.1038/nbt.2122. [DOI] [PubMed] [Google Scholar]
- 29.Bahn J.H., Lee J.H., Li G., Greer C., Peng G., Xiao X. Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res. 2012;22:142–150. doi: 10.1101/gr.124107.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peters J. The role of genomic imprinting in biology and disease: an expanding view. Nat. Rev. Genet. 2014;15:517–530. doi: 10.1038/nrg3766. [DOI] [PubMed] [Google Scholar]
- 31.Proudhon C., Bourc'his D. Identification and resolution of artifacts in the interpretation of imprinted gene expression. Brief. Funct. Genomics. 2010;9:374–384. doi: 10.1093/bfgp/elq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lykke-Andersen S., Jensen T.H. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015;16:665–677. doi: 10.1038/nrm4063. [DOI] [PubMed] [Google Scholar]
- 33.Kim J.I., Ju Y.S., Park H., Kim S., Lee S., Yi J.H., Mudge J., Miller N.A., Hong D., Bell C.J., et al. A highly annotated whole-genome sequence of a Korean individual. Nature. 2009;460:1011–1015. doi: 10.1038/nature08211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berg T., Hegelund Myrback T., Olsson M., Seidegard J., Werkstrom V., Zhou X.H., Grunewald J., Gustavsson L., Nord M. Gene expression analysis of membrane transporters and drug-metabolizing enzymes in the lung of healthy and COPD subjects. Pharmacol. Res. Perspect. 2014;2:e00054. doi: 10.1002/prp2.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forbes S.A., Beare D., Gunasekaran P., Leung K., Bindal N., Boutselakis H., Ding M., Bamford S., Cole C., Ward S., et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805–D811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E., et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cibulskis K., Lawrence M.S., Carter S.L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E.S., Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Picardi E., Pesole G. REDItools: high-throughput RNA editing detection made easy. Bioinformatics. 2013;29:1813–1814. doi: 10.1093/bioinformatics/btt287. [DOI] [PubMed] [Google Scholar]
- 39.Mayba O., Gilbert H.N., Liu J., Haverty P.M., Jhunjhunwala S., Jiang Z., Watanabe C., Zhang Z. MBASED: allele-specific expression detection in cancer tissues and cell lines. Genome Biol. 2014;15:405. doi: 10.1186/s13059-014-0405-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mudvari P., Kowsari K., Cole C., Mazumder R., Horvath A. Extraction of molecular features through exome to transcriptome alignment. J. Metabolomics Syst. Biol. 2013;22:1. doi: 10.13188/2329-1583.1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mudvari P., Movassagh M., Kowsari K., Seyfi A., Kokkinaki M., Edwards N.J., Golestaneh N., Horvath A. SNPlice: Splice-modulating SNPs from RNA-sequencing data. Bioinformatics. 2014;31:1191–1198. doi: 10.1093/bioinformatics/btu804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leon-Novelo L.G., McIntyre L.M., Fear J.M., Graze R.M. A flexible Bayesian method for detecting allelic imbalance in RNA-seq data. BMC Genomics. 2014;15:920. doi: 10.1186/1471-2164-15-920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang K., Li J.B., Gao Y., Egli D., Xie B., Deng J., Li Z., Lee J.H., Aach J., Leproust E.M., et al. Digital RNA allelotyping reveals tissue-specific and allele-specific gene expression in human. Nat. Methods. 2009;6:613–618. doi: 10.1038/nmeth.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Degner J.F., Marioni J.C., Pai A.A., Pickrell J.K., Nkadori E., Gilad Y., Pritchard J.K. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics. 2009;25:3207–3212. doi: 10.1093/bioinformatics/btp579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Main B.J., Bickel R.D., McIntyre L.M., Graze R.M., Calabrese P.P., Nuzhdin S.V. Allele-specific expression assays using Solexa. BMC Genomics. 2009;10:422. doi: 10.1186/1471-2164-10-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fontanillas P., Landry C.R., Wittkopp P.J., Russ C., Gruber J.D., Nusbaum C., Hartl D.L. Key considerations for measuring allelic expression on a genomic scale using high-throughput sequencing. Mol. Ecol. 2010;19(Suppl. 1):212–227. doi: 10.1111/j.1365-294X.2010.04472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Satya R.V., Zavaljevski N., Reifman J. A new strategy to reduce allelic bias in RNA-Seq readmapping. Nucleic Acids Res. 2012;40:e127. doi: 10.1093/nar/gks425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skelly D.A., Johansson M., Madeoy J., Wakefield J., Akey J.M. A powerful and flexible statistical framework for testing hypotheses of allele-specific gene expression from RNA-seq data. Genome Res. 2011;21:1728–1737. doi: 10.1101/gr.119784.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fogarty M.P., Xiao R., Prokunina-Olsson L., Scott L.J., Mohlke K.L. Allelic expression imbalance at high-density lipoprotein cholesterol locus MMAB-MVK. Hum. Mol. Genet. 2010;19:1921–1929. doi: 10.1093/hmg/ddq067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tuch B.B., Laborde R.R., Xu X., Gu J., Chung C.B., Monighetti C.K., Stanley S.J., Olsen K.D., Kasperbauer J.L., Moore E.J., et al. Tumor transcriptome sequencing reveals allelic expression imbalances associated with copy number alterations. PLoS One. 2010;5:e9317. doi: 10.1371/journal.pone.0009317. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.