

Abstract
Uncovering genetic variation through resequencing is limited by the fact that only sequences with similarity to the reference genome are examined. Reference genomes are often incomplete and cannot represent the full range of genetic diversity as a result of geographical divergence and independent demographic events. To more comprehensively characterize genetic variation of pigs (Sus scrofa), we generated de novo assemblies of nine geographically and phenotypically representative pigs from Eurasia. By comparing them to the reference pig assembly, we uncovered a substantial number of novel SNPs and structural variants, as well as 137.02-Mb sequences harboring 1737 protein-coding genes that were absent in the reference assembly, revealing variants left by selection. Our results illustrate the power of whole-genome de novo sequencing relative to resequencing and provide valuable genetic resources that enable effective use of pigs in both agricultural production and biomedical research.
Sus scrofa (i.e., pig or swine) is of enormous agricultural importance and also an attractive model for biomedical research and applications. There are over 730 distinct pig breeds worldwide, of which two thirds reside in Europe and China (Chen et al. 2007), whose diverse phenotypes are shaped by the combined effects of local adaptation and artificial selection (Ai et al. 2015). Efforts have been made to characterize the genetic variation that underlies this phenotypic diversity using resequencing data and the genome of the European domestic Duroc pig as a reference (Groenen et al. 2012; Rubin et al. 2012; Choi et al. 2015; Moon et al. 2015). Nonetheless, resequencing is limiting in terms of capturing genetic variation and assessing gaps and misassigned regions of the reference genome (Weisenfeld et al. 2014). In contrast, multiple de novo assemblies of pig genomes from different regions and breeds promise a more accurate and comprehensive understanding of genetic variation within this species (Besenbacher et al. 2015; Chaisson et al. 2015b). Among populations of plants (coccolithophores [Read et al. 2013], Arabidopsis thaliana [Gan et al. 2011], soybean [Li et al. 2014], and rice [Zhang et al. 2014]), animals (mosquitoes [Neafsey et al. 2015] and macaques [Yan et al. 2011]), and even modern humans (Li et al. 2010a), a surprisingly large amount of variation has been uncovered by de novo assemblies.
To advance the characterization of the genetic diversity of pigs, we generated de novo assemblies of nine geographically and phenotypically representative individuals from Eurasia. Combining this resource with genome assembly of the Tibetan wild boar (Li et al. 2013), we carried out in-depth comparisons between 10 de novo assemblies and the reference genome. We uncovered a substantial number of single nucleotide polymorphisms (SNPs) and structural variants, as well as hundreds of millions of base pairs that are not present in the reference genome, including thousands of protein-coding genes that are either missing or fragmented in the reference genome, which contain potentially important genetic information pertaining to porcine evolution.
Results
De novo genome assemblies of nine pig breeds
We sequenced the genomes of nine female individuals from nine diverse breeds (five originated in Europe and four originated in China) to an average of ∼100-fold coverage (∼229.5 gigabase [Gb]) using Illumina sequencing technology and a whole-genome shotgun strategy (Supplemental Fig. S1; Supplemental Table S1). The genomes were independently assembled using SOAPdenovo (Supplemental Methods; Li et al. 2010b), which yielded contig N50 sizes of 28.99 to 42.66 kilobases (kb), scaffold N50 sizes of 1.26 to 2.45 megabases (Mb), and a total of 2.45 to 2.49 Gb of ungapped sequences that exhibited genomic features similar to those of the reference assembly (Supplemental Figs. S2–S7; Supplemental Tables S2–S6; Groenen et al. 2012). We also improved the available genome assembly of the Tibetan wild boar (Li et al. 2013) by increasing the contig N50 size from 20.69 kb to 22.54 kb and the ungapped genome assembly size from 2.43 Gb to 2.44 Gb (Supplemental Table S4).
Discovery and characterization of SNPs
We identified 8.86–15.95 million (M) SNPs in individual pig genomes using an assembly-versus-assembly approach (Supplemental Methods). These SNPs were consistent with more than 98% SNPs identified from the Illumina's porcine 60K Genotyping BeadChip (v.2) (Supplemental Table S7) and covered most SNPs identified by resequencing as implemented in SAMtools (Li et al. 2009) (98.78%) and GATK (McKenna et al. 2010) (97.65%) and 3.12–5.40 M SNPs (33.46%–35.25%) in divergent regions that failed to be cataloged by these algorithms, where unassembled short reads are difficult to be mapped (Fig. 1; Supplemental Figs. S8, S9).
Figure 1.
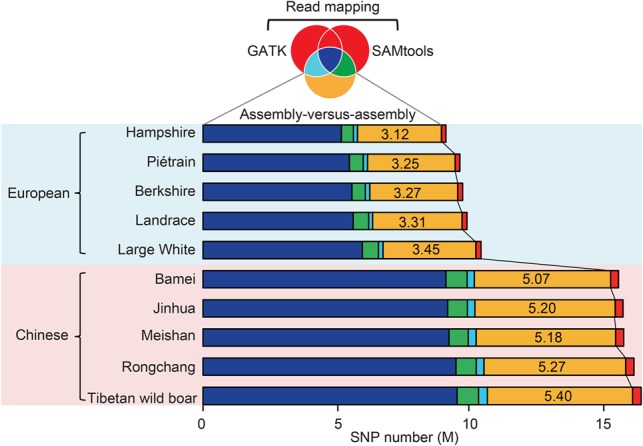
Comparison of SNP calling between the assembly-versus-assembly method and resequencing approaches based on read mapping. The Venn diagram with colors corresponding to the bar chart shows the sharing of identified SNPs among the assembly-versus-assembly method and two resequencing algorithms as implemented in SAMtools and GATK. An average of 4.25 M SNPs per breed were specifically identified by the assembly-versus-assembly method (marked as yellow), while only 0.24 k SNPs per breed were categorized by resequencing approaches (marked as red). A significant fraction of the detected SNPs by SAMtools (8.11 M per individual) and GATK (7.77 M per individual) was coincident (7.41 M; or 91.24% of SAMtools and 95.34% of GATK) (Supplemental Fig. S8).
Extensive intercontinental genomic divergence was reflected by the significantly larger amount of variation of Chinese pigs when compared to the reference Duroc genome of European origin (15.14–15.95 M SNPs; the Ts/Tv ratio: 2.13 to 2.15) than that between European pigs and Duroc (8.86–10.14 M SNPs; the Ts/Tv ratio: 1.95 to 1.99) (P < 10−16, Mann–Whitney U test) (Fig. 2A,B; Supplemental Fig. S10), attributable to considerable divergence time between European and Asian lineages (at least 1 million yr) and their independent domestication in multiple locations across Eurasia in the past ∼10,000 yr (Larson et al. 2005; Groenen et al. 2012; Frantz et al. 2013).
Figure 2.

Genomic variation between Chinese and European pigs. (A) Geographic locations of the original pig breeds. The Duroc (donor of the reference genome; it is denoted by a star) and Hampshire pigs were developed mainly in North America but originated in Europe. (B) Neighbor-joining phylogenetic tree, number of SNPs, transition/transversion ratio (Ts/Tv), heterozygous SNP ratio, patterns of regions of homozygosity (ROHs), and length and number of indels in the 10 breeds (left to right). Violin plots of the heterozygous SNP ratio and Ts/Tv ratio were generated using nonoverlapping 1-Mb windows (the medians are shown). For ROH, the circled area indicates the total length of ROHs in each breed. (C) Pairwise genomic similarity of Chinese and European pigs by identical score (IS) values within each 10-kb window across the genome (n = 259,511).
We also observed higher genomic diversity of Chinese pigs than European pigs, reflected by the former's higher heterozygous SNP ratio (2.17 × 10−3 to 2.69 × 10−3 vs. 0.94 × 10−3 to 1.63 × 10−3) and lower homozygosity (382 regions of homozygosity [ROHs] with a total size of 107.5 Mb vs. 907 ROHs totaling 289.9 Mb per assembly) (P < 10−16, Mann–Whitney U test) (Fig. 2B; Supplemental Figs. S11, S12). Principal component analysis (PCA) and identity score (IS) analysis of pairwise breed genomes also recapitulated these findings (Fig. 2C; Supplemental Fig. S13A). This may be a reflection of the fact that European origin breeds have undergone intense selection in inbred commercial lines for economical traits, while Chinese breeds experienced weaker selection in scattered, individual farms and exhibited relatively weak linkage disequilibrium (LD) (Supplemental Fig. S13B; White 2011). Another possible explanation is that European wild boars (ancestors of European domestic pigs) may have suffered more pronounced population bottlenecks during the last glacial maximum (∼20,000 yr ago) compared to their Asian counterparts (Bosse et al. 2012; Groenen et al. 2012).
We pooled the SNPs of 10 breeds into a nonredundant set of 33.60 M sites that account for ∼81.25% of the estimated repertoire of SNPs in the pig (Supplemental Fig. S14; Supplemental Table S8; Supplemental Methods), of which 6.34 M (18.87% of 33.60 M) SNPs were considered to be novel based on their absence in the pig dbSNP (Build 143) entries (Supplemental Fig. S15). Compared with synonymous SNPs (122.44 k), missense SNPs (83.39 k) exhibited greater diversity among breeds (77.44% of the estimated repertoire compared to 80.61%), accounted for a larger proportion of breed-specific (and thus rare) SNPs (32.42% compared to 30.18%), and had a higher ratio of homozygous to heterozygous SNPs (0.37 compared to 0.32) (Supplemental Figs. S14, S16), which may be associated with breed-specific adaptation.
Maps of structural variation
We detected 161.45–279.98 k insertions (15.99–23.07 Mb in length) and 137.89–269.55 k deletions (3.61–5.63 Mb in length) in individual genomes against the reference genome (Fig. 2B; Supplemental Table S9; Supplemental Methods). More than 80% of the insertions and deletions (indels) were 1–10 bp in length, and there was also a relatively high number of indels ∼300 bp in length, due to the enrichment of indels of tRNAGlu-derived short interspersed element (SINE/tRNAGlu) (Supplemental Fig. S17; Ai et al. 2015). Repetitive elements (38.05% of the genome) comprised ∼52.73% of indels, which are an important source of structural variation in the pig genome. Moreover, the SINE/tRNAGlu (290.47 Mb and containing 18.09% of indels) showed higher incidence of indels than the predominant long interspersed elements (LINE/L1) (636.50 Mb and containing 15.48% of indels) (Supplemental Fig. S18).
The indels appeared to be regulated by selection: most indels were located in intergenic regions (72.20%–74.14%), the indel ratio was lower in the coding sequences than in introns (Supplemental Fig. S19), and more conserved genes showed fewer structural variants (Supplemental Fig. S20). We observed an enrichment of short indels (1–15 bp in length) in coding sequences (414 of 1582, or 26.17%) that were multiples of 3 bp, which is expected to preserve the reading frame, and identified 1152 frameshift mutations in 947 genes (Supplemental Fig. S21; Supplemental Table S10), which mainly represented the cellular functions of ‘binding of nucleoside, ATP, and cation’ and ‘neuron development’ (Supplemental Table S11). As with the SNPs, distribution of indels across the genome also reflected a deep phylogenetic split between European and Chinese pigs and higher genetic variability of Chinese pigs than European pigs (Supplemental Fig. S22; Bosse et al. 2012; Groenen et al. 2012).
Signatures of diversifying selection in pig breeds
To uncover genetic variation underlying phenotypic diversity of pigs, we identified breed-specific signatures left by diversifying selection using a relative homozygous SNP density (RSD) algorithm (Supplemental Methods; Atanur et al. 2013). We identified 493 separate genomic regions of 20–150 kb (a total of 20.10 Mb and containing 308 genes) to be under selection (FDR < 0.05) (Fig. 3A; Supplemental Table S12). These putative selected regions also exhibited significantly strong LD and lower negative Tajima's D-values (P < 10−16, Mann–Whitney U test) (Supplemental Fig. S23), and distinct phylogenetic relationships compared to the genomic background (Supplemental Fig. S24).
Figure 3.

Identification of breed-specific selective sweeps. (A) Number of homozygous SNPs in breed-specific selected regions. Of 74.21 k homozygous SNPs in 20.10 Mb selected regions, 65.75 k (88.60%) were unique to a particular breed, which was highly concentrated in a small fraction (0.79%) of the genome and likely contributed to diversifying selection. (B) Selective sweep regions identified in the Rongchang pig. (Top panels, top half) Genes residing within or in the vicinity (±5 kb) of the selected regions are presented for each chromosome and ordered according to their locations. (Top panels, lower half) Degree of haplotype sharing of selected regions in pairwise comparisons among the 10 breeds. Homozygous SNP frequencies in individual breeds were used to calculate identity scores in 10-kb windows. Boxes (left) indicate pairwise comparison presented on that row (E, European pigs; C, Chinese pigs) according to the color assigned to each pig breed (right). Heat map colors indicate identity scores. (Middle panel) Percentage stacked column showing RSD values in the Rongchang-specific selected regions across 10 breeds sequenced. Rongchang showed predominantly higher RSD values than other breeds, indicating that only this breed has SNPs compared to the reference genome in this region. (Bottom half) RSD in 10-kb windows for Rongchang plotted along chromosomes. Black lines indicate selected regions (FDR < 0.05). Nine selected genes orthologous to the mammalian fat deposition genes are marked in red.
Most homozygous SNPs (88.60%) in the selected regions were unique to a particular breed (Fig. 3A), exhibiting a lower degree of haplotype sharing with other breeds than pairwise between other breeds (Fig. 3B; Supplemental Fig. S25). These private SNPs were highly concentrated in a small number of discrete genomic regions (0.79% of the genome) and may be associated with phenotypes described by standard breed criteria (Wang et al. 2011): typically, nine (out of 49, or 18.37%; P = 0.004, χ2 test) and six (out of 59, or 10.17%; P = 0.491, χ2 test) genes within or in the vicinity of the selected regions in the fatty Rongchang and Jinhua pigs were orthologous to well-established mammalian fat deposition genes (Fig. 3B; Supplemental Fig. S25A; Kunej et al. 2013), including factors involved in the regulation of feed intake and energy homeostasis (CEP120, GABRA2, NPPA, NPY1R, and NYP5R), lipid metabolism (ABCC4, ANGPT2, LRPAP1, and PRKAG2), and indicators of obesity-induced hypertension, inflammatory signaling, and insulin resistance (ADD1, HSPD1, MMP2, PIK3R4, RAE1, and TBCA) (Supplemental Table S13). In contrast to highly inbred European pigs that have undergone selection for lean growth (high protein and low fat content; lean meat percentage of the carcass ranging between 63%–73%) as a response to demands for reduced calorie intake in modern society, Chinese pigs have been selected for extreme fatness all along (typical lean meat percentage is under 45%) (Supplemental Fig. S1), driven by the demand for energy-rich food in developing countries until ∼10 yr ago (Wang et al. 2011).
We also identified 16 (31.37%; P = 8.21 × 10−11, χ2 test) out of 51 genes with strong selective sweep signals in the Tibetan wild boar (Supplemental Fig. S25B; Supplemental Table S13) that were likely driven by the harsh and hypoxic environment of the Tibetan plateau and might have a role in the formation of characteristic phenotypes, such as an insulating layer formed by hard skin and long, dense hair, and larger lungs and hearts (Li et al. 2013).
Identifying missing sequences of the reference pig genome
There are considerable unidentified regions (289.24 Mb of 2.81 Gb, or 10.29%) in the reference pig assembly (Sscrofa10.2) (Groenen et al. 2012), of which 266.15 Mb (91.92%) is composed of 5317 gaps of at least 50 kb long (Supplemental Fig. S26). To recover such missing genetic information, we retrieved ∼9.17 G ‘orphan reads’ for which neither end mapped to the reference genome (Supplemental Fig. S27) and relocalized them to their respective assemblies of origin. Consequently, we identified 83.8 k sequences of ≥500 bp (137.02 Mb in length) that were missing in the reference genome (Table 1; Supplemental Table S14). Only a small portion of missing sequences was considered to be insertions (∼0.91 Mb) or copy number gains (∼4.16%) (Supplemental Tables S14, S15; Supplemental Methods). Compared with whole assemblies, these missing sequences exhibited a similar heterozygous SNP ratio (2.67 × 10−3 vs. 2.56 × 10−3; P = 0.623, Mann–Whitney U test) but significantly higher GC content (43.07% vs. 41.41%; P < 10−16, Mann–Whitney U test) and repeat ratio (47.57% vs. 38.38%; P < 10−16, Mann–Whitney U test) (Supplemental Fig. S28).
Table 1.
Summary of missing sequences and genes of the reference genome (Sscrofa10.2)
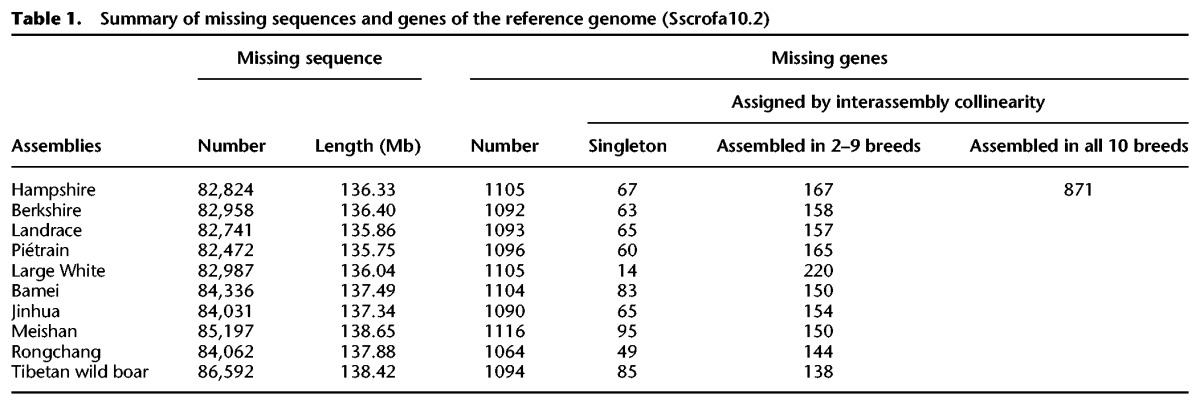
Most sequences missing in the reference genome were common between different assemblies, as most orphan reads (95.04%) could cross-align to missing sequences of other assemblies with coverage (97.10% with depth ≥4 per base) comparable to that against their respective assemblies (mapping ratio = 95.83% and coverage = 99.51%) (Supplemental Fig. S29). Pairwise similarity of the orphan reads and the missing sequences among 10 breeds revealed a clear distinction between European and Asian pigs, as well as a relatively higher genetic variability in Chinese pigs than in European pigs (Supplemental Fig. S30; Bosse et al. 2012; Groenen et al. 2012), suggesting that these common sequences, which were absent in the reference genome, may be important sources of pig diversity and contain biologically meaningful information.
We were also able to fill in 71.37% (3795 of 5317) of the gaps in the reference genome with missing sequences of at least one breed (Supplemental Fig. S27; Supplemental Tables S14, S16; Supplemental Methods). These filled missing sequences were highly collinear across 10 breeds and exhibited a similar distribution over the reference assembly gaps (average of pairwise Pearson's r = 0.89, P < 10−16) (Supplemental Fig. S31). Typical examples are shown in Supplemental Figure S32.
Recovery of missing genes
Of the average 20,782 protein-coding genes (87.13% were supported by evidence of transcription) predicted in each of the 10 assemblies (Supplemental Figs. S33–S35; Supplemental Table S17; Supplemental Methods), we found an average of 1096 (5.27%) genes to be embedded or almost completely contained (>50% to >90% overlap of gene length, respectively) in the missing sequences of the reference assembly (Table 1; Supplemental Table S18), which we referred to as ‘missing’ genes (Kidd et al. 2010; Genovese et al. 2013).
To check whether these predicted missing genes are likely to be functional, we compared their conservation level across 19 mammalian genomes and found that they generally exhibited similar identity (81.55% vs. 83.60%) and coverage (96.32% vs. 97.37%) as annotated genes (Supplemental Fig. S36). Coding sequences of missing genes were enriched at a high cross-species (human, cow, and sheep) identity level (>90%), also consistent with the sequence identity distribution of well-annotated coding sequences of the reference genome (Supplemental Fig. S37). We then retrieved ∼0.59 G orphan reads against the reference genome from each of 96 paired-end RNA-seq libraries (7–10 libraries for each breed) and mapped them onto the missing genes in their respective assemblies (Supplemental Fig. S38A,B). Consequently, an average of 91.51% (1003 of 1096) missing genes in each assembly showed log2-transformed FPKM expression values (denoted as fragments per kb of transcript per Mb orphan reads) greater than 0.3 in at least one library (Supplemental Fig. S38C), suggesting that a considerable number of missing genes are functional and biologically important.
To determine the collinear relationships of missing genes among 10 breeds, we separately aligned the protein sequences of nine assemblies to the assembly of the Large White breed, which had the longest scaffold N50 size (2.45 Mb). Using the MCScanX toolkit (Wang et al. 2012), we found that 10,313 of 10,959 (94.10%) missing genes in all 10 assemblies belonged to 1091 interassembly collinear gene models, of which 871 (79.84%) models were present in all 10 assemblies (Table 1; Supplemental Tables S18, S19). There were a total of 646 missing genes (14–95 per assembly) assembled in only a single breed, which could be found in other assemblies when orphan reads from short-insert (180 and 500 bp) libraries were used for mapping (coverage 94.05% at least 1× depth), suggesting that the absence of these singleton genes from other assemblies is likely an artifact of fragmentation or misassignment in short read assembly (Supplemental Fig. S39; Alkan et al. 2011; Chaisson et al. 2015b).
Together with the longest gene model of 1091 interassembly collinear genes and 646 singleton genes, we obtained 1737 missing gene models (Table 1). Aligning these missing genes to RefSeq proteins of pig, human, cow, and mouse yielded 1731 (99.65%) hits in at least one species (Supplemental Table S19), of which 359 (20.66%) missing genes could not be aligned to any known RefSeq proteins of pig, indicating that these genes have not been characterized in pig. Among hits that matched functionally classified proteins, the most abundant were members of olfactory receptors (65 hits, P = 1.60 × 10−12, χ2 test), G-protein coupled receptors (104 hits, P = 9.81 × 10−6, χ2 test), and those involved in neurological system processes (112 hits, P = 4.26 × 10−6, χ2 test) (Supplemental Table S20), which are known to be rapidly evolving between species (Mainland et al. 2014). We also recovered genes corresponding to economically important traits that are valuable for future functional analyses and improvements of pig as an important livestock species, such as genes related to pork production (74 of 1515 fat deposition genes [Kunej et al. 2013], or 4.88%) and disease resistance (76 of 1517 genes annotated with the GO: 0002376; immune system process, or 5.01%) (Supplemental Table S19). Typical examples are shown in Supplemental Figure S40.
Selection in missing genes
To reveal variants left by selection, we measured pairwise the extent of population differentiation of the coding SNPs in the missing genes between Chinese wild boars (32.57 k coding SNPs) and seven Chinese domestic populations (23.02 k coding SNPs per population) using the FDIST approach as implemented in Arlequin (Supplemental Figs. S41, S42; Supplemental Table S21; Excoffier and Lischer 2010). A total of 605 nonredundant coding SNPs embedded in 328 missing genes were found to be under directional selection in seven Chinese domestic populations (FDR < 0.05, FDIST test) (Supplemental Fig. S43; Supplemental Table S22), which also exhibited significantly lower Q-values in a Bayesian test (Foll and Gaggiotti 2008) and FST values in a ‘model-free’ global FST test when compared to other unselected loci (P < 10−16, Mann–Whitney U test) (Supplemental Fig. S44). The missing genes under selection in seven Chinese domestic populations were commonly enriched for biological processes related to ‘binding of actin, calcium ion, and cytoskeletal protein’ (Supplemental Fig. S45A). Intriguingly, 71 genes harboring 110 selected coding SNPs of domestic Erhualian pigs (one of the most prolific pig breeds known) (Wang et al. 2011) belonged predominantly to fertility-related categories, such as ‘sexual reproduction’ (seven genes: ADAM20, AKT1, GMCL1, MICALCL, NOTCH1, SPIN4, and SPTBN4; P = 0.001) and ‘placenta development’ (three genes: AKT1, RXRA, and VWF; P = 0.012), which may underlie the breed's markedly larger litters (∼3 to 5 more piglets per litter) (Supplemental Fig. 45B).
The expression of missing genes under selection also showed remarkably higher tissue specificity, reflected by the lower Shannon entropy (H) values (a measure of the specificity of gene expression across tissues) (Schug et al. 2005) than unselected missing genes (1.98 vs. 2.37 per gene; P < 10−16, Mann–Whitney U test) (Supplemental Fig. S46). As opposed to constitutive genes that are ubiquitously expressed and essential for basic cellular functions, tissue-specific genes are usually associated with the development of generally desirable traits, such as disease resistance, muscle growth, fat deposition, and reproduction, and thus are more likely prone to be shaped by selection.
None of the selected coding SNPs was a nonsense mutation (resulting in premature stop codons in transcribed mRNAs) (Supplemental Table S22), supporting the idea that gene inactivation did not play a prominent role during pig domestication and consistent with the results from screens in chickens (Rubin et al. 2010), rabbits (Carneiro et al. 2014), and pigs (based on reference genome) (Rubin et al. 2012). Compared to synonymous substitutions, missense substitutions showed significantly lower genetic differentiation (global Fst, 0.05 compared to 0.10 per locus; P < 10−16, Mann–Whitney U test) between Chinese wild boars and domestic pigs (Supplemental Fig. S47). Nonetheless, there were still 127 genes harboring selected missense mutations, which were overrepresented in the highly variable olfactory receptor family (12 genes; P = 0.02, χ2 test) (Supplemental Table S22; Mainland et al. 2014). Of these, three missense mutations embedded in two genes related to the development of obesity were of interest: the closely linked Asn566-His (T1,696-G) and Ser578-Cys (G1,733-C) substitutions (D′ = 1, r2 = 0.975) found in ALPK3 (alpha kinase 3) (Fig. 4; Supplemental Fig. S48), and a Thr18-Ile (C53-T) substitution in PKD1L2 (polycystin 1 like 2 [gene/pseudogene]) (Supplemental Fig. S49). These three missense mutations exhibited significant selection signals (FDR < 0.05, FDIST test) between Chinese wild boars and one of seven domestic populations (Min and Erhualian, respectively) but were nearly fixed in the more genetically homogeneous European/North American domestic pigs, possibly as a result of stronger selective pressure in Western societies, although larger sample sizes, intercontinental genetic discrepancy of pig genomes, and functional analyses are required to validate the nonneutrality of these genes.
Figure 4.
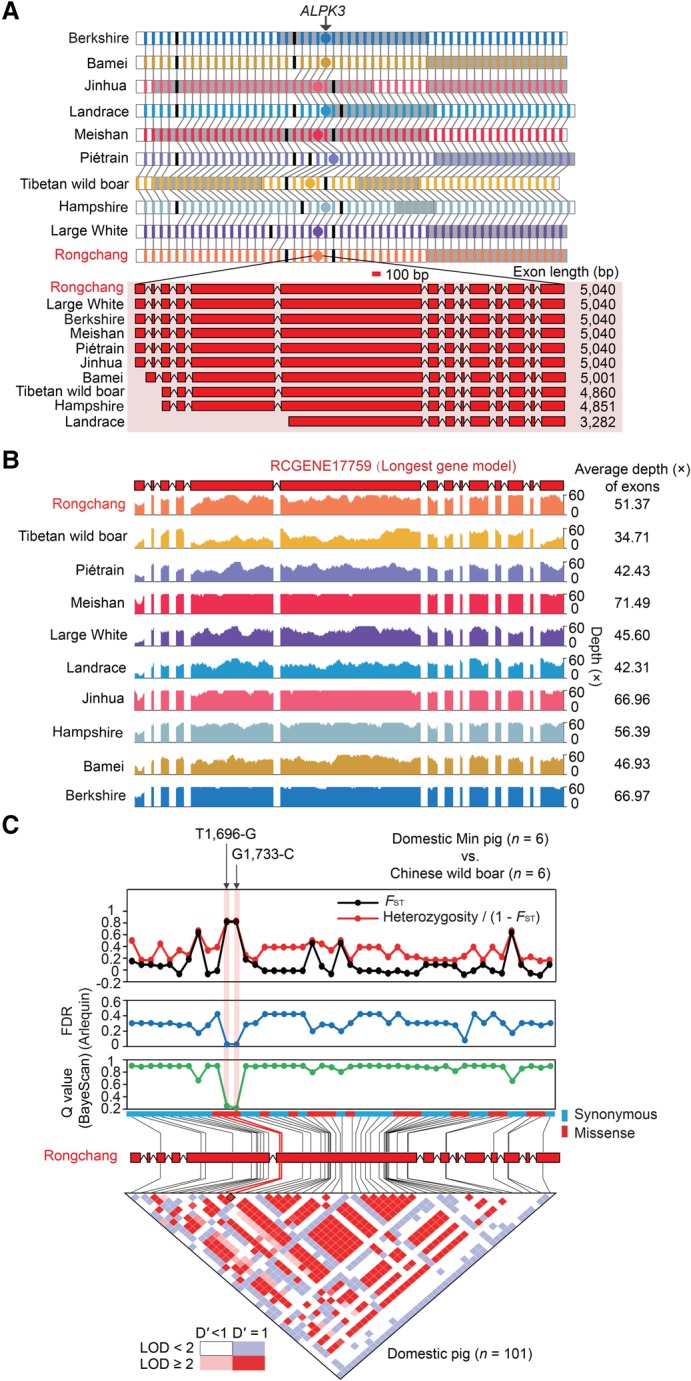
Details of assembled ALPK3 gene and selected variants. (A) Structure of assembled ALPK3. (Top panel) The interassembly collinear genes (colored rectangles) among 10 assemblies are linked by gray lines, and the genes not present in all 10 assemblies are marked in black. ALPK3 is denoted by a circle. Different scaffolds are shown as alternating white and gray backgrounds. (Bottom panel) Comparison of structure of ALPK3 among the 10 assemblies. Boxes and lines indicate exons and introns, respectively. (B) Coverage and depth for the longest gene model of ALPK3 (Gene ID: RCGENE17759) by cross-mapping reads from paired-end DNA libraries (insert sizes of 180 and 500 bp) of the 10 assemblies. The higher coverage depth (≥30×) suggests slightly different structures of ALPK3, which is attributable to limitations of short read assembly; as such, the longest gene model is considered more reliable and used for subsequent analyses. (C) Two selected missense mutations (T1,696-G and G1,733-C) in ALPK3 between Chinese wild boars (n = 6) and domestic Min pigs (n = 6). (Top panels) FST and heterozygosity/(1−FST), FDR (Arlequin), and Q-values (BayeScan) are plotted for 45 coding SNPs (18 missenses and 27 synonymous mutations). (Bottom panels) LD pattern of 45 SNPs in 101 domestic pigs from China (n = 41), North America (n = 12), and Europe (n = 48). Squares shaded in pink or red indicate significant LD between SNP pairs (bright red indicates pairwise D′ = 1), white squares indicate no evidence of significant LD, and blue squares indicate pairwise D′ = 1 without statistical significance. The adjacent T1,696-G and G1,733-C are closely linked (D′ = 1, r2 = 0.975, LOD = 41.6).
ALPK3 plays a role in cardiomyocyte differentiation; knockout of this gene in mice is associated with marked hypertrophic and dilated forms of cardiomyopathy (Van Sligtenhorst et al. 2012). ALPK3 shows the strongest evidence of positive selection in the polar bear, which has a lipid-rich diet throughout life (Liu et al. 2014). Selection of ALPK3 in domestic pigs suggests that potential protection against the chronically deleterious effects of a ‘diabetogenic’ environment (high calorie, atherogenic diet, and little physical exercise) on the cardiovascular system may be favorable (Gerstein and Waltman 2006; Koopmans and Schuurman 2015). PKD1L2 is primarily associated with fatty acid synthase in the skeletal muscle fiber; its overexpression in mice provokes myofiber atrophy and suppressed lipogenesis (Mackenzie et al. 2009). The triglycerides accumulated between or within myofibers represent a large energy source (contributes up to 20% of total energy turnover during physical exercise in human) (Roepstorff et al. 2005). Selection of PKD1L2 is likely related to the relatively weak athletic performance of domestic pigs compared to wild boars due to limited active space in pig farms.
Discussion
We describe an assembly-versus-assembly approach that relies on multiple independently assembled genomes for improving the power of variant detection, as opposed to the currently dominant resequencing approach. This catalog of variants, including SNPs, indels, and common and rare variants is a valuable resource for further investigation of the genetic makeup of porcine phenotypic diversity and adaptive evolution. We show that high-quality de novo assembly of individual genomes followed by comparison with the reference sequence is necessary for identifying novel genetic variation across geographical ranges and different evolutionary histories. Such experimental design is increasingly affordable with the advances in sequencing technology (Zook and Salit 2015), especially long-read sequencing (Chaisson et al. 2015a) and single-molecule mapping (Koren et al. 2012) technologies.
Interpretation of the consequences of genetic variation has typically relied on reference sequences, relative to which genes and variants are annotated and examined. However, we recovered hundreds of millions of base pairs that were not present in the pig reference genome, including thousands of protein-coding genes that are either missing or fragmented in the reference genome, which harbor abundant variants associated with economic traits that are likely subjected to artificial selection. These newly recovered genes can now be incorporated into genotyping platforms and expression microarrays to facilitate their functional characterization. Recovered sequences missing from the reference genome could also be the source of genetic signals that have been ascertained by linkage, association, and copy number variation studies but not yet mapped to causal mutations.
Methods
De novo sequencing and assembly of pig genomes
We sequenced the genomes of nine geographically and phenotypically representative pig breeds using Illumina sequencing technology and a whole-genome shotgun strategy (Fig. 2A; Supplemental Fig. S1). Short-insert (180 and 500 bp) and long-insert (2, 5, 6, and 10 kb) DNA libraries were paired-end sequenced on the Illumina HiSeq 2500 platform (Supplemental Fig. S2; Supplemental Table S1). We independently assembled nine genomes using SOAPdenovo (Li et al. 2010b), which is a de Bruijn graph algorithm–based de novo genome assembler (Supplemental Methods). We performed repeat annotation for 10 breed assemblies and the reference genome using the same pipeline (Supplemental Figs. S5, S6; Supplemental Methods).
SNP and indel calling using an assembly-versus-assembly method
We took advantage of an assembly-versus-assembly approach to identify candidate variants and further filtered out spurious variants by aligning short sequencing reads (Supplemental Methods). In brief, we first extracted candidate SNPs and small- and intermediate-scale indels (1–50 kb) in 10 assemblies by pairwise gapped alignment of the 10 assemblies and the reference genome assembly (Sscrofa10.2) using the LASTZ program. Then, the paired-end short-insert reads (180 and 500 bp) were separately aligned to the 10 assembled genomes and the reference genome using the BWA software (v.0.7.12) (Li and Durbin 2009). We filtered spurious SNPs and determined the heterozygous or homozygous mutations (depth ≥ 10) using SAMtools (v.1.3) (Li et al. 2009). With regard to indels, we eliminated spurious indel calls based on the calculation of read coverage for each indel locus with different criteria for indels ≤50 bp or >50 bp (Li et al. 2011).
Identification of selected regions using the RSD algorithm
To identify signatures of diversifying selection of pig breeds, relative homozygous SNP density (RSD) in nonoverlapping 10-kb windows across the reference genome was calculated for each individual using a previously reported methodology (Supplemental Methods; Atanur et al. 2013).
RNA-seq and data processing
The 92 strand-specific RNA libraries (7–10 tissue libraries for each of 10 individuals, which were used for de novo genome assemblies) were sequenced on the Illumina HiSeq 2500 platform (Supplemental Methods). High-quality reads were mapped to their respective de novo assemblies (Supplemental Figs. S35, S46) or the reference genome (Supplemental Fig. S38) using TopHat (v.2.1.0) (Trapnell et al. 2009). Cufflinks (v.2.2.1) (Trapnell et al. 2012) was used to quantify gene expression.
Discovery of missing sequences and missing genes
We retrieved ‘orphan reads’ from paired-end DNA libraries with insert sizes of 180 and 500 bp for each of 10 breeds, where both ends of the read cannot be uniquely mapped to the reference genome (Supplemental Fig. S27). We relocalized these orphan reads to their respective assemblies. Sequences (≥500 bp in length) absent from the public reference genome assembly that were mapped by at least four orphan reads per base were considered ‘missing sequences’ (Kidd et al. 2010).
To identify genes embedded in the missing sequences, we conducted annotation of protein-coding genes in the 10 assemblies separately, using a combination of evidences from reference assembly-guided approach, the ab initio- and homology-based methods, as well as RNA-seq data (Supplemental Figs. S33, S34; Supplemental Table S17; Supplemental Methods). We considered genes that showed >50% overlap of the gene length with missing sequences to be either missing or fragmented in the reference genome and referred to them as ‘missing genes.’
Determination of interassembly collinear genes
The protein sequences of genes in the nine assemblies were separately queried against the protein sequences of the Large White assembly, which had the longest scaffold N50 size (∼2.45 Mb), using BLASTp with an E-value cutoff of 10−5 and restricting the output to a maximum of five hits per gene to serve as input for the MCScanX algorithm (Wang et al. 2012), which was used to detect and classify high-confidence collinear blocks of coding genes (Supplemental Tables S18, S19).
Detecting coding SNPs in missing genes under selection
To test whether the recovered genes missing in the reference genome were under selection, we retrieved ∼365.55 Gb orphan reads against the reference genome from 117 publicly available pig genomes (Ai et al. 2015; Choi et al. 2015; Moon et al. 2015) and aligned them to the intact scaffolds harboring missing genes across the 10 breed assemblies (∼636.38 Mb per assembly) (Supplemental Fig. S41). Of these, six wild boars and 41 domestic pigs belonging to seven populations in China that have high-coverage depth (27.29× of the reference genome, 14.43× of missing gene embedded scaffolds by 3.91 Gb orphan reads per individual) (Ai et al. 2015) were used to test for differentiation and possibly selection (Supplemental Fig. S42). The remaining 70 individuals (including 10 Korean wild boars and 60 European/North American domestic pigs) with intermediate coverage (15.87× of the reference genome, 6.99× of missing gene embedded scaffolds by 2.60 Gb orphan reads per individual) (Choi et al. 2015; Moon et al. 2015) were used to investigate the patterns of selected loci (Supplemental Fig. S41).
We measured pairwise the extent of population differentiation of the coding SNPs in the missing genes between Chinese wild boars and seven Chinese domestic populations using the FDIST approach as implemented in Arlequin (v.3.5.2.2) (Supplemental Fig. S43; Supplemental Table S21; Supplemental Methods; Excoffier and Lischer 2010). We also measured pairwise global FST values (Supplemental Fig. S44A) and performed a Bayesian test using the program BayeScan (v.2.1) (Supplemental Fig. S44B; Foll and Gaggiotti 2008) for every gene to detect highly differentiated SNPs between populations.
Data access
The nine pigs and Tibetan wild boar BioProjects are accessible at NCBI BioProject (http://www.ncbi.nlm.nih.gov/bioproject) under accession numbers PRJNA309108 and PRJNA186497, respectively. The assembled whole-genome sequences have been submitted to NCBI GenBank (http://www.ncbi.nlm.nih.gov/genbank) under accession numbers LUXQ00000000.1 (Meishan), LUXR000000 00.1 (Rongchang), LUXS00000000.1 (Hampshire), LUXT000000 00.1 (Landrace), LUXU00000000.1 (Piétrain), LUXV00000000.1 (Bamei), LUXW00000000.1 (Berkshire), LUXX00000000.1 (Large White), LUXY00000000.1 (Jinhua), and AORO00000000.2 (Tibetan wild boar, v.2). The unassembled sequencing reads of nine pigs and Tibetan wild boar have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession numbers SRP068560 and SRA065461, respectively. All RNA-seq reads and the genotyping data of the Illumina's porcine 60K Genotyping BeadChip (v.2) have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE77776 and GSE83910, respectively. SNPs and small indels (1–50 bp) identified using an assembly-versus-assembly method have been submitted to NCBI dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/) under assay numbers ss2137144068, ss2590667644–ss2624264572 (SNPs), and ss2137144058–ss2137297824, and ss2586846515–ss2590667643 (indels, discontinuous). Large indels (>50 bp) identified using an assembly-versus-assembly method have been submitted to NCBI dbVar (https://www.ncbi.nlm.nih.gov/dbvar) under accession number nstd138.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (31530073, 31522055, 31472081, 31372284, 31402046, and 31401073), the National Special Foundation for Transgenic Species of China (2014ZX0800950B and 2014ZX08006-003), the National Program for Support of Top-notch Young Professionals, the Program for Innovative Research Team of Sichuan Province (2015TD0012), the Specialized Research Fund of the Ministry of Agriculture of China (NYCYTX-009), the Program for Changjiang Scholars and Innovative Research Team in University (IRT13083), the National High Technology Research and Development Program of China (863 Program) (2013AA102502), the Science and Technology Support Program of Sichuan (Pig breeding-16ZC2850), the Fund of Fok Ying-Tung Education Foundation (141117), the National Key Technology R&D Program of China (2011BAD28B01), the Chongqing Fund of Application and Development (CSTC 2013YYKFC80003), the Modern Agricultural Industry Technology System (CARS-36), and the Chongqing Foundation of Agricultural Development (12404 and 14409).
Author contributions: Mingz. L., S.T., J.W., R.L., and X.L. led the experiments and designed the analytical strategy. L.C., D.L., A.J., Yingk. L., S.S., L.Z., Y.J., and L.B. performed animal work and prepared biological samples. L.J., J.M., X.W., Zongg. L., S.Z., and Z.J. constructed the DNA and RNA libraries and performed sequencing. Mingz. L., S.T., Yu.L. Q.T., Hongf.L., and T.C. designed the bioinformatics analysis process. Yu L., X.Z., Y.F., Haif. L., D.W., Zongw.L., and H.Z. performed the genome assembly and annotation. S.T., Mingz. L., L.C., Yan L., C.L., and G.W. performed the variation calling. Mingz. L., S.T., Y.G., C.L., Z.G., G.T., and J.Z. identified missing sequences and missing genes. S.T., Mingz. L., Q.T., X.Z., Q.P., M.M., and C.Y. performed selective sweep analyses. Mingz. L., L.C., R.L., and X.L. wrote the paper. Ming. L., J.W., V.N.G., and S.Z. revised the paper.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.207456.116.
Freely available online through the Genome Research Open Access option.
References
- Ai H, Fang X, Yang B, Huang Z, Chen H, Mao L, Zhang F, Zhang L, Cui L, He W, et al. 2015. Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing. Nat Genet 47: 217–225. [DOI] [PubMed] [Google Scholar]
- Alkan C, Sajjadian S, Eichler EE. 2011. Limitations of next-generation genome sequence assembly. Nat Methods 8: 61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanur SS, Diaz AG, Maratou K, Sarkis A, Rotival M, Game L, Tschannen MR, Kaisaki PJ, Otto GW, Ma MC, et al. 2013. Genome sequencing reveals loci under artificial selection that underlie disease phenotypes in the laboratory rat. Cell 154: 691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besenbacher S, Liu S, Izarzugaza JM, Grove J, Belling K, Bork-Jensen J, Huang S, Als TD, Li S, Yadav R, et al. 2015. Novel variation and de novo mutation rates in population-wide de novo assembled Danish trios. Nat Commun 6: 5969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosse M, Megens HJ, Madsen O, Paudel Y, Frantz LA, Schook LB, Crooijmans RP, Groenen MA. 2012. Regions of homozygosity in the porcine genome: consequence of demography and the recombination landscape. PLoS Genet 8: e1003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro M, Rubin CJ, Di Palma F, Albert FW, Alfoldi J, Barrio AM, Pielberg G, Rafati N, Sayyab S, Turner-Maier J, et al. 2014. Rabbit genome analysis reveals a polygenic basis for phenotypic change during domestication. Science 345: 1074–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Huddleston J, Dennis MY, Sudmant PH, Malig M, Hormozdiari F, Antonacci F, Surti U, Sandstrom R, Boitano M, et al. 2015a. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517: 608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Wilson RK, Eichler EE. 2015b. Genetic variation and the de novo assembly of human genomes. Nat Rev Genet 16: 627–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Baxter T, Muir WM, Groenen MA, Schook LB. 2007. Genetic resources, genome mapping and evolutionary genomics of the pig (Sus scrofa). Int J Biol Sci 3: 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JW, Chung WH, Lee KT, Cho ES, Lee SW, Choi BH, Lee SH, Lim W, Lim D, Lee YG, et al. 2015. Whole-genome resequencing analyses of five pig breeds, including Korean wild and native, and three European origin breeds. DNA Res 22: 259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Lischer HE. 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10: 564–567. [DOI] [PubMed] [Google Scholar]
- Foll M, Gaggiotti O. 2008. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180: 977–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz LA, Schraiber JG, Madsen O, Megens HJ, Bosse M, Paudel Y, Semiadi G, Meijaard E, Li N, Crooijmans RP, et al. 2013. Genome sequencing reveals fine scale diversification and reticulation history during speciation in Sus. Genome Biol 14: R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan X, Stegle O, Behr J, Steffen JG, Drewe P, Hildebrand KL, Lyngsoe R, Schultheiss SJ, Osborne EJ, Sreedharan VT, et al. 2011. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 477: 419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Handsaker RE, Li H, Altemose N, Lindgren AM, Chambert K, Pasaniuc B, Price AL, Reich D, Morton CC, et al. 2013. Using population admixture to help complete maps of the human genome. Nat Genet 45: 406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein HC, Waltman L. 2006. Why don't pigs get diabetes? Explanations for variations in diabetes susceptibility in human populations living in a diabetogenic environment. Can Med Assoc J 174: 25–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenen MA, Archibald AL, Uenishi H, Tuggle CK, Takeuchi Y, Rothschild MF, Rogel-Gaillard C, Park C, Milan D, Megens HJ, et al. 2012. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 491: 393–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Sampas N, Antonacci F, Graves T, Fulton R, Hayden HS, Alkan C, Malig M, Ventura M, Giannuzzi G, et al. 2010. Characterization of missing human genome sequences and copy-number polymorphic insertions. Nat Methods 7: 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopmans SJ, Schuurman T. 2015. Considerations on pig models for appetite, metabolic syndrome and obese type II diabetes: from food intake to metabolic disease. Eur J Pharmacol 759: 231–239. [DOI] [PubMed] [Google Scholar]
- Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, Wang Z, Rasko DA, McCombie WR, Jarvis ED, et al. 2012. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol 30: 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunej T, Jevsinek Skok D, Zorc M, Ogrinc A, Michal JJ, Kovac M, Jiang Z. 2013. Obesity gene atlas in mammals. J Genomics 1: 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson G, Dobney K, Albarella U, Fang M, Matisoo-Smith E, Robins J, Lowden S, Finlayson H, Brand T, Willerslev E, et al. 2005. Worldwide phylogeography of wild boar reveals multiple centers of pig domestication. Science 307: 1618–1621. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Li Y, Zheng H, Luo R, Zhu H, Li Q, Qian W, Ren Y, Tian G, Li J, et al. 2010a. Building the sequence map of the human pan-genome. Nat Biotechnol 28: 57–63. [DOI] [PubMed] [Google Scholar]
- Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, et al. 2010b. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20: 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zheng H, Luo R, Wu H, Zhu H, Li R, Cao H, Wu B, Huang S, Shao H, et al. 2011. Structural variation in two human genomes mapped at single-nucleotide resolution by whole genome de novo assembly. Nat Biotechnol 29: 723–730. [DOI] [PubMed] [Google Scholar]
- Li M, Tian S, Jin L, Zhou G, Li Y, Zhang Y, Wang T, Yeung CK, Chen L, Ma J, et al. 2013. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat Genet 45: 1431–1438. [DOI] [PubMed] [Google Scholar]
- Li YH, Zhou G, Ma J, Jiang W, Jin LG, Zhang Z, Guo Y, Zhang J, Sui Y, Zheng L, et al. 2014. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat Biotechnol 32: 1045–1052. [DOI] [PubMed] [Google Scholar]
- Liu S, Lorenzen ED, Fumagalli M, Li B, Harris K, Xiong Z, Zhou L, Korneliussen TS, Somel M, Babbitt C, et al. 2014. Population genomics reveal recent speciation and rapid evolutionary adaptation in polar bears. Cell 157: 785–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie FE, Romero R, Williams D, Gillingwater T, Hilton H, Dick J, Riddoch-Contreras J, Wong F, Ireson L, Powles-Glover N, et al. 2009. Upregulation of PKD1L2 provokes a complex neuromuscular disease in the mouse. Hum Mol Genet 18: 3553–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainland JD, Keller A, Li YR, Zhou T, Trimmer C, Snyder LL, Moberly AH, Adipietro KA, Liu WL, Zhuang H, et al. 2014. The missense of smell: functional variability in the human odorant receptor repertoire. Nat Neurosci 17: 114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon S, Kim TH, Lee KT, Kwak W, Lee T, Lee SW, Kim MJ, Cho K, Kim N, Chung WH, et al. 2015. A genome-wide scan for signatures of directional selection in domesticated pigs. BMC Genomics 16: 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey DE, Waterhouse RM, Abai MR, Aganezov SS, Alekseyev MA, Allen JE, Amon J, Arca B, Arensburger P, Artemov G, et al. 2015. Highly evolvable malaria vectors: the genomes of 16 Anopheles mosquitoes. Science 347: 1258522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read BA, Kegel J, Klute MJ, Kuo A, Lefebvre SC, Maumus F, Mayer C, Miller J, Monier A, Salamov A, et al. 2013. Pan genome of the phytoplankton Emiliania underpins its global distribution. Nature 499: 209–213. [DOI] [PubMed] [Google Scholar]
- Roepstorff C, Vistisen B, Kiens B. 2005. Intramuscular triacylglycerol in energy metabolism during exercise in humans. Exerc Sport Sci Rev 33: 182–188. [DOI] [PubMed] [Google Scholar]
- Rubin CJ, Zody MC, Eriksson J, Meadows JR, Sherwood E, Webster MT, Jiang L, Ingman M, Sharpe T, Ka S, et al. 2010. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 464: 587–591. [DOI] [PubMed] [Google Scholar]
- Rubin CJ, Megens HJ, Martinez Barrio A, Maqbool K, Sayyab S, Schwochow D, Wang C, Carlborg O, Jern P, Jorgensen CB, et al. 2012. Strong signatures of selection in the domestic pig genome. Proc Natl Acad Sci 109: 19529–19536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug J, Schuller WP, Kappen C, Salbaum JM, Bucan M, Stoeckert CJ Jr. 2005. Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol 6: R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Sligtenhorst I, Ding ZM, Shi ZZ, Read RW, Hansen G, Vogel P. 2012. Cardiomyopathy in α-kinase 3 (ALPK3)-deficient mice. Vet Pathol 49: 131–141. [DOI] [PubMed] [Google Scholar]
- Wang LY, Wang AG, Wang LX, Li K, Yang GS, He RG, Qian L, Xu NY, Huang RH, Peng ZZ, et al. 2011. Animal genetic resources in China: pigs (ed. China National Commission of Animal Genetic Resources), pp. 2–16. China Agricultural Press, Beijing. [Google Scholar]
- Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H, et al. 2012. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40: e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenfeld NI, Yin S, Sharpe T, Lau B, Hegarty R, Holmes L, Sogoloff B, Tabbaa D, Williams L, Russ C, et al. 2014. Comprehensive variation discovery in single human genomes. Nat Genet 46: 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White S. 2011. From globalized pig breeds to capitalist pigs: a study in animal cultures and evolutionary history. Environ Hist 16: 94–120. [Google Scholar]
- Yan G, Zhang G, Fang X, Zhang Y, Li C, Ling F, Cooper DN, Li Q, Li Y, van Gool AJ, et al. 2011. Genome sequencing and comparison of two nonhuman primate animal models, the cynomolgus and Chinese rhesus macaques. Nat Biotechnol 29: 1019–1023. [DOI] [PubMed] [Google Scholar]
- Zhang QJ, Zhu T, Xia EH, Shi C, Liu YL, Zhang Y, Liu Y, Jiang WK, Zhao YJ, Mao SY, et al. 2014. Rapid diversification of five Oryza AA genomes associated with rice adaptation. Proc Natl Acad Sci 111: E4954–E4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zook JM, Salit M. 2015. Advancing benchmarks for genome sequencing. Cell Systems 1: 176–177. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.