

Abstract
Mismatch repair (MMR) is one of the main systems maintaining fidelity of replication. Differences in correction of errors produced during replication of the leading and the lagging DNA strands were reported in yeast and in human cancers, but the causes of these differences remain unclear. Here, we analyze data on human cancers with somatic mutations in two of the major DNA polymerases, delta and epsilon, that replicate the genome. We show that these cancers demonstrate a substantial asymmetry of the mutations between the leading and the lagging strands. The direction of this asymmetry is the opposite between cancers with mutated polymerases delta and epsilon, consistent with the role of these polymerases in replication of the lagging and the leading strands in human cells, respectively. Moreover, the direction of strand asymmetry observed in cancers with mutated polymerase delta is similar to that observed in MMR-deficient cancers. Together, these data indicate that polymerase delta (possibly together with polymerase alpha) contributes more mismatches during replication than its leading-strand counterpart, polymerase epsilon; that most of these mismatches are repaired by the MMR system; and that MMR repairs about three times more mismatches produced in cells during lagging strand replication compared with the leading strand.
Replication is a very accurate process. Its fidelity is achieved through three main components: base selectivity of polymerases, proofreading activity of their exonuclease domains, and repair of mismatches that escaped proofreading by the mismatch repair (MMR) system (Kunkel 2009). Studies in yeast indicate that the effectiveness of each of these steps depends on the mismatch type and that MMR compensates for the infidelity of polymerases (Kunkel 2011; St Charles et al. 2015). The classical model of the eukaryotic replication fork (Larrea et al. 2010) suggests a division of labor in replication of the leading and lagging strands among the major DNA polymerases, with polymerase epsilon (Pol epsilon) replicating the leading strand and polymerases alpha (Pol alpha) and delta (Pol delta) replicating the lagging strand, with the possible exception of replication origins and other specific regions where Pol delta may contribute to replication of both strands (Yeeles et al. 2017). Under this model, the asymmetry in mutation rates between the leading and the lagging DNA strands may arise due to differences in fidelity of polymerases replicating these strands. In yeasts, different replicative polymerases possess different biases in the types of mutations they introduce, leading to differences in mismatch types between the leading and the lagging strands (St Charles et al. 2015).
The strand asymmetry of mutations is also observed in MMR-deficient cancers (Haradhvala et al. 2016; Morganella et al. 2016). As MMR is primarily a coreplicative process (Hombauer et al. 2011; Liao et al. 2015), we hypothesized that this asymmetry is due to a joint effect of the differences in rates of mismatches produced by replicative polymerases on the leading and on the lagging strands, and differences in number of mutations repaired by the MMR between the two strands. To investigate this question, we employed data from patients with inherited biallelic MMR deficiency (bMMRD) and somatic mutations in one of the two major replicative polymerases, Pol epsilon (mutated Pol epsilon) or Pol delta (mutated Pol delta).
Results
Stand-specific mutational patterns in cancers with mutated Pol epsilon or Pol delta
Mutations in replicative polymerases are frequent in cancers with inherited bMMRD and result in a hypermutable phenotype. In these patients, the fidelity of the damaged polymerase is decreased by a factor of 100 to 1000, and most mutations are produced by it (Korona et al. 2011; Henninger and Pursell 2014; Erson-Omay et al. 2015; Shlien et al. 2015). We found that the relative frequencies of mutation types were distorted in these cancers compared with tumors without mutations in polymerases (Fig. 1). bMMRD samples with mutations in Pol epsilon are strongly enriched in C → T mutations, especially in the GpCpG context, and C → A mutations in the NpCpT context (Supplemental Fig. S1). We compared the mutational spectra of bMMRD tumors with mutated Pol epsilon to the mutational signatures from the COSMIC database (http://cancer.sanger.ac.uk/cosmic/signatures) (Supplemental Table S1). The mutational spectrum was similar to signatures 6, 15, and 20, which are known to be reflective of MMR deficiency (Supplemental Table S1), and also to signature 14 (cosine distance = 0.74) (Supplemental Table S1). In the COSMIC database, the only signature attributed to Pol epsilon deficiency was signature 10. The etiology of signature 14 was previously unknown, although it has been observed in samples with increased mutation rate and replicative asymmetry (Tomkova et al. 2017). To resolve this, we employed additional data on samples with mutated Pol epsilon and with or without somatic disruption of the MMR from The Cancer Genome Atlas (TCGA) database. As expected, the spectrum of bMMRD samples with mutated Pol epsilon is more similar to that of TCGA samples with deficient MMR (cosine distance = 0.78) than with intact MMR (cosine distance = 0.43) (Supplemental Fig. S2). We found that the mutational spectrum of TCGA samples strongly depended on the MMR status: the spectrum of samples with intact MMR matched well with signature 10 (cosine distance = 0.92), while the spectrum of samples with defective MMR matched with signature 14 (cosine distance = 0.97). Therefore, signature 14 is the signature of mutated Pol epsilon not corrected by the MMR.
Figure 1.
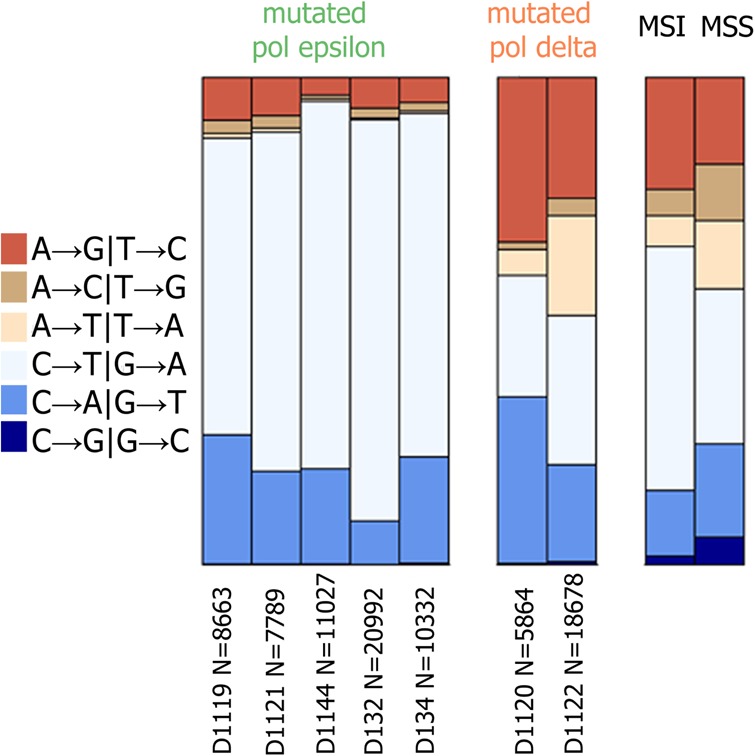
Mutation frequencies in bMMRD cancers with subsequent mutations in Pol epsilon or Pol delta. Data for seven exomes of bMMRD cancer (five mutated Pol epsilon and two mutated Pol delta). Relative frequencies of single-nucleotide substitutions are shown irrespective of the strand; data from TCGA database for MMR-deficient (MSI) and MMR-proficient (MSS) samples without mutations in Pol epsilon and Pol delta are shown for comparison. N is the number of mutations observed in each sample.
We then used mutation data from the bMMRD samples to ask if mismatches produced by human polymerases are biased toward one mismatch from the complementary pair and to determine the preferred mismatch. By using an approach that determines the replication fork polarity (FP) as the derivative of the replication timing (RT) (Baker et al. 2012; Haradhvala et al. 2016; Morganella et al. 2016; Seplyarskiy et al. 2016), we predicted for each genomic region whether the reference strand is replicated more frequently as leading (FP > 0) or lagging (FP < 0). Briefly, FP values reflect the ratio of the frequencies of passages of the replication fork in forward and reverse directions relative to the reference strain. We then compared the rates of complementary mutations between the leading and the lagging strands in cancers with mutated Pol epsilon and mutated Pol delta (Fig. 2A,B; Supplemental Fig. S3).
Figure 2.

Replication asymmetry in bMMRD samples with mutations in replicative polymerases. (A) A schematic representation of estimation of the ratio of the mutation rates for complementary mutations. In the example shown, the frequency of the C-dT mismatch, resulting in the C → A mutation, is 1.5 times higher than the rate of the complementary G → T mutation when Pol epsilon is mutated and four times higher when Pol delta is mutated. Leftmost and rightmost bins together correspond to the 20% of the genome for which we can identify with the highest confidence that the reference strand is replicated primarily as lagging or leading, respectively. (B) The ratio of the mutation rates for complementary mutations (vertical axis) as a function of the propensity of the replication fork to replicate the reference strand as lagging or leading (horizontal axis). x-axis bins correspond to the percentiles of the replication timing derivative. Vertical bars represent 95% confidence intervals. Asterisks indicate significance of the deviation from one at the rightmost (or leftmost) bin. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. D corresponds to nucleotides A, G, or T. H corresponds to nucleotides A, C, or T. Note the logarithmic vertical axis.
We find that each polymerase usually produces one of the two complementary mismatches with a higher frequency (Fig. 2B). This effect changes monotonically with FP values. The largest difference is observed in the most extreme replication fork direction bins, which correspond to the genomic regions where we could predict FP with the highest confidence (Seplyarskiy et al. 2016). In what follows, we use the values in these bins for our comparisons and model; when all data were used, the observed asymmetry was weaker as expected, but its direction was the same (Supplemental Table S2). The direction of the asymmetry observed for mutated Pol epsilon for all substitution types are consistent with prior experimental results (Shinbrot et al. 2014).
Moreover, for two complementary pairs of mutations (C → A/G → T and T → G/A → C mutations), the biases associated with Pol epsilon and Pol delta were the opposite: Pol epsilon preferentially produced mismatches resulting in these mutations on the leading strand, while Pol delta produced them on the lagging strand (Fig. 2B), in line with the observations in yeast (Shcherbakova et al. 2003; Fortune et al. 2005; Lujan et al. 2014). Similar asymmetry patterns in samples with inactivating mutations in MMR system and Pol epsilon were observed in other available cancer data sets: whole genomes of uterine corpus endometrial carcinoma (UCEC) and colon adenocarcinoma (COAD), as well as in another independent data set of bMMRD glioblastoma cancers (Supplemental Figs. S4–S6; Erson-Omay et al. 2015). Moreover, these trends are consistent between individual samples (Supplemental Table S3), which shows that this asymmetry is specific to the mutational processes rather than cancer sample or cancer type.
Mutational spectra and strand asymmetries in MMR-deficient cancers
To investigate the properties of MMR, we compared COAD and UCEC cancers with functional and dysfunctional MMR pathways. MMR inactivation leads to abundant small insertions and deletions (indels) at simple sequence repeats. The resulting microsatellite instability (MSI) phenotype can be used to identify MMR deficiency. We used experimental data from TCGA database where cancers were characterized as microsatellite stable (MSS) or unstable. Inactivation of MMR can be caused by mutations in MMR genes, and for some of the samples, we were able to find somatic nonsense and frame-shift mutations in genes of the MMR system (Supplemental Table S4). However, MMR inactivation is more often caused by epigenetic alterations that change the expression of MMR genes, for example, methylation of the MLH1 gene promoter (Simpkins et al. 1999), or even driven by inactivation of genes not directly involved in MMR (Li et al. 2013). Therefore, we confirmed MMR deficiency in MSI cancers by analyzing the resulting mutational spectra. In all MSI samples, we observed high load of the mutational signatures of defective DNA MMR (signatures 6, 15, 20, 26 in Supplemental Table S5; http://cancer.sanger.ac.uk/cosmic/signatures), thus confirming MMR deficiency. The mutational spectra of COAD and UCEC cancers are somewhat distinct (Supplemental Fig. S7a,b). In many MSI samples of UCEC, we also observed the clock-like signature 5 (Alexandrov et al. 2015; http://cancer.sanger.ac.uk/cosmic/signatures) and other mutational signatures not related to MMR deficiency (Supplemental Table S5), indicating that the mutational patterns in UCEC MSI cancers are confounded by MMR-independent mutational processes. Among the MSS samples, a high fraction of the TpCpN → G and TpCpN → T mutations (corresponding to signatures 2 and 13; http://cancer.sanger.ac.uk/cosmic/signatures) was observed in UCEC but not in COAD samples; otherwise, the MSS spectra of the two cancer types were very similar. Signatures 2 and 13 are the mutational signatures of the APOBEC activity (Alexandrov et al. 2013); we excluded samples with these signatures (Supplemental Table S6) from further analysis.
We asked which types of substitutions are corrected by the MMR more effectively in humans. For this, we compared the mutational spectra of MMR-deficient and MMR-proficient cancers (Fig. 3A). The efficiency of the MMR of a particular mismatch type can be obtained as the ratio of the rates of the corresponding mutation in MMR-deficient and MMR-proficient cells. MMR corrects all types of mutations in all contexts (Fig. 3B), with a particularly high efficiency for mismatches resulting in the following mutations: C → T in the GpCpN context, A → G in all contexts, and C → A in the CpCpN (Fig. 2), in line with previous studies (Supek and Lehner 2015).
Figure 3.
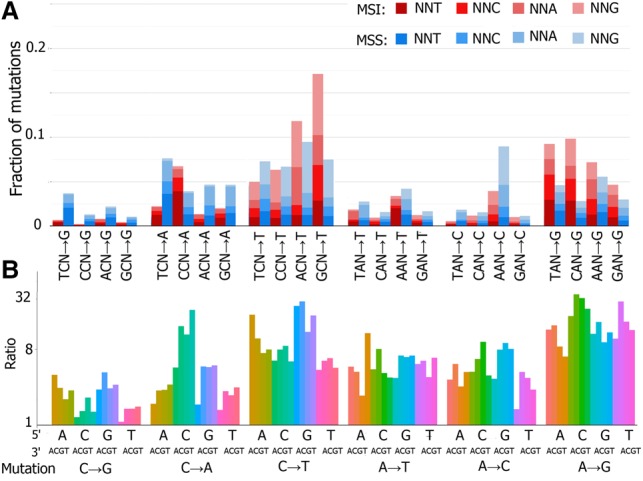
Comparison of mutational spectra of MMR-deficient and MMR-proficient cancers. Complementary mutation types were pooled. Data for MSI (n = 10) and MSS (n = 22) colon adenocarcinoma samples and uterine corpus endometrial carcinoma were pooled. Data for whole-genome sequencing. (A) Relative frequencies of the 96 mutation types (all possible mutation types in all possible tri-nucleotide contexts) in MSI and MSS cancers. (B) Ratio of the rates of each substitution in MSI and MSS cancers. Note the logarithmic vertical axis.
Similar to the analysis of samples with mutated polymerases, we then compared the rates of complementary mutations between the leading and the lagging strands for somatic mutations in MSS cancers and MSI cancers, as well as for germline mutations inferred from human polymorphisms (Fig. 4) or human–chimpanzee divergence data (Supplemental Fig. S8). As above, the below results are based on the 20% of the genome with the most robust estimation of the FP; the asymmetries in the rest of the genome were similar (Supplemental Table S7).
Figure 4.
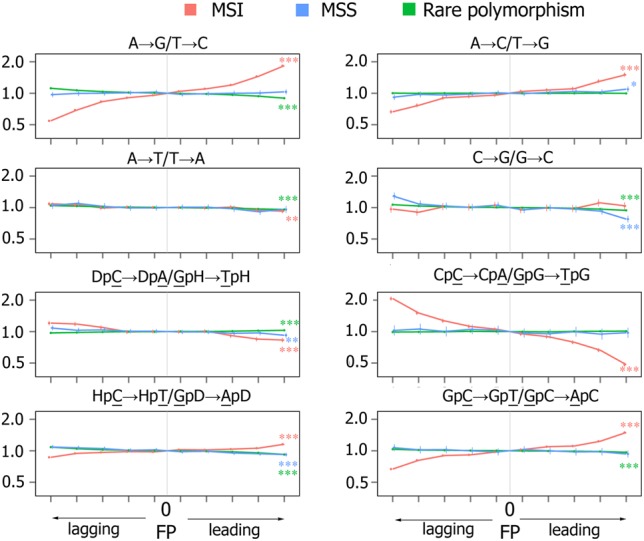
Strand asymmetry of mutations. Panels show ratios of the mutation rates for complementary mutations in 10 MSI (three COAD and seven UCEC) and 22 MSS (eight COAD and 14 UCEC) cancers and in rare human polymorphisms. Whole-genome data used. Axes and notations are as in Figure 2B.
In rare polymorphisms, the replication asymmetry between complementary mutations is much weaker, in line with previous results (Chen et al. 2011). The small observed asymmetry for some mutation types is likely due to non-replication-related errors. The best-studied example is the C → T mutation, which is enriched on the lagging strand in the germline mainly due to the activity of APOBEC family proteins (Seplyarskiy et al. 2017). The lack of strong asymmetry in germline mutations and in MSS cancers indicates that replication-associated mutational biases between the two strands in MMR-proficient cancer and germline cells are weak. In contrast, in MSI cancers where the MMR system is not proficient, we observe a strongly biased distribution of complementary mutations between the leading and the lagging DNA strands. A particularly strong (1.5- to 1.8-fold) asymmetry is observed for mutations that correspond to mismatches effectively repaired by the MMR (A → G, CpCpN → A, and GpCpN → T). The asymmetry is small or nearly absent for mutation types depleted in mutational spectra of MSI cancers (A → T, C → G, DpCpN → A, HpCpN → T), with the exception of the A → C mutation, where the asymmetry is 1.5-fold. For the A → C mutation, the asymmetry was high, although this mutation is not assigned to the mutational signature of MMR deficiency (http://cancer.sanger.ac.uk/cosmic/signatures). The rate of this mutation in MSI cancers was particularly elevated in CpApG and GpApB (B corresponds to nucleotides C, G, or T) contexts (Fig. 3B; Supplemental Fig. S9a), and its asymmetry in these contexts is also significantly higher than in all other contexts (Supplemental Fig. S9b). Thus, the A → C mutations in some contexts appear to be a minor mutational signature of the MMR.
Conceivably, the observed effect of the FP could be confounded by differences in RT between genomic regions. MMR is known to act more effectively in early replicating regions (Supek and Lehner 2015), and this could contribute to the observed asymmetry. Indeed, we found that the bins with the highest absolute values of FP are slightly biased toward early replicating regions (Supplemental Fig. S10). However, this bias cannot explain the observed asymmetry, because a similar level of asymmetry is observed in early and late replicating regions (Supplemental Table S8).
Separate analyses of UCEC and COAD cancer types and separate analysis of individual samples provide concordant results (Supplemental Tables S9, S10), confirming that the asymmetry is mainly determined not by the cancer type but by the (in)activity of the MMR system, although the observed minor differences between samples may likely reflect an admixture of MMR-independent mutational processes. The asymmetry is very similar if we analyze only intergenic regions (Supplemental Fig. S11), implying that it is not associated with transcription. We also reproduced our results in an independent data set of exome sequences of MSI and MSS cancers from TCGA involving a larger number of samples (Supplemental Fig. S12), separating them by cancer type: COAD, stomach adenocarcinoma (STAD), and UCEC. Again, the asymmetries observed were similar and concordant between cancer types (Supplemental Table S9) and samples (Supplemental Table S10).
MMR corrects single-nucleotide insertions and deletions (indels) with the highest effectiveness among all single nucleotide errors. In MMR-deficient cells, indels in homopolymer tracts and dinucleotide tandem repeats are the most frequent type of mutations. As in yeast (Lujan et al. 2015), in humans deletions are more common than insertions in MMR-deficient cells, and their rate increases with the homopolymer tract length. We studied the most frequent deletions type, deletions of A or T in corresponding homopolymer tracts. We observe a small asymmetry for deletions (Supplemental Fig. S13), supporting the conjecture that the MMR activity differs between the two strands.
Thus, the strand biases observed in MMR-deficient cancers are robust. In MMR-proficient cells, these biases have to be compensated by the MMR. MMR corrects more errors on one of the two strands, thus equalizing the mutation rate between strands.
Replication-based asymmetry in MMR-deficient cancers matches that in cancers with mutated polymerase delta
To better understand this balance between mutation and repair biases, we compare the direction of the mutational strand asymmetry in the extreme FP bins in MSI cancers and in cancers with mutated polymerases. Notably, the asymmetry in mutated Pol delta cancers is similar to that in MSI cancers with wild-type polymerases (Fig. 5; Supplemental Fig. S14), suggesting that Pol delta is also the main contributor to the mutations when it is intact. We also obtained very similar results using the newly available experimental data on FP obtained by sequencing of Okazaki fragments (Petryk et al. 2016; Supplemental Fig. S15). Furthermore, the contexts of mutations of MMR-deficient cancers are also more similar to those in cancers with mutated Pol delta than that in cancers with mutated Pol epsilon (Supplemental Table S11).
Figure 5.
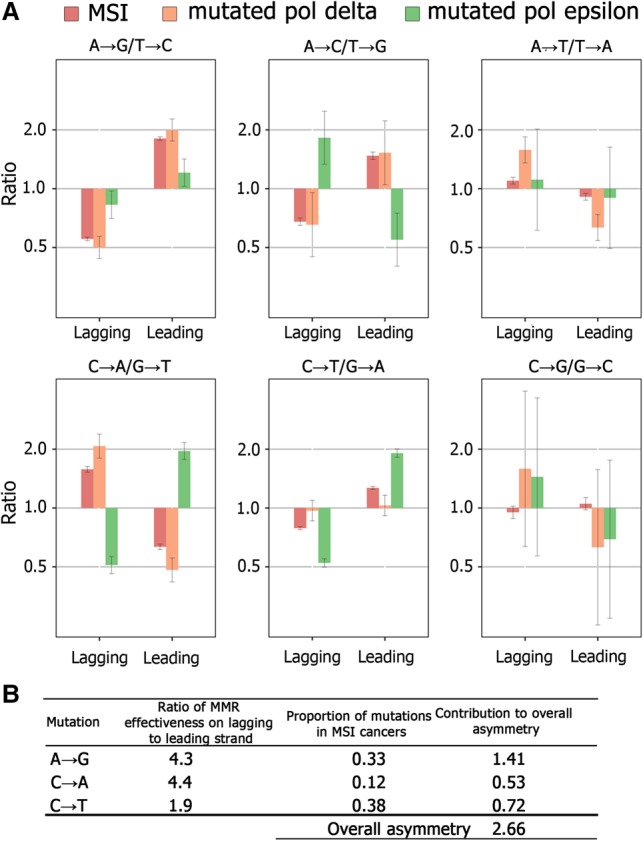
Strand asymmetry of mutations in MSI cancers and in bMMRD cancers with mutated Pol epsilon and Pol delta cancers. (A) The asymmetry for complementary mutations in the 20% of the genome where replication asymmetry could be determined with the highest confidence (corresponding to bins 1 and 10 in Figs. 2B, 4). Error bars, 95% confidence intervals. (B) Model-estimated ratio of MMR effectiveness on the lagging and the leading strands.
We designed a simple model using the asymmetry observed in cancers with mutated Pol epsilon and mutated Pol delta to estimate the contributions of these polymerases to mutagenesis (see Methods). It assumes that (1) all mutations in cancers with mutated Pol epsilon (mutated Pol delta) arise due to mismatches produced during leading (respectively, lagging) strand replication; (2) each of these polymerases independently contributes some fraction of mutations; (3) the frequencies of these mutations are not affected by the MMR system in MSI cancers; and (4) the direction of the replication fork in the 20% of the genome where replication asymmetry could be determined with the highest confidence is known exactly. We applied this model to the three mutation types with substantially elevated frequencies in MSI spectra, that is, those efficiently corrected by the MMR (Fig. 3): A → G, C → A and C → T. For these mutation types, we found that the contribution of Pol delta to the asymmetry observed in MSI cancers was 1.9- to 4.4-fold higher than the contribution of Pol epsilon (Supplemental Table S12). Accounting for the frequencies of the corresponding mutations, the overall contribution of Pol delta to mutagenesis was approximately threefold greater than that of Pol epsilon (Fig. 5B). This implies that the main driver of the asymmetry in MSI cancers are the mutations introduced by Pol delta.
Discussion
While previous studies have revealed the asymmetry in the substitution rates between the leading and lagging strand, its cause remained elusive. One reason for this was the difficulty in distinguishing between mutations resulting from mismatches in complementary strands. For example, an excess of C → A mutations on the lagging strand is equally consistent with an excess of G → T mutations on the leading strand. Here, we aim to resolve this by relating the observed asymmetries to the mutational signatures of polymerases. In particular, for the C → A/G → T mutation, we observed that neither of the two major polymerases introduces many G-dA mismatches (which would lead to G → T mutations); instead, both polymerases demonstrate an excess of C-dT mismatches (which would lead to C → A mutations). Thus, the above pattern more likely results from an excess of C → A mutations on the lagging strand than of G → T on the leading strand.
As MMR is primarily a coreplicative process (Hombauer et al. 2011; Liao et al. 2015), most mutations in MMR-deficient cancers are replicative errors. Therefore, the strand biases observed in MSI cancers reveal the biases of corresponding polymerases without the confounding factor of MMR. We show that the strand bias associated with MSI is largely concordant with the strand bias observed in cancers with mutated Pol delta for different mutation types, implying that the asymmetry in MSI cancers is likely due to the higher prevalence of mutations introduced by Pol delta as it replicates the lagging strand.
This analysis is subject to several caveats. First, we assumed that the ratio of error rates among complementary mutations measured for mutated polymerases is the same as that of wild-type polymerases. This is true as long as the magnitude of the strand bias primarily reflects the selectivity of nucleotide incorporation during DNA synthesis. At least for the C → A versus G → T mutations, the error rate estimated from a lactase array for wild-type Pol epsilon confirms this (Shinbrot et al. 2014). Second, we assumed that the frequencies at which mismatches are incorporated into DNA are independent of the functionality of the MMR. This is a parsimonious assumption, and we are unaware of any data falsifying it. Third, our approach allows predicting only the preferential direction of the replication fork; therefore, the magnitude of asymmetry may be underestimated, especially if it is very strong (Seplyarskiy et al. 2016). These caveats, however, are unlikely to affect our conclusions qualitatively.
Our analysis relies on the classical eukariotic model of replication fork, where each strand is replicated by its own polymerase. This model has been challenged recently. According to an alternative model, Pol delta is responsible for the synthesis of both the leading and the lagging DNA strands, while the exonuclease activity of Pol epsilon is involved in the correction of mismatches produced by Pol delta during leading strand replication (Johnson et al. 2015). However, this new model contradicts much of the available data and is highly controversial (Burgers et al. 2016; Lujan et al. 2016). Most previous experimental studies confirm the classical model, including experiments with mutant polymerases (Pursell et al. 2007; Kunkel and Burgers 2008; Nick McElhinny et al. 2008; Johnson et al. 2015) or incorporation of ribonucleotides (Clausen et al. 2015; Johnson et al. 2015), where specific mutations were observed on the corresponding strands; experiments investigating the association of proteins with leading and lagging strands of DNA replication forks (Yu et al. 2014); and biochemical experiments of assembly and stabilization of replication complexes (Georgescu et al. 2014, 2015; Langston et al. 2014). Moreover, recent evidence suggests that Pol epsilon does not proofread errors made by Pol delta (Flood et al. 2015). This model (Johnson et al. 2015) also contradicts our data, as it does not predict the opposite strand biases we observe in cancers with mutated Pol epsilon and Pol delta (Fig. 2B). Furthermore, it predicts no asymmetry in MMR-deficient cancers with mutated Pol epsilon because, under this model, both strands are replicated by Pol delta and repaired by Pol epsilon. Conversely, our data are in perfect agreement with the classical model, which assumes that different strands are replicated by different polymerases.
Another recent study suggests that Pol delta can replicate both DNA strands near replication origins (Yeeles et al. 2017). We believe that these results cannot affect our inferences, as they suggest that Pol delta replicates both strands only at replication origins, while the bulk of the genome is replicated according to the classical model. This is also consistent with our results: If most of the genome was replicated by Pol delta alone, we would not observe the opposite patterns of asymmetry between Pol epsilon and Pol delta.
According to the classical model, alongside Pol delta, polymerase alpha is also involved in replication of the lagging strand, with Pol delta repairing the mismatches introduced by it (Pavlov et al. 2006). A recent study also proposed that rapid reassociation of DNA-binding proteins can prevent Pol delta–mediated displacement of Pol alpha–synthesized DNA (Reijns et al. 2015). This could increase mutation rate on the lagging strand due to unrepaired errors produced by error-prone Pol alpha. Therefore, Pol alpha may contribute to the patterns observed in cancers with mutated Pol delta. We are unaware of human data that could allow us to distinguish between the contributions of Pol alpha and Pol delta. However, in yeasts with mutations in Pol alpha, the strand asymmetry is qualitatively similar to that in yeasts with mutated Pol delta (Lujan et al. 2014), suggesting that both enzymes contribute to the excess of mutations on the lagging strand. More generally, factors including those discussed above can affect our quantitative estimates for the extent of the asymmetry introduced and repaired for different mutation types.
Mutational spectra differ between the MMR-proficient and MMR-deficient cancers with somatic mutations in Pol epsilon or delta cancers (Supplemental Fig. S16). However, the level of replication asymmetry was similar among all cancers with mutated Pol epsilon or mutated Pol delta (Supplemental Table S13). Therefore, like in yeasts (Lujan et al. 2014), while the MMR may change the mutational spectra, it is insufficient to compensate for the radical asymmetries introduced by mutated Pol epsilon and mutated Pol delta.
Since the strand asymmetry observed in MMR-proficient cells, including the germline, is weak, the strand biases introduced by polymerases have to be compensated by the MMR. As more mismatches are incorporated on the lagging strand, this also implies that the MMR repairs more mismatches on the lagging strand. This can have two explanations. First, the MMR efficiency could differ between the strands, so that it is more likely to repair a mismatch on the lagging than on the leading strand. Second, MMR could have equal efficiency between strands, preserving the asymmetries of coreplicative mutations, but could radically reduce their numbers, removing the overall mutational asymmetries by increasing the fraction of symmetric non-coreplicative mutations. It should be possible to distinguish between these two models by comparing the effect of MMR deficiency between Pol epsilon and Pol delta mutated cells. If MMR is more efficient on the lagging strand, MMR deficiency should lead to a more radical increase in the mutation rate in Pol delta mutated strains, compared with Pol epsilon mutated cells. In yeast, MMR inactivation has a twofold stronger effect on the mutation rate in Pol delta mutated strains (Lujan et al. 2014), consistent with this expectation.
In humans, data on MMR-proficient cancers with mutated polymerases could resolve this issue. Unfortunately, sufficient data are currently available only for cancers with mutated Pol epsilon, but not with mutated Pol delta. Still, if the MMR has a weaker effect on the mismatches on the leading strand, the status of the MMR should affect the mutation rate in cancers where most mutations are caused by mismatches introduced by Pol epsilon to a lesser extent compared with cancers where both polymerases have comparable contribution. In line with this expectation, the mutation rate differs between MMR-proficient and MMR-deficient cancers by a factor of about nine, while the same ratio for cancers with mutated Pol epsilon is only approximately two (Supplemental Fig. S17). Therefore, the available data suggest that MMR not only removes more mismatches but also does it more efficiently, on the lagging strand.
In summary, we show that the polymerase error rate is higher during lagging strand replication, that this asymmetry is primarily due to mutations produced by Pol delta (and probably Pol alpha) on the lagging strand, and that a higher number and, likely, a higher fraction of these mismatches is removed by the MMR on the lagging strand (Fig. 6). This is in agreement with the biochemical property of the MMR to preferentially eliminate mismatched nucleotides on the DNA strand containing the nick (Pluciennik et al. 2010). As the lagging strand is replicated in Okazaki fragments, their ends could represent a signal of the nascent strand for the MMR, facilitating its recruitment to this strand (Lujan et al. 2012). Our observations are strongly concordant with experiments in yeasts (Lujan et al. 2014), indicating that basic principles of MMR are conserved between yeasts and humans. While our results reflect the properties of the MMR in somatic cells, they are likely to be similar in germline cells. In the absence of MMR, the asymmetry in the numbers and types of mismatches produced during replication of leading and lagging strands in germline cells would lead not only to an increase of the genomic mutation rate but also to bias the local nucleotide content. From the evolutionary point of view, the concordance between the biases in introduction and repair of mismatches helps reduce the genomic mutation rate and prevent accumulation of local strand biases in nucleotide composition.
Figure 6.
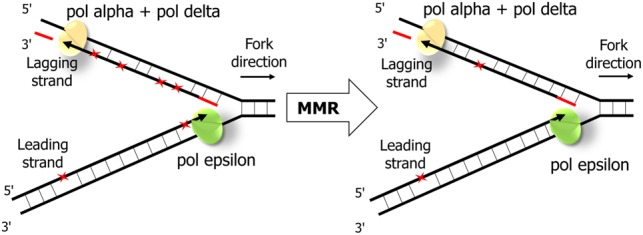
The schematic representation of MMR effectiveness during the leading and the lagging strand replication. While mismatches (red asterisks) are introduced more frequently during replication of the lagging strand by Pol delta, MMR corrects more mismatches on the lagging strand.
Methods
Mutation data
We used the following previously published data: (1) somatic mutations for whole-exome sequences of MSI (n = 159) and MSS (n = 782) cancers and for whole-genome sequences of MSI (n = 11) and MSS (n = 27) cancers from the data portal of The Cancer Genome Atlas (TCGA; https://portal.gdc.cancer.gov/); (2) BAM files with aligned reads for ultra-hypermutated cancers with inherited bMMRD and somatic mutation in Pol epsilon or Pol delta (Shlien et al. 2015); (3) VCF files for hypermutated cancers with inherited homozygous mutation in the MSH2 gene (MMR system) and somatic mutation in Pol epsilon (Erson-Omay et al. 2015); (4) human–chimpanzee–orangutan multiple alignment from the UCSC Genome Browser (https://genome.ucsc.edu/); and (5) human polymorphism data from 1000 Genomes Project. Substitution rates were calculated as the number of substitutions of a particular type divided by the number of target sites. For asymmetry analyses of the MSI and MSS TCGA data, cancers with mutations in replicative polymerases were excluded. For analysis of the interspecies data, we obtained mutations in the human line after its divergence from the chimpanzee by maximum parsimony, using orangutan as the outgroup.
Identification of somatic mutations in ultra-hypermutated cancers
Somatic mutations were identified using MuTect (v. 1.1.4) (Cibulskis et al. 2013) under the default parameters. Mutations were then filtered against common single-nucleotide polymorphisms (SNPs) found in dbSNP and against the Catalog of Somatic Mutations in Cancer (COSMIC database).
Leading vs. lagging strand asymmetry
The derivative of the RT at the position of the mutation was used as a proxy for the probability that the reference strand is replicated as leading or lagging in the current position, as described previously (Seplyarskiy et al. 2016). RT for lymphoblastoid cell lines was used (Koren et al. 2012). The genome was categorized by these values into 10 equal bins, with the low value of the derivative corresponding to the propensity of the DNA segment to be replicated as lagging; high value, as leading (Supplemental Fig. S18). For each bin, the numbers of substitutions and target sites were calculated. Each substitution was counted twice: as a substitution on the reference sequence with the corresponding derivative of the RT and as a complementary substitution with the inverse derivative. Thus, each plot of substitution asymmetry (Figs. 2B, 4) is symmetric with respect to zero. Score confidence intervals were obtained for the relative risk in a 2×2 table. For separate analysis of late and early replicating regions, the genome was separated in two categories by the RT value (25% of the genome with the highest and the lowest RT values).
Okazaki fragments sequencing data
We downloaded data for Okazaki fragments sequencing for lymphoblastoid cell line GM06990 (Petryk et al. 2016). We used RFD values to identify whether reference strand is replicated primarily as leading (low RFD) or lagging (high RFD). We subdivided the genome into 10 equal bins according to the RFD values (Supplemental Fig. S19).
Indel analyses
We used data on single-nucleotide deletions in poly(A) and poly(T) tracts for MSI cancer genomes, for tracts with length six to eight identical nucleotides where enough deletions were found for analysis.
APOBEC enrichment
APOBEC enrichment was counted for each sample as ratio of C → K mutation rates in TpCpW and VpCpW contexts. Weights of mutational signature were calculated using the R-package “deconstructSigs” (Rosenthal et al. 2016).
Model for mutational biases between strands
We calculated the ratios of the mutation rates using the following logic. From Figure 5, for each type of mutation A → B, we obtained the ratio of its rate r(A → B) and the rate of its complement r(A′ → B′) on the leading strand. then
| (1) |
where α is the fraction of mutations A → B and A′ → B′ that are produced by Pol epsilon on the leading strand; 1 − α is the fraction of such mutations that are produced by Pol delta on the lagging strand; xɛ and 1 − xɛ are the fractions of mutations A → B and A′ → B′, respectively, produced by Pol epsilon; xδ and 1 − xδ are the fractions of mutations A → B and A' → B', respectively, produced by Pol delta; and xMMR and 1 − xMMR are the fractions of mutations A → B and A' → B', respectively, on the leading strand in MSI cancers.
For example, consider mutation C → A/G → T. From Figure 5, xMMR/(1 − xMMR) is 0.63. As the C → A/G → T ratio for the leading strand in mutated Pol epsilon cancers is 1.96 (Fig 5), the fraction of C → A mutations produced by Pol epsilon is xɛ = 0.66, and the fraction of G → T mutations produced by Pol epsilon is 1 − xɛ = 0.34. Similarly, as the C → A/G → T ratio for the lagging strand in mutated Pol delta cancers is 2.07, the fraction of C → A mutations produced by Pol delta is xδ = 0.67, and the fraction of G → T mutations produced by Pol delta is 1 − xδ = 0.33. From equation 1, (1 − α)/α = 4.39. In other words, for this mutation type, Pol delta produces mismatches leading to this mutation type on the lagging strand approximately four times more often than Pol epsilon produces them on the leading strand. As no strand bias is observed in MSS cancers, the repair bias by MMR has to be exactly inverse, repairing 4 times as many mutations produced by Pol delta than by Pol epsilon. The results for the two other mutation types are calculated similarly (Supplemental Table S12). The overall strand asymmetry of MMR was calculated as the mean asymmetry across the five mutation types, weighted by the proportions of each mutation in MSI cancers.
Supplementary Material
Acknowledgments
We thank Shamil Sunyaev, Pasha Mazin, and Sonya Garushyants for useful discussion. This work was performed at IITP RAS and supported by the Russian Science Foundation grant no. 14-50-00150.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.219915.116.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, Stratton MR. 2015. Clock-like mutational processes in human somatic cells. Nat Genet 47: 1402–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker A, Audit B, Chen C-L, Moindrot B, Leleu A, Guilbaud G, Rappailles A, Vaillant C, Goldar A, Mongelard F, et al. 2012. Replication fork polarity gradients revealed by megabase-sized U-shaped replication timing domains in human cell lines. PLoS Comput Biol 8: e1002443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgers PMJ, Gordenin D, Kunkel TA. 2016. Who is leading the replication fork, Pol ɛ or Pol δ? Mol Cell 61: 492–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-L, Duquenne L, Audit B, Guilbaud G, Rappailles A, Baker A, Huvet M, d'Aubenton-Carafa Y, Hyrien O, Arneodo A, et al. 2011. Replication-associated mutational asymmetry in the human genome. Mol Biol Evol 28: 2327–2337. [DOI] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. 2013. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31: 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen AR, Lujan SA, Burkholder AB, Orebaugh CD, Williams JS, Clausen MF, Malc EP, Mieczkowski PA, Fargo DC, Smith DJ, et al. 2015. Tracking replication enzymology in vivo by genome-wide mapping of ribonucleotide incorporation. Nat Struct Mol Biol 22: 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erson-Omay EZ, Çağlayan AO, Schultz N, Weinhold N, Omay SB, Özduman K, Köksal Y, Li J, Serin Harmancı A, Clark V, et al. 2015. Somatic POLE mutations cause an ultramutated giant cell high-grade glioma subtype with better prognosis. Neuro Oncol 17: 1356–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood CL, Rodriguez GP, Bao G, Shockley AH, Kow YW, Crouse GF. 2015. Replicative DNA polymerase δ but not ɛ proofreads errors in cis and in trans. PLoS Genet 11: e1005049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortune JM, Pavlov YI, Welch CM, Johansson E, Burgers PMJ, Kunkel TA. 2005. Saccharomyces cerevisiae DNA polymerase δ: high fidelity for base substitutions but lower fidelity for single- and multi-base deletions. J Biol Chem 280: 29980–29987. [DOI] [PubMed] [Google Scholar]
- Georgescu RE, Langston L, Yao NY, Yurieva O, Zhang D, Finkelstein J, Agarwal T, O'Donnell ME. 2014. Mechanism of asymmetric polymerase assembly at the eukaryotic replication fork. Nat Struct Mol Biol 21: 664–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu RE, Schauer GD, Yao NY, Langston LD, Yurieva O, Zhang D, Finkelstein J, O'Donnell ME. 2015. Reconstitution of a eukaryotic replisome reveals suppression mechanisms that define leading/lagging strand operation. eLife 4: e04988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haradhvala NJ, Polak P, Stojanov P, Covington KR, Shinbrot E, Hess JM, Rheinbay E, Kim J, Maruvka YE, Braunstein LZ, et al. 2016. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell 164: 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henninger EE, Pursell ZF. 2014. DNA polymerase ɛ and its roles in genome stability. IUBMB Life 66: 339–351. [DOI] [PubMed] [Google Scholar]
- Hombauer H, Srivatsan A, Putnam CD, Kolodner RD. 2011. Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science 334: 1713–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RE, Klassen R, Prakash L, Prakash S. 2015. A Major role of DNA polymerase δ in replication of both the leading and lagging DNA strands. Mol Cell 59: 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Polak P, Nemesh J, Michaelson JJ, Sebat J, Sunyaev SR, McCarroll SA. 2012. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet 91: 1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korona DA, Lecompte KG, Pursell ZF. 2011. The high fidelity and unique error signature of human DNA polymerase ɛ. Nucleic Acids Res 39: 1763–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA. 2009. Evolving views of DNA replication (in)fidelity. Cold Spring Harb Symp Quant Biol 74: 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA. 2011. Balancing eukaryotic replication asymmetry with replication fidelity. Curr Opin Chem Biol 15: 620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Burgers PM. 2008. Dividing the workload at a eukaryotic replication fork. Trends Cell Biol 18: 521–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston LD, Zhang D, Yurieva O, Georgescu RE, Finkelstein J, Yao NY, Indiani C, O'Donnell ME. 2014. CMG helicase and DNA polymerase form a functional 15-subunit holoenzyme for eukaryotic leading-strand DNA replication. Proc Natl Acad Sci 111: 15390–15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrea AA, Lujan SA, Nick McElhinny SA, Mieczkowski PA, Resnick MA, Gordenin DA, Kunkel TA. 2010. Genome-wide model for the normal eukaryotic DNA replication fork. Proc Natl Acad Sci 107: 17674–17679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Mao G, Tong D, Huang J, Gu L, Yang W, Li G-M. 2013. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell 153: 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Schroeder JW, Gao B, Simmons LA, Biteen JS. 2015. Single-molecule motions and interactions in live cells reveal target search dynamics in mismatch repair. Proc Natl Acad Sci 112: E6898–E6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan SA, Williams JS, Pursell ZF, Abdulovic-Cui AA, Clark AB, Nick McElhinny SA, Kunkel TA. 2012. Mismatch repair balances leading and lagging strand DNA replication fidelity. PLoS Genet 8: e1003016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan SA, Clausen AR, Clark AB, MacAlpine HK, MacAlpine DM, Malc EP, Mieczkowski PA, Burkholder AB, Fargo DC, Gordenin DA, et al. 2014. Heterogeneous polymerase fidelity and mismatch repair bias genome variation and composition. Genome Res 24: 1751–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan SA, Clark AB, Kunkel TA. 2015. Differences in genome-wide repeat sequence instability conferred by proofreading and mismatch repair defects. Nucleic Acids Res 43: 4067–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan SA, Williams JS, Kunkel TA. 2016. Eukaryotic genome instability in light of asymmetric DNA replication. Crit Rev Biochem Mol Biol 51: 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganella S, Alexandrov LB, Glodzik D, Zou X, Davies H, Staaf J, Sieuwerts AM, Brinkman AB, Martin S, Ramakrishna M, et al. 2016. The topography of mutational processes in breast cancer genomes. Nat Commun 7: 11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Gordenin DA, Stith CM, Burgers PMJ, Kunkel TA. 2008. Division of labor at the eukaryotic replication fork. Mol Cell 30: 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov YI, Frahm C, Nick McElhinny SA, Niimi A, Suzuki M, Kunkel TA. 2006. Evidence that errors made by DNA polymerase α are corrected by DNA polymerase δ. Curr Biol 16: 202–207. [DOI] [PubMed] [Google Scholar]
- Petryk N, Kahli M, d'Aubenton-Carafa Y, Jaszczyszyn Y, Shen Y, Silvain M, Thermes C, Chen C-L, Hyrien O. 2016. Replication landscape of the human genome. Nat Commun 7: 10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluciennik A, Dzantiev L, Iyer RR, Constantin N, Kadyrov FA, Modrich P. 2010. PCNA function in the activation and strand direction of MutLα endonuclease in mismatch repair. Proc Natl Acad Sci 107: 16066–16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pursell ZF, Isoz I, Lundström E-B, Johansson E, Kunkel TA. 2007. Yeast DNA polymerase ɛ participates in leading-strand DNA replication. Science 317: 127–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijns MAM, Kemp H, Ding J, de Procé SM, Jackson AP, Taylor MS. 2015. Lagging-strand replication shapes the mutational landscape of the genome. Nature 518: 502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. 2016. deconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol 17: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seplyarskiy VB, Soldatov RA, Popadin KY, Antonarakis SE, Bazykin GA, Nikolaev SI. 2016. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res 26: 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seplyarskiy VB, Andrianova MA, Bazykin GA. 2017. APOBEC3A/B-induced mutagenesis is responsible for 20% of heritable mutations in the TpCpW context. Genome Res 27: 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova PV, Pavlov YI, Chilkova O, Rogozin IB, Johansson E, Kunkel TA. 2003. Unique error signature of the four-subunit yeast DNA polymerase ɛ. J Biol Chem 278: 43770–43780. [DOI] [PubMed] [Google Scholar]
- Shinbrot E, Henninger EE, Weinhold N, Covington KR, Goksenin AY, Schultz N, Chao H, Doddapaneni H, Muzny DM, Gibbs RA, et al. 2014. Exonuclease mutations in DNA polymerase ɛ reveal replication strand specific mutation patterns and human origins of replication. Genome Res 24: 1740–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlien A, Campbell BB, de Borja R, Alexandrov LB, Merico D, Wedge D, Van Loo P, Tarpey PS, Coupland P, Behjati S, et al. 2015. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat Genet 47: 257–262. [DOI] [PubMed] [Google Scholar]
- Simpkins SB, Bocker T, Swisher EM, Mutch DG, Gersell DJ, Kovatich AJ, Palazzo JP, Fishel R, Goodfellow PJ. 1999. MLH1 promoter methylation and gene silencing is the primary cause of microsatellite instability in sporadic endometrial cancers. Hum Mol Genet 8: 661–666. [DOI] [PubMed] [Google Scholar]
- St Charles JA, Liberti SE, Williams JS, Lujan SA, Kunkel TA. 2015. Quantifying the contributions of base selectivity, proofreading and mismatch repair to nuclear DNA replication in Saccharomyces cerevisiae. DNA Repair 31: 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Lehner B. 2015. Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature 521: 81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkova M, Tomek J, Kriaucionis S, Schuster-Böckler B. 2017. Widespread impact of DNA replication on mutational mechanisms in cancer. bioRxiv 10.1101/111302. [DOI] [Google Scholar]
- Yeeles JTP, Janska A, Early A, Diffley JFX. 2017. How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol Cell 65: 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Gan H, Han J, Zhou Z-X, Jia S, Chabes A, Farrugia G, Ordog T, Zhang Z. 2014. Strand-specific analysis shows protein binding at replication forks and PCNA unloading from lagging strands when forks stall. Mol Cell 56: 551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.