

Abstract
Cholangiocarcinoma (CCA) is a fatal disease with a five-year survival of <30%. For a majority of patients chemotherapy is the only therapeutic option, and virtually all patients relapse. Gemcitabine is the frontline agent for treatment of CCA. Patients treated with gemcitabine monotherapy survive ~8 months. Combining this agent with cisplatin increases survival by ~3 months, but neither regimen produces durable remissions. The molecular etiology of this disease is poorly understood.
To facilitate molecular characterization and development of effective therapies for CCA, we established a panel of patient-derived xenograft (PDX) models of CCA. We used two of these models to investigate the anti-tumor efficacy and mechanism of action of the bromodomain inhibitor JQ1, an agent that has not been evaluated for the treatment of CCA.
The data show that JQ1 suppressed the growth of the CCA PDX model CCA2, and demonstrate that growth suppression was concomitant with inhibition of c-Myc protein expression. A second model (CCA1) was JQ1-insensitive, with tumor progression and c-Myc expression unaffected by exposure to this agent. Also selective to CCA2 tumors, JQ1 induced DNA damage and apoptosis, and downregulated multiple c-Myc transcriptional targets that regulate cell cycle progression and DNA repair. These findings suggest that c-Myc inhibition and several of its transcriptional targets may contribute to the mechanism of action of JQ1 in this tumor type. We conclude that BET inhibitors such as JQ1 warrant further investigation for the treatment of CCA.
Keywords: cholangiocarcinoma, bromodomain inhibitor, JQ1, patient-derived xenograft models (PDX), c-Myc
INTRODUCTION
Cholangiocarcinoma (CCA), a neoplasm of epithelial cells lining bile ducts, originates in any portion of the biliary tree (1). Tumors located within the bile ducts of the liver are designated as intra-hepatic CCA (IHCC); tumors arising in the biliary tree of the peripancreatic region are termed extra-hepatic CCA (EHCC). The incidence of IHCC is increasing worldwide, while the incidence of EHCC has remained stable or has decreased (2). Approximately 30% of CCA patients present with disease amenable to surgical resection, the only curative treatment for CCA. The remaining 70% present with advanced or metastatic disease, and undergo systemic chemotherapy with gemcitabine or gemcitabine and cisplatin. This combination extends median overall survival from ~8 months for gemcitabine alone to ~11 months, but is not curative (3).
To facilitate evaluation of novel therapies for CCA, we developed five CCA patient-derived xenograft (PDX) models from primary human tumor specimens, and verified that early passage tumors in murine hosts retain specific molecular, histologic, and genetic characteristics of their human tumors of origin. The models were derived from four independent EHCC specimens and from a metastatic lesion of a patient with IHCC. These are the first PDX models for EHCC and metastatic IHCC.
While the molecular etiology of CCA is incompletely understood, several key characteristics common to this tumor type have been identified. These include mutations in codons 12 and 13 of the KRAS gene, as well as elevated levels of epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2) and the oncogenic transcription factor c-Myc (4,5). KRAS encodes a GTPase of the Ras family. Gain-of-function mutations of KRAS occur in 23–50% of IHCC and 30–40% of EHCC tumors. These mutations constitutively activate the Ras pathway and accelerate tumor progression (6). A subset of CCA tumors (13–15%) harbors gain-of-function mutations in the EGFR gene, and this growth factor receptor is overexpressed in 25–31% of IHCC and 21–58% of EHCC tumors (7–10). Detectable EGFR expression has been identified as a prognostic factor for tumor recurrence (5). Similarly, while HER2 is overexpressed less frequently than EGFR (0.9–8.5% of CCA tumors), its overexpression is associated with poor prognosis (5). The oncogenic transcription factor c-Myc is a basic helix-loop-helix zipper protein that complexes with Max to bind to consensus E-box enhancer sequences. c-Myc regulates transcription of gene products that control diverse cell functions that include proliferation, apoptosis, and cell cycle progression (11). c-Myc is expressed in 94% (59/63) of EHCC tumors and 42% (10/24) of IHCC tumors (4,12). Experimental downregulation of c-Myc expression decreases the invasiveness of CCA cells in vitro suggesting that c-Myc contributes to progression of this tumor type (13).
Since a majority of CCA tumors overexpress c-Myc and the bromodomain and extraterminal domain (BET) inhibitor JQ1 downregulates expression of c-Myc in some tumor types, we sought to determine if JQ1 had anti-tumor activity in preclinical CCA models. JQ1 inhibits the activity of BET family members (BRD2, BRD3, BRD4, and BRDT) (14). This family of proteins binds to acetylated lysine residues of histones. Interaction of BET proteins, a component of BET-dependent transcriptional complexes, with acetylated lysine residues at specific genetic loci regulates the association of these complexes with genetic loci and, therefore, regulates transcription of gene products that depend on this mechanism for expression (14,15). JQ1 binds to the recognition pocket for acetylated lysine residues of BET proteins, predominantly BRD4, thereby competitively inhibiting BET-histone binding and recruitment of transcriptional complexes to genomic loci (14). JQ1 suppresses tumor growth in preclinical models of multiple tumors types, but has not been evaluated in preclinical CCA models (14,16–18).
MATERIALS AND METHODS
Ethics Statement
Human subject and animal use were approved by the Institutional Review Board (IRB) and Institutional Animal Care and Use Committee (IACUC) of the University of Alabama at Birmingham (Birmingham, AL). All patients who provided tumor specimens gave informed consent.
Establishment of CCA PDX models
As published previously, primary human tissue not needed for diagnosis was confirmed to contain tumor cells (LNC), and implanted subcutaneously into immunocompromised mice within 1–6 hours of resection (19). PDX models were deemed viable if F1 tumors demonstrated progressive growth. Five of five specimens (CCA1-5) met these criteria. In addition to tissue specimens implanted, specimens were also embedded in paraffin (FFPE), snap frozen, or archived in DMSO as viable tissue.
Mutational status of codons 12 of KRAS
Detailed methods have been published previously (19). Briefly, DNA was isolated from F0 (primary tumor) and F1 generation tissue using a DNA/RNA extraction kit (Epicentre, Madison, WI, USA). Concentration and quality of nucleic acids were determined with an ND-1000 spectrophotometer and Nanodrop 3.0.1 software (Nanodrop, Coleman Technologies, Inc., Wilmington, DE, USA). PCR reactions were done with 400 ng DNA, using conditions and primers as published to generate a 214bp reaction product that included codons 12 of the KRAS gene (19). Following gel electrophoresis and DNA extraction (Fermentas-Fisher Scientific, Savana, GA, USA), PCR products were sequenced by the Center for AIDS Research (CFAR) DNA sequencing core at UAB. Electropherograms were analyzed using Finch TV (version1.4.0; www.geospiza.com).
Histologic Analysis: Hematoxylin-Eosin (H&E) staining
Slides containing thin sections of FFPE specimens were stained with hematoxylin and counterstained with eosin using standard methods (19).
Immunohistochemistry (IHC)
IHC was performed using published standard methods (18). Tissue was incubated with primary antibody overnight to detect human c-Myc (AHO0062) (Invitrogen, Fredrick, MD, USA), EGFR (Santa Cruz Biotech, Santa Cruz, CA, USA), HER2, LCK and MSH2 (Cell Signaling, Danvers, MA, USA), Chk1 and E2F1 (Bethyl Laboratories, Montgomery, TX, USA), BRCA2 (Santa Cruz Biotech, Santa Cruz, CA, USA) or TEK (Proteintech, Rosemont, IL, USA). Antibody binding was detected using immPRESS™ secondary reagents (Vector Laboratories, Burlington, CA, USA) and DAB High Contrast chromogen (Scytek Laboratories, Logan, UT, USA) according to manufacturers’ instructions. Slides were counterstained with Harris Hematoxylin (Fisher Scientific, Suwanee, GA, USA), and photomicrographs taken using an Olympus BH-2 microscope with DP70 camera and DPS-BSW v3.1 software (Center Valley, PA, USA). Expression indices (EI) were calculated as percent of tumor cells expressing detectable protein (1–100) multiplied by staining intensity (1-weak, 2-moderate, 3-strong), with the highest possible score of 300 (20).
Nanostring gene expression analysis
Total RNA (400ng) was isolated from frozen specimens by Triazol-chloroform, and analyzed in the UAB NanoString Laboratory (http://www.uab.edu/medicine/radonc/en/nanostring) using nCounter Analysis System (NanoString Technlogies, Seattle, WA, USA) as described previously (18).
Drug efficacy studies
We prepared JQ1 according to published procedures (14). When subcutaneous tumors reached ~100–200 mm3, tumor bearing mice (7–10 tumors/group) received 50 mg/kg JQ1 or vehicle (vehicle control, VC) intraperitoneally daily for 20 days. Tumors were measured with Vernier calipers (Fowler/Slyvac, Newtown, MA, USA) twice weekly, and tumor volumes calculated using the equation v = (π/6)d3. Twenty-four hours after completion of treatment, mice were euthanized and tumor tissue fixed in formalin and paraffin embedded (FFPR) or frozen in liquid nitrogen and archived.
Statistics
Unless analyzed by NanoString (above), tumor volumes were compared by two-way analysis of variance followed by Bonferroni post test (GraphPad Prism 5.0). Values presented equal mean ± S.E.M. P<0.05 was considered significant.
RESULTS
Clinical characteristics of patients from whom specimens were obtained to develop PDX models
We obtained CCA tumor specimens from each of five consented patients undergoing standard of care resection or biopsy. Of the five specimens, four (CCA1-4) were obtained from primary tumor loci, and were EHCC subtype and Stage IIA or IIB (Table 1). All four EHCC specimens were obtained by Whipple procedure which involves partial pancreatic resection. The specimen from which CCA5 was derived was obtained at biopsy of a metastatic peritoneal lesion, and is IHCC subtype and Stage IV. Tumor differentiation status ranged from well differentiated (G1) to moderately (G2) or poorly differentiated (G3). Microscopic evaluation confirmed lymph node involvement and perineural invasion in three of the four EHCC patients. Limited clinical information was available for the CCA5 tumor of origin.
Table 1. Clinical characteristics of tumor from which CCA PDX models were derived.
The tumor tissue used to generate PDX models were obtained from consented patients undergoing surgical resection (CCA1-CCA4) or biopsy (CCA5) as part of standard of care for treatment of CCA.
| ID | TNM1 | Stage | Grade | Sub Class | LN2 met | L.I3 | P.I.4 | pT5 (cm) |
|---|---|---|---|---|---|---|---|---|
| CCA1 | pT2N1 | IIB | G3 | EHCC | Y | Y | Y | 1.8 |
| CCA2 | pT3N0 | IIA | G2-G3 | EHCC | N | Y | N | 3.5 |
| CCA3 | pT3N1 | IIB | G3 | EHCC | Y | Y | Y | 1.3 |
| CCA4 | pT3N1 | IIB | G1 | EHCC | Y | N | Y | 1.2 |
| CCA5 | met6 | IV | n/a | IHCC | n/a | n/a | n/a | n/a |
TNM staging system. T represents the size and extent of the primary tumor; N represents the number of lymph nodes involved; M indicates presence of metastasis.
Lymph node metastases present: Yes/No
Lymphovascular invasion
Perineural invasion
Size of primary tumor
Metastases
n/a: not available
All CCA specimens produced viable PDXs that retained histological characteristics of the primary tumors from which they were derived
CCA1-4 surgical specimens were implanted subcutaneously into immunocompromised mice, within an hour of resection. Tumor progression was monitored weekly. For model CCA5, the interval between surgery and implantation was ~6 hours. Growth rates for CCA1, CCA2 and CCA3, allowed for transplantation of F1 generation tumors within 9–16 weeks (Figure S1a). CCA4 and CCA5 tumors grew more slowly, with F1 tumors transplanted at 32–37 weeks. CCA3 F1 generation tumors were viable but highly vascularized and bloody, making precise quantitation of tumor size difficult. All five models were readily evaluable regarding histology of F1 tumors.
We compared the histologic characteristics of FFPE preparations of F0 and F1 tumors, to determine whether the model tumors retained the histology of the tumors of origin. In CCA tumors, cells that line biliary ducts are usually low cuboidal, with a relatively high nuclear-cytoplasmic (N:C) ratio, as seen for CCA1, CCA2 and CCA3 (Figure S1b). Additional histologic and morphologic features and comparison between F0 and F1 tumors are detailed in Supplementary data for each model (Figure S2).
The histopathological analysis of each of the F1 generation PDXs demonstrates that each model retains defining morphology and histology of the primary tumor from which it was derived. We did observe minor discrepancies in some of the models, such as relatively small differences in amount of peritumoral stroma or in differentiation status. A plausible explanation for the decrease in peritumoral stroma in the PDX is the increase in tumor cellularity composing the PDX. This type of change in stromal content has been documented previously in other types of PDX models including pancreatic cancer, lung cancer, and breast cancer models (18,21,22). Overall, PDX models retain predominant characteristics of tumors of origin.
PDX models retain the mutational status of KRAS codons 12 of primary tumors of origin
Mutations in the KRAS gene are among the most frequent genetic alterations in human cancers, with mutations in this gene estimated to occur in 17–25% of solid tumors (23). KRAS mutations typically (>90%) occur as point mutations in codons 12 and/or 13, and these mutations result in constitutive activation of Ras/Raf/Mek/Erk or PI3K signaling pathways that promote cell growth and survival (24).
Human-specific primers were used to amplify a 214 base pair genomic sequence that included codons 12 of KRAS (Figure 1a). No PCR product was obtained with murine DNA from mouse normal pancreas (NP-mouse), confirming the specificity of primer set for human KRAS. Three of the five primary tumors and their corresponding PDX models contained mutations in the sequence encoding codon 12 (Figure 1b). CCA2 and CCA3 harbor a G/A (GGT → GAT) transition resulting in substitution of aspartic acid (D) for the normal glycine (G). CCA4 possessed a G/T (GGT → GTT) transversion resulting in substitution of valine (V) for glycine (G). The apparent attenuation of several peaks in the tracings for CCA3 and CCA4 most likely reflects a low ratio of tumor cell:desmoplastic cell components in the samples received from resection. Surgical specimens from which CCA3 and CCA4 models were derived contained <30% tumor cells. Specimens from which CCA1, CCA2 and CCA5 models were derived contained >50% tumor cells. We detected no mutations in codon 12 in CCA1 or CCA5 F0 or F1 specimens. The data show that PDX models retain the KRAS status of the tumors from which they were derived (Table 2).
Figure 1. PDX models of CCA retain the KRAS sequence in codons 12 of the primary tumors from which they were derived.
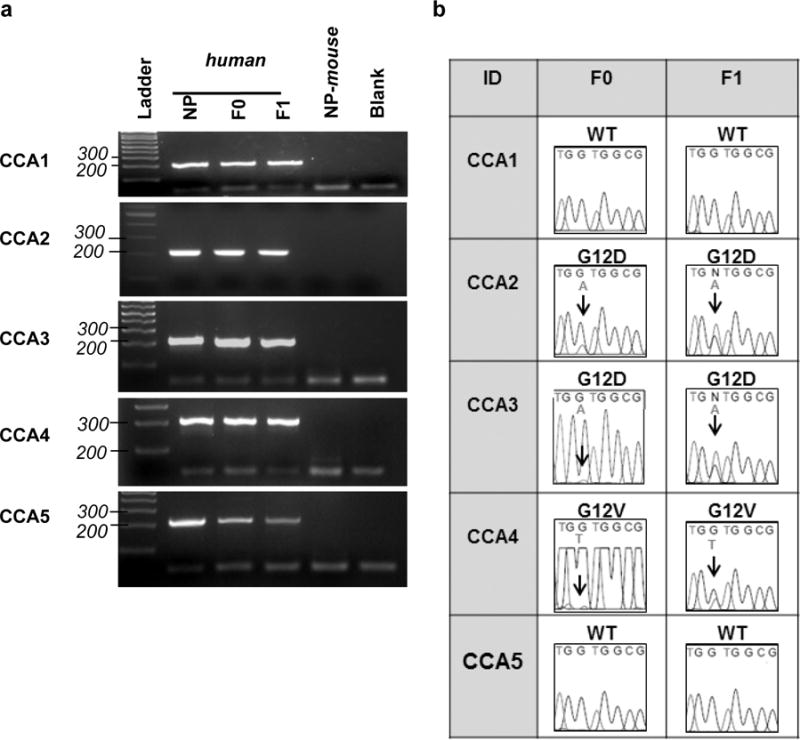
(a) Representative PCR products from genomic DNA, using human specific KRAS primers that amplify a region of the KRAS gene encoding codons 12. NP-mouse = mouse normal pancreas DNA as template. Blank = no DNA control. (b) KRAS codon 12 sequence analyzed by Sanger Sequencing. PCR products were gel extracted, purified, and sequenced by the UAB Heflin Center Genomic Core. Results were visualized using Finch TV (detailed in Methods).
Table 2. PDX models of CCA retain the KRAS sequence in codons 12 and 13 of the primary tumors from which they were derived.
The sequence of codon 12 of the KRAS gene was determined for F0 and F1 tumors.
| Specimen ID | KRAS codon 12 sequence change (F0) | Mutation conserved in PDX (F1) | Amino acid substitution |
|---|---|---|---|
| CCA1 | GGT | Yes | WT |
| CCA2 | GGT → GAT | Yes | G12D |
| CCA3 | GGT → GAT | Yes | G12D |
| CCA4 | GGT → GTT | Yes | G12V |
| CCA5 | GGT | Yes | WT |
Immunohistochemical analysis for expression of genes reported to contribute to progression of CCA tumors: EGFR, HER2 and c-Myc
As stated above, an elevated level of expression of EGFR, HER2 or c-Myc has been reported to support CCA tumor progression or to be associated with poor prognosis (4,5). Immunohistochemical (IHC) analyses demonstrated that EGFR, HER2 and c-Myc were expressed at similar levels in PDX tumors and tumors of origin (Figure S2a). IHC expression data are summarized and expression indices presented below each image and detailed in supplementary data (Figure S2).
CCA1
The F0 primary tumor tissue shows moderate to strong staining for EGFR, while F1 PDX tissue has a moderate staining pattern. EGFR localizes to cell membranes in F0 and F1 tissue. F0 and F1 tumors express minimal to undetectable HER2 and moderate levels of c-Myc.
CCA2
F0 and F1 specimens are characterized by minimal to moderate patchy membranous EGFR. HER2 is undetectable in both F0 and F1 specimens. The F0 and F1 PDX show weak nuclear and moderate cytoplasmic expression for c-Myc protein.
CCA3
F0 and F1 specimens display similar moderate membranous staining for EGFR. F0 and F1 tissues have similar moderate basolateral membranous staining patterns for HER2. Both the F0 and F1 tissues have weak to undetectable nuclear c-Myc, with fewer than 10% of tumor cells showing positive reactivity.
CCA4
F0 and F1 tumor cells show strong basolateral expression of EGFR. HER2 expression is not detectable in either specimen. F0 tissue shows discrete moderate nuclear and cytoplasmic expression of c-Myc, while tumor cells in F1 specimens have minimal to moderate nuclear staining for c-Myc with increased staining for cytoplasmic c-Myc.
CCA5
Both F0 primary and F1 PDX tissues show mild to moderate cytoplasmic staining for EGFR, and also mild membranous reactivity. Both specimens are negative for HER2 expression. The F0 tumor tissue displays weak immunoreactivity for nuclear c-Myc, with concurrent mild cytoplasmic staining. The F1 PDX also shows weak nuclear staining for c-Myc, with no cytoplasmic staining.
IHC data demonstrated no detectable c-Myc or HER2 in normal pancreas, and a moderate level of EGFR expression (Figure S2b). IHC data also indicate that 3/5 PDX models showed moderate levels of c-Myc (CCA1, 2 and 4) and 3/5 models (CCA1, 3 and 4) showed ~1.6- to 1.9-fold higher levels of EGFR than normal pancreas. One model (CCA3) showed a weak to moderate level of HER2. HER2 is expressed at similar levels in F0 and F1 tumors, while levels of expression of EGFR and c-Myc varied by >10% in the PDX compared to the original corresponding tumor. Overall, the data demonstrate that first generation PDX tumors (F1) retained expression patterns of their primary tumors of origin (F0) for c-Myc, EGFR, and HER2, and that each tumor expressed at least one key protein associated with CCA tumor progression.
The bromodomain inhibitor JQ1 inhibited tumor growth selectively in JQ1-sensitive UAB-CCA2 tumors
Published work demonstrates that downregulation of c-Myc in CCA cells inhibits the invasiveness of this type of tumor cell in vitro, that JQ1 downregulates c-Myc expression in some tumor types, and that c-Myc expression may be BET-dependent (13,14,25,26). As CCA1 and CCA2 models had similar growth rates and similar endogenous levels of c-Myc, we evaluated the effect of JQ1 on tumor growth and on c-Myc expression in these two models (Figure 2 and Table 3). If inhibition of c-Myc is an independent indicator of JQ1 efficacy in CCA tumors, we would expect inhibition of c-Myc to occur concomitantly with inhibition of tumor growth.
Figure 2. JQ1 inhibited CCA2 tumor growth in vivo.
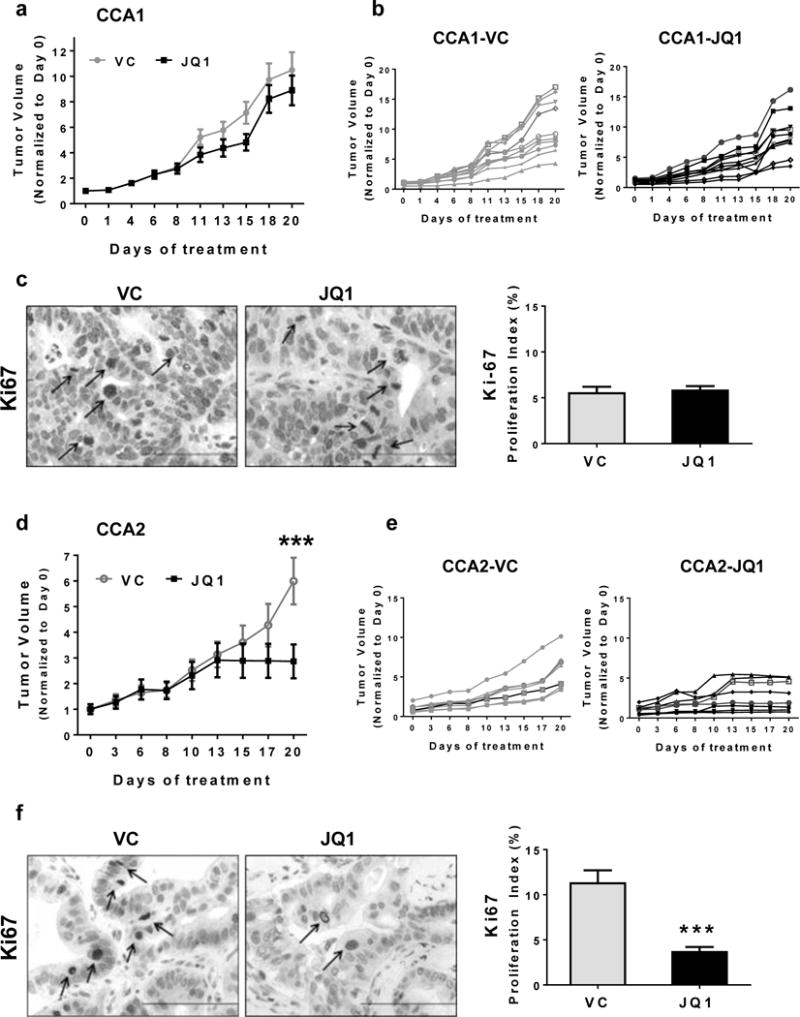
Tumor-bearing mice were treated with JQ1 (50 mg/kg) or vehicle (VC) daily for 20 days. Average tumor volume (a, d) or individual tumor volumes (b, e) for CCA1 or CCA2 tumor volumes (mm3) were normalized to tumor volume on Day 0. Tumor tissue was harvested 24 hours after completion of treatment, and immunohistochemistry (IHC) done to detect the proliferation marker Ki67 (c. f). IHC data were quantitated and results are reported as proliferation indices, calculated as percent of nuclei with grade 1–3 level of detectable Ki67 protein. Scale bar = 10μm.
Table 3. Fold change in expression of genes downregulated by ≥2.4-fold by JQ1 in CCA2 tumors, compared with CCA1 tumors.
Exposure to JQ1 in vivo downregulated expression of 24 genes by ≥2.4-fold in the relatively sensitive CCA2 tumors. In contrast, JQ1 downregulated only 2 genes to this extent (FAS and FGF2) in a subset of 16 of these 24 genes in CCA1 tumors. These data indicate inhibition of expression of FAS and FGF2 are likely not associated with sensitivity to JQ1 and that expression of BRCA2, WEE1, CDK6, CHEK1, CDK4, MMP9, LFT3, PPARG, E2F1, MSH2, TP53, FGFR2, MYCN, and MYC may be BET-dependent in CCA tumors.
| CCA2 | CCA1 | ||||||
|---|---|---|---|---|---|---|---|
| Gene | Accession # | JQ1 | VC | Fold Change | JQ1 | VC | Fold Change |
| FAS | NM_000043.3 | 13.23 | 31.45 | −2.38 | 32.41 | 82.71 | −2.55 |
| BRCA2 | NM_000059.3 | 41.73 | 102.2 | −2.45 | 1.97 | 1.97 | 1 |
| WEE1 | NM_003390.2 | 7.13 | 17.69 | −2.48 | 508.96 | 467.08 | 1.09 |
| CDK6 | NM_001259.5 | 132.33 | 334.13 | −2.53 | 115.55 | 189.77 | −1.64 |
| MYBL2 | NM_002466.2 | 115.02 | 294.82 | −2.56 | Not included | ||
| CHEK1 | NM_001274.2 | 91.61 | 237.82 | −2.6 | 498.43 | 414.43 | 1.20 |
| FGR | NM_001042729.1 | 4.07 | 10.81 | −2.65 | Not included | ||
| CDK4 | NM_000075.2 | 66.16 | 192.61 | −2.91 | 572.19 | 560.1 | 1.02 |
| MYCL1 | NM_001033082.1 | 7.13 | 21.62 | −3.03 | Not included | ||
| CTGF | NM_001901.2 | 492.66 | 1505.53 | −3.06 | Not included | ||
| CDH11 | NM_001797.2 | 224.95 | 691.84 | −3.08 | Not included | ||
| MMP9 | NM_004994.2 | 27.48 | 85.5 | −3.11 | 6.65 | 9.87 | −1.48 |
| FGF2 | NM_002006.4 | 27.48 | 88.45 | −3.22 | 45.29 | 144.14 | −3.18 |
| FLT3 | NM_004119.1 | 3.05 | 9.83 | −3.22 | 1.17 | 1 | 1.17 |
| PPARG | NM_138711.3 | 160.83 | 518.88 | −3.23 | 501.94 | 512.72 | −1.02 |
| E2F1 | NM_005225.1 | 37.66 | 122.84 | −3.26 | 215.07 | 248.57 | −1.16 |
| MSH2 | NM_000251.1 | 63.11 | 213.25 | −3.38 | 704.5 | 579.41 | 1.22 |
| TP53 | NM_000546.2 | 131.31 | 479.57 | −3.65 | 127.25 | 107.28 | 1.19 |
| FGFR2 | NM_000141.4 | 111.97 | 410.78 | −3.67 | 123.74 | 133.61 | −1.08 |
| TEK | NM_000459.2 | 16.29 | 62.89 | −3.86 | Not included | ||
| LCK | NM_001042771.1 | 17.3 | 71.74 | −4.15 | Not included | ||
| MYCN | NM_005378.4 | 25.45 | 130.7 | −5.14 | 65.2 | 59.02 | 2.25 |
| MYC | NM_002467.3 | 238.19 | 1375.81 | −5.78 | 448.08 | 578.53 | −1.29 |
| TIMP3 | NM_000362.4 | 352.19 | 2565.89 | −7.29 | Not included | ||
We administered the well-tolerated regimen of 50 mg/kg JQ1 daily for 20 days and monitored the growth of tumors in mice treated with vehicle alone (VC) or JQ1 (Figures 2a/2d) (14,16). Tumor volumes were normalized to Day 0, with the average tumor volume for each group designated as 1.0. Figures 2b/2e demonstrate individual tumor growth in each group. We also conducted IHC analyses for the proliferation marker Ki-67 in tumor tissue.
The regimen of JQ1 used did not inhibit CCA1 tumor growth (Figures 2a/2b), and Ki-67 immunostaining demonstrated that JQ1 had no effect on expression of this proliferation marker in this model (Figure 2c). In contrast, JQ1 inhibited the growth of CCA2 tumors (Figures 2d/2e), evident as a >50% decrease in tumor volume on the final day of treatment; and the Ki-67 index reflected the JQ1-mediated inhibition of tumor progression as a ~70% reduction in the percentage of tumor cells expressing this proliferation marker (Figure 2f). The time course of quantitated values of tumor volumes and P values comparing these volumes calculated using GraphPad Prism software are shown in Table S1. For comparison, we evaluated the efficacy of gemcitabine 100mg/kg weekly for ~16 weeks in our model (A.L. Miller and K.J. Yoon, unpublished observation). We observed that gemcitabine induced partial tumor regressions within initial 6 weeks of treatment (~ 50% decrease in tumor volume), but tumors regrew in spite of continued gemcitabine treatment. This observation with our preclinical model reflects clinical observations in that while gemcitabine is the most effective agent for CCA and sometimes induces temporary remissions, relapse is virtually inevitable. Addition of other conventional agents such as cisplatin to gemcitabine regimens induces only an incremental increase in duration of survival.
Given that JQ1 inhibits BET proteins and that BET-associated transcriptional complexes regulate expression of a myriad of genes, we assessed the in vivo effect of JQ1 on a panel of PanCancer genes (14,15). By comparing expression profiles of CCA2 tumor tissue from JQ1- and VC-treated mice we identified RNAs downregulated by JQ1, using the NanoString nCounter platform of 230 PanCancer genes.
JQ1 decreased the expression in vivo of twenty-four genes by ≥ 2.4-fold
Heatmapping showed that JQ1 decreased the expression of RNA encoding 24 genes by ≥ 2.4-fold, including RNA encoding MYC family members c-MYC, MYCN and MYCL1 which decreased by 5.78-, 5.14- and 3.03-fold, respectively (Figure 3a, Table 3). Expression of each of these MYC family members has been reported to be BRD4-dependent in some tumor types (14,17,27). A subset of the other 21 genes affected are direct (TP53, MSH2, E2F1, CDK4, CDK6, BRCA2, FAS) or indirect (CHEK1) transcriptional targets of c-Myc (28–32). Whether expression of the remaining 13 genes depends on c-Myc in this tumor type is unknown. The fold change in 16 of the 24 genes downregulated by ≥ 2.4-fold by JQ1 in CCA2 tumors was compared with the fold downregulation of these genes in CCA1 tumors (Table 3). This comparison revealed that cell cycle regulatory proteins Chk1 and E2F1 were selectively downregulated in CCA2 tumors, suggesting that inhibition of Chk1 and E2F1 expression contributes to the cytotoxicity of JQ1. (Gene products downregulated by ≥ 2.4-fold by JQ1in CCA1 tumors are shown in Table S2.)
Figure 3. JQ1 downregulated c-Myc and its downstream targets in a JQ1-sensitive CCA2 model.
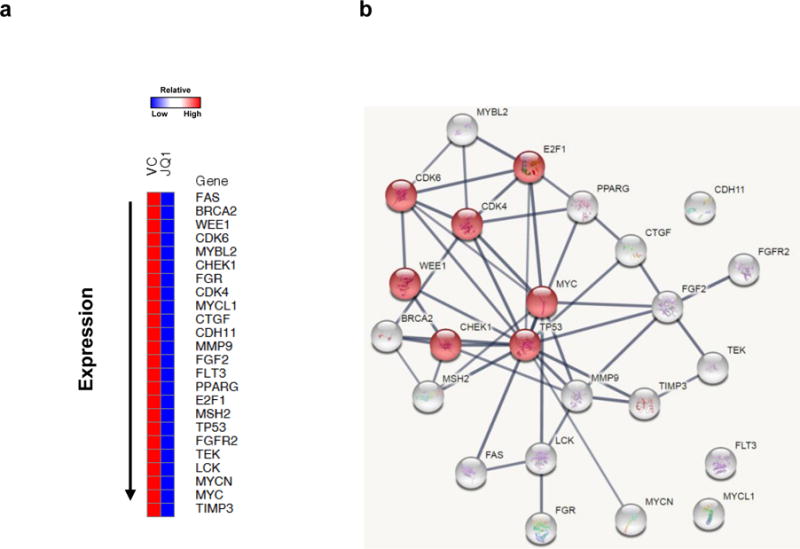
(a) JQ1 inhibited c-Myc and cell cycle related gene products in CCA2 tumors. Genes downregulated ≥ 2.4-fold by NanoString analyses are displayed by heatmap. (b) Twenty-four genes downregulated ≥ 2.4-fold were entered into the STRING database and a protein-protein interaction network generated. The red nodes indicate proteins involved in cell cycle regulation.
STRING v10 analysis (Search Tool for the Retrieval of Interacting Genes/Proteins) database (www.string-db.org) identified pathways likely to be impacted by JQ1-induced downregulation of these 24 gene products. Results of this analysis are presented graphically in Figure 3b, with individual genes represented as nodes. Lines connecting various nodes are based on confidence scores, with darker lines denoting higher confidence scores. Confidence scores represent probabilities (performance predictions) that connected proteins are functionally linked (KEGG mapping) (33). Seven of the 24 downregulated genes contribute to cell cycle regulation (MYC, TP53, CHEK1, WEE1, CDK4, CDK6, E2F1 [red nodes]) (false discovery rate; FDR = 9.71×10−9), five are involved in the p53 signaling pathway (CDK4, CDK6, CHEK1, FAS, TP53) (FDR = 4.37×10−7), seven are involved in PI3K-AKT signaling (CDK4, CDK6, FGF2, FGFR2, MYC, TEK, TP53) (FDR=1.82×10−6) (Table S3), and one contributes to DNA repair (BRCA2).
We then reasoned that if inhibition of c-Myc expression by JQ1 comprised a primary mechanism of action of JQ1 or an independent indicator of JQ1 efficacy, JQ1-sensitive CCA2 tumors exposed to this BET inhibitor would have lower levels of c-Myc than VC-treated tumors, and JQ1 would not affect c-Myc expression in CCA1 tumors which were unaffected by JQ1 (14–16).
JQ1 altered tumor histology and downregulated expression of c-Myc protein and multiple c-Myc transcriptional targets selectively in the JQ1-sensitive CCA2 PDX model
Histopathological analysis (H&E) demonstrated no gross differences in differentiation status or cytological features (N:C ratio, nuclear polarity, or nuclear pleomorphism) between VC- and JQ1-treated CCA1 tumors (Figure 4). CCA2 tumors exposed to JQ1 displayed a decrease in N:C ratio, an increase in peritumoral stroma, and a decrease in overall tumor cellularity, reflecting the decreased tumor cell viability of this JQ1-sensitive model.
Figure 4. H&E stained sections of tumors exposed in vivo to vehicle (VC) or JQ1 demonstrate JQ1-induced changes in histology.
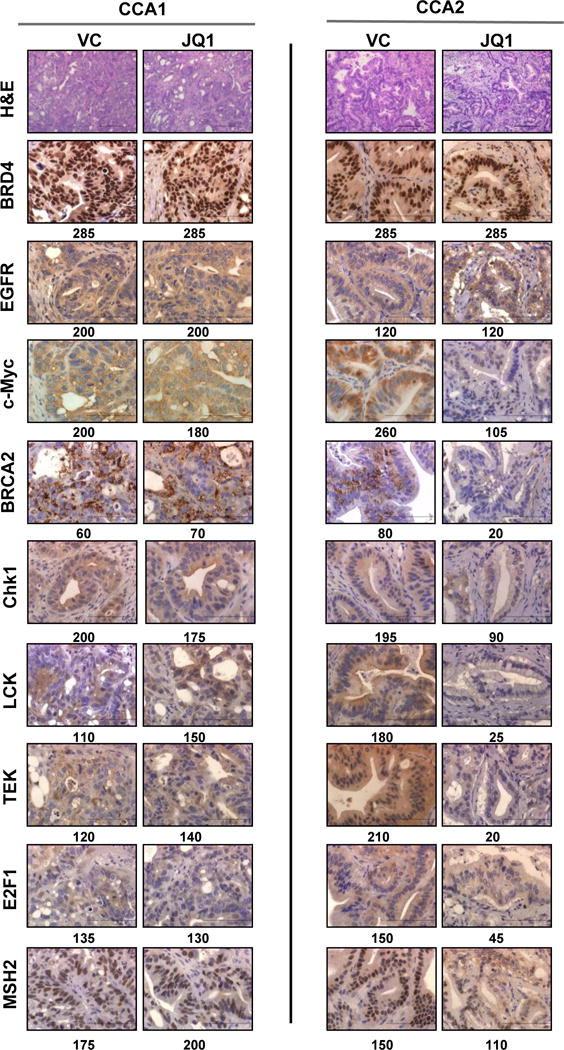
JQ1 downregulated c-Myc, BRCA2, Chk1, LCK, TEK, E2F1, and MSH2 protein expression in CCA2 tumors, but not in CCA1 tumors. JQ1 did not inhibit BRD4 or EGFR expression in either tumor model. Total expression index values are shown below each image. Scale bar = 20μm (H&E) or 10μM (all others). Expression indices for BRD4, EGFR, c-Myc, BRCA2, Chk1, LCK, TEK, E2F1, and MSH2 were calculated as described in Materials and Methods and are shown in Table S4.
JQ1 did not affect c-Myc protein level in CCA1 tumors: EI of 200 in control-treated tumors versus 180 in JQ1-treated tumors)(Figure 4, Table S4). In contrast, JQ1-sensitive CCA2 tumors expressed ~2.5-fold less c-Myc protein than tumors from VC-treated mice (EI of 260 vs 105) at completion of therapy. If we consider cytoplasmic c-Myc to represent nascent protein, then CCA2 tumors exposed to JQ1 expressed ~5-fold less c-Myc (EI of 170 vs 30) than VC-treated tumors (Figure 4, Table S4). These data indicate that JQ1 concomitantly suppressed tumor growth and c-Myc expression selectively in CCA2 PDX tumors, in agreement with Nanostring analyses showing that tumors harvested from JQ1-treated mice expressed ~5.7-fold less c-Myc RNA than tumors from control mice. Consistent with the established mechanism of action of JQ1, JQ1 did not alter BRD4 expression as assessed by IHC analysis (Figure 4). Additional IHC images and expression indices for BRD4 expression in CCA3-CCA5 are shown in Figure S3.
As an indirect indication of c-Myc function, we determined whether specific transcriptional targets of c-Myc were also downregulated. IHC data showed that JQ1 selectively inhibited expression in CCA2 tumors of Chk1 by ~2-fold and BRCA2 by ~4-fold (Figure 4, Table S4), each of which has been reported to be regulated by c-Myc (28,29,34). IHC analyses also demonstrated downregulation of four additional proteins that were identified by NanoString to be downregulated in CCA2 (Figure 4, Table S4): JQ1 inhibited expression of LCK, TEK, E2F1 and MSH2 proteins by 1.4- to ~10-fold. In contrast, JQ1 did not affect BRD4 and EGFR expression in CCA1 or CCA2 tumors. We performed immunoblots to corroborate IHC results. These analyses were consistent with IHC data and showed that JQ1 inhibited expression of TEK, LCK, Chk1, and MSH2 in CCA2 tumors (Figure S4). As mentioned above, exposure to JQ1 in vivo downregulated expression of 24 genes by ≥ 2.4-fold in sensitive CCA2 tumors, but downregulated only 2 genes to this extent (FAS and FGF2) in the subset of 16 of these genes in CCA1 tumors (Table 3). The data suggest that inhibition of expression of FAS and FGF2 are unlikely to be associated with sensitivity to JQ1 and that expression of CHEK1, BRCA2, WEE1, CDK6, CDK4, MMP9, FLT3, PPARG, E2F1, MSH2, TP53, FGFR2, MYCN, and MYC may be BET-dependent in CCA tumors. We propose that proteins selectively downregulated in the more drug-sensitive CCA2 model may contribute to the cytotoxicity of JQ1.
JQ1 induced DNA damage and apoptosis in CCA2 tumors
Based on observations in the literature that BET proteins are essential to the DNA damage response, we hypothesized that JQ1 would induce DNA damage and further apoptosis (35). To address this hypothesis, we performed IHC staining for the DNA double strand break marker phospho-H2AX (Ser-139-ɣH2AX) and the apoptosis indicators cleaved caspase 3 and cleaved PARP (Figure 5) (36). IHC results indicated increased levels of ɣH2AX, cleaved caspase 3 and cleaved PARP in JQ1-sensitive CCA2 tumors and no change in levels of these proteins in CCA1 tumors. JQ1-induced DNA damage in vivo has not been reported previously. Quantitation of IHC results demonstrated that JQ1-treated CCA2 tumor specimens had a greater percentage of ɣH2AX positive cells and higher apoptotic indices than VC-treated CCA1 specimens (P<0.0001) (Figures 5a/5b/5c).
Figure 5. JQ1 induced DNA damage and apoptosis in CCA2 PDX tumors in vivo.
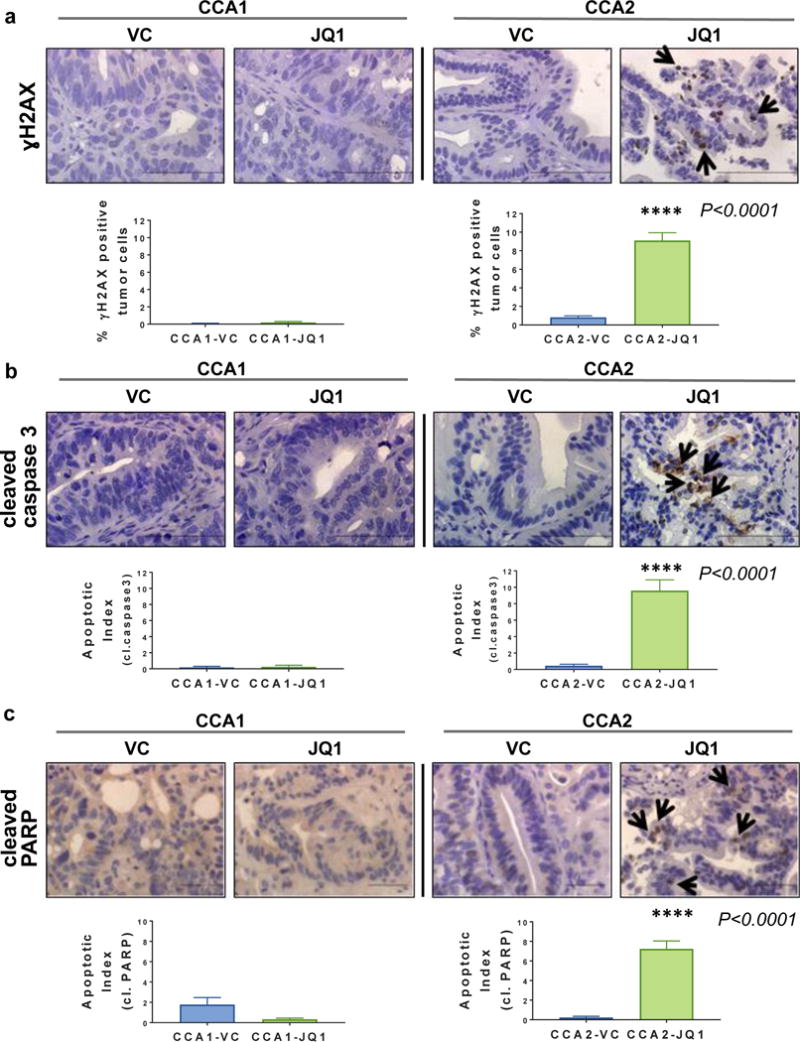
(a) Tissue sections were immunostained to detect ɣH2AX as an indicator of DNA damage or (b) cleaved caspase 3 or (c) cleaved PARP as an indicator of apoptosis, each of which is involved in DNA damage response. VC = vehicle control. Scale bar = 10μm. Quantitation of γ-H2AX immunostaining comparing data from CCA1 vs CCA2 tumors is shown in graphs below images. Apoptotic Indices were calculated by counting the number of apoptotic cells per total number of tumor cells (50) and shown as graphs. ****P<0.0001
In summary, the data show that selective to CCA2 tumors, JQ1 inhibited tumor growth, induced DNA damage and apoptosis, and inhibited expression of c-Myc and c-Myc transcriptional targets associated with cell cycle regulation and DNA repair. The data suggest that the inhibition of expression c-Myc and selected downstream transcriptional targets may contribute to the mechanism of action of JQ1 in JQ1-sensitive CCA tumors.
DISCUSSION
Cholangiocarcinoma (CCA) is a uniformly fatal disease (1). To evaluate potential novel therapies for CCA, we developed and characterized five independently derived PDX models from primary EHCC and metastatic IHCC tumor specimens (CCA1-5), the first such models reported. The models retained specific genetic, histologic, and molecular characteristics of tumors of origin. Using two of these models to evaluate the efficacy of the BET bromodomain inhibitor JQ1, the data showed that inhibition of tumor growth was associated with inhibition of c-Myc protein expression, as has been reported for some other types of solid tumors (14,25,37,38). The data also show the novel observations that the BET inhibitor JQ1 concomitantly and selectively suppressed tumor growth, induced DNA damage, and inhibited Chk1, BRCA2, LCK, TEK, E2F1 and MSH2 protein expression in the JQ1-sensitive in vivo model.
An emerging literature documents recent interest in the use of BET bromodomain inhibitors for treating hematologic and solid malignancies (39). BET inhibitors bind directly to proteins that harbor BET bromodomains (14). A consequence of this binding is the inhibition of association of BET-dependent transcription complexes to a profile of genetic loci and inhibition of expression of gene products dependent on this transcription mechanism (15). As c-Myc contributes to the phenotype of many tumor types and its transcription is thought to be BET protein-dependent, early studies postulated that the primary mechanism of action of JQ1 was indirect inhibition of c-Myc expression (14–16). Interestingly, recent data indicate that gene products dependent on BET-regulated transcription may be cell type specific (18,40). In multiple myeloma, B-cell acute lymphoblastic leukemia, and medulloblastoma cells, JQ1 inhibited expression of c-Myc in vitro and in vivo (16,25,41,42). A recent study from our lab demonstrated that JQ1 did not inhibit c-Myc expression in pancreatic ductal adenocarcinoma PDX models in vivo (18). The current study demonstrates that JQ1 inhibits c-Myc selectively in a drug-sensitive CCA tumor model.
c-Myc is expressed in up to 94% of CCA tumors (12). Experimental downregulation of c-Myc decreases the invasiveness of QBC939 bile duct carcinoma cells in vitro and inhibits CCA tumor progression in a mouse model of chronic cholestasis (13,43). Conversely, upregulation of c-Myc supported cholestatic cholangiocarcinogenesis in vivo. c-Myc is estimated to regulate ~15% of human gene products, with diverse functions that include cell cycle checkpoint and DNA damage response proteins (Chk1), cyclin dependent kinases (CDK4/6), and DNA repair proteins (BRCA2) (29–31). Consistent with the proposed mechanism of action of BET inhibitors, JQ1 inhibited expression of c-Myc and multiple c-Myc transcriptional targets, but also inhibited gene products whose expression is c-Myc-independent such as FOSL1 (40). Our findings that JQ1 induces DNA damage and apoptosis in vivo is consistent with a recent in vitro study demonstrating that JQ1 induced apoptosis and delayed the DNA damage response in leukemic cells by interfering with the association of the BET protein BRD4 and p53 (44).
While the observed difference in sensitivity of independently derived CCA1 and CCA2 tumors to JQ1 is undoubtedly multifactorial, several observed differences between the two models are consistent with published reports linking a given molecular characteristic with JQ1 sensitivity. Since c-Myc regulates expression of many genes, multiple c-Myc targets could contribute to the efficacy of JQ1, and the expression profile comparison between treated and untreated CCA1 and CCA2 tumors (Tables 3/S2/S3) represents a first step toward identifying such proteins. Our hypothesis is that the efficacy of JQ1 involves simultaneous downregulation of expression of multiple transcriptional targets of c-Myc. More specifically, we postulate that simultaneous downregulation of the c-Myc transcriptional targets Chk1 and E2F1 is essential for JQ1-induced growth inhibition of CCA2 tumors. This postulate is based on: 1) our observation that JQ1 inhibited these two transcripts in CCA2 but not CCA1 tumors, 2) data in the literature demonstrating that each of these proteins regulate cell cycle transition from G2 to M or G1 to S; and 3) that inhibition of expression or dysfunction of these proteins decreases tumor cell proliferation (45–48). Further, CCA1 tumors express wild type KRAS; CCA2 tumors harbor a G12D KRAS mutation. Notably, Shimamura et al. suggested that non-small cell lung cancer cells harboring KRAS mutations were more sensitive to JQ1 than cells expressing the wild type protein (49). Work to directly address the effect of KRAS status on the anti-tumor efficacy of JQ1 is ongoing.
In summary, we established five novel PDX models of CCA that retain specific genetic and histologic characteristics of primary tumors of origin. JQ1 selectively inhibited the growth of CCA2 compared to CCA1 tumors and concomitantly inhibited expression of c-Myc and several of its known transcriptional targets including Chk1, BRCA2, E2F1 and MSH2 in CCA2 tumors. JQ1 also induced DNA damage and apoptosis in vivo. Induction of DNA damage in vivo by JQ1 has not been demonstrated previously. The observed induction of DNA damage suggests that JQ1 may enhance the efficacy of agents that are selectively toxic to tumor cells deficient in DNA repair. We conclude that BET inhibitors such as JQ1 warrant further investigation for the treatment of CCA.
Supplementary Material
Acknowledgments
The authors are indebted to the cholangiocarcinoma patients who agreed to participate in this study. This project was supported by NIH grant R21 CA205501, American Cancer Society-Institutional Research Grant (IRG-60-001-50), and UAB/UMN SPORE in pancreatic cancer (P50 CA101955).
Financial Support: This project was supported by NIH grant R21 CA205501 (KJY), American Cancer Society-Institutional Research Grant (IRG-60-001-50) (KJY), and UAB/UMN SPORE in pancreatic cancer pilot grant (P50 CA101955) (KJY).
References
- 1.Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. 2014;383:2168–79. doi: 10.1016/S0140-6736(13)61903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chong DQ, Zhu AX. The landscape of targeted therapies for cholangiocarcinoma: current status and emerging targets. Oncotarget. 2016;7:46750–67. doi: 10.18632/oncotarget.8775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. The New England journal of medicine. 2010;362:1273–81. doi: 10.1056/NEJMoa0908721. [DOI] [PubMed] [Google Scholar]
- 4.Tokumoto N, Ikeda S, Ishizaki Y, Kurihara T, Ozaki S, Iseki M, et al. Immunohistochemical and mutational analyses of Wnt signaling components and target genes in intrahepatic cholangiocarcinomas. International journal of oncology. 2005;27:973–80. [PubMed] [Google Scholar]
- 5.Yoshikawa D, Ojima H, Iwasaki M, Hiraoka N, Kosuge T, Kasai S, et al. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. British journal of cancer. 2008;98:418–25. doi: 10.1038/sj.bjc.6604129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Dell MR, Huang JL, Whitney-Miller CL, Deshpande V, Rothberg P, Grose V, et al. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 2012;72:1557–67. doi: 10.1158/0008-5472.CAN-11-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gwak GY, Yoon JH, Shin CM, Ahn YJ, Chung JK, Kim YA, et al. Detection of response-predicting mutations in the kinase domain of the epidermal growth factor receptor gene in cholangiocarcinomas. Journal of cancer research and clinical oncology. 2005;131:649–52. doi: 10.1007/s00432-005-0016-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harder J, Waiz O, Otto F, Geissler M, Olschewski M, Weinhold B, et al. EGFR and HER2 expression in advanced biliary tract cancer. World journal of gastroenterology: WJG. 2009;15:4511–7. doi: 10.3748/wjg.15.4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leone F, Cavalloni G, Pignochino Y, Sarotto I, Ferraris R, Piacibello W, et al. Somatic mutations of epidermal growth factor receptor in bile duct and gallbladder carcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:1680–5. doi: 10.1158/1078-0432.CCR-05-1692. [DOI] [PubMed] [Google Scholar]
- 10.Yang X, Wang W, Wang C, Wang L, Yang M, Qi M, et al. Characterization of EGFR family gene aberrations in cholangiocarcinoma. Oncology reports. 2014;32:700–8. doi: 10.3892/or.2014.3261. [DOI] [PubMed] [Google Scholar]
- 11.Nasi S, Ciarapica R, Jucker R, Rosati J, Soucek L. Making decisions through Myc. FEBS letters. 2001;490:153–62. doi: 10.1016/s0014-5793(01)02118-4. [DOI] [PubMed] [Google Scholar]
- 12.Voravud N, Foster CS, Gilbertson JA, Sikora K, Waxman J. Oncogene expression in cholangiocarcinoma and in normal hepatic development. Human pathology. 1989;20:1163–8. doi: 10.1016/s0046-8177(89)80006-1. [DOI] [PubMed] [Google Scholar]
- 13.Li ZR, Wu YF, Ma CY, Nie SD, Mao XH, Shi YZ. Down-regulation of c-Myc expression inhibits the invasion of bile duct carcinoma cells. Cell biology international. 2011;35:799–802. doi: 10.1042/CBI20110099. [DOI] [PubMed] [Google Scholar]
- 14.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–34. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–17. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer discovery. 2013;3:308–23. doi: 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia PL, Miller AL, Kreitzburg KM, Council LN, Gamblin TL, Christein JD, et al. The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene. 2015 doi: 10.1038/onc.2015.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia PL, Council LN, Christein JD, Arnoletti JP, Heslin MJ, Gamblin TL, et al. Development and histopathological characterization of tumorgraft models of pancreatic ductal adenocarcinoma. PloS one. 2013;8:e78183. doi: 10.1371/journal.pone.0078183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Briasoulis E, Tsokos M, Fountzilas G, Bafaloukos D, Kosmidis P, Samantas E, et al. Bcl2 and p53 protein expression in metastatic carcinoma of unknown primary origin: biological and clinical implications. A Hellenic Co-operative Oncology Group study. Anticancer research. 1998;18:1907–14. [PubMed] [Google Scholar]
- 21.DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nature medicine. 2011;17:1514–20. doi: 10.1038/nm.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moro M, Bertolini G, Tortoreto M, Pastorino U, Sozzi G, Roz L. Patient-derived xenografts of non small cell lung cancer: resurgence of an old model for investigation of modern concepts of tailored therapy and cancer stem cells. Journal of biomedicine & biotechnology. 2012;2012:568567. doi: 10.1155/2012/568567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arrington AK, Nelson RA, Falor A, Luu C, Wiatrek RL, Fakih M, et al. Impact of medical and surgical intervention on survival in patients with cholangiocarcinoma. World journal of gastrointestinal surgery. 2013;5:178–86. doi: 10.4240/wjgs.v5.i6.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends in biochemical sciences. 2014;39:91–100. doi: 10.1016/j.tibs.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:912–25. doi: 10.1158/1078-0432.CCR-13-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16669–74. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki K, Yamamoto K, Arakawa Y, Yamada H, Aiba K, Kitagawa M. Antimyeloma activity of bromodomain inhibitors on the human myeloma cell line U266 by downregulation of MYCL. Anti-cancer drugs. 2016 doi: 10.1097/CAD.0000000000000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoglund A, Nilsson LM, Muralidharan SV, Hasvold LA, Merta P, Rudelius M, et al. Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:7067–79. doi: 10.1158/1078-0432.CCR-11-1198. [DOI] [PubMed] [Google Scholar]
- 29.Luoto KR, Meng AX, Wasylishen AR, Zhao H, Coackley CL, Penn LZ, et al. Tumor cell kill by c-MYC depletion: role of MYC-regulated genes that control DNA double-strand break repair. Cancer Res. 2010;70:8748–59. doi: 10.1158/0008-5472.CAN-10-0944. [DOI] [PubMed] [Google Scholar]
- 30.Menssen A, Hermeking H. Characterization of the c-MYC-regulated transcriptome by SAGE: identification and analysis of c-MYC target genes. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:6274–9. doi: 10.1073/pnas.082005599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffman B, Liebermann DA. Apoptotic signaling by c-MYC. Oncogene. 2008;27:6462–72. doi: 10.1038/onc.2008.312. [DOI] [PubMed] [Google Scholar]
- 32.He C, Jiang H, Geng S, Sheng H, Shen X, Zhang X, et al. Expression and prognostic value of c-Myc and Fas (CD95/APO1) in patients with pancreatic cancer. International journal of clinical and experimental pathology. 2014;7:742–50. [PMC free article] [PubMed] [Google Scholar]
- 33.von Mering C, Jensen LJ, Snel B, Hooper SD, Krupp M, Foglierini M, et al. STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic acids research. 2005;33:D433–7. doi: 10.1093/nar/gki005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, et al. Genomic targets of the human c-Myc protein. Genes & development. 2003;17:1115–29. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Floyd SR, Pacold ME, Huang Q, Clarke SM, Lam FC, Cannell IG, et al. The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature. 2013;498:246–50. doi: 10.1038/nature12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Molecular cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 37.Feng Q, Zhang Z, Shea MJ, Creighton CJ, Coarfa C, Hilsenbeck SG, et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell research. 2014;24:809–19. doi: 10.1038/cr.2014.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wyce A, Degenhardt Y, Bai Y, Le B, Korenchuk S, Crouthame MC, et al. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget. 2013;4:2419–29. doi: 10.18632/oncotarget.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nature reviews Drug discovery. 2014;13:337–56. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- 40.Lockwood WW, Zejnullahu K, Bradner JE, Varmus H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:19408–13. doi: 10.1073/pnas.1216363109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Venkataraman S, Alimova I, Balakrishnan I, Harris P, Birks DK, Griesinger A, et al. Inhibition of BRD4 attenuates tumor cell self-renewal and suppresses stem cell signaling in MYC driven medulloblastoma. Oncotarget. 2014;5:2355–71. doi: 10.18632/oncotarget.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ott CJ, Kopp N, Bird L, Paranal RM, Qi J, Bowman T, et al. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood. 2012;120:2843–52. doi: 10.1182/blood-2012-02-413021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang H, Li TW, Peng J, Tang X, Ko KS, Xia M, et al. A mouse model of cholestasis-associated cholangiocarcinoma and transcription factors involved in progression. Gastroenterology. 2011;141:378–88. 88 e1–4. doi: 10.1053/j.gastro.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stewart HJ, Horne GA, Bastow S, Chevassut TJ. BRD4 associates with p53 in DNMT3A-mutated leukemia cells and is implicated in apoptosis by the bromodomain inhibitor JQ1. Cancer medicine. 2013;2:826–35. doi: 10.1002/cam4.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bryant C, Rawlinson R, Massey AJ. Chk1 inhibition as a novel therapeutic strategy for treating triple-negative breast and ovarian cancers. BMC cancer. 2014;14:570. doi: 10.1186/1471-2407-14-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ravi D, Beheshti A, Abermil N, Passero F, Sharma J, Coyle M, et al. Proteasomal Inhibition by Ixazomib Induces CHK1 and MYC-Dependent Cell Death in T-cell and Hodgkin Lymphoma. Cancer Res. 2016;76:3319–31. doi: 10.1158/0008-5472.CAN-15-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shats I, Deng M, Davidovich A, Zhang C, Kwon JS, Manandhar D, et al. Expression level is a key determinant of E2F1-mediated cell fate. Cell death and differentiation. 2017 doi: 10.1038/cdd.2017.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shats I, Gatza ML, Liu B, Angus SP, You L, Nevins JR. FOXO transcription factors control E2F1 transcriptional specificity and apoptotic function. Cancer Res. 2013;73:6056–67. doi: 10.1158/0008-5472.CAN-13-0453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shimamura T, Chen Z, Soucheray M, Carretero J, Kikuchi E, Tchaicha JH, et al. Efficacy of BET bromodomain inhibition in Kras-mutant non-small cell lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:6183–92. doi: 10.1158/1078-0432.CCR-12-3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bressenot A, Marchal S, Bezdetnaya L, Garrier J, Guillemin F, Plenat F. Assessment of apoptosis by immunohistochemistry to active caspase-3, active caspase-7, or cleaved PARP in monolayer cells and spheroid and subcutaneous xenografts of human carcinoma. The journal of histochemistry and cytochemistry: official journal of the Histochemistry Society. 2009;57:289–300. doi: 10.1369/jhc.2008.952044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.