

Abstract
Every second year, the community experiment “Critical Assessment of Techniques for Structure Prediction” (CASP) is conducting an independent blind assessment of structure prediction methods, providing a framework for comparing the performance of different approaches and discussing the latest developments in the field. Yet, developers of automated computational modeling methods clearly benefit from more frequent evaluations based on larger sets of data. The “Continuous Automated Model EvaluatiOn (CAMEO)” platform complements the CASP experiment by conducting fully automated blind prediction assessments based on the weekly pre-release of sequences of those structures, which are going to be published in the next release of the PDB Protein Data Bank. CAMEO publishes weekly benchmarking results based on models collected during a four-day prediction window, on average assessing ca. 100 targets during a time frame of five weeks. CAMEO benchmarking data is generated consistently for all participating methods at the same point in time, enabling developers to benchmark and cross-validate their method’s performance, and directly refer to the benchmarking results in publications. In order to facilitate server development and promote shorter release cycles, CAMEO sends weekly email with submission statistics and low performance warnings. Many participants of CASP have successfully employed CAMEO when preparing their methods for upcoming community experiments. CAMEO offers a variety of scores to allow benchmarking diverse aspects of structure prediction methods. By introducing new scoring schemes, CAMEO facilitates new development in areas of active research, e.g. modeling quaternary structure, complexes or ligand binding sites.
Keywords: CAMEO, CASP, protein structure modeling, protein structure prediction, model quality assessment, continuous evaluation, model confidence, benchmarking, oligomeric assessment, ligand binding site accuracy
2. Introduction
Research projects in the biomedical sciences often leverage insights from protein structure predictions when direct experimental structure information is not available. Recent examples range from data driven protein design[1] to combining genetic diversity studies with comparative structural analyses[2] to elucidating the structure and function of the nucleopore complex protein Nup82 in a hybrid modeling approach[3] and contributions to the structure elucidation in the Zika-dengue virus antibody cross-neutralization[4]. Routine application of models in research projects requires fully automated, robust, reliable, and accurate modeling pipelines[5], and for “real-world modeling” cases, the usability of a structure prediction method depends on many factors. However, the prediction performance of modeling tools reported in the literature is often based on different background information, diverse target data sets, and distinct evaluation metrics, making quantitative comparisons between methods impossible. This well-known problem is successfully addressed by regular independent blind assessments in the form of the community experiment “Critical Assessment of Techniques in Structure Prediction” (CASP)[6–8]. CASP is organized every two years, assessing methods based on approximately 100 prediction targets, and culminates in a meeting, where researchers compare the performance of the various approaches and discuss latest developments. Yet, developers of automated server methods clearly benefit from more frequent benchmarking on larger data sets in between CASP seasons, as offered by the “Continuous Automated Model EvaluatiOn (CAMEO)” platform[9].
CAMEO was inspired by previous attempts to establish automated evaluations[10, 11] with the conceptual difference, that CAMEO conducts fully automated prediction assessments based on the pre-release of sequences[12] of those structures which are going to be published by the PDB Protein Data Bank[13] the following Wednesday, i.e. the evaluation is based on blind predictions of unpublished 3D structures. By selecting about 20 prediction targets per week, a significant volume of ~100 benchmarking targets is reached within five weeks. The short evaluation cycles have been relied on by many algorithm and score developers when preparing for the CASP experiment. CAMEO benchmarking is performed across all participating servers at the same point in time, i.e. all methods have access to the same background information such as template information in PDB or protein sequences in UniProt[14]. Benchmarking results (models, reference structures, scores) are publicly available to document a method’s historic performance (e.g. in publications) and can be used as training data for further methods development.
CAMEO significantly facilitates the development of modern protein structure prediction approaches: methods are transparently assessed by a variety of scores established by the community, each representing different aspects of structure prediction. Overall model accuracy is measured e.g. by lDDT[15], CADscore[16], TM-score[17], GDT-HA[18, 19], MaxSub score[20]. Notably, measures which require superposition of a model onto the target structure are prone to fail for multi-domain proteins or cases of large domain rearrangements. In CASP this problem is addressed by splitting the target structures into actual assessment units (AUs) [21] by human intervention with the support of computational approaches[22, 23]. However, such manual effort is not feasible for unsupervised assessment on a weekly basis within CAMEO. Superposition-free measures, which are robust with respect to domain movements, are therefore the methods of choice for fully automated mode of operation[15, 16, 24]. Besides overall structure similarity, other aspects evaluated are accuracy of predicted ligand binding sites based on lDDT, oligomeric state accuracy based on quaternary state scores (QS-score)[25], or reliability of local model confidence estimates (“model B-factor”).
CAMEO offers web-based views to assess both overall server performance using aggregated scores and as well as results on individual targets for in-depth inspection. While results for servers registered as “public servers” are visible for anyone, new methods can be registered as “development servers”. Results for development servers are only visible to other developers in anonymized form, allowing to informally benchmark a new algorithm with other state-of-the-art methods. The status of a method can be changed from “development server” to “public” when the testing phase is successfully concluded, thereby making the benchmarking results publicly visible.
CAMEO sends out weekly summary email to server developers, listing the predicted as well as the missed targets. A “performance alert” warns developers about individual predictions scoring significantly lower than those of other methods, often facilitating the identification of specific problems and limitations of new methods.
CAMEO is an open platform inviting the community to participate by suggesting alternative scoring approaches, evaluation schemes and new categories. CAMEO currently supports the categories “3D Structure Prediction” (3D) and “Model Quality Estimation” (QE). A new category for evaluating residue -residue contact predictions is currently being established, and the 3D Structure Prediction category is being extended to allow for assessment of heteromeric complexes and ligand conformations.
3. Materials and Methods
Selection of Prediction Targets
The weekly target set for CAMEO is compiled from each PDB pre-release by clustering all sequences of entries in the PDB with a 99% sequence identity threshold employing cd-hit[26]. Protein sequences with less than 30 amino acid residues are excluded. For each sequence we run BLAST[16] against a database of PDB entries and exclude those sequences exhibiting more than 85% sequence identity and at least 70% coverage to any known experimental structure. The remaining entries define the total available number of targets respecting the original PDB pre-release order apart from those entries now missing. The first 20 targets are then selected to comprise the weekly target set effectively limiting the computational load imposed by CAMEO.
Baseline Predictions
In order to being able to monitor algorithmic improvements of modeling methods over time, CAMEO uses baseline servers as null models, where the computational pipelines are kept constant while the underlying databases are updated weekly. The “NaiveBlast” (CAMEO 3D) baseline uses BLAST for searching the sequences of released PDB entries for templates. For each target, it selects the first BLAST hit (if any) as template to build a model using MODELLER (v9.2) applying default parameters[27]. The model is then trimmed to match the residues covered by the template. The baseline server “naivePSIBLAST” (CAMEO QE) assumes that conserved regions of a protein model are of higher quality than divergent regions. It searches the most recent version of the NCBI NR database with PSI-BLAST[28] using three iterations with the target sequence as query applying an e-value threshold of 1e-10. The sequence conservation estimate Ci for a given residue i is derived from the position specific information content in the PSSM (Position Specific Scoring Matrix) according to formula (1).
| (1) |
with Ii representing the individual information content for a given residue i. The individual estimates Ci are then set as model confidence estimates for all atoms in the respective residues.
The “BaselinePotential” server (CAMEO QE) implements a classical distance based statistical potential as described by Sippl and coworkers[29]. Statistics have been extracted for pairwise distances between all chemically distinguishable heavy atoms in the 20 naturally occurring amino acids. Histograms have been built with a bin size of 0.5Å and maximal distance of 10Å, neglecting all interactions from residues being closer than four in sequence. The underlying data is composed of a non-redundant set of experimentally determined protein structures (2995 culled chains from the PISCES webserver[30] with max pairwise sequence identity of 20% and X-Ray resolution better than 1.6Å). The resulting potential functions are applied on all pairwise interactions and per residue scores are estimated by averaging all outcomes of interactions a residue is involved in. A subsequent sequential smoothing applies a Gaussian filter with a standard deviation of four residues to reduce noise. To avoid amino acid specific biases, a linear model is trained for all 20 naturally occurring amino acids to predict per-residue lDDT scores.
Local installation of model quality assessment tools
In general, predictions are submitted to CAMEO by servers maintained by the respective methods developers. The following computational tools were available for download and have been installed and used locally on the CAMEO server: Dfire (Version 1.1)[31], Prosa 2003[29], ProQ2[32] and Verify3d[33].
Numerical Scores for Structure Assessment
CAMEO applies a variety of numerical scores for assessing different aspects of modeling. The local distance difference test (lDDT) is an all-atom superposition-independent score based on comparing interaction distances observed in the prediction to the corresponding ones in the reference based on a 15 Å cutoff. If the difference is within a set threshold the interaction is counted as preserved. The final value is the average fraction of preserved interactions at four interaction thresholds at 0.5Å, 1Å, 2Å and 4Å. lDDT assigns low scores to residues with large stereochemical deviations and physically impossible close contacts. The lDDT-BS score (Local Distance Difference Test - Binding Site, Fig. 1) is the average of the individual lDDT local scores (applying an inclusion radius of 10Å) of those residues which form a binding site on the respective target. This analysis is limited on experimental structure including a biologically relevant ligand. Here, a binding site is defined as the set of amino acid residues in the reference protein structure which have at least one atom within a 4.0 Å radius of any atom of the ligand, for ions a radius of 3.0 Å is applied. When calculating the lDDT-BS score, all chains in the model are considered and the best scoring combination is retained. In case a target protein entry consists of several oligomeric assemblies, the best score across all assemblies and their individual chain combinations is reported. When a binding site is located at the interface of an oligomeric structure and the prediction does not match the oligomeric state of the target, the lDDT-BS is not calculated and the prediction is treated as non-existent. Fig. 1 was created with OpenStructure[34].
FIGURE 1.
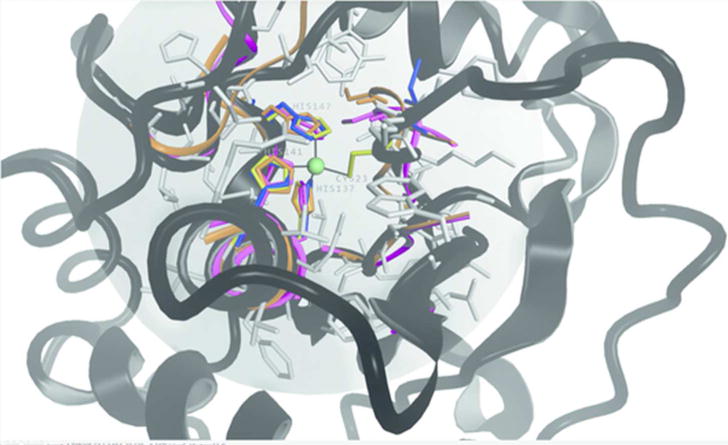
Illustration of the lDDT-BS analysis for target 2016-07-30_00000063_1 (Myroilysin, PDB ID 5CZW, black cartoon). For the evaluation a reference residue set is created based on any residue in a 3 Å radius from the Zinc ion (light green sphere). The lDDT is calculated based on a 10 Å inclusion radius (grey sphere, grey sticks). Zinc coordinating residues CYS23, HIS137, HIS141 and HIS147 are shown as yellow sticks. Residues of the structure predictions matching the reference set are displayed both in ribbon and sticks in orange for SWISS-MODEL, in blue IntFOLD4-TS and in magenta Sparks-X. All predictions reproduced the Histidine residues with little variation from the reference structure, while they showed a much greater deviation for Cysteine 23.
The QS-score[25] is assessing the correctness of the predicted quaternary structure by considering the assembly interface as a whole. The QS-score expresses the fraction of shared interface contacts (residues on different chains with a Cβ-Cβ distance < 12 Å) between two assemblies, i.e. model and reference structure. To unambiguously identify the residues of all protein chains in complexes, QS-score initially determines a mapping between equivalent polypeptide chains of the compared structures by exploiting internal symmetries where possible. A QS-score close to 1 translates to very similar interfaces, matching stoichiometry and a majority of identical interfacial contacts. A QS-score close to 0 indicates a radically diverse quaternary structure, probably different stoichiometry and potentially representing alternative binding conformations. QS-score is suitable for comparing homo- or hetero-oligomers with identical or different stoichiometry, alternative relative orientations of chains, and distinct amino acid sequences (i.e. homologous complexes).
Evaluation of Model Confidence
Following CASP standards, models are expected to indicate atomic error estimates in Å in the B-factor column. Reliability of error estimates is evaluated with a Receiver-Operator Characteristics (ROC) Area Under the Curve (AUC) analysis on a per residue level, where residues with a local lDDT value of higher or equal 0.6 are classified as predicted correctly. Note that the ROC AUC is undefined for extreme models with all residues classified as “correct” or “incorrect”. Some servers invert the quality scale and thus their data has to be inverted when evaluating the model confidence (see Fig. S2 for more details). Fig. 2 was created with the ROCR package[35].
FIGURE 2.
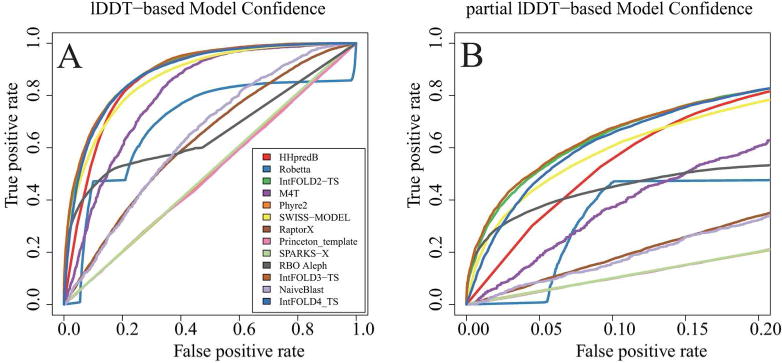
Model confidence ROC plot based on pooling all residues from all predictions across a 3-months timeframe matching the CASP12 prediction season, applying a classification threshold of 60 lDDT. All public servers at the time are shown.
Quality Estimation of Protein Structure Predictions
The Cα distances have been extracted from the data provided by the CASP12 assessors and originate from the LGA output without domain splits as performed in the CASP12 “Estimate of Model Accuracy” category.
4. Results & Discussion
a. Target Set and Target Difficulty
CAMEO is based on the pre-release of amino acid sequences by the PDB, which currently consist of ca. 180-240 new entries weekly, out of which on average 20 per week are selected as CAMEO prediction targets. Since the start of the project, 5116 targets were evaluated in the 3D structure category during a timeframe of 290 weeks. Unlike in CASP, targets for the automated weekly CAMEO assessment are not split into “assessment units” to avoid manual intervention for defining the domain boundaries, which would be required especially for difficult cases[21]. Instead, preferentially superposition independent scores which are robust to domain movements are used. Based on the prediction results, targets are classified as “Hard” if the averaged lDDT over all model-1 predictions of all servers is smaller than 50, “Easy” targets are those with an averaged lDDT higher than 75, and “Medium” targets in between (see Fig. S1).
For comparing results from CAMEO with CASP12 in this manuscript, an analysis date range was chosen to match the CASP12 prediction season (2016-05-01 - 2016-07-31) amounting to 250 targets, randomly selected during 13 weeks from 2334 protein sequences of 1072 experimental structures determined by NMR or X-RAY diffraction released by the PDB. The CAMEO target set used in this study contained 75 “Hard” targets, 129 “Medium” and 46 “Easy” targets. For comparison, 77 prediction targets (96 assessment units after splitting) formed the basis for the assessment in CASP12, which were assigned to three categories based on difficulty and average server performance. This resulted in 38 template based modeling targets (TBM), 39 free modeling domains (FM) and 19 domains in the mixed FM/TBM category[23].
b. Comparing CAMEO and CASP12 Structure Prediction Assessment
The CAMEO philosophy consists of offering a variety of scores, allowing users to rank methods by different aspects depending on their specific scientific interests. For target evaluations in the 3D category, scores that are invariant to domain movements and applicable to multidomain proteins such as lDDT based scores, CADscore and QS-score are most useful and shown in webpages displaying averaged values. For completeness, also superposition-dependent measures (such as GDT-HA, TM-score, RMSD or MaxSub) are provided for individual targets. It is important to note that many groups run anonymous servers in CAMEO for method development in preparation for CASP, resulting in the respective public server version performing seemingly worse in CAMEO than in CASP12. In this paper, we can only compare the results of the publicly accessible servers that are present in both CASP12 and CAMEO, albeit these may represent slightly different versions. Nevertheless, when comparing methods performance in CASP12 and CAMEO, matching rankings of the top methods are obtained.
Comparing Servers Based on lDDT
The top 3 servers participating both in CAMEO and CASP12 ranked by average lDDT are in identical order Robetta (Baker)[36] in the lead closely followed by RaptorX (Xu)[37–39] and IntFOLD4-TS (McGuffin)[40]. In both rankings (Table 1 and Table 2) SPARKS-X(Zhou)[41] is outperforming the Floudas Server(“Princeton_template”, Floudas)[42] albeit in CAMEO by only 0.7 lDDT units. HHPredB (Soeding)[43] shows a significantly worse performance in CAMEO, as there were no predictions submitted during 3 weeks due to technical issues, RBO Aleph(Brock)[44] suffered from an error introduced at the beginning of CASP, which the team discovered when analyzing CAMEO data, but was only corrected after the busy CASP12 season was concluded. Although the average lDDT values are considerably lower in CASP12 as in CAMEO (table 2), this does not translate to worse models for CASP12 targets modelled by the same servers. The CAMEO and CASP12 ranking are largely comparable – the difference in absolute scores rather reflects the overall difference in target difficulty distribution in the two data sets as discussed above. The standard deviations observed originate in the variation found for hard targets, as opposed to the easy targets, where the standard deviations are significantly smaller (see Table S1).
Table 1.
CASP12 ranking based on lDDT averaging across all targets for model-1.
| Server Name | lDDT (all targets) |
|---|---|
| BAKER-ROSETTASERVER | 49.3±18.56 |
| RaptorX | 46.1±18.35 |
| IntFOLD4 | 42.5±18.51 |
| HHPred0 | 40.5±18.56 |
| RBO_Aleph | 39.2±16.88 |
| ZHOU-SPARKS-X | 34.1±18.06 |
| FLOUDAS_SERVER | 31.7±17.54 |
Table 2.
CAMEO lDDT based ranking for the time frame 2016-05-01 - 2016-07-30. Only “public” servers at the time are shown, the underlying individual target sets may differ and thus cannot result in an absolute performance measure.
| Server Name | lDDT (all Targets) |
|---|---|
| Robetta | 65.3 ±16.45 |
| RaptorX | 63.8±16.57 |
| IntFOLD3-TS** | 62.2±17.53 |
| IntFOLD4-TS | 62.0 ±16.32 |
| SWISS-MODEL | 56.5±22.49 |
| SPARKS-X | 56.3±18.25 |
| Princeton_template | 55.6±15.68 |
| IntFOLD2-TS** | 55.1±17.47 |
| HHpredB* | 47.6±17.41 |
| M4T** | 45.1±16.74 |
| Phyre2** | 44.5 ±23.135 |
| NaiveBLAST** | 43.3±25.59 |
| RBO Aleph* | 38.6±16.28 |
method had technical problems during the CASP12 season, leading to suboptimal or missing data.
Method is not reflecting the current development and shown in CAMEO for historic comparison.
c. Assessment Scores specific to CAMEO
i. Ligand binding site quality
Application of models in life science research projects such as protein engineering or functional characterization often focus on ligand binding sites. For overall good models, the accuracy of binding site details does not necessarily correlate with overall model quality[45], and the assessment of binding site quality in models is therefore a relevant measure. The reference site is defined based on biologically relevant ligands present in the target structure by all residues within 4.0 Å radius of any ligand atom, in the case of ions 3.0Å is applied. This reference set is compared to the corresponding residues in the predictions by determining the atomic lDDT score on this substructure (lDDT-BS). Ligand binding site accuracy evaluations are grouped based on the PDB ligand classification in four classes: ionic (I), organic (O), short nucleotides (N) and short peptides (P) from two to ten bases or residues, respectively. Note that this measure does not rely on ligands actually being present in the predictions, and currently only two structure prediction servers in CAMEO actually aim to model ligands in their predictions, notably SWISS-MODEL[46] and IntFOLD4-TS[40]. Table 3 shows the ranking based on the ligand binding site quality, where the top 5 methods produce acceptable binding sites on average.
Table 3.
Averaged ligand binding site quality scores for the servers registered as public in CAMEO during the CASP12 time frame, the underlying individual target sets may differ and thus do not represent an absolute performance measure.
| Server Name | lDDT BS (all targets) |
|---|---|
| IntFOLD4-TS | 67.8±24.58 |
| IntFOLD3-TS** | 67.3±24.57 |
| SWISS-MODEL | 66.9±29.01 |
| RaptorX | 66.6±22.45 |
| Robetta | 66.5±22.51 |
| SPARKS-X | 65.0±22.98 |
| IntFOLD2-TS** | 62.3±25.83 |
| M4T** | 58.5±23.79 |
| Princeton_template | 56.1±20.83 |
| HHPredB* | 56.0±24.43 |
| Phyre2** | 55.5±30.56 |
| NaiveBlast** | 53.2±27.49 |
| RBO Aleph* | 40.4±25.22 |
method had technical problems during the CASP12 season, leading to suboptimal or missing data.
Method is not reflecting the current development and shown in CAMEO for historic comparison.
Applications of models for biomedical research often require correct representation of the interactions between a protein and bound molecules such as cofactors, substrates or inhibitors, which serves as incentive for CAMEO development.
Target 2016-07-30_00000063_1 (Myroilysin, PDB ID 5CZW[47], Fig. 1) illustrates the details of the lDDT-BS analyses. Myroilysin is a new bacterial member of the M12A family of metzincin metallopeptidases and activated by a cysteine-switch mechanism. The Cysteine and three Histidines are coordinating a Zinc ion. For the Histidines the structure predictions concur with the experimental structure, for the Cysteine they exhibited more variation.
Quaternary Structure Assessment
The majority of proteins across species are biologically active in form of higher-order quaternary structure assemblies rather than as monomers. CAMEO has recently added the first set of scores assessing quaternary states. “QS-score”[25] assesses the correctness of the complex stoichiometry and interface geometry and provides a mapping between the protein chains in the model and reference complex. Based on this chain mapping, MM-align scores are computed, measuring the overall structural similarity. Currently only Robetta and SWISS-MODEL are submitting homo-oligomers to CAMEO for evaluation. We observed that more groups submitted oligomer assemblies in CASP12, yet these oligomers were not all created by fully automated pipelines as would be required for joining CAMEO. As these CASP12 methods get automated and reproducible, we expect a significant increase in servers predicting oligomers in CAMEO in the future. While current modeling servers are focusing on homo-oligomers, a significant part of the potential CAMEO targets from the weekly PDB release are hetero-complexes. Therefore, future versions of CAMEO will include hetero-complexes as evaluation targets.
ii. Response time
From a practical perspective, users of structure prediction servers are interested in the most accurate prediction in the shortest time frame possible. Depending on the number of proteins to be modelled and the type of application, the optimal balance between speed and accuracy may vary. CAMEO measured the response time servers in CAMEO require from submission to completing their predictions and send them back for evaluation by email. This time frame is limited by the five day prediction window based on the release cycle of the PDB and represents a demanding requirement for some structure prediction servers employing algorithms that involve extensive sampling. Please note that results are subject to undisclosed priority scheduling of CAMEO submissions are directly dependent on the performance of the compute cluster employed. The average user experience may differ, although we encourage CAMEO participants to treat CAMEO submissions equal to those from public server users.
The fastest servers take around 15 - 20 min to complete a model on average (Table 4). HHPredB was fastest outperforming SWISS-MODEL by 6 min, both well below 30min. Currently, RaptorX strikes the best balance between time spent on modeling and the observed model quality by lDDT (Table 2). HHpredB and SWISS-MODEL are returning models below 30min on average but their distance to RaptorX is ~ 6-7 lDDT units both in CASP and CAMEO (3-4 lDDT units for the latest CAMEO data). HHpredB is one of the fastest algorithms, albeit missing some data in both time frames analyzed here (Tables 4 and 5). This translates to inconsistent CAMEO performance, rather than the method as such being slower in CAMEO.
Table 4.
Average response time from submission of the sequence to reception of structure prediction by CAMEO. Please note that results are subject to undisclosed priority scheduling of CAMEO submissions are directly dependent on the performance of the compute cluster employed.
| Server Name | Avg. response time (hh:mm:ss) |
|---|---|
| HHpredB* | 00:21:38 |
| SWISS-MODEL | 00:27:35 |
| SPARKS-X | 01:29:17 |
| Phyre2** | 02:14:08 |
| Princeton_template | 03:22:15 |
| NaiveBLAST** | 04:07:31 |
| M4T** | 08:20:48 |
| RaptorX | 13:53:17 |
| RBO Aleph* | 16:12:39 |
| IntFOLD2-TS** | 28:09:28 |
| IntFOLD3-TS** | 28:23:14 |
| Robetta | 29:03:28 |
| IntFOLD4-TS | 36:42:55 |
method had technical problems during the CASP12 season, leading to suboptimal or missing data.
Method is not reflecting the current development and shown in CAMEO for historic comparison.
Table 5.
Response times and selected scores for a common subset based on data from the last three months (2017-03-24 - 2017-06-17) in CAMEO.
| Server Name | Avg. response time (hh:mm:ss) | lDDT | lDDT-BS |
|---|---|---|---|
| SWISS-MODEL | 00:15:11 | 63.9 | 72.4 |
| Phyre2** | 00:38:27 | 42.9 | 55.9 |
| NaiveBLAST** | 01:00:05 | 49.2 | 55.9 |
| PRIMO_BST_CL | 01:08:22 | 45.6 | 50.0 |
| PRIMO | 01:12:26 | 45.6 | 50.0 |
| PRIMO_BST_3D | 01:21:09 | 44.4 | 49.6 |
| SPARKS-X | 01:51:03 | 59.7 | 66.1 |
| PRIMO_HHS_CL | 02:10:46 | 33.0 | 41.5 |
| PRIMO_HHS_3D | 02:24:50 | 32.4 | 41.4 |
| Princeton_TEMPLATE | 03:05:11 | 57.7 | 53.8 |
| RBO Aleph | 10:36:44 | 49.0 | 52.1 |
| RaptorX | 10:57:31 | 67.5 | 68.5 |
| M4T** | 11:08:04 | 47.6 | 53.4 |
| IntFOLD4-TS | 20:00:47 | 57.6 | 60.3 |
| HHpredB | 25:09:48 | 64.7 | 67.1 |
| IntFOLD3-TS** | 25:33:26 | 66.0 | 70.2 |
| IntFOLD2-TS** | 29:58:10 | 50.8 | 50.2 |
| Robetta | 34:08:11 | 69.4 | 67.6 |
Method is not reflecting the current development and is kept in CAMEO for historic reasons.
Table 5 shows current CAMEO data relating the response time to modeling performance. Currently among the fasted servers are SWISS-MODEL, Phyre2[48], PRIMO[49], SPARKS-X and Princeton_template all staying below 4 hours to return predictions of high to medium quality. The longest modeling times on average are observed for IntFOLDx-TS and Robetta, yet these servers are consistently delivering high quality models. RaptorX and RBO Aleph are returning models well below 12h, with RaptorX currently in the top three ranked servers according to lDDT (all atoms).
iii. Model confidence estimates
Any prediction has its limitations. For scientists utilizing predictions for structure guided work in place of experimental structure information, it is paramount to realize in which aspects a model is not faithfully representing the “real” protein structure, which translates to estimating local residue- wise model confidence. In CAMEO 3D the model confidence estimates are assessed against lDDT by employing Receive-Operator Characteristics (ROC, Fig. 2). For this analysis, residues extracted from all model-1 predictions of a given server in a particular time frame are pooled together, assuming that estimates are scaled between targets and interpretable as absolute values. The results over the CAMEO time frame 2016-05-01 - 2016-07-30 reveal large discrepancies between individual servers (see also Fig. S2). IntFOLD4-TS[40] was most consistently assigning correct model confidence estimates, closely followed by HHPredB and SWISS-MODEL. Scatter plots of the individual server data are given in the supplementary material. Princeton_template and SPARKS-X are not assigning any estimates and the baseline server NaiveBlast is using default MODELLER B-factors output.
For reference, we have included the ROC AUC values in Table 6, where we corroborate a clear lead tied by IntFOLD4_TS[40], SWISS-MODEL and HHPredB. The three servers are performing significantly better than any other server listed in providing realistic local model confidence estimates.
Table 6.
ROC AUC values of the pooled model confidence analysis based on lDDT (2016-05-01 – 2016-07-30). Note that not all methods provide confidence estimates in the server versions registered with CAMEO.
| Server Name | ROC AUC | pROC AUC (0.0-0.2) |
|---|---|---|
| IntFOLD3-TS** | 0.90 | 0.12 |
| IntFOLD2-TS** | 0.89 | 0.12 |
| IntFOLD4-TS | 0.89 | 0.12 |
| HHPredB | 0.87 | 0.10 |
| SWISS-MODEL | 0.87 | 0.11 |
| M4T** | 0.80 | 0.07 |
| Robetta | 0.70 | 0.06 |
| RBO Aleph | 0.66 | 0.08 |
| NaiveBlast** | 0.64 | 0.03 |
| RaptorX | 0.63 | 0.03 |
| SPARKS-X | 0.51 | 0.02 |
| Princeton_template | 0.50 | 0.02 |
| Phyre2** | 0.50 | 0.02 |
Method is not reflecting the current development and is kept in CAMEO for historic reasons.
d. Model Quality Assessment Category - CAMEO QE
As mentioned in the previous chapter, not all modeling servers are providing reliable local confidence estimates for their predictions. Model quality assessment tools offer an alternative assessment independent of the modeling method and are employed routinely in molecular modeling projects.
CAMEO QE, thus, offers a weekly evaluation of commonly used model quality assessment tools in a separate category. The evaluation is based on the models harvested from CAMEO 3D during the first 24h of a new evaluation cycle. These are submitted to the participating QE servers and results are required to be send back by email within three days. CAMEO QE focuses on all-atom local error estimates reflecting the use case of investigating the quality with a particular scientific question in mind by individual domains or residue segments of a given protein as each might differ substantially in quality.
Figure 3 displays the ROC AUC based assessment of the residue-wise error estimates across the 3-months data set (2016-05-01 - 2016-07-30), where the base line consisted of a PSIBLAST based method. Important to note that historically well-performing tools such as Verify3D[33], Prosa[29] and Dfire[31] today are clearly outperformed by newer methods such as ModFOLD4[32, 50], ModFOLD6[51], VoroMQA[52], QMEANDisCo and ProQ2[32].
FIGURE 3.
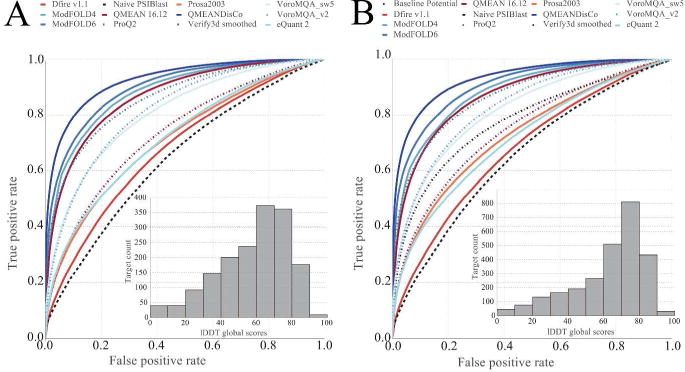
Panel A - ROC analysis of the residue-wise error estimates of the public methods active in CAMEO during 2016-05-01 - 2016-07-30. Historically well-performing tools such as Verify3D, Prosa and Dfire are outperformed by newer methods. Panel B - public methods currently available in CAMEO. New methods are constantly emerging, such as QMEANDisCo, eQuant2[53] and ModFOLD6, with QMEANDisCo currently being in narrow lead over ModFOLD6. The insets show the lDDT distribution of the underlying 3D models serving as targets for the QE category.
CAMEO employs the all-atom lDDT as the target value in the evaluation of model quality assessment tools, ensuring a robust automated assessment for multi-domain proteins. In contrast, superposition based Cα distances are employed in CASP[54], resulting in significantly different rankings. This prompted us to directly compare the Cα distance based approach with the residue-wise all-atom lDDT scores as they are used in the CAMEO QE.
Fig. 4 (panel A) illustrates the differences when basing QE assessment on lDDT or superposition based Cα distances. For example, in case of multi-domain targets such as T0920 with the specific example of model TS220_1 (Fig. 4, panel B), the result of domain movement are residues classified as correctly modelled in terms of lDDT (local lDDT > 60.0) but wrong in terms of Cα distance (d > 3.8 Å). Also, Cα distance based scores neglect 90% of the interatomic interactions of atoms in a protein structure (Fig. 4, panel C). These are crucial when assessing high quality models, where the main differences between models are the correctness of the stereochemistry or atomic interactions such as electrostatic interactions as well as hydrogen bonds. Also, the dependence on a single superposition renders the Cα distance based scores less reproducible especially for low quality models, where different tools can create different global superpositions. For example, Model TS313_1 for target T0869 (Fig. 4, panel D) displays the largest matching helix superposed by LGA, classifying all of its residues as correctly modelled (27% of all residues in the model) neglecting the unnatural environment of that helix. In contrast lDDT has not classified any of the residues as correctly modelled (local lDDT < 60.0).
FIGURE 4.
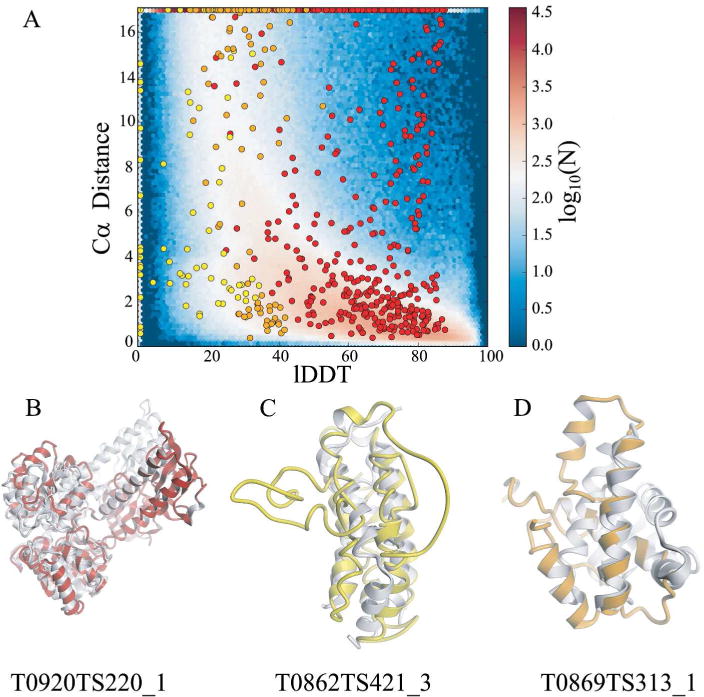
Illustration of the limitations of superposition based Cα distances in estimating model accuracy prediction. Panel A - Cα distances are compared to the corresponding local lDDT values for three exemplary quality predictions from CASP12. Cases of high Cα deviations contrasting high quality assigned by the all-atom lDDT values are indicated as red data points at the top right of the graph, and short Cα distances that contrast with low lDDT values are indicated as yellow and orange data points in the lower left area of the plot. The following examples illustrate reasons for these discrepancies: Panel B - the underlying global superposition fails for large domain movements and multi-domain proteins (red circles in panel A). Panel C – 90% of the atomic interactions are missing by focusing on Cα atoms, limiting in particular the assessment of high quality models. Panel D - evaluation of residue neighborhoods are implicitly excluded when considering Cα atoms only. lDDT assigns low scores to residues with large stereochemical deviations and physically impossible close contacts (e.g. yellow data points at lDDT value of 0.0, translating to unphysically positioned backbone and side chain atoms. The background image in panel A represents the data for all QE-stage2 submissions in CASP12.
We would like to emphasize that beyond the extremes shown in Fig. 4, these observations are valid for both, low quality and high quality models (Fig. S3). For continuous assessment the sensitivity of local superposition independent scores such as lDDT are crucial to correct for bad sidechain orientations and invalid stereochemistry. This cannot be achieved by a backbone only evaluation and directly contributes to the different rankings of local quality estimation methods in CAMEO and CASP.
Confidence estimates are undoubtedly crucial for conveying a model’s utility, and independently assessing a model’s quality remains a very important task until all modeling methods provide reliable confidence estimates. Cα distances are only useful for the hardest modeling cases, when judging the overall quality of the predicted fold. High quality predictions require superposition-independent assessment methods and the inclusion of all atomic interactions in the assessment. Due to the different nature of the assessment, the top performing methods in CAMEO are not matching those observed in CASP12.
Conclusion & Outlook
CAMEO deviates from the scoring methods used in CASP due to the requirement for unsupervised operation, but also to foster new algorithmic improvements by introducing new aspects in the assessment such as oligomers. Providing a broad spectrum of scores in contrast to a single ranking allows methods developers, reviewers and users to compare methods on such aspects which matter most to their specific interest in a transparent and independent way.
CAMEO enables evaluations based on common subsets of targets as well as pairwise head-to-head comparisons, producing publicly available publication ready data. The functionality to register new developments as anonymous servers alongside the public ones allowed new developments to reach maturity before announcing them publicly. Many groups have intensely used CAMEO for preparation of CASP experiments, e.g. by registering multiple development servers and comparing them in real time to the productive or historic versions, using the web-based analysis tools as well as the downloadable data provided by CAMEO. The result of this orthogonal assessment of different aspects is an overall increased robustness of the methods.
Prediction of quaternary structures remains a challenge in structure prediction[55] as observed both in CAMEO and CASP12. Currently, most methods require human intervention and only very few servers can automatically predict quaternary structure[25] CAMEO supports new algorithmic developments by providing new scoring schemes for developers, such as “QS-score” that was recently added to CAMEO, alongside with a growing number of other oligomeric state assessing scores[56].
Besides evaluating predictions for model accuracy, CAMEO evaluates the response time for each server, which is a crucial factor from a user perspective. Response time evaluation is obviously linked to availability of infrastructure resources, priority queuing and concurrent use by other processes and we should emphasize that a “slow” method in CAMEO is not necessarily intrinsically slow, but the servers may be at the limit of their capacity.
In summary, CAMEO has continuously added categories, scores and web-based analysis tools to serve the community in efficiently improving existing and developing new prediction algorithms. CAMEO aims at promoting methods producing biologically relevant models in particular concerning ligands, cofactors and oligomeric states.
Future developments in CAMEO will target the evaluation of structure prediction of hetero-oligomers, ligand pose evaluation, and contact prediction, addressing areas of active research in the area of computational structural biology.
Supplementary Material
Acknowledgments
We would like to thank for the invaluable feedback that we obtained from observers and participants alike, in particular the Schwede group members for testing the latest CAMEO releases. We would like to thank the RCSB PDB for publishing the pre-release data openly and responding promptly to questions. We would like to thank the community for their support, their new scores and prediction methods. We would like to thank Tobias B. Thüring for contributing technical scripts. We are grateful to the sciCORE team for providing excellent support and computational resources.
Funding
SIB Swiss Institute of Bioinformatics toward the development of CAMEO, OpenStructure and the use of sciCORE computing infrastructure. NIH and National Institute of General Medical Sciences (U01 GM093324-01) partially to CAMEO; Funding from ELIXIR EXCELERATE to CAMEO. Funding for open access charge: SIB Swiss Institute of Bioinformatics.
Footnotes
Author contributions:
JH manages the project, implemented the initial 3D category workflow, the 3D scores API, an early website prototype, analyzed the data, added scores and prepared the manuscript; MB developed and implemented QS score provided as external script; GS contributed the graphs to Figure 4, the discussion of evaluating Cα distances vs lDDT and the “BaselinePotential” method; DB is running the QE and 3D workflows, developed the current web interface and database queries; RG develops the contact prediction category; SR co-developed the web interface; AB developed the QE workflow and initial full stack of the web platform, added new scores to CAMEO 3D, implemented baseline servers and the initial CAMEO CP support; KM developed an early prototype; TS conceived the CAMEO concept, supervised the implementation, and edited the manuscript.
Conflict of interest
None.
References
- 1.Rocklin GJ, et al. Global analysis of protein folding using massively parallel design, synthesis, and testing. Science. 2017;357(6347):168–175. doi: 10.1126/science.aan0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ucarli C, et al. Genetic diversity at the Dhn3 locus in Turkish Hordeum spontaneum populations with comparative structural analyses. Scientific reports. 2016;6:20966. doi: 10.1038/srep20966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernandez-Martinez J, et al. Structure and Function of the Nuclear Pore Complex Cytoplasmic mRNA Export Platform. Cell. 2016;167(5):1215–1228 e25. doi: 10.1016/j.cell.2016.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barba-Spaeth G, et al. Structural basis of potent Zika-dengue virus antibody cross-neutralization. Nature. 2016;536(7614):48–53. doi: 10.1038/nature18938. [DOI] [PubMed] [Google Scholar]
- 5.Schwede T, et al. Outcome of a workshop on applications of protein models in biomedical research. Structure. 2009;17(2):151–9. doi: 10.1016/j.str.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moult J, et al. A large-scale experiment to assess protein structure prediction methods. Proteins. 1995;23(3):ii–v. doi: 10.1002/prot.340230303. [DOI] [PubMed] [Google Scholar]
- 7.Moult J, et al. Critical assessment of methods of protein structure prediction: Progress and new directions in round XI. Proteins. 2016;84(Suppl 1):4–14. doi: 10.1002/prot.25064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moult J, et al. Critical Assessment of Methods of Protein Structure Prediction (CASP) - Round XII. Proteins. 2017 doi: 10.1002/prot.25415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haas J, et al. The Protein Model Portal–a comprehensive resource for protein structure and model information. Database : the journal of biological databases and curation. 2013;2013:bat031. doi: 10.1093/database/bat031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rost B, Eyrich VA. EVA: large-scale analysis of secondary structure prediction. Proteins. 2001;(Suppl 5):192–9. doi: 10.1002/prot.10051. [DOI] [PubMed] [Google Scholar]
- 11.Bujnicki JM, et al. LiveBench-1: continuous benchmarking of protein structure prediction servers. Protein science : a publication of the Protein Society. 2001;10(2):352–61. doi: 10.1110/ps.40501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berman H, et al. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007;35:D301–3. doi: 10.1093/nar/gkl971. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rose PW, et al. The RCSB Protein Data Bank: new resources for research and education. Nucleic Acids Res. 2013;41:D475–82. doi: 10.1093/nar/gks1200. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ihssen J, et al. Increased efficiency of Campylobacter jejuni N-oligosaccharyltransferase PglB by structure-guided engineering. Open biology. 2015;5(4):140227. doi: 10.1098/rsob.140227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mariani V, et al. lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics. 2013;29(21):2722–8. doi: 10.1093/bioinformatics/btt473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olechnovic K, Kulberkyte E, Venclovas C. CAD-score: a new contact area difference-based function for evaluation of protein structural models. Proteins. 2013;81(1):149–62. doi: 10.1002/prot.24172. [DOI] [PubMed] [Google Scholar]
- 17.Xu J, Zhang Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 2010;26(7):889–95. doi: 10.1093/bioinformatics/btq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zemla A. LGA: A method for finding 3D similarities in protein structures. Nucleic acids research. 2003;31(13):3370–4. doi: 10.1093/nar/gkg571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kopp J, et al. Assessment of CASP7 predictions for template-based modeling targets. Proteins. 2007;69(Suppl 8):38–56. doi: 10.1002/prot.21753. [DOI] [PubMed] [Google Scholar]
- 20.Siew N, et al. MaxSub: an automated measure for the assessment of protein structure prediction quality. Bioinformatics. 2000;16(9):776–85. doi: 10.1093/bioinformatics/16.9.776. [DOI] [PubMed] [Google Scholar]
- 21.Kinch LN, et al. CASP 11 target classification. Proteins. 2016;84(Suppl 1):20–33. doi: 10.1002/prot.24982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng H, et al. ECOD: an evolutionary classification of protein domains. PLoS computational biology. 2014;10(12):e1003926. doi: 10.1371/journal.pcbi.1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abriata LA, et al. Definition and classification of evaluation units for tertiary structure prediction in CASP12 facilitated through semi-automated metrics. Proteins. 2017 doi: 10.1002/prot.25403. [DOI] [PubMed] [Google Scholar]
- 24.Olechnovic K, Venclovas C. The use of interatomic contact areas to quantify discrepancies between RNA 3D models and reference structures. Nucleic acids research. 2014;42(9):5407–15. doi: 10.1093/nar/gku191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bertoni M, et al. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Scientific reports. 2017;7(1):10480. doi: 10.1038/s41598-017-09654-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–9. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 27.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. Journal of molecular biology. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 28.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research. 1997;25(17):3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sippl MJ. Recognition of errors in three-dimensional structures of proteins. Proteins. 1993;17(4):355–62. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- 30.Wang G, Dunbrack RL., Jr PISCES: a protein sequence culling server. Bioinformatics. 2003;19(12):1589–91. doi: 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- 31.Zhang C, et al. A knowledge-based energy function for protein-ligand, protein-protein, and protein-DNA complexes. Journal of medicinal chemistry. 2005;48(7):2325–35. doi: 10.1021/jm049314d. [DOI] [PubMed] [Google Scholar]
- 32.Uziela K, Wallner B. ProQ2: estimation of model accuracy implemented in Rosetta. Bioinformatics. 2016;32(9):1411–3. doi: 10.1093/bioinformatics/btv767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eisenberg D, Luthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods in enzymology. 1997;277:396–404. doi: 10.1016/s0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 34.Biasini M, et al. OpenStructure: an integrated software framework for computational structural biology. Acta crystallographica. Section D. Biological crystallography. 2013;69(Pt 5):701–9. doi: 10.1107/S0907444913007051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sing T, et al. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21(20):3940–1. doi: 10.1093/bioinformatics/bti623. [DOI] [PubMed] [Google Scholar]
- 36.Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic acids research. 2004;32:W526–31. doi: 10.1093/nar/gkh468. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peng J, Xu J. RaptorX: exploiting structure information for protein alignment by statistical inference. Proteins. 2011;79(Suppl 10):161–71. doi: 10.1002/prot.23175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kallberg M, et al. Template-based protein structure modeling using the RaptorX web server. Nature protocols. 2012;7(8):1511–22. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGuffin LJ, et al. IntFOLD: an integrated server for modelling protein structures and functions from amino acid sequences. Nucleic acids research. 2015;43(W1):W169–73. doi: 10.1093/nar/gkv236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McGuffin LJ, et al. Accurate template-based modeling in CASP12 using the IntFOLD4-TS, ModFOLD6, and ReFOLD methods. Proteins. 2017 doi: 10.1002/prot.25360. [DOI] [PubMed] [Google Scholar]
- 41.Yang Y, et al. Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of query and corresponding native properties of templates. Bioinformatics. 2011;27(15):2076–82. doi: 10.1093/bioinformatics/btr350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khoury GA, et al. Princeton_TIGRESS 2.0: High refinement consistency and net gains through support vector machines and molecular dynamics in double-blind predictions during the CASP11 experiment. Proteins. 2017;85(6):1078–1098. doi: 10.1002/prot.25274. [DOI] [PubMed] [Google Scholar]
- 43.Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–8. doi: 10.1093/nar/gki408. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mabrouk M, et al. RBO Aleph: leveraging novel information sources for protein structure prediction. Nucleic acids research. 2015;43(W1):W343–8. doi: 10.1093/nar/gkv357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kryshtafovych A, et al. Evaluation of the template-based modeling in CASP12. Proteins. 2017 doi: 10.1002/prot.25425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Biasini M, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic acids research. 2014;42:W252–8. doi: 10.1093/nar/gku340. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu D, et al. Myroilysin Is a New Bacterial Member of the M12A Family of Metzincin Metallopeptidases and Is Activated by a Cysteine Switch Mechanism. The Journal of biological chemistry. 2017;292(13):5195–5206. doi: 10.1074/jbc.M116.758110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kelley LA, et al. The Phyre2 web portal for protein modeling, prediction and analysis. Nature protocols. 2015;10(6):845–58. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hatherley R, et al. PRIMO: An Interactive Homology Modeling Pipeline. PLoS One. 2016;11(11):e0166698. doi: 10.1371/journal.pone.0166698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McGuffin LJ, Buenavista MT, Roche DB. The ModFOLD4 server for the quality assessment of 3D protein models. Nucleic acids research. 2013;41:W368–72. doi: 10.1093/nar/gkt294. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maghrabi AHA, McGuffin LJ. ModFOLD6: an accurate web server for the global and local quality estimation of 3D protein models. Nucleic acids research. 2017 doi: 10.1093/nar/gkx332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olechnovic K, Venclovas C. VoroMQA: Assessment of protein structure quality using interatomic contact areas. Proteins. 2017;85(6):1131–1145. doi: 10.1002/prot.25278. [DOI] [PubMed] [Google Scholar]
- 53.Bittrich S, F.H.a.D.L. Advanced Technologies for Data Mining and Knowledge Discovery. Springer; 2016. eQuant - A Server for Fast Protein Model Quality Assessment by Integrating High-Dimensional Data and Machine Learning. Beyond Databases, Architectures and Structures; p. 14. [Google Scholar]
- 54.Elofsson A, et al. Methods for estimation of model accuracy in CASP12. Proteins. 2017 doi: 10.1002/prot.25395. [DOI] [PubMed] [Google Scholar]
- 55.Lafita A, et al. Assessment of protein assembly prediction in CASP12. Proteins. 2017 doi: 10.1002/prot.25408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mukherjee S, Zhang Y. MM-align: a quick algorithm for aligning multiple-chain protein complex structures using iterative dynamic programming. Nucleic acids research. 2009;37(11):e83. doi: 10.1093/nar/gkp318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.