

Abstract
Designed peptides that fold autonomously to specific conformations in aqueous solution are useful for elucidating protein secondary structural preferences. For example, autonomously folding model systems have been essential for establishing the relationship between α-helix length and α-helix stability, which would be impossible to probe with α-helices embedded in folded proteins. Here, we use designed peptides to examine the effect of strand length on antiparallel β-sheet stability. α-Helices become more stable as they grow longer. Our data show that a two-stranded β-sheet (“β-hairpin”) becomes more stable when the strands are lengthened from five to seven residues, but that further strand lengthening to nine residues does not lead to further β-hairpin stabilization for several extension sequences examined. (In one case, all-threonine extension, there may be an additional stabilization on strand lengthening from seven to nine residues.) These results suggest that there may be an intrinsic limit to strand length for most sequences in antiparallel β-sheet secondary structure.
Most proteins must fold to a specific three-dimensional shape to perform their biological functions. There is great interest in identifying the factors that determine native conformations; however, despite considerable study, it is not yet possible to predict tertiary folding patterns on the basis of primary structure. A few secondary structures (especially α-helix and β-sheet) recur throughout known protein structures, and understanding the forces that control conformational preferences within the common secondary structures should contribute to our understanding of conformational preferences at tertiary and quaternary levels. The α-helix has been very carefully scrutinized because there are well-established design principles for creating synthetic peptides that adopt α-helical secondary structure in the absence of a specific tertiary context (1–7). Until recently, the lack of autonomously folding β-sheet model systems made it impossible to conduct analogous studies with this secondary structure (8). In the past several years, however, a number of short peptides (9–24 residues) that display double- or triple-stranded antiparallel β-sheet conformations in aqueous solution have been reported (9–11). These model systems provide thermodynamic (12–23) and kinetic (24) insights on β-sheet folding behavior. [Solvent-exposed β-sheets in specific tertiary contexts have provided a complementary approach for elucidation of β-sheet conformational preferences (25, 26)]. Here, we show how small designed peptides can be used to assess an aspect of β-sheet stability that has not previously been addressed experimentally.
α-Helices become more stable as the length of the helix increases (5–7). This length-dependent effect on conformational stability arises because helix initiation is thermodynamically unfavorable but helix propagation is favorable, at least for some residues (1, 2). Analogous length-dependent stabilization is observed for double-helical nucleic acid conformations (27) and for unnatural oligomers that adopt helical secondary structures (28). Length-dependent stabilization is a more complex issue for sheet secondary structure than for a helix: two dimensions must be considered for a sheet (20), whereas only one dimension (along the helix axis) is important for a helix (1, 5–7). Fig. 1 shows the two dimensions in which protein β-sheet could display length-dependent stabilization, for propagation from a two-stranded antiparallel sheet (a “β-hairpin”): along the strand direction (Fig. 1a) and perpendicular to the strand direction (Fig. 1b). Each dimension can be evaluated independently because of our ability to design short peptides that display β-sheet conformations containing a predetermined number of strands with defined lengths (9–11). We have previously obtained evidence for length-dependent stabilization perpendicular to the strand direction (Fig. 1b) in a designed triple-stranded β-sheet that folds in water (20). Similar observations have been reported for a three-stranded β-sheet that folds in aqueous methanol (13) and more recently for a three-stranded design that folds in water (22). However, little or no length-dependent stabilization perpendicular to the strand direction was detected in water for an alternative three-stranded β-sheet design (23). Here we focus on the question of length-dependent stabilization along the strand direction (Fig. 1a).
Figure 1.

The two dimensions of potential length-dependent cooperativity in antiparallel β-sheet: (a) along the strand direction; (b) perpendicular to the strand direction.
Materials and Methods
Peptide Synthesis.
All linear peptides other than DP-TT2 were prepared and purified as described previously (29). DP-TT2 was synthesized on methylbenzhydrylamine resin by using the in situ neutralization/O-benzotriazol-1-yl-N,N,N,′N′-tetramethyluronium hexafluorophosphate activation protocol for Boc-Solid-phase peptide synthesis (30). Dimethylformamide was used as coupling solvent for most of DP-TT2, but DMSO was used for the last five residues.
NMR.
NMR experiments were performed as described earlier. Most data were acquired on a Varian INOVA 500 MHz spectrometer at 277 K; additional nuclear Overhauser effect (NOE) spectroscopy and rotating-frame Overhauser effect spectroscopy (ROESY) data were obtained for DP and c(DP)2 by using a Bruker (Billerica, MA) AVANCE 750 MHz spectrometer, and for DP-TT2 by using a Varian 600 MHz spectrometer at 277 K. Pulsed-field gradient phase-sensitive heteronuclear sequential quantum correlation (HSQC) experiments were carried out by using the “gHSQC” pulse sequence provided by Varian. A spectral window of 27,000 Hz was used for the carbon-13 dimension, which was externally referenced to 2,2-dimethyl-2-silapentane-5-sulfonate.
NOE-Restrained Dynamics.
Simulations were performed for DP-TT2 by using the program dyana (31). NOE restraints were derived from NOE spectroscopy and ROESY data. Only NOEs between nonadjacent and turn-defining residues were used as restraints for dynamics analysis. NOE intensities were qualitatively assigned to be strong, medium, weak, or very weak, and assigned constraints of 3, 4, 5, and 6 Å, respectively; a total of 32 restraints were used for DP-TT2. Restraints were checked by using the “distance check” function within dyana, which showed there were no “lonely” or possibly misassigned NOEs that could unduly influence the final conformation. dyana was used to generate 500 random structures, which were subsequently annealed. The best 10 structures (fewest restraint violations) were selected.
Aggregation Studies.
Sedimentation equilibrium experiments were carried out at 4°C on a Beckman Coulter Optima XL-A instrument. The peptides were studied at their respective NMR sample concentrations and at lower concentrations in H2O/D2O (9:1 ratio), 100 mM sodium deuteroacetate buffer, with pH adjusted to 3.8 with NaOH (pH measurements were not corrected for isotope effects). Revolution speeds of 44 K/min and 56 K/min were used. Data were analyzed by using the igor pro program (Wavemetrics, Lake Oswego, OR). Peptides were found to be monomeric in all cases.
Results
Our approach to probing the effects of strand length on β-sheet stability is based on a designed 12-residue peptide (DP; Fig. 2) that we have previously shown to adopt a β-hairpin conformation in aqueous solution (14, 16). DP contains a central d-Pro-Gly segment to induce formation of a “mirror image” β-turn (type I′ or II′) (32–36); the local right-handed twist of such turns is compatible with the right-handed twist of the strands in a β-sheet (37). Although d-proline is not a proteinogenic residue, structural analysis of 12-mer DP via two-dimensional NMR shows that this peptide adopts a native-like two-stranded antiparallel β-sheet conformation in aqueous solution (14, 16). Ornithine, one of the strand residues in DP, is not found in natural proteins; however, the presence of this residue does not diminish the biological relevance of our results, because we have shown that the analogue of DP containing lysine in place of ornithine behaves identically to DP (16). The folded state of DP, like that of other short peptides, is not fully populated in aqueous solution at accessible temperatures; the β-hairpin conformation of DP is ≈68% populated at 4°C (16).
Figure 2.

Structures of peptides used in this study.
To determine whether antiparallel β-sheet becomes more stable as the strands grow longer, we compared the folded state population of DP with the populations of longer peptides that contain the sequence of DP at their cores (e.g., DP-TT and DP-TT2; Fig. 2). NMR methods (38–41) were used to monitor β-sheet population at strand residues in the core, which are common to all of the peptides. The extension increment, two residues on each strand, is dictated by the fact that there are two types of strand residue in a double-stranded antiparallel β-sheet: residues that form cross-strand hydrogen bonds (“hydrogen bonded”) and residues that direct their backbone hydrogen-bonding sites away from the other strand (“nonhydrogen bonded”). Thus, extension by two residues in each strand is necessary to generate a regular antiparallel β-sheet series in which all members have the same type of interstrand pairing at the termini (hydrogen bonded, in our case). Threonine was selected for the first set of extensions because this residue has a high β-sheet propensity (42–44), but it is not very hydrophobic, in contrast to other β-sheet-prone residues. Analytical ultracentrifugation indicated that none of these peptides aggregates under the conditions of the NMR measurements.
The NMR data for DP, DP-TT, and DP-TT2 summarized in Fig. 3 indicate qualitatively that both the 12-residue core and the threonine extension segments engage in intramolecular β-sheet interactions. The chemical shifts of α-protons (δαH) are very sensitive to secondary structure; residues in β-sheets tend to display downfield-shifted δαH, and residues in α-helices tend to display upfield-shifted δαH, relative to the unstructured state (“random coil”) (45, 46). Fig. 3 presents ΔδαH [=δαH (obs) − δαH (random coil)] for the strand residues in each peptide. We follow the standard practice of using random coil δαH values determined with very short peptides (e.g., Gly-Gly-Xxx-Ala) (47). Segments containing three or more ΔδαH values >+0.1 are considered to be β-strands (45, 46). By this qualitative criterion, β-sheet formation extends from the turn to the penultimate residue in each strand for DP, DP-TT, and DP-TT2.
Figure 3.

ΔδαH = observed δαH − random coil δαH for the strand residues DP (filled bars), DP-TT (open bars) and DP-TT2 (striped bars), 1 mM each in aqueous (9:1 H2O/D2O) sodium deuteroacetate buffer, pH 3.8 (uncorrected), 4°C. The reported random coil δαH value for lysine was used for ornithine (random coil δαH values from ref. 47). No data are shown for the N-terminal residue of each peptide because this terminus is uncapped. For DP-TT2, δαH of Thr-17 and Thr-18 could not be unambiguously assigned; the ΔδαH values shown for these two residues are based on the average of the two observed δαH values (4.54 and 4.72 ppm). Chemical shifts were externally referenced to 2,2-dimethyl-2-silapentane-5-sulfonate (DSS).
ROSEY (39, 40) data indicate that the core segments of DP-TT and DP-TT2 adopt β-hairpin conformations analogous to that observed for DP. As previously reported (16), DP displays numerous interstrand NOEs, all of which are consistent with the expected β-hairpin folding pattern. Many of the corresponding NOEs are also observed for the central 12 residues of the longer peptides (summarized graphically for DP-TT2 in Fig. 4). In each case, a set of NOEs is observed between the Tyr and Leu residues that should be directly across from one another in the expected β-hairpin conformation. For DP, we observe also a set of NOEs between Tyr-2 and Lys-9, a “diagonal” pairing that reflects the right-handed twist commonly displayed by β-sheets (37). DP-TT displays a set of NOEs for an additional diagonal pairing, between Thr-2 and Leu-13, and DP-TT2 displays an analogous set of NOEs for diagonal pairing between Thr-4 and Leu-15. NMR data were used to generate the solution structure of DP-TT2 with the program dyana (31). Among the 10 best structures rms deviation = 2.54 ± 0.82 Å (backbone atoms only). Disorder is evident toward the end of each strand, but the β-hairpin conformation is well-defined over the central residues (rms deviation = 0.79 ± 0.40 Å among the 10 best structures for Arg-5 through Gln-16 of DP-TT2). Neither DP-TT nor DP-TT2 displayed any NOE evidence of an alternative folded conformation.
Figure 4.

NOEs between nonadjacent residues observed in NOE spectroscopy and ROESY analysis for 1 mM DP-TT2 in H2O/D2O (9:1 vol/vol) or in D2O, 4°C, 100 mM sodium deuterioacetate buffer, pH 3.8 (uncorrected), 200 ms mixing time. The solid arrows indicate NOEs between protons in the backbone and the dashed arrows indicate networks of NOEs between side chains. The backbone Hα-Hα NOE between Thr-4 and Thr-17 and NOEs between Thr side chains could not be observed because of resonance overlap.
Our experimental strategy requires that the folding behavior of DP conform to a two-state model, unfolded vs. β-hairpin, and that it be possible to quantify the folding equilibrium. We have established both of these features in previous studies of DP. α-Proton chemical shift data (δαH) are used to determine β-hairpin population (16) because, as discussed above, this NMR parameter is very sensitive to secondary structure. This strategy allows us to monitor population at several independent sites along the peptide backbone. We found earlier (16) that only strand residues in hydrogen-bonded positions (Val-3, Val-5, Orn-8, and Ile-10 of DP; highlighted in Fig. 2) are suitable for population analysis. Aromatic sidechains, e.g., Tyr-2 of DP, seem to interfere with quantification on the basis of strand residues in nonhydrogen-bonded positions, the α-protons of which are directed toward the other strand in the folded state (16, 18).
Population analysis of DP and related peptides based on δαH data requires that one know δαH for the limiting states, unfolded (δU) and folded (δF), because conformational interconversion is rapid on the NMR timescale. Replacing d-Pro with l-Pro completely abolishes β-hairpin formation (14, 16, 18, 34, 36), and the l-Pro diastereomer of DP provides an excellent representation of the completely unfolded state of DP (i.e., an excellent source of δU values). Closing off the open end of DP with a second d-Pro-Gly segment generates a cyclic peptide, c(DP)2, which has a very high β-sheet population (i.e., an excellent source of δF values) (16). The β-sheet population (Pβ) of DP at a given strand residue can be calculated based on three chemical shift measurements (Eq. 1),
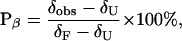 |
where δobs is δαH for the residue of interest in DP, δU is δαH for the residue of interest in the l-Pro diastereomer of DP, and δF is δαH for the residue of interest in cyclic peptide c(DP)2. We use the same δU and δF values for population analysis of DP-TT and DP-TT2, because the threonine extensions are well-separated from the four hydrogen-bonded strand residues used to determine β-hairpin population. β-Hairpin formation by DP is a two-state process, as demonstrated by the uniform temperature dependence of β-hairpin population determined at the four indicator residues, Val-3, Val-5, Orn-8, and Ile-10 (ref. 29). In the analysis below, we have assumed that two-state behavior is displayed by DP-TT and DP-TT2 and other extended peptides, at least in the core segments that correspond to the 12 residues of DP.
Table 1 shows β-hairpin populations calculated at the four indicator residues of DP, DP-TT, and DP-TT2 at 4°C. Also shown are differences in β-hairpin stability (ΔΔG) between DP and the longer peptides. ΔΔG values were calculated for each of the four indicator residues on the basis of the population data and a two-state folding model; the four residue-specific ΔΔG values were averaged to obtain the ΔΔG values shown in Table 1. For each peptide, the β-hairpin populations deduced at the four different residues are reasonably consistent. The apparent variation in deduced population within individual peptides could arise from real differences in the extent of β-hairpin folding at each position and/or from systematic error associated with the choice of reference peptides. Both sources of variation should be mitigated in the calculation of ΔΔG because comparison of specific residues in homologous peptides should lead to cancellation of systematic effects. Ultimately, it is not the numbers themselves but the qualitative implications of the trends that are of greatest interest.
Table 1.
β-Hairpin population calculated at each indicator residue (listed from N to C terminus) from δαH data, according to Eq. 1
| Peptide | Val #1 Pop. | Val #2 Pop. | Orn Pop. | Ile Pop. | Average ΔΔG (kcal/mol) |
|---|---|---|---|---|---|
| DP | 70% | 76% | 65% | 59% | — |
| DP-TT | 80% | 84% | 76% | 72% | −0.29 ± 0.03 |
| DP-TT2 | 85% | 92% | 79% | 80% | −0.53 ± 0.21 |
| DP-ST | 78% | 88% | 74% | 70% | −0.29 ± 0.17 |
| DP-ST2 | 78% | 80% | 71% | 69% | −0.17 ± 0.10 |
| DP-TA | 89% | 104% | 85% | 78% | −0.59 ± 0.14 |
| DP-TA2 | 83% | 92% | 82% | 74% | −0.50 ± 0.24 |
| DP-ST/Y | 81% | 92% | 82% | 74% | −0.48 ± 0.25 |
| DP-ST/Y2 | 78% | 84% | 71% | 70% | −0.22 ± 0.10 |
The % β-hairpin values have an uncertainty of ±2–4%, which arises from the ±0.01 ppm uncertainty in δαH. ΔΔG for each 16- or 20-mer, relative to 12-mer DP, was calculated for each residue in the usual way [(Keq for β-hairpin formation = (δobs − δU)/(δF − δobs); ΔG = −RTInKeq; ΔΔG = ΔG (16- or 20-mer) − ΔG (12-mer)]. The uncertainty in ΔΔG was calculated by using the standard deviation in ΔΔG for the four indicator residues in each peptide and Student's t-test at the 95% confidence level (this procedure treats each residue as an independent reporter of peptide ΔΔG).
The average ΔΔG value for DP-TT in Table 1 indicates that extending the strands of DP from five to seven residues causes a significant enhancement of β-hairpin stability. However, extending the strands from seven to nine residues gives a more ambiguous result. The ΔΔG value for DP-TT2 does not differ from the ΔΔG value for DP-TT within the uncertainty of the measurements, but the deduced β-hairpin population increases at each of the four indicator residues of DP-TT2 relative to DP-TT. This result raises the possibility that little or no additional stabilization is achieved by strand lengthening from seven to nine residues. We compared three other NMR parameters for DP, DP-TT, and DP-TT2, CαH–CαH NOE intensities, Cα chemical shifts, and N-H chemical shifts, to determine whether the trend reflected by the δαH-derived ΔΔG values is reliable. These comparisons strengthened our confidence in the δαH-based approach.
Where possible, we estimated β-hairpin population via CαH–CαH NOE intensities for nonhydrogen bonded residue pairs directly across from one another (e.g., Tyr-2 and Leu-11 in DP). This approach starts with the assumption that the CαH–CαH distance in the folded state is 2.3 Å, which corresponds to ideal antiparallel β-sheet. In addition, it is assumed that the CαH–CαH distance in the unfolded state is too large to allow any NOE. This strategy is inherently problematic because of the inverse sixth power dependence of NOE intensity on H–H separation, which magnifies the effect of errors in starting assumptions. For DP, the Tyr-2 CαH–Leu-11 CαH NOE intensity implies ≈50% β-hairpin population, which is in reasonable agreement with the 68 ± 8% β-hairpin population deduced from δαH data under the same conditions. Proximity between CαH chemical shifts and the solvent resonance prevented estimation of the β-hairpin population for DP from the Glu-4 CαH–Lys-9 CαH NOE intensity. This problem also prevented population estimation from either the Tyr-4 CαH–Leu-13 CαH NOE or the Glu-6 CαH–Lys-11 CαH NOE of DP-TT. However, both the Tyr-6 CαH–Leu-15 CαH and the Glu-8 CαH–Lys-13 CαH NOE intensities could be analyzed quantitatively for DP-TT2, and both indicated ≈60% β-hairpin population, which is in reasonable agreement with the 84 ± 6% β-hairpin population deduced for DP-TT2 from the δαH data. Most importantly, the NOE-based population analysis is consistent with the δαH-based analysis in showing that strand lengthening (DP to DP-TT2) leads to enhanced β-hairpin population.
13Carbon chemical shifts of α carbons (δαc) are sensitive to secondary structure, with residues in β-sheet showing upfield shifts, and residues in α-helices showing downfield shifts relative to random coil (48). Fig. 5 shows Δδαc [= δαc(obs) − δαc(random coil)] data for the strand residues common to DP, DP-TT, DP-TT2, and c(DP)2. The δαc (random coil) values were obtained from the l-Pro diastereomer of DP. For each peptide, Δδαc < 0 for all but one or two strand residues. This trend supports our conclusion that each peptide displays a high degree of β-sheet folding. Variations from the expected behavior at Tyr and Leu indicate that Δδαc measurements report on more than the secondary structural environment of the residue in question, which is also true of ΔδαH measurements. The qualitative trends among DP, DP-TT, DP-TT2, and c(DP)2 support our conclusion that β-sheet population increases along this series.
Figure 5.

ΔδαC data for the common core residues of DP (diagonally striped bars), DP-TT (solid bars), DP-TT2 (horizontally striped bars), and c(DP)2 (open bars). The δαC random coil values were obtained from the l-Pro diastereomer of DP. No data are shown for Arg-1 because in the random coil reference peptide (and in DP), this residue has an α-amino group rather than an α-amido group. Conditions for NMR experiment are as described in Fig. 3.
We used Eq. 1 to calculate β-hairpin population from δαc data at 4°C for DP, DP-TT, and DP-TT2, by using the l-Pro diastereomer of DP to provide δU and c(DP)2 to provide δF. These calculations involved the four indicator residues used for the δαH-based population estimates, Val-3, Val-5, Orn-8, and Ile-10 of DP and the analogous residues of DP-TT and DP-TT2. For DP, the δαc data suggested 57 ± 13% β-hairpin population (vs. 68 ± 8% suggested by the δαH data), for DP-TT, the δαc data suggested 65 ± 6% β-hairpin population (vs. 78 ± 5% suggested by the δαH data), and for DP-TT2, the δαc data suggested 76 ± 5% β-hairpin population (vs. 84 ± 6% suggested by the δαH data). Thus, the δαc approach consistently implies a moderately lower β-hairpin population than does the δαH approach, but the length-dependent trend is comparable in the two data sets. [The uncertainty at the level of average β-hairpin population, calculated from either δαH or δαc data, is greater than the uncertainty calculated for average ΔΔG (Table 1), because at least some systematic error is eliminated in the ΔΔG calculations, as pointed out above.]
1H chemical shifts of amide protons (δNH) are sensitive to intramolecular hydrogen bond formation in well-folded proteins, with δNH values for NH internally hydrogen bonded downfield relative to δNH values for NH exposed to solvent (49). The supporting information provides ΔδNH [=δNH (obs) − δNH (random coil)] data for the strand residues common to DP, DP-TT, DP-TT2, and c(DP)2. The δNH (random coil) values were obtained from the l-Pro diastereomer of DP. ΔδNH > 0 for most residues in most peptides, but the ΔδNH values are largest for residues that are expected to participate in interstrand hydrogen bonds in the folded state (Val-3, Val-5, and Ile-10 in DP, and analogous residues plus Gln in the other peptides). Thus, even though the β-hairpin conformations of DP, DP-TT, and DP-TT2 are not fully populated, these systems appear to follow the ΔδNH trend observed in folded proteins (49). The general trends among the ΔδNH data for the internally hydrogen bonded residues across the series DP, DP-TT, DP-TT2 are consistent with the ΔΔG data in indicating that β-hairpin population increases as the strands grow longer.
Four independent NMR parameters, ΔδαH, CαH–CαH NOE intensities, Δδαc and ΔδNH, support our conclusion that lengthening the five-residue strands of DP to generate DP-TT and DP-TT2 leads to an increase in β-hairpin stability as measured at core residues common to all three peptides. The data suggest additional stabilization from further lengthening beyond seven-residue strands (from DP-TT to DP-TT2), although this conclusion is more ambiguous (Table 1). We therefore examined additional extension sequences.
The design of the second extension series, DP-ST and DP-ST2 (Fig. 2), was based on the observation that serine has the second-highest β-sheet propensity (after threonine) for a strand at the outer edge of a β-sheet (44). The ΔΔG data show that, as for DP-TT, 16-mer DP-ST forms a β-hairpin that is significantly more stable than the β-hairpin formed by 12-mer DP (Table 1). However, ΔΔG data for DP-ST2 indicate that lengthening the β-hairpin strands from seven to nine residues does not provide any additional stability. This conclusion was supported by ΔδNH data (published as supporting information on the PNAS web site, www.pnas.org).
We considered two alternative explanations for the discontinuous behavior clearly displayed in the ST series, i.e., for the observation that lengthening the strands from five to seven residues stabilizes the β-hairpin, whereas further strand lengthening to nine residues provides little or no further stabilization. Hypothesis 1: Length-dependent stabilization occurs in antiparallel β-sheet, but a competing length-dependent effect destabilizes the β-sheet conformation when strands grow long enough Hypothesis 2: There is an energetically favorable interaction in the β-hairpin conformations of DP-ST and DP-ST2 between the first extension increment and the core segment, but no analogous interaction between the second extension increment and “inner” part of DP-ST2. ROESY data for DP-ST and DP-ST2 suggest an extension/core interaction that would be consistent with the second explanation: the diagonal side-chain–side-chain pairing between a threonine residue in the first extension increment of the N-terminal strand (Thr-2 in DP-ST or Thr-4 in DP-ST2) and the leucine residue in the C-terminal strand of the core (Leu-13 in DP-ST or Leu-15 in DP-ST2). In contrast, no diagonal NOEs were detected in DP-ST2 between Thr-2 (in the second extension increment) and Thr-17 (in the first extension increment). (The same pattern of side-chain NOEs was observed in the DP-TT/DP-TT2 series.) We have previously shown through mutational analysis of DP that diagonal interstrand side-chain–side-chain interactions can contribute to the stability of antiparallel β-sheet (29); therefore, the stability trend among DP, DP-ST, and DP-ST2 could arise if the diagonal interactions between Thr-2 and Leu-13 in DP-ST and between Thr-4 and Leu-15 in DP-ST2 contributed to net β-hairpin stability.
To distinguish between the two hypotheses outlined above, we examined two additional 16-mer/20-mer sets containing the DP core (Fig. 2). In DP-TA and DP-TA2, the nonhydrogen-bonded residues of the extension have been changed to alanine, relative to DP-TT and DP-TT2. We assume that the alanine side chain is so short that interactions with side chains on an adjacent strand will not contribute significantly to β-sheet stability (50). Therefore, if the enhanced stability of the β-hairpin conformations of DP-TT and DP-ST relative to the β-hairpin conformation of DP arises from diagonal interaction between Thr-2 and Leu-13 in DP-TT or DP-ST (hypothesis 2), then the β-hairpin conformation of DP-TA should be less stable than the β-hairpin conformation of DP-TT or DP-ST. Table 1 shows that the β-hairpin conformation of DP-TA is comparable in stability to the β-hairpin conformation of DP-TT or DP-ST and that further extension to DP-TA2 does not lead to a significant increase in β-hairpin stability relative to DP-TA. (These conclusions for the TA series are supported by δNH data, which are published as supporting information on the PNAS web site, www.pnas.org) These results suggest that hypothesis 2 is incorrect and that the increase in β-hairpin stability observed for 16-mers relative to 12-mer DP reflects an intrinsic effect of β-hairpin length (hypothesis 1).
Extension series ST/Y (Fig. 2) provides a complementary test of the competing hypotheses presented above, because this extension sequence is expected to allow stabilizing diagonal side-chain–side-chain interactions both between extension and core, and, for the 20-mer, within the core. The ST/Y extension contains serine in the hydrogen-bonded positions; the N-terminal extension has threonine in the nonhydrogen-bonded positions, and the C-terminal extension has tyrosine in the nonhydrogen-bonded positions. This design allows diagonal interactions between Thr-2 and Leu-13, in the extension and core, respectively, of DP-ST/Y, and between Thr-4 and Leu-15, in the extension and core, respectively, of DP-ST/Y2. These interactions are analogous to the Thr/Leu diagonal interactions in DP-TT, DP-TT2, DP-ST, and DP-ST2. The new extension also allows a diagonal interaction between two residues in the extension portions of 20-mer DP-ST/Y2, Thr-2 and Tyr-17. Side-chain–side-chain NOEs consistent with all three of these diagonal interactions were observed. For each of these diagonal pairings, both side chains are sufficiently long that we expect a favorable contribution from the diagonal interaction(s) to overall β-hairpin stability. Population analysis indicates that the behavior in the ST/Y series is comparable to that seen in the ST and TA extension series: the β-hairpin conformation of 16-mer DP-ST/Y is significantly more stable than the β-hairpin conformation of 12-mer DP, but the β-hairpin conformation of 20-mer DP-ST/Y2 does not differ significantly in terms of stability from the β-hairpin conformation of 16-mer DP-ST/Y. These conclusions are supported by ΔδNH data for the ST/Y series (see supporting information, www.pnas.org). These results provide further evidence against hypothesis 2.
As an additional test of the competing hypotheses, we examined a set of β hairpins on the basis of a less stable core sequence, the Tyr-2 → Ala variant of DP. As indicated in the supporting information (www.pnas.org), this alteration leads to a considerable diminution of β-hairpin stability. For two different extension series, TA and ST/Y, the trend follows that observed for extension of DP: the 16-mer is significantly more stable than the 12-mer, but the 20-mer is not more stable than the 16-mer.
Discussion
Our results suggest that there is an intrinsic limit on strand length in antiparallel β-sheets, at least for some sequences. Starting from a two-stranded β-sheet containing five-residue strands, we find a consistent increase in conformational stability on lengthening the strands to seven residues for four different extension sequences, but no further increase on further strand lengthening to nine residues for three of the four extension sequences. In the fourth case, all-threonine extensions, there may be additional stabilization on strand lengthening from seven to nine residues.
Why should there be a discontinuous effect of strand lengthening on antiparallel β-sheet stability? One explanation for this behavior is that as the strand length grows, so does a propensity within each strand to form α-helix. [Proline and glycine are strong helix breakers, so it is very unlikely that an α-helix would propagate through the central turn segment (42, 43)]. At some point, the intrastrand interactions that favor the helix begin to counterbalance the interstrand interactions that stabilize the β-hairpin. Because α-helical folding causes α-proton chemical shifts to move upfield relative to the random coil position, in contrast to the downfield shift induced by β-sheet folding (45, 46), the δαH-based population analysis is very sensitive to relatively small changes in the α-helical propensity on strand lengthening. Alternatively, our observations could be explained by an increase in random coil population as the strands grow longer. However, it is not clear to us why the population of the random coil state should increase with increasing strand length, in contrast to the expectation of increasing α-helix population with increasing length, if the sequence is conducive. (The distinctive behavior of the all-threonine extensions in our studies may reflect threonine's combination of high β-sheet propensity and low α-helix propensity.)
The existence of intrinsic limits on β-strand length for a majority of possible strand sequences is consistent with the results of a statistical survey of protein crystal structures, which indicate that the prevalence of β-strands decreases steadily as the strands grow longer (51, 52). In contrast, α-helix prevalence increases as the number of residues grows to four or five. α-Helix prevalence declines at greater lengths, but the decline is less precipitous than for β-strands (51, 52). The insights provided by our model study are important for understanding how natural proteins fold, for designing new proteins (53), and for elucidating pathologically important β-sheet aggregation processes (54, 55).
Supplementary Material
Acknowledgments
We thank Profs. Tom Record and Fleming Crim and all three reviewers for helpful comments. This research was supported by the National Science Foundation (NSF) (CHE-9820952) and the National Institutes of Health (NIH) (GM61238). H.E.S. was supported in part by a National Research Service Award (T32 GM08923). J.F.E. was supported by a Fellowship from the Ministerio de Educacion y Cultura (Spain) and the Fulbright Commission. NMR and mass spectrometers were purchased in part with support from NIH and NSF. The analytical ultracentrifuge is part of the University of Wisconsin Biophysics Instrumentation Facility.
Abbreviations
- NOE
nuclear Overhauser effect
- ROESY
rotating frame Overhauser effect spectroscopy
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates reported in this paper have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1JY9).
References
- 1.Qian H, Schellman J A. J Phys Chem. 1992;96:3987–3994. [Google Scholar]
- 2.Chakrabartty A, Baldwin R L. Adv Protein Chem. 1995;46:141–176. [PubMed] [Google Scholar]
- 3.Baldwin R L, Rose G D. Trends Biochem Sci. 1999;24:26–33. doi: 10.1016/s0968-0004(98)01346-2. [DOI] [PubMed] [Google Scholar]
- 4.Wallimann P, Kennedy R J, Kemp D S. Angew Chem Int Ed Engl. 1999;38:1290–1292. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1290::AID-ANIE1290>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 5.Scholtz J M, Qian H, York E J, Stewart J M, Baldwin R L. Biopolymers. 1991;31:1463–1470. doi: 10.1002/bip.360311304. [DOI] [PubMed] [Google Scholar]
- 6.Rohl C A, Scholtz J M, York E J, Stewart J M, Baldwin R L. Biochemistry. 1992;31:1263–1269. doi: 10.1021/bi00120a001. [DOI] [PubMed] [Google Scholar]
- 7.Zimm B H, Doty P, Iso K. Proc Natl Acad Sci USA. 1959;45:1601–1607. doi: 10.1073/pnas.45.11.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nesloney C L, Kelly J W. Bioorg Med Chem. 1996;4:739–766. doi: 10.1016/0968-0896(96)00051-x. [DOI] [PubMed] [Google Scholar]
- 9.Searle M S. J. Chem. Soc. Perkin Trans. 2001. 21011–21020. [Google Scholar]
- 10.Serrano L. Adv Protein Chem. 2000;53:49–85. doi: 10.1016/s0065-3233(00)53002-2. [DOI] [PubMed] [Google Scholar]
- 11.Gellman S H. Curr Opin Chem Biol. 1998;2:717–724. doi: 10.1016/s1367-5931(98)80109-9. [DOI] [PubMed] [Google Scholar]
- 12.Santiveri C M, Rico M, Jimenez M A. Protein Sci. 2000;9:2151–2160. doi: 10.1110/ps.9.11.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maynard A J, Sharman G J, Searle M S. J Am Chem Soc. 1998;120:1996–2007. [Google Scholar]
- 14.Stanger H E, Gellman S H. J Am Chem Soc. 1998;120:4236–4237. [Google Scholar]
- 15.Andersen N H, Dyer R B, Fesinmeyer R M, Gai F, Liu Z, Neidigh J W, Tong H. J Am Chem Soc. 1999;121:9879–9880. [Google Scholar]
- 16.Syud F, Espinosa J F, Gellman S H. J Am Chem Soc. 1999;121:11577–11578. [Google Scholar]
- 17.Honda S, Kobayashi N, Munekata E. J Mol Biol. 2000;295:269–278. doi: 10.1006/jmbi.1999.3346. [DOI] [PubMed] [Google Scholar]
- 18.Espinosa J F, Gellman S H. Angew Chem Int Ed. 2000;39:2330–2333. doi: 10.1002/1521-3773(20000703)39:13<2330::aid-anie2330>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 19.Carulla N, Woodward C, Barany G. Biochemistry. 2000;39:7927–7937. doi: 10.1021/bi992927l. [DOI] [PubMed] [Google Scholar]
- 20.Schenck H L, Gellman S H. J Am Chem Soc. 1998;120:4869–4870. [Google Scholar]
- 21.Kortemme T, Ramírez-Alvarado M, Serrano L. Science. 1998;281:253–256. doi: 10.1126/science.281.5374.253. [DOI] [PubMed] [Google Scholar]
- 22.Griffiths-Jones S R, Searle M S. J Am Chem Soc. 2000;122:8350–8356. [Google Scholar]
- 23.de Alba E, Santoro J, Rico M, Jiménez M A. Protein Sci. 1999;8:854–865. doi: 10.1110/ps.8.4.854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muñoz V, Thompson P A, Hofrichter J, Eaton W A. Nature (London) 1997;390:196–198. doi: 10.1038/36626. [DOI] [PubMed] [Google Scholar]
- 25.Smith C K, Regan L. Acc Chem Res. 1997;30:153–161. [Google Scholar]
- 26.Mayo K H, Ilyina E. Protein Sci. 1998;7:358–368. doi: 10.1002/pro.5560070216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Engel J, Schwarz G. Angew Chem Int Ed. 1970;9:389–400. doi: 10.1002/anie.197003891. [DOI] [PubMed] [Google Scholar]
- 28.Wang X, Espinosa J F, Gellman S H. J Am Chem Soc. 2000;122:4821–4822. [Google Scholar]
- 29.Syud F A, Stanger H E, Gellman S H. J Am Chem Soc. 2001;123:8667–8677. doi: 10.1021/ja0109803. [DOI] [PubMed] [Google Scholar]
- 30.Schnölzer M, Alewood P, Jones A, Alewood D, Kent S B H. Int J Peptide Protein Res. 1992;40:180–187. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- 31.Guntert P, Mumenthaler C, Wüthrich K. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 32.Sibanda B L, Blundell T L, Thornton J M. J Mol Biol. 1989;206:759–777. doi: 10.1016/0022-2836(89)90583-4. [DOI] [PubMed] [Google Scholar]
- 33.Gunasekaran K, Ramakrishnan C, Balaram P. Protein Eng. 1997;10:1131–1141. doi: 10.1093/protein/10.10.1131. [DOI] [PubMed] [Google Scholar]
- 34.Haque T S, Little J C, Gellman S H. J Am Chem Soc. 1996;118:6975–6985. [Google Scholar]
- 35.Karle I L, Awasthi S K, Balaram P. Proc Natl Acad Sci USA. 1996;93:8189–8193. doi: 10.1073/pnas.93.16.8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Struthers M D, Cheng R P, Imperiali B. Science. 1996;271:342–345. doi: 10.1126/science.271.5247.342. [DOI] [PubMed] [Google Scholar]
- 37.Chothia C. J Mol Biol. 1973;75:295–302. doi: 10.1016/0022-2836(73)90022-3. [DOI] [PubMed] [Google Scholar]
- 38.Dyson H J, Wright P E. Annu Rev Biophys Biophys Chem. 1991;20:519–538. doi: 10.1146/annurev.bb.20.060191.002511. [DOI] [PubMed] [Google Scholar]
- 39.Bax A, Davis D G. J Magn Reson. 1985;65:355–360. [Google Scholar]
- 40.Bothner-by A A, Stephens R L, Lee J M, Warren C D, Jeanloz R W. J Am Chem Soc. 1984;106:811–813. [Google Scholar]
- 41.Wüthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley; 1986. [Google Scholar]
- 42.Chou P Y, Fasman G D. Biochemistry. 1973;13:211–222. doi: 10.1021/bi00699a001. [DOI] [PubMed] [Google Scholar]
- 43.Muñoz V, Serrano L. Proteins Struct Funct Genet. 1994;20:301–311. doi: 10.1002/prot.340200403. [DOI] [PubMed] [Google Scholar]
- 44.Minor D L, Kim P S. Nature (London) 1994;371:264–267. doi: 10.1038/371264a0. [DOI] [PubMed] [Google Scholar]
- 45.Wishart D S, Sykes B D, Richards F M. J Mol Biol. 1991;222:311–333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- 46.Wishart D S, Sykes B D, Richards F M. Biochemistry. 1992;31:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- 47.Bundi A, Wüthrich K. Biopolymers. 1979;18:285–297. [Google Scholar]
- 48.Spera S, Bax A. J Am Chem Soc. 1991;113:5490–5492. [Google Scholar]
- 49.Wagner G, Pardi A, Wüthrich K. J Am Chem Soc. 1983;105:5948–5949. [Google Scholar]
- 50.Yang A-S, Honig B. J Mol Biol. 1995;252:366–376. doi: 10.1006/jmbi.1995.0503. [DOI] [PubMed] [Google Scholar]
- 51.Kabsch W, Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 52.Dill K A. Biochemistry. 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 53.DeGrado W F, Summa C M, Pavone V, Nastri F, Lombardi A. Annu Rev Biochem. 1999;68:779–819. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 54.Dobson C M. Trends Biochem Sci. 1999;24:329–332. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 55.Kelly J W. Proc Natl Acad Sci USA. 1998;95:930–932. doi: 10.1073/pnas.95.3.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.