

Abstract
HIV infection causes a disruption of gut-associated lymphoid tissue, driving a shift in the composition of gut microbiota. A deeper understanding of the metabolic changes and how they affect the interplay with the host is needed. Here, we assessed functional modifications of HIV-associated microbiota by combining metagenomic and metatranscriptomic analyses. The transcriptionally active microbiota was well-adapted to the inflamed environment, overexpressing pathways related to resistance to oxidative stress. Furthermore, gut inflammation was maintained by the Gram-negative nature of the HIV-associated microbiota and underexpression of anti-inflammatory processes, such as short chain fatty acid biosynthesis or indole production. We performed co-occurrence and metabolic network analyses that showed relevance in the microbiota structure of both taxonomic and metabolic HIV-associated biomarkers. The Bayesian network revealed the most determinant pathways for maintaining the structure stability of the bacterial community. In addition, we identified the taxa’s contribution to metabolic activities and their interactions with host health.
Introduction
HIV infection is characterized by a profound Th17 cell depletion in the gut-associated lymphoid tissue (GALT) and by a subsequent mucosa breakdown, leading to a dysregulation of immune-epithelial barrier that allows translocation of bacterial products to the blood. Further, the enhanced microbial translocation causes systemic immune activation resulting in a chronic inflammatory state. Additionally, HIV infection induces a shift in the gut microbiota composition that is characterized by an enrichment of pro-inflammatory Gram-negative bacteria and potential pathogens [1–10]. Interestingly, HIV-associated dysbiosis has been related to systemic immune activation, microbial translocation, and blood T cell activation [11, 12]. However, the effect of potential functional shifts on human health has been largely overlooked. Previously, we showed that some metabolic pathways increased in HIV-infected subjects and were correlated with the extent of immune activation and bacterial translocation [6]. Recently, using metaproteomic and meta-metabolomic approaches, we detected an association between the optimal response to antiretroviral therapy (ART) and the active fraction of the gut microbiome, suggesting a functional role for the microbiota in the immunological response to ART [13, 14]. Nevertheless, few works have been addressed on the transcriptional regulation of the gut microbiota metagenome [15–17] and to our knowledge, no studies on the gene expression level of the HIV-associated microbiota have been performed. In this work, we used a combination of metagenomic and metatranscriptomic analyses to obtain key information about microbiota metabolism, assessing which predicted genes are expressed in the community and to what extent. We found that the HIV-associated community is adapted to oxidative stress and did not express pathways related to anti-inflammatory metabolic processes.
The gut microbiome is a complex community with highly interacting microbial species that maintain a close interplay with the host. Network analysis is a powerful tool that is used to study tightly interlinked biological systems and has been used to measure the importance of a species or genes within a community by identifying keystone elements to elucidate coexistence patterns spanning from pairs of microbial taxa in several ecosystems as well as to determine their contribution to specific conditions, such as health status or disease [5, 18–28].
In the present study, ecological network analysis revealed that HIV-associated microbiota established a stable community in which overrepresented species are important network hubs. Similarly, enzymatic network analysis showed that HIV infection caused dramatic changes in the metabolic structure of the microbiota. Finally, the integration of functional, taxonomic, and clinical data in a Bayesian network provided insight into connections between bacterial metabolism and HIV immunopathogenesis.
Materials and methods
Study cohort and clinical features
We used baseline fecal samples from a previously described cohort [5] that included HIV-infected subjects: viremic untreated (VU) (n = 12), immunological ART responders (n = 18), and non-responders (n = 9) (IR and INR, ≥350 and <350 CD4+ T cell counts after 2 years of viral suppression, respectively), with unmatched HIV uninfected individuals (n = 15) as controls. The markers of inflammation and bacterial translocation, thymic function, and T cell activation were determined [5]. This study was approved by the Ethics Committee at both clinical centers (University Hospital Clínico San Carlos [approval number 11/284] and University Hospital Ramón y Cajal). All participants gave informed consent prior to the initiation of study procedures.
Total DNA and RNA extraction and sequencing
Total DNA and RNA extraction and the construction of the sequencing libraries are described in the methods in the online supplementary information. Sequencing was performed at the Centre for Public Health Research (FISABIO-Salud Pública, Valencia, Spain). All of the sequences have been deposited in the EBI database under the number PRJEB23871.
Initial processing, assembly, annotation, and bioinformatic analysis
Raw paired-end reads from metagenomics (DNAseq) and metatranscriptomics (RNAseq) were trimmed for adapters and transposase sequences and filtered to remove artifacts and low-quality sequences as described in the supplementary methods. Reads matching the human genome were discarded, and the paired-end reads were joined to obtain longer paired-end sequences using pandaseq tool (v4.0.3) [29] (parameters: -N -l 50 -o 10).
The metagenomic paired-end sequences were assembled by Ray-Meta [30] (version 2.3.1 parameters: -k 31 -minimum-contig-length 300) [30]. The K-mer size that maximized the N50 value was selected for the Ray-Meta assembler. Similarly, metatranscriptomic paired-end sequences were assembled using the Trinity assembler [31] to obtain functional contigs. Next, the open reading frame (ORF) prediction for metagenomics and metatranscriptomics contigs (and sequences not assembled into the contigs) was performed using MetaGeneMark software [32] (prokaryotic version 3.25) and the TransDecoder.LongOrfs package [33], respectively. All ORFs were translated into amino acids and clustered using USEARCH software (v8.1.1831 parameters: -id 0.95 -threads 2 -strand both --query_cov 0.9). The resulting non-redundant database (hereafter ORFaanr) was compared to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [34] using rapsearch2 software for functional annotation at the gene family (KO) and pathway (ko) levels. The functional assignation criteria were based on the ORF best-hit, prioritizing the e-value, followed by the bit-score, percentage of identity and, finally, alignment length. The relative abundance of each metagenomic ORF was calculated by mapping all of the reads against the nucleotide sequences of the ORFaanr database using the soap aligner (version 2.21) and soap.coverage script (2.7.7) [35, 36] according to the pipeline of Nielsen et al. [37]. For taxonomic assignment, we mapped both metagenomics and metatranscriptomic paired-end sequences against an in-house non-redundant genome database (Genomeclustdb) (see the supplementary information).
To identify taxa and metabolic pathways as biomarkers, we applied the linear discriminant analysis (LDA) effect size (LEfSe) algorithm [38].
To correlate the bacterial taxa to the metabolic pathways involved in HIV-associated dysbiosis, we performed a generalized linear model (GLM) by setting the pathway biomarkers as the response variable and the species matrix as the predictors. Then, we mapped all of the reads from each pathway biomarkers against the reference genomes of the species biomarkers (see supplementary methods).
To deeply study the metabolic pathways involved in the inflammation produced by HIV infection, we conducted a sensitive search of remote gene homologs and species that could contain them in the metagenomic and metatranscriptomic data. Sensitive detection was performed using hmmsearch software and blastp against the ORFaanr database (see supplementary information).
A full description of the methods used for ecological, clustering, ordination, and correlation analyses are described in the supplementary information.
Analysis of gene expression
The transcript abundance was measured by mapping the metatranscriptomic sequences against the ORFaanr using the RSEM tool implemented in the rsem-synthesis-reference-transcripts algoritm. To normalize the transcript expression values by the sequencing depth and gene length, we calculated the fragments per kilobase of target transcript length per million reads mapped (FPKM).
As described by Franzosa et al. [15], to detect over- or underexpressed genes, we calculated the log RNA/DNA abundance ratio for each KO in all samples. A gene was over- or underexpressed if the log RNA/DNA abundance ratio was significantly different from zero by applying the t-student test using the Benjamini–Hochberg correction (BH-adjusted P value <0.01).
Ecological and metabolic networks
The ecological and metabolic networks were estimated using the taxonomic and functional data obtained from metagenomics and metatranscriptomics for HIV-associated microbiota. The consistency of these estimates was tested using the Erdős–Rényi model (Supplementary Table 1).
The ecological network obtained included all species present at least in 70% of the samples and whose average relative abundance was above 0.01%. The correlation matrix was estimated using the SparCC.py script (parameters:-i 10) [39]. Statistical support was achieved by performing 1000 bootstrap resamplings using the script MakeBootstraps.py [39]. A significant correlation required a BH-adjusted P value below 0.01 and an absolute value of the correlation coefficient above 0.1. The co-occurrence network was estimated using the igraph R package (function “graph.adjacency” mode “undirected”) by removing all loops and unconnected nodes (igraph functions “simplify” and “delete_vertices”).
The metabolic network was created using the KEGGgraph R package. First, we downloaded the KGML files from the KEGG website for the corresponding pathways in which the previously identified KOs from the metagenome (DNA-KO) and metatranscriptome (RNA-KO) were involved. The list of selected metabolic pathways was then parsed using the R function “parseKGML2Graph,” and the network was assembled using the “mergeKEGGgraphs” function (edgemode = “directed”). The total network was then plotted using the igraph R package (function plot.igraph).
The methods to determine the topological properties of networks (small-world effect, connectivity distribution, and modularity); betweenness, degree, and eigenvector centrality indexes; and network fragmentation are described in the methods in the online supplementary information.
Multiomic Bayesian network
A multiomic Bayesian network (BN) was estimated using the metagenome, metatranscriptome, and metabolome data from the HIV+ infected subjects to predict the effect of microbiota on the markers of innate and T cell activation variables, thymic function, and bacterial translocation. An interesting property of BN is that it can be dissected in Markov blankets (MB). An MB of a node contains all of the variables needed to predict the behavior of that node. The methods, algorithms, and statistics used to construct BN and MB are described in the methods in the online supplementary information.
Results
HIV-associated metagenome
Figure 1a presents a cluster configuration in which most HIV+ subjects showed clearly distinct gene content (KO) compared to control subjects (ADONIS test, P value = 0.001). This difference remained significant when the HIV− group was compared against each of the other three different HIV+ groups (ADONIS test, P value = 0.001) (Supplementary Figure 1a). To address the genes and pathways that consistently explain the differences between the groups, we used the LEfSe tool. We found that 34 KEGG pathways (ko) (Fig. 1b) and 186 KEGG orthology groups (KO) (Supplementary Figure 1b) were significantly different between the HIV+ and control subjects, hereafter referred as pathway (ko) or gene (KO) biomarkers. HIV+ bacterial communities presented an increase in pro-inflammatory pathways, such as lipopolysaccharide (LPS) biosynthesis (ko00540) and pathways related to infectious diseases (ko05111 and ko05120). Interestingly, two pathways (ko00250 and ko00908) that may be involved in the resistance to oxidative stress showed a significantly higher abundance in the HIV+ group. However, the HIV gene content was depleted of pathways related to signal transduction and membrane transport (Fig. 1b).
Fig. 1.

Comparison of the microbiota gene composition between HIV+ and HIV− groups from metagenomics. a NMDS analysis of the KO composition. Red ellipses represent a cluster mainly composed of HIV-infected subjects; blue ellipses represent a cluster mainly composed by uninfected subjects. The cluster configuration was validated using the ADONIS test (P value = 0.001). b Linear discriminative analysis (LDA) effect size LEfSe analysis of pathways (ko) between HIV-infected (red) and uninfected (blue) individuals. LDA scores (log10) for the most discriminative pathways in HIV− subjects were represented on the negative scale; LDA-positive scores indicated enriched pathways in HIV+ patients
We also observed significant differences between each HIV subgroup and the control group, with a core of metabolic pathways with a high abundance of KOs related to the resistance to oxidative stress (Supplementary Figure 1c, d, e).
The taxonomic assignment retrieved from the metagenomes also revealed significant differences in microbiota composition between groups (Supplementary Figure 2a). HIV+-associated microbiota were characterized by an increase in species from the genera Prevotella, Acidaminococcus, and Streptococcus and a decrease of commensal species, such as Bacteroides, Bifidobacterium, Akkermansia, Odoribacter, and Alistipes (Supplementary Figure 2b).
Metatranscriptome of HIV-associated microbiota
We observed that 964 KOs from the metagenome, DNA-KO, were not present in the metatranscriptomic data. This difference could be due to transcriptional regulation of the metagenome, which could be reflected in the metatranscriptome. On other hand, 157 KOs from the metatranscriptome, RNA-KO, were missing in the metagenome data set. These RNA-KOs could correspond to transcripts of low-abundant but transcriptionally active bacteria. Although this study is focused on major functions and species, which are well represented, a limitation in sequencing depth of both, metagenome and metatranscriptome, should also be considered when interpreting our results. Moreover, we found a good correlation between the DNA-KO and RNA-KO abundance (r = 0.84, P value = 2.2e−16). To address the difference in the functional profiles, we calculated, based on Franzosa et al. [15], the Bray–Curtis dissimilarity and evenness indexes for DNA-KOs and RNA-KOs in the HIV+ and HIV− groups. We found, in both groups, that the RNA-KO composition was significantly more variable than DNA-KO composition (Fig. 2a) but the DNA-KO and RNA-KO evenness index was similar (Fig. 2b). Thus, the differences observed in the DNA-KO and RNA-KO profiles could be explained by transcriptional regulation of the metagenome based on the environmental requirement. Additionally, the comparison of both indexes between the HIV+ and HIV− groups revealed a lower dissimilarity index of HIV-associated microbiota that could reflect the functional adaptation to the HIV environment (Fig. 2a). Moreover, HIV-specific functions could be preferentially expressed, reducing the evenness of this group (Fig. 2b). Clustering analysis showed that RNA-KOs were statistically distinct in the HIV-infected individuals compared to the controls (ADONIS test, P value = 0.006) and across all subgroups (ADONIS test, P value = 0.001) (Fig. 3a and Supplementary Figure 3a). In the LEfSe analysis (Fig. 3b and Supplementary Figure 3a), HIV-associated microbiota showed a significantly higher abundance of transcripts, mainly related to the stress response (ko04141, ko00521, ko00730, and ko00053) and a depletion of the highly discriminative transcripts related to anti-inflammatory metabolic processes, such as butanoate metabolism (ko00650), propanoate metabolism (ko00640), or fatty acid metabolism (ko00071), supporting our previous findings that indicated abnormalities in SCFA production [5].
Fig. 2.

Diversity comparisons between metagenomic and metatranscriptomic data. a Bray–Curtis index of the KO composition. b Pielou’s evenness index of the KO composition. HIV+ (red) and control (blue) subjects. Group pairwise comparison was set using the Wilcoxon signed-rank test
Fig. 3.
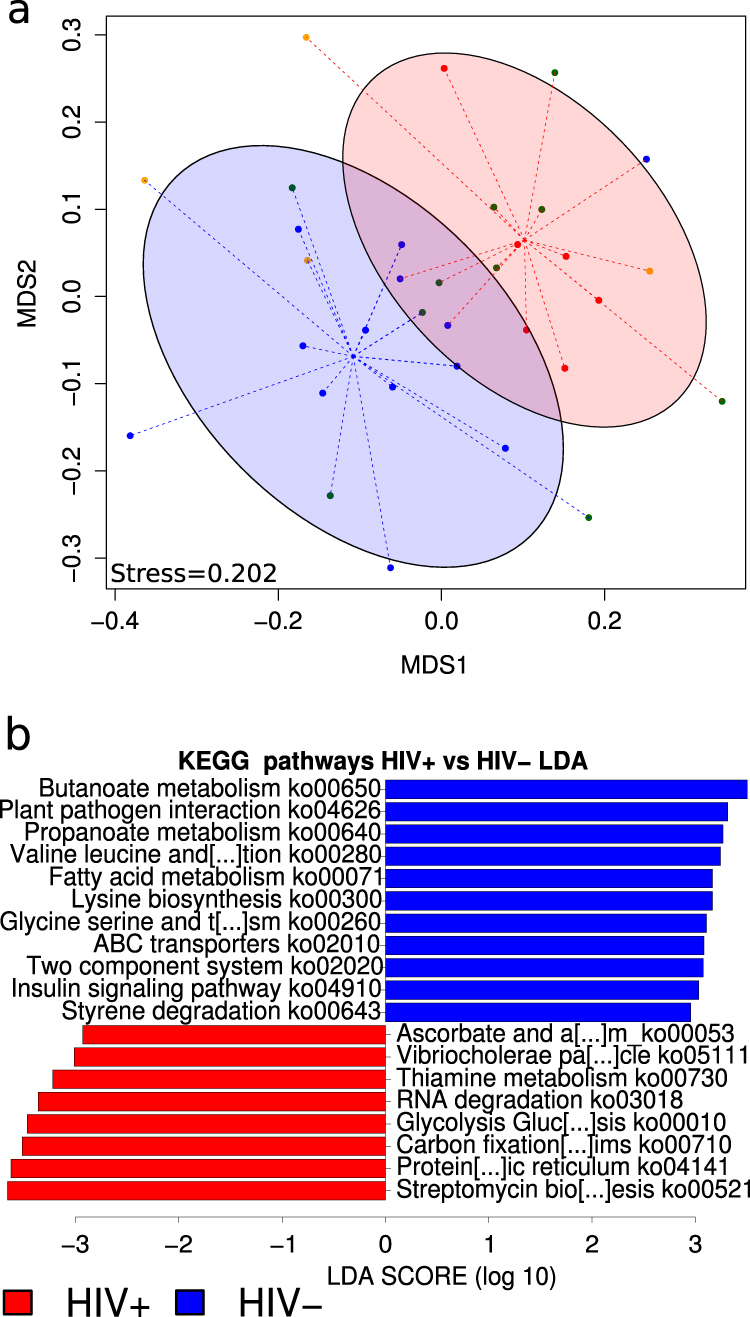
Comparison of microbiota gene composition between HIV-infected and uninfected subjects from metatranscriptomics. a NMDS analysis of the KO composition. b Linear discriminative analysis (LDA) effect size LEfSe analysis of pathways (ko) between HIV+ (red) and HIV− (blue) subjects. LDA scores (log10) for most discriminative pathways in HIV uninfected subjects are represented on the positive scale, whereas LDA-negative scores indicated enriched pathways in HIV-infected individuals
To determine the genes that were differentially expressed (over or under), we calculated the log RNA/DNA abundance ratio for each one in both HIV+ and HIV− groups. Thus, we found that 49.08% of the total genes were differentially expressed in the HIV-infected group and 40.61% in the controls. We also found that 13.89% and 11.90% of the differentially expressed genes were overexpressed in HIV+ and HIV− groups, respectively. Figure 4 illustrates the heatmap of the DNA-KO and RNA-KO biomarkers that were significantly over- or underexpressed in the microbiota of HIV-infected and healthy individuals. In HIV-infected subjects, anti-inflammatory metabolic pathways, such as the propanoate (ko00640) and butanoate (ko00650) pathways, were underexpressed. By contrast, genes related to stress resistance mechanisms (ko00730, ko00521, and ko4141) were overexpressed. Thus, the transcriptional profile of the HIV-associated microbiota indicated that preferential expression of the metabolic pathways was able to attenuate oxidative stress, which could be caused by local inflammation.
Fig. 4.

Heatmap of relative gene expression of pathway biomarkers. Hierarchical clustering representation of gene expression ratio (|log10 RNA/DNA| > 0). Only pathways that were significantly over- or underexpressed (BH-adjusted P value ≤0.05) and were significantly differentially expressed between HIV+ and HIV− subjects (Wilcoxon test, BH-adjusted P value ≤0.05) were included in the analysis. Overrepresented pathways in HIV+ subjects are in red, blue indicates overrepresented pathways in uninfected individuals. VU viremic untreated subjects (red); IR immunological responders (green); INR immunological non-responders (orange) and HIV− subjects (blue) were represented as tips of the column’s cladogram. The brown to purple gradient represents relative gene expression level
Unlike the microbiota composition obtained from the metagenome, the one retrieved from the metatranscriptomes was dominated by species of the Firmicutes phylum (mean 77.68 ± 14.27% Firmicutes and 17 ± 12.17% Bacteroidetes) and archeon species belonging to the genus Methanobrevibacter (Supplementary Figure 4). Moreover, the transcriptionally active microbiota of the HIV+ subjects was significantly different from those of the HIV− individuals (ADONIS test, P value = 0.005), in which Prevotella, Acidaminococcus, Coprobacillus, and Streptococcus differentially increased (Supplementary Figure 5).
Bacterial role in HIV-associated dysbiosis
GLM analysis revealed significant positive correlations between the bacterial taxa and pathway biomarkers for the HIV+ group (Supplementary Table 2). We assessed whether the species biomarkers carried the genes involved in their correlated pathways or promoted the growth of other members of the microbiota responsible for metabolic functions. We found that HIV species biomarkers presented the genes involved in their related metabolic pathways. We also observed that the different species of Prevotella had genes involved in almost all of the HIV pathway biomarkers (Supplementary Table 3), suggesting a major role for this genera in HIV pathogenesis.
Microbial metabolism of dietary tryptophan and choline
To study the role of HIV-associated microbiota in the pro-inflammatory kynurenine pathway, we conducted a sensitive search of homolog genes in the metagenomic and metatranscriptomic data sets. We found the genes and transcripts of three of the five steps of this metabolic route and they were present in very low abundance (Fig. 5a). The missing enzymes were those that catabolize oxygen-dependent reactions, such as the enzymes of the first step, indole amine 2,3-dioxygenase (IDO1) or tryptophan 2,3-dioxygenase, and 3-hydroxyanthranilate 3,4-dioxygenase. Thus, 3-hydroxyanthranilate was mainly transformed into 3-methoxyanthranilate by a methylation reaction catalyzed by a wide-range of bacterial or human methyltransferases (Fig. 5a). In addition, the species that were able to degrade tryptophan via the kynurenine pathway were a minor bacterial group that mainly belonged to the Proteobacteria phylum (Supplementary Table 4). More importantly, none of the species significantly correlated with the kynurenine/tryptophan ratio determined in the same set of samples (Supplementary Table 4).
Fig. 5.

Metabolism of dietary tryptophan and choline. Remote gene homolog detection from (a) metabolic routes of IDO1 tryptophan catabolism, (b) tryptophan fermentation into indole and (c) fermentation of choline and L-carnitine to TMA. Cyan shows enzymes and reaction flow arrows from one metabolite to the next in the metabolic pathway. Bar plots represent relative abundance of the gene and transcripts for each enzyme in each step from VU (red), IR (green), INR (orange), and HIV− (blue) subjects. Statistical tests between relative abundance of the four groups of the cohort was performed using the Kruskal–Wallis test. Pink shows the metabolite 3-hydroxyanthranilate. VU viremic untreated, IR immunological responder, INR immunological non-responder
The anaerobic gut environment favors non-oxidative degradation of tryptophan in indole and derivatives via bacterial tryptophanase. Due to the anti-inflammatory characteristics of these catabolites, we investigated the gene and transcript contents involved in tryptophan fermentation in HIV+ and HIV− microbiota (Fig. 5b). The expression level of the tryptophanase gene was higher, although not significant, in HIV− subjects than in HIV+ individuals (Supplementary Figure 6). The main bacterial species involved in tryptophan fermentation belonged to Prevotella, Acidaminococcus, and Clostridium genera (data not shown).
Trimethylamine (TMA) is a bacterial metabolite of choline fermentation via choline TMA-lyase that is converted in the liver to trimethylamine-N-oxide (TMAO). Both metabolites are related to cardiovascular disease and atherosclerosis in both the general population and HIV-infected subjects. Thus, we searched for the homolog gene of the cutC (Choline TMA Lyase) and cutD genes (Choline TMA−Lyase activating protein) in the metagenomes and metatranscriptomes of HIV− and HIV+ individuals (Fig. 5c). We did not find significant differences among the groups (Supplementary Figure 6), in congruence with the non-differential plasma TMAO concentrations described previously [5]. We found few homolog genes that catabolized the transformation of L-carnitine in TMA (Fig. 5c).
Ecological and functional networks of HIV-associated bacterial community
To study ecological and functional interactions in the HIV-associated bacterial community, we estimated the co-occurrence and metabolic networks in which nodes (species or metabolic functions) are pairwise-connected by lines (edges or links) (Fig. 6a and Supplementary Figure 7). The resulting networks met the properties of a biological network with respect to the connectivity distribution and small-world effect (Supplementary Table 5).
Fig. 6.

Co-occurrence ecologic network. a Co-occurrence network inferred from correlation matrix obtained using SparCC algorithm (two-sided pseudo P value ≤0.001 based on bootstrapping of 1000 repetitions <0.01 and a coefficient >0.1) with species present in at least 70% of samples. Nodes represent species belonging to Firmicutes (cyan), Bacteroides (orange), Actinobacteria (pink), and Proteobacteria (green) phyla. The size of the node was scaled to the logarithm of its degree of centrality, and only nodes belonging to quantile 95 were labeled. Edges represent positive correlations (blue) and negative correlations (red) for each pair of species. Colored polygons represent a cluster community (modules) defined by the walktrap community algorithm. b Barplot representation of phylum of each species for the 20 modules defined in the ecological network
The co-occurrence network presented a high degree of centrality (average of the numbers of edges per species) and high modularity coefficient (Supplementary Table 5). We found more negative interactions (57%) than co-occurrence relationships (43%). We determined 20 modules representing link-dense areas separated by regions of low connectivity and they contained at least three different species but the biggest module presented 34 species (module size). These clusters were dominated by Firmicutes and Bacteroidetes phyla and, to a lesser extent, by Actinobacteria and Proteobacteria. Interestingly, in the modules with members of Bifidobacteria, there are rarely species of Proteobacteria, indicating competence between these two phyla (Fig. 6b).
The betweenness centrality of a species, which measures its relevance in the community structure, allows identification of the highest connected bacterial taxa in the network, referred to as hub-species. Thus, Prevotella copri, an important HIV+ species biomarker, appeared to be an important hub of the HIV+ microbial community as well as other commensal bacteria (Supplementary Figure 8). Complementary to the betweenness centrality, the eigenvector centrality measures the importance of a node based on the connectivity degree of the node to which it was connected. As several bacteria are directly linked to the highly connected hub-species, we found that the eigenvector centrality coefficient revealed more hub-species than those observed with betweenness centrality, such as species related to SCFA production (Supplementary Figure 8). Moreover, we found a significant positive correlation between the hub-species and LDA score of the species biomarkers (Supplementary Figure 9). These results indicated that the bacterial species that characterized the HIV-associated microbiota (species biomarkers with a high LDA score) were the highest connected nodes and therefore responsible for the network structure.
We also applied a network approach to the bacterial metabolism of the HIV-associated microbiota (Supplementary Figure 7). In the enzymatic network, the nodes (3700 nodes) represented the enzymes and were connected by directed edges (31725 edges) if they catalyzed successive reactions (product/substrate). This network showed a high degree of compartmentalization with a high modularity coefficient and fragmentation index, as has been found in other metabolic network studies [22]. To assess the relevance of the enriched and depleted DNA- and RNA-KO biomarkers found for the HIV-associated microbiota, we mapped them in the metabolic network. We observed that their betweenness, degree, and eigenvector centrality indexes were higher than those of the rest of the enzymes in the network (Supplementary Figure 10). These results indicated the KO biomarkers were the centralcore of the metabolic network, revealing its relevance in the metabolism of the HIV-associated microbiota (Supplementary Figure 7).
HIV-associated dysbiosis and host health: butanoate Markov blanket
To study the overall effect of HIV-associated dysbiosis on HIV immunopathogenesis, we integrated the metagenomics, metatranscriptomics, metabolomics, and clinical variables data [5, 13, 14] for the HIV+ subjects in a Bayesian network (Supplementary Figure 11). The network contained 190 nodes and 548 links, being the clinical variables and to a less extent the DNA-kos, those that showed higher degree centrality (Supplementary Figure 12a and Supplementary Table 6). The butanoate pathway retrieved from metatranscriptomics (RNA-butanoate metabolism) also showed a high number of direct links to the clinical variables. Moreover, Prevotella species, such as P copri, P. sp. oral taxon 299, P. melaninogenica and P. salivae, were central nodes in the BN.In a BN, the Markov blankets (MB) of a node A contains the set of neighboring nodes, corresponding to its parents, the set of nodes that posses a direct edge to the node A; its children, the set of nodes from which the node A has a direct edge, and other nodes that posses a direct edge to the node A children’s nodes. All together predict the behavior of the node A. In order to identify the nodes that are related to a large number of variables, we estimated the MB of all the nodes in the BN. We found that the MB of the metabolic pathways obtained from metagenomics and/or metatranscriptomics have the higher size (number of nodes) (Supplementary Figure 12b), with a considerable number of links to the clinical variables (Supplementary Figure 12c). The MB of the RNA-butanoate metabolism (hereafter butanoate MB) included a greater number of nodes, namely, 89 nodes, 16 of which were clinical variables (Fig. 7). Butanoate MB possessed direct links, with systemic markers related to inflammation (hs-CRP), bacterial translocation (BPI), endothelial dysfunction (ADMA), and coagulation (D-dimers). We also detected significant positive correlations with metabolic pathways related to propanoate (ko00640) and fatty acid metabolism (ko00071) and a significant negative correlation with the Nadir CD4+ T cell counts. In the butanoate MB, P. copri appeared as a central node and was correlated positively with important HIV-associated pathways related to resistance to oxidative stress (ko00250 and ko00900) and amino acid metabolism (ko00400 and ko00473) while this bacteria correlated negatively with lipoic acid metabolism (ko00785), an antioxidant pathway. The CD4+ T cell counts showed high connectivity but most links were with different types of unknown metabolites. The bile salt muricholic acid, which was overrepresented in HIV-infected subjects, showed positive correlations with CD4+ T cell counts. We also found that the oleanane triterpene-related metabolite and various membrane-structural lipids showed significant negative correlations with the immune activation markers, especially the percentage of HLA-DR+CD38+CD4+ T cells and HLA-DR+CD38+CD8+ T cells. Interestingly, the IR group showed the highest abundance of such metabolites among HIV-infected individuals (Kruskal–Wallis P value = 0).
Fig. 7.

Butanoate Markov blanket. Subgraph of “multiomic” BN composed of metagenomic (blue nodes), metatranscriptomic (green nodes), metabolomic (pink circle nodes), and clinical variables (golden square nodes) from HIV+ subjects. Data (metagenomic and metatranscriptomic) include the relative abundance of the species (circles) and pathway (squares). Nodes are labeled in blue for variables overrepresented in HIV− subjects or red for variables that were overrepresented in HIV+ subjects. Arrows indicate conditional dependencies between variables. The Spearman correlation coefficient is represented by the arrow’s color: blue for a significant positive correlation (BH-adjusted P value <0.1), red for a significant negative correlation (BH-adjusted P value <0.1), and gray for a non-significant correlation
Discussion
In the present study, we integrated metagenomics, metatranscriptomics, and metabolomics from HIV-associated microbiota to gain a deep understanding of how the microbial community contributes to inflammation in HIV infection.
Combining metagenomics and metatranscriptomics, we observed that the metagenome was subjected to transcriptional regulation giving a more variable functional profile, in accordance with Franzosa et al. [15]. Thus, a considerable fraction of transcripts (49.08%) was differentially expressed in HIV-associated microbiota. However, HIV infection had an important impact on the gene expression profile, which was shaped by metabolic functions, allowing adaptation to an inflammatory environment. In recent studies, changes in the gene expression profiles of the gut microbial community in response to environmental conditions, such as xenobiotics or colitis, have also been described [17, 40]. Notably, HIV-associated microbiota have a significantly higher abundance of pathways and metabolites related to resistance to oxidative stress. For taxonomic composition, we found that most transcriptionally active bacteria belonged to the Firmicutes phylum, unlike the bacterial community structure described from metagenomic studies that was enriched in Gram-negative bacteria, such as Prevotella, Acidaminococcus (belonging to Firmicutes, but Gram-negative bacteria), Desulfovibrio, or Succinivibrio [1, 2, 4, 6, 9, 10, 13]. These results indicated that bacterial taxa such as Streptococcus, Leuconostoc, Anaerostipes, and Blautia, although not abundant, play an important role in metabolism. Moreover, the archeon Methanobrevibacter smithii emerged as a transcriptionally active member of the microbiota, in agreement with a previous study [15]. These species play an important role in the efficient degradation of polysaccharides by consuming the residual hydrogen derived from bacterial fermentation. Studies on xenobiotics metabolism in mice also indicated significantly higher expression from the Firmicutes phylum than from Bacteroidetes [41]. However, species of Prevotella, Acidaminococcus, Bacteroides, and Streptococcus were identified as distinctive members of HIV-associated microbiota in both metagenomics and metatranscriptomic data sets. In fact, Prevotella and Acidominococcus species presented a gene content that was involved in all of the pathways that characterized the dysbiotic metabolism of HIV-associated microbiota like those related to oxidative stress resistance. In addition, the genes involved in pathogenesis processes were more abundant, as seen in other inflammatory disease [42]. Moreover, enrichment of genes related to LPS biosynthesis was detected in metagenomic analysis, but no differential expression was found. As LPS constitutes the external part of the outer membrane of Gram-negative bacteria, gene expression of the LPS biosynthesis pathway would respond to bacterial overgrowth rather than to an inflammatory environment. However, LPS, as a microbe-associated molecular pattern, can be recognized by the Toll-like receptor 4 of various immune cells that promote an inflammatory response. Thus, inflammation induced by HIV infection could be enhanced by the high number of Gram-negative bacteria, such as Prevotella, Fermentimonas, Acidaminococcus, Megasphaera, Bibersteinia, or Pseudomonas, that are characteristic of the HIV-associated microbiota [1, 2, 4, 9, 10, 43–45]. Another important factor in the maintenance of systemic inflammation is the depletion of anti-inflammatory bacteria, such as Bacteroides, and decrease of species related to SCFA biosynthesis [5]. Strikingly, the metagenomic data set was found enriched in genes that were involved in butanoate and propanoate metabolism but were underexpressed in HIV-associated microbiota. Altogether, the metabolic profile of HIV-associated microbiota was characterized by overexpression of the pathways related to resistance to oxidative stress and underexpression of well-known anti-inflammatory SCFA biosynthesis pathways. Therefore, this bacterial community was well-adapted to gut inflammation caused by HIV infection, which is in turn maintained by a high abundance of Gram-negative bacteria composed of HIV-associated microbiota.
IDO1 involved in tryptophan catabolism through the kynurenine pathway is correlated with epithelial barrier disruption and bacterial translocation in HIV infection [46, 47]. Nevertheless, we found neither the IDO1 gene nor its expression in the bacterial metagenomic and metatranscriptomic data sets from HIV-infected individuals. However, Serrano-Villar et al. [13] recently described a statistical increase in the abundance of 3-hydroxyanthranilic acid in the gut metabolome of HIV patients. As human IDO1 is upregulated in HIV infection [46], metabolic complementation of the bacterial pathway could occur, leading to accumulation of 3-hydroxyanthranilic acid. Similar microbial-mammalian co-metabolism has been described for choline fermentation [48]. In anaerobiosis, the gut microbiota metabolizes tryptophan to indole and others derivatives, such as indole-3-acetic acid or indole-3-aldhehyde. These metabolites play a beneficial role in human health, activating the aryl hydrocarbon receptor (AHR) and hence increasing interleukin 22 secretion, which is involved in various anti-inflammatory processes. In HIV infection, impaired expression of the bacterial tryptophanase gene would have repercussions in tryptophan metabolism and tryptophan could be oxidized via the kynurenine pathway by HIV+ subjects. Lamas et al. [49] described a reduction in tryptophan metabolites in IBD patients that was associated with lower AHR activation and an increase in kynurenine level. Further studies are needed to address specific host–microbe complementation and the mechanisms involved.
The gut microbiome is a complex community whose members are highly connected to maintain its stability [19, 20, 22, 50, 51]. The HIV-associated ecological network has a high degree of connectivity and various discriminative biomarker species, mainly Prevotella copri, appear to be essential in the ecological network structure. The exclusion interaction between Bifidobacteria and Proteobacteria species could be due to environmental modifications caused by HIV infection. Likewise, the KO biomarkers associated with HIV infection, which have a high degree of centrality, were the core of the metabolic network of the HIV-associated community. Thus, HIV infection causes dramatic changes in the metabolic structure of the gut microbiota, losing and gaining important central metabolic enzymes. However, in other gut dysbiosis, the highly abundant enzymes tend to be located at the periphery of the metabolic network [22]. Finally, the multiomic Bayesian network showed interactions among bacteria, pathways, metabolites, and immune activation markers within the HIV-associated microbiome. The dissection of HIV-associated BN into MB revealed that metabolic pathways, specifically butanoate biosynthesis, were the most determinant elements to maintain the microbiome structure and interplay with the host. In the butanoate MB, we also detected associations between the important taxonomic biomarker, P. copri, and several pathways increased in HIV-infected participants, such as alanine aspartate and glutamate metabolism, which can confer resistance to oxidative stress. The microbial metabolome also interacted with various inflammation markers in the butanoate MB. In particular, the immune activation markers %CD8+HLA−DR+CD38+T cells and %CD4+HLA−DR+CD38+ T cells correlated negatively with several membrane-structural lipids and cholesterol glucuronide-related metabolites. However, deeper metabolomic analyses are needed to identify the metabolites that interact with the adaptive immune system.
This work is the first study on HIV-associated metabolism that combines metagenomics and metatranscriptomics. We detected transcriptionally active bacteria that overexpressed genes related to resistance to oxidative stress as a response to the inflamed environment and underexpressed the anti-inflammatory pathways. The network approaches identified bacterial taxa and microbial metabolic pathways that have a high impact on human health. These findings could guide the development of new therapies to improve clinical outcomes by tackling the microbiome.
Electronic supplementary material
Acknowledgements
This work was supported by grants to AM from the Spanish Ministry of Economy and Competitiveness (projects SAF 2012-31187, SAF2013-49788-EXP, and SAF2015-65878-R), the Carlos III Institute of Health (projects PIE14/00045 and AC15/00022), and the Generalitat Valenciana (project PrometeoII/2014/065) and was co-financed by FEDER. This work was supported by the Instituto de Salud Carlos III (Plan Estatal de IþDþi 2013–2016, project PI15/00345 and the Spanish AIDS Research Network (RD16/0025/0001 project) and co-financed by the European Development Regional Fund “A way to achieve Europe” (ERDF). JFV-C was supported by a CONACYT-SECITI fellowship, México. C.B.; DR and MF were funded by the Spanish Ministry of Economy and Competitiveness (CTQ2014-55279-R and BIO2014- 54494-R). SSV was supported by a grant from the Spanish Ministry of Science and Innovation (Contratos Juan Rodés, ECC/1051/2013). SM was funded by the Hospital Universitario Ramón y Cajal. VE was funded by the Hospital Clínico San Carlos. MJG was supported by a grant from the Fomento de la Investigación Sanitaria y Biomédica de la Comunidad Valenciana (FISABIO) (UGP-14-116).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Contributor Information
Andrés Moya, Phone: +34 96 1925951, Email: andres.moya@uv.es.
María José Gosalbes, Phone: +34 961925965, Email: maria.jose.gosalbes@uv.es.
Electronic supplementary material
The online version of this article (10.1038/s41396-018-0151-8) contains supplementary material, which is available to authorized users.
References
- 1.Ling Z, Jin C, Xie T, Cheng Y, Li L, Wu N, et al. Alterations in the fecal microbiota of patients with HIV-1 infection: An Observational Study in A Chinese Population. Nat Publ Gr. 2016;6:30673. doi: 10.1038/srep30673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lozupone Ca, Li M, Campbell TB, Flores SC, Linderman D, Gebert MJ, et al. Alterations in the gut microbiota associated with HIV-1 infection. Cell Host Microbe. 2013;14:329–39. doi: 10.1016/j.chom.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lozupone CA, Rhodes ME, Neff CP, Fontenot AP, Campbell TB, Palmer BE. HIV-induced alteration in gut microbiota. Gut Microbes. 2014;5:562–70. doi: 10.4161/gmic.32132. [DOI] [PubMed] [Google Scholar]
- 4.Lozupone CA, Rhodes ME, Neff CP, Fontenot AP, Campbell TB, Palmer BE. HIV-induced alteration in gut microbiota: driving factors, consequences, and effects of antiretroviral therapy. Gut Microbes. 2014;5:562–70. doi: 10.4161/gmic.32132. [DOI] [PubMed] [Google Scholar]
- 5.Serrano-Villar S, Vázquez-Castellanos JF, Vallejo A, Latorre A, Sainz T, Ferrando-Martínez S, et al. The effects of prebiotics on microbial dysbiosis, butyrate production and immunity in HIV-infected subjects. Mucosal Immunol. 2017;10:1279–93. doi: 10.1038/mi.2016.122. [DOI] [PubMed] [Google Scholar]
- 6.Vazquez-Castellanos J, Serrano-Villar S, Latorre A, Artacho A, Madrid N, Vera M, et al. Altered metabolism of gut microbiota contributes to chronic immune activation in HIV-infected individuals. Mucosal Immunol. 2015;8:760–72. doi: 10.1038/mi.2014.107. [DOI] [PubMed] [Google Scholar]
- 7.Vujkovic-Cvijin I, Dunham RM, Iwai MC, Maher SS, Albright RG, Broadhurst MJ, Hernandez RD, et al. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci Transl Med. 2013;5:193ra91. doi: 10.1126/scitranslmed.3006438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dillon S, Lee E, Kotter C, Austin G. An altered intestinal mucosal microbiome in HIV-1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal Immunol. 2014;7:983–94. doi: 10.1038/mi.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zilberman-Schapira G, Zmora N, Itav S, Bashiardes S, Elinav H, Elinav E. The gut microbiome in human immunodeficiency virus infection. BMC Med. 2016;14:4–11. doi: 10.1186/s12916-016-0625-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ellis C, Ma ZM, Mann S, Al. E. Molecular characterization of stool microbiota in HIV-infected subjects by panbacterial and order-level 16S ribosomal DNA (rDNA) quantification and correlations with immune activation. J Acquir Immune Defic Syndr. 2011;57:363–70. doi: 10.1097/QAI.0b013e31821a603c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monaco CL, Gootenberg DB, Zhao G, Handley SA, Ghebremichael MS, Lim ES, et al. Altered virome and bacterial microbiome in human immunodeficiency virus-associated acquired immunodeficiency syndrome. Cell Host Microbe. 2016;19:311–22. doi: 10.1016/j.chom.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dinh DM, Volpe GE, Duffalo C, Bhalchandra S, Tai AK, Kane AV, et al. Intestinal microbiota, microbial translocation, and systemic inflammation in chronic HIV infection. J Infect Dis. 2015;211:19–27. doi: 10.1093/infdis/jiu409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serrano-Villar S, Rojo D, Martinez-Martinez M, Deusch S, Vazquez-Castellanos JF, Bargiela R, et al. Gut bacteria metabolism impacts immune recovery in HIV-infected individuals. eBioMedicine. 2016;8:203–16. doi: 10.1016/j.ebiom.2016.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serrano-Villar S, Rojo D, Martinez M, Deusch S, Vazquez-Castellanos J, Bargiela R, et al. HIV infection results in metabolic alterations in the gut microbiota different from those induced by other diseases. Sci Rep. 2016;6:26192. doi: 10.1038/srep26192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franzosa EA, Morgan XC, Segata N, Waldron L, Reyes J, Earl AM, et al. Relating the metatranscriptome and metagenome of the human gut. PNAS. 2014;111:E2329–38. doi: 10.1073/pnas.1319284111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bikel S, Valdez-Lara A, Cornejo-Granados F, Rico K, Canizales-Quinteros S, Soberon X, et al. Combining metagenomics, metatranscriptomics and viromics to explore novel microbial interactions: towards a systems-level understanding of human microbiome. Comput Struct Biotechnol J. 2015;13:390–401. doi: 10.1016/j.csbj.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ilott NE, Bollrath J, Danne C, Schiering C, Shale M, Adelmann K, et al. Defining the microbial transcriptional response to colitis through integrated host and microbiome profiling. ISME J. 2016;10:2389–404. doi: 10.1038/ismej.2016.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–80. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faust K, Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol. 2012;10:538–50. doi: 10.1038/nrmicro2832. [DOI] [PubMed] [Google Scholar]
- 20.Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J, et al. Microbial co-occurrence relationships in the human microbiome. PLoS Comput Biol. 2012;8:e1002606. doi: 10.1371/journal.pcbi.1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, et al. Human genetics shape the gut microbiome. Cell. 2014;159:789–99. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greenblum S, Turnbaugh PJ, Borenstein E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. PNAS. 2011;109:594–9. doi: 10.1073/pnas.1116053109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kittelmann S, Seedorf H, Walters WA, Clemente JC, Knight R, Gordon JI, et al. Simultaneous amplicon sequencing to explore co-occurrence patterns of bacterial, archaeal and eukaryotic microorganisms in rumen microbial communities. PLoS ONE. 2013;8:e47879. doi: 10.1371/journal.pone.0047879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oakley BB, Morales CA, Line J, Berrang ME, Meinersmann RJ, Tillman GE, et al. The poultry-associated microbiome: network analysis and farm-to-fork characterizations. PLoS ONE. 2013;8:e57190. doi: 10.1371/journal.pone.0057190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517:205–8. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Widder S, Besemer K, Singer GA, Ceola S, Bertuzzo E, Quince C, et al. Fluvial network organization imprints on microbial co-occurrence networks. PNAS. 2014;111:12799–804. doi: 10.1073/pnas.1411723111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zimmerman AE, Martiny AC, Allison SD. Microdiversity of extracellular enzyme genes among sequenced prokaryotic genomes. ISME J. 2013;7:1187–99. doi: 10.1038/ismej.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eiler A, Heinrich F, Bertilsson S. Coherent dynamics and association networks among lake bacterioplankton taxa. ISME J. 2012;6:330–42. doi: 10.1038/ismej.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinforma. 2012;13:31. doi: 10.1186/1471-2105-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boisvert S, Raymond F, Godzaridis E, Laviolette F, Corbeil J. Ray meta: scalable de novo metagenome assembly and profiling. Genome Biol. 2012;13:R122. doi: 10.1186/gb-2012-13-12-r122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–12. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu W, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Research. 2010;38:e132. doi: 10.1093/nar/gkq275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hass B. TransDecoder (find coding regions within transcripts). http://transdecoder.github.io/. 2015.
- 34.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu S, Fang L, Xu X. Using SOAP aligner for short reads alignment. Curr Protoc Bioinforma. 2013;44:1–17. doi: 10.1002/0471250953.bi1111s44. [DOI] [PubMed] [Google Scholar]
- 36.Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. 2014;32:834–41. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 37.Nielsen HB, Almeida M, Juncker AS, Rasmussen S, Li J, Sunagawa S, et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotechnol. 2014;32:822–8. doi: 10.1038/nbt.2939. [DOI] [PubMed] [Google Scholar]
- 38.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Friedman J, Alm EJ. Inferring correlation networks from genomic survey data. PLoS Comput Biol. 2012;8:e1002687. doi: 10.1371/journal.pcbi.1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maurice CF, Haiser HJ, Turnbaugh PJ. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell. 2013;152:39–50. doi: 10.1016/j.cell.2012.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ursell LK, Knight R. Xenobiotics and the human gut microbiome: metatranscriptomics reveal the active players. Cell Metab. 2013;17:317–8. doi: 10.1016/j.cmet.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13:R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mutlu Ea, Keshavarzian A, Losurdo J, Swanson G, Siewe B, Forsyth C, et al. A compositional look at the human gastrointestinal microbiome and immune activation parameters in HIV infected subjects. PLoS Pathog. 2014;10:e1003829. doi: 10.1371/journal.ppat.1003829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. elife. 2013;2:e01202. doi: 10.7554/eLife.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–57. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Favre D. Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci Transl Med. 2010;2:32ra36. doi: 10.1126/scitranslmed.3000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sandler NG, Douek DC. Microbial translocation in HIV infection: causes, consequences and treatment opportunities. Nat Rev Microbiol. 2012;10:655–66. doi: 10.1038/nrmicro2848. [DOI] [PubMed] [Google Scholar]
- 48.Russell WR, Hoyles L, Flint HJ, Dumas ME. Colonic bacterial metabolites and human health. Curr Opin Microbiol. 2013;16:246–54. doi: 10.1016/j.mib.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 49.Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, Da Costa G, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med. 2016;22:598–605. doi: 10.1038/nm.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT, et al. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–14. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.The NIH HMP Working Group. The NIH Human Microbiome Project. Genome Res. 2009;19:2317–23. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.