

Abstract
Mutation rate variation has been under intense investigation for decades. Despite these efforts, little is known about the extent to which environmental stressors accelerate mutation rates and influence the genetic load of populations. Moreover, most studies on stressors have focused on unicellular organisms and point mutations rather than large-scale deletions and duplications (copy number variations [CNVs]). We estimated mutation rates in Daphnia pulex exposed to low levels of environmental stressors as well as the effect of selection on de novo mutations. We conducted a mutation accumulation (MA) experiment in which selection was minimized, coupled with an experiment in which a population was propagated under competitive conditions in a benign environment. After an average of 103 generations of MA propagation, we sequenced 60 genomes and found significantly accelerated rates of deletions and duplications in MA lines exposed to ecologically relevant concentrations of metals. Whereas control lines had gene deletion and duplication rates comparable to other multicellular eukaryotes (1.8 × 10−6 per gene per generation), the presence of nickel and copper increased these rates fourfold. The realized mutation rate under selection was reduced to 0.4× that of control MA lines, providing evidence that CNVs contribute to mutational load. Our CNV breakpoint analysis revealed that nonhomologous recombination associated with regions of DNA fragility is the primary source of CNVs, plausibly linking metal-induced DNA strand breaks with higher CNV rates. Our findings suggest that environmental stress, in particular multiple stressors, can have profound effects on large-scale mutation rates and mutational load of multicellular organisms.
Germline mutations provide the raw material for evolutionary change, but also the genetic variation associated with heritable diseases. Because spontaneous mutations are more often harmful or neutral than beneficial, the accumulation of mutations in the genome has important fitness consequences (Baer et al. 2007; Lynch 2010a). The frequency at which mutations are generated, as well as the environmental triggers and selective forces influencing mutation rates are therefore fundamental to biology. Accurately measuring the mutation rate, however, poses a considerable challenge due to the infrequent nature of mutations and the action of natural selection, which eliminates many deleterious mutations to bias the sample of observed mutations. Mutation accumulation (MA) experiments have been particularly effective for directly measuring mutation rates because repeated bottlenecks reduce the effect of selection, allowing all but the most deleterious mutations to accrue over multiple generations (Halligan and Keightley 2009). The comparison of MA experiments with a population experiencing selection can then be used to infer the fitness consequences of new mutations and their contribution to mutational load, ideally by using large populations started with organisms of the same genetic background to eliminate the impact of genotype on mutation rates (Baer et al. 2005; Ness et al. 2015). Studies that conduct whole-genome sequencing of MA lines have begun to evaluate the extent to which mutation rates vary across taxa and within species (Schrider et al. 2013; Ness et al. 2015), but few have compared these rates with a population under selection, let alone using the same genetic lineage for this comparison (Flynn et al. 2017). Furthermore, empirical evidence on the factors underlying mutation rate variation is limited relative to our theoretical understanding (Baer et al. 2007), including the contribution of different environmental conditions, and the long-term effects of highly mutagenic environments (Lynch 2016).
It is established that various exogenous and endogenous stresses induce both DNA breaks and somatic mutations. Stress affecting genome stability or DNA repair can contribute not only to point mutations but also to copy number variations (CNVs)—genetic deletions, duplications, and insertions—which can encompass genes and have relevant consequences in cancer and genetic diseases (Helleday et al. 2014; Sudmant et al. 2015; Carvalho and Lupski 2016). However, experimental fitness assays have provided indirect and contradictory findings concerning the effects of stress on the accumulation of germline mutations (Goho and Bell 2000; Joyner-Matos et al. 2011). Moreover, although stress-induced mutations in unicellular organisms have received some attention, very few genetic studies have directly investigated the heritable consequences of environmental stresses on the rate of mutations across generations in multicellular organisms (Sharp and Agrawal 2012; Bull et al. 2018). The mutational consequences and evolutionary responses to stress in unicellular organisms cannot necessarily be generalized to multicellular organisms because of several key differences, including the higher efficacy of natural selection and lower mutation rates generally seen in unicellular organisms and the occurrence of somatic mutations in multicellular organisms that can influence individual fitness without being inherited (Lynch 2010b). There is currently limited empirical data on the evolutionary impact of environmental stress on germline mutational spectra in multicellular organisms. In eukaryotes, genetic screens of tandem repeats in either germlines or in parents and their offspring have revealed elevated mutation rates upon exposure to air pollution, tobacco smoke, and metals (Somers et al. 2002; Rogstad et al. 2003; Marchetti et al. 2011). Similarly, higher frequencies of CNVs and INDELs were reported in offspring after parent irradiation (Adewoye et al. 2015). Even scarcer are genomic approaches that utilize MA experiments to assess the variation in mutation rates across environments after multiple generations. MA experiments have revealed that a stressful high temperature increased the rate of short tandem repeats in Caenorhabditis elegans measured after 100 generations (Matsuba et al. 2013), and Arabidopsis thaliana grown under salinity stress accumulated about twice as many short insertions and deletions (INDELs) than control lines after only 10 generations (Jiang et al. 2014). However, only one of these past studies surveyed CNVs, which have distinct mutational mechanisms (Lam et al. 2010) that could be more readily induced by stress. It remains unclear whether CNV rates over multiple generations differ across environments and whether they contribute to mutational load.
In this study, we directly estimate genome-wide mutation rates including point mutations, INDELs, and large-scale duplications and deletions under metal stressors in Daphnia. This is one of the first studies to estimate large-scale mutation rates under different environmental conditions and under contrasting selection regimes using a single genetic background. Our approach combines two long-term experiments seeded with the same ancestral Daphnia lineage: one MA experiment in which selection was minimized and one non-MA population under selection maintained for the entire duration of the MA experiment. This unique design allowed us to directly infer the selective effects on mutations. Additionally, we perform a sequence analysis at mutational breakpoints to inform on the potential source of large-scale mutations and the causes of rate variation across environmental conditions.
Results
Mutation accumulation after 100 generations
We sequenced 60 Daphnia pulex genomes including nine MA lines exposed to copper (Cu), 9 MA lines exposed to nickel (Ni), 9 MA lines exposed to a mixture of nickel and copper (NiCu), 24 MA lines maintained in controlled benign conditions (Con), and nine non-MA isolates randomly chosen from a population evolving under selection in benign conditions for the same duration as the MA experiment (Fig. 1). The consensus genotype of all MA lines was used to infer the genotype of their common ancestor and the mutations accumulated in each sample. Mutation filtering was calibrated to reduce false positives based on the validation of randomly selected variant calls using PCR and Sanger sequencing (Methods). After filtering, we detected a total of 916 de novo single-nucleotide mutations and small (1–50 bp) INDELs, as well as 776 deletions and 406 duplications larger than 500 bp (Fig. 2; Supplemental Tables S1, S2). Duplications typically doubled the locus copy number, whereas deletions typically had half the number of reads (Supplemental Fig. S1). Genomes with more deletions tended to have more duplications (Pearson's R = 0.57, P < 0.001), but the number and total length of CNVs per genome were not associated with overall depth of coverage (R2= 0.03, P = 0.09 and R2= 0.02, P = 0.12, respectively). Further, increasing the coverage of two randomly selected MA lines (C01 and C35) did not affect the detection of CNVs.
Figure 1.
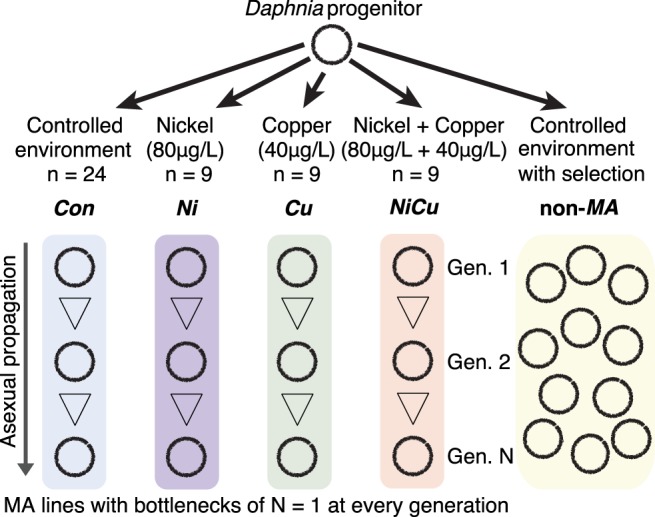
Experimental design. An obligate parthenogenetic Daphnia pulex progenitor was used to seed both a mutation accumulation (MA) experiment propagated in four different environments for an average of 103 generations as well as a non-MA population with selection and competition.
Figure 2.

Number of mutations per 100 Mbp per generation detected in each genome. Mutations include single-nucleotide mutations (SNMs), small (<50 bp) insertions and deletions (INDELs), and large-scale (>500 bp) duplications and deletions. The number of generations used for non-MA isolates was inferred from a life history experiment.
A total of 243 deletions and 130 duplications overlapped single-copy genes, giving rise to a total of 300 “gene CNVs,” including 180 gene deletions and 139 gene duplications (e.g., Fig. 3A,B). In addition, there were 177 “partial gene CNVs” that included 136 partially deleted genes and 50 partially duplicated genes (e.g., Fig. 3C,D; Supplemental Table S3). Many multicopy genes were found among CNVs, but these were excluded from our gene CNV analysis to limit biases from reads mapping to multiple genomic positions due to the highly duplicated nature of the reference genome (Colbourne et al. 2011; Keith et al. 2016).
Figure 3.

Relative read depth at four CNV loci. Read depth (log2 coverage) between genomes with a large-scale mutation (genomic deletions and duplications) and the average of all other MA lines is shown, in which a ratio above zero (blue) indicates the focal genome has more coverage than average, and a ratio below zero (red) indicates the focal genome has less coverage. (A) Duplication in the NiCu line C376 overlapping an uncharacterized single-copy gene (DAPPUDRAFT_325978). (B) Deletion in Con line C40 overlapping an uncharacterized single-copy gene (DAPPUDRAFT_114421) and several multiple-copy genes. (C) A duplication in C376 and (D) a deletion in C40 that lead to a partial gene CNV in the mre11 gene (DAPPUDRAFT_188962) and a neighboring uncharacterized gene (DAPPUDRAFT_227378).
Metal stress can increase large-scale mutation rates
A subset of MA lines in our experiment was exposed to metals (copper and nickel) that are prominent environmental stressors in aquatic habitats (Yan et al. 2016). Variation in the mutation rate can arise if cellular stressors due to metals perturb DNA replication, increase DNA damage, or alter DNA repair (Baer et al. 2007). The rates of single-nucleotide mutations and INDELs as well as transition/transversion ratios were similar between Con lines and the average of all metal-exposed lines (Fig. 4). The highest rate of single-nucleotide mutations was observed in lines exposed to nickel, but this was not significantly higher compared to Con lines. In contrast, two of the metal-exposed lines (Ni and NiCu) had significantly greater rates of CNVs (sum of duplications and deletions) than Con lines after Bonferroni correction (Fig. 5A); the average error and SEM of CNVs per genome per generation was 0.15 (SEM 0.09) for Con lines, whereas Ni lines had 1.4× higher rates with an average of 0.20 (SEM 0.06) per genome per generation (Mann–Whitney U test, P = 0.007), and NiCu lines experienced 3.0× higher rates with an average rate of 0.43 (SEM 0.16; Mann–Whitney U test, P = 0.002). Cu lines had an average of 0.19 CNVs per genome per generation, but this was not significantly higher than Con lines (Mann–Whitney U test, P = 0.069). Although deletion rates were significantly greater in each of the metal-exposed lines compared to Con lines after Bonferroni correction (all with Mann–Whitney U test, P < 0.01), only the metal mixture NiCu had significantly higher duplication rates (Mann–Whitney U test, P < 0.01). An important caveat is that there is high variance in the number of CNVs detected within treatments including several samples with zero CNVs; although the Mann–Whitney U test is a nonparametric test, it might fail in the presence of ties. Controlling for the number of sites analyzed, the overall rates of CNVs per called site per generation were 6.5 (SEM 4.1) × 10−10 for Con, 8.9 (SEM 2.8) × 10−10 for Ni, 8.2 (SEM 4.2) × 10−10 for Cu, and 19.1 (SEM 0.7) × 10−10 for NiCu. The elevated CNV rates observed in metal lines remained after accounting for sample size differences across treatments using random permutations (Supplemental Fig. S2). CNV rates were not correlated with generation time (Pearson's R < 0.001, P = 0.99).
Figure 4.
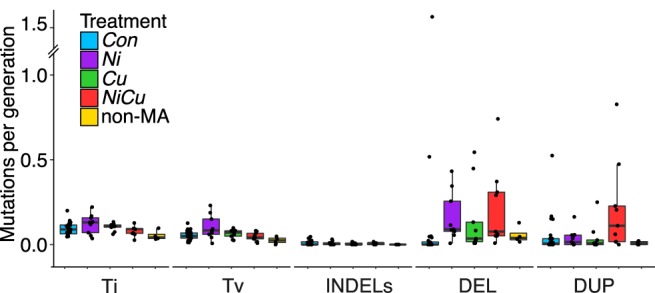
Number of mutations per 100 Mbp per generation across treatments and experiments. Box plots indicate mutation rates for transitions (Ti), transversions (Tv), small (<50 bp) insertions and deletions (INDELs), and large-scale (>500 bp) deletions (DEL) and duplications (DUP). Each sample is represented by a data point, and the number of generations used for non-MA isolates was inferred from a life history experiment.
Figure 5.

CNV rates and lengths, with and without selection. (A) Mean CNV rates (sum of duplications and deletions per nucleotide per generation) and standard errors among MA treatment groups showing significant differences among treatments: (*) P < 0.01; (**) P < 0.005. A comparison with non-MA isolates given various relative propagation rates compared to MA lines is shown, with a dotted line indicating the relative propagation rate in non-MA genomes to reach the same mutation rates as in control MA lines. We estimated the relative propagation rate based on a life history experiment (indicated with an arrow). (B) Box plot of CNV length distributions across treatments. (C) The percentage length of CNV regions that overlap genes, in which only a single non-MA isolate has genes deleted, and two more isolates had a gene duplication.
Extensive levels of gene deletions and duplications
To investigate whether the effect of metal stress also extends to functional regions of the genome, we evaluated the impact of CNVs on single-copy genes. As with overall CNV rates, the rate of gene deletions and gene duplications (per gene per generation) varied across samples and treatments (Table 1; Supplemental Table S1). The mean deletion and duplication rates overlapping genes (both partially and completely) in Con lines were 2.1 (SEM 1.4) × 10−6 and 0.9 (SEM 0.4) × 10−6, respectively. The combined rate amounts to 3.1 (SEM 1.7) × 10−6 CNVs per gene per generation. Ni lines had higher gene deletion (4.0 [SEM 2.1] × 10−6) and gene duplication (1.8 [SEM 1.4] × 10−6) rates, but these were not significant after Bonferroni correction. Similar results were found for Cu lines with a gene deletion rate of 2.7 (SEM 1.3) × 10−6 and a gene duplication rate of 1.1 (SEM 0.7) × 10−6. In contrast, NiCu lines had gene CNV rates four times higher than Con lines at 12.4 × 10−6 (Mann–Whitney U test, P < 0.001), with a gene deletion rate almost threefold higher at 6.4 (SEM 2.4) × 10−6 and a gene duplication rate more than sixfold higher at 6.0 (SEM 2.6) × 10−6. Even after taking the average of only the nine Con lines with the highest CNV rates, NiCu lines still had a higher mean.
Table 1.
Mean CNV, DEL, and DUP rates ± standard errors for the whole genome and for single-copy genes per generation for each treatment

CNVs do not always encompass entire gene sequences, giving rise to partial gene deletions and duplications as well as complete gene CNVs. After separating these two categories, we found that rates of partial gene CNVs were generally within one order of magnitude from the rates of complete gene CNVs (Supplemental Table S1), similar to what has been found in earlier studies of Drosophila melanogaster and Caenorhabditis elegans (Lipinski et al. 2011; Schrider et al. 2013). Overall, partial gene CNVs exhibited the same general patterns as complete gene CNVs, with significantly higher rates in NiCu lines (Mann–Whitney U test, P < 0.001) (Supplemental Fig. S3). When only considering complete gene CNVs, Con lines had an average rate of 1.8 (SEM 1.0) × 10−6. This was not statistically different compared to both Ni lines at 3.9 (SEM 2.0) × 10−6 and Cu lines at 2.5 (SEM 1.3) × 10−6. In contrast, the NiCu lines had rates three times higher than Con lines (7.8 × 10−6; Mann–Whitney U test, P = 0.023). These results indicate that chronic exposure to sublethal levels of a metal mixture can substantially increase the rate at which large-scale heritable mutations arise in genomes and affect genes.
CNV breakpoint analysis suggests a preponderance of error-prone double-strand break repair
Whole-genome sequencing enables nucleotide-resolution breakpoint analysis, which uses sequence information surrounding the start and end of CNVs to infer the mechanism of mutation formation such as nonallelic homologous recombination (NAHR) and nonhomologous recombination (NHR) (Lam et al. 2010; Cardoso-Moreira et al. 2012). Using this approach, we found that almost every duplication (92%) and deletion (96%) was associated with CNV formation mechanisms other than NAHR, suggesting a major role for NHR (Supplemental Table S2). NHR consists of error-prone pathways of DNA break repair such as nonhomologous end-joining (NHEJ), and its predominance in Daphnia is more pronounced than what has been found in humans (Lam et al. 2010), but similar to findings in Drosophila (Cardoso-Moreira et al. 2012; Zichner et al. 2013). Further breakpoint analyses were carried out for deletions with high confidence breakpoints (Methods), revealing that almost half (49%) of the NHR events displayed short regions of DNA sequence homology (microhomology stretches), which is more frequent than expected based on random permutations (P = 0.002) and is a characteristic feature of NHR (Lam et al. 2010). We found that NHR events tended to have high DNA flexibility, with significantly lower helix stability (Mann–Whitney U test, P = 0.018) and lower GC content (Mann–Whitney U test, P = 0.016) compared to other formation mechanisms (Supplemental Fig. S4). This is in line with previous findings in humans, suggesting that NHR mechanisms such as NHEJ are often associated with fragile genomic regions susceptible to double-strand breaks (Lam et al. 2010). Most CNVs overlapping in multiple MA lines have different breakpoints (50%–83%) suggesting independent recurrent CNVs that could represent deletion hotspots such as those previously reported in Daphnia (Xu et al. 2011). We found no significant differences in CNV formation mechanisms between Con lines and any of the metal treatments. The two MA lines with the highest rates of CNVs (Con-C40 and NiCu-C376) had a CNV overlapping the mre11 gene (Fig. 3; Supplemental Table S3), a key player in DNA damage response and double-strand break repair. The expression level of this gene has important consequences on the choice of DNA repair pathway and its efficiency (Rass et al. 2009). An unbalanced copy number of mre11 could alter expression of the gene and reduce DNA repair fidelity, thereby increasing genomic CNV rates over time. We do not know, however, when the mutations to mre11 occurred during the experiment.
Selection against CNVs in non-MA isolates
The elevated mutation rates detected among MA lines exposed to metals reflect heritable mutations that arise in nearly selection-free conditions, whereas purifying selection is expected to purge many new deleterious mutations. To evaluate the influence of selection on the rates and spectra of mutations in our experiment, we compared the MA lines with isolates from the population propagated under selection and seeded from the same original progenitor lineage. Given that Con MA lines and the non-MA population were propagated under identical environmental conditions, the underlying rate of mutation was expected to be the same while the amount of mutations accumulated was expected to differ due to selection. Over the same period of time as the MA experiment, the non-MA isolates accumulated 60% fewer mutations than Con lines, both in terms of small-scale mutations and CNVs (Supplemental Table S1). In contrast to MA lines that have each acquired mutations independently, there were three single-nucleotide mutations and two deletions detected in multiple non-MA isolates shared by descent. However, no mutations (point mutations or large-scale mutations) were shared in all isolates, suggesting that their last common ancestor is the original progenitor from the start of our experiment (Supplemental Fig. S5). Accounting for the shared mutations among lineages and their genealogy, the average number of accumulated single-nucleotide mutations and CNVs in the non-MA population was three times lower than the average of MA lines. To achieve the same rate of CNVs as in Con MA lines, the non-MA isolates would need to have still been at generation 26 by the time Con lines had already reached generation 100 (Fig. 5A). This very low propagation rate is however highly unlikely given that we found no difference between non-MA isolates and Con MA lines in either life span (mean of 46 d versus 43 d; t(14.6) = 1.69, P = 0.11) or age of first reproduction (mean of 11.5 d versus 11.5 d; t(14.7) = 0.18, P = 0.86). Based on mean life span and age of first reproduction measured in life history experiments (Bull et al. 2018), we calculated that the average non-MA isolate would have reached between 62 and 75 generations, with the slowest lineage at generation 30 (Supplemental Methods). Late reproduction was assigned more weight in calculating generation time, which would underestimate the number of generations if selection favored faster reproduction in the population. Nevertheless, an estimate of 62 generations still gives a mean realized rate of CNVs at least twice as low as MA lines (Table 1). Selection removing spontaneous CNVs and single-nucleotide mutations is likely responsible for the lower mutational incidence among non-MA isolates, as well as the lower variance across independent non-MA lineages compared to MA lines (Supplemental Table S1).
Given the uncertainty of the exact generation numbers in the non-MA population, characteristics of CNVs intersecting functional regions were also used to evaluate whether isolates experienced selection. We found that the proportion of CNVs that overlap single-copy genes is at least three times lower in non-MA compared to MA genomes, whereas the mean length of CNVs was not different (P > 0.05) (Fig. 5B,C). Furthermore, all but two gene CNVs in the non-MA experiment were found in a single isolate (CC9) within a 100-kb region deleting seven single-copy genes (Supplemental Fig. S6). Finally, there was a single partial CNV gene among non-MA genomes, whereas MA lines had rates of partial gene CNVs comparable to rates of complete gene CNVs (Supplemental Table S1; Supplemental Fig. S3). These findings reveal a very low realized mutation rate affecting genes and the apparent efficiency of selection under constant benign conditions, suggesting that the elevated CNV mutation rates induced by stress increase mutation burden.
Discussion
Comparable rates of gene deletion and duplication across organisms in benign conditions
The rates of gene deletion and duplication that we calculated using 24 MA lines of Daphnia pulex (3.1 × 10−6, or 1.8 × 10−6 for complete gene CNVs) are within an order of magnitude of those calculated using eight MA lines of Saccharomyces cerevisiae (5.5 × 10−6) (Lynch et al. 2008), 10 MA lines of Caenorhabditis elegans (3.4 × 10−7) (Lipinski et al. 2011), and eight MA lines of Drosophila melanogaster (1.1 × 10−6) (Schrider et al. 2013). Our rate is, however, much lower than recent single-copy gene estimates calculated from 20 MA lines of C. elegans (3.0 × 10−5) (Konrad et al. 2018) and from seven total MA lines derived from two different genetic backgrounds of D. pulex (5.4 × 10−5) (Keith et al. 2016). The different rates from our study and this latter Daphnia study, which implement similar approaches and the same CNV detection software, could be partially due to the different sample sizes analyzed because we found a negative association between the number of CNVs detected and the number of genomes included in the analysis (Supplemental Fig. S7). Using permutations to randomly sample the same number of genomes used in Keith et al. (2016), we reached a similar rate of 1.2 × 10−5 gene CNVs for single-copy genes among Con lines. However, our PCR validations confirmed the presence of false positives when analysis was performed on only a subset of our samples rather than our full data set (Supplemental Table S4). The lower mutation rate estimate based on the full data set is more similar to estimates from other model organisms and appears to be less susceptible to false positives in our data, although Keith et al. (2016) also had high validation rates for their data set. Another difference between the two studies that could contribute to the different rates is the depth of coverage, although we found that CNV detection was not correlated with sequencing depth and that average CNV rates were unaffected after doubling the coverage of two randomly chosen MA lines. Differences between studies might also simply reflect the prominent interindividual variation in mutation rates, (e.g., Konrad et al. 2018). Importantly, our study focuses on the relative mutation rates among treatments of a single genetic background, and the rate of CNVs under a metal mixture (NiCu) remains higher than Con lines regardless of the number of samples included in our analysis (either 5, 10, 20, 30, or 40 samples). Our study describes variation in genome-wide rates of CNVs under different environmental conditions, as well as under different selection regimes using the same genetic background.
Effects of nickel and copper on mutation rates
The extent of DNA damage induced by stressors is generally proportional to the exposure dose, whereas the mutational outcomes of this damage depend on DNA repair processes (Langie et al. 2015). Furthermore, multiple stressors such as metal mixtures can have different biological impacts than individual stressors, due to complex interactions (Altshuler et al. 2011; Langie et al. 2015). Our results illustrate that exposure to low but ecologically relevant levels of a metal mixture (nickel + copper) is associated with the highest CNV mutation rate in our experiment, with triple the rates of CNVs and quadruple the rates of gene CNVs compared to controls, whereas copper and nickel alone had a moderate to no measurable effect. Because the NiCu treatment had an overall greater concentration of metals (80 µg/L of nickel + 40 µg/L of copper) than either of the single metal treatments (either 80 µg/L of nickel or 40 µg/L of copper), the increase in CNV rates could be also attributable to a combination of exceeding a critical dose-response threshold and/or to the exposure to a metal mixture. However, the similar rates of small-scale mutations across treatments suggests that DNA replication error rates and/or base and excision repair pathways were comparatively unaffected by metal stress, at least in the germline. This is perhaps surprising given that cellular stress can induce somatic point mutations and metals can impair excision repair pathways (Langie et al. 2015). In plants, both small-scale and large-scale mutations show higher rates under environmental stress, but different stressors have different effects: Whereas high levels of salinity doubles the heritable rate of short INDELs and transversions (Jiang et al. 2014), various other abiotic stresses preferentially affect homologous recombination frequency compared to point mutations and microsatellite instability both in somatic cells and through transgenerational changes (Yao and Kovalchuk 2011), an effect plausibly contributing to stress-induced CNVs (DeBolt 2010). Despite the conserved DNA repair and recombination pathways across taxa as diverged as plants and humans, species-specific duplications or deletions of genes in these pathways could contribute to differences in the repair mechanisms used in response to DNA damage (Singh et al. 2010).
Genetic mechanisms underlying CNVs
The rate of mutations depends on a combination of factors including the amount of DNA damage and the efficacy of the DNA repair machinery, which can both vary under different genetic conditions and environments (Baer et al. 2007; Sharp and Agrawal 2016). DNA damage can be repaired using a multitude of alternative DNA damage response pathways, some of which are more error-prone than others. For example, the two main competing pathways for repairing DNA double-strand breaks are homologous recombination (HR) that uses a copy from a homologous template, and a more error-prone nonhomologous recombination (NHR) process called nonhomologous end-joining (NHEJ) that ligates the ends of broken DNA (Ciccia and Elledge 2010; Lam et al. 2010; Carvalho and Lupski 2016). A high propensity for DNA damage or for error-prone repair pathways can elevate mutation rates, but whether these are influenced by environmental stressors such as metals remains largely unknown.
Given the particularly high proportion of CNVs associated with NHR across all treatments, we hypothesize that the elevated CNV rate under exposure to a metal mixture is caused by an increase in double-strand breaks in the germline, leading to greater opportunities for recombination and DNA repair errors producing CNVs. This elevated mutation rate can increase mutational load and trigger deleterious phenotypic effects. Environmental stressors have previously been linked to increases in germline DNA strand breaks that are potentially caused by an increase in reactive oxygen species due to stress (Yauk et al. 2008). Additionally, metals can increase the incidence of sequence insertions at repaired double-strand break sites by NHEJ, proposed to be caused by interference with enzymatic processes of the proteins involved in NHEJ (Morales et al. 2016). This combination of an increase in both strand breaks and DNA repair errors could explain the higher rates of CNVs when exposed to nickel and copper, but this would need to be tested. Although HR also leads to repair errors (Rodgers and McVey 2016), organisms that primarily repair DNA via more error-prone mechanisms might be predisposed to greater CNV rate variation and amplified effects when faced with environmental stressors. We cannot rule out the possibility that nonallelic homologous recombination (NAHR) also contributes to differences in mutation rates because our study focused on genomic regions with single-copy genes, likely underestimating the full impact of NAHR, which is an important source of recurrent CNVs occurring in segmental duplications (Gu et al. 2008). Future mutation accumulation studies focusing on double-strand break repair under metal stress would help elucidate the underlying genetic mechanisms contributing to CNV rate variation.
An alternative explanation for an increase in mutation rates is that environmental stressors alter DNA repair fidelity. For example, different metals and exposure doses have been shown to differentially modulate the way cells repair double-strand breaks, alternating between HR and the more error-prone NHEJ, two competing repair pathways (Morales et al. 2016). Stressful conditions in general can cause a shift to error-prone double-strand break repair (Ponder et al. 2005), and lower physiological condition has been shown to lead to more mutations via changes in DNA repair pathways with different fidelity (Wang and Agrawal 2012; Sharp and Agrawal 2016). Contrary to these previous findings, we did not observe differences in the mechanism of CNV formation among treatments, potentially because CNVs under benign conditions are already associated with error-prone pathways. Instead, our results suggest that a mixture of nickel and copper induces more frequent germline DNA strand breakage in Daphnia. This finding has important evolutionary consequences particularly in taxa that propagate asexually (either cyclically or obligately) like Daphnia. Previous studies conducted on Daphnia propagated asexually under benign conditions documented high rates of loss of heterozygosity (due to gene conversion, deletion, and ameiotic recombination) that can contribute to decreasing overall fitness (Omilian et al. 2006; Xu et al. 2011; Keith et al. 2016; Flynn et al. 2017). Studying mutation rates in other organisms with different underlying genome architecture, propensity for mechanisms of DNA repair, and general levels of DNA repair fidelity would further illuminate the extent to which these genomic attributes either promote or dampen the mutagenic effects of metals in germlines.
Methods
Daphnia mutation accumulation experiment
To assess mutation rate variation under different environmental conditions, we conducted a mutation accumulation (MA) experiment using a total of 51 independent lines of Daphnia pulex over an average of 103 generations (Fig. 1). Twenty-four replicate MA lines were propagated in benign soft-water media as described in Flynn et al. (2017), herein labeled as MA controls (Con). In addition, nine nickel-exposed MA lines (Ni) were maintained in 80 µg/L of nickel, nine copper-exposed MA lines (Cu) were maintained in 40 µg/L of copper, and nine MA lines were maintained in a mixture of nickel and copper (NiCu; 80 µg/L of nickel + 40 µg/L of copper). These sublethal concentrations of metals did not elicit a measurable difference in mortality, average brood size, or time to first clutch, and are comparable to Daphnia habitats that experienced decades of contamination of copper and nickel in the Sudbury, Canada, area (Yan et al. 2016). Each MA line was propagated using single progeny descendants every generation, and all were seeded with a single Daphnia obligate parthenogenetic progenitor. The ancestral progenitor for all MA lines was collected from Canard Pond (Latitude 42°12′′, Longitude −82°98′′) in Windsor, Ontario, Canada. All MA lines were maintained at 18°C with a humidity of 70% and a 12-h light/dark cycle. MA lines were fed ad libitum with a mixture of algae (Ankistrodesmus sp., Scenedesmus sp., and Pseudokirchneriella sp). Backups for MA lines were maintained in case of mortality or sterility of the focal individual, and were used in ∼6% of transfers with an average of once every 16 generations per line. Although this introduces some level of selection against lethal and sterility-causing mutations that could lead to underestimating mutation rates, the frequency of backup lines used across treatments was not significantly different. All 24 Con samples were used in a previous study (Flynn et al. 2017).
Daphnia population under selection
To evaluate the effect of selection on mutation rates, a large non-MA population seeded from the same ancestral progenitor as the MA lines was allowed to propagate without induced population bottlenecks for the duration of the MA experiment. Thus, whereas the MA lines experienced minimal selection, the non-MA population evolved with selection. The non-MA population was maintained in a 15-L tank under the same conditions as the Con MA lines with identical media, temperature, and lighting conditions. Feeding was performed twice a week using the same mixture of algae as the MA lines. Six isolates were randomly chosen for sequencing when the Con MA lines had reached an average of 101 generations (1368 d of propagation) and were also used in a previous study (Flynn et al. 2017). Three additional isolates were sequenced after an average of 136 generations (1642 d of propagation) (Supplemental Methods). The census size of the population at the earlier time point was estimated to be between 100 and 250 (Flynn et al. 2017). Although natural population size fluctuations probably occurred, the lack of fixed mutations (i.e., shared across all non-MA isolates versus the ancestor) and the few shared mutations observed provides little evidence for severe population bottlenecks (Supplemental Fig. S5). Future studies involving highly replicated non-MA populations would be needed to assess the extent of stochastic allele frequency variation among different populations.
Sample processing and sequencing
Tissue collection, library preparation, and sequencing followed the approach described in Flynn et al. (2017). Tissue was collected from three to five clonal individuals per line raised in a sterile medium. During 48 h prior to isolating DNA, animals were fed sterile Sephadex beads 10 times a day to eliminate food content from the gut, while being treated with antibiotics to reduce microbial contamination. DNA was extracted following the cetyltrimethylammonium bromide method (Doyle and Doyle 1987). DNA samples were quantified with PicoGreen Quant-iT and were diluted to 2.5 ng/µL. We adopted a library preparation protocol derived from the standard Illumina Nextera approach that was optimized to reduce the use of reagents (Baym et al. 2015). Samples were dual indexed (one index at the 3′ end and another index at the 5′ end) such that each sample had a unique index combination per sequencing lane. Libraries were cleaned and short products removed with Beckman Coulter AMPure XP beads. Libraries were then normalized, pooled into three groups, and run on a total of five lanes of Illumina HiSeq 100 bp and 150 bp paired-end reads at Genome Quebec. Adapter sequences were removed, and overlapping sequences merged from FASTQ files using SeqPrep (https://github.com/jstjohn/SeqPrep). For each of the sequencing lanes, reads were mapped against the Daphnia pulex reference genome (Colbourne et al. 2011) using the short read alignment tool BWA v0.7.10 (Li and Durbin 2009). After alignment, reads were cleaned and sorted, and duplicates were removed with Picard tools v1.123 (https://broadinstitute.github.io/picard). Resulting BAM files were used for estimating depth of coverage, achieving an average of 13× coverage. Two randomly selected MA lines (C01 and C35) were intentionally sequenced to a higher depth to test the effect of doubling the sequence coverage on mutation rates. All analyses were carried out twice, once before the increase in coverage of the two samples, and once after the increase in coverage. This increase in coverage did not affect the recovery of CNVs, nor the estimated mutation rate of single nucleotides (Flynn et al. 2017).
Small-scale variant calling
Single-nucleotide mutations and INDELs were called using GATK v.3.3.0 (McKenna et al. 2010), first using HaplotypeCaller to assign putative genotypes for each individual separately, followed by GenotypeGVCFs to refine variant calling over all samples simultaneously. Variants were filtered using GATK based on various quality and alignment metrics including variant quality, mapping quality, and strand bias (QD < 2, QUAL < 50, FS > 60, MQ < 40, MQRankSum < −12.5, ReadPosRankSum < −8 for single-nucleotide mutations, and QD < 2, QUAL < 50, FS > 200, ReadPosRankSum < −20 for INDELs). To further prevent false positive variant calls from the sequencing data, we excluded nonnuclear sites, repeat masked regions, sites without read coverage from each sample, and regions with overall depth lower than expected (average 6×) or greater than twice the expected coverage (average 26×). These filtering steps were informed by both follow-up inspection of mapped reads in a genome browser and Sanger sequencing of single-nucleotide mutations and INDELs called at various filtering stages and with different read depths as described in Flynn et al. (2017). We retained ∼25% of the reference genome as callable sites for identifying single-nucleotide mutations and INDELs. As expected, all MA lines had unique mutation profiles despite allowing shared mutations among lines. We did not identify shared single-nucleotide mutations across MA lines or any signature of potential contamination across lines propagated in isolation. The raw sequence data can be found in SRA (PRJNA341529).
Large deletions and duplications
Four different CNV detection programs were initially run for determining putative deletions and duplications utilizing read depth, split-read and/or paired-end approaches. Read depth analysis was performed using CNVnator v0.3 (Abyzov et al. 2011) with a bin size of 500 bp to uncover putative deletions and duplications for each sample compared to the reference genome. Another read depth approach called CNV-seq v0.2-8 (Xie and Tammi 2009) was used that compares pairwise samples. CNV-seq was run using a sliding 250 bp window on every pairwise comparison between MA lines (i.e., all pairwise combinations among the 51 Con, Ni, Cu, and NiCu samples), and between each non-MA isolate and every MA line (but not non-MA isolates with one another since they can share CNVs by descent). CNVs were called if four consecutive windows had a log2 depth of coverage difference above 0.44 or below 0.6, which requires a coverage ratio >1.36 or <0.66, respectively. CNVs detected in every pairwise comparison were identified for each sample, followed by the merging of CNVs within 10 kb of one another to represent a single CNV, to overcome the majority of assembly gaps and repetitive regions (Keith et al. 2016). Paired-end read mapping and soft-clipped split-reads were also used to infer structural variants using SoftSV v1.4 (Bartenhagen and Dugas 2016). Because paired reads of short fragments overlap one another, inhibiting the ability to detect CNVs, SoftSV was also analyzed after trimming all reads to 50 bp. Trimming the ends of paired reads can theoretically permit independent mapping of each paired read by removing overlapping sequences, thereby improving chances of detecting CNVs. In addition to the three tools mentioned above, we used a simple in-house read depth approach to estimate CNVs among genes in individual lines as follows. For each gene from each sample, read depth was standardized by the total read depth of the respective sample to enable comparisons across lines, and read depth was centered to two to approximate diploid copy numbers. We compared all MA lines with one another and with non-MA isolates using the deviation of normalized read depth among samples to identify candidate gene duplications and gene deletions. At a diploid locus, we would expect mutants with a deletion to have at least half as much coverage as nonmutant lines, and mutants with a duplication to have at least twice as much coverage as nonmutant lines. Due to variability in read depth coverage, we used slightly less stringent thresholds while still requiring mutants to be outliers based on 1.5× interquartile ranges. Genes were considered as deleted if the MA line with the lowest standardized read depth was less than 0.66× compared to all other MA lines, while being an outlier with at least 0.5 fewer absolute copies. Genes were considered as duplicated if the MA line with the highest standardized read depth was more than 1.4× compared to all other MA lines, while being an outlier with at least 0.5 more absolute copies. Based on the overlaps of CNVs detected from all four methods, CNV-seq had an overwhelmingly higher proportion of gene CNVs overlapping our read depth method (up to 10-fold more than both CNVnator and SoftSV) and also shared the highest proportion of pairwise concordant CNVs among the three implemented tools. Combined with the fact that CNV-seq was also used in a recent analysis among other Daphnia MA lines and had high validation rates (Keith et al. 2016), we decided to solely rely on the results of CNV-seq. To evaluate the effects of sample size on CNV detection, we repeated our CNV analyses and rate estimates using random sampling of 5, 10, 20, 30, and 40 genomes. Whereas absolute rate estimates differed, the relative rates between treatments were not affected.
To identify CNVs shared by descent as well as mutation hotspots, we allowed overlapping (shared) CNVs among samples (e.g., two samples with deletions versus all other samples but not between one another). However, we did not allow shared mutations to occur in >50% of lines, which could be due to differences between the ancestor and the reference genome. Overlapping CNVs in non-MA isolates were interpreted as shared by descent, and shared CNVs among MA lines were considered as recurrent CNVs (potentially hotspots). CNVs with an average depth of coverage below 6× were removed. Protein-coding genes that intersected with remaining duplications or deletions (with a minimum 5% of their length) were considered as putative gene CNVs (>95% length overlap were considered as “complete” gene CNVs as opposed to partial gene CNVs).
Mutation validations
Sanger sequencing of randomly selected single-nucleotide mutations and INDELs confirmed 21 out of 25 mutations as described in Flynn et al. (2017). Long-range PCR amplification of CNVs was performed to validate the presence or absence of large-scale mutations in the putative mutant sample and two other independent MA lines. Primer pairs were designed based on the ancestral progenitor's sequence around inferred breakpoints from randomly selected CNV loci, in addition to one CNV overlapping the mre11 gene, two CNVs found in multiple samples, and five CNVs that were excluded after filtering (Supplemental Table S4). Our PCR approach successfully verified 12 out of 14 CNV tests, and confirmed all (four out of four) putative CNVs that were called with fewer samples (but not detected after increasing the number of sample comparisons) were false positives.
CNV rate calculation
Duplications and deletions were evaluated using only the scaffolds that contained one of the 10,673 “single-copy” protein-coding genes in Daphnia to reduce the impact of mismapping against the highly duplicated reference genome (Colbourne et al. 2011; Keith et al. 2016). Single-copy genes were determined as genes without any duplicates in the Daphnia reference genome by identifying paralogs using EnsemblMetazoa v30. The number of sites kept for analysis and used to calculate mutation rates was 113,196,346 bp (57% of the reference genome), with 8699 single-copy protein-coding genes found on 1313 scaffolds. CNVs that had an average coverage below 6× across all samples were removed. The duplication and deletion rates per genome were estimated using the formula μ = n/T, where n equals the number of duplication or deletion events and T is the number of generations that a sample was propagated. Because all samples are compared over the same genomic regions, these rates can be used to compare treatments. CNV rates per genome per nucleotide were compared across studies and were calculated using μ = n/(2 × L × T), in which L is the total number of loci (nucleotides) analyzed. For mutation rates of gene duplications and deletions, n was the number of gene CNVs and L was the number of single-copy protein-coding genes analyzed as mentioned above.
The nine non-MA isolates were sampled at two time points: six when MA Con lines reached an average of 101 generations (1368 d), and three more when MA Con lines reached an average of 136 generations (1642 d). Due to potentially overlapping generations, the non-MA population likely achieved lower mean generations than MA lines. To estimate the average number of generations, 10 non-MA isolates and 10 Con MA lines with seven replicate offspring from each focal mother were used in a life history experiment (Bull et al. 2018; Supplemental Methods). The average generation time of the population was estimated based on the mean age at first reproduction and longevity, and weighted by average clutch sizes. To compare realized rates of CNVs in the non-MA population that has likely faced greater selective pressures than the MA lines, we calculated the realized mutation rates taking into account the genealogy and shared mutations among lineages (Supplemental Methods). Moreover, we used a range of generations in the denominator to represent propagation rates up to five times slower relative to MA lines (i.e., relative propagation rates from 0.2 to 1), encompassing the average and lower-bound generation estimates.
Breakpoint detection and CNV formation mechanisms
Breakpoint analysis of CNVs was performed to infer the molecular mechanism of deletion and duplication formation separately. These approaches compare the nucleotide sequences surrounding the ends (breakpoints) of CNVs with expected genomic signatures of different CNV formation mechanisms, including the identification of sequence identity across breakpoint junctions (Lam et al. 2010). For each deletion, reads that mapped around putative breakpoint boundaries were assembled using TIGRA-SV v0.4 (Chen et al. 2014). Assembled contigs were then realigned to the reference to define breakpoints using AGE v0.4 (Abyzov and Gerstein 2011). Filtering was performed to assign breakpoints with high confidence based on the comparison between the sequence alignment and the predicted deletion region; contig alignments were required to have at least 95% sequence identity including flanking regions, as well being within 4 kb of the estimated breakpoint ranges from CNV-seq and overlapping at least 50% of the estimated range. When multiple alternative breakpoints were found, we selected the ones closest to the estimated range. Breakpoints were estimated for each CNV, and deletion formation mechanisms (such as NHR) were inferred for 43% of deletions using BreakSeq v1.3 (Lam et al. 2010) as well as the DNA flexibility, DNA helix stability, and GC content at breakpoints. For duplications, breakpoints were examined for the presence of sequence homology to evaluate the potential contribution of homology-based mechanisms such as NAHR. Regions surrounding each duplication breakpoint (spanning 25% of the CNV length at each end plus 500 bp outside the CNV) were compared to identify homologous blocks with a minimum of 85% sequence identity and a minimum length of 50 bp, the same thresholds as those used in BreakSeq for NAHR classifications of deletions.
Data access
Raw sequencing data from this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA341529. Sanger trace files from this study have been submitted to the NCBI Trace Archive (https://trace.ncbi.nlm.nih.gov/Traces/trace.cgi) under trace identifier numbers 2344447466–2344447601.
Supplementary Material
Acknowledgments
We thank all the students who contributed to maintaining the MA lines over the past five years. We thank T. Crease, D. Denver, B. Fryer, D. Haffner, R. Vergilino, K. Millette, N. Yan, J. Witt, G. Zhang, and M. Dutton for helpful suggestions during the project. This project was supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) CREATE training program on Aquatic Ecosystem Health (397997-2011), an NSERC Discovery Grant (341423-2012), and a Canada Research Chair (237170) to M.E.C. J.M.F. was supported by an NSERC Undergraduate Summer Research Award.
Author contributions: F.J.J.C. and M.E.C. conceived and designed the project. J.M.F. and J.K.B. prepared the samples for sequencing. J.M.F. and F.J.J.C. designed the analytical approach and performed the PCR validations. J.K.B. performed the life history experiment. F.J.J.C. analyzed the data and wrote the paper. All authors contributed to and approved the final manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.234724.118.
References
- Abyzov A, Gerstein M. 2011. AGE: defining breakpoints of genomic structural variants at single-nucleotide resolution, through optimal alignments with gap excision. Bioinformatics 27: 595–603. 10.1093/bioinformatics/btq713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abyzov A, Urban AE, Snyder M, Gerstein M. 2011. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21: 974–984. 10.1101/gr.114876.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adewoye AB, Lindsay SJ, Dubrova YE, Hurles ME. 2015. The genome-wide effects of ionizing radiation on mutation induction in the mammalian germline. Nat Commun 6: 6684 10.1038/ncomms7684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler I, Demiri B, Xu S, Constantin A, Yan ND, Cristescu ME. 2011. An integrated multi-disciplinary approach for studying multiple stressors in freshwater ecosystems: Daphnia as a model organism. Integr Comp Biol 51: 623–633. 10.1093/icb/icr103 [DOI] [PubMed] [Google Scholar]
- Baer CF, Shaw F, Steding C, Baumgartner M, Hawkins A, Houppert A, Mason N, Reed M, Simonelic K, Woodard W, et al. 2005. Comparative evolutionary genetics of spontaneous mutations affecting fitness in rhabditid nematodes. Proc Natl Acad Sci 102: 5785–5790. 10.1073/pnas.0406056102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer CF, Miyamoto MM, Denver DR. 2007. Mutation rate variation in multicellular eukaryotes: causes and consequences. Nat Rev Genet 8: 619–631. 10.1038/nrg2158 [DOI] [PubMed] [Google Scholar]
- Bartenhagen C, Dugas M. 2016. Robust and exact structural variation detection with paired-end and soft-clipped alignments: SoftSV compared with eight algorithms. Brief Bioinform 17: 51–62. 10.1093/bib/bbv028 [DOI] [PubMed] [Google Scholar]
- Baym M, Kryazhimskiy S, Lieberman TD, Chung H, Desai MM, Kishony R. 2015. Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS One 10: e0128036 10.1371/journal.pone.0128036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull JK, Flynn JM, Chain FJJ, Cristescu ME. 2018. Fitness and genomic consequences of chronic exposure to low levels of copper and nickel in Daphnia pulex mutation accumulation lines. G3 (Bethesda) 10.1534/g3.118.200797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso-Moreira M, Arguello JR, Clark AG. 2012. Mutation spectrum of Drosophila CNVs revealed by breakpoint sequencing. Genome Biol 13: R119 10.1186/gb-2012-13-12-r119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Lupski JR. 2016. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet 17: 224–238. 10.1038/nrg.2015.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Chen L, Fan X, Wallis J, Ding L, Weinstock G. 2014. TIGRA: a targeted iterative graph routing assembler for breakpoint assembly. Genome Res 24: 310–317. 10.1101/gr.162883.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204. 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, Oakley TH, Tokishita S, Aerts A, Arnold GJ, Basu MK, et al. 2011. The ecoresponsive genome of Daphnia pulex. Science 331: 555–561. 10.1126/science.1197761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBolt S. 2010. Copy number variation shapes genome diversity in Arabidopsis over immediate family generational scales. Genome Biol Evol 2: 441–453. 10.1093/gbe/evq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19: 11–15. [Google Scholar]
- Flynn JM, Chain FJJ, Schoen DJ, Cristescu ME. 2017. Spontaneous mutation accumulation in Daphnia pulex in selection-free vs. competitive environments. Mol Biol Evol 34: 160–173. 10.1093/molbev/msw234 [DOI] [PubMed] [Google Scholar]
- Goho S, Bell G. 2000. Mild environmental stress elicits mutations affecting fitness in Chlamydomonas. Proc R Soc B 267: 123–129. 10.1098/rspb.2000.0976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Zhang F, Lupski JR. 2008. Mechanisms for human genomic rearrangements. Pathogenetics 1: 4 10.1186/1755-8417-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halligan DL, Keightley PD. 2009. Spontaneous mutation accumulation studies in evolutionary genetics. Annu Rev Ecol Evol Syst 40: 151–172. 10.1146/annurev.ecolsys.39.110707.173437 [DOI] [Google Scholar]
- Helleday T, Eshtad S, Nik-Zainal S. 2014. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet 15: 585–598. 10.1038/nrg3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Mithani A, Belfield EJ, Mott R, Hurst LD, Harberd NP. 2014. Environmentally responsive genome-wide accumulation of de novo Arabidopsis thaliana mutations and epimutations. Genome Res 24: 1821–1829. 10.1101/gr.177659.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner-Matos J, Bean LC, Richardson HL, Sammeli T, Baer CF. 2011. No evidence of elevated germline mutation accumulation under oxidative stress in Caenorhabditis elegans. Genetics 189: 1439–1447. 10.1534/genetics.111.133660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith N, Tucker AE, Jackson CE, Sung W, Lucas Lledó JI, Schrider DR, Schaack S, Dudycha JL, Ackerman M, Younge AJ, et al. 2016. High mutational rates of large-scale duplication and deletion in Daphnia pulex. Genome Res 26: 60–69. 10.1101/gr.191338.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konrad A, Flibotte S, Taylor J, Waterston RH, Moerman DG, Bergthorsson U, Katju V. 2018. Mutational and transcriptional landscape of spontaneous gene duplications and deletions in Caenorhabditis elegans. Proc Natl Acad Sci 115: 7386–7391. 10.1073/pnas.1801930115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam HY, Mu XJ, Stütz AM, Tanzer A, Cayting PD, Snyder M, Kim PM, Korbel JO, Gerstein MB. 2010. Nucleotide-resolution analysis of structural variants using BreakSeq and a breakpoint library. Nat Biotechnol 28: 47–55. 10.1038/nbt.1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langie SA, Koppen G, Desaulniers D, Al-Mulla F, Al-Temaimi R, Amedei A, Azqueta A, Bisson WH, Brown DG, Brunborg G, et al. 2015. Causes of genome instability: the effect of low dose chemical exposures in modern society. Carcinogenesis 36(Suppl 1): S61–S88. 10.1093/carcin/bgv031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski KJ, Farslow JC, Fitzpatrick KA, Lynch M, Katju V, Bergthorsson U. 2011. High spontaneous rate of gene duplication in Caenorhabditis elegans. Curr Biol 21: 306–310. 10.1016/j.cub.2011.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2010a. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci 107: 961–968. 10.1073/pnas.0912629107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2010b. Evolution of the mutation rate. Trends Genet 26: 345–352. 10.1016/j.tig.2010.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2016. Mutation and human exceptionalism: our future genetic load. Genetics 202: 869–875. 10.1534/genetics.115.180471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Sung W, Morris K, Coffey N, Landry CR, Dopman EB, Dickinson WJ, Okamoto K, Kulkarni S, Hartl DL, et al. 2008. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci 105: 9272–9277. 10.1073/pnas.0803466105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti F, Rowan-Carroll A, Williams A, Polyzos A, Berndt-Weis ML, Yauk CL. 2011. Sidestream tobacco smoke is a male germ cell mutagen. Proc Natl Acad Sci 108: 12811–12814. 10.1073/pnas.1106896108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuba C, Ostrow DG, Salomon MP, Tolani A, Baer CF. 2013. Temperature, stress and spontaneous mutation in Caenorhabditis briggsae and Caenorhabditis elegans. Biol Lett 9: 20120334 10.1098/rsbl.2012.0334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales ME, Derbes RS, Ade CM, Ortego JC, Stark J, Deininger PL, Roy-Engel AM. 2016. Heavy metal exposure influences double strand break DNA repair outcomes. PLoS One 11: e0151367 10.1371/journal.pone.0151367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness RW, Morgan AD, Vasanthakrishnan RB, Colegrave N, Keightley PD. 2015. Extensive de novo mutation rate variation between individuals and across the genome of Chlamydomonas reinhardtii. Genome Res 25: 1739–1749. 10.1101/gr.191494.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omilian AR, Cristescu ME, Dudycha JL, Lynch M. 2006. Ameiotic recombination in asexual lineages of Daphnia. Proc Natl Acad Sci 103: 18638–18643. 10.1073/pnas.0606435103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponder RG, Fonville NC, Rosenberg SM. 2005. A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol Cell 19: 791–804. 10.1016/j.molcel.2005.07.025 [DOI] [PubMed] [Google Scholar]
- Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS. 2009. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol 16: 819–824. 10.1038/nsmb.1641 [DOI] [PubMed] [Google Scholar]
- Rodgers K, McVey M. 2016. Error-prone repair of DNA double-strand breaks. J Cell Physiol 231: 15–24. 10.1002/jcp.25053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogstad SH, Keane B, Collier MH. 2003. Minisatellite DNA mutation rate in dandelions increases with leaf-tissue concentrations of Cr, Fe, Mn, and Ni. Environ Toxicol Chem 22: 2093–2099. 10.1002/etc.5620220919 [DOI] [PubMed] [Google Scholar]
- Schrider DR, Houle D, Lynch M, Hahn MW. 2013. Rates and genomic consequences of spontaneous mutational events in Drosophila melanogaster. Genetics 194: 937–954. 10.1534/genetics.113.151670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp NP, Agrawal AF. 2012. Evidence for elevated mutation rates in low-quality genotypes. Proc Natl Acad Sci 109: 6142–6146. 10.1073/pnas.1118918109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp NP, Agrawal AF. 2016. Low genetic quality alters key dimensions of the mutational spectrum. PLoS Biol 14: e1002419 10.1371/journal.pbio.1002419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Roy S, Choudhury SR, Sengupta DN. 2010. DNA repair and recombination in higher plants: insights from comparative genomics of Arabidopsis and rice. BMC Genomics 11: 443 10.1186/1471-2164-11-443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers CM, Yauk CL, White PA, Parfett CL, Quinn JS. 2002. Air pollution induces heritable DNA mutations. Proc Natl Acad Sci 99: 15904–15907. 10.1073/pnas.252499499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Fritz MH, et al. 2015. An integrated map of structural variation in 2,504 human genomes. Nature 526: 75–81. 10.1038/nature15394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AD, Agrawal AF. 2012. DNA repair pathway choice is influenced by the health of Drosophila melanogaster. Genetics 192: 361–370. 10.1534/genetics.112.143321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Tammi MT. 2009. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics 10: 80 10.1186/1471-2105-10-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Omilian AR, Cristescu ME. 2011. High rate of large-scale hemizygous deletions in asexually propagating Daphnia: implications for the evolution of sex. Mol Biol Evol 28: 335–342. 10.1093/molbev/msq199 [DOI] [PubMed] [Google Scholar]
- Yan ND, Bailey J, McGeer JC, Manca MM, Keller WB, Celis-Salgado MP, Gunn JM. 2016. Arrive, survive and thrive: essential stages in the re-colonization and recovery of zooplankton in urban lakes in Sudbury, Canada. J Limnol 75: 4–14. 10.4081/jlimnol.2016.1226 [DOI] [Google Scholar]
- Yao Y, Kovalchuk I. 2011. Abiotic stress leads to somatic and heritable changes in homologous recombination frequency, point mutation frequency and microsatellite stability in Arabidopsis plants. Mutat Res 707: 61–66. 10.1016/j.mrfmmm.2010.12.013 [DOI] [PubMed] [Google Scholar]
- Yauk C, Polyzos A, Rowan-Carroll A, Somers CM, Godschalk RW, Van Schooten FJ, Berndt ML, Pogribny IP, Koturbash I, Williams A, et al. 2008. Germ-line mutations, DNA damage, and global hypermethylation in mice exposed to particulate air pollution in an urban/industrial location. Proc Natl Acad Sci 105: 605–610. 10.1073/pnas.0705896105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zichner T, Garfield DA, Rausch T, Stütz AM, Cannavó E, Braun M, Furlong EE, Korbel JO. 2013. Impact of genomic structural variation in Drosophila melanogaster based on population-scale sequencing. Genome Res 23: 568–579. 10.1101/gr.142646.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.