

Abstract
Background
Long thought “relics” of evolution, not until recently have pseudogenes been of medical interest regarding regulation in cancer. Often, these regulatory roles are a direct by-product of their close sequence homology to protein-coding genes. Novel pseudogene-gene (PGG) functional associations can be identified through the integration of biomedical data, such as sequence homology, functional pathways, gene expression, pseudogene expression, and microRNA expression. However, not all of the information has been integrated, and almost all previous pseudogene studies relied on 1:1 pseudogene–parent gene relationships without leveraging other homologous genes/pseudogenes.
Results
We produce PGG families that expand beyond the current 1:1 paradigm. First, we construct expansive PGG databases by (i) CUDAlign graphics processing unit (GPU) accelerated local alignment of all pseudogenes to gene families (totaling 1.6 billion individual local alignments and >40,000 GPU hours) and (ii) BLAST-based assignment of pseudogenes to gene families. Second, we create an open-source web application (PseudoFuN [Pseudogene Functional Networks]) to search for integrative functional relationships of sequence homology, microRNA expression, gene expression, pseudogene expression, and gene ontology. We produce four “flavors” of CUDAlign-based databases (>462,000,000 PGG pairwise alignments and 133,770 PGG families) that can be queried and downloaded using PseudoFuN. These databases are consistent with previous 1:1 PGG annotation and also are much more powerful including millions of de novo PGG associations. For example, we find multiple known (e.g., miR-20a-PTEN-PTENP1) and novel (e.g., miR-375-SOX15-PPP4R1L) microRNA-gene-pseudogene associations in prostate cancer. PseudoFuN provides a “one stop shop” for identifying and visualizing thousands of potential regulatory relationships related to pseudogenes in The Cancer Genome Atlas cancers.
Conclusions
Thousands of new PGG associations can be explored in the context of microRNA-gene-pseudogene co-expression and differential expression with a simple-to-use online tool by bioinformaticians and oncologists alike.
Keywords: pseudogenes, database, functional prediction, gene regulation, network analysis, high-performance computing, graphics processing unit, competing endogenous RNA
Background
Pseudogenes were previously considered unimportant relics of evolution that played an unclear role in biological processes [1]. However, more pseudogenes have been discovered to be involved in gene regulation [2–4]. These regulatory relationships between pseudogenes and genes have increasingly been explored, such as the transcriptional regulation of PTEN by pseudogene PTENP1 in several cancer conditions [5]. PTEN acts as a tumor suppressor gene, which is underexpressed in gastric cancer. However, by overexpressing PTENP1 in gastric cancer, both PTEN underexpression and cell proliferation are mitigated via the regulatory relationship between PTEN and PTENP1 [6]. Relationships between these pseudogenes and their parent genes have been found to play critical roles indicating functional potentials of these pseudogenes [7, 8]. This point can most clearly be seen in the importance of the role that sequence homology between pseudogenes and coding genes plays in competing endogenous RNA (ceRNA) networks [9, 10]. In ceRNA networks the pseudogenes act as decoy targets for the microRNAs (miRNAs) targeting a protein-coding gene. In short, researchers have made huge strides in understanding pseudogenes from genomic variation to functional potentials [11, 12], and from “deciphering” the mechanism of ceRNA networks [13] to experimental validation [14].
With this progress, there has been renewed interest in pseudogenes, especially in relation to cancer [15]. This interest has even uncovered biomarkers in human cancer including but not limited to SUMO1P3 upregulation as a diagnostic biomarker in gastric cancer and OCT4-pg4 expression as a prognostic biomarker in hepatocellular carcinoma [16–18]. Pseudogene expression has been used to stratify tumor subtypes in seven distinct cancer types [19]. However, owing to the close sequence homology between pseudogenes and their parent genes, identifying the expression profile unique to a pseudogene or highly homologous gene can be challenging. Efforts have been made to address these technical challenges in estimating pseudogene expression using modified alignment and quantification techniques [20]. Perhaps more intriguing is that pseudogenes can be somatically acquired in cancer development effectively “representing a new class of mutations” [21, p.1] that can be either activating or inactivating mutations which function as an on/off switch [22]. Specific pseudogenes have been implicated in specific cancers. For example, FTH1 regulates tumorigenesis in prostate cancer [23], TP73-AS1 regulates proliferation in esophageal squamous cell carcinoma [24], and pseudogenes NKAPP1, MSTO2P, and RPLP0P2 are associated with poor prognosis in lung adenocarcinoma [25].
For these reasons, having a complete understanding of these pseudogene-gene (PGG) relationships is important. While studying these relationships, a common conception is to only consider the pseudogenes in relation to their parent genes with highest homology [7–9, 26]. There have also been pioneering studies probing pseudogene functions through aligning them to parent proteins (corresponding to the parent genes) and then to parent protein domains [7, 27, 28].
The conventional idea of single parent genes may not be comprehensive enough to model the complex phylogenetic relationships involving multiple genes and pseudogenes in a homolog family. While pseudogenes diverged from their parent genes distantly in the past, only the daughter protein-coding genes other than the original parent gene may now exist. The result is that aligning to the true phylogenetic parent gene itself may not be possible. For this reason, we advocate the use of homologous gene families rather than single parent genes to compare against pseudogenes. By viewing the homologies as a weighted network instead of a single scalar value, we believe that new relationships can be uncovered.
We build the PGG family databases using two methods: (i) CUDAlign [29] based local alignment of all pseudogenes to gene families (totaling 1.6 billion individual local alignments and >40,000 graphics processing unit [GPU] hours). By aligning all pseudogenes to all gene families (CUDAlign), we can study underlying sequence homology and more easily set cutoffs to assign pseudogenes to gene families. (ii) Basic Local Alignment and Search Tool (BLAST) [30] based assignment of pseudogenes to gene families. This provides a fast heuristic search option. BLAST derivative methods have been commonly used to find parent genes in previous pseudogene studies [31, 32]. Using these two methods, we show that these pseudogenes are usually assigned to the gene family of their parent genes but are often not exclusively so. Besides, most pseudogenes can be categorized into processed pseudogenes and unprocessed pseudogenes depending on whether they came from retrotranscription of messenger RNAs [11, 33, 34]. We take these differences into account using both of our methods (CUDAlign and BLAST).
Furthermore, we make these data publicly downloadable from GitHub [35]. We also created an R Shiny web application called PseudoFuN (Pseudogene Functional Networks) [36] that supports querying the PGG databases, interactive visualization and functional analysis of the PGG networks, and visualization of PGG co-expression and miRNA binding (including binding prediction with Miranda [37], PicTar [38], and TargetScan [39]) using The Cancer Genome Atlas (TCGA) and GTEx (Genotype-Tissue Expression) Project–derived public data [20, 40, 41]. Besides, we provide another interactive web application hosted by the Ohio Supercomputer Center (OSC), which supports querying novel sequences against any of our PGG databases and visualization of the resulting PGG networks.
The PGG databases can be used to study pseudogene-gene-miRNA co-expression indicative of ceRNA networks across the entire TCGA. With these diverse tools provided by PseudoFuN, it is possible to generate hypotheses regarding (i) the regulatory roles of pseudogenes across tumor and normal tissue, (ii) PGG relationships through de novo reassignment of pseudogenes to gene families, and (iii) functional annotation of pseudogenes. We expect these databases and tools to have more use in cancer studies.
Methods
Construction of PGG database
To generate these gene families, we use two methods: (i) CUDAlign-based local alignment of pseudogenes against consensus sequences representing gene families and (ii) BLAST-based search of pseudogene sequences against all gene sequences (Fig. 1). These two approaches can be thought of as heuristic but different processes. The local sequence alignment approach is heuristic in that only two gene sequences are used from each gene family to reduce the search space. These sequences are the most similar and representative sequences to all the other gene sequences in the family. The BLAST-based approach is heuristic in that not all sequences are fully aligned during the process due to the seed-and-extend steps of BLAST [42]. The result is that not every relationship between pseudogene and gene family is recorded, which is an advantage in runtime but a disadvantage in studying underlying sequence homology.
Figure 1:

Workflow for both CUDAlign and BLAST databases. Left side PGG families are produced using the BLAST matches. Right side PGG families are produced using the PGG family alignment matrix with percentile cutoffs using CUDAlign.
CUDAlign-based local alignment of gene families
Gene homolog families were generated using the Ensembl biomart gene homolog database [43, 44]. The pairs of homologous genes were separated into connected components using the Python networkx package [45]. These connected component sub-graphs are considered gene families in this study. To reduce the number of alignments that needed to be performed, we selected consensus genes from each family that would be used to represent the entire family.
The consensus sequences were selected by aligning every member of the gene family to every other member using local alignment with CUDAlign [29]. The two members of the family with the largest sum alignment scores across all other family members were selected as the consensus sequences to increase the number of candidate sequences. If only one member existed in the family, then that member was the consensus sequence. Using the list of these consensus sequences we then aligned every consensus sequence to every pseudogene in the human genome GRCh38 annotated by GENCODE Release 25 (GENCODEv25) [46].
Specifically the pseudogenes are split up into processed, unprocessed, and other (unclear whether processed or unprocessed) on the basis of their mechanisms of formation [47]. We performed different alignment procedures for processed and unprocessed pseudogenes. The processed pseudogenes were aligned to all consensus gene transcripts with the highest local alignment score recorded. The unprocessed pseudogenes were aligned to the full genomic sequences of each of the consensus genes with the highest local alignment score recorded. Theoretically unprocessed pseudogenes can align to both exonic and intronic regions of DNA, while processed pseudogenes can only align to exonic regions. In our previous database we did not perform this two-procedure strategy in part to reduce the runtime of the problem [48]. These changes make the database much more complete and biologically relevant. The other pseudogenes were aligned to both the transcripts and the genomic sequence recording the highest score.
These scores, one for each combination of pseudogene to gene family, were stored for further analysis. Pseudogenes were assigned to families using a cutoff score (i.e., percentiles of the alignment scores per PGG alignment matrix) and a maximum number of assignments (i.e., the top four alignments above a cutoff). If greater than the top four alignments were used, the PGG families were too large to calculate the pairwise alignment matrix. The resulting sets of pseudogenes and genes are called PGG families. This method was used to allow a pseudogene to be assigned multiple families as well as prevent pseudogenes from being assigned families if their alignment score was low. We used the 99th percentile cutoff (corresponding alignment score 54), 99.9th percentile cutoff (135), and the 99.99th percentile cutoff (198) to generate three resultant databases named CUDAlign54, CUDAlign135, and CUDAlign198, respectively. A fourth database that is less stringent, CUDAlign18, is also included in the web applications using a 97.5th percentile cutoff (18). All these flavors of databases are available for search in our web applications.
BLAST-based generation of PGG families
In contrast to the local alignment of every combination of pseudogene to gene family, PGG families were also created by assigning the pseudogenes to the family containing its closest BLAST search match. This approach was used to contrast with the CUDAlign method, which uses up to the top four matches. The pseudogenes were separated into processed, unprocessed, and other. Then, all genes in the GENCODE Release 25 annotation were used to generate genomic, transcript, and combined BLAST databases (BlastDB). The processed pseudogenes would be BLAST searched against transcript BlastDB, unprocessed against the genomic sequence BlastDB, and the other pseudogenes were BLAST searched against the combined genomic/transcript BlastDB. The pseudogene was assigned to the gene family containing the best match from the BLAST search.
Comparison between PGG families and pseudogene–parent gene pairs
We also conduct a comparison to the Pseudogene.org resource [49]. In this comparison, we consider pseudogenes and parent gene pairs from the Pseudogene.org psiDr [31] database (old) [50] and on GENCODE Release 10 from the Pseudogene.org psiCube [11] database (new) [51]. From our databases, we consider every combination of pseudogene to gene within a PGG family as a pair (e.g., a family with 3 genes and 2 pseudogenes would have 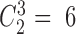 pairs). Because we have multiple flavors of PGG databases including the BLAST-based version and the CUDAlign-based versions, we compare the intersections between two Pseudogene.org versions and our BLAST/CUDAlign-based versions. We show the intersections of PGG pairs in Venn diagrams.
pairs). Because we have multiple flavors of PGG databases including the BLAST-based version and the CUDAlign-based versions, we compare the intersections between two Pseudogene.org versions and our BLAST/CUDAlign-based versions. We show the intersections of PGG pairs in Venn diagrams.
Development of PseudoFuN web applications
Aside from generating different flavors of the PGG databases, we assemble them into an online R Shiny application called PseudoFuN [36], which supports gene and pseudogene symbol queries against our PGG databases, generates dynamic networks, produces gene ontology [52] (GO) tables, and provides additional functional analysis features (Table 1). The functionalities, such as calculating the gene co-expression for any resultant PGG network in any of the TCGA [53] cancer types, are important for ceRNA network hypothesis generation in human cancers. More information can be found in the README file and tutorial on the PseudoFuN website.
Table 1:
Summary of features that are freely available at the PseudoFuN website
| PseudoFuN feature | Additional description |
|---|---|
| Interactive visualization of PGG family networks including the query pseudogene/gene | Users can query any single gene or pseudogene symbol, e.g., PTENP1. Nodes are colored by sub-clusters within the network. |
| Functional enrichment analysis of PGG family | Functional enrichment can be conducted on the genes within the PGG family on Biological Process, Molecular Function, or Cellular Components annotations. The GO functional enrichment is calculated with (i) Fisher exact test [55], (ii) Kolmogorov-Smirnov Classic [56], or (iii) Kolmogorov-Smirnov Elim [56]. |
| Genomic loci mapping of PGG family | The genes in the PGG family can be mapped back to the genome using a circus plot to identify potential loci of interest. |
| Data download for all of the figures | Users can also download results including (i) the DPgE table for all pseudogenes in the selected cancer, (ii) the gene and pseudogene expression, (iii) miRNA correlation table. |
| Links to other gene databases for more information | By directly clicking the node in the network, users can open the GeneCards and Ensembl websites [43, 57] for detailed gene information. |
| Gene/pseudogene co-expression analysis across the entire TCGA | Once a PGG family has been identified the gene/pseudogene co-expression matrix is calculated across 1 of the 32 available TCGA cancer types. |
| Tumor vs normal DE of genes/pseudogenes across all TCGA cancer types | The gene/pseudogene DE is calculated for all members of the selected PGG family. There is also an option to run DE on a specified cancer for all pseudogenes, which can be viewed or downloaded as a table. |
| Predicted miRNA targets involved in the PGG families across all TCGA cancer types | The miRNA targets involved in the selected cancer and PGG family are displayed to show which miRNAs could regulate the PGG family members using the miRNA correlation tables from TCGA. |
| DPgE analysis | Differential pseudogene expression is calculated for each of the pseudogenes in TCGA cancers using dreamBase expression information [20]. The online tool allows for manipulation and download of the table. |
DE: differential expression; DPgE: differential pseudogene expression.
Additionally we created another web application hosted by the OSC OnDemand [54] platform. This application has multiple functionalities including the query of Ensembl gene ID or a novel sequence against one selected flavor of our databases. For each of these features we provide a simple-to-use interface that allows users to select which database to query, allows download of the query hits, and allows users to interactively explore the PGG family networks including GO information.
Use cases in multiple cancers
Furthermore, three use cases are provided to show the potential utility of PseudoFuN to researchers and oncologists looking for functional relationships between pseudogenes, genes, and miRNAs. Use Case I validates known PGG functional relationships. Use Case II identifies high-confidence novel miRNA-pseudogene-gene relationships. Use Case III is primarily focused on agreement with a validation study. We focused on pseudogenes/genes that were differentially expressed (DE) in low RARG/low TACC1/high miR-96 compared to the reverse in prostate cancer cell lines and also DE in our PGG networks in TCGA prostate cancer samples.
Results
Local alignment of gene families
We performed 1.6 billion local alignments between all pseudogenes and all gene family consensus sequences. The process required >40,000 GPU hours on the Oakley cluster at the OSC. The highest scores for each gene family and pseudogene were stored in a 17,273 × 26,754 matrix of pseudogene-to-gene-family alignment scores (∼462 million elements). From this matrix, we are able to explore global PGG family homology relationships and assign pseudogenes to ≥1 gene families with high sequence homology.
As one might expect, the number of pseudogenes with high alignments (defined as above a percentile threshold) to many gene families is relatively low. It can be seen that the majority of pseudogenes will align to one gene family in the CUDAlign databases (Fig. 2). We evaluate alignment of pseudogenes to genes using the Smith-Waterman local pairwise alignment score [58] between a pseudogene and a gene. These scores indicate the highest score possible for two sequences based on their specific dynamic programing matrix, which is solved by the Smith-Waterman algorithm. The cutoffs we use, 18, 54, 135, and 198, indicate the 97.50th, 99.0th, 99.90th, and 99.99th percentiles of alignment scores in our alignment matrix between all pseudogenes and consensus sequences. Another feature of note is that there are some pseudogenes that align to many gene families (e.g., nine pseudogenes, UBE2Q2P1, RP11-313J2.1, TPTEP1, BMS1P1, CTD-2245F17.3, SCAND2P, GTF2IP7, WHAMMP3, and IGLV3-2, have alignment scores >54 in 15,000 gene families and 571 pseudogenes [see Supplementary Table 2] have alignment scores >54 in 1,000 gene families).
Figure 2:

The number pseudogenes that align to gene families. The x-axis is the number of gene families, which have an alignment score above a specified cutoff (the different colored lines). The y-axis is the number of pseudogenes with an alignment score higher than the cutoff to the number of gene families on the x-axis. The inset gray box is a closer view of the low-range gene family numbers (1–10) to show higher-resolution patterns.
In contrast to previous belief in single PGG homology, some pseudogenes are related to many genes. It is worth considering that these high-homology pseudogenes (e.g., FTLP10 with 3,006 gene family pairwise alignments over a 54 threshold) may play a role in regulating major biological processes [59] and disease [60]. Of the nine highest homology pseudogenes (Supplementary Table 2), one, RP11-313J2.1, is a zinc finger pseudogene and two, CTD-2245F17.3 and SCAND2P, are located in the promoters of zinc finger genes. Four pseudogenes in the nine highest homology pseudogenes ( RP11-313J2.1, CTD-2245F17.3, SCAND2P, and WHAMMP3) also have 92–96% sequence identity with zinc finger genes (ZNF72P, ZNF518A, ZNF37A, and ZNF788P/ ZNF20, respectively) when BLAST searched against the human genome. Of the 571 highest homology pseudogenes (Supplementary Table 2), we found 27 zinc finger pseudogenes. Using EnrichR [61] we identified enrichment in GO Molecular Function GO:0 004430 1-phosphatdylinositol 4-kinase activity (Fisher exact test P-value = 0.001), and enrichment for GO Biological Process GO:00 70475 rRNA base methylation (Fisher exact test P-value = 0.003). In the ARCHS4 database [62] 324 transcription factors were significantly co-expressed (Benjamini-Hochberg adjusted Fisher exact test P-value < 0.05) with members of the 571 highest homology pseudogenes. Of those 324 transcription factors, 228 were zinc finger genes. These findings show that the highest homology pseudogenes, like zinc finger genes, likely contain repetitive elements that align to many genomic loci.
BLAST generation of PGG families
The BLAST-generated database was larger than the CUDAlign-generated databases, with 68,578 total connections. This database was also much simpler to compute because it was not an exhaustive search. These conclusions make it a simple method for quickly estimating the pseudogene-to-gene relationships.
Direct comparison to pseudogene parents
We compare our databases to the previous pseudogene–parent gene databases retrieved from Pseudogene.org resources (Fig. 3). This shows that our methods reconstruct most of the pseudogene–parent gene relationships identified by Pseudogene.org. The overall consistency of our databases (BLAST and CUDAlign) with both Pseudogene.org databases (new and old) was 75% (i.e., all our databases combined). Individually, the BLAST-based database contained 61% of the Pseudogene.org relationships (both new and old) and the CUDAlign 54 cutoff contained 60% of the Pseudogene.org relationships (both new and old). Our databases also generated a larger pool of possible interactions. It is worth noting that 391 pseudogenes and 152 genes in the new Pseudogene.org (GENCODE Release 10) are not present in the GENCODE Release 25 annotation used in our analysis. These genes and pseudogenes together account for 1,030 edges that were used in our comparison. Accounting for these differences in the annotation, we are able to reconstruct 85% of the PGG relationships in the new Pseudogene.org database. Because these associations were generated without prior PGG relationship information and the annotations have changed slightly since Pseudogene.org, our methods prove to independently identify known and unknown PGG relationships at a high rate.
Figure 3:

Comparison of database members. The top six plots are comparisons between the CUDAlign databases using different cutoffs, the BLAST database, and the Pseudogene.org parent genes. The bottom row shows intra-database comparisons, left: Pseudogene.org, middle: CUDAlign database of different alignment score cutoffs, right: relative size of all databases.
Development of a pseudogene query tool
The R Shiny application is a comprehensive hypothesis-generating tool that is freely available on the internet [36]. This tool provides a wide array of functionality that a researcher can access quickly and download results as the raw data for more in-depth analysis. These features are outlined in detail in Table 1.
Use cases: Assisting functional study of ceRNA networks in cancer
To illustrate the utility of our databases and tools we present three use cases.
Use Case I: to validate known PGG relationships, we first identified 31 benchmark PGG relationships from three studies [ 15, 16, 23] and query our databases. These studies represent prominent regulatory pseudogenes in cancers by established laboratories. We query a gene/pseudogene name one at a time, and PseudoFuN will return the top PGG network(s) that contain the query (Table 2). In general, we found that our databases together were able to identify 87% of the benchmarking cases (Table 2) and the CUDAlign versions were able to identify 65% of the benchmarking cases. Perhaps most importantly, three of the cases identified by CUDAlign ( ATP8A2, CXADR, PERP) were not identified by the more traditional BLAST approach (Table 2), showing that consensus sequence alignment can identify some overlooked relationships. Next, individual benchmark cases were evaluated in more detail (Supplementary Fig. 2).
Table 2:
Benchmarking analysis of PseudoFuN databases
| Gene | BlastDB | CUDAlign18 | CUDAlign54 | CUDAlign135 | CUDAlign198 | PMID |
|---|---|---|---|---|---|---|
| PTEN | Yes | No | No | No | No | 26442270 |
| TUSC | No | No | No | No | No | 26442270 |
| INTS6 | Yes | No | No | No | No | 26442270 |
| OCT4 | Yes | Yes | Yes | Yes | Yes | 26442270 |
| HMGA1 | Yes | Yes | Yes | Yes | Yes | 26442270 |
| CYP4Z1 | No | No | No | No | No | 26442270 |
| BRAF | Yes | No | No | No | No | 26442270 |
| KLK4 | No | No | No | No | No | 22726445 |
| ATP8A2 | No | Yes | Yes | No | No | 22726445 |
| CXADR | No | Yes | Yes | Yes | Yes | 22726445 |
| CALM2 | Yes | Yes | Yes | Yes | Yes | 22726445 |
| TOMM40 | Yes | Yes | Yes | Yes | Yes | 22726445 |
| NONO | Yes | Yes | Yes | Yes | Yes | 22726445 |
| PERP | No | Yes | Yes | Yes | Yes | 22726445 |
| DUSP8 | Yes | Yes | No | No | No | 22726445 |
| YES1 | Yes | Yes | No | No | No | 22726445 |
| GJA1 | Yes | No | No | No | No | 22726445 |
| AURKA | Yes | Yes | Yes | Yes | Yes | 22726445 |
| RHOB | No | No | No | No | No | 22726445 |
| HMGB1 | Yes | Yes | Yes | Yes | Yes | 22726445 |
| EIF4A1 | Yes | Yes | No | No | No | 22726445 |
| EIF4H | Yes | Yes | Yes | Yes | Yes | 22726445 |
| SNRP6 | Yes | Yes | Yes | Yes | Yes | 22726445 |
| RAB1 | Yes | No | No | No | No | 22726445 |
| VDAC1 | Yes | Yes | No | No | No | 22726445 |
| RCC2 | Yes | No | No | No | No | 22726445 |
| PTMA | Yes | Yes | Yes | Yes | Yes | 22726445 |
| NDUFA9 | Yes | Yes | Yes | Yes | Yes | 22726445 |
| CES7 | Yes | No | No | No | No | 22726445 |
| EPCAM | Yes | Yes | Yes | Yes | Yes | 22726445 |
| FTH1 | Yes | Yes | Yes | Yes | Yes | 29240947 |
| Hits | 24/31 | 20/31 | 16/31 | 15/31 | 15/31 | |
| Total hits | 27/31 |
Genes indicate the gene with which the pseudogenes are associated in the literature. BLAST and CUDAlign columns indicate the specific databases. PMID indicates the literature from which the PGG relationship was derived. Benchmark totals are included at the bottom of the table.
PTENP1 is a processed pseudogene homologous to PTEN, a tumor suppressor gene. PTENP1 is selectively lost in cancer and may regulate PTEN expression as a miRNA decoy target [5, 6]. We have observed differential co-expression patterns of PGG families in tumor vs normal tissue for PTENP1 network in multiple cancers including breast cancer (Supplementary Fig. 3B and C). We identified known miRNAs (hsa-miR-93 targets PTEN in breast cancer [63]) targeting PTEN PGG network nodes, providing insights into ceRNA regulation (Supplementary Fig. 3D). These insights are important because some pseudogenes competitively bind to miRNAs and thus regulate gene expression. We also identify hsa-miR-103a-3p, known to regulate PTEN in endometrial [64] and colorectal cancers [65], in breast cancer (Supplementary Fig. 3D). The miRNA hsa-miR-20a, known to regulate PTEN by the ceRNA mechanism in prostate cancer [66], was also identified in breast cancer. The ceRNA network regulatory relationship is governed by effect modulation of miRNA on gene expression by pseudogene expression (Supplementary Fig. 1A, C, E). This leads to a correlation between pseudogene (miRNA decoy targets) and gene (miRNA targets) expression (Supplementary Fig. 1D), where pseudogenes and homologous genes competitively bind to miRNAs. KRAS-KRASP1 regulatory network was also identified by our database (Supplementary Fig. 2). KRAS and KRASP1 are known to be involved in ceRNA network regulation [5, 10, 66]. PseudoFuN query of KRAS identified co-expression patterns in prostate cancer consistent with ceRNA network regulation by hsa-miR-145, a known modulator of KRAS in prostate cancer [67]. The FTH1 query also resulted in the identification of pseudogenes (FTH1P2, FTH1P8, FTH1P11, FTH1P16) that regulate FTH1 in prostate cancer [23] as well novel miRNAs that may be involved in ceRNA network regulation of FTH1 in prostate cancer. GBP1 is an IFN-α–induced transcript that is involved in immune response in prostate cancer [68]. The GBP1-involved PGG network also contained the pseudogene GBP1P1, which may have a ceRNA regulatory role in breast cancer [69] and in some neurodegenerative diseases [70].
Use Case II: We wanted to identify possible gene-miRNA relationships of interest within our database. We chose to study these relationships with respect to miR-96, a known cancer regulator miRNA in prostate cancer [71]. Through DE analysis between tumors in the TCGA-PRAD cohort with lower expression of RARG and TACC1 (also a miR-96 target) and high expression of miR-96 (low RARG/low TACC1/high miR-96), compared to the reverse, we previously identified that altered SOX15 gene expression is significantly associated with worse disease-free survival. We visualized expression patterns of SOX15 PGG families, and corresponding miRNA associations, where miR-96 is included as a validation.
Interestingly we identified the pseudogene PPP4R1L as a potential member of a SOX15 ceRNA network (Fig. 4A). PPP4R1L and SOX15 are both significantly DE between tumor and normal controls (Bonferroni-corrected P-value = 3.42 × 10−7, 2.01 × 10−14, respectively; Fig. 4E). PPP4R1L and SOX15 are significantly co-expressed (Pearson correlation coefficient = 0.51, P-value < 2.2 × 10−16) in tumor tissue but much less correlated in normal controls in prostate cancer (Pearson correlation coefficient = 0.24, P-value = 0.09; Fig. 4B and C). Positively correlated expression is an assumption when determining ceRNA network relationships [72] (Supplementary Fig. 1). Both SOX15 and PPP4R1L are likely regulated by hsa-miR-375 based on the TCGA prostate cancer dataset. hsa-miR-375 is associated with docetaxel resistance in prostate cancer [73, 74] and PPP4R1L knock-down in HeLa cells induces taxol resistance [75]. These findings are intriguing because taxol and docetaxel are closely related chemical compounds. PPP4R1L is also located in a region associated with high mutation rates in cancer cell lines [75], which could be indicative of mutational “on/off switches” in pseudogene regulation.
Figure 4:

PseudoFuN online output for SOX15 PGG family. A, Interactive graph visualization of the SOX15 PGG network. B, TCGA prostate co-expression matrix for SOX15 PGG family genes and pseudogenes across normal samples. C, TCGA prostate co-expression matrix for SOX15 PGG family genes and pseudogenes across tumor samples. D, Negatively correlated miRNAs for all members of the SOX15 PGG family. E, Differential gene and pseudogene expression for tumor and normal samples for each member of the SOX15 PGG family in the prostate cancer TCGA dataset. FPKM: fragments per kilobase million.
Use Case III: We were most interested in the DE genes (and related pseudogenes) that both appeared in our PGG database and were contained in networks with genes DE in low RARG/low TACC1/high miR-96 compared to vice versa. We searched the DE genes in our PGG database and identified the top networks with enriched number of DE genes. As a result, parent genes HTR7, CNN2, MSN, and TAGLN2 are DE; they generate pseudogenes, which are specifically expressed in prostate cancer samples [16]. These four parent genes are also detected in our five top PGG families involving miR-96–regulated (direct or indirect) DE genes. We identified HTR7P1 pseudogene in the same PGG family as HTR7 gene, which is potentially regulated by hsa-miR-607 and hsa-miR-3654 in the TCGA prostate cancer dataset (Supplementary Fig. 4). Eleven CNN2 pseudogenes (CNN2P1-CCN2P4, CNN2P6-CNN2P12) were identified in the CNN2 PGG family along with TAGLN2 and TAGLN2P1. TAGLN2P1is DE between the tumor and normal samples in the prostate dataset (Supplementary Fig. 5; Bonferroni-corrected P-value = 6.23 × 10−4). MSN and MSNP1 were in the same PGG family and hsa-miR-96 potentially regulates MSN in the TCGA prostate cancer dataset (Supplementary Fig. 5). In addition, although our DE genes were detected from prostate cancer, we further compared them with DE pseudogenes identified in four other cancer types and we observed interesting results (see Supplementary Materials—Potential regulatory roles in cancer).
Discussion
We identify 133,770 PGG families that have significant potential to reveal important information about regulatory PGG relationships in health and disease. Within these families we identify both new and existing regulatory networks that contain pseudogenes such as PTENP1, KRAS1P, FTH1P8/11/16, and GBP1P1 (Fig. 4). Because all genes and all pseudogenes are included in our database, there are thousands of opportunities to identify new regulatory relationships. These thousands of opportunities can be easily stratified using gene name, pseudogene name, and cancer type. Our PseudoFuN web application makes it a simple and intuitive process to query pseudogenes (or genes) to identify which gene families they may be regulating as well as the functions that are attributed to the members of the network. We also have an application hosted by the OSC that allows the querying of novel sequences against our database.
From these networks, we can also identify possible relationships of DE pseudogenes in various cancers. For instance, both PPP4R1L pseudogene and SOX15 are DE in prostate cancer and associated with hsa-miR-375. These types of relationships should be further evaluated along with more complex regulation with multiple miRNAs, pseudogenes, and genes. It is experimentally shown that SOX15is regulated by hsa-miR-96 [71] and it may be important to include hsa-miR-96 in the hsa-miR-375-SOX15-PPP4R1L potential ceRNA network. Aside from PGG family-specific differential pseudogene expression, the PseudoFuN application allows for comprehensive differential pseudogene expression (DPgE) analysis in any of the TCGA cancer datasets.
The use of this database also has utility in integrative analysis where the databases can be used as a mask for other data modalities. Some examples would be using the nodes (genes and pseudogenes) in each of the PGG families as groups in gene expression experiments. Similarly, these groups could be used for feature reduction when visualizing data. We hope researchers can use these relationships we have identified to reduce large numbers of candidate associations down to numbers that can be easily validated and generate new candidates when querying novel sequences. For instance, miRNA-gene pairs filtered through the sets of PGG families would identify high-priority ceRNA candidates.
Conclusions
We generate multiple large databases of PGG family relationships and the tools to study them for use by biomedical researchers. These databases are more comprehensive than previous PGG databases by including many more homology relationships in PGG families, thus more powerful for experiment validation and knowledge discovery. These databases are useful in identifying PGG regulatory relationships in 32 cancer types and show high similarity with known PGG relationships. Aside from the known relationships we identify many unknown relationships. Furthermore, these databases and associated analyses can be easily accessed online or through the OSC OnDemand platform, allowing for novel hypotheses to be assessed quickly by biomedical researchers. We find evidence of both known regulatory PGG relationships and novel hypothesized relationships that we plan to validate. PseudoFuN is a comprehensive, dynamic tool that allows any bioinformatician or oncologist to find novel regulatory pseudogenes within their cancer or gene network of interest.
Availability of supporting data and materials
We have made the PGG family data publicly downloadable from GitHub [35]. We also created an R Shiny web application called PseudoFuN [36] that supports querying the PGG databases, interactive visualization and functional analysis of the PGG networks, and visualization of PGG co-expression and miRNA binding. Apache License 2.0 is associated with PseudoFuN (R Shiny web application). These data are also available in GigaDB [76]. In addition, we provide another interactive web application hosted on Ohio Supercomputer Center (OSC) OnDemand, which supports querying novel sequences against any of our PGG databases and visualization of the resulting PGG networks.
Availability of supporting source code and requirements
Project name: PseudoFuN
Project home page: https://github.com/yanzhanglab/PseudoFuN_app, https://github.com/OSC/pseudofun, https://integrativeomics.shinyapps.io/pseudofun_app/
Operating system: platform independent
Programming language: R, Python, JavaScript
Other requirements: not applicable
License: CC, MIT
OSC OnDemand application access: contact yan.zhang@osumc.edu.
Additional files
There is an additional Supplementary Materials file containing additional information on the data and additional analyses. It includes the following figures and tables:
Supplementary Figure 1. Example of ceRNA network regulation of gene expression. A, A graphical view of how pseudogene expression can regulate gene expression. B, A cellular view of ceRNA network regulation. C, Equations used to model the correlation between gene and pseudogene expression in a ceRNA network. D, The distribution of the PGG correlations based on the models in C. E, The effect that pseudogene expression has on the miRNA-induced change in gene expression.
Supplementary Figure 2. PseudoFuN online output for PTEN PGG family. A, Interactive graph visualization of the PTEN PGG network. B, TCGA prostate co-expression matrix for PTEN PGG family genes and pseudogenes across normal samples. C, TCGA prostate co-expression matrix for PTEN PGG family genes and pseudogenes across tumor samples. D, Negatively correlated miRNAs for all members of the PTEN PGG family. E, Differential gene and pseudogene expression for tumor and normal samples for each member of the PTEN PGG family in the prostate cancer TCGA dataset.
Supplementary Figure 3. PseudoFuN online output for HTR7 PGG family. A, Interactive graph visualization of the HTR7 PGG network. B, TCGA breast cancer co-expression matrix for HTR7 PGG family genes and pseudogenes across normal samples. C, TCGA breast cancer co-expression matrix for HTR7 PGG family genes and pseudogenes across tumor samples. D, Negatively correlated miRNAs for all members of the HTR7 PGG family in breast cancer. E, Differential gene and pseudogene expression for tumor and normal samples for each member of the HTR7 PGG family in the breast cancer TCGA dataset.
Supplementary Figure 4. PseudoFuN online output for CNN2/TAGLN2 PGG family. A, Interactive graph visualization of the CNN2/TAGLN2 PGG network. B, TCGA prostate co-expression matrix for CNN2/TAGLN2 PGG family genes and pseudogenes across normal samples. C, TCGA prostate co-expression matrix for CNN2/TAGLN2 PGG family genes and pseudogenes across tumor samples. D, Negatively correlated miRNAs for all members of the CNN2/TAGLN2 PGG family. E, Differential gene and pseudogene expression for tumor and normal samples for each member of the CNN2/TAGLN2 PGG family in the prostate cancer TCGA dataset.
Supplementary Figure 5. PseudoFuN online output for MSN PGG family. A, Interactive graph visualization of the MSN PGG network. B, TCGA prostate co-expression matrix for MSN PGG family genes and pseudogenes across normal samples. C, TCGA prostate co-expression matrix for MSN PGG family genes and pseudogenes across tumor samples. D, Negatively correlated miRNAs for all members of the MSN PGG family. E, Differential gene and pseudogene expression for tumor and normal samples for each member of the MSN PGG family in the prostate cancer TCGA dataset.
Supplementary Figure 6. The PGG families in our network with the most DE genes after miR-96 treatment. The line weights indicate the sequence homology between members of the PGG family. Red nodes indicate miR-96 targets. Yellow nodes with names indicate other genes contained in the PGG family. Yellow nodes without names are pseudogenes contained within the network.
Supplementary Figure 7. The user interface of the OSC OnDemand web application. A, Main query page where a user can search either sequences or Ensembl gene IDs. B, Representative output of one of the gene searches. This includes an interactive network and the GO information.
Supplementary Figure 8. GBP1P1 DE in TCGA prostate cancer (information retrieved from Han et al.).
Supplementary Table 1. DE parent gene/pseudogenes potentially regulated by miR-96 in prostate cancer vs TCGA-derived DE pseudogenes.
Abbreviations
BLAST: Basic Local Alignment and Search Tool; ceRNA: competing endogenous RNA; DE: differential expression/differentially expressed; DPgE: differential pseudogene expression; FPKM: fragments per kilobase million; GO: gene ontology; GPU: graphics processing unit; miRNA: microRNA; OSC: Ohio Supercomputer Center; PGG: pseudogene-gene; PseudoFuN: Pseudogene Functional Networks; TCGA: The Cancer Genome Atlas.
Competing interests
The authors declare that they have no competing interests.
Funding
This work is partially supported by NIH-NLM MIDAS Training Fellowship (4T15LM011270-05) and NIH-NLM Ruth L. Kirschstein Predoctoral Individual National Research Service Award (1F31LM013056-01) awarded to Travis Johnson. It is also supported by The Ohio State University Startup Funds and OSU Comprehensive Cancer Center Support Grant (P30CA016058) to Yan Zhang.
Author contributions
T.S.J., S.L., Z.H., and Y.Z. performed data analyses. T.S.J., E.F., and Z.H. developed the web applications. Y.Z. and T.S.J. conceived and initiated this project. Y.Z. and K.H. supervised the project. M.J.C. provided experimental data. All authors contributed to biological interpretation. T.S.J., Y.Z., M.J.C., and S.D.L. wrote the manuscript. All authors read and approved the manuscript.
Supplementary Material
10/21/2018 Reviewed
11/10/2018 Reviewed
2/10/2019 Reviewed
ACKNOWLEDGEMENTS
Kun Huang acknowledges the support from Indiana University Precision Health Initiative. Yan Zhang acknowledges the support from the Department of Biomedical Informatics at OSU. The authors thank the Ohio Supercomputer Center (OSC) for providing computing resources.
References
- 1. Vanin EF. Processed pseudogenes: characteristics and evolution. Annu Rev Genet. 1985;19:253–72. [DOI] [PubMed] [Google Scholar]
- 2. Mighell AJ, Smith NR, Robinson PA, et al.. Vertebrate pseudogenes. FEBS Lett. 2000;468:109–14. [DOI] [PubMed] [Google Scholar]
- 3. Pink RC, Wicks K, Caley DP, et al.. Pseudogenes: pseudo-functional or key regulators in health and disease?. RNA. 2011;17:792–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chan JJ, Tay Y. Noncoding RNA:RNA regulatory networks in cancer. Int J Mol Sci. 2018;19:E1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Poliseno L, Salmena L, Zhang J, et al.. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang R, Guo Y, Ma Z, et al.. Long non-coding RNA PTENP1 functions as a ceRNA to modulate PTEN level by decoying miR-106b and miR-93 in gastric cancer. Oncotarget. 2017;8:26079–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lam HY, Khurana E, Fang G, et al.. Pseudofam: the pseudogene families database. Nucleic Acids Res. 2009;37:D738–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zheng D, Gerstein MB.. A computational approach for identifying pseudogenes in the ENCODE regions. Genome Biol. 2006;7(Suppl 1):S13.1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. An Y, Furber KL, Ji S. Pseudogenes regulate parental gene expression via ceRNA network. J Cell Mol Med. 2017;21:185–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Poliseno L, Pandolfi PP. PTEN ceRNA networks in human cancer. Methods. 2015;77–78:41–50. [DOI] [PubMed] [Google Scholar]
- 11. Sisu C, Pei B, Leng J, et al.. Comparative analysis of pseudogenes across three phyla. Proc Natl Acad Sci U S A. 2014;111:13361–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Y, Li S, Abyzov A, et al.. Landscape and variation of novel retroduplications in 26 human populations. PLoS Comput Biol. 2017;13:e1005567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cesana M, Daley GQ. Deciphering the rules of ceRNA networks. Proc Natl Acad Sci U S A. 2013;110:7112–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chiu HS, Martinez MR, Bansal M, et al.. High-throughput validation of ceRNA regulatory networks. BMC Genomics. 2017;18:418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Poliseno L, Marranci A, Pandolfi PP. Pseudogenes in human cancer. Front Med (Lausanne). 2015;2:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kalyana-Sundaram S, Kumar-Sinha C, Shankar S, et al.. Expressed pseudogenes in the transcriptional landscape of human cancers. Cell. 2012;149:1622–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mei D, Song H, Wang K, et al.. Up-regulation of SUMO1 pseudogene 3 (SUMO1P3) in gastric cancer and its clinical association. Med Oncol. 2013;30:709. [DOI] [PubMed] [Google Scholar]
- 18. Wang L, Guo ZY, Zhang R, et al.. Pseudogene OCT4-pg4 functions as a natural micro RNA sponge to regulate OCT4 expression by competing for miR-145 in hepatocellular carcinoma. Carcinogenesis. 2013;34:1773–81. [DOI] [PubMed] [Google Scholar]
- 19. Han L, Yuan Y, Zheng S, et al.. The Pan-Cancer analysis of pseudogene expression reveals biologically and clinically relevant tumour subtypes. Nat Commun. 2014;5:3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zheng LL, Zhou KR, Liu S, et al.. dreamBase: DNA modification, RNA regulation and protein binding of expressed pseudogenes in human health and disease. Nucleic Acids Res. 2018;46:D85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cooke SL, Shlien A, Marshall J, et al.. Processed pseudogenes acquired somatically during cancer development. Nat Commun. 2014;5:3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shukla R, Upton KR, Munoz-Lopez M, et al.. Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell. 2013;153:101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chan JJ, Kwok ZH, Chew XH, et al.. A FTH1 gene:pseudogene:microRNA network regulates tumorigenesis in prostate cancer. Nucleic Acids Res. 2018;46:1998–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zang W, Wang T, Wang Y, et al.. Knockdown of long non-coding RNA TP73-AS1 inhibits cell proliferation and induces apoptosis in esophageal squamous cell carcinoma. Oncotarget. 2016;7:19960–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wei Y, Chang Z, Wu C, et al.. Identification of potential cancer-related pseudogenes in lung adenocarcinoma based on ceRNA hypothesis. Oncotarget. 2017;8:59036–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Milligan MJ, Lipovich L. Pseudogene-derived lncRNAs: emerging regulators of gene expression. Front Genet. 2014;5:476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bateman A, Birney E, Durbin R, et al.. The Pfam protein families database. Nucleic Acids Res. 2000;28:263–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Finn RD, Mistry J, Schuster-Bockler B, et al.. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34:D247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jain C, Kumar S. Fine-grained GPU parallelization of pairwise local sequence alignment. In: 21st International Conference on High Performance Computing (HiPC), Dona Paula, India, 2014. IEEE, 2015. [Google Scholar]
- 30. Soroceanu L, Matlaf L, Khan S, et al.. Cytomegalovirus immediate-early proteins promote stemness properties in glioblastoma. Cancer Res. 2015;75:3065–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pei B, Sisu C, Frankish A, et al.. The GENCODE pseudogene resource. Genome Biol. 2012;13:R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Z, Carriero N, Zheng D, et al.. PseudoPipe: an automated pseudogene identification pipeline. Bioinformatics. 2006;22:1437–9. [DOI] [PubMed] [Google Scholar]
- 33. Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–5. [DOI] [PubMed] [Google Scholar]
- 34. Baertsch R, Diekhans M, Kent WJ, et al.. Retrocopy contributions to the evolution of the human genome. BMC Genomics. 2008;9:466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y. 2018. PseudoFuN GitHub. https://github.com/yanzhanglab/PseudoFuN_app. Accessed 15 June 2018. [Google Scholar]
- 36. Johnson TS, Li S, Franz E, et al.. PseudoFuN 2018. https://integrativeomics.shinyapps.io/pseudofun_app/. [Google Scholar]
- 37. Miranda KC, Huynh T, Tay Y, et al.. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–17. [DOI] [PubMed] [Google Scholar]
- 38. Krek A, Grun D, Poy MN, et al.. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. [DOI] [PubMed] [Google Scholar]
- 39. Agarwal V, Bell GW, Nam JW, et al.. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Grossman RL, Heath AP, Ferretti V, et al.. Toward a shared vision for cancer genomic data. N Engl J Med. 2016;375:1109–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carithers LJ, Moore HM. The Genotype-Tissue Expression (GTEx) Project. Biopreserv Biobank. 2015;13:307–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Altschul SF, Gish W, Miller W, et al.. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. [DOI] [PubMed] [Google Scholar]
- 43. Zerbino DR, Achuthan P, Akanni W, et al.. Ensembl 2018. Nucleic Acids Res. 2018;46:D754–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ensembl. Ensembl Biomart. 2018. uswest.ensembl.org/biomart/martview.
- 45. Hagberg AA, Schult DA, Swart PJ. Exploring network structure, dynamics, and function using NetworkX. In: Proceedings of the 7th Python in Science Conference (SciPy 2008), Pasadena, CA, 2008.
- 46. Harrow J, Frankish A, Gonzalez JM, et al.. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Echols N, Harrison P, Balasubramanian S, et al.. Comprehensive analysis of amino acid and nucleotide composition in eukaryotic genomes, comparing genes and pseudogenes. Nucleic Acids Res. 2002;30:2515–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Johnson TS, Li S, Kho JR, et al.. Network analysis of pseudogene-gene relationships: from pseudogene evolution to their functional potentials. Pac Symp Biocomput. 2018;23:536–47. [PMC free article] [PubMed] [Google Scholar]
- 49. Karro JE, Yan Y, Zheng D, et al.. Pseudogene.org: a comprehensive database and comparison platform for pseudogene annotation. Nucleic Acids Res. 2007;35:D55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. pseudogenes.org: psiDr. pseudogene.org/psidr/similarity.dat.
- 51. pseudogenes.org: psiCube. http://pseudogene.org/psicube/
- 52. Ashburner M, Ball CA, Blake JA, et al.. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weinstein JN, Collisson EACancer Genome Atlas Research Network; et al.; Cancer Genome Atlas Research Network The Cancer Genome Atlas pan-cancer analysis project. Nat Genet. 2013;45:1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hudak D, Johnson D, Chalker A, et al.. Open OnDemand: a web-based client portal for HPC centers. J Open Source Softw. 2018;3:622. [Google Scholar]
- 55. Fisher RA. Tests of significance in harmonic analysis. Proc R Soc London A. 1929;125:54. [Google Scholar]
- 56. Alexa A, Rahnenfuhrer J. Gene set enrichment analysis with topGO 2018. https://bioconductor.org/packages/release/bioc/vignettes/topGO/inst/doc/topGO.pdf. [Google Scholar]
- 57. Stelzer G, Rosen N, Plaschkes I, et al.. The GeneCards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinformatics. 2016;54:1.30.1–33. [DOI] [PubMed] [Google Scholar]
- 58. Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147:195–7. [DOI] [PubMed] [Google Scholar]
- 59. Carmona U, Li L, Zhang L, et al.. Ferritin light-chain subunits: key elements for the electron transfer across the protein cage. Chem Commun (Camb). 2014;50:15358–61. [DOI] [PubMed] [Google Scholar]
- 60. Wu T, Li Y, Liu B, et al.. Expression of ferritin light chain (FTL) is elevated in glioblastoma, and FTL silencing inhibits glioblastoma cell proliferation via the GADD45/JNK pathway. PLoS One. 2016;11:e0149361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kuleshov MV, Jones MR, Rouillard AD, et al.. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lachmann A, Torre D, Keenan AB, et al.. Massive mining of publicly available RNA-seq data from human and mouse. Nat Commun. 2018;9:1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li N, Miao Y, Shan Y, et al.. MiR-106b and miR-93 regulate cell progression by suppression of PTEN via PI3K/Akt pathway in breast cancer. Cell Death Dis. 2017;8:e2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guo C, Song WQ, Sun P, et al.. LncRNA-GAS5 induces PTEN expression through inhibiting miR-103 in endometrial cancer cells. J Biomed Sci. 2015;22:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Geng L, Sun B, Gao B, et al.. MicroRNA-103 promotes colorectal cancer by targeting tumor suppressor DICER and PTEN. Int J Mol Sci. 2014;15:8458–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yang C, Wu D, Gao L, et al.. Competing endogenous RNA networks in human cancer: hypothesis, validation, and perspectives. Oncotarget. 2016;7:13479–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cui SY, Wang R, Chen LB. MicroRNA-145: a potent tumour suppressor that regulates multiple cellular pathways. J Cell Mol Med. 2014;18:1913–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Persano L, Moserle L, Esposito G, et al.. Interferon-alpha counteracts the angiogenic switch and reduces tumor cell proliferation in a spontaneous model of prostatic cancer. Carcinogenesis. 2009;30:851–60. [DOI] [PubMed] [Google Scholar]
- 69. Welch JD, Baran-Gale J, Perou CM, et al.. Pseudogenes transcribed in breast invasive carcinoma show subtype-specific expression and ceRNA potential. BMC Genomics. 2015;16:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Costa V, Esposito R, Aprile M, et al.. Non-coding RNA and pseudogenes in neurodegenerative diseases: “the (un)usual suspects.”. Front Genet. 2012;3:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Long MD, Singh PK, Russell JR, et al.. The miR-96 and RARgamma signaling axis governs androgen signaling and prostate cancer progression. Oncogene. 2019;38(3):421–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Xu J, Feng L, Han Z, et al.. Extensive ceRNA-ceRNA interaction networks mediated by miRNAs regulate development in multiple rhesus tissues. Nucleic Acids Res. 2016;44:9438–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Costa-Pinheiro P, Ramalho-Carvalho J, Vieira FQ, et al.. MicroRNA-375 plays a dual role in prostate carcinogenesis. Clin Epigenetics. 2015;7:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wang Y, Lieberman R, Pan J, et al.. miR-375 induces docetaxel resistance in prostate cancer by targeting SEC23A and YAP1. Mol Cancer. 2016;15:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat Cell Biol. 2005;7:591–600. [DOI] [PubMed] [Google Scholar]
- 76. Johnson TS, Li S, Franz E, et al.. Supporting data for “PseudoFuN: Deriving functional potentials of pseudogenes from integrative relationships with genes and microRNAs across 32 cancers.”. Gigascience Database. 2019. 10.5524/100577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
10/21/2018 Reviewed
11/10/2018 Reviewed
2/10/2019 Reviewed