

Abstract
STUDY QUESTION
Are genetic effects on endometrial gene expression tissue specific and/or associated with reproductive traits and diseases?
SUMMARY ANSWER
Analyses of RNA-sequence data and individual genotype data from the endometrium identified novel and disease associated, genetic mechanisms regulating gene expression in the endometrium and showed evidence that these mechanisms are shared across biologically similar tissues.
WHAT IS KNOWN ALREADY
The endometrium is a complex tissue vital for female reproduction and is a hypothesized source of cells initiating endometriosis. Understanding genetic regulation specific to, and shared between, tissue types can aid the identification of genes involved in complex genetic diseases.
STUDY DESIGN, SIZE, DURATION
RNA-sequence and genotype data from 206 individuals was analysed and results were compared with large publicly available datasets.
PARTICIPANTS/MATERIALS, SETTING, METHODS
RNA-sequencing and genotype data from 206 endometrial samples was used to identify the influence of genetic variants on gene expression, via expression quantitative trait loci (eQTL) analysis and to compare these endometrial eQTLs with those in other tissues. To investigate the association between endometrial gene expression regulation and reproductive traits and diseases, we conducted a tissue enrichment analysis, transcriptome-wide association study (TWAS) and summary data-based Mendelian randomisation (SMR) analyses. Transcriptomic data was used to test differential gene expression between women with and without endometriosis.
MAIN RESULTS AND THE ROLE OF CHANCE
A tissue enrichment analysis with endometriosis genome-wide association study summary statistics showed that genes surrounding endometriosis risk loci were significantly enriched in reproductive tissues. A total of 444 sentinel cis-eQTLs (P < 2.57 × 10−9) and 30 trans-eQTLs (P < 4.65 × 10−13) were detected, including 327 novel cis-eQTLs in endometrium. A large proportion (85%) of endometrial eQTLs are present in other tissues. Genetic effects on endometrial gene expression were highly correlated with the genetic effects on reproductive (e.g. uterus, ovary) and digestive tissues (e.g. salivary gland, stomach), supporting a shared genetic regulation of gene expression in biologically similar tissues. The TWAS analysis indicated that gene expression at 39 loci is associated with endometriosis, including five known endometriosis risk loci. SMR analyses identified potential target genes pleiotropically or causally associated with reproductive traits and diseases including endometriosis. However, without taking account of genetic variants, a direct comparison between women with and without endometriosis showed no significant difference in endometrial gene expression.
LARGE SCALE DATA
The eQTL dataset generated in this study is available at http://reproductivegenomics.com.au/shiny/endo_eqtl_rna/. Additional datasets supporting the conclusions of this article are included within the article and the supplementary information files, or are available on reasonable request.
LIMITATIONS, REASONS FOR CAUTION
Data are derived from fresh tissue samples and expression levels are an average of expression from different cell types within the endometrium. Subtle cell-specifc expression changes may not be detected and differences in cell composition between samples and across the menstrual cycle will contribute to sample variability. Power to detect tissue specific eQTLs and differences between women with and without endometriosis was limited by the sample size in this study. The statistical approaches used in this study identify the likely gene targets for specific genetic risk factors, but not the functional mechanism by which changes in gene expression may influence disease risk.
WIDER IMPLICATIONS OF THE FINDINGS
Our results identify novel genetic variants that regulate gene expression in endometrium and the majority of these are shared across tissues. This allows analysis with large publicly available datasets to identify targets for female reproductive traits and diseases. Much larger studies will be required to identify genetic regulation of gene expression that will be specific to endometrium.
STUDY FUNDING/COMPETING INTEREST(S)
This work was supported by the National Health and Medical Research Council (NHMRC) under project grants GNT1026033, GNT1049472, GNT1046880, GNT1050208, GNT1105321, GNT1083405 and GNT1107258. G.W.M is supported by a NHMRC Fellowship (GNT1078399). J.Y is supported by an ARC Fellowship (FT180100186). There are no competing interests.
Keywords: endometrium, RNA-sequencing, gene expression, endometriosis, expression quantitative trait loci (eQTL), genetic regulation, tissue specific
Introduction
Genetic effects on transcriptional regulation underlie the pathogenic mechanisms of many human traits and diseases (Peters et al., 2016; Ongen et al., 2017; Gamazon et al., 2018). Genetic variants that regulate gene expression, termed expression quantitative trait loci (eQTLs) can be shared between tissues or can be tissue specific (Consortium et al., 2017). Consequently, to investigate genetic contributions to disease mechanisms, we must investigate the tissue, and ultimately individual cell types, relative to a disease to understand the genetic contribution to pathogenesis.
The endometrium, as the inner most layer of the uterus is a vitally important reproductive tissue, integral for fertility and implicated in many reproductive disorders. It undergoes cyclical changes largely driven by hormonal regulation and changes in cellular composition (Evans et al., 2016). Our previous studies have shown endometrial gene expression and methylation is influenced by genetic variation (Powell et al., 2016; Fung et al., 2017; Fung et al., 2018; Mortlock et al., 2019) and suggest these genetic variants contribute to the susceptibility to reproductive disorders. The specific genes that are influenced by these genetic variants and that contribute to disease pathogenesis however remain to be elucidated.
Endometriosis, characterised by endometrial-like tissue that form lesions outside the uterus is a common reproductive disorder affecting 6–10% of reproductive aged women and it is believed to stem from endometrial tissue (Bulletti et al., 2010; Giudice, 2010). A recent genome-wide association study (GWAS) identified 27 genomic loci associated with endometriosis (Rahmioglu et al., 2018) and our previous studies showed that expression of critical endometrial target genes and methylation of CpG sites are altered in genomic regions associated with endometriosis susceptibility (Powell et al., 2016; Fung et al., 2017; Fung et al., 2018; Mortlock et al., 2019). This suggests that susceptibility to endometriosis is mediated by changes in endometrial gene expression and methylation under the control of genetic risk factors. The underlying mechanisms increasing endometriosis susceptivity at many of these genetic regions is not yet clear.
RNA sequencing (RNA-seq) is a powerful gene expression technique that quantitates individual RNA transcripts with a broader dynamic range than microarray technology, which captures only 30% of the data available in RNA-seq (Mortazavi et al., 2008; Wang et al., 2009; Zhao et al., 2014). The aim of this study was therefore to extend our understanding of the genetic regulation of transcription in endometrium by analysing paired-end total RNA sequence data from 206 endometrial samples, characterize the eQTLs in the endometrium and their similarity to other tissues, and determine their association with susceptibility genes for reproductive traits and diseases, such as endometriosis.
Materials and Methods
Tissue enrichment analysis
To identify the tissue types that could be associated with endometriosis, we performed cell-type enrichment analysis using the method outlined in Finucane et al. (2018). We used the summary statistics from the endometriosis meta-analysis conducted by Sapkota et al. (2017), which contained 17 045 endometriosis cases and 191 596 controls. Gene expression data were obtained from the GTEx project (Consortium, 2015, Consortium et al., 2017) and the Franke Lab (Pers et al., 2015). The GTEx dataset contains gene expression data from RNA-Seq analysis of 53 different tissues or cell types from 8550 human samples. The Franke Lab gene expression data comes from microarray analysis of 152 distinct tissues and cells types from 37 427 human samples. The different tissues or cell types were classified into nine groups for the visualisation, including adipose, blood or immune, cardiovascular, central nervous system, digestive, liver, musculoskeletal-connective, pancreas and other.
For each gene in all of the distinct tissue or cell-type gene expression datasets, we calculated the t-statistic, a measure of tissue specific gene expression. Within each tissue or cell type, the t-statistics for each gene was ranked and only the top 10% were used in the next step of the analysis. From each gene, we added a 100-kb window on either side of the transcribed region to construct the genome annotation and then performed stratified linkage disequilibrium (LD) score regression on the endometriosis GWAS summary statistics to test whether the disease heritability was enriched in loci containing genes with the highest expression in particular tissues. Tissue or cell types with FDR < 0.05 were classified as significantly enriched.
Sample collection
Woman of European ancestry and reproductive age were recruited from clinics at the Royal Women’s Hospital (RWH) and Melbourne IVF Clinic (IVF) in Melbourne, Australia. A total of 206 women were included in the study, consisting of 184 RWH gynaecology patients and 22 IVF patients. RWH patients underwent investigative laparoscopic surgery in response to pathological symptoms and/or infertility during which endometrial tissue samples were extracted by curettage. A detailed clinical history and surgical and pathological results were obtained for each participant in RWH sample set.
The endometriosis status was recorded following surgical diagnosis at laparoscopy for women from RWH, or self-reported for women from the IVF clinic. A histological assessment of each endometrial biopsy was performed by an experienced pathologist to categorise samples into seven different menstrual cycle stages including menstrual (M), early-proliferative (EP), mid-proliferative (MP), late-proliferative (LP), early-secretory (ES), mid-secretory (MS) and late-secretory (LS). We excluded samples that were from women of non-European ancestry or who underwent hormonal treatment and tissues that showed abnormality on histopathology or with ambiguous diseases status or cycle stage. A summary of samples and associated clinical detail is provided in Table I. From each individual, we collected whole blood samples prior to surgery and endometrial tissue. The endometrial tissue was stored in RNAlater (Life Technologies, USA) at −80°C for later RNA extraction, while whole blood was prepared for DNA isolation.
Table I. Patient numbers and information.
| RWH | IVF | Total number of samples | |
|---|---|---|---|
| Endometriosis assessment | Surgically confirmed | Self-reported | |
| Number of samples | 184 | 22 | 206 |
| Endometriosis status | |||
| Case | 135 | 8 | 143 |
| Control | 49 | 14 | 63 |
| Stage of cycle | |||
| Menstrual (M) | 14 | 0 | 14 |
| Early-proliferative (EP) | 5 | 0 | 5 |
| Mid-proliferative (MP) | 71 | 1 | 72 |
| Late-proliferative (LP) | 21 | 1 | 22 |
| Early-secretory (ES) | 21 | 10 | 31 |
| Mid-secretory (MS) | 31 | 10 | 41 |
| Late-secretory (LS) | 21 | 0 | 21 |
Number of patients recruited from the RWH and Melbourne IVF Clinic (IVF) and associated endometriosis and stage of menstrual cycle information.
Ethics approval and consent to participate
The study was approved by the Royal Women’s Hospital Human Research Ethics Committee (Projects 11–24 and 16–43), the Melbourne IVF Human Research Ethics Committee (Project 05–11) and the University of Queensland. Informed consent was obtained from all participants.
Genotyping
DNA samples from each of the 206 individuals were genotyped on HumanCoreExome or Infinium PsychArray chips (Illumina, USA). Quality control (QC) was performed in PLINK according to the protocol outlined in Fung et al. (2018). Following QC, a total of 282 625 SNPs (hg19) were phased using Shapelt V2 and taken forward to imputation using the haplotype reference consortium reference panel (version r1.1 2016) on the Michigan Imputation Server. SNPs with low imputation quality (R2 < 0.8), missing rate > 5%, minor allele frequency (MAF) < 1 × 10–4, and Hardy–Weinberg equilibrium < 1 × 10−6 after imputation were removed. The remaining SNP positions were lifted-over to the Ensembl genome build 38 (GRCh38) using CrossMap v.0.2.8. SNPs failing to lift-over were assigned to their new GRCh38 position manually based on dbSNP151 GRCh38 patch release 7 (GRCh38.p7), leaving 6 230 993 SNPs for further analysis.
RNA extraction
Total RNA was isolated from endometrial tissue using the Allprep DNA/RNA Mini Kit (Qiagen, CA) as per the manufacturer’s instructions. RNA quality was checked using the Bioanalyzer 2100 (Agilent Technologies, CA) and RNA concentration was measured using the NanoDropND-6000 (Thermo Fisher Scientific, USA). All samples were high quality with an RNA integrity number greater than 8.
RNA sequencing
The RNA samples were treated with Turbo DNA-free kit (Thermo Fisher Scientific, USA) prior to RNA-seq library generation. Stranded RNA-seq libraries were prepared using the Illumina TruSeq Stranded Total RNA Gold protocol which includes ribosomal depletion (Illumina, USA). Libraries were pooled and sequenced to a mean depth of 37 490 673 reads (178 samples; 75 bp pair-ended reads) on the Illumina HiSeq 4000 and 40 818 062 reads (28 samples; 120 bp pair-ended reads) on the Illumina HiSeq 2000 (Ilumina, USA). Raw sequencing reads were quality checked using FastQC v0.11.7(Andrews, 2010) and MultiQC v1.6(Ewels et al., 2016). Low quality reads and contaminating HiSeq Illumina adapter sequences were trimmed using Trimmomatic v0.36(Bolger et al., 2014).
Trimmed reads were aligned against the human reference genome (Ensembl Homo sapiens GRCh38 release 84) using HISAT2 v2.0.5. Transcript assembly was performed using StringTie v1.3.1(Pertea et al., 2015, 2016) and the Ensembl Homo sapiens GRCh8 release 91 reference assembly. Reads mapping to each known transcript were directly counted in StringTie to generate transcript-, exon- and intron-level expression matrices in ‘fragments per kilobase of transcript per million mapped reads’ units for each individual. Raw gene count matrices were also produced using a Python script provided by StringTie.
Normalisation of RNA-Seq counts
Genes expressed at a low level, i.e. genes with counts per million (CPM) <0.22 (~10 counts) and expressed in <90% of the samples, were removed. Raw gene counts were normalized for composition bias and total raw reads (library size) using the Trimmed Mean of M (TMM) (Fadista et al., 2014; Taneera et al., 2015; Seo et al., 2016) method in the edgeR R package v3.22.3 (Robinson et al., 2010). Normalized counts were converted to CPM and log2 transformed (log2-CPM).
eQTL analysis
RNA-seq counts and genotype data from the 206 individuals was used to test association between genotype and gene expression. A total of 17 022 genes expressed in >90% samples were included in the analysis. Expression values were TMM-normalized, converted and log2 transformed (log2-CPM) as described above. Individual level genotype data for 6 230 993 SNPs was also included in the analysis. The cis-eQTL analysis was carried out using a linear regression model in the MatrixeQTL R package v2.2(Shabalin, 2012). cis-eQTLs were defined as SNPs located within ±250 kb from gene start and stop position. Batch effects (lanes within the same flow-cell and between flow-cells), stage of menstrual cycle and endometriosis diseases status where included in the model as covariates. Comparisons with SNPs on a different chromosome to the associated gene, were classified as trans-eQTLs. The trans-eQTL analysis was also performed using MatrixeQTL with the same covariates, this time setting a MAF threshold of >0.05 leaving 4 922 014 SNPs to be included in the analysis. To identify independent cis-eQTL signals, we performed a conditional analysis using the COJO method in GCTA (Yang et al., 2011; Yang et al., 2012), including the effect of the eSNP (SNP associated with gene expression) with the smallest P-value as a covariate in the model for each of the 444 genes with Bonferroni significant eQTLs.
Correlation with endometrial, GTEx and eQTLGen eQTLs
To evaluate whether the genetic effects on gene expression in endometrial tissue also occurred in other tissues, we correlated eQTL effects with 48 tissues from the GTEx v7 project (Supplementary Table SI) (Consortium, 2015; Consortium et al., 2017) and the blood eQTL dataset from eQTLGen Consortium (eQTLGen) consisting of 31 684 individuals (Võsa et al., 2018). We used the rb method developed by Qi et al. (2018) to estimate the correlation between genetic effects at top cis-eQTLs whilst accounting for eQTL effect estimation errors (Qi et al., 2018). Briefly, we used the top significant brain eQTLs (PeQTL < 5 × 10−8) from the religious orders study and memory and ageing project (ROSMAP) (Ng et al., 2017) as a reference to avoid ascertainment bias. Subsequently top ROSMAP cis-eQTLs present in both the endometrium eQTL dataset and the GTEx tissue eQTL dataset being tested were used in the effect size (ES) correlation analysis. Genome positions of endometrial eQTLs from this study were converted from the GRCh38 assembly back to the GRCh37 assembly for direct comparison with ROSMAP, GTEx and eQTLGen datasets.
Overlap of genetic regulation of gene expression in endometrium with other tissues from the GTEx and eQTLGen database
To identify potential endometrial specific cis-eQTLs, we examined the overlap of Bonferroni significant, independent endometrial cis-eQTLs with cis-eQTLs in the 48 different tissue types in the GTEx (Supplementary Table SI) and eQTLGen datasets (Võsa et al., 2018). cis-eQTLs were matched between tissues based on the same eSNP and gene associations including eSNPs in linkage with the lead eSNP (r2 > 0.8). The direction of effect was also considered for those with the same eSNP.
Differential expression
The limma R package was used to analyse genome-wide differential gene expression following the removal of genes expressed at a low level. Only genes with a minimum of 10 counts and expressed in at least 90% of samples were analysed for differential expression. We performed three distinct differential gene expressions comparisons: (i) between endometriosis cases and controls, (ii) between cases and controls in the mid-proliferative stage and (iii) between cases and controls in the mid-secretory stage. We fitted batch effect (flow-cell and lane) as covariates in all three models and corrected for stage of menstrual cycle in model 1. The normalized counts were transformed using the voom (Law et al., 2014) function in limma before being fitted to the linear model. The eBayes method was used to contrast between groups. Resulting P-values were adjusted using Benjamini–Hochberg with a significance threshold of 0.05. Only genes passing FDR < 0.05 were categorized as being significantly differentially expressed genes.
Association between transcription and disease
GWAS SNPs
We assessed the overlap of endometrial eQTLs and endometriosis associated SNPs. To date, 27 loci have been associated with endometriosis based on a GWAS meta-analysis across different populations (Sapkota et al., 2017; Rahmioglu et al., 2018). To identify overlaps between endometrial cis-eQTLs and endometriosis risk loci, we analysed LD between significant cis-eQTL (FDR < 0.05) eSNPs and the lead risk SNPs within the 27 risk loci. eQTLs with r2 > 0.8, as calculated using LDlink (Machiela and Chanock, 2015), were defined as being in LD with the related risk SNP. We also assessed the overlap with SNPs associated with various other traits in the GWAS catalogue.
Transcriptome-wide association study
In the absence of gene expression data from large cohorts, new powerful statistical approaches allow us to impute gene expression and conduct a transcriptome-wide association study (TWAS). The resulting association statistics reflect underlying relationships between gene expression and disease risk contributing to the identification of target genes underlying complex traits. Transcriptome-integrated genetic association resource (TIGAR) is a software tool used to impute transcriptomic data and perform TWAS using summary-level GWAS data. We used the Train Dirichlet process regression imputation model in TIGAR to estimate cis-eQTL ES in endometrium using the 206 RNA-seq samples with matched imputed genotype data. Using estimates from the training model as weights, we conducted a TWAS using endometriosis GWAS summary-statistics from the Sapkota et al. (2017) meta-analysis. Details of the training and association models can be found in Nagpal et al. (2019). To investigate multiple genes in significantly associated loci, we tested for a correlation between the predicted gene expression in each loci. Gene expression was imputed using estimated effects sizes from the training model and individual genotype data from 5186 individuals belonging to the QIMRHCS cohort (Nyholt et al., 2012; Sapkota et al., 2017). Gene expression for each individual was predicted by matrix multiplication between estimated ES and additive genotype (Xg) (Predicted_GE = ES*Xg). The Pearson correlation was calculated between the predicted expression of each gene pair in a locus using the cor function in the R environment.
Summary data-based Mendelian randomization
To assess the association between genetic variant, gene expression and trait, we performed summary data-based Mendelian randomization (SMR) (Zhu et al., 2016). The SMR was conducted by integrating the summary eQTL data and the GWAS meta-analysis from Sapkota et al. (2017). Associations passing the SMR test and with a PHEIDI of >0.05/(number of genes passing the SMR test) were considered significant. Due to the limited power of our eQTL analysis, the presence of both secondary eQTL signals and secondary signals in multiple endometriosis risk loci, we also conducted a multi-SNP-based SMR to use information from all significant SNPs in each region (Wu et al., 2018). To further avoid confounding effects of multiple signals, reduce multiple testing burden and leverage the power of the large GWAS, we conducted a modified SMR analysis in which the trait (endometriosis) was treated as the exposure and the gene expression within each GWAS locus (2 MB either side of top SNP) was treated as the outcome. Using this modified SMR approach the top GWAS SNPs are selected for testing based on their association with endometriosis. To increase power, we repeated the SMR analyses using the large eQTLGen blood eQTL dataset as a proxy (Võsa et al., 2018). Using available GWAS summary statistics for age at menopause (Day et al., 2015) and epithelial ovarian cancer (Phelan et al., 2017), we conducted an SMR analysis using endometrial eQTLs against these traits.
Results
Enrichment of genes in endometriosis risk loci in reproductive tissues
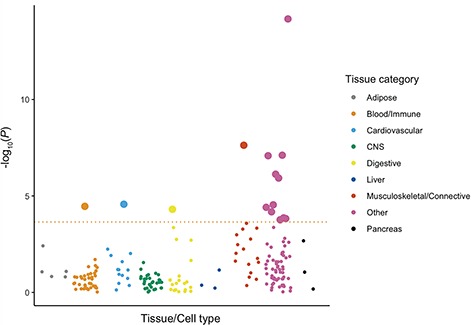
Understanding the role of genetic regulation of gene expression in complex traits and diseases is more powerful when measuring genetic effects in disease relevant tissues. Using summary statistics from Sapkota et al. (2017) and gene expression data from a range of tissues and cell types, we show a significant enrichment for genes annotated to endometriosis-associated loci in female reproductive tissues, myometrium, arteries, serum and digestive tissue (Table II, Fig. 1). This suggests effects of regulation of gene expression on endometriosis risk loci are more likely to be detected in reproductive tissues including the endometrium.
Table II. Cell-types and tissues enriched for genes at endometriosis risk loci.
| Tissue/Cell type | Tissue Category | P-value |
|---|---|---|
| Fallopian_Tube | Other | 6.96E-07 |
| A05.360.319.679.690.Myometrium | Musculoskeletal/Connective | 4.86E-04 |
| Uterus | Other | 8.14E-04 |
| A05.360.319.679.Uterus | Other | 8.35E-04 |
| Cervix_Ectocervix | Other | 2.18E-03 |
| Cervix_Endocervix | Other | 2.66E-03 |
| A07.231.114.Arteries | Cardiovascular | 1.03E-02 |
| A05.360.319.Genitalia..Female | Other | 1.06E-02 |
| A15.145.846.Serum | Blood/Immune | 1.15E-02 |
| A06.407.Endocrine.Glands | Other | 1.21E-02 |
| Esophagus_Muscularis | Digestive | 1.34E-02 |
| A05.360.Genitalia | Other | 1.53E-02 |
| A05.360.319.679.490.Endometrium | Other | 2.10E-02 |
| A06.407.312.Gonads | Other | 2.17E-02 |
| A05.360.319.114.630.Ovary | Other | 2.31E-02 |
Numbers preceding tissues correspond to the National Institute of Health (NIH) medical subject heading (MeSH) tree structure numbers used to label and distinguish tissues in the Franke lab dataset.
Figure 1. Multi-tissue enrichment analysis results for endometriosis risk loci. Each dot represents a tissue or cell type from either the GTEx dataset (total N = 8550) or the Franke lab dataset (total N = 37 427) and each colour represents a different tissue category. Tissues or cell types passing the FDR cut off (FDR < 0.05) with a –log10 P-value < 3.65 are shown as large dots.
eQTLs in endometrium from RNA-Seq
Following assembly and quantification of RNA-seq reads in endometrial samples from 206 European women, we identified 29 791 genes expressed (CPM > 0.22; ~10 counts) in at least two samples. On average samples expressed 25 980 genes. When restricting the gene set to only genes expressed in >90% of samples, we retained 17 022 Ensembl genes. The 12 769 genes only expressed in 1–90% of samples were expressed in varying proportions of samples reflecting the complex structure of gene expression in endometrium previously reported (Fung et al., 2018).
Integrating data for 6 230 993 (4 922 014 for trans) genotyped and imputed SNPs with the RNA-seq data from the 206 European women (17 022 genes expressed in >90% samples), and following Bonferroni correction for multiple testing, we detected genetic effects on gene expression in endometrium for 444 sentinel cis-eQTLs (P < 2.57 × 10−9) and significant trans-eQTLs for 30 genes (P < 5.97 × 10−13) (Table III) (Supplementary Fig. S1a and b). An additional 22 independent secondary cis-eQTL signals were detected following conditional analysis whereby the association test was rerun for each gene conditioning on the effect of the primary sentinel SNP. We identified novel cis-eQTLs, following Bonferroni correction, for 327 genes not previously reported as significant in endometrium. The eQTL dataset generated in this study is available at http://reproductivegenomics.com.au/shiny/endo_eqtl_rna/.
Table III. Number of cis and trans-eQTLs identified in endometrial tissue.
| eQTLs | Pass Bonferroni | Pass FDR (< 0.05) * | ||
|---|---|---|---|---|
| eQTLs | Unique genes | eQTLs | Unique genes | |
| Total cis-eQTL | 40 227 | 444 | 207 071 | 3726 |
| Total trans-eQTL | 1344 | 30 | 12 647 | 1369 |
*The number of cis-eQTLs and trans-eQTLs that pass the less stringent Benjamini-Hochberg threshold of FDR <0.05 is included for comparison (Pcis < 5.32 × 10−4; Ptrans < 4.73 × 10−8)
When comparing these results to our previous endometrium microarray study (Fung et al., 2018), we find 75% of Bonferroni significant eQTLs identified by RNA-Seq were nominally significant (P < 0.05) in the microarray data with ES highly correlated (R = 0.75). This reflects the high correlation between the average expression of each gene in endometrial tissue measured by RNA-Seq versus microarray (R = 0.902 ± 0.01) (Supplementary Fig. S2a–e) and a high correlation of gene expression within samples (R = 0.789 ± 0.101) (Supplementary Fig. S3). There were 28 FDR significant trans-eGenes (genes with a trans-eQTL) replicated between the two endometrial datasets.
Between tissue correlations
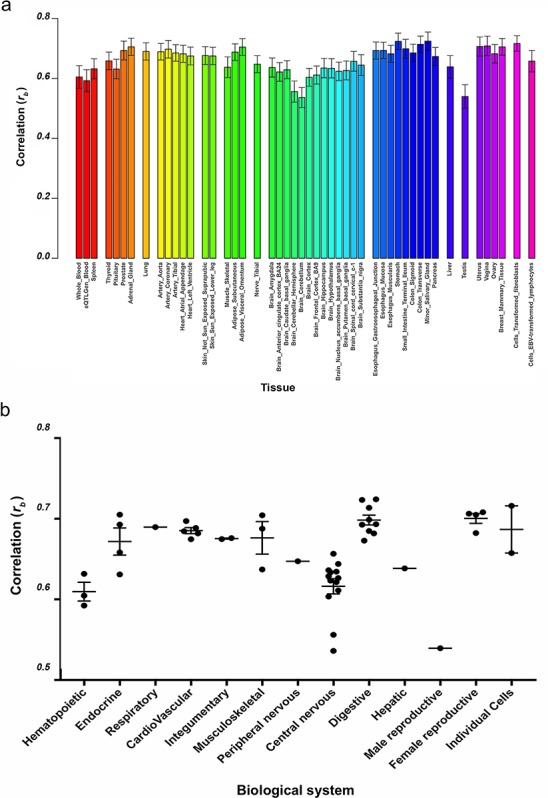
Overall, we observed good global correlation in genetic effects on gene expression between tissues, suggesting that many eQTLs exhibit more general shared effects. The correlation in eQTL effects (rb) between endometrium and the 48 tested tissues ranged from rb = 0.54 with brain cerebellum to rb = 0.72 with minor salivary gland (Fig. 2a, Supplementary Table SII), where rb represents the correlation of eQTL effects between tissues accounting for estimation errors in the eQTL effects (Qi et al., 2018). Female reproductive tissues including vagina, uterus, breast mammary tissue and ovary all had correlations above 0.68 (Fig. 2a and b). We observed large correlations between genetic effects in endometrium and digestive tissues such as the salivary gland (rb = 0.72), stomach (rb = 0.72) and colon (rb = 0.71) and individual cell types such as fibroblasts (rb = 0.72) (Fig. 2a and b). A lower correlation was observed with central nervous system tissues and hematopoietic tissues (Fig. 2a and b). The genetic effects on gene expression in endometrium had the highest correlation with tissue derived from the endoderm (average rb = 0.69) (Supplementary Fig. S4), compared to tissue derived from the other germ cell layers.
Figure 2. Correlation of cis-effects on gene expression between tissues. a) Correlation of cis-effects (rb) between endometrium and 48 tissues from GTEx and eQTLGen blood. Each tissue is represented by a different colour and grouped according to biological system. b) Correlation of cis-effects (rb) between endometrium and 48 tissues grouped into biological systems.
Shared and tissue specific eQTLs
A total of 305 Bonferroni significant endometrial eQTLs were also reported in at least one other GTEx tissue, with an average of 18 reported in any single tissue. The number of endometrial eQTLs reported in each GTEx tissue was highly correlated with the sample size (correlation = 0.88). When comparing endometrial eQTLs with the 48 GTEx tissues, we found a large proportion showed high tissue specificity, observed in only three tissues or fewer (174 sentinel eQTLs). Alternatively, the eQTLs found in more than three tissues were commonly observed across most tissues (185 sentinel eQTLs in >24 tissues) (Supplementary Fig. S5). However, when checking for overlap with the much larger eQTLGen blood dataset (n = 31 684) (Võsa et al., 2018), 71.6% of endometrial eQTLs were also reported in blood. The large overlap between eQTLs identified in endometrium and blood is likely due to the large sample size in the eQTLGen dataset which has sufficient power to detect eQTLs with much smaller effects. We identified 68 sentinel cis-eQTLs that may be specific to endometrium (Supplementary Table SIII), although a large proportion (85%) of the cis-eQTLs in endometrium were also reported in other tissues.
Association with reproductive traits and pathologies
Endometriosis
Differential gene expression between endometriosis cases (n = 143) and controls (n = 63) was tested using limma in R. Following correction for stage of cycle, no genes were significantly differentially expressed. Differences between cases and controls within proliferative and secretory stages with the largest sample sizes was also tested. No significant differences were observed between cases (n = 56) and controls (n = 16) in the mid-proliferative stage. Nominal differences in expression between cases (n = 27) and controls (n = 14) were observed for 43 genes in the mid-secretory stage (Table IV). Expression differences for these 43 genes however, were no longer significant following correction for the multiple testing of the three different comparisons, suggesting that these may represent false positives or that there is insufficient power to detect subtle genome-wide significant effects in our current dataset.
Table IV. Differentially expressed genes.
| Ensembl ID | Gene Name | Log Fold Change | P-Value | Adjusted P-Value |
|---|---|---|---|---|
| ENSG00000180730 | SHISA2 | 1.44 | 1.70E-06 | 3.04E-02 |
| ENSG00000204116 | CHIC1 | 0.61 | 5.21E-06 | 3.81E-02 |
| ENSG00000154864 | PIEZO2 | 1.22 | 6.41E-06 | 3.81E-02 |
| ENSG00000101311 | FERMT1 | −1.60 | 1.16E-05 | 4.64E-02 |
| ENSG00000108370 | RGS9 | 1.08 | 2.32E-05 | 4.64E-02 |
| ENSG00000259865 | AL390728.6 | 0.95 | 2.84E-05 | 4.64E-02 |
| ENSG00000160214 | RRP1 | −0.41 | 3.51E-05 | 4.64E-02 |
| ENSG00000171791 | BCL2 | 0.65 | 3.78E-05 | 4.64E-02 |
| ENSG00000075213 | SEMA3A | 1.49 | 4.11E-05 | 4.64E-02 |
| ENSG00000173681 | BCLAF3 | 0.39 | 4.15E-05 | 4.64E-02 |
| ENSG00000251273 | LINC02228 | 0.45 | 4.54E-05 | 4.64E-02 |
| ENSG00000105755 | ETHE1 | −0.76 | 4.83E-05 | 4.64E-02 |
| ENSG00000171121 | KCNMB3 | 0.76 | 4.86E-05 | 4.64E-02 |
| ENSG00000155657 | TTN | 0.96 | 5.21E-05 | 4.64E-02 |
| ENSG00000183671 | GPR1 | 1.65 | 5.49E-05 | 4.64E-02 |
| ENSG00000279377 | AC003973.3 | 1.45 | 5.51E-05 | 4.64E-02 |
| ENSG00000147465 | STAR | −2.21 | 5.53E-05 | 4.64E-02 |
| ENSG00000096968 | JAK2 | 0.58 | 5.64E-05 | 4.64E-02 |
| ENSG00000189056 | RELN | 1.48 | 6.14E-05 | 4.64E-02 |
| ENSG00000162302 | RPS6KA4 | −0.47 | 6.76E-05 | 4.64E-02 |
| ENSG00000197056 | ZMYM1 | 0.42 | 6.81E-05 | 4.64E-02 |
| ENSG00000163751 | CPA3 | 3.44 | 7.19E-05 | 4.64E-02 |
| ENSG00000165338 | HECTD2 | 0.53 | 7.26E-05 | 4.64E-02 |
| ENSG00000182253 | SYNM | 0.80 | 7.33E-05 | 4.64E-02 |
| ENSG00000131653 | TRAF7 | −0.40 | 7.35E-05 | 4.64E-02 |
| ENSG00000151164 | RAD9B | 0.89 | 7.50E-05 | 4.64E-02 |
| ENSG00000188906 | LRRK2 | 1.23 | 7.70E-05 | 4.64E-02 |
| ENSG00000156284 | CLDN8 | 1.05 | 8.05E-05 | 4.64E-02 |
| ENSG00000173950 | XXYLT1 | −0.41 | 8.44E-05 | 4.64E-02 |
| ENSG00000233251 | AC007743.1 | 1.28 | 8.55E-05 | 4.64E-02 |
| ENSG00000151229 | SLC2A13 | 0.87 | 8.70E-05 | 4.64E-02 |
| ENSG00000114120 | SLC25A36 | 0.42 | 8.85E-05 | 4.64E-02 |
| ENSG00000137077 | CCL21 | 2.42 | 9.01E-05 | 4.64E-02 |
| ENSG00000148334 | PTGES2 | −0.45 | 9.19E-05 | 4.64E-02 |
| ENSG00000165731 | RET | 1.27 | 9.50E-05 | 4.64E-02 |
| ENSG00000243709 | LEFTY1 | −2.14 | 9.73E-05 | 4.64E-02 |
| ENSG00000188321 | ZNF559 | 0.53 | 9.81E-05 | 4.64E-02 |
| ENSG00000148600 | CDHR1 | 1.27 | 1.01E-04 | 4.64E-02 |
| ENSG00000136261 | BZW2 | −0.52 | 1.01E-04 | 4.64E-02 |
| ENSG00000119383 | PTPA | −0.42 | 1.14E-04 | 4.96E-02 |
| ENSG00000164128 | NPY1R | 1.73 | 1.14E-04 | 4.96E-02 |
| ENSG00000137871 | ZNF280D | 0.41 | 1.17E-04 | 4.96E-02 |
Genes differentially expressed between women with and without endometriosis in endometrium in the mid-secretory stage of the menstrual cycle. Genes with a positive fold change are upregulated in cases and those with a negative fold change are downregulated in cases.
We performed a transcriptome-wide association analysis to identify gene expression associated with endometriosis risk. Gene expression and genotype data from the 206 samples was used to estimate the weighted effect of each SNP on each cis-gene and combined with summary-level endometriosis GWAS data (Sapkota et al., 2017) to impute gene expression and perform a TWAS. Using a transcriptome-wide significance threshold of 3.28 × 10−6, we identified 252 genes associated with endometriosis located at 39 independent loci (Fig. 3, Table V, Supplementary Table SIV). Five of these loci harboured genome-wide significant SNPs associated with endometriosis including; rs1903068 on chromosome 4 near kinase insert domain receptor (KDR), rs12700667 on chromosome 7 near LOC100506236, rs10090060 on chromosome 8 near ganglioside induced differentiation associated protein 1 (GDAP1), rs1802669 on chromosome 10 near MLLT10 Histone Lysine Methyltransferase DOT1L Cofactor (MLLT10) and rs4762326 on chromosome 12 near vezatin (VEZT) (Table V). The remaining loci all included nominally significant SNPs from the GWAS ranging from P = 2.64 × 10−3–4.40 × 10−7 (Table V). Many implicated regions contain multiple significant genes whose predicted gene expression was, in most cases, correlated (Supplementary Fig. S6). The most significant gene at each locus is presented in Table V. This analysis highlights genes potentially involved in endometriosis pathogenesis previously associated with endometriosis risk and provides stronger support for novel regions showing only nominal significance in GWAS results.
Figure 3. Association between gene expression in endometrium and endometriosis. Manhattan plot showing the strength of association between endometriosis and gene expression in endometrium. Each point on the plot represents a gene and alternating colours distinguish different chromosomes. The red line represents the transcriptome-wide Bonferroni significance cut off of P < 3.28 × 10 – 6.
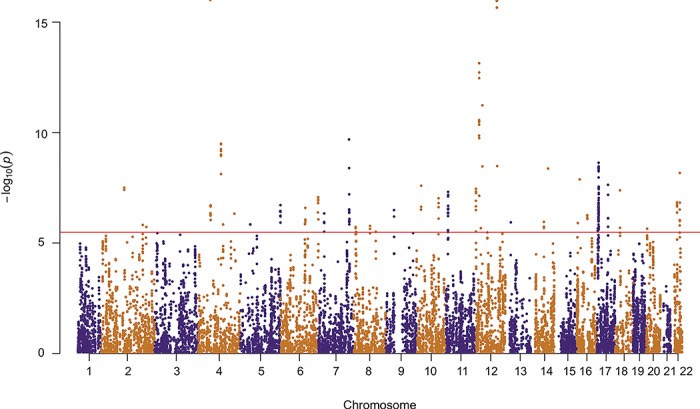
Table V. Significant TWAS genes.
| TWAS Region | Top SNP in region (±1 MB) from 2017 Endometriosis GWAS | |||||||
|---|---|---|---|---|---|---|---|---|
| CHR | Start | End | Top Gene | Top SNP | CHR | BP | Effect | P_value |
| 2 | 107 313 787 | 107 503 564 | ST6GAL2 | rs1516201 | 2 | 108 303 027 | 0.088 | 5.99E-05 |
| 2 | 190 744 335 | 191 068 210 | C2orf88 | rs1241158 | 2 | 191 725 653 | −0.171 | 5.08E-04 |
| 2 | 206 858 445 | 206 951 027 | INO80D | rs112822178 | 2 | 206 151 383 | −0.221 | 5.22E-05 |
| 4 | 55 095 264 | 57 194 791 | PDGFRA | rs1903068 | 4 | 56 008 477 | 0.100 | 1.04E-11 |
| 4 | 103 552 660 | 103 940 896 | LRRC37A15P | rs12498897 | 4 | 104 934 629 | −0.058 | 2.74E-04 |
| 4 | 113 066 553 | 113 116 412 | FAM241A | rs13116274 | 4 | 112 133 135 | −0.049 | 8.67E-04 |
| 4 | 164 031 225 | 164 088 073 | NAF1 | rs10007601 | 4 | 164 934 874 | −0.060 | 3.58E-04 |
| 5 | 44 745 002 | 44 828 694 | AC093297.2 | rs13186320 | 5 | 44 302 177 | 0.063 | 2.64E-03 |
| 5 | 179 660 143 | 180 288 286 | ZFP62 | rs6877489 | 5 | 179 677 545 | 0.053 | 4.87E-04 |
| 6 | 110 567 131 | 112 254 939 | FYN | rs11153311 | 6 | 112 009 325 | 0.092 | 3.92E-06 |
| 6 | 168 841 831 | 170 584 692 | C6orf120 | rs9460235 | 6 | 170 391 393 | −0.171 | 4.24E-05 |
| 7 | 26 191 860 | 26 413 949 | NFE2L3 | rs12700667 | 7 | 25 901 639 | 0.095 | 9.08E-10 |
| 7 | 138 145 079 | 140 177 035 | TRIM24 | rs28469460 | 7 | 139 378 750 | 0.054 | 3.66E-04 |
| 8 | 8 859 657 | 9 009 084 | ERI1 | rs13261266 | 8 | 9 356 565 | −0.080 | 7.62E-06 |
| 8 | 10 962 201 | 10 967 236 | AF131215.5 | rs756038 | 8 | 11 336 781 | −0.054 | 1.43E-04 |
| 8 | 74 884 672 | 74 897 118 | TMEM70 | rs78103255 | 8 | 75 311 331 | 0.087 | 4.40E-07 |
| 8 | 100 973 164 | 101 143 496 | RGS22 | rs2721973 | 8 | 101 492 473 | 0.060 | 2.58E-05 |
| 9 | 37 120 536 | 37 436 987 | ZCCHC7 | rs67952628 | 9 | 37 669 203 | −0.103 | 1.32E-03 |
| 10 | 21 068 902 | 21 814 611 | SKIDA1 | rs7084454 | 10 | 21 821 274 | 0.065 | 9.06E-06 |
| 10 | 101 370 282 | 101 491 857 | SLC25A28 | rs2495704 | 10 | 102 434 157 | −0.112 | 2.11E-05 |
| 11 | 8 703 958 | 9 550 071 | TMEM41B | rs118135101 | 11 | 9 576 348 | −0.085 | 3.59E-04 |
| 12 | 73 725 | 772 872 | CCDC77 | rs525631 | 12 | 335 010 | 0.048 | 7.79E-04 |
| 12 | 14 518 610 | 15 750 333 | ERP27 | rs66716825 | 12 | 15 554 246 | 0.067 | 1.48E-05 |
| 12 | 22 778 009 | 22 843 599 | ETNK1 | rs7307965 | 12 | 23 132 669 | 0.055 | 2.92E-04 |
| 12 | 29 542 227 | 29 937 692 | TMTC1 | rs10743670 | 12 | 29 857 902 | 0.044 | 1.62E-03 |
| 12 | 94 542 499 | 96 794 338 | LTA4H | rs4762326 | 12 | 95 668 951 | 0.079 | 2.20E-09 |
| 13 | 24 995 064 | 25 086 948 | PARP4 | rs2057561 | 13 | 26 059 265 | −0.065 | 1.91E-05 |
| 14 | 59 655 364 | 59 972 128 | DAAM1 | rs4542561 | 14 | 59 883 922 | −0.098 | 4.88E-06 |
| 14 | 78 708 734 | 80 330 762 | NRXN3 | rs61976091 | 14 | 79 079 685 | −0.108 | 1.89E-04 |
| 16 | 4 239 375 | 4 292 081 | SRL | rs224215 | 16 | 3 301 360 | 0.067 | 1.92E-05 |
| 16 | 12 756 919 | 12 897 874 | CPPED1 | rs112606877 | 16 | 12 939 765 | 0.069 | 1.88E-04 |
| 16 | 46 614 466 | 47 735 434 | VPS35 | rs11863453 | 16 | 47 464 948 | −0.051 | 1.20E-03 |
| 17 | 4 067 201 | 4 269 923 | UBE2G1 | rs2585274 | 17 | 5 125 249 | 0.063 | 1.47E-05 |
| 17 | 6 779 954 | 8 286 531 | DNAH2 | rs62059792 | 17 | 7 437 665 | −0.066 | 1.46E-04 |
| 17 | 48 260 650 | 48 450 575 | COL1A1 | rs9907631 | 17 | 49 216 162 | −0.064 | 1.19E-03 |
| 18 | 18 526 867 | 19 105 378 | ROCK1 | rs112763730 | 18 | 18 666 368 | 0.123 | 3.91E-04 |
| 20 | 5 080 486 | 5 093 749 | TMEM230 | rs439007 | 20 | 5 024 928 | −0.064 | 1.85E-05 |
| 22 | 29 083 731 | 29 453 475 | ZNRF3 | rs9614041 | 22 | 30 123 029 | −0.104 | 2.60E-04 |
| 22 | 41 220 539 | 41 636 938 | EP300 | rs34503826 | 22 | 40 833 762 | −0.049 | 4.25E-04 |
GWAS data is from Sapkota et al. (2017). All positions are based on the hg19 genome version.
Loci associated with endometriosis and the most significant GWAS SNPs within these regions.
Functional annotation of genetic variants associated with complex traits and diseases is another valuable method to identify target genes and prioritise them for further study. Previous studies have reported eQTLs associated with endometriosis on chromosome 1 (Powell et al., 2016) and 12 (Holdsworth-Carson et al., 2016). In this study, we identified eQTLs for two genes, VEZT and FYVE, RhoGEF and PH domain containing 6 (FGD6) overlapping the GWAS signal for endometriosis risk locus on chromosome 12 (Table VI). SMR is a method for testing whether the eQTLs and GWAS signals overlap by chance or have some causal association. Implementation of a standard SMR analysis (Zhu et al., 2016) found no significant associations between genetic variants, endometrial gene expression and endometriosis risk. The standard implementation for SMR analysis tests only the most significant eQTL at each locus and assumes only one causal variant is associated with both gene expression and the trait. To assess the possibility that additional independent eQTLs and GWAS signals in each region contribute to heterogeneity and dilute the signal, we conducted a multi-SNP-based SMR (SMR-multi) (Wu et al., 2018). This increases the power to detect a pleiotropic signal by including multiple SNPs within each individual cis-eQTL locus. There were no signals that passed the SMR-multi analysis.
Table VI. eQTLs with eSNPs associated with endometriosis risk.
| Ensembl_ID | SNP | Chr | BP | Statistic | Beta | P-Value | FDR | Gene_ID |
|---|---|---|---|---|---|---|---|---|
| ENSG00000180263 | rs12320196 | 12 | 95 251 609 | 5.073 | 0.233 | 9.52E-07 | 2.63E-04 | FGD6 |
| ENSG00000028203 | rs12320196 | 12 | 95 251 609 | 4.556 | 0.111 | 9.47E-06 | 1.95E-03 | VEZT |
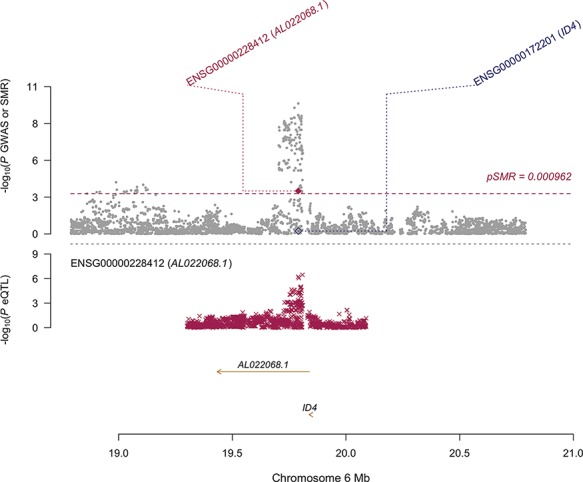
A modified SMR selecting the most significant endometriosis-associated GWAS SNPs as the instrument, thereby treating endometriosis as the exposure, reduced the multiple testing burden and detected three signals that pass both the SMR test and the HEIDI (HEterogeneity In Depedent Instruments) test; FGD6, VEZT (Fig. 4; Table VII) and AL022068.1 (Fig. 5; Table VII). The HEIDI test is used to distinguish independent overlapping signals and the same SNP influencing expression and disease risk. SNPs with low HEIDI P-values have a higher probably of being independent overlapping signals and are subsequently disregarded. The expression of both FGD6 and VEZT was also significantly associated with endometriosis in the TWAS analysis.
Figure 4. SMR locus plot of the VEZT/FGD6 locus. In the top plot, grey dots represent P-values for SNPs reported in Sapkota et al.’s (2017) GWAS meta-analysis for endometriosis and diamonds represent the P-values for probes from the reverse SMR test. Crosses in the middle and bottom plots represent the eQTL P-values of SNPs associated with expression of VEZT and FGD6 in endometrium respectively. Positions are in hg38.
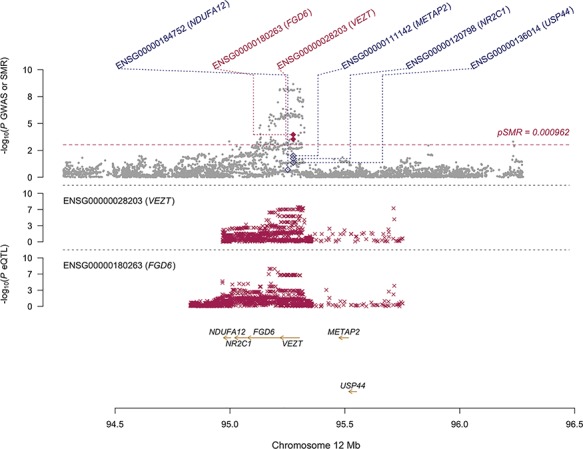
Table VII. Modified SMR results in endometrium.
| Gene | SNP | Chr | A1 | b_eQTL | p_eQTL | b_GWAS | p_GWAS | b_SMR | p_SMR | p_HEIDI |
|---|---|---|---|---|---|---|---|---|---|---|
| FGD6 | rs4762326 | 12 | T | 0.233 | 9.52E-07 | 0.079 | 2.20E-09 | 2.966 | 1.09E-04 | 5.12E-02 |
| VEZT | rs4762326 | 12 | T | 0.111 | 9.47E-06 | 0.079 | 2.20E-09 | 1.410 | 2.89E-04 | 2.46E-01 |
| AL022068.1 | rs760794 | 6 | T | 0.252 | 7.25E-05 | 0.085 | 1.79E-10 | 2.959 | 6.15E-04 | 1.47E-01 |
P-value significance thresholds:
PSMR < 9.62E-04
PHEIDI > 0.017
Results of modified SMR analysis using endometrial eQTLs and endometriosis summary statistics.
Figure 5. SMR locus plot of the AL022068.1 locus. In the top plot, grey dots represent P-values for SNPs reported in Sapkota et al.’s (2017) GWAS meta-analysis for endometriosis and diamonds represent the P-values for probes from the reverse SMR test. Crosses in the bottom plot represent the eQTL P-values of SNPs associated with expression AL022068.1 in endometrium. Positions are in hg38.
To increase power, we performed the same SMR analysis using the larger eQTLGen blood eQTL dataset as a proxy. Whilst this increases the power to detect genetic variants that effect gene expression consistently in blood and endometrium, we lose power to detect endometrium specific signals. Using the standard SMR approach, we detected one association between rs2473290 and the cell division cycle 42 (CDC42) gene that passed the SMR test (PSMR = 5.77 × 10−10), but did not pass the HEIDI test (PHEIDI = 3.74 × 10−5). We repeated the SMR analysis after conditioning the CDC42 eQTLs and GWAS on the lead SNPs from an additional three independent CDC42 eQTL signals. The CDC42 association remained significant and sat just under the threshold for the HEIDI test (PSMR = 1.44 × 10−6; PHEIDI = 1.11 × 10−2).
Like that performed for endometrial eQTLs, we also conducted an SMR-multi analysis using the eQTLGen data to check if heterogeneity results from multiple independent eQTL signals at this locus. Using all SNPs in each region resulted in an SMR-multi p-value (PSMR-multi = 7.45 × 10−10) for CDC42 similar to that observed from the standard SMR test (Supplementary Fig. S7). However, we detected a second association between rs11801382 and long intergenic non-protein coding RNA 339 (LINC00339) that failed to pass the standard SMR test, but passed the SMR-multi test (PSMR-multi = 2.73 × 10−10, PSMR = 5.64 × 10−2) (Supplementary Fig. S7). The modified SMR test using the eQTLGen data found 18 associations that passed the SMR test (PSMR < 2.78 × 10−4) including LINC00339, CDC42, FGD6 and VEZT (Table VIII). However, none of the 18 pass the HEIDI test and therefore we could not distinguish between independent signals or one SNP affecting expression and disease risk in these regions.
Table VIII. Modified SMR results in blood.
| Gene | SNP | Chr | A1 | b_eQTL | p_eQTL | b_GWAS | p_GWAS | b_SMR | p_SMR | p_HEIDI |
|---|---|---|---|---|---|---|---|---|---|---|
| LINC00339 | rs12037376 | 1 | A | −0.658 | 3.27E-310 | 0.147 | 8.87E-17 | −4.475 | 2.58E-16 | 2.63E-15 |
| CDC42 | rs12037376 | 1 | A | 0.361 | 1.88E-243 | 0.147 | 8.87E-17 | 2.455 | 6.96E-16 | 1.96E-15 |
| SRD5A3 | rs1903068 | 4 | A | 0.095 | 2.90E-29 | 0.100 | 1.04E-11 | 0.953 | 5.98E-09 | 4.62E-09 |
| NDUFA12 | rs4762326 | 12 | T | 0.115 | 3.38E-47 | 0.079 | 2.20E-09 | 1.462 | 3.27E-08 | 1.95E-10 |
| FGD6 | rs4762326 | 12 | T | 0.126 | 2.91E-26 | 0.079 | 2.20E-09 | 1.595 | 1.89E-07 | 8.78E-10 |
| RMND1 | rs1971256 | 6 | T | −0.164 | 1.06E-53 | −0.089 | 3.74E-08 | 1.839 | 2.18E-07 | 1.28E-05 |
| CCDC170 | rs1971256 | 6 | T | 0.145 | 8.63E-41 | −0.089 | 3.74E-08 | −1.625 | 3.60E-07 | 1.62E-06 |
| NR2C1 | rs4762326 | 12 | T | −0.071 | 4.54E-19 | 0.079 | 2.20E-09 | −0.903 | 6.73E-07 | 2.38E-09 |
| PAX8-AS1 | rs10167914 | 2 | A | 0.092 | 3.74E-15 | −0.111 | 1.10E-09 | −0.832 | 1.46E-06 | 8.40E-06 |
| CLOCK | rs1903068 | 4 | A | 0.058 | 1.28E-11 | 0.100 | 1.04E-11 | 0.585 | 1.60E-06 | 4.88E-05 |
| ATIC | rs1250244 | 2 | C | 0.120 | 1.54E-19 | −0.102 | 8.93E-08 | −1.176 | 4.17E-06 | 6.82E-05 |
| HSPG2 | rs12037376 | 1 | A | 0.112 | 4.56E-08 | 0.147 | 8.87E-17 | 0.759 | 4.90E-06 | 2.43E-12 |
| PSD4 | rs10167914 | 2 | A | −0.056 | 8.65E-11 | −0.111 | 1.10E-09 | 0.511 | 8.91E-06 | 1.26E-07 |
| RAP1GAP | rs12037376 | 1 | A | 0.055 | 5.10E-07 | 0.147 | 8.87E-17 | 0.376 | 1.71E-05 | 5.78E-11 |
| SRD5A3-AS1 | rs1903068 | 4 | A | 0.117 | 3.83E-08 | 0.100 | 1.04E-11 | 1.171 | 1.90E-05 | 1.26E-07 |
| NBPF3 | rs12037376 | 1 | A | 0.063 | 9.56E-07 | 0.147 | 8.87E-17 | 0.430 | 2.42E-05 | 1.50E-13 |
| PAX8 | rs10167914 | 2 | A | 0.050 | 6.58E-09 | −0.111 | 1.10E-09 | −0.455 | 2.65E-05 | 6.00E-05 |
| VEZT | rs4762326 | 12 | T | 0.046 | 6.62E-09 | 0.079 | 2.20E-09 | 0.589 | 3.12E-05 | 1.80E-07 |
P-value significance thresholds:
PSMR < 2.78E-04
PHEIDI > 2.78E-03
Results of modified SMR analysis using eQTLGen blood eQTLs and endometriosis GWAS summary statistics.
Reproductive traits
Endometrial eQTLs also provide a valuable resource to functionally annotate genetic variants associated with other reproductive traits and diseases. We tested for overlap between independent Bonferroni significant eQTLs and SNPs associated with traits in the GWAS Catalog. We identified genetic variants that regulate endometrial gene expression and were associated with 288 traits and diseases (P < 5 × 10−8) (Supplementary Table SV). This included various reproductive traits such as age of menarche onset, age of menopause onset, polycystic ovary syndrome, ovarian cancer and breast cancer. Two signals passed both the SMR and HEIDI test for age at menopause. These included loci on chromosomes 17 (rs2175957), associated with expression of Neighbor of BRCA1 LncRNA 2 (NBR2) (PSMR = 2.27 × 10−7, PHEIDI = 5.23 × 10−2) and chromosome 20 (rs11699690), associated with expression of Copine 1 (CPNE1) (PSMR = 8.16 × 10−7, PHEIDI = 8.51 × 10−2) (Supplementary Fig. S8). Three signals passed both the SMR and HEIDI tests for epithelial ovarian cancer including two on chromosome 17; rs80028338 associated with expression of Leucine Rich Repeat Containing 37A (LRRC37A) (PSMR = 7.51 × 10−10, PHEIDI = 3.44 × 10−1) and rs17665188 associated with expression of Leucine Rich Repeat Containing 37 Member A2 (LRRC37A2) (PSMR = 1.5 × 10−9, PHEIDI = 6.68 × 10−1) (Supplementary Fig. S9a). The third signal was located on chromosome 8; rs76837345 associated with expression of Charged Multivesicular Body Protein 4C (CHMP4C) (PSMR = 3.25 × 10−6, PHEIDI = 9.73 × 10−2) (Supplementary Fig. S9b). No signals passed the SMR test for epithelial endometrioid ovarian cancer.
Discussion
We analysed genetic regulation of gene expression in the endometrium to determine how this relates to regulation in other human tissues, and whether genetic risk factors for endometriosis act through genetic effects on endometrial gene expression. The majority of common genetic effects on disease risk are located in non-coding regions of the genome and most likely act through regulation of gene expression in relevant pathogenic tissues (Consortium et al., 2017; Gamazon et al., 2018). We studied endometrial tissue because it is not represented in international projects like GTEx (Consortium et al., 2017) and is a probable source of cells that initiate endometriosis lesions (Sampson, 1927; Anglesio et al., 2017; Noë et al., 2018; Suda et al., 2018). The transport of endometrial cells to the peritoneal cavity by retrograde menstruation as a cause of endometriosis was first proposed by Sampson (1927) and is supported by recent studies of somatic mutations in endometrium and endometriosis lesions (Anglesio et al., 2017; Noë et al., 2018; Suda, et al., 2018). Our results show that the expression of genes located in genomic regions associated with endometriosis are significantly enriched in female reproductive tissues including uterus and endometrium, supporting this approach.
We first analysed genetic effects on gene expression (eQTLs) in endometrium and compared the eQTL profiles with tissues in GTEx (Consortium et al., 2017) and large eQTL studies in blood. Generating an endometrial eQTL dataset from genotype and RNA-seq data, rather than our previous microarray data (Fung et al., 2018) allowed a more accurate comparison with eQTLs reported in the GTEx (Consortium et al., 2017). The large proportion (71.6%) of endometrial eQTLs also reported in the eQTLGen blood dataset highlights the potential power of using large datasets as a proxy for tissue shared eQTLs. Correlation in eQTL effects between endometrium and other tissues ranged from 0.54 to 0.72 with high correlations in eQTL effects between endometriosis and other reproductive tissues (vagina, uterus, breast, ovary). The relatively high correlations in eQTL effects is consistent with shared eQTL effects reported in the GTEx data (Consortium et al., 2017; Ongen et al., 2017), with more shared effects observed among tissues with greater biological similarity, for example among the reproductive tissues (Consortium et al., 2017; Ongen et al., 2017). Lower correlations between endometrium and blood in GTEx, endometrium and testis, and endometrium and brain is consistent with previous reports showing eQTLs in whole blood and testis have a high degree of tissue specificity (Ongen et al., 2017).
The highest correlation of eQTL effects between endometrium and GTEx was observed with tissues of the digestive system. The underlying biology that leads to shared eQTLs between tissue is not yet clear. Digestive tract tissue is composed of an epithelial cell lining, endocrine epithelial glandular structures and mesenchymal derived support cells that function to secrete compounds required for tissue digestion and gut homeostasis (Okumura and Takeda, 2017). The endometrium also has an epithelial lining with endocrine epithelial glandular structures and an endocrine secretory function designed to facilitate embryo implantation (Hempstock et al., 2004), supported by mesenchymal derived stromal cells, surrounded by the smooth muscle of the myometrium. While many differences in the secreted substances exist, the molecular mechanisms to perform these roles may require similar gene regulation.
In contrast to the high correlation in cis-eQTL effects between tissues, no endometrial trans-eGenes are reported in GTEx tissues and only three Bonferroni significant trans-eGenes in endometrium are reported in the eQTLGen blood dataset (Võsa et al., 2018). These findings are consistent with trans-eQTLs being more tissue specific (Grundberg et al., 2012; Kirsten et al., 2015; Consortium et al., 2017). The number of trans-genes identified in our study relative to sample size is similar to trans-eQTL mapping for testis in GTEx which had the highest number of reported trans-eGenes (n = 35) (Consortium et al., 2017). The larger number of trans-eQTLs in testis and endometrium may reflect the importance of trans-SNPs in regulating gene expression in reproductive tissues. Larger eQTL studies in endometrium would be required to have sufficient power to accurately investigate trans-acting genetic regulation of transcription.
We next analysed association between endometriosis and endometrial gene regulation. Differential expression analysis found no genome-wide significant differences in gene expression between endometriosis cases and controls following correction for multiple testing, consistent with our previous reports in eutopic endometrium (Fung et al., 2017, 2018). An alternative approach combining gene expression and genotype data uses a powerful statistical method to impute gene expression from our eQTL data in a larger sample and conduct a TWAS. TWAS methods have been applied to identify functional loci in prostate cancer, obesity-related traits, Alzheimer’s, Crohn’s disease, diabetes and rheumatoid arthritis (Gusev et al., 2016; Mancuso et al., 2018; Hu et al., 2019; Nagpal et al., 2019). We conducted the first TWAS analysis for endometriosis using our endometrial gene expression data and identified 39 genomic regions associated with endometriosis. Five of the loci associated with endometriosis in the TWAS contain GWAS SNPs previously associated with the disease, including the VEZT locus, KDR locus, GDAP1 locus and the MLLT10 locus. The association between expression at the VEZT locus and endometriosis risk highlighted in the TWAS has been reported previously (Sapkota et al., 2017; Fung et al., 2018), and is supported by SMR analyses in this study. The other regions are novel. These have not been reported as genome-wide significant but all have nominal evidence of association in the GWAS studies. TWAS association does not imply causation but rather predicts differential gene expression between endometriosis cases and controls, with or without biological consequence. The correlation between predicted expression of genes at each genomic region limits the resolution to identify single target genes. Instead the method highlights potential candidates that may warrant further investigation.
Several signals for genetic risk factors for endometriosis from GWAS (Sapkota et al., 2017) and eQTLs map to the same regions of the genome. The overlapping signals are observed in different data sets and the overlap can occur by chance. Therefore, we used implementations of the SMR test to formally evaluate overlap in the signals and exclude chance overlap. We identified eQTLs in endometrium in which the same variant on chromosome 12 was associated with expression of both VEZT and FGD6, as well as risk of endometriosis. Several studies have reported associations between genetic variants at 12q22 locus near VEZT and increased risk of endometriosis (Nyholt et al., 2012; Rahmioglu et al., 2015; Holdsworth-Carson et al., 2016; Matalliotakis et al., 2017) and both genes were significantly associated with endometriosis in our TWAS analysis. The modified SMR analysis for expression in both endometrium and blood provides strong evidence that the same causal variant influences VEZT and FGD6 expression and endometriosis risk. Both VEZT and FGD6 play a role in plasma membrane, cell adhesion and cytoskeletal remodelling, all of which are important for development of endometriotic lesions (Guo et al., 2011; Holdsworth-Carson et al., 2016). Increased expression of VEZT has been reported in endometriosis cases, as has increased expression of epidermal growth factor receptor (EFGR) that is associated with expression of FGD6 (Ejskjær et al., 2009; Meola et al., 2010; de Graauw et al., 2014). Both genes have also been associated with expression of CDC42, another endometriosis risk gene (Powell et al., 2016) responsible for cell division, growth and migration (Miao et al., 2013; Steenblock et al., 2014).
We observed a potential causal association between CDC42 expression and endometriosis. CDC42 passed the SMR test in blood and, following conditional analysis for multiple eQTL signals in the locus, came very close to passing the heterogeneity test. The multi-SNP SMR analysis identified a second variant associated with the expression of nearby LINC00339 and endometriosis risk. These results are consistent with our previous studies where genetic regulation of LINC00339 has been associated with endometriosis (Powell et al., 2016; Fung et al., 2018). Our results provide further support that genetic effects on endometriosis risk act through altered gene expression of VEZT and FGD6 on chromosome 12 (Holdsworth-Carson et al., 2016; Powell et al., 2016) and through LINC00339 and possibly CDC42 on chromosome 1 (Powell et al., 2016).
Using the standard SMR approach, we identified pleiotropic and potential causal associations between gene expression and age at menopause and ovarian cancer. The eQTLs for NBR2 and CPNE1 that were associated with age of menopause are reported in all 48 GTEx tissues but with varying ES. NBR2, which is located close to the tumour suppressor BRAC1, has been shown to have similar function to that of a tumour suppresser-regulating AMPK (Liu et al., 2016). The largest genetic effect for NBR2 was observed in ovary. We also identified eQTLs associated with ovarian cancer; one for LRRC37A was only reported in endometrium and the larger eQTLGen dataset with low expression across most GTEx tissues with the exception of testis. Interestingly, the eQTL for its paralog LRRC37A2 was reported in 47 GTEx tissues, again with varying ES. Another signal passing the ovarian cancer SMR was for CHMP4, which is known as a prime candidate for epithelial ovarian cancer susceptibility due to its role in cell cycle regulation and regulation by TP53 (Pharoah et al., 2013).
Our study has important limitations. This is the largest study of genetic effects on gene expression in endometrium but it is small and lacks power in comparison with much larger eQTL studies in blood. Identifying and recruiting tissue donors is challenging due to the invasiveness of sampling, therefore limiting our ability to collect tissue on a large scale. Our analysis is conducted in fresh endometrial tissue consisting of multiple cell types. Consequently, expression levels are an average of expression from different cell types within the samples, which may mask smaller cell specific effects and identify only those large enough to be observed at a tissue level. Changes in cellular composition and cell activity across the cycle will contribute to variation in transcription across the cycle and between samples. We corrected for stage of the menstrual cycle in our analyses that will include changes in cellular composition across the cycle. Characterizing genetic regulation in individual cell types within the endometrium may also be important to understand the functional effects of disease risk variants. Future studies of expression in different cell types may identify novel cell-specific eQTLs if separation of the cells does not disrupt gene regulation and the studies have sufficient power. Techniques such as single-cell RNA-Seq offer an innovative solution to measure expression from individual cell populations; however, this technique would also introduce new practical, economic and computational challenges.
In conclusion, generation of an endometrial eQTL dataset using RNA-Seq and genome-wide genotyping data identified 327 novel genetic effects on transcription in endometrium. The ability to compare this dataset with publicly available eQTL datasets in GTEx and eQTLGen has identified high correlations in genetic effects between endometrium and both reproductive and digestive tissues and has allowed us to identify 68 endometrial cis-eQTLs not observed in other tissues. Analysis of genetic effects on gene expression in endometrium provide further evidence that genetic risk factors for endometriosis act through expression of VEZT, FGD6, CDC42 and LINC00339. This was supported by the TWAS analysis with association for the VEZT/FGD6 locus and endometriosis risk. The TWAS also identified a further 38 genomic regions harbouring potential target genes for functional follow-up. Expanding our knowledge of the genetic regulation in endometrium and integrating our data with publically available datasets creates an important resource to identify gene targets regulating female reproductive traits and diseases.
Supplementary Material
Acknowledgements
We thank the women who participated in the study, research nurses Ranita Charitra, Tracy Middleton and Irene Bell, who recruited and consented all the endometrial biopsy patients at the RWH, and the surgeons and anaesthetists who collected tissue and blood samples.
Authors’ roles
S.M, R.I.K, J.N.F, P.A.W.R and G.W.M designed the study with input from the other authors. S.M, R.I.K, J.N.F, J.E.G, W.T.T and S.J.H-C coordinated data collection and QC of data, with support, input and oversight from G.G, F.Y, R.R, T.Q, J.Y, M.H, B.M, P.A.W.R and G.W.M. Data analysis was performed by S.M, R.I.K, R.R and F.Y and was interpreted by all authors. S.W.L built the web browser for summary results. S.M, R.I.K, B.M and G.W.M drafted the report with input from all other authors. The final report has been critically revised and approved by all authors.
Funding
National Health and Medical Research Council (NHMRC) under project grants GNT1026033, GNT1049472, GNT1046880, GNT1050208, GNT1105321, GNT1083405 and GNT1107258; NHMRC Fellowship (GNT1078399) to G.W.M.; Australian Research Council (ARC) Fellowship (FT180100186) to J.Y.
Conflict of interest
The authors declare that they have no conflicts of interest.
References
- Andrews S. Fast QC: a quality control tool for high throughput sequence data, 2010
- Anglesio MS, Papadopoulos N, Ayhan A, Nazeran TM, Noë M, Horlings HM, Lum A, Jones S, Senz J, Seckin T et al. . Cancer-associated mutations in endometriosis without cancer. N Engl J Med 2017;376:1835–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulletti C, Coccia ME, Battistoni S, Borini A. Endometriosis and infertility. J Assist Reprod Genet 2010;27:441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium G. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015;348:648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium G, Aguet F, Brown AA, Castel SE, Davis JR, He Y, Jo B, Mohammadi P, Park Y, Parsana P et al. . Genetic effects on gene expression across human tissues. Nature 2017;550:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day FR, Ruth KS, Thompson DJ, Lunetta KL, Pervjakova N, Chasman DI, Stolk L, Finucane HK, Sulem P, Bulik-Sullivan B et al. . Large-scale genomic analyses link reproductive aging to hypothalamic signaling, breast cancer susceptibility and BRCA1-mediated DNA repair. Nat Genet 2015;47:1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graauw M, Cao L, Winkel L, Van Miltenburg M, Le Dévédec SE, Klop M, Yan K, Pont C, Rogkoti V, Tijsma A. Annexin A2 depletion delays EGFR endocytic trafficking via cofilin activation and enhances EGFR signaling and metastasis formation. Oncogene 2014;33:2610. [DOI] [PubMed] [Google Scholar]
- Ejskjær K, Sorensen BS, Poulsen SS, Mogensen O, Forman A, Nexo E. Expression of the epidermal growth factor system in eutopic endometrium from women with endometriosis differs from that in endometrium from healthy women. Gynecol Obstet Invest 2009;67:118–126. [DOI] [PubMed] [Google Scholar]
- Evans J, Salamonsen LA, Winship A, Menkhorst E, Nie G, Gargett CE, Dimitriadis E. Fertile ground: human endometrial programming and lessons in health and disease. Nat Rev Endocrinol 2016;12:654. [DOI] [PubMed] [Google Scholar]
- Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016;32:3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadista J, Vikman P, Laakso EO, Mollet IG, Esguerra JL, Taneera J, Storm P, Osmark P, Ladenvall C, Prasad RB et al. . Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc Natl Acad Sci 2014;111:13924–13929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane HK, Reshef YA, Anttila V, Slowikowski K, Gusev A, Byrnes A, Gazal S, Loh P-R, Lareau C, Shoresh N et al. . Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat Genet 2018;50:621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung JN, Girling JE, Lukowski SW, Sapkota Y, Wallace L, Holdsworth-Carson SJ, Henders AK, Healey M, Rogers PAW, Powell JE et al. . The genetic regulation of transcription in human endometrial tissue. Hum Reprod 2017;32:893–904. [DOI] [PubMed] [Google Scholar]
- Fung JN, Mortlock S, Girling JE, Holdsworth-Carson SJ, Teh WT, Zhu Z, Lukowski SW, McKinnon BD, McRae A, Yang J et al. . Genetic regulation of disease risk and endometrial gene expression highlights potential target genes for endometriosis and polycystic ovarian syndrome. Sci Rep 2018;8:11424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon ER, Segrè AV, van de Bunt M, Wen X, Xi HS, Hormozdiari F, Ongen H, Konkashbaev A, Derks EM, Aguet F et al. . Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat Genet 2018;50:956–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudice LC. Clinical practice: endometriosis. N Engl J Med 2010;362:2389–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundberg E, Small KS, Hedman ÅK, Nica AC, Buil A, Keildson S, Bell JT, Yang T-P, Meduri E, Barrett A et al. . Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet 2012;44:1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Jing C, Li L, Zhang L, Shi Y, Wang J, Liu J, Li C. Down-regulation of VEZT gene expression in human gastric cancer involves promoter methylation and miR-43c. Biochem Biophys Res Commun 2011;404:622–627. [DOI] [PubMed] [Google Scholar]
- Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BWJH, Jansen R, de Geus EJC, Boomsma DI, Wright FA et al. . Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet 2016;48:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempstock J, Cindrova-Davies T, Jauniaux E, Burton GJ. Endometrial glands as a source of nutrients, growth factors and cytokines during the first trimester of human pregnancy: a morphological and immunohistochemical study. Reprod Biol Endocrin RB&E 2004;58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdsworth-Carson SJ, Fung JN, Luong HTT, Sapkota Y, Bowdler LM, Wallace L, Teh WT, Powell JE, Girling JE, Healey M et al. . Endometrial vezatin and its association with endometriosis risk. Hum Reprod 2016;31:999–1013. [DOI] [PubMed] [Google Scholar]
- Hu Y, Li M, Lu Q, Weng H, Wang J, Zekavat SM, Yu Z, Li B, Gu J, Muchnik S et al. . A statistical framework for cross-tissue transcriptome-wide association analysis. Nat Genet 2019;51:568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsten H, Al-Hasani H, Holdt L, Gross A, Beutner F, Krohn K, Horn K, Ahnert P, Burkhardt R, Reiche K et al. . Dissecting the genetics of the human transcriptome identifies novel trait-related trans-eQTLs and corroborates the regulatory relevance of non-protein coding loci†. Hum Mol Genet 2015;24:4746–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014;15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Xiao Z-D, Han L, Zhang J, Lee S-W, Wang W, Lee H, Zhuang L, Chen J, Lin H-K et al. . LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nat Cell Biol 2016;18:431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 2015;31:3555–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso N, Gayther S, Gusev A, Zheng W, Penney KL, Kote-Jarai Z, Eeles R, Freedman M, Haiman C, Pasaniuc B et al. . Large-scale transcriptome-wide association study identifies new prostate cancer risk regions. Nat Commun 2018;9:4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matalliotakis M, Zervou MI, Matalliotaki C, Rahmioglu N, Koumantakis G, Kalogiannidis I, Prapas I, Zondervan K, Spandidos DA, Matalliotakis I et al. . The role of gene polymorphisms in endometriosis. Mol Med Rep 2017;16:5881–5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meola J, e Silva JCR, Dentillo DB, da Silva JWA, Veiga-Castelli LC, de Souza Bernardes LA, Ferriani RA, de Paz CCP, Giuliatti S, Martelli L. Differentially expressed genes in eutopic and ectopic endometrium of women with endometriosis. Fertil Steril 2010;93:1750–1773. [DOI] [PubMed] [Google Scholar]
- Miao R, Guo X, Zhi Q, Shi Y, Li L, Mao X, Zhang L, Li C. VEZT, a novel putative tumor suppressor, suppresses the growth and tumorigenicity of gastric cancer. PLoS One 2013;8:e74409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 2008;5:621. [DOI] [PubMed] [Google Scholar]
- Mortlock S, Restuadi R, Levien R, Girling JE, Holdsworth-Carson SJ, Healey M, Zhu Z, Qi T, Wu Y, Lukowski SW et al. . Genetic regulation of methylation in human endometrium and blood and gene targets for reproductive diseases. Clin Epigenetics 2019;11:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagpal S, Meng X, Epstein MP, Tsoi LC, Patrick M, Gibson G, De Jager PL, Bennett DA, Wingo AP, Wingo TS et al. . TIGAR: an improved Bayesian tool for transcriptomic data imputation enhances gene mapping of complex traits. Am J Hum Genet 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng B, White CC, Klein H-U, Sieberts SK, McCabe C, Patrick E, Xu J, Yu L, Gaiteri C, Bennett DA et al. . An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat Neurosci 2017;20:1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noë M, Ayhan A, Wang T-L, Shih I-M. Independent development of endometrial epithelium and stroma within the same endometriosis. J Pathol 2018;245:265–269. [DOI] [PubMed] [Google Scholar]
- Nyholt DR, Low S-K, Anderson CA, Painter JN, Uno S, Morris AP, MacGregor S, Gordon SD, Henders AK, Martin NG et al. . Genome-wide association meta-analysis identifies new endometriosis risk loci. Nat Genet 2012;44:1355–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura R, Takeda K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp Mol Med 2017;e338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongen H, Brown AA, Delaneau O, Panousis NI, Nica AC, Consortium GT, Dermitzakis ET. Estimating the causal tissues for complex traits and diseases. Nat Genet 2017;49:1676. [DOI] [PubMed] [Google Scholar]
- Pers TH, Karjalainen JM, Chan Y, Westra H-J, Wood AR, Yang J, Lui JC, Vedantam S, Gustafsson S, Esko T et al. . Biological interpretation of genome-wide association studies using predicted gene functions. Nat Commun 2015;6:5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea G, Leek J, Salzberg S. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 2016;11:1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea G, Antonescu C, Chang T-C, Mendell J, Salzberg S. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 2015;33:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JE, Lyons PA, Lee JC, Richard AC, Fortune MD, Newcombe PJ, Richardson S, Smith KGC. Insight into genotype-phenotype associations through eQTL mapping in multiple cell types in health and immune-mediated disease. PLoS Genet 2016;12:e1005908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pharoah PDP, Tsai Y-Y, Ramus SJ, Phelan CM, Goode EL, Lawrenson K, Buckley M, Fridley BL, Tyrer JP, Shen H et al. . GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat Genet 2013;45:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan CM, Kuchenbaecker KB, Tyrer JP, Kar SP, Lawrenson K, Winham SJ, Dennis J, Pirie A, Riggan MJ, Chornokur G et al. . Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat Genet 2017;49:680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JE, Fung JN, Shakhbazov K, Sapkota Y, Cloonan N, Hemani G, Hillman KM, Kaufmann S, Luong HT, Bowdler L et al. . Endometriosis risk alleles at 1p36.12 act through inverse regulation of CDC42 and LINC00339. Hum Mol Genet 2016;25:5046–5058. [DOI] [PubMed] [Google Scholar]
- Qi T, Wu Y, Zeng J, Zhang F, Xue A, Jiang L, Zhu Z, Kemper K, Yengo L, Zheng Z et al. . Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat Commun 2018;9:2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmioglu N, Banasik K, Christofidou P, Danning R, Galarneau G, Giri A, MacGregor S, Mortlock S, Sapkota Y, Schork AJ et al. . Large-scale genome-wide association meta-analysis of endometriosis reveals 13 novel loci and genetically-associated comorbidity with other pain conditions. bioRxiv 2018. [Google Scholar]
- Rahmioglu N, Montgomery GW, Zondervan KT. Genetics of endometriosis. Womens Health 2015;11:577–586. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson JA. Metastatic or embolic endometriosis, due to the menstrual dissemination of endometrial tissue into the venous circulation. Am J Pathol 1927;3. [PMC free article] [PubMed] [Google Scholar]
- Sapkota Y, Steinthorsdottir V, Morris AP, Fassbender A, Rahmioglu N, De Vivo I, Buring JE, Zhang F, Edwards TL, Jones S et al. . Meta-analysis identifies five novel loci associated with endometriosis highlighting key genes involved in hormone metabolism. Nat Commun 2017;8:15539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo M, Kim K, Yoon J, Jeong JY, Lee H-J, Cho S, Kim H. RNA-seq analysis for detecting quantitative trait-associated genes. Sci Rep 2016;6:24375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics 2012;28:1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenblock C, Heckel T, Czupalla C, Santo AIE, Niehage C, Sztacho M, Hoflack B. The Cdc42 guanine nucleotide exchange factor FGD6 coordinates cell polarity and endosomal membrane recycling in osteoclasts. J Biol Chem 2014;289:18347–18359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda K, Nakaoka H, Yoshihara K, Ishiguro T, Tamura R, Mori Y, Yamawaki K, Adachi S, Takahashi T, Kase H et al. . Clonal expansion and diversification of cancer-associated mutations in endometriosis and Normal endometrium. Cell Rep 2018;24:1777–1789. [DOI] [PubMed] [Google Scholar]
- Taneera J, Fadista J, Ahlqvist E, Atac D, Ottosson-Laakso E, Wollheim CB, Groop L. Identification of novel genes for glucose metabolism based upon expression pattern in human islets and effect on insulin secretion and glycemia. Hum Mol Genet 2015;24:1945–1945. [DOI] [PubMed] [Google Scholar]
- Võsa U, Claringbould A, Westra H-J, Bonder MJ, Deelen P, Zeng B, Kirsten H, Saha A, Kreuzhuber R, Kasela S et al. . Unraveling the polygenic architecture of complex traits using blood eQTL meta-analysis. bioRxiv 2018. [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009;10:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zeng J, Zhang F, Zhu Z, Qi T, Zheng Z, Lloyd-Jones LR, Marioni RE, Martin NG, Montgomery GW et al. . Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat Commun 2018;9:918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Ferreira T, Morris AP, Medland SE, Genetic Investigation of ATC, Replication DIG, Meta-analysis C, PAF M, Heath AC, Martin NG et al. . Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet 2012;44:369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011;88:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Fung-Leung W-P, Bittner A, Ngo K, Liu X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS One 2014;9:e78644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, Montgomery GW, Goddard ME, Wray NR, Visscher PM et al. . Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 2016;48:481–487. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.