

Apoptotic cell death can be an efficient defense reaction of mammalian cells infected with obligate intracellular pathogens; the host cell dies and the pathogen cannot replicate. While this is well established for viruses, there is little experimental support for such a concept in bacterial infections. All Chlamydiales are obligate intracellular bacteria, and different species infect vastly different hosts. Chlamydia trachomatis infects human epithelial cells; Parachlamydia acanthamoebae replicates in amoebae.
KEYWORDS: apoptosis, Chlamydia, Parachlamydia, cell defense, epithelial cells
ABSTRACT
Apoptotic cell death can be an efficient defense reaction of mammalian cells infected with obligate intracellular pathogens; the host cell dies and the pathogen cannot replicate. While this is well established for viruses, there is little experimental support for such a concept in bacterial infections. All Chlamydiales are obligate intracellular bacteria, and different species infect vastly different hosts. Chlamydia trachomatis infects human epithelial cells; Parachlamydia acanthamoebae replicates in amoebae. We here report that apoptosis impedes growth of P. acanthamoebae in mammalian cells. In HeLa human epithelial cells, P. acanthamoebae infection induced apoptosis, which was inhibited when mitochondrial apoptosis was blocked by codeletion of the mediators of mitochondrial apoptosis, Bax and Bak, by overexpression of Bcl-XL or by deletion of the apoptosis initiator Noxa. Deletion of Bax and Bak in mouse macrophages also inhibited apoptosis. Blocking apoptosis permitted growth of P. acanthamoebae in HeLa cells, as measured by fluorescence in situ hybridization, assessment of genome replication and protein synthesis, and the generation of infectious progeny. Coinfection with C. trachomatis inhibited P. acanthamoebae-induced apoptosis, suggesting that the known antiapoptotic activity of C. trachomatis can also block P. acanthamoebae-induced apoptosis. C. trachomatis coinfection could not rescue P. acanthamoebae growth in HeLa; in coinfected cells, C. trachomatis even suppressed the growth of P. acanthamoebae independently of apoptosis, while P. acanthamoebae surprisingly enhanced the growth of C. trachomatis. Our results show that apoptosis can be used in the defense of mammalian cells against obligate intracellular bacteria and suggest that the known antiapoptotic activity of human pathogenic chlamydiae is indeed required to permit their growth in human cells.
INTRODUCTION
Apoptosis is a form of regulated, or programmed, cell death. During apoptosis, a signaling program is initiated that leads to the death of the cell, accompanied by characteristic biochemical and morphological features. In terms of infectious biology, apoptosis is often discussed as a means to combat infection (reviewed in reference 1). Apoptosis can affect inflammation and the immune response through both death of immune cells and immunomodulating effects of dying cells (2). Through cell loss due to apoptosis, infected tissue may be altered in its structure and composition. Most obviously, apoptosis can frustrate the endeavor of obligate intracellular organisms to replicate. Viruses especially depend on the integrity of the infected cell, and apoptotic cell death can prevent viral replication (3). It has been speculated that apoptosis even may have evolved to combat viral infection (4) and that apoptosis of an infected cell can block the generation of viral progeny, which has been reported over 25 years ago (5).
Presumably because of this contingency, it has generally been assumed that the same must be the case for obligate intracellular bacteria. Indeed, the idea is obvious that such bacteria will also be vulnerable to apoptosis of the infected cell. Chlamydia is the best-investigated example of this category. The chlamydiae infecting mammalian cells—members of the family of Chlamydiaceae and the genus Chlamydia—can, where investigated, all inhibit apoptosis of the infected cell, i.e., a Chlamydia-infected cell resists a stimulus that would cause apoptosis in a noninfected cell (6, 7). It has also been shown that the antiapoptotic activity of Chlamydia trachomatis can be overcome by direct, massive activation of the mitochondrial apoptotic pathway (high-level overexpression of the proapoptotic Bcl-2 family Bim) and that this experimental manipulation blocks replication of the bacteria (8). All this suggests that apoptosis can be used as a defense reaction against chlamydial infection and that Chlamydia needs its antiapoptotic activity to keep the infected cell alive. However, to be conclusive, it will have to be shown that infection with Chlamydia is interpreted by the infected cell as a signal to activate the apoptosis apparatus and that only the chlamydial antiapoptotic activity prevents cell death. While a recent study identifies “sublethal” activity of the apoptosis apparatus in human cells infected with C. trachomatis (providing some support for the view that the cell interprets chlamydial infection as a proapoptotic signal) (9), there are substantial experimental gaps in this line of argument.
In the order Chlamydiales, there is, in addition to the Chlamydiaceae, an ever-increasing number of families whose members especially differ in terms of host tropism (10). The family Parachlamydiacea infects amoebae and is considered endosymbionts of these protozoa (11). Two species of the family, Protochlamydia amoebophila and Parachlamydia acanthamoebae, have been investigated in several studies for their ability to infect metazoan cells, and apoptosis induction has been tested in these studies. P. acanthamoebae has been found to induce apoptosis in macrophages (12) but not in pneumocytes, fibroblasts (13), or HEp-2 epithelioid tumor cells (14). One study found that P. acanthamoebae was able to replicate in various amoebae but not in a panel of four human cell lines of epithelial, lymphocyte, and myeloid origin (15). P. amoebophila has been reported to trigger apoptosis in HEp-2 cells (14). In insect cells, both P. acanthamoebae and P. amoebophila induced apoptosis, and chemical caspase inhibition blocked cell death and enabled growth of the bacteria (16). What insect cell apoptosis means for mammalian apoptosis is difficult to say because the signaling pathways are different; in insect cells, caspase activation is the result of the loss or inactivation of the ubiquitin ligase Diap1 (17), and this pathway is unimportant in mammalian apoptosis.
We here used P. acanthamoebae to test the hypothesis that apoptosis can act as a defense mechanism against Chlamydiales in human cells. Our results show that P. acanthamoebae induces mitochondrial apoptosis in HeLa cells but can grow when apoptosis is genetically prevented. They illustrate that apoptosis can act as a defense reaction against infection with obligate intracellular bacteria and suggest that human pathogenic chlamydiae were compelled to acquire the ability to inhibit apoptosis during their evolutionary association with animals, developing a dedicated apoptosis machinery.
RESULTS
P. acanthamoebae infection induces mitochondrial apoptosis in infected mammalian cells.
We here used HeLa cells, the standard cell to study chlamydial growth; cell biology of chlamydial infection in vitro is best understood in HeLa, and HeLa cells have the complete functional apparatus of apoptosis. We infected the HeLa cell cultures with 25 P. acanthamoebae cells per one HeLa cell (we will refer to this as multiplicity of infection [MOI] of 25) (see Fig. S5D for a titration on [apoptosis-resistant] HeLa cells). When HeLa cells were infected with P. acanthamoebae, substantial apoptotic cell death became visible between 24 and 48 h. Infected cells showed activation of caspase-3, a hallmark of the downstream apoptotic pathway, as identified by the presence of cleaved (activated) caspase-3 (Fig. 1A, Fig. S2A) and by the activation of a caspase-3/7 fluorescence reporter (Fig. 1B). Apoptosis was observed from an MOI of about 5 and increased at higher infectious doses (Fig. S1A and B). In a few cells, P. acanthamoebae was detectable by fluorescence in situ hybridization (FISH) at 24 and 48 h postinfection (p.i.) (Fig. S5A to C). This may mean that a few cells escaped apoptosis and P. acanthamoebae was able to show some growth in these cells. These data show that P. acanthamoebae induces apoptosis in HeLa cells in a relatively slow process, suggesting that growth of the bacteria was the process to start apoptosis.
FIG 1.

P. acanthamoebae infection induces mitochondrial apoptosis in HeLa cells. HeLa cells were infected with P. acanthamoebae and analyzed for cell death and caspase-3 activation. (A) HeLa CTRL or Bax/Bak-deficient cells were analyzed for activation of caspase-3 by flow cytometry 24 and 48 h after infection. (B) DEVD (caspase-3 reporter cell line) and DEVG (control cell line) were infected for 24 h to analyze caspase activation. (C) CTRL and mutant cell lines (Bax/Bak or Noxa deficient; Bcl-XL overexpressing) were infected with P. acanthamoebae and stained for active caspase-3 after 24 h. (D) HeLa CTRL and Bax/Bak-deficient cells were infected with P. acanthamoebae (24, 48, or 72 h) and stained with a live/dead stain to determine percentage of dead cells in the population. (E) CTRL and mutant cell lines (Bax/Bak or Noxa deficient; Bcl-XL overexpressing) were infected with P. acanthamoebae and stained for active caspase-3 after 24 h and 48 h of incubation at 30°C. (F) DEVD and DEVG were infected for 24 h to analyze caspase activation (incubation at 30°C). The MOI was 25 in all experiments. p.c., positive control, treatment with Mcl-1 inhibitor S63845 (500 nM) and ABT-737 (1 μM) for 1 h; h.-t., heat treated (1 min at 95°C). Data are shown as means/standard errors of the mean (SEM) of at least three independent experiments. Experiments for positive controls were done separately. *, P < 0.05 (unpaired two-tailed t test) between control and mutant cells (A and C to E) or P < 0.05 (paired two-tailed t test) between uninfected and infected cells (B and F).
Caspase-3 is active in all cases of apoptosis and can be activated by two separately acting upstream “initiator” caspases, caspase-8 and caspase-9 (18). Caspase-8 regulates death receptor-dependent apoptosis, whereas caspase-9 is active during mitochondrial apoptosis. Mitochondrial apoptosis appears to be the much more common form of apoptosis, and the sublethal signals in the apoptosis apparatus that are triggered by C. trachomatis infection use the mitochondrial apoptosis pathway (9). We therefore tested whether P. acanthamoebae also induced apoptosis through this pathway. We infected HeLa cells deficient in the two Bcl-2 family effector proteins Bax and Bak. The presence of either Bax or Bak is essential to mitochondrial apoptosis (19). We further used HeLa cells overexpressing the antiapoptotic Bcl-2 family member Bcl-XL; this expression also blocks mitochondrial apoptosis (20). Both Bax/Bak-deficient and Bcl-XL-overexpressing HeLa cells were protected against apoptosis induced by infection with P. acanthamoebae (Fig. 1A and C, Fig. S2). Treatment of Bcl-XL-overexpressing cells with a specific Bcl-XL inhibitor reversed the protection (Fig. S3A).
To obtain information on the apoptotic signals involved, we also generated HeLa cells deficient in the proapoptotic Bcl-2 family member Noxa, which antagonizes the antiapoptotic protein Mcl-1 (21) and is involved in the apoptotic response to viral infection (22). Noxa-deficient cells also showed a reduced apoptotic response to the infection with P. acanthamoebae (Fig. 1C, Fig. S2C). These results establish that P. acanthamoebae infection induces apoptosis through the mitochondrial pathway upon infection of HeLa cells. Although significantly and substantially less apoptosis was observed, there was still some caspase activation in a P. acanthamoebae-infected population of Bax/Bak-deficient HeLa cells. Cells lacking Bax and Bak are completely resistant to mitochondrial apoptosis, while extrinsic apoptosis can still kill the cells through caspase-8 (19, 23). Our data therefore suggest that, in addition, caspase-8-dependent apoptosis is triggered to a smaller extent in these conditions.
We further analyzed cell death (rather than only apoptosis) in infected cells by staining for plasma membrane integrity. Cell death was increasingly induced over 72 h with slower kinetics than caspase activation. This is to be expected since caspase activity during apoptosis precedes cell death. Cell death was again strongly reduced in Bax/Bak-deficient HeLa cells when compared to control HeLa cells (Fig. 1D). When Bcl-XL-expressing HeLa cells were infected with P. acanthamoebae in the presence of the Bcl-XL inhibitor, cell death was restored (Fig. S3B). This suggests that the cell death observed is at least mostly due to caspase activation and apoptosis. When P. acanthamoebae were heat inactivated, or when the antibiotic rifampin was included, apoptosis upon P. acanthamoebae infection was strongly reduced (Fig. S1D), suggesting that bacterial protein synthesis was required. It has been reported earlier that P. acanthamoebae can grow to some extent in HEp-2 cells at 30°C (24). We therefore tested the level of apoptosis induction at this temperature and found that apoptosis induction was slower but occurred to a substantial extent at 48 h (Fig. 1E and F). The results were similar for cell death at 37°C versus 30°C (Fig. S4).
Macrophages are not typical host cells of human pathogenic chlamydiae. However, mammalian macrophages serve as host cells for Legionella, an environmental pathogen that naturally infects amoebae. Because amoebae are also natural hosts to P. acanthamoebae, we did similar experiments as before with macrophages. We chose a system where murine macrophages are differentiated from “conditionally immortalized” progenitor lines. Progenitors are transduced to express a Hoxb8 oncogene that is active only in the presence of estrogen and can be turned off by its withdrawal. We chose this system to be able to use Bax/Bak-deficient cells. Most of Bax/Bak double-deficient mice die shortly after birth (25), making the recovery of macrophages very challenging. Hoxb8-derived macrophages, however, are basically indistinguishable from primary macrophages (26). We infected these macrophages with P. acanthamoebae and measured caspase-3-activation. Infection led to the activation of caspase-3 in about a third of infected cells over 24 h, and the lack of Bax and Bak protected the cells to a significant degree, although its effect was not as strong as the one observed in HeLa cells (Fig. 2A). When we analyzed cell death, we found that the lack of Bax and Bak protected the cells strongly at earlier time points (Fig. 2B). Therefore, P. acanthamoebae induces cell death in murine Hoxb8-derived macrophages, and a substantial part of this cell death is due to mitochondrial apoptosis.
FIG 2.
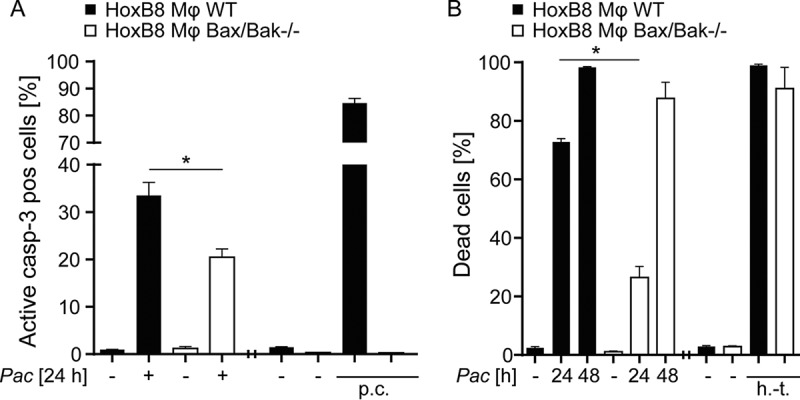
Induction of partly mitochondrial apoptosis by P. acanthamoebae infection in macrophages. HoxB8 macrophages were infected with P. acanthamoebae for 24 or 48 h. (A) HoxB8 macrophages (wild type [WT] or Bax/Bak deficient) were infected for 24 h and analyzed by flow cytometry to detect active caspase-3. (B) HoxB8 macrophages (WT or Bax/Bak deficient) were infected for 24 or 48 h and analyzed by flow cytometry to detect cell death by live/dead staining. The MOI was 2.5. p.c., positive control, treatment with Mcl-1-inhibitor S63845 (3 μM) and ABT-737 (5 μM) for 4 h; h.-t., heat treated (1 min at 95°C). Experiments for positive controls were done separately. Data are shown as means/SEM of at least three independent experiments *, P < 0.05 (unpaired two-tailed t test) between control and mutant cells.
Protection against apoptosis permits growth of P. acanthamoebae in mammalian cells.
We first tested for the presence of P. acanthamoebae using FISH of infected cells. In infected HeLa control cells, there were few positive signals after 24 and after 48 h (Fig. S5A to C), indicative of limited growth of the bacteria. However, FISH showed an increasingly stronger signal over a period of 48 h in Bax/Bak-deficient HeLa cells infected with P. acanthamoebae (Fig. S5A and C). A similar pattern was seen in HeLa cells overexpressing Bcl-XL (Fig. S5B) and in HeLa cells lacking Noxa (Fig. S5C). We titrated P. acanthamoebae on Bax/Bak-deficient HeLa cells. The MOI of 25 (determined in amoebae) resulted in detectable infection in about 70% of the HeLa cells (Fig. S5D). These results suggested that P. acanthamoebae was indeed able to infect HeLa cells if mitochondrial apoptosis was blocked experimentally.
To test for bacterial protein synthesis, we measured the incorporation of l-azidohomoalanine (AHA) by the bacteria. AHA is incorporated into newly synthesized proteins, and the labeled proteins can be made fluorescent using Click-iT chemistry. As shown in Fig. 3 and Fig. S6A, a clear AHA signal was obtained in infected Bax/Bak-deficient HeLa cells, indicating that P. acanthamoebae was able to perform protein synthesis. Addition of the antibiotic rifampin strongly reduced the signal, although some residual fluorescence was still detected, suggesting that the antibiotic had a strong effect on bacterial protein synthesis (in susceptible bacteria, rifampin blocks mRNA synthesis). Cycloheximide, an inhibitor of eukaryotic translation, had no effect on bacterial protein synthesis (Fig. 3). At higher resolution, P. acanthamoebae-infected cells showed multiple small fluorescent spots, which may reflect individual bacteria (Fig. S6 and S7). Bacterial structures that very likely represent replicating P. acanthamoebae were observed inside cytoplasmic vacuoles of Bax/Bak-deficient HeLa cells by electron microscopy (Fig. S9). When apoptosis is blocked, P. acanthamoebae is thus able to synthesize proteins in infected HeLa cells but is sensitive to rifampin.
FIG 3.

Detection of protein synthesis in P. acanthamoebae during infection of Bax/Bak-deficient HeLa cells. HeLa cells deficient in Bax and Bak were infected with P. acanthamoebae (MOI of 25) for 48 h. Cells were labeled with AHA at time point 47 h p.i. for 1 h, and newly synthesized proteins were detected using a Click-iT reaction. The Click-iT-reaction products are shown in green; blue gives a Hoechst 33342 counterstain to visualize bacterial and host DNA. Some aliquots were treated with cycloheximide (100 ng/ml) and/or rifampin (10 ng/μl) 2 h prior to labeling. Scale bars, 50 μm. Arrows indicate P. acanthamoebae. Pictures are representative of at least three independent experiments.
We next investigated bacterial growth by measuring genome equivalents of P. acanthamoebae in infected HeLa cells. Between 24 and 48 h, there was a small, insignificant increase in P. acanthamoebae DNA in infected HeLa cells. Significant growth was, however, measured in all apoptosis-deficient HeLa cells identified earlier (Bax/Bak-deficient, Bcl-XL-overexpressing, and Noxa-deficient cells) (Fig. 4A, Fig. S8). Treatment of Bcl-XL-overexpressing HeLa cells with the Bcl-XL inhibitor reduced growth of P. acanthamoebae (Fig. S3C).
FIG 4.
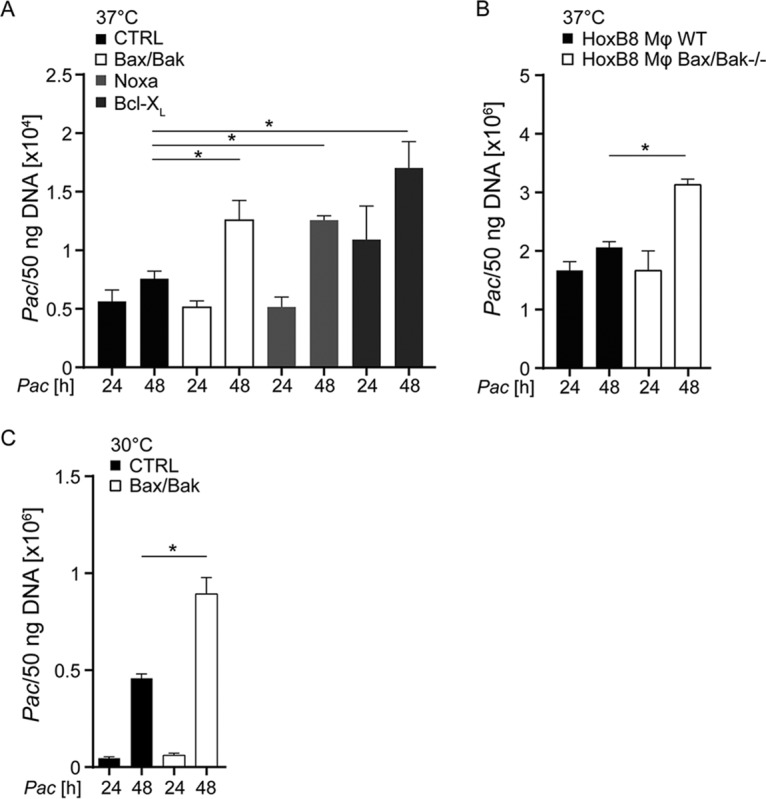
Detection of P. acanthamoebae replication in epithelial cells and HoxB8 macrophages. HeLa cells (CTRL, Bax/Bak deficient, Bcl-XL overexpressing, or Noxa deficient) (A) or HoxB8 macrophages (WT or Bax/Bak deficient) (B) were infected with P. acanthamoebae at 37°C. (C) HeLa cells (CTRL or Bax/Bak deficient) were infected with P. acanthamoebae at 30°C. The MOI was 25 in all experiments. DNA was isolated 24 or 48 h p.i. and quantified by qPCR. Detectable P. acanthamoebae genomic DNA equivalents per 50 ng of total DNA isolated from each culture are shown. In addition, the presence of the human hypoxanthine phosphoribosyltransferase (HPRT) gene was quantified in the DNA samples, showing very little variation from the DNA measurements (Fig. S8). Data are means/SEM of at least three independent experiments. *, P < 0.05 (unpaired two-tailed t test) between control and mutant cells.
In macrophages, there seemed to be some replication of the bacteria taking place; the increase in P. acanthamoebae genomic DNA was significantly higher in Bax/Bak-deficient macrophages (Fig. 4B). This indicates that inhibition of apoptosis permits replication of the bacteria in mammalian cells. When cultures were incubated at 30°C, growth of P. acanthamoebae was enhanced, but the difference between control and Bax/Bak-deficient cells was similar to the results from cultures at 37°C (Fig. 4C, Fig. S8B).
We then infected control or Bax/Bak-deficient HeLa cells, extracted the bacteria after 24 or 48 h, and used these extractions to reinfect amoeba cells; 24 h later, P. acanthamoebae DNA was quantified in amoeba cultures. There was a slight increase in the numbers of bacteria replicating in amoebae when we compared bacterial preparations taken after 24 and 48 h of HeLa control cell infection. This increase was, however, significantly greater when Bax/Bak-deficient HeLa cells were used for the initial infection (Fig. 5). These results show that more infectious bacteria can be isolated from Bax/Bak-deficient than from control HeLa cells after 48 than after 24 h; P. acanthamoebae thus is able to complete its developmental cycle and to produce infectious progeny in HeLa cells where apoptosis is inhibited.
FIG 5.
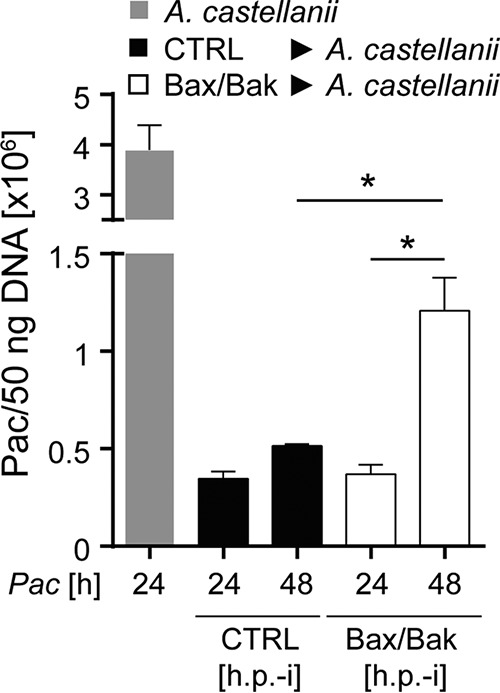
Generation of infectious progeny of P. acanthamoebae in Bax/Bak-deficient HeLa cells. HeLa CTRL or Bax/Bak-deficient cells were infected with P. acanthamoebae. After 24 h or 48 h, cultures were mechanically disrupted, and the suspension was used to infect amoeba (Neff). Twenty-four hours after amoebal infection, cultures were lysed, DNA was extracted, and P. acanthamoebae DNA was quantified by qPCR. For comparison, A. castellanii was infected with an MOI of 1 for 24 h. P. acanthamoebae genomic DNA per 50 ng of total DNA isolate is shown. Data are means/SEM of at least three independent experiments. *, P < 0.05 (paired two-tailed t test).
C. trachomatis can inhibit P. acanthamoebae-induced apoptosis.
Because C. trachomatis has strong antiapoptotic activity and P. acanthamoebae grows only in HeLa when apoptosis is inhibited, we tested whether coinfection with C. trachomatis inhibits P. acanthamoebae-induced apoptosis and permits growth of P. acanthamoebae in control HeLa cells. Infection with C. trachomatis indeed had a moderate but significant effect in reducing apoptosis induced by P. acanthamoebae infection in control but not in Bax/Bak-deficient HeLa cells (which have lower apoptosis rates anyway) (Fig. 6A). However, coinfection with C. trachomatis failed to promote the growth of P. acanthamoebae. Rather, coinfection with P. acanthamoebae significantly enhanced the growth of C. trachomatis. C. trachomatis significantly reduced the growth of P. acanthamoebae both in control and, more strongly, in Bax/Bak-deficient HeLa cells (Fig. 6B). This was unexpected. C. trachomatis is certainly better adapted to growth in human cells than P. acanthamoebae, but why the presence of P. acanthamoebae promotes growth of C. trachomatis is not easily explained.
FIG 6.

Coinfection with C. trachomatis inhibits apoptosis induced by P. acanthamoebae, but coinfection with P. acanthamoebae supports growth of C. trachomatis. HeLa (CTRL or Bax/Bak-deficient) cells were infected with C. trachomatis (MOI of 1) and/or P. acanthamoebae (MOI of 25 or MOI of 1) and analyzed at 48 h p.i. for apoptosis induction (A) and bacterial intracellular growth (B). (B, Left) C. trachomatis detection. (B, Right) P. acanthamoebae detection. Bacterial presence was quantified as genome equivalents per extracted total DNA). Data are means/SEM of at least three independent experiments. *, P < 0.05 (paired two-tailed t test).
DISCUSSION
These results show that apoptosis can be used by mammalian cells to block the replication of Chlamydiales and suggest that human pathogenic chlamydiae depend on their ability to inhibit apoptosis for their infection of humans.
Amoeba-dwelling Parachlamydiaceae are commonly found in the environment. A recent study of water from 48 Swiss domestic samples identified Chlamydiales in 35 of them, and about half of them contained Parachlamydiacea (27). It has been suggested that P. acanthamoebae may be an agent of pneumonia and miscarriage, and a low percentage of humans carry antibodies to P. acanthamoebae, suggestive of a previous infection (28, 29). P. acanthamoebae infection may well be underreported, but the current consensus is that it is, despite its high prevalence in the environment, not a very common agent of human infection. There are other mechanisms that may preclude human infection with P. acanthamoebae, especially the inability to evade an immune response, but the inability to escape host cell apoptosis may suffice to preclude infection of human cells.
Our results establish that mitochondrial apoptosis is the main pathway of apoptosis induction by P. acanthamoebae in HeLa cells and macrophages, although there was, somewhat surprisingly, still activation of caspase-3 in the absence of mitochondrial apoptosis. Nonmitochondrial apoptosis is typically implemented through caspase-8-activation downstream of death receptors. In the case of HeLa cell infection, this would require the upregulation of a ligand for a death receptor, such as tumor necrosis factor (TNF) secretion (which would then signal in an autocrine/paracrine fashion through the TNF receptor). Which ligand-receptor pair may mediate this smaller proapoptotic effect of P. acanthamoebae infection is not clear. Apoptosis in most cells was inhibited by blocking mitochondrial apoptosis. Mitochondrial apoptosis is mediated by the Bcl-2 family of proteins, with pro- and antiapoptotic family members jointly inducing or inhibiting apoptosis. Although an additional effect of a third protein, Bok, has been proposed, it is generally accepted in the field that all mitochondrial apoptosis requires Bax or Bak, and the combined loss of the two proteins therefore prevents mitochondrial apoptosis (19). Bcl-XL can bind to and inhibit most, perhaps all, proapoptotic Bcl-2-proteins, and overexpression of Bcl-XL therefore is expected to have a similar effect to the loss of Bax and Bak.
How the upstream signal to undergo apoptosis is conveyed to mitochondria, and how the Bcl-2 family integrates these signals to reach a decision to induce cell death, is generally poorly understood. For instance, the chemical staurosporine is very commonly used to induce mitochondrial apoptosis, but the signals upstream of mitochondria are basically unknown. We have found a contribution from one of the triggers of mitochondrial apoptosis, the Bcl-2 subfamily of BH3-only proteins, Noxa. Noxa-deficient HeLa cells showed reduced apoptosis upon P. acanthamoebae infection. Noxa is one of the trigger proteins of apoptosis within the Bcl-2-family (this group is known as BH3-only proteins) and specifically inactivates the antiapoptotic protein Mcl-1. We tested Noxa because a role of Noxa during viral infection had been found previously (22). How proapoptotic stimuli are transmitted to mitochondria is still a mystery in many cases. This role of Noxa provides a first step in understanding this upstream signaling. It seems possible that a Noxa-Mcl-1 axis does function more generally in the recognition of intracellular pathogens.
The next question is how P. acanthamoebae is recognized by the infected cells. The cell needs receptors to pick up the presence of the bacteria. Macrophages have a broader pattern of such potential receptors, especially Toll-like receptors (TLR), and a previous report has found that TLR2 and TLR4 contribute to cytokine production by macrophages upon recognition of P. acanthamoebae (30). HeLa cells have a narrower set of receptors and express little or no TLR. As far as is known, C. trachomatis is recognized by HeLa cells through cytosolic pattern recognition receptors, namely, the receptor for peptidoglycan fragments, Nod1 (31), and the receptor for cytosolic DNA, cyclic GMP-AMP synthase (cGAS) (32). Notably, all of these receptors have a principal proapoptotic capacity, even though this activity normally seems to be balanced by other signaling events during infection (for NOD1, there is only one report of proapoptotic activity, which is experimentally somewhat indirect [33]; the cGAS/STING signaling axis has been shown to be able to induce apoptosis directly [34]). It is therefore a likely possibility that these receptors are involved also in bacterial recognition and then in apoptosis induction. Proapoptotic signals of C. trachomatis infection have been identified, which stay sublethal during infection of HeLa cells (35). It seems a plausible scenario that both C. trachomatis and P. acanthamoebae have proapoptotic potential but that the signals are tempered by the C. trachomatis-dependent antiapoptotic activity. Lack of such activity in P. acanthamoebae may make the difference between apoptosis induction and inhibition.
Myeloid cells often show a different response from epithelial cells to infection with pathogens. In many cases, macrophages are better at controlling intracellular growth, and this is also the case for Chlamydia. Although some growth can be detected and depends on polarization of the macrophages used (36), macrophages are not as permissive to replication of Chlamydia as are epithelial cells (37). There are, however, exceptions to this dichotomy; for instance, Legionella can grow very well in human macrophages. An intriguing parallel is that Legionella, like P. acanthamoebae, evolved to live in protozoa, not in human cells. We found that although P. acanthamoebae grew substantially better in apoptosis-resistant macrophages, there was some growth also in wild-type macrophages. Perhaps this reflects a similarity between the physiological host cell and mammalian phagocytes.
Coinfection with C. trachomatis reduced P. acanthamoebae growth in HeLa cells. This may perhaps be simply explained by the better adaptation of C. trachomatis, which may result in the deprivation of P. acanthamoebae of essential host cell-derived nutrients. It is possible that the recognition of P. acanthamoebae by the host cell leads to the activation of signaling pathways that support the growth of C. trachomatis. Perhaps C. trachomatis even cannibalizes on P. acanthamoebae; P. acanthamoebae may contain building blocks that are costly and laborious to synthesize for C. trachomatis. In any case, this seems an interesting example of bacterial interplay.
Genome analyses show that Chlamydiales originated from a common predecessor, which lived at a time when only protozoan eukaryotes existed (10). The simplest line of argument is therefore that this progenitor was adapted to unicellular hosts, which have no known apoptosis system. The chlamydial line staying with protozoa, like P. acanthamoebae, has no need for apoptosis inhibition, while the line that ended up as human pathogenic bacteria, like C. trachomatis, had to deal with the developing apoptotic defense. Accordingly, C. trachomatis is able to counter apoptosis, but P. acanthamoebae is not.
MATERIALS AND METHODS
Cell lines and cell culture.
HeLa229 cells (ATCC CCL-2.1) were cultured under standard conditions (37°C and 5% CO2) in Gibco RPMI 1640 medium (Thermo Fisher Scientific; catalog no. 61870044) containing 10% (vol/vol) fetal bovine serum (FBS) (Sigma-Aldrich; catalog no. F7524). For generation of gene-deficient cells, CRISPR/Cas9 genome editing was used by transducing the cells with the lentiviral vector lentiCRISPR v2 (Addgene) (41 ) followed by selection with puromycin (InvivoGen). A guide RNA (gRNA) targeting the Noxa locus was used (TCGAGTGTGCTACTCAACTC).
Murine HoxB8-immortalized macrophage progenitor cells were generated from bone marrow of wild-type or Bax/Bak homozygous deficient mice. Cells were transduced with a retrovirus driving expression of an estrogen receptor-Hoxb8 fusion protein, followed by selection of the cells in the presence of the granulocyte-macrophage colony-stimulating factor (GM-CSF, produced in B16 cells) as reported (26). Cells were cultured in RPMI 1640, without l-glutamine, with 2.0 g/liter NaHCO3, very low endotoxin (PAN-Biotech P04-17525), with 10% (vol/vol) FBS (heat inactivated, 56°C for 1 h; Thermo Fisher Scientific; catalog no. 10270106 [lot no. 41Q7141K]), 1% GM-CSF, and 1 μM β-estradiol (Sigma-Aldrich; catalog no. E2758). To differentiate progenitor cells into macrophages, 106 cells were seeded into a 10-cm non-surface-treated cell culture dish (petri dish, BD Falcon) in VLE-RPMI containing 10% (vol/vol) FBS (heat inactivated) and 1% GM-CSF (differentiation medium; no estrogen). Cells differentiated for 7 days, and macrophages were harvested using Accutase (Sigma-Aldrich; catalog no. A6964) for 10 min incubating at 37°C. Cells were resuspended in the differentiation medium, and 5 × 105 cells per well were seeded in non-surface-treated 12-well-plates (Corning; catalog no. 351143) for infections.
The amoeba Acanthamoeba castellanii Neff strain (kindly provided by Matthias Horn, Vienna) was cultured at room temperature in TSY medium (Trypticase soy broth with yeast extract) containing 30 g/liter Trypticase soy broth (BD Biosciences; catalog no. 211768) and 10 g/liter yeast extract (BD Biosciences; catalog no. 212750) with a pH of 7.3.
Infectious agents and infections.
The C. trachomatis lymphogranuloma venereum (LGV) strain L2 was kindly provided by Agathe Subtil, Paris, and the P. acanthamoebae strain UV-7 was kindly provided by Matthias Horn, Vienna. HeLa229 cells were infected (3 × 105 cells in suspension [1 ml]) and seeded in 6-well plates. HoxB8 macrophages (5 × 105 per well) were seeded in non-surface-treated 12-well plates infected 3 h later with the indicated MOI values for the indicated times. Infected cells were incubated under standard conditions.
Caspase-3 reporter cells.
Caspase-3 activation was detected as reported by flow cytometry using the fluorescence resonance energy transfer (FRET) reporter cell lines DEVD and DEVG (9). The reporter cells express a FRET construct, and loss of the FRET signal indicates activation of caspase-3.
Detection of protein synthesis.
Bacterial translational activity was detected using Click-iT-labeling of proteins. We seeded 2 × 104 HeLa229 cells per well into a μ-Slide 8-well slide (Ibidi; catalog no. 80826). Cells were infected with P. acanthamoebae (MOI of 25). A Click-iT-reaction was performed as described previously following the manufacturer’s instructions (Thermo Fisher Scientific; catalog no. C10269) (38). Briefly, 2 h before harvesting, cells were incubated in methionine- and cystine-free medium (Dulbecco modified Eagle medium [DMEM]; Thermo Fisher Scientific; catalog no. 21013024). Rifampin (10 ng/μl; Sigma-Aldrich; catalog no. R3501) and cycloheximide (100 ng/μl; Sigma-Aldrich; catalog no. 01810) were added to some samples. AHA (25 μM; Thermo Fisher Scientific; catalog no. C10102) in methionine- and cystine-free medium was added 1 h before harvesting. Cells were fixed with 4% paraformaldehyde (PFA) and permeabilized with 0.2% Triton X-100, followed by a blocking step in 3% bovine serum albumin (BSA) in PBS. Alexa Fluor 488 alkyne (Thermo Fisher Scientific; catalog no. 10267) was used to label the newly synthesized proteins by binding to the integrated AHA. The cells were counterstained with Hoechst 33342 (Sigma-Aldrich; catalog no. B2261) for the detection of the bacterial DNA and the host cell nucleus. Cells were covered with mounting medium (Ibidi; catalog no. 50001) and stored at 4°C. Incorporation of labeled amino acids was detected using fluorescence microscopy. A BZ 9000 microscope (Keyence), together with the corresponding software BZ-II Viewer and BZ-II Analyzer, was used to acquire and visualize the pictures. Z-stack pictures were taken using a confocal microscope (Zeiss; LSM 880) to analyze the three-dimensional structure of the infected cell. Pictures were processed using the software ZEN blue and ZEN black (Zeiss).
Fluorescence in situ hybridization.
The 16S rRNA of bacteria was labeled by hybridization with a Cy5-coupled probe (TGCTCCCCTTGCTTTCG) (metabion) and observed by fluorescence microscopy (Keyence). We seeded 2 × 104 infected HeLa229 or Neff cells or 5 × 104 macrophages (infected 3 h after seeding) per well into μ-Slide 8-well slides. After infection with the indicated MOI for the indicated time points and incubation at 37°C and 5% CO2, the slides were washed once with Dulbecco's phosphate-buffered saline (DPBS) and fixed by adding 100 μl 4% PFA (Morphisto; order no. 11762.00250) per well. Ethanol permeabilization was performed by incubating the cells in 50% (vol/vol), 80% (vol/vol), and 100% (vol/vol) ethanol, followed by drying at room temperature (RT). Hybridization took place at 46°C (using a hybridization oven) and in 100 μl per well of hybridization buffer (0.01% (vol/vol) SDS (Carl Roth; order no. CN30.2), 25% (vol/vol) formamide (Carl Roth; order no. P040.1), 900 mM NaCl (Carl Roth; order no. 9265.2), and 20 mM Tris-HCl (pH 8.0) (catalog no. B2261 and 6331.3, respectively; Sigma-Aldrich) in Milli-Q water containing the Cy5-labeled probe (1 pmol/μl). The slides were incubated for 2 h in the dark, followed by incubation in prewarmed washing buffer (0.01% (vol/vol) SDS, 25% (vol/vol) formamide, 150 mM NaCl, 20 mM Tris-HCl (pH 8.0), and 5 mM EDTA (Sigma-Aldrich; catalog no. E5134-250G) in Milli-Q water at 46°C for 30 min. Ice-cold Milli-Q water was added to the cells after removing the washing buffer, and the nuclei were stained with Hoechst 33342. Cells were covered with mounting medium and stored at 4°C in the dark until microscopy.
Flow cytometry.
The infections were performed as described above. As a positive control, HeLa cells were treated with the combination of Mcl-1 inhibitor S68345 (Selleck Chemicals; catalog no. S8383) and ABT-737 (Selleck Chemicals; catalog no. S1002-10) as indicated. Cells were harvested by trypsinization (trypsin; Thermo Fisher; catalog no. 25300096) and collected after stopping the trypsin reaction with 10% (vol/vol) FBS containing medium, followed by washing three times with DPBS (Life Technologies; catalog no. 14190169), centrifugation (1,500 × g for 5 min), and fixation with 4% PFA for 15 min at RT and methanol (VWR; catalog no. 20864.320) for at least 30 min at −20°C. Anti-cleaved caspase-3 (Asp175) antibody (Cell Signaling Technologies; catalog no. 9661S) was added, diluted 1:500 in dilution buffer (DPBS with 1% [vol/wt] BSA [Serva; catalog no. 11930.04]), and incubated at RT for 2 h. Secondary antibodies (anti-rabbit IgG-DyLight647 [Dianova; catalog no. 711-605-152] and anti-rabbit IgG-DyLight488 [Dianova; catalog no. 711-454-152]) were added after removing the primary antibody and incubated for 1 h at RT. Cells were analyzed by flow cytometry on a fluorescence-activated cell sorter (FACS) Calibur (BD Biosciences). Data were analyzed with the software FlowJo version 10.4.
Live/dead staining.
To detect cell death after infection, the far-red Live/Dead fixable dead-cell stain kit (Life Technologies; catalog no. L10120) was used. For positive controls, HeLa cells were heat treated (1 min at 95°C) directly after harvesting and before staining with the Live/Dead staining kit. The experiments were performed as suggested by the manufacturer.
Monitoring of bacterial growth.
To detect intracellular bacterial growth of P. acanthamoebae and C. trachomatis during infections, DNA of infected cells was isolated at the indicated time points, and quantitative PCR (qPCR) was performed. The DNA was isolated using the genomic DNA isolation kit obtained from Invitrogen (PureLink genomic DNA minikit; catalog no. K182002). For qPCR, 50 ng of isolated DNA was used and mixed with SYBR selected master mix (Thermo Fisher Scientific; catalog no. 4472918). For C. trachomatis detection, the published primers specific for the 16S gene were used (Ctr16sF, TCGAGAATCTTTCGCAATGGAC, and Ctr16sR, CGCCCTTTACGCCCAATAAA [39]), and for P. acanthamoebae, detection primers specific for their 16S gene were used (PacF, CTCAACTCCAGAACAGCATTT, and PacR, CTCAGCGTCAGGAATAAGC [40]). We used primers specific for the human HPRT gene (hHPRT_1, ACCACCGTGTGTTAGAAAAG, and hHPRT_2, AAAGGGAACTGCTGACAAAG) to quantify the host cell gene. The PCR was performed using the LightCycler 480 II (Roche), and threshold cycle (CT) values were calculated using the software provided (LightCycler 480 software release 1.2.1.62 SP3 [Roche]). Calculated CT values were used to determine the amount of bacterial DNA in 50 ng DNA isolation by using the equation of the standard curve, which was generated beforehand. For this, serial dilution of purified bacterial DNA was used, and CT values against the DNA amount are shown.
Inhibition of Bcl-XL in Bcl-XL-overexpressing HeLa229 cells.
Infection was performed as described previously for HeLa cells. The Bcl-XL-overexpressing cells were infected for 48 h at 37°C. Twenty-four hours prior to harvesting, cells were additionally treated with Bcl-XL inhibitor A-1155463 (Selleck Chemicals; catalog no. S7800) (5 to 15 μM), followed by active caspase-3 detection, determination of dead cells per population, and P. acanthamoebae growth detection via qPCR as described in the respective sections.
Reinfection of Acanthamoeba.
Infection was performed as described above for HeLa229 cells (control [CTRL] and Bax/Bak deficient). Cells were then trypsinized and washed several times by centrifugation (1,500 × g for 5 min) to eliminate free-swimming bacteria. Cells were resuspended in TSY medium (500 μl) and broken up with glass beads (Carl Roth; catalog no. A556.1). Two hundred fifty microliters of the suspension (after additional centrifugation [1,500 × g for 5 min] to remove cell waste) were used to infect amoeba cells for 24 h.
Electron microscopy.
HeLa cells were seeded on glass coverslips and infected with P. acanthamoebae (MOI of 25). After 48 h, cells were fixed for 30 min at room temperature in 4% paraformaldehyde plus 0.05% glutaraldehyde (Sigma-Aldrich; catalog no. G7776-10ML). Slides were stored in 1 M potassium dihydrogen orthophosphate (KH2PO4; Carl Roth; catalog no. 3904.2) buffer.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by a grant of the Deutsche Forschungsgemeinschaft to G.H. We thank Matthias Horn, Vienna, for providing P. acanthamoebae and for advice concerning P. acanthamoebae propagation.
D.B. and O.K. performed experiments and contributed data. G.H. designed and supervised the study. D.B. and G.H. wrote the manuscript.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Hacker G. 2018. Apoptosis in infection. Microbes Infect 20:552–559. doi: 10.1016/j.micinf.2017.10.006. [DOI] [PubMed] [Google Scholar]
- 2.Leist M, Jaattela M. 2001. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol 2:589–598. doi: 10.1038/35085008. [DOI] [PubMed] [Google Scholar]
- 3.Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G. 2008. Viral control of mitochondrial apoptosis. PLoS Pathog 4:e1000018. doi: 10.1371/journal.ppat.1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vaux DL, Hacker G. 1995. Hypothesis: apoptosis caused by cytotoxins represents a defensive response that evolved to combat intracellular pathogens. Clin Exp Pharmacol Physiol 22:861–863. doi: 10.1111/j.1440-1681.1995.tb01951.x. [DOI] [PubMed] [Google Scholar]
- 5.Clem RJ, Fechheimer M, Miller LK. 1991. Prevention of apoptosis by a baculovirus gene during infection of insect cells. Science 254:1388–1390. doi: 10.1126/science.1962198. [DOI] [PubMed] [Google Scholar]
- 6.Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, Greenberg AH, Zhong G. 1998. Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome C release and caspase activation. J Exp Med 187:487–496. doi: 10.1084/jem.187.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhong Y, Weininger M, Pirbhai M, Dong F, Zhong G. 2006. Inhibition of staurosporine-induced activation of the proapoptotic multidomain Bcl-2 proteins Bax and Bak by three invasive chlamydial species. J Infect 53:408–414. doi: 10.1016/j.jinf.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 8.Ying S, Pettengill M, Latham ER, Walch A, Ojcius DM, Hacker G. 2008. Premature apoptosis of Chlamydia-infected cells disrupts chlamydial development. J Infect Dis 198:1536–1544. doi: 10.1086/592755. [DOI] [PubMed] [Google Scholar]
- 9.Brokatzky D, Dorflinger B, Haimovici A, Weber A, Kirschnek S, Vier J, Metz A, Henschel J, Steinfeldt T, Gentle IE, Hacker G. 2019. A non-death function of the mitochondrial apoptosis apparatus in immunity. EMBO J 38:e100907. doi: 10.15252/embj.2018100907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horn M, Collingro A, Schmitz-Esser S, Beier CL, Purkhold U, Fartmann B, Brandt P, Nyakatura GJ, Droege M, Frishman D, Rattei T, Mewes HW, Wagner M. 2004. Illuminating the evolutionary history of chlamydiae. Science 304:728–730. doi: 10.1126/science.1096330. [DOI] [PubMed] [Google Scholar]
- 11.Konig L, Wentrup C, Schulz F, Wascher F, Escola S, Swanson MS, Buchrieser C, Horn M. 2019. Symbiont-mediated defense against Legionella pneumophila in amoebae. mBio 10:e00333-19. doi: 10.1128/mBio.00333-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greub G, Mege JL, Raoult D. 2003. Parachlamydia acanthamoebae enters and multiplies within human macrophages and induces their apoptosis. Infect Immun 71:5979–5985. doi: 10.1128/iai.71.10.5979-5985.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casson N, Medico N, Bille J, Greub G. 2006. Parachlamydia acanthamoebae enters and multiplies within pneumocytes and lung fibroblasts. Microbes Infect 8:1294–1300. doi: 10.1016/j.micinf.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 14.Ito A, Matsuo J, Nakamura S, Yoshida A, Okude M, Hayashi Y, Sakai H, Yoshida M, Takahashi K, Yamaguchi H. 2012. Amoebal endosymbiont Protochlamydia induces apoptosis to human immortal HEp-2 cells. PLoS One 7:e30270. doi: 10.1371/journal.pone.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayashi Y, Nakamura S, Matsuo J, Fukumoto T, Yoshida M, Takahashi K, Mizutani Y, Yao T, Yamaguchi H. 2010. Host range of obligate intracellular bacterium Parachlamydia acanthamoebae. Microbiol Immunol 54:707–713. doi: 10.1111/j.1348-0421.2010.00265.x. [DOI] [PubMed] [Google Scholar]
- 16.Sixt BS, Hiess B, Konig L, Horn M. 2012. Lack of effective anti-apoptotic activities restricts growth of Parachlamydiaceae in insect cells. PLoS One 7:e29565. doi: 10.1371/journal.pone.0029565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steller H. 2008. Regulation of apoptosis in Drosophila. Cell Death Differ 15:1132–1138. doi: 10.1038/cdd.2008.50. [DOI] [PubMed] [Google Scholar]
- 18.Hengartner MO. 2000. The biochemistry of apoptosis. Nature 407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 19.Youle RJ, Strasser A. 2008. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 20.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. 2010. The BCL-2 family reunion. Mol Cell 37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. 2005. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 22.Eitz Ferrer P, Potthoff S, Kirschnek S, Gasteiger G, Kastenmüller W, Ludwig H, Paschen SA, Villunger A, Sutter G, Drexler I, Häcker G. 2011. Induction of Noxa-mediated apoptosis by modified vaccinia virus Ankara depends on viral recognition by cytosolic helicases, leading to IRF-3/IFN-beta-dependent induction of pro-apoptotic Noxa. PLoS Pathog 7:e1002083. doi: 10.1371/journal.ppat.1002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Czabotar PE, Lessene G, Strasser A, Adams JM. 2014. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 24.Yamane C, Yamazaki T, Nakamura S, Matsuo J, Ishida K, Yamazaki S, Oguri S, Shouji N, Hayashi Y, Yoshida M, Yimin, Yamaguchi H. 2015. Amoebal endosymbiont Parachlamydia acanthamoebae Bn9 can grow in immortal human epithelial HEp-2 cells at low temperature; an in vitro model system to study chlamydial evolution. PLoS One 10:e0116486. doi: 10.1371/journal.pone.0116486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR, Thompson CB. 2000. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol Cell 6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang GG, Calvo KR, Pasillas MP, Sykes DB, Hacker H, Kamps MP. 2006. Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat Methods 3:287–293. doi: 10.1038/nmeth865. [DOI] [PubMed] [Google Scholar]
- 27.Lienard J, Croxatto A, Gervaix A, Levi Y, Loret JF, Posfay-Barbe KM, Greub G. 2017. Prevalence and diversity of Chlamydiales and other amoeba-resisting bacteria in domestic drinking water systems. New Microbes New Infect 15:107–116. doi: 10.1016/j.nmni.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greub G. 2009. Parachlamydia acanthamoebae, an emerging agent of pneumonia. Clin Microbiol Infect 15:18–28. doi: 10.1111/j.1469-0691.2008.02633.x. [DOI] [PubMed] [Google Scholar]
- 29.Ammerdorffer A, Stojanov M, Greub G, Baud D. 2017. Chlamydia trachomatis and chlamydia-like bacteria: new enemies of human pregnancies. Curr Opin Infect Dis 30:289–296. doi: 10.1097/QCO.0000000000000369. [DOI] [PubMed] [Google Scholar]
- 30.Roger T, Casson N, Croxatto A, Entenza JM, Pusztaszeri M, Akira S, Reymond MK, Le Roy D, Calandra T, Greub G. 2010. Role of MyD88 and Toll-like receptors 2 and 4 in the sensing of Parachlamydia acanthamoebae. Infect Immun 78:5195–5201. doi: 10.1128/IAI.00786-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buchholz KR, Stephens RS. 2008. The cytosolic pattern recognition receptor NOD1 induces inflammatory IL-8 during C. trachomatis infection. Infect Immun 76:3150–3155. doi: 10.1128/IAI.00104-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Yeruva L, Marinov A, Prantner D, Wyrick PB, Lupashin V, Nagarajan UM. 2014. The DNA sensor, cyclic GMP-AMP synthase, is essential for induction of IFN-beta during Chlamydia trachomatis infection. J Immunol 193:2394–2404. doi: 10.4049/jimmunol.1302718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inohara N, Koseki T, del Peso L, Hu Y, Yee C, Chen S, Carrio R, Merino J, Liu D, Ni J, Núñez G. 1999. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J Biol Chem 274:14560–14567. doi: 10.1074/jbc.274.21.14560. [DOI] [PubMed] [Google Scholar]
- 34.Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, Radtke F, Ablasser A. 2017. Signalling strength determines proapoptotic functions of STING. Nat Commun 8:427. doi: 10.1038/s41467-017-00573-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brokatzky D, Dorflinger B, Haimovici A, Weber A, Kirschnek S, Vier J, Metz A, Henschel J, Steinfeldt T, Gentle IE, Hacker G. 2019. A non-death function of the mitochondrial apoptosis apparatus in immunity. EMBO J 38:e102325. doi: 10.15252/embj.2018100907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gracey E, Lin A, Akram A, Chiu B, Inman RD. 2013. Intracellular survival and persistence of Chlamydia muridarum is determined by macrophage polarization. PLoS One 8:e69421. doi: 10.1371/journal.pone.0069421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun HS, Eng EW, Jeganathan S, Sin AT, Patel PC, Gracey E, Inman RD, Terebiznik MR, Harrison RE. 2012. Chlamydia trachomatis vacuole maturation in infected macrophages. J Leukoc Biol 92:815–827. doi: 10.1189/jlb.0711336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohmer M, Tzivelekidis T, Niedenfuhr N, Volceanov-Hahn L, Barth S, Vier J, Borries M, Busch H, Kook L, Biniossek ML, Schilling O, Kirschnek S, Hacker G. 2018. Infection of HeLa cells with Chlamydia trachomatis inhibits protein synthesis and causes multiple changes to host cell pathways. Cell Microbiol 21:e12993. doi: 10.1111/cmi.12993. [DOI] [PubMed] [Google Scholar]
- 39.Goldschmidt P, Rostane H, Sow M, Goepogui A, Batellier L, Chaumeil C. 2006. Detection by broad-range real-time PCR assay of Chlamydia species infecting human and animals. Br J Ophthalmol 90:1425–1429. doi: 10.1136/bjo.2006.096420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Casson N, Posfay-Barbe KM, Gervaix A, Greub G. 2008. New diagnostic real-time PCR for specific detection of Parachlamydia acanthamoebae DNA in clinical samples. J Clin Microbiol 46:1491–1493. doi: 10.1128/JCM.02302-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.