

Summary
To discover regulatory elements driving the specificity of gene expression in different cell types and regions of the developing human brain, we generated an atlas of open chromatin from nine dissected regions of the mid-gestation human telencephalon, as well as microdissected upper and deep layers of the prefrontal cortex. We identified a subset of open chromatin regions (OCRs), termed predicted regulatory elements (pREs), that are likely to function as developmental brain enhancers. pREs showed temporal, regional, and laminar differences in chromatin accessibility, and were correlated with gene expression differences across regions and gestational ages. We identified two functional de novo variants in a pRE for autism risk gene SLC6A1, and demonstrated using CRISPRa that this pRE regulates SCL6A1. Additionally, mouse transgenic experiments validated enhancer activity for pREs proximal to FEZF2 and BCL11A. Thus, this atlas serves as a resource for decoding neurodevelopmental gene regulation in health and disease.
~19,000 enhancers defined in nine regions of the developing human telencephalon
Chromatin dynamics correlate with sequence motifs and spatiotemporal gene expression
Identified cortical layer-specific enhancers and validated a Layer 5 FEZF2 enhancer
Genetic variants from patients alter activity of an enhancer for an autism risk gene
Graphical Abstract
“In Brief”:
A high-resolution atlas of regulatory elements driving regional, temporal, and laminar gene expression programs in the developing human telencephalon reveals enhancers and genetic variants regulating human disease genes.
Introduction
The development of the human telencephalon, the seat of cognition and consciousness, requires the stepwise generation of regions and cell types, long-distance migrations of cells and axons, and the formation of precise connections (J. Rubenstein and Rakic 2013). This spatiotemporal precision is mirrored in gene expression patterns across the telencephalon, which are orchestrated by transcription factors (TFs) binding to diverse classes of proximal and distal regulatory elements. Genomic regulatory elements (REs) play an important role in forebrain development (Visel et al. 2007, 2013; Dickel et al. 2018), and identifying REs specific to cell types and brain regions is an important step towards elucidating the transcriptional mechanisms underlying human brain development and interpreting genetic risk variants associated with neurodevelopmental disorders.
Chromatin at transcriptionally active REs is typically nucleosome-depleted and accessible to nuclease digestion in assays such as ATAC-seq (Buenrostro et al. 2015). This technology enables mapping the dynamics of the chromatin accessibility landscape that accompanies, and likely determines, gene expression changes during brain development. A recent study used this technique to map chromatin accessibility changes across human cortical neuron differentiation (de la Torre-Ubieta et al. 2018). Our work expands on this study by 1) assaying anatomically distinct cortical regions, as well as sub-cortical regions of the telencephalon that have not been analyzed previously, 2) tracking changes between early and late midfetal development, 3) dissecting differences in accessibility between cortical laminae, and 4) identifying thousands of candidate enhancers specific to these processes.
We generated an atlas of the chromatin accessibility landscape of the midfetal human telencephalon. Midfetal development is a critical time for diversification, exhibiting high regional variation in gene expression which declines in late fetal development as well as postnatally (M. Li et al. 2018). ATAC-seq was performed on nuclei extracted from samples of six cortical regions and the basal ganglia anlage (the three ganglionic eminences) dissected from intact specimens. We identified statistically significant OCRs, generated a list of predicted regulatory elements (pREs) from these OCRs, and validated enhancer activities using luciferase transcription and transgenic enhancer assays. Spatiotemporal differences in gene expression across the developing telencephalon were associated with differential chromatin accessibility at pREs. We further identified pREs that may drive laminar gene expression differences and the development of upper and deep layers of the cortex, and identified a Layer 5-specific enhancer for FEZF2. Motif analysis uncovered TFs that may bind pREs and drive enhancer specificity.
To investigate the application of this resource to decoding genetic variation in human neurodevelopmental disorders, we analyzed de novo variants identified from whole genome sequencing (WGS) of autism spectrum disorder (ASD) cases (An et al 2018). We identified functional mutations in a pRE located in an intron of high confidence ASD and epilepsy risk gene SLC6A1, whose function as an enhancer we validated by CRISPRa. Thus, in addition to enumerating open chromatin regions and candidate enhancers that drive regional and cellular identity in the developing human telencephalon, this atlas also provides a framework for predicting the cell types and brain regions impacted by non-coding genetic risk variants during development.
Results
Identifying open chromatin regions in the mid-gestation human telencephalon
We performed ATAC-seq on fresh, intact samples of mid-gestation (14–19 gestation weeks [gw], measured from the last menstrual period) human fetal telencephalon, including six regions dissected from cortical anlage: dorsal lateral prefrontal cortex (PFC), motor, primary somatosensory cortex (S1), temporal cortex, parietal cortex, and primary visual cortex (V1); and three subcortical regions (basal ganglia anlage): medial, lateral, and caudal ganglionic eminences (MGE, LGE, CGE) (Figure 1A). Based on mouse fate mapping experiments, the MGE generates pallidal neurons and interneurons that integrate into the cortex and striatum, the LGE generates striatal medium spiny neurons, and the CGE generates subpallial amygdala neurons and cortical interneurons (J. L. R. Rubenstein and Campbell 2013). The regional dissections spanned the full thickness of the telencephalic wall, from the ventricular zone to the meninges. Thus, all samples included progenitors and immature neurons, glia, blood vessels, and meninges. Twenty-five ATAC-seq libraries were generated as described (Buenrostro et al. 2015) and sequenced using Illumina Hiseq 2500 to a median depth of 16 million post-QC reads per sample. Reads were mapped to the human genome build GRCh37 using the ENCODE pipeline (Lee et al. 2016) and were highly enriched at transcription start sites (Figure S1A) and overlapped with DNase hypersensitivity loci (Roadmap Epigenomics Consortium et al. 2015) (mean 58.8% within universal DHS peaks; Table S1).
Figure 1: Defining open chromatin regions (OCRs) in the fetal human telencephalon.
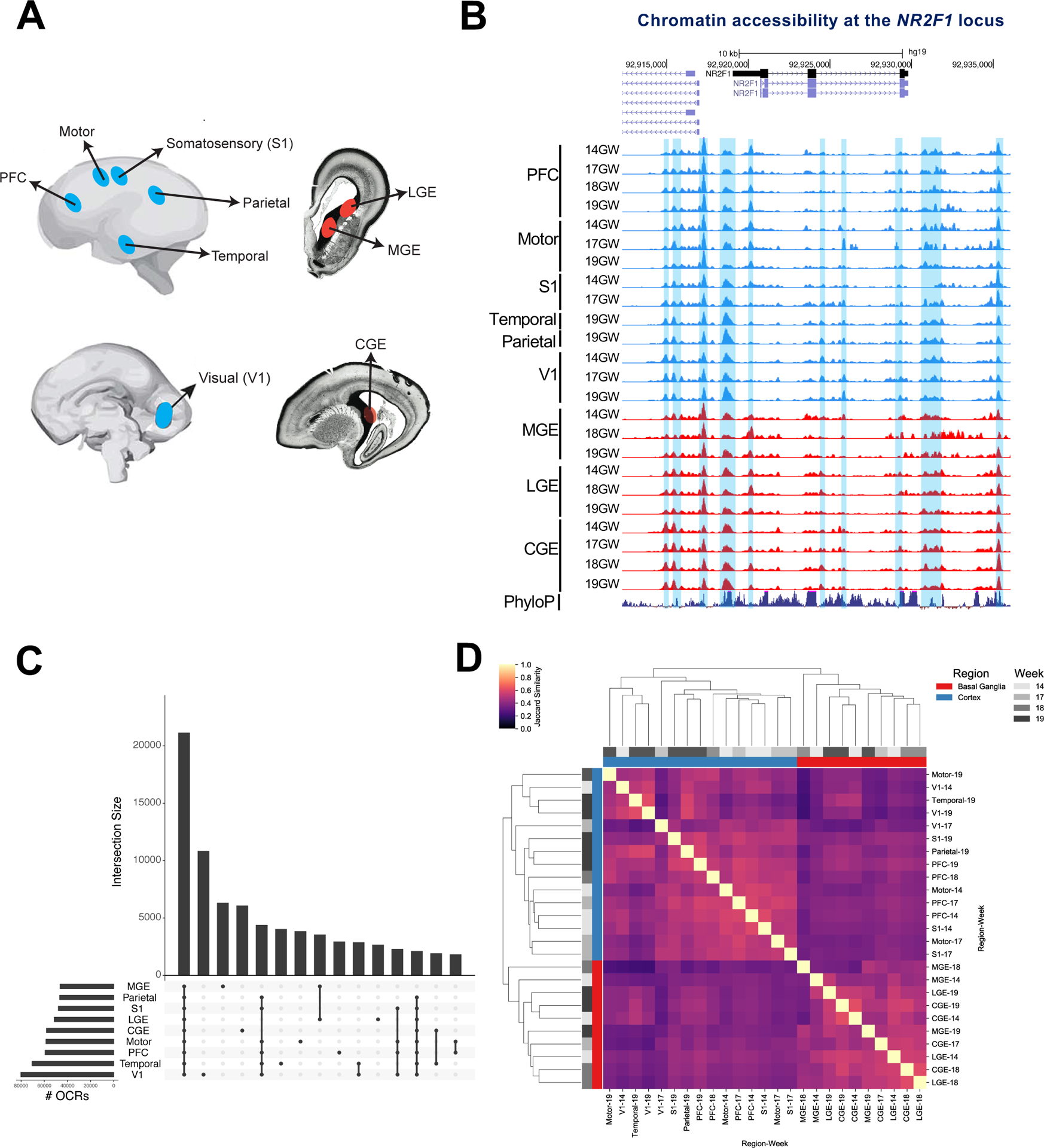
1A) Schematic of fetal tissue collection.
1B) ATAC-seq reads from all fetal samples collected mapped to the NR2F1 locus. Blue shaded regions are OCRs. Y axis scale is 0 to 10.
1C) OCR intersections and # OCRs pooled across samples for each region.
1D) Heat map of jaccard similarity of OCRs in all samples.
The ENCODE pipeline identified statistically significant open chromatin regions (OCRs) in twenty-four ATAC-seq libraries that passed stringent quality filtering (Figure 1B, Methods). We obtained a median of 61,497 high-confidence OCRs per sample (Table S1), totaling 130,131 unique OCRs merged across all telencephalic regions. Roughly 16% of merged OCRs are shared between all assayed regions, while 23% are shared between all assayed cortical regions. This leaves many OCRs that are specific to one region, e.g. primary visual cortex, MGE, and CGE (Figure 1C). Clustering samples by OCR similarity shows two main groups representing the cortex and basal ganglia (Figure 1D), and within cortex samples cluster by anatomical proximity. In concordance with other human DNase hypersensitivity data, 96% of OCRs overlapped with fetal brain DNase peaks (Meuleman et al. 2019).
Predicting developmental brain enhancers
Chromatin accessibility is elevated at known forebrain enhancers and reduced at enhancers for other tissues: examples of a forebrain enhancer (hs433) and a limb enhancer (hs72) are shown in Figures 2A and 2B. To identify which OCRs are likely to function as neurodevelopmental enhancers, we developed a model using a machine learning algorithm trained on candidate enhancers tested in vivo (Methods). Specifically, we included all human sequences from the VISTA Enhancer Browser database (Visel et al. 2007) with strong embryonic brain enhancer activity as true positives, compared to sequences that showed activity in non-brain tissues or weak/no activity in VISTA as true negatives. Each positive and negative sequence was annotated with functional genomics data and the sub-sequences (k-mers) it contains. The algorithm then learned from these features how to distinguish the positive brain enhancer regions from the negative control regions (Figure S1B).
Figure 2: Predicting neurodevelopmental enhancers.
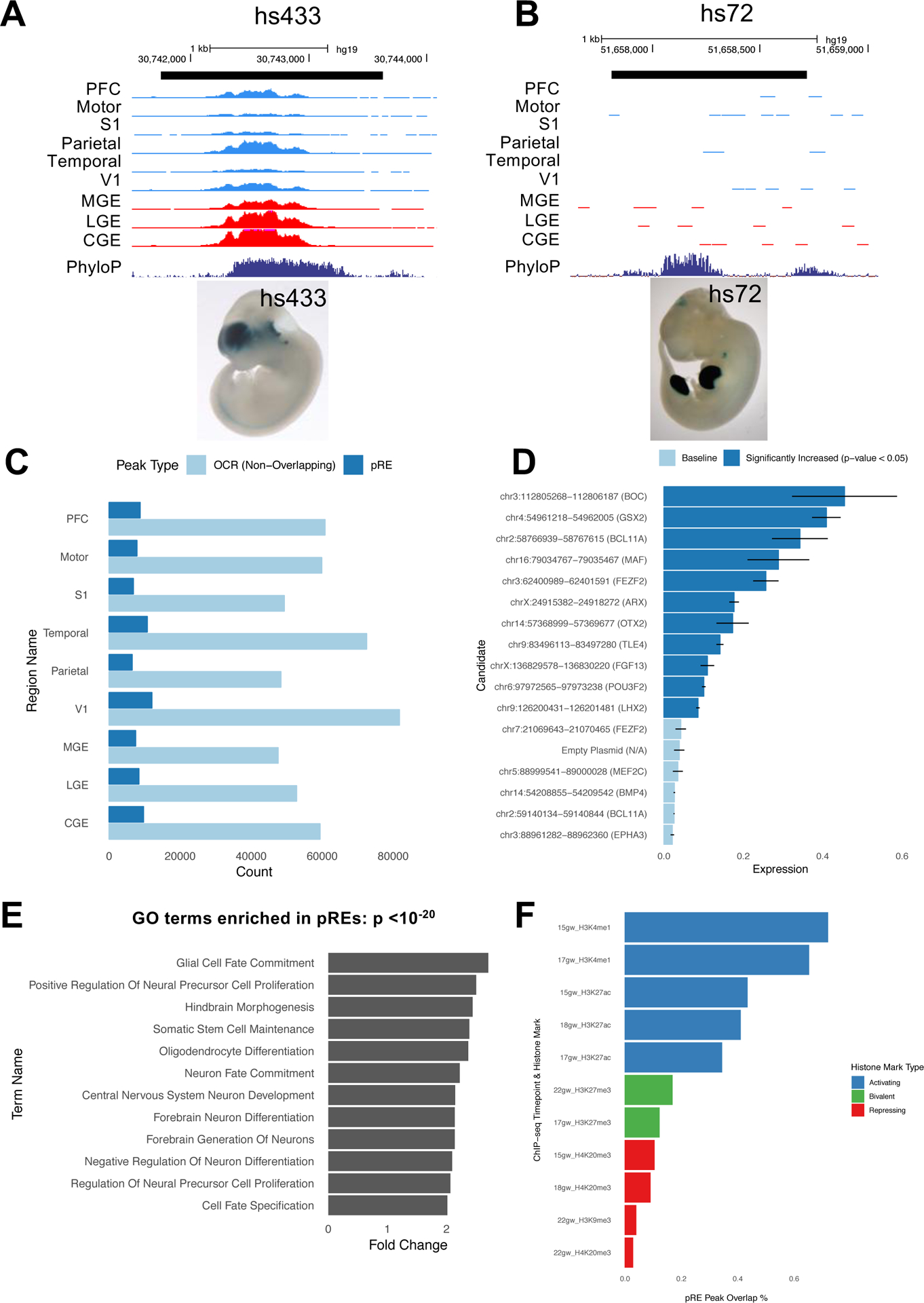
2A) ATAC-seq reads, combined across multiple samples per brain region, at VISTA brain enhancer hs433. Y axis scale is 0 to 500. E11.5 enhancer transgenic mouse hs433 is shown and lacZ is stained in blue (from enhancer.lbl.gov website).
2B) ATAC-seq reads, combined across multiple samples per brain region, at VISTA limb enhancer hs72. Y axis scale is 0 to 500. E11.5 enhancer transgenic mouse hs72 is shown and lacZ expression is stained in blue (from enhancer.lbl.gov website).
2C) Fraction of OCRs that are pREs in each brain region.
2D) Mean firefly luciferase levels in human neuroblastoma cells, normalized to Renilla. Fifteen pREs (chromosomal locations and nearest gene indicated) were cloned upstream of a minimal promoter and firefly luciferase ORF. Dark blue bars indicate pREs with increased luciferase signal activity compared to empty vector; error bars indicate standard error across four replicate experiments. ARX VISTA enhancer hs119 is included as a positive control for comparison.
2E) Gene ontology terms enriched in pREs with GREAT analysis, complete list in Table S2C.
2F) The percent of OCRs in PFC that overlap different histone modification peaks from midfetal PFC (15gw-22gw), subtracted from the percent of pREs in PFC that overlap different histone modification peaks.
Applying the resulting model to 103,829 OCRs that do not overlap promoters (Methods), we identified a subset of 19,151 predicted regulatory elements (pREs) (Table S2A) comprising 18.4% of all OCRs (Figure 2C, median 6,918 pREs per brain region). We expect these pREs consist primarily of distal enhancers, though other cis-regulatory elements may be also be included. We tested the ability of 15 pREs with high prediction scores (>0.85) to regulate transcription using a transfection assay with a luciferase readout. Ten pREs (66%) were validated by this assay in a mitotically active human brain derived BE(2)-C cell line (Figure 2D); zero of eight randomly selected OCRs that are not pREs were transcriptionally active (Figure S1C).
Gene ontology analysis of the nearest genes to pREs using GREAT shows enrichment of neurodevelopmental terms such as “central nervous system development”, “neuron differentiation”, and “forebrain generation of neurons” (Figure 2E), while no terms are enriched for genes proximal to non-pRE OCRs. pREs are more highly conserved than OCRs, but not as conserved as the training set of VISTA enhancers, which were originally selected for having exceptionally high conservation scores (Visel et al. 2007) (Figure S1D).
To study the histone modification landscape of pREs and OCRs, we generated ChIP-seq data (Methods) for multiple activating and repressive histone marks from midfetal PFC tissue at various time points (15gw-22gw). pREs in the PFC more often overlap activating marks such as H3K27ac (55% overlap), a mark of active enhancers (Creyghton et al. 2010), compared to OCRs (47% overlap). They also less frequently overlap repressive histone modifications H3K27me3, H3K9me3, and H4K20me3 (Figures 2F and S1E, Tables S2D and S2E).
To track OCRs that are activated or inactivated across development, we analyzed ATAC-seq and ChIP-seq data from early (14gw and 15gw) and late (18gw) midfetal PFC. We found OCRs that don’t change accessibility are concentrated at TSSs and contain sequence motifs for promoter-binding transcription factors (Figure S2A). OCRs that gain or lose accessibility across mid-fetal development are distal from TSSs and are enriched for sequence motifs for MEF2 family TFs (gained accessibility) and CTCF (lost accessibility) (Figure S2A). These dynamic OCRs are more likely to be pREs (16% of gains, 19% of losses) than are stable OCRs (11%) (p-value < 2.2e-16, binomial test). Further, the subset of OCRs where changes in accessibility during development are accompanied by gain or loss of the H3K27ac mark are enriched for sequence motifs of TFs that regulate cortical development, such as BCL11A, MEF2A-D, NEUROD1, NEUROG2 and TCF4 (gain of accessibility and H3K27ac), and NUR77 (loss of accessibility and H3K27ac) (Figures S2A, S2B, and S2C).
Identifying region-specific predicted enhancers
We next associated differences in gene expression with regional differences in chromatin accessibility at pREs. We used single-cell RNA-seq data from 14–19gw MGE, PFC, and V1 samples (Nowakowski et al. 2017). A subset of the specimens used to generate the RNA-seq data were also used to generate our ATAC-seq libraries and thus are well-matched. We identified 1,800 differentially expressed (DE) genes pairwise between the MGE, PFC, and V1. pREs with differential chromatin accessibility were highly enriched for DE genes (odds ratios > 3, Figure 3A). We identified 510 differentially accessible pREs that were associated with DE genes between cortex (PFC or V1) and MGE (Tables S3A and S3B). For example, MBIP is more highly expressed in MGE compared to PFC and V1 (3-fold change, q-value 1.2*10−16), while KCNQ3 has the opposite expression pattern (4-fold change, q-value 1.3*10−13). Both MBIP and KCNQ3 have accompanying differences in chromatin accessibility at proximal pREs (Figures 3B and 3C). These results are supported by in situ hybridization experiments (Tucker et al. 2008; Diez-Roux et al. 2011) (Figure S3A).
Figure 3: Regional differences in chromatin accessibility at pREs.
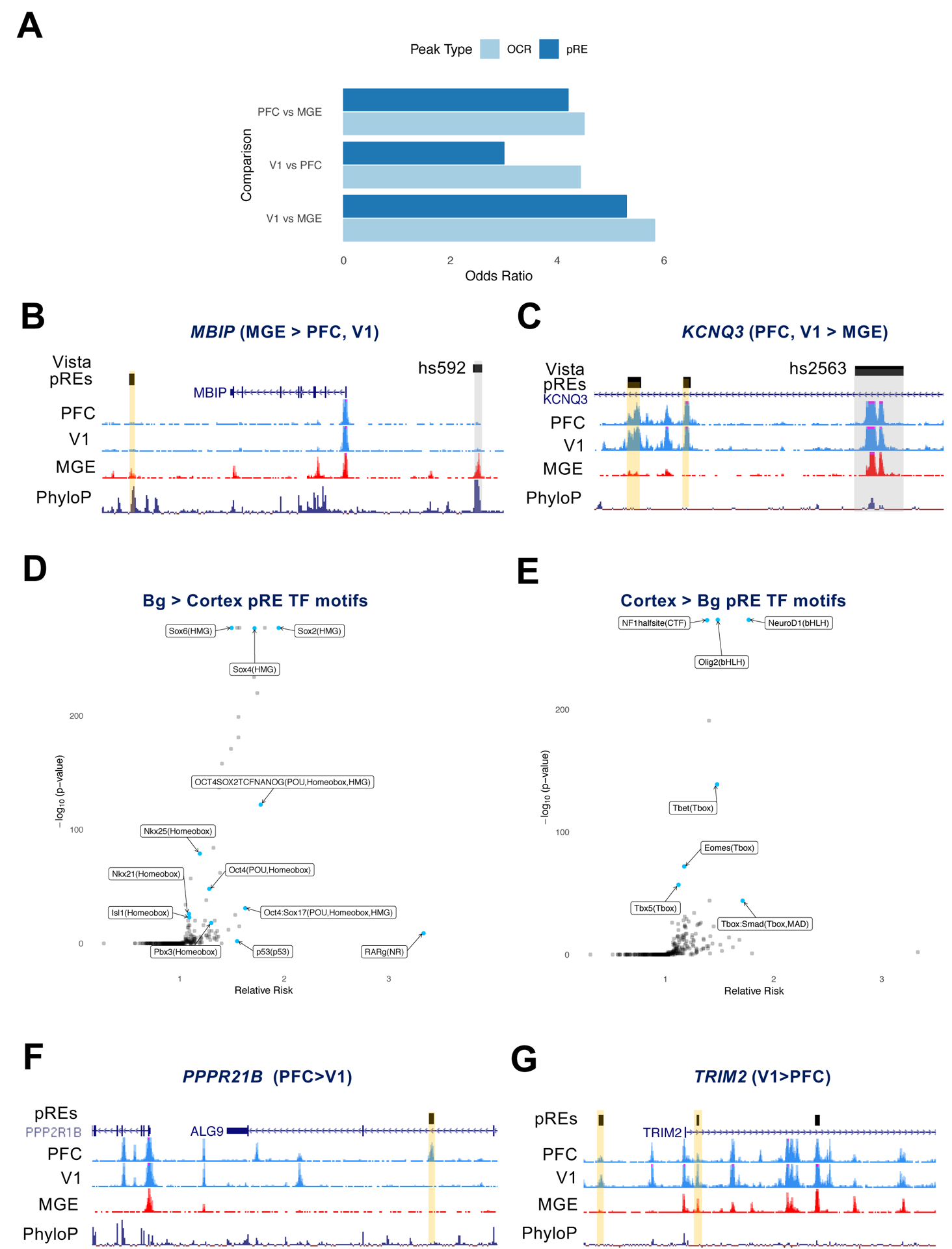
3A) Odds ratio for the co-occurence of genes that were differentially expressed and proximal to differentially accessible chromatin (100 kb), computed between pairs of brain regions and reported separately for OCRs and pREs.
3B,C) ATAC-seq reads combined from multiple samples in PFC, V1, and MGE. pREs are highlighted in yellow, VISTA enhancers in grey. The nearby genes are differentially expressed between the regions indicated. Y axis scale is 0 to 50 for ATAC-seq tracks.
3D) Effect size and significance of TF motifs enriched in basal ganglia-specific pREs compared to cortex-specific pREs.
3E) Effect size and significance of TF motifs enriched in cortex-specific pREs compared to basal ganglia-specific pREs.
3F,G) ATAC-seq reads combined from multiple samples in PFC, V1, and MGE. pREs are highlighted in yellow. The nearby genes are differentially expressed between the regions indicated. Y axis scale is 0 to 50 for ATAC-seq tracks.
Focusing on regional differences within the telencephalon, we identified 6,942 pREs (36.2%) that were cortex specific and 3,462 pREs (18%) that were basal ganglia specific (Table S2A). Here, we define “specific” as a pRE that has no statistically significant ATAC-seq signal in the other brain regions in our data. In order to explore potential upstream regulators of pREs, we looked for enrichment of TF binding motifs in these region-specific pREs. Basal ganglia-specific pREs were enriched for several motifs including SOX TFs, with 9.9% containing the composite motif for OCT4-SOX2, a sequence that promotes MGE enhancer function in mice (Sandberg et al. 2018) (Figure 3D). Cortex-specific pREs were enriched for motifs of TFs that have well known functions in the developing cortex, including NEUROD1, OLIG2, NF1, and the T-BOX family members (e.g. TBR1 and TBR2/Eomes) (Figure 3E).
To discover elements likely driving rostral-caudal differences in gene expression within the cortex, we identified 79 pREs with differential accessibility between PFC and V1 that were proximal to genes differentially expressed between these same two regions (Table S3C). For example, PPP2R1B is more highly expressed rostrally in PFC (1.2 fold change, q-value 0.019), while TRIM2 is more highly expressed caudally in V1 (1.7 fold change, q-value 0.047) which is supported by in situ hybridization experiments (Diez-Roux et al. 2011) (Figure S3A). Both gene loci are proximal to differentially accessible pREs (Figures 3F and 3G).
To identify pREs that are accessible in only one telencephalic sub-region, we combined OCRs across timepoints within each region, identified OCRs exclusive to a single brain region, and filtered the OCR list to those that were also pREs (Figure S3B, Table S2A). The resulting 5,318 region-specific pREs (27.7% of total) differed in their genomic distributions compared to other pREs, more often overlapping intronic and intergenic regions (Figure S3C).
Identifying cortical developmental stage-specific pREs
Since dramatic differences are observed in gene expression over developmental time in the fetal brain (M. Li et al. 2018) we sought to quantify changes in chromatin accessibility between the early and late stages of midfetal cortical development. Combining samples from the PFC and motor cortex, we identified 1,330 pREs specific to 14gw and 3,559 specific to 19gw in frontal cortex samples. GREAT analysis of early midfetal specific pREs yielded gene ontology terms for processes occurring during early cortical development such as “cerebral cortex radially oriented cell migration” and “neural precursor cell proliferation”. Late midfetal specific pREs were enriched for processes occurring with neuronal maturation such as “axon extension”, “long term synaptic potentiation”, “neurotransmitter secretion”, and “neuron fate commitment” (Figures 4A and 4B, Table S4A).
Figure 4: Temporal differences in chromatin accessibility at cortical pREs.
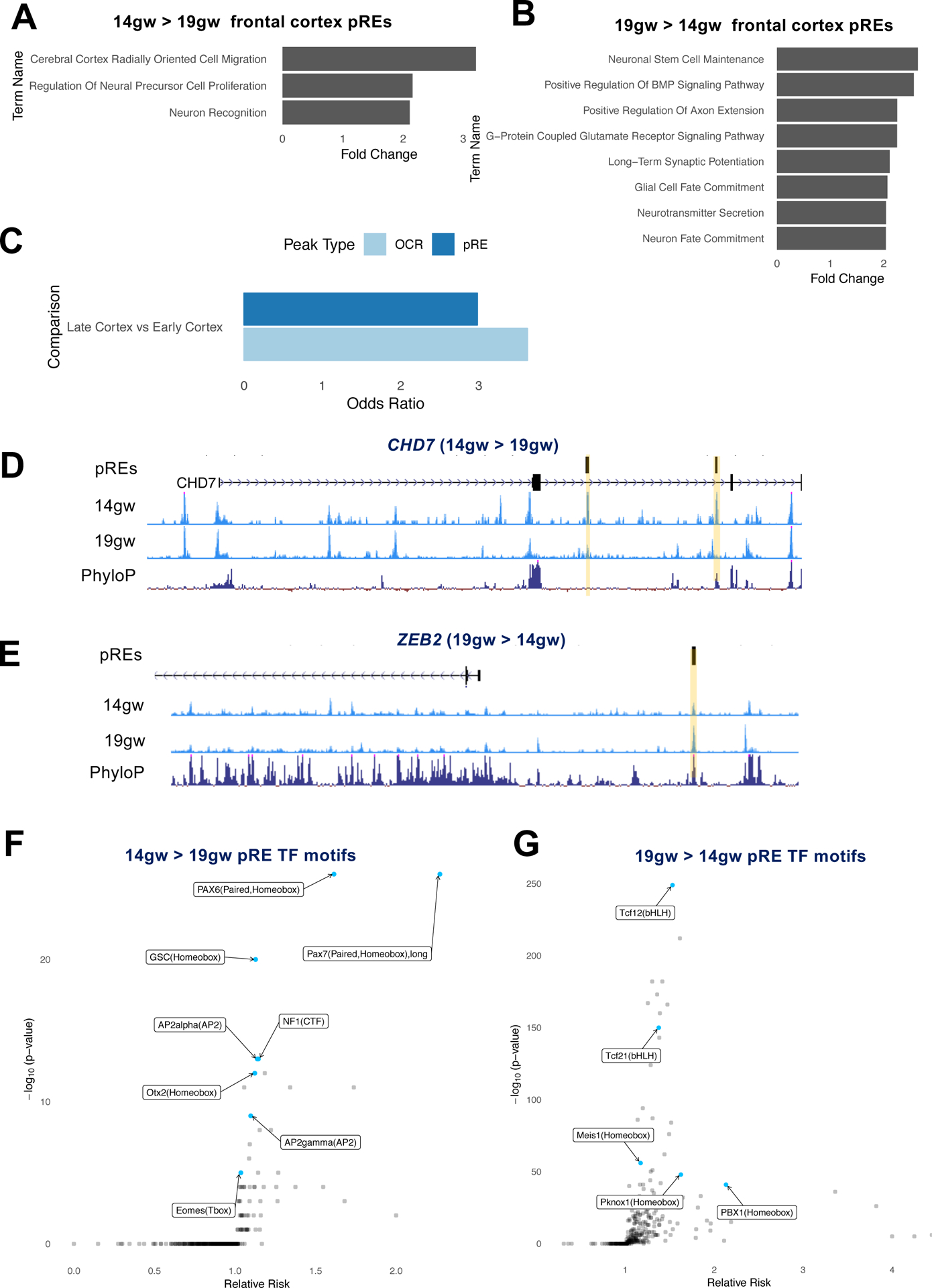
4A) GREAT analysis of functions associated with 14gw-specific pREs from frontal cortex tissues (combined PFC and motor samples).
4B) GREAT analysis of functions associated with 19gw-specific pREs from frontal cortex tissues.
4C) Genes differentially expressed in frontal cortex between 14gw and 19gw are enriched for 14gw- and 19gw-specific pREs and OCRs.
4D,E) ATAC-seq reads from 14gw and 19gw frontal cortex pooled samples. pREs are highlighted in yellow. The nearby genes are differentially expressed at 14gw and 19gw, respectively. Y axis scale is 0 to 50.
4F,G) Effect size and significance of TF motifs enriched in 14gw frontal cortex-specific pREs compared to 19gw-specific pREs, and vice versa.
To associate these differences in pREs with gene expression changes across midgestation, we integrated 14gw and 19gw pREs with single-cell RNA-seq data from age-matched samples (Nowakowski et al. 2017). 14gw- and 19gw-specific pREs in frontal cortex were enriched for genes differentially expressed between those ages (Figure 4C). We identified 203 age-specific pREs that were proximal to differentially expressed genes between 14gw and 19gw (Table S4B). For instance, the gene CHD7 (Vissers et al. 2004), which is associated with CHARGE syndrome and is expressed in cortical progenitors in the ventricular zone (Figure S4A) (Diez-Roux et al. 2011), is more highly expressed in 14gw cortex compared to 19gw (2-fold change, q-value 0.0009). CHD7 also contains two intronic pREs that are specific to early but not late frontal cortex (Figure 4D). In contrast is ZEB2, which is expressed in immature cortical excitatory neurons (Seuntjens et al. 2009). ZEB2 is more highly expressed in 19gw cortex (3-fold change, q-value 0.0027) and is likewise proximal to a pRE that is specific to late but not early frontal cortex development (Figure 4E).
Analysis of TF motifs in 14gw cortical pREs found enrichment of homeodomain TF motifs (e.g. PAX6), while motifs for BHLH TFs (e.g. TCF12) and homeobox TFs (PBX1) are enriched in 19gw cortical pREs (Figures 4F and 4G). Enrichment of these motifs may reflect the changing cellular makeup of the tissue: PAX6 is specifically expressed in cortical progenitors in the ventricular zone which are more abundant early in cortical development, while PBX1 is highly expressed in neurons that are more populous in later stages (D. D. M. O’Leary, Chou, and Sahara 2007; Golonzhka et al. 2015). Motif analysis of 422 14gw pREs that lose H3K27ac along with chromatin accessibility by 19gw shows enrichment of TF motifs associated with neuronal progenitors or stem cell state, including LHX2, NUR77, OCT4-SOX2-TCF-NANOG, SOX21, SOX21, SOX3 and SOX10 (Figure S4B).
Identifying putative enhancers for deep and superficial cortical projection neurons
Upper and deep layer excitatory projection neurons of the cortex have been predicted (Willsey et al. 2013; Parikshak et al. 2013) and shown (Velmeshev et al. 2019) to be altered in ASD. The regulatory elements that drive the specification of cortical projection neuron subtypes have not been identified in humans, although conserved enhancers have been studied in mice (Shim et al. 2012). We reasoned that candidate cortical neuron subtype enhancers could be identified by integrating our pREs with ATAC-seq data from upper and deep cortical layers.
We microdissected the upper and deep layers of the cortical plate of PFC from fresh 18gw and 19gw hemispheres along with whole PFC dissections that span from the ventricular zone to the pia (Figure 5A). We then performed ATAC-seq and identified 71,628 upper layer and 57,008 deep layer OCRs at an Irreproducible Discovery Rate of 10% across three replicates (Figure 5B). Of these, 5,517 OCRs were specific to deep layers and 20,867 were specific to upper layers of PFC (Table S5A).
Figure 5: Laminar differences in chromatin accessibility in the developing PFC.
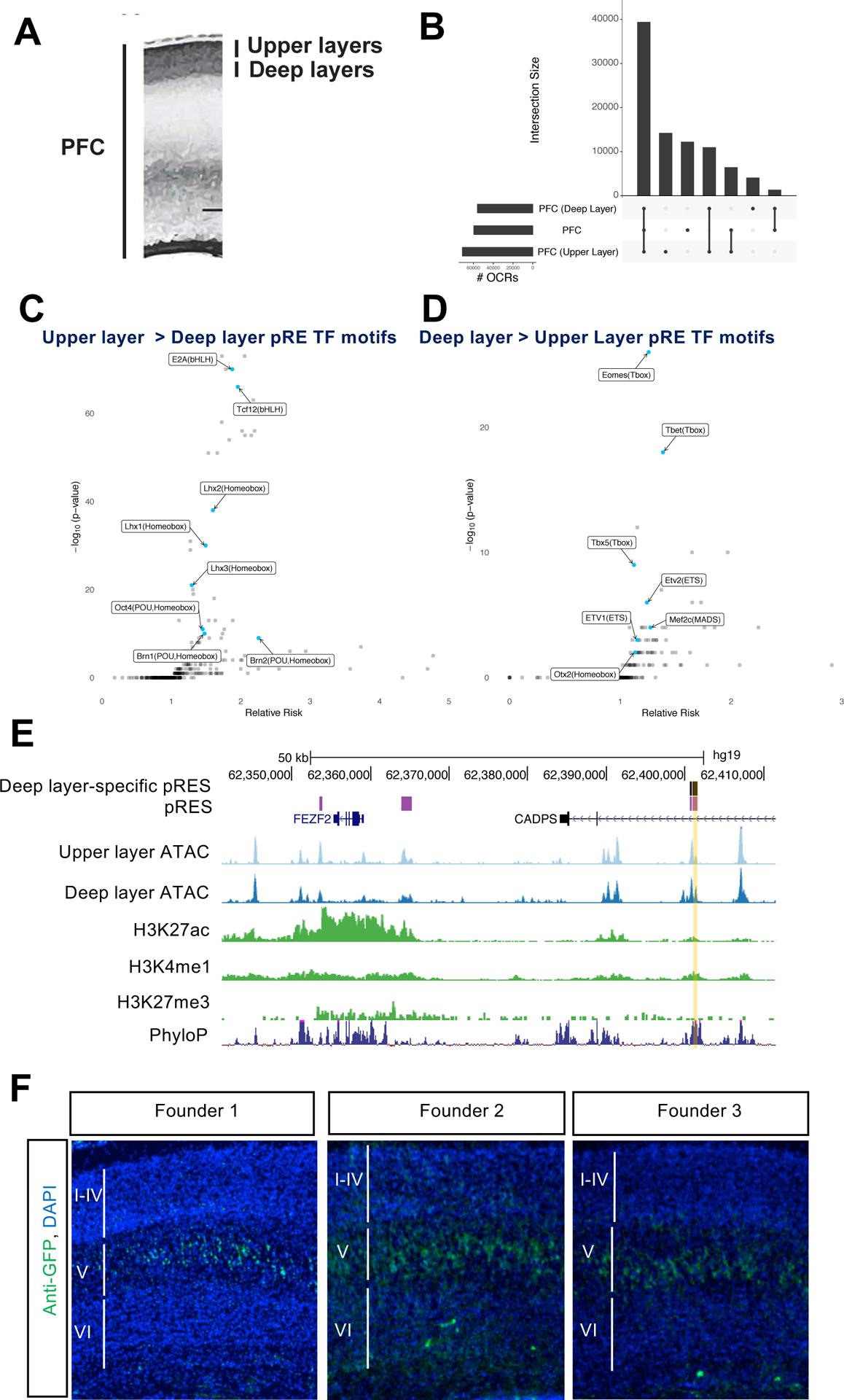
5A) Schematic of micro-dissection of upper and deep layers of cortex, for 18gw and 19gw ATAC-seq, and dissection of whole PFC, from germinal zone to cortical plate.
5B) OCR intersections and numbers of OCRs pooled across samples for upper layers, deep layers, and whole PFC.
5C) TF motifs enriched in upper layer-specific pREs.
5D) TF motifs enriched in deep layer-specific pREs.
5E) ATAC-seq reads combined from multiple samples from upper layer and deep layer microdissections of PFC at 18gw and 19gw, nearby Layer 5 expressed gene FEZF2. The yellow shaded pRE is called an OCR only in deep layer samples, was positive in luciferase assay (Figure 2D) and was tested by transgenic enhancer mouse assay. ChIP-seq reads are combined from multiple samples of whole midfetal PFC for H3K27ac, H3K4me1, H3K27me3.
5F) FEZF2 enhancer transgenic mice. Postnatal day 2 sections with anti-GFP immunostaining in green and DAPI in blue. Cortical Layer 5 specific expression is indicated in three transgenic founders.
To investigate whether OCR differences between deep and superficial PFC layers reflect gene regulatory differences, we analyzed TF motifs in the layer-specific OCRs that overlapped our original pREs. TF motif analysis of 2,382 upper layer-specific pREs showed enrichment for LHX (LIM HOMEODOMAIN), BRN (POU HOMEODOMAIN), and BHLH family motifs (Figure 5C). These motifs are bound by transcription factors whose expression is elevated in superficial layers of the mouse and human cortex, including LHX2, LHX5, BRN1, BRN2, and BHLHB5 (Sestan and State 2018). Similarly, motif analysis of 461 deep layer-specific pREs showed enrichment for T-box and ETS family TFs (Figure 5D). These motifs are bound by TBR1, ETV1 and ETV2, transcription factors whose expression is elevated in deep layers (Sunkin et al. 2013). Thus, the motif enrichment of TFs expressed in superficial or deep layer cortical neurons suggests that differences in chromatin accessibility at layer-specific pREs arise from different cell types residing in different cortical layers. In addition, this assay may also identify motifs for TFs associated with maturing neurons that are migrating through these cortical layers (Table S5B).
To identify pREs that may be driving laminar differences in gene expression, we linked pREs with markers of upper and deep layer neurons obtained from analysis of single-cell RNA-seq of human midfetal cortex (Nowakowski et al. 2017). Upper and deep layer neuron marker genes were enriched for proximity to upper and deep layer-specific pREs (odds ratios 6.44 and 2.22 respectively, p-value < 0.05). For example, the superficial layer marker gene ADCYAP1R1 is proximal to an upper layer-specific pRE, while deep layer marker gene PPFIA2 has an intronic pRE with deep layer-specific chromatin accessibility (Figures S5 and S5B).
We validated one of these deep layer-specific pREs proximal to FEZF2 (Figure 5E), a well studied cortical TF expressed in Layer 5 that specifies sub-cerebral projection neurons (B. Chen, Schaevitz, and McConnell 2005; J.-G. Chen et al. 2005; B. Chen et al. 2008; Molyneaux et al. 2005). This previously undescribed pRE was chosen based on its deep-layer-specific chromatin accessibility (Figure 5E), high enhancer prediction score (>0.85), and its activity in a luciferase transcription assay (Figure 2D). We cloned the pRE into the CT2IG GFP reporter vector (Silberberg et al. 2016) and generated transgenic mice by pronuclear injection. Three founders showed Layer 5 specific GFP expression in the neocortex at postnatal day 2 (Figure 5F), and one showed Layer 5 and Layer 6 specific expression (not shown). This enhancer will facilitate the molecular elucidation of transcriptional mechanisms that drive layer-specific gene expression, and can be used as a tool to drive expression in Layer 5 pyramidal neurons.
At 18gw, the upper cortex layers consist of Layer 2–5 neurons while deep layers consist of Layer 5–6 and subplate neurons (Nowakowski et al. 2016). To assign pREs to distinct cortical neuron subtypes, we used cells purified from mouse CRE lines that label Layer 5 subcortical projection neurons and Layer 6 corticothalamic projection neurons using Rbp4-CRE and Ntsr1-CRE BAC transgenic mice, respectively (Gong et al. 2003). We crossed the CRE mice to Intact GFP reporter mice (Mo et al. 2015) and performed ATAC-seq on nuclei purified by fluorescence-activated cell sorting (FACS) from the rostral third of the cortex at postnatal day 2 (Methods). After calling ATAC-seq peaks and lifting over to the human genome, we found 7,124 (12.5%) and 7,025 (12.3%) of human midfetal deep layer OCRs intersect mouse Layer 5 and Layer 6 OCRs, respectively. Similarly, 393 (7.5%) and 413 (7.9%) of human midfetal deep layer pREs overlap mouse Layer 5 and Layer 6 OCRs, respectively (Table S5A). Some conserved pREs are differentially accessible across Layers 5 and 6; for example, we identified an upper layer pRE for the canonical upper layer gene SATB2 that overlaps Layer 5 but not Layer 6 neuron OCRs identified in neonatal mouse (Figure S5C).
Candidate regulatory elements for neurodevelopmental disorder genes
BCL11A is a gene associated with developmental delay (DD) and ASD (Sanders et al. 2015)(Sanders et al. 2015; Satterstrom et al. 2020)(Sanders et al. 2015) that encodes a transcriptional regulator in brain and blood cells. There are 23 pREs within one megabase of the BCL11A locus with diverse patterns of accessibility across telencephalic regions (Figure 6A). We validated one of these for enhancer function using transgenic mice generated as above, choosing a pRE based on high enhancer prediction score (>0.85) and luciferase transcription enhancer activity (Figure 2D, labeled BCL11A). This pRE is an OCR in the basal ganglia as well as cortical regions (Figure 6A). Like the endogenous BCL11A gene, two transgenic founders showed GFP expression in cortex, striatum, and hippocampus at postnatal day 2 (Figures 6B and S6A).
Figure 6: Identifying pREs that regulate neurodevelopmental disorder genes.
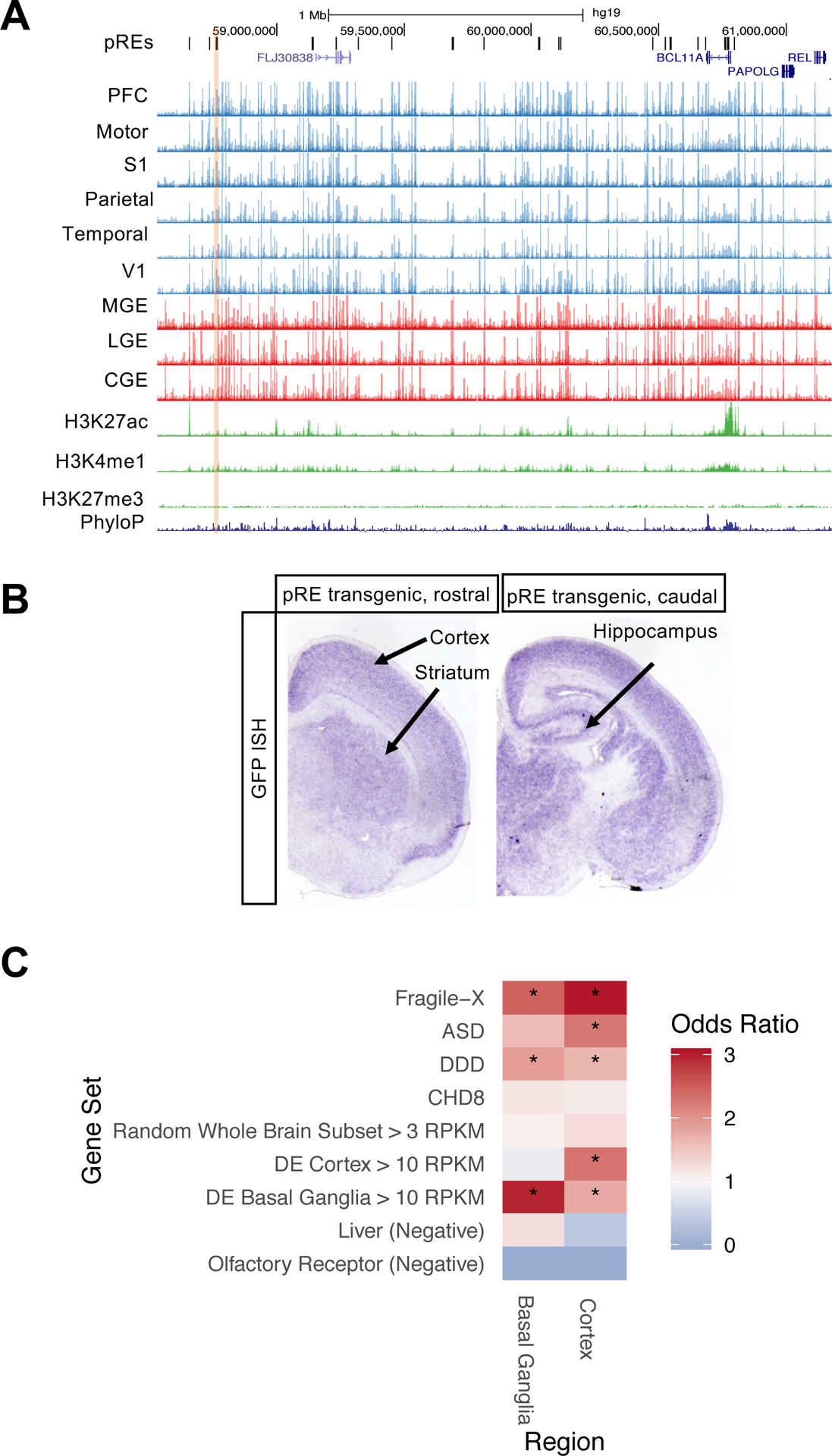
6A) ATAC-seq reads, combined across multiple samples per brain region, at pREs proximal to BCL11A on chromosome 2. The yellow shaded pRE was positive in luciferase assay (FIgure 2D) and was tested by transgenic enhancer mouse assay. Y axis scale is 0 to 500.
6B) Postnatal day 2 coronal sections of BCL11A enhancer transgenic mouse with GFP reporter expression in purple (RNA in situ hybridization for GFP). Cortical, striatal, and hippocampal expression is indicated.
6C) Gene set enrichment analysis for genes proximal (within 50 kb) to cortex- and basal-ganglia specific OCRs. Gene sets included ASD genes (Sanders et al. 2015), biological targets of Fragile X Mental Retardation protein (FMRP) (Darnell et al. 2011), biological targets of ASD gene CHD8 (Cotney et al. 2015), and developmental delay disorder (DD) genes (Deciphering Developmental Disorders Study 2015). See Methods for description of control gene sets.
Using a gene set enrichment analysis, we asked whether neurodevelopmental disorder genes such as BCL11A are enriched for candidate REs that are region-specific. In contrast to our previous analyses using pREs, OCRs were used here due to the limited number of region-specific pREs. We started with sets of genes associated with neurodevelopmental disorders including ASD (Sanders et al. 2015) and DD (Deciphering Developmental Disorders Study 2017). Similar to previous studies (An et al. 2018), genes were included that interact transcriptionally or physically with genes in these sets such as loci bound by the ASD gene product CHD8 (Cotney et al. 2015), and proteins that interact with Fragile X mental retardation protein FMRP (Darnell et al. 2011). FMRP targets and DD genes were significantly enriched in both cortex (q-values 1.9*10−21 and 4.4*10−3) and basal ganglia (q-values 2.6*10−5 and 2.4*10−2), while ASD genes were significantly enriched in cortex (q-value 2*10−3) (Figure 6C). We repeated this analysis for sub-regions of cortex and the basal ganglia and found neurodevelopmental disorder gene sets enriched in specific sub-regions (Figures S6B and S6C, Tables S6A and S6B).
Function altering de novo point mutations in a pRE that regulates ASD gene expression
To further investigate the potential roles of pREs in human neurodevelopmental disorders, we studied de novo variants identified by WGS of 1,902 quartet families (An et al. 2018), composed of an individual diagnosed with ASD, an unaffected sibling and both parents. The category-wide association study (CWAS) framework (Werling et al. 2018) divides the genome into over 50,000 categories defined by functional annotations, conservation across species, gene-defined regions, or proximity to genes implicated in ASD, each of which is tested for enrichment of de novo variants in cases vs. controls. Correcting for the categories tested, no single non-coding category has reached significance. However, a de novo risk score (DNRS) to assess risk across multiple categories implicated promoter regions (An et al. 2018). Adding the pREs to the CWAS analysis did not yield a statistically significant result after multiple-testing correction. However, intronic pREs near ASD-associated genes showed a strong trend towards enrichment for cases vs. controls (Figure S7).
The gene SLC6A1, encoding the neuronal GABA transporter GAT-1, is associated with ASD (Sanders et al. 2015) (Sanders et al. 2015; Satterstrom et al. 2020)(Sanders et al. 2015) and myoclonic atonic epilepsy/absence seizures with developmental delay (Heyne et al. 2018). De novo variants from two individuals with ASD but no seizures were identified by WGS (An et al. 2018) and mapped to an intronic pRE near SLC6A1 (Figure 7A, Tabls S7). This pRE has increased chromatin accessibility in the basal ganglia compared to cortex, and in frontal cortical tissues it shows increased accessibility at later midfetal ages and is a 19gw-specific pRE. The mouse homologous region is an OCR in neonatal cortical Layer 6, but not Layer 5, neurons (Figure 7B). We tested the function of the two de novo variants in this pRE, and found they significantly reduced enhancer activity using a luciferase transcription assay in neuroblastoma cells (Figure 7C).
Figure 7: Functional de novo ASD variants in a SLC6A1 enhancer.
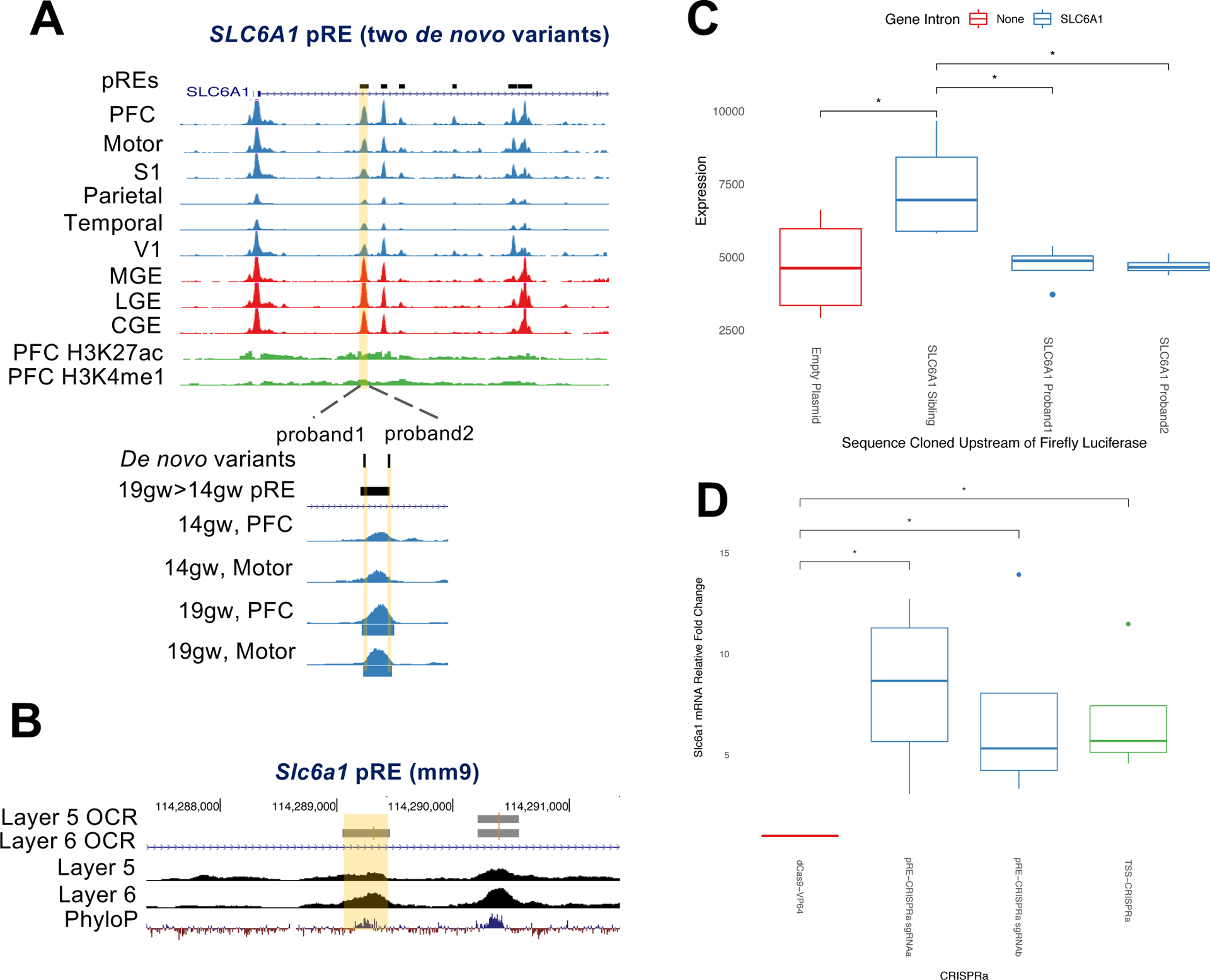
7A) ATAC-seq reads, combined across multiple samples per brain region, at high confidence ASD and epilepsy risk gene SLC6A1. An intronic pRE (highlighted in yellow) contains two de novo variants from separate ASD probands, highlighted in yellow below and labeled “proband 1” and “proband 2.” ATAC-seq reads from frontal cortical samples (PFC and motor cortex) at 14gw and 19gw show increased accessibility of this pRE at 19gw, and peak calling shows it is a 19gw specific OCR (called OCRs are blue bars beneath ATAC-seq tracks). H3K27ac and H3K4me1 ChIP-seq reads in green are from 15gw PFC.
7B) ATAC-seq reads at mouse gene locus Slc6a1, from purified populations of Layer 5 and Layer 6 neurons. Layer 5 and Layer 6 called OCRs are indicated. The homologous region to the human pRE that contains two de novo ASD patient mutations is highlighted yellow.
7C) Mean firefly luciferase levels in human neuroblastoma cells, normalized to Renilla, testing enhancer activity of SLC6A1 pRE and the functional effects of two proband point mutations in the pRE on luciferase expression levels. Error bars indicate standard error across four replicate experiments.
7D) CRISPRa in mouse primary cortical neurons targeting the Slc6a1 pRE (sgRNAa and sgRNAb, blue) and promoter (TSS, green). The results are the mRNA fold increased normalized to Actb using DDCT method. The mean fold increase over untransfected control (dCas9-VP64, red) from four independent experiments and two technical replicates is represented. *p<0.01 (ANOVA, Tukey test).
We used CRISPRa (Gilbert et al. 2013) to test whether the pRE is an enhancer for SLC6A1. We designed two sgRNAs targeting the homologous mouse element in the intron of Slc6a1 (Figure 7B), and one targeting the promoter. We generated lentiviral constructs and infected primary cortical cultures from dCAS9-VP64/+ P0 mouse pups. These neurons express dCAS9 fused to four copies of the VP16 transcriptional activator (Matharu et al. 2019) (Methods). dCAS9-VP64 neurons infected with Slc6a1 pRE sgRNAs up-regulated Slc6a1 mRNA levels five- to ten-fold, and the promoter sgRNA up-regulated expression five-fold (Figure 7D). These experiments provide evidence that this element, containing function-altering de novo mutations identified from two individual ASD patients, is an enhancer for SLC6A1. Further, this pRE provides a potential target for gene therapy to rescue haploinsufficiency in patients with deleterious SLC6A1 loss of function mutations.
Discussion
We generated an atlas of the chromatin accessibility landscape across nine regions of the mid-gestation human telencephalon and predicted regulatory elements within open chromatin. A substantial proportion of these elements show regional, temporal, and laminar differences in chromatin accessibility that correlate with differences in single-cell RNA-seq expression patterns (Nowakowski et al. 2017), ontology terms of nearby genes, and TF binding sites. We identified enhancers of genes associated with neurodevelopmental disorders, including validated enhancers of BCL11A and SLC6A1. Two functional de novo mutations from separate individuals with ASD were identified in an intronic pRE of ASD and epilepsy risk gene SLC6A1, whose enhancer function we demonstrated using CRISPRa in cortical neurons. These findings suggest that dynamically utilized pREs across the mid-gestation telencephalon may be involved in the numerous neurodevelopmental processes this tissue undergoes in health and disease.
This annotated collection of ATAC-seq and ChIP-seq data is searchable by area of interest with the following fields: (1) Gene and Locus. Tables S2A and S2B list the upstream and downstream genes of all predicted pREs and OCRs, respectively. (2) Brain Region. Table S2A organizes all pREs by the brain region in which they overlap OCRs, and notes the pREs specific to basal ganglia, cortex, and pREs specific to just one telencephalic sub-region. Table S3 lists pREs proximal to genes differentially expressed between MGE, PFC, and V1, whose chromatin accessibility is specific to each pairwise comparison. (3) Histone modification landscape. Tables S2D and S2E annotate pREs and OCRs based on overlap with activating and repressive histone modification peaks from PFC ChIP-seq data. (4) Cortical Laminae. pREs for upper and deep layer cortical neurons in fetal PFC, which are enriched for upper and deep layer TF motifs, are listed in Table S5 and annotated for TF motifs which are present in each pRE. This table also notes which pREs are conserved OCRs across mouse neonatal Layer 5 and 6 cortical neurons. (5) Cortical developmental stage. pREs for early and late midfetal frontal cortex are listed in Table S4. (6) Cell Type. Table S6C provides genes that are expressed exclusively in particular cell types in the developing telencephalon and the region specific OCRs near those loci. (7) Human disorders. Table S6B provides region specific OCRs that are proximal to ASD-associated genes, developmental delay genes, FMRP targets, and CHD8 targets. (8) De Novo Non-coding Variants. Table S7A lists pREs that contain more than one ASD patient de novo mutation (An et al. 2018).
Transgenic mouse experiments have illustrated the exquisite spatiotemporal and cell type-specific activity of neurodevelopmental enhancers afforded by combinatorial binding of TFs expressed in graded, overlapping patterns in the brain (Silberberg et al. 2016; Pattabiraman et al. 2014; Visel et al. 2013; Erwin et al. 2014). pREs contain motifs for T-box family and Nkx family TFs (Figures 3D and 3E), which specify pallial and subpallial structures, respectively. Interestingly, the composite OCT4/SOX2 motif was enriched in basal ganglia specific pREs, whose function has been demonstrated in a mouse MGE enhancer of Tcf12 (Sandberg et al. 2018). Within pREs that were differentially accessible over cortical ages or cortical laminae, we identified motifs for TFs that have been well-studied in mice, indicating their importance to human brain development. The pREs provided here (Table S2) may be integrated with mouse ChIP-seq studies of these and other TFs to indicate which loci of interest may be functional enhancers in different regions of the developing human brain.
This pRE atlas can also be integrated with ATAC-seq data from other species. For instance, we integrated ATAC-seq data generated from mouse neonatal cortical Layer 5 and 6 purified neurons and attributed chromatin accessibility in 573 pREs to these specific cortical neuronal subtypes (Figure 5, Table S5A). The pREs provided here are also a resource for curating a subset of loci accessible in mouse cell types which are likely to be conserved human neurodevelopmental enhancers.
We emphasize that pREs are predictions based on transgenic mouse enhancer assays and described by diverse sequence and functional genomics features. We validated enhancer activity of twelve pREs using luciferase assays, transgenic mice, and CRISPRa (Figures 2, 5, 6, and 7). However, further work is needed to test their function in vivo and whether they can be used therapeutically.
Implications for human genetics and disorders
This atlas has various implications to the field of neurogenetics, and is likely to be particularly relevant to ASD and other early onset neurodevelopmental disorders. Studies of later onset syndromes such as schizophrenia and bipolar disorder have focused on the association of common non-coding alleles having small effects, while progress in ASD genetics has mainly come from studies of rare and de novo mutations in the genic portion of the genome. Such studies have identified dozens of large effect genes, with the predominant genomic mechanism involving putative loss-of-function heterozygous de novo mutations (Sanders et al. 2015; Satterstrom et al. 2020). Moreover, whole genome sequencing (WGS), which led to the identification of the rare SLC6A1 enhancer mutations described here, has not yet proven successful in identifying specific rare non-coding mutations contributing to ASD as study cohorts are still markedly underpowered to accomplish this goal (Werling et al. 2018; An et al. 2018).
The demonstration that rare non-coding de novo mutations mapping near SLC6A1 fall within a bona-fide functional regulatory element and alter enhancer function prima facie expands our understanding of the contribution of the non-coding genome to risk. More broadly, the findings point to the critical importance of the data types generated in our study for the interpretation of WGS studies of ASD and other related neurodevelopmental disorders. The ability to segment the non-coding genome into highly relevant functional elements will be an essential precursor to increasing the power of WGS generally. With regard to ASD, the question of when and where the large effect mutations discovered via exome sequencing are acting is a critically important one (State and Šestan 2012; Willsey et al. 2013; Sestan and State 2018). As mutations in regulatory elements have the potential to convey far more spatiotemporal information compared to loss-of-function heterozygous alleles in the genes they regulate, the mapping and functional assessment of rare non-coding mutations has the potential to link ASD risk with specific cell types, brain regions, and temporal epochs.
Moreover, identifying the regulatory elements of genes implicated in neurodevelopmental disorders is essential for elucidating the transcriptional circuitry organizing their expression. Intronic pREs of ASD genes that contain case mutations, such as the intronic SLC6A1 pRE (Figures 7 and S7), may be important developmental enhancers for those genes and may provide both an avenue to illuminate the pathobiology of ASD, but also to provide tractable opportunities to modify expression as a future therapy (Matharu et al. 2019).
Similarly, identifying the downstream REs controlled by ASD risk genes, many of which encode transcription regulators or chromatin modifiers (De Rubeis et al. 2014; Cotney et al. 2015), is critical in understanding how mutations alter expression programs impacting brain development and function. Indeed, ASD genes and FMRP targets appear to be under precise epigenetic control, as they are significantly enriched for region-specific OCRs (Figure 6C, S6B, and S6C). These OCRs may be sensitive to epigenetic alterations that occur in the brain when ASD (Katayama et al. 2016; Cotney et al. 2015) or FMRP (Korb et al. 2017) genes lose function, and may indicate which brain regions are specifically impacted by resulting chromatin changes.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact, John Rubenstein (john.rubenstein@ucsf.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Raw ATAC-seq and ChIP-seq sequencing data has been deposited to dbGAP (phs002033.v1.p1). Processed bigwigs, peaks, and OCR/pRE annotations have been deposited to GEO (GSE149268).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Developing Human Brain Samples
De-identified tissue samples were obtained with patient consent in strict observance of the legal and institutional ethical regulations. Protocols were approved by the Human Gamete, Embryo, and Stem Cell Research Committee, and the Institutional Review Board at the University of California, San Francisco. Fresh fetal brain samples were obtained from elective terminations, with no karyotype abnormalities or genetic conditions reported, and transported in freshly made Cerebral Spinal Fluid on ice (CSF). Samples ranged from 14gw to 21gw in age and included male and female sexes (see Table S1 for sample metadata). All dissections and experiments were performed within two hours of tissue acquisition. Dissections of each brain region included the entire telencephalic wall, from the ventricular zone to the meninges, except for experiments performed on upper and deep cortical layers where the cortical plate from the PFC was microdissected under a microscope.
Animal Models
All procedures and animal care were approved and performed in accordance with National Institutes of Health and the University of California San Francisco Laboratory Animal Research Center (LARC) guidelines, UCSF IACUC approval number AN180174–02. Ntsr1-CRE or Rbp4-CRE (Gong et al. 2003) homozygous male mice were crossed to Intact flox/flox females (Mo et al. 2015). The Ntsr1-CRE mouse strain used for this research project, B6.FVB(Cg)-Tg(Ntsr1-cre)GN220Gsat/Mmucd, RRID:MMRRC_030648-UCD, was obtained from the Mutant Mouse Resource and Research Center (MMRRC) at University of California at Davis, an NIH-funded strain repository, and was donated to the MMRRC by MMRRC at UCD, University of California, Davis. Made from the original strain (MMRRC:032081) donated by Nathaniel Heintz, Ph.D., The Rockefeller University, GENSAT <https://protect2.fireeye.com/url?k=c19cddd3–9ddce8ed-c19cface-0cc47ad9c120–2678b1e782f452c7&u=www.gensat.org/> and Charles Gerfen, Ph.D., National Institutes of Health, National Institute of Mental Health. The Rbp4-CRE mouse strain used for this research project, STOCK Tg(Rbp4-cre)KL100Gsat/Mmucd, RRID:MMRRC_031125-UCD, was obtained from the Mutant Mouse Resource and Research Center (MMRRC) at University of California at Davis, an NIH-funded strain repository, and was donated to the MMRRC by MMRRC at UCD, University of California, Davis. Made from the original strain donated by Nathaniel Heintz, Ph.D., The Rockefeller University, GENSAT <https://protect2.fireeye.com/url?k=c19cddd3–9ddce8ed-c19cface-0cc47ad9c120–2678b1e782f452c7&u=www.gensat.org/> and Charles Gerfen, Ph.D., National Institutes of Health, National Institute of Mental Health.
Transgenic Animal Models
Transgenic mice were generated and bred in CD1 background. To clone the putative BCL11A and FEZF2 enhancers into the CT2IG vector, the following primers were used:
| Bcl11a_Forward | ttttgaattcAAAAGAGAAAATGCGTTTCCAG |
| Bcl11a_Reverse | tttttggcgcgccTTGGAGGAAAAGGCTATCCA |
| Fezf2_Forward | ttttgaattcCACACTGATTGTGGCACATTTT |
| Fezf2_Reverse | ttttggcgcgccCGCATTCTGAAGCACTGAGA |
For the BCL11A pRE, a 960 bp amplicon from human genomic DNA was obtained using Phusion HF DNA Polymerase and gel purified; a 822bp amplicon was gel purified for the FEZF2 pRE. Amplicons were digested with EcoRI and AscI restriction enzymes, and ligated into linearized Hsp68-CreERT2-IRES-GFP vector (Visel et al. 2013). Sanger sequencing confirmed insertion of the pRE into the vector and linearized vector was submitted for pronuclear injection at the Gladstone Transgenic Core Facility.
Founders were screened for the transgene using the following genotyping primers:
| pCT2IG_geno_F | CCACCATATTGCCGTCTTTT |
| pCT2IG_geno_R | GAACTTCAGGGTCAGCTTGC |
Three positive BCl11A founders and four positive FEZF2 founders were bred to wildtype mice, and F1 generation mice were analyzed at postnatal day 2.
Primary Cell Cultures
The H11P3CAG-dCas9-VP64 (dCas9-VP64) mouse was a gift from Nadav Ahituv (Matharu et al. 2019). Homozygous H11P3CAG-dCas9-VP64 mice were crossed to wildtype mice. Cortical cultures were grown by dissecting the whole neocortex from 12 neonatal heterozygous mice. Male and female mice were included. Cortical cultures were prepared as follows: dissociation of tissue in Papain (Worthington), plating cells onto wells prepared sequentially with Poly-L-Lysine (10 μg/mL) and Laminin (5 μg/mL), in DMEM culture media (1% N2, 1% B-27 w/o Vitamin A, 10% Fetal Bovine Serum, 1% penicillin, streptomycin, glutamine) at a concentration of 3–5 × 106 cells /well.
Cell lines
Human brain-derived neuroblastoma cells BE(2)-C from ATCC were passaged three times and grown to confluency in a 1:1 mixture of Eagle’s Minimum Essential Medium and F12 Medium with 10% fetal bovine serum.
METHOD DETAILS
ATAC-seq library generation from human samples
From each dissection, intact nuclei were isolated by manually douncing the tissue twenty times in 1mL Buffer 1 (300mM sucrose, 60mM KCl, 15mM NaCl, 15mM Tris-HCl, pH 7.5, 5mM MgCl2, 0.1mM EGTA, 1mM DTT, 1.1mM PMSF, Protease inhibitors) on ice using a loose pestle douncer, and then lysed on ice for 10 minutes after adding 1mL Buffer 2 (300mM sucrose, 60mM KCl, 15mM NaCl, 15mM Tris-HCl, pH 7.5, 5mM MgCl2, 0.1mM EGTA, 0.1% NP-40, 1mM DTT, 1.1mM PMSF, Protease inhibitors). During these ten minutes, nuclei were counted using trypan blue and 50,000 nuclei were spun down at 7,000rpm for ten minutes at 4C. Nuclei were resuspended in 25uL Tagmentation buffer, 22.5 uL Nuclease Free H20, and 2.5 uL Tagmentation Enzyme from Nextera DNA Library Prep Kit, gently mixed, and placed in 37C water bath for thirty minutes. The tagmentation reaction was stopped by MinElute PCR purification and DNA was eluted in 10uL Nuclease Free water. ATAC-seq library generation was performed using Illumina barcode oligos as described (Buenrostro et al 2015), for 8–11 cycles PCR using NEBNext High Fidelity 2x PCR master mix. The number of cycles was empirically determined for each library by qPCR. Libraries were bioanalyzed using Agilent High Sensitivity DNA Kit, pooled together and sequenced on Hiseq 2500 using paired end sequencing.
ATAC-seq library generation from mouse samples
Nuclei from individual mouse brains were isolated as above using 1mL Buffer 1, douncing gently with loose pestle on ice, and lysing in Buffer 2. After centrifugation, nuclei were resuspended in PBS with 4% Fetal Calf Serum and taken to FACSAria fluorescent cell sorter. 50,000 GFP positive nuclei were isolated, spun down at 7,000rpm at 4C, and resuspended in Tagmentation reaction from Nextera DNA Library Prep Kit, and placed in 37C water bath for thirty minutes. Tagmentation was stopped by MinElute PCR purification and DNA was eluted in 10uL Nuclease Free water. ATAC-seq libraries were prepared as above, bioanalyzed on Agilent High Sensitivity DNA kit, and sequenced on Hiseq 2500 using paired end sequencing.
ChIP-seq library generation
Samples were acquired as for ATAC-seq above. All dissections and downstream experiments were performed within two hours of tissue acquisition. Dissections of each brain region included the entire telencephalic wall, from the ventricular zone to the meninges.
From each dissection, intact nuclei were isolated by manually douncing the tissue twenty times in 1mL Buffer 1 (300mM sucrose, 60mM KCl, 15mM NaCl, 15mM Tris-HCl, pH 7.5, 5mM MgCl2, 0.1mM EGTA, 1mM DTT, 1.1mM PMSF, 50mM Sodium Butyrate, EDTA-free Protease inhibitors) on ice using a loose pestle douncer, and then lysed on ice for 10 minutes after adding 1mL Buffer 2 (300mM sucrose, 60mM KCl, 15mM NaCl, 15mM Tris-HCl, pH 7.5, 5mM MgCl2, 0.1mM EGTA, 0.1% NP-40, 1mM DTT, 1.1mM PMSF, 50mM Sodium Butyrate, EDTA-free Protease inhibitors). During this ten minutes, nuclei were counted using trypan blue and 500,000 nuclei were spun down at 7,000rpm for ten minutes at 4C. Nuclei were resuspended in 0.250mL MNase buffer (320mM sucrose, 50mM Tris-HCl, pH 7.5, 4mM MgCl2, 1mM CaCl2, 1.1mM PMSF, 50mM Sodium Butyrate) and incubated in a 37C water bath with 2 microliters Micrococcal Nuclease enzyme (NEB) for eight minutes. Micrococcal Nuclease digestion was stopped by adding 10 microliters 0.5M EDTA, and chromatin was spun down for 10 minutes 10,000rpm 4C. Soluble fraction “S1” supernatant was saved at 4C overnight, and “S2” fraction was dialyzed overnight in 250uL dialysis buffer at 4C (1mm Tris-HCl pH 7.5, 0.2mM EDTA, 0.1mM PMSF, 50mM Sodium Butyrate, Protease Inhibitors). Next day S1 and S2 fractions were combined, 50 microliters were saved as input, and Chromatin immunoprecipitation was set up in ChIP buffer: 50mM Tris, pH 7.5, 10mM EDTA, 125 mM NaCl1, 0.1% Tween. 250m M Sodium Butyrate was supplemented for H3K27ac ChIPs. The following antibodies were used for ChIP: H3K27ac (Millipore, cma309), H3K4me1 (Abcam, ab8895), H3K27me3 (Millipore, 07–449), H4K20me3 (Abcam, ab9053), and H3K9me3 (Abcam, ab8898). 1 microliter of antibody was added to 1mL chromatin in ChIP buffer and incubated overnight at 4C rotating. Protein A and Protein G beads (10 microliters for each ChIP) were blocked overnight in 700uL ChIP buffer, 20 uL yeast tRNA (20mg/mL), and 300uL BSA (10mg/mL). Beads were washed three times for five minutes on ice in Wash buffer 1 (50 mM Tris, pH 7.5 , 10mM EDTA , 125mM NaCl, 0.1% Tween-20, with protease inhibitors and 5mM sodium butyrate) and three times in Wash buffer 2 (50 mM Tris, pH 7.5 , 10mM EDTA , 175mM NaCl, 0.1% NP-40, with protease inhibitors and 5mM sodium butyrate), and ChIP DNA was eluted in elution buffer at 37C and purified by phenol chloroform extraction and ethanol precipitation. Sequencing lIbraries were made using Nugen Ovation Ultralow V2 kit and quantified by Agilent High Sensitivity DNA kit on the Agilent bioanalyzer.
Luciferase Assay
Primers in Table S7B were used to amplify pREs and OCRs from human genomic DNA, then cloned into the minimal promoter pGl4.23 luciferase vector (Promega) using SacI and XhoI restriction sites (underlined) in the vector’s multiple cloning site.
Confluent neuroblastoma cells were transfected in four 96-well plates with luciferase vectors (predicted enhancer-pGL4.23 or empty vector pGL4.23) and pRL renilla vector. Two days later, cells were lysed and luciferase levels detected using the Promega dual reporter luciferase assay kit. Luciferase levels were normalized to Renilla and averaged across four replicate experiments.
Transgenic Mouse Analysis
Postnatal day 2 mice were perfused with 4% paraformaldehyde, whole brain was dissected, and postfixed overnight in 4% paraformaldehyde, and transferred to 30% sucrose overnight. 20 micron thick cryosections were obtained and either in situ hybridization or immunofluorescence experiments were performed. In situ hybridization using GFP RNA probe was performed as described (Sandberg et al. 2018). In situs were developed at 37C and were imaged two days later. Immunofluorescence was performed using rabbit anti-GFP antibody (Abcam) at 1:500 dilution.
Testing function of de novo variants in pREs
The following primers were used to amplify the SLC6A1 pRE from human genomic DNA. The amplicon was then cloned into the minimal promoter pGl4.23 luciferase vector (Promega) using SacI and XhoI restriction sites (underlined) in the vector’s multiple cloning site.
| Forward primer | Reverse primer |
|---|---|
| ctggccggtacctgagctTTTTAGCAGGTTGGTTCAGCATA | cagatcttgatatcctcgagTCTGGCTCTCATTCAAGGAACC |
The pRE was tested for enhancer activity by luciferase assay in BE(2)-C as described above. To introduce point mutations into these pREs, we used site directed mutagenesis. Phusion PCR was performed using the Slc6a1-pGl4.23 vector as template and the following two primers to introduce the de novo point mutation (capitalized) into the pRE sequence. All variants were taken from An et al. 2018 supplemental data.
| pRE in ASD gene intron w/point mutation | Inverse PCR primer (forward) | Inverse PCR primer (reverse) |
|---|---|---|
| Slc6a1 proband1 | tgcAcagttcatacagccaaga | ctgTgcatgcaaagccaagagg |
| Slc6a1 proband2 | cccGaacttggagctagacagg | gttCggggagggggctgctctg |
Luciferase levels were compared between empty vector, unmutated (sibling) pRE, and pREs containing the above mutations. Four replicate experiments in BE (2)-C cells were performed and analyzed as described above.
CRISPRa in primary neuronal cultures
We designed two sgRNAs against the mouse SLC6A1 pRE homologous sequence (hg19: chr3:11,041,081–11,042,183, mm10:chr6:114288804–114289713) using the CHOPCHOP tool (Labun et al. 2019) and designed an sgRNA against the mouse SLC6A1 promoter using the BROAD GPP CRISPRa designer tool (Doench et al. 2016). The following sgRNAs were annealed and cloned into U6-stuffer-longTracer-GFP lentivirus vector:
| Slc6a1 pRE sgRNAa | CCCTCCCGAGACTAATGGCT |
| Slc6a1 pRE sgRNAa | CGACACCCTCCCGAGACTAA |
| Slc6a1 TSS | CACGGACAAGCCCCGCCTAG |
sgRNA lentivirus was produced in 293T cells through transfection of packaging plasmids psPax2, pmD2G (Addgene) and sgRNA lentivirus vector. Concentrated virus was used to infect primary cortical neuronal cultures derived from neonatal dCAS9-VP64/+ mice upon seeding. RNA was purified from culture 5 days later using Qiagen’s RNEasy Plus Mini kit, DNase treated using Turbo DNAse I (Ambion), reverse transcribed to cDNA using Superscript III, and assayed for target gene expression by qRT-PCR using Maxima SYBR Green / ROX qPCR master mix. Optimal qRT-PCR primer concentration was determined by the standard curve method, and for each condition relative Slc6a1 mRNA levels were normalized to Actb using the DDCT method and averaged across technical replicates and four biological replicates.
| beta_actin_F | ATGTGGATCAGCAAGCAGGA |
| beta_actin_R | AGGGTGTAAAACGCAGCTCAG |
| slc6a1_F | GACAGCCAGTTCTGTACCGT |
| slc6a1_R | GCAATGAAGAGTTCACGGCG |
QUANTIFICATION AND STATISTICAL ANALYSIS
The hg19 reference genome was used in all analyses due to the integration of many diverse datasets, with GRCh38 coordinates lifted over to hg19 where necessary. Analyses were conducted using python, pandas (McKinney 2012), R (R Core Team 2018), bioconductor (Huber et al. 2015), and bedtools2 (Quinlan and Hall 2010).
Peak Calling
Paired-end reads were aligned and peaks were called using the ENCODE ATAC-seq pipeline with default parameters (Lee et al. 2016). The pipeline produces multiple sets of peak calls, including those generated by macs2 (Zhang et al. 2008) and the Irreproducible Discovery Rate (IDR) package for R (Q. Li 2014). The latter was used to select a smaller, more confident set of peaks that are likely to be consistent across biological replicates (Q. Li et al. 2011). Peaks overlapping ENCODE blacklisted regions were removed (Amemiya, Kundaje, and Boyle 2019). The pipeline created pseudo-replicates where a portion of reads are held-out in order to estimate a threshold for calling consistent peaks.
Peak Merging
For each (region, timepoint) combination, peaks separated by up to 100 bp were merged into a single peak to reduce variation in peak calls attributable to coverage differences (bedtools merge −d 100). The union of peaks across all samples was intersected with itself to identify peaks overlapping by at least 75% (bedtools intersect –f 0.75 −e). These overlaps were converted to a matrix with peak coordinates as rows and (region, timepoint) combinations as columns. A (region, timepoint) column was assigned a 1 when one of its peaks overlapped a row’s peak, otherwise a 0 was assigned.
To identify region-specific peaks, columns corresponding to the same brain region were merged: a 1 was assigned if a peak was called for any timepoint in that region, otherwise a 0 was assigned. This resulted in a new matrix with peaks as rows and regions as columns. Region-specific peaks were then those peaks with a 1 in a single column (region) and 0s in all other columns (regions).
Regulatory Element Prediction
Elastic Net and Random Forest classifiers (Pedregosa et al. 2011) were trained on and generated predictions for OCRs merged across all brain regions. Training labels were generated using OCRs in combination with VISTA enhancers. OCRs were labeled positive if they overlapped validated developmental brain enhancers, or labeled negative if they overlapped validated non-brain enhancers or enhancers that failed to validate. Features were generated for each OCR including 5-mers (counts of all 5 consecutive base pairs occurring within the peak), binary overlaps with ChIP-seq peaks and average peak methylation from Roadmap Epigenomics (Roadmap Epigenomics Consortium et al. 2015), binary overlaps with either fragment of statistically significant Hi-C loops (Won et al. 2016; Rao et al. 2014), and average evolutionary conservation across the region (Siepel et al. 2005; Pollard et al. 2010). For each peak without a training label, a continuous-valued prediction was generated corresponding to the model’s confidence that the OCR is a developmental brain enhancer. OCRs were considered pREs if either algorithm’s prediction was above 0.5, representing 19,151 out of 103,829 OCRs (18.4%) after merging across samples and excluding promoter overlaps (1500 bp upstream, 500 bp downstream from a GENCODE v19 TSS (Frankish et al. 2019).
TF Motif Enrichment
The findMotifsGenome.pl script provided by Homer (Heinz et al. 2010) was used to identify TF motifs enriched in pREs that overlapped OCRs in one or more brain regions, depending on the analysis. The set of genomic regions used as foreground and background are provided below:
Cortex versus Basal Ganglia
Foreground: OCRs present in at least one of the cortical regions (pfc, motor, parietal, somato, temporal) and overlapping a pRE
Background: OCRs absent in all basal ganglia regions (cge, mge, lge) and overlapping a pRE
Cortex
Foreground: OCRs present only in the cortical region of interest (pfc, motor, parietal, somato, or temporal) and overlapping a pRE
Background: OCRs open in more than one cortical region and overlapping a pRE
Basal Ganglia
Same as for cortex, but with basal ganglia regions (cge, mge, lge)
PFC Deep Layer versus Upper Layer
Foreground: OCRs present only in PFC deep layer neurons and overlapping a pRE
Background: OCRs present only in PFC upper layer neurons and overlapping a pRE
Early versus Late Frontal Cortex
Foreground: OCRs present in at least one frontal cortex region (pfc, motor) at 14gw and overlapping a pRE
Background: OCRs absent in all frontal cortex regions at 19gw
Association of Changes in Gene Expression with Chromatin State
Single cell RNA-seq data was downloaded for mge, pfc, and v1 brain regions (Nowakowski et al. 2017). MAST (Finak et al. 2015) identified differentially expressed genes between each pair of brain regions (V1 vs. PFC, V1 vs. MGE, PFC vs. MGE, FDR 5%). For each pair, differentially accessible pREs were associated with nearby genes using GREAT (McLean et al. 2010). Proximal associations used default parameters (5 kb upstream and 1 kb downstream of the TSS) while distal associations were restricted to a 100 kb window around the TSS. For a pair of brain regions, R’s fisher.test function estimated the odds ratio of differentially expressed genes over non-differentially expressed genes being linked to a uniquely open chromatin region. This same analysis was performed using genes differentially expressed between early and late frontal cortex timepoints, and again for genes differentially expressed between PFC upper and deep layers. These analyses were performed independently for OCRs and pREs.
Peak Annotation
Genomic annotations were computed (Cavalcante and Sartor 2017) for both region-specific and non-specific pREs. The amount of each overlap (in bp) was scaled by the total length of the pRE.
TSS Enrichment
TSS enrichment was calculated using the approach of the ATAqC code in the ENCODE pipeline (Lee et al. 2016) in combination with RefSeq TSS annotations (N. A. O’Leary et al. 2016). Each TSS was extended by 2kb in both directions and tiled with 10bp non-overlapping windows. For each sample, coverage over these windows was calculated using sambamba (Tarasov et al. 2015). The mean coverage for each window was computed over all TSSs. A background value was calculated using the mean coverage of the 10 most flanking bins, corresponding to TSS offsets −2000 to −1900 and +1900 to +2000. The raw coverage at each bin was divided by this background value. The maximum ratio of coverage to background over all bins was used as the enrichment value.
Disease Gene Enrichment in RS OCRs
Disease gene sets were obtained (An et al. 2018) and annotated with transcription start site(s) from GENCODE v19 (Frankish et al. 2019). To increase power for the primary analysis, OCRs were separated into cortex- or basal ganglia-specific OCRs.
To test if a gene set was more associated with region-specific open chromatin than expected by chance, logistic regression (R’s glm function with family = binomial) was performed using the inverse distance from a gene’s TSS to the closest regional OCR as a covariate, and the presence or absence of the gene in the set as the dependent variable. Distance was capped at 1 megabase. One-sided p-values were computed from glm’s t-statistic using R’s pnorm function with lower_tail = False.
Negative control gene sets were used including liver (Subramanian et al. 2005), olfactory receptor (Rouillard et al. 2016), and a size-matched set of genes expressed in whole brain (Hawrylycz et al. 2012).
Analysis of Relative Risk of De Novo ASD Mutations in Enhancers
Annotated de novo ASD mutations (An et al. 2018) were intersected with OCRs and pREs to annotate each mutation as having those as functional annotations. CWAS was conducted as described in An et al., using OCRs and PREs in addition to their non-coding annotation categories.
Supplementary Material
Table S2: pRE and OCR annotations, related to Figure 2 and Figure S2.
Table S2A: pRE genomic coordinates, conservation score, nearest genes, telencephalic regions which overlap an OCR, pRE prediction score.
Table S2B: OCR genomic coordinates, conservation score, nearest genes, telencephalic regions which overlap an OCR, pRE prediction score.
Table S2C: GREAT analysis of pREs.
Table S2D: pRE genomic coordinates, overlaps with histone ChIp-seq peaks from PFC.
Table S2E: OCR genomic coordinates, overlaps with histone ChIP-seq peaks from PFC.
Table S3: Region-specific pREs proximal to differentially expressed genes between PFC, V1, MGE. Related to Figure 3 and Figure S3.
Table S3A: MGE and V1 specific pREs proximal to differentially expressed genes between MGE and V1.
Table S3B: MGE and PFC specific pREs proximal to differentially expressed genes between MGE and PFC.
Table S3C: V1 and PFC specific pREs proximal to differentially expressed genes between V1 and PFC.
Table S4: Temporal differences in chromatin accessibility at cortical pREs, related to Figure 4 and Figure S4.
Table S4A: GREAT analysis of early (14gw) and late (19gw) frontal cortex pREs. Table S4B: 14gw and 19gw specific pREs proximal to differentially expressed genes between the two timepoints.
Table S5: Layer specific OCRs, related to Figure 5 and Figure S5.
Table S5A: OCRs annotated whether they overlap upper layer or deep layer OCRs.
Table S5B: Upper layer OCRs annotated by whether they contain motifs for TFs expressed in immature neurons, or mature superficial neurons, or both.
Table S6: OCRs proximal to genes related to neurodevelopmental disorders or markers of particular cell types in the developing telencephalon, related to Figure 6 and Figure S6.
Table S6A: Percentage of genes per gene set located within 50kb of a region-specific open chromatin region. Fragile-X (FMRP) has a high percentage of genes proximal to RS OCRs, and is enriched across all regions.
Table S6B: Region-specific OCRs proximal to disease genes (ASD, DD, FMRP targets, and CHD8 targets).
Table S6C: Region-specific OCRs proximal to marker genes of different cell types in the developing telencephalon (Nowakowski et al 2017).
Table S7: pREs containing de novo variants and luciferase assay cloning oligonucleotides.
Table S7A: pREs that contain more than one de novo ASD patient variant (An et al 2018), related to Figure 7 and Figure S7.
Table S7B: Oligonucleotide sequences of forward and reverse primers used to amplify pREs and OCRs from human genomic DNA, then cloned into the minimal promoter pGl4.23 luciferase vector (Promega) using SacI and XhoI restriction sites (underlined) in the vector’s multiple cloning site. Related to Figure 2 and Figure S1.
Figure S1: Predicting neurodevelopmental enhancers, related to Figure 1
S1A) Enrichment of ATAC-seq reads centered at RefSeq TSSs, calculated using the ENCODE standard (Methods). Coverage is normalized using the average raw coverage at regions flanking the TSS (offsets from −2000 to −1900 and +1900 to +2000, indicated by the blue vertical lines) as background, resulting in a median TSS signal to background ratio of 42.3 across samples.
S1B) The ten largest and smallest model coefficients (L2-penalized) from a logistic regression classifier trained using various epigenetic and sequence features (Methods) to distinguish between developmental brain enhancers versus non-brain enhancers (ex: heart, limb) and failed enhancer candidates validated by VISTA. These coefficients represent features most predictive of the positive (brain enhancer, blue) and negative (non-brain enhancer and failed enhancers, red) classes. Predictive positive features include open chromatin and H3K27ac in neural progenitors (N2), H3K27ac in hESC cells with neural markers, conserved TF binding sites, and other open chromatin and activating histone marks in neural cell types.
S1C) Mean firefly luciferase levels in human neuroblastoma cells, normalized to Renilla. Eight randomly selected OCRs (blue bars) that are not pREs were cloned upstream of a minimal promoter and firefly luciferase ORF in the pGL4.23 vector. The green bar indicates the only sequence tested, a pRE proximal to GSX2, which had increased luciferase activity compared to empty vector (red bar). Error bars indicate standard error across two replicate experiments.
S1D) Phastcons and PhyloP scores (100-way alignments, vertebrate conservation) of pREs, OCRs, and VISTA enhancers.
S1E) Percent of PFC pREs that overlap called peaks from 11 histone ChIP-seq datasets from midfetal PFC (ages 15gw-22gw). Histone marks are classified based on their association with activating, repressing, or bivalently regulating gene regulatory elements and transcription.
Figure S2: Tracking OCR activation and inactivation across midfetal development, related to Figure 1
S2A) The following sets of PFC OCRs are binned according to orientation and distance to gene TSS in the human genome: OCRs that do not change chromatin accessibility between 14gw and 18gw in the PFC, OCRs that gained accessibility from 14gw to 18gw, and OCRs that lost accessibility from 14gw to 18gw. Below, TF motif analysis of sets of PFC OCRs that change chromatin accessibility from 14gw to 18gw, and subsets of those OCRs where increase or decrease in chromatin accessibility is accompanied by gain or loss of H3K27ac enrichment, respectively. The top significant TF motifs are shown for OCRs where there was no change in accessibility, while all the significant (p<0.05) TF motifs are listed for all other sets of OCRs.
S2B) Example of an OCR (highlighted in yellow) proximal to the EOMES/TBR2 gene locus, which loses both chromatin accessibility and H3K27ac in the PFC between 14gw and 18gw.
S2C) Example of OCRs (highlighted in yellow) proximal to the SEMA3A gene locus, which gain both chromatin accessibility and H3K27ac in the PFC between 14gw and 18gw.
Figure S3: Regional differences in chromatin accessibility at pREs, related to Figure 3
S3A) In situ hybridization for Kcnq3, Pppp2r1b, and Trim2 mRNA (purple) in wildtype murine embryos at E14.5, from Eurexpress database (www.eurexpress.org) (Diez-Roux et al. 2011). Kcnq3 is expressed in the cortex (both rostral and caudal) and not the basal ganglia (bg), Pppp2r1b is expressed in rostral cortex, and Trim2 is expressed in caudal cortex.
S3B) Summary of region-specific OCRs and pREs identified in this study.
S3C) Annotation of genomic features at region-specific pREs compared to pREs shared across multiple brain regions.
Figure S4: Temporal differences in chromatin accessibility at pREs, related to Figure 4
S4A) In situ hybridization for Chd7 mRNA (purple) in wildtype murine embryos at E14.5, from Eurexpress database (www.eurexpress.org) (Diez-Roux et al. 2011). Expression in the ventricular zone (VZ) of cortex is indicated.
S4B) TF motif analysis of 422 PFC pREs that lose chromatin accessibility as well as H3K27ac peaks from 14gw to 18gw. All significant (p<0.05) TF motifs are listed.
Figure S5: Laminar differences in chromatin accessibility in the developing PFC, related to Figure 5.
S5A,B) ATAC-seq reads combined from multiple samples from upper layer and deep layer microdissections of PFC at 18gw and 19gw, mapped to A) upper and B) deep layer marker genes, respectively. Layer-specific pREs are indicated in yellow. Y-axis scale is 0 to 50.
S5C) ATAC-seq reads combined from multiple samples from upper layer and deep layer microdissections of PFC at 18gw and 19gw, nearby upper layer gene SATB2. Highlighted in yellow are two pREs that are upper-layer specific (not OCR in deep layer). One of these two is also a conserved OCR in purified populations of Layer 5 neurons in Rbp4-CRE transgenic mice, but not a conserved layer 6 neuron OCR in Ntsr1-CRE transgenic mice. Y-axis scale is 0 to 50.
Figure S6: Identifying pREs that regulate neurodevelopmental disorder genes, related to Figure 6.
S6A) Postnatal day 2 sections of BCL11A enhancer transgenic mouse, a separate founder line than the one shown in Figure 6B. GFP reporter expression in purple (RNA in situ hybridization for GFP). Cortical, striatal, and hippocampal expression is indicated. S6B) Enrichment analysis of genes with a TSS proximal (within 50 kb) to region-specific OCRs. ASD genes (Sanders et al. 2015), biological targets of Fragile X Mental Retardation protein (FMRP) (Darnell et al. 2011), biological targets of ASD gene CHD8 (Cotney et al. 2015), and developmental delay (DDD) genes (Deciphering Developmental Disorders Study 2015) were compared to liver (Subramanian et al. 2005) and olfactory receptor (Rouillard et al. 2016) gene sets.
S6C) Enrichment analysis of genes having a TSS within a set maximum distance of region-specific OCRs, using multiple distance thresholds from 5 kb to 100 kb in increments of 5kb. A general trend for higher enrichment is seen for more proximal distance thresholds, though these points are not always statistically significant (indicated by point shape) due to the reduced number of region-specific OCRs for testing.
Figure S7: CWAS approach for testing enrichment of ASD case vs. control de novo variants, related to Figure 7.
Category wide association study (CWAS) on de novo ASD mutations from 1,902 quartet families (An et al. 2018) using genome annotations such as conservation scores, gene proximity to neurodevelopmental disorder genes, OCRs and pREs. Although pRE-containing categories did not cross the threshold for statistical significance (p-value = 7.5*10−6 after Bonferroni correction for 6,711 effective tests), they had higher relative risks (labeled and highlighted in grey; ASD=ASD gene locus) than the same categories using OCRs rather than pREs (labeled in red x’s and highlighted in grey). This demonstrates the value of our machine learning approach for identifying a subset of OCRs related to ASD.
Acknowledgments
This work was supported by the research grants to JLR from: Nina Ireland, NIH grants R01NS099099 and R01MH049428 and to KSP from: NIMH R01 MH109907 and NIMH U01 MH116438 and to ARK from: NIMH U01MH105989. ATAC-seq and ChIP-seq datasets for early neural differentiation timepoints were provided by the Ahituv lab (Inoue et al. 2018).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
JLR and ARK are cofounders, stockholders, and currently on the scientific board of Neurona, a company studying the potential therapeutic use of interneuron transplantation.
References
- Amemiya Haley M., Kundaje Anshul, and Boyle Alan P.. 2019. “The ENCODE Blacklist: Identification of Problematic Regions of the Genome.” Scientific Reports 9 (1): 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Joon-Yong, Lin Kevin, Zhu Lingxue, Werling Donna M., Dong Shan, Brand Harrison, Wang Harold Z., et al. 2018. “Genome-Wide de Novo Risk Score Implicates Promoter Variation in Autism Spectrum Disorder.” Science 362 (6420). 10.1126/science.aat6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro Jason D., Wu Beijing, Chang Howard Y., and Greenleaf William J.. 2015. “ATAC-Seq: A Method for Assaying Chromatin Accessibility Genome-Wide.” Current Protocols in Molecular Biology 109 (1): 21.29.1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalcante Raymond G., and Sartor Maureen A.. 2017. “Annotatr: Genomic Regions in Context.” Bioinformatics 33 (15): 2381–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Bin, Schaevitz Laura R., and McConnell Susan K.. 2005. “Fezl Regulates the Differentiation and Axon Targeting of Layer 5 Subcortical Projection Neurons in Cerebral Cortex.” Proceedings of the National Academy of Sciences of the United States of America 102 (47): 17184–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Bin, Wang Song S., Hattox Alexis M., Rayburn Helen, Nelson Sacha B., and McConnell Susan K.. 2008. “The Fezf2-Ctip2 Genetic Pathway Regulates the Fate Choice of Subcortical Projection Neurons in the Developing Cerebral Cortex.” Proceedings of the National Academy of Sciences of the United States of America 105 (32): 11382–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Jie-Guang, Rasin Mladen-Roko, Kwan Kenneth Y., and Sestan Nenad. 2005. “Zfp312 Is Required for Subcortical Axonal Projections and Dendritic Morphology of Deep-Layer Pyramidal Neurons of the Cerebral Cortex.” Proceedings of the National Academy of Sciences of the United States of America 102 (49): 17792–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotney Justin, Muhle Rebecca A., Sanders Stephan J., Liu Li, Willsey A. Jeremy, Niu Wei, Liu Wenzhong, et al. 2015. “The Autism-Associated Chromatin Modifier CHD8 Regulates Other Autism Risk Genes during Human Neurodevelopment.” Nature Communications 6 (March): 6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton Menno P., Cheng Albert W., Welstead G. Grant, Kooistra Tristan, Carey Bryce W., Steine Eveline J., Hanna Jacob, et al. 2010. “Histone H3K27ac Separates Active from Poised Enhancers and Predicts Developmental State.” Proceedings of the National Academy of Sciences of the United States of America 107 (50): 21931–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell Jennifer C., Van Driesche Sarah J., Zhang Chaolin, Sharon Hung Ka Ying, Mele Aldo, Fraser Claire E., Stone Elizabeth F., et al. 2011. “FMRP Stalls Ribosomal Translocation on mRNAs Linked to Synaptic Function and Autism.” Cell 146 (2): 247–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study. 2015. “Large-Scale Discovery of Novel Genetic Causes of Developmental Disorders.” Nature 519 (7542): 223–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study. 2017. “Prevalence and Architecture of de Novo Mutations in Developmental Disorders.” Nature 542 (7642): 433–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubeis De, Silvia Xin He, Goldberg Arthur P., Poultney Christopher S., Samocha Kaitlin, Cicek A. Erucment, Kou Yan, et al. 2014. “Synaptic, Transcriptional and Chromatin Genes Disrupted in Autism.” Nature 515 (7526): 209–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickel Diane E., Ypsilanti Athena R., Pla Ramón, Zhu Yiwen, Barozzi Iros, Mannion Brandon J., Khin Yupar S., et al. 2018. “Ultraconserved Enhancers Are Required for Normal Development.” Cell 172 (3): 491–99.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez-Roux Graciana, Banfi Sandro, Sultan Marc, Geffers Lars, Anand Santosh, Rozado David, Magen Alon, et al. 2011. “A High-Resolution Anatomical Atlas of the Transcriptome in the Mouse Embryo.” PLoS Biology 9 (1): e1000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench John G., Fusi Nicolo, Sullender Meagan, Hegde Mudra, Vaimberg Emma W., Donovan Katherine F., Smith Ian, et al. 2016. “Optimized sgRNA Design to Maximize Activity and Minimize off-Target Effects of CRISPR-Cas9.” Nature Biotechnology 34 (2): 184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwin Genevieve D., Oksenberg Nir, Truty Rebecca M., Kostka Dennis, Murphy Karl K., Ahituv Nadav, Pollard Katherine S., and Capra John A.. 2014. “Integrating Diverse Datasets Improves Developmental Enhancer Prediction.” PLoS Computational Biology 10 (6): e1003677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak Greg, Andrew McDavid Masanao Yajima, Deng Jingyuan, Gersuk Vivian, Shalek Alex K., Slichter Chloe K., et al. 2015. “MAST: A Flexible Statistical Framework for Assessing Transcriptional Changes and Characterizing Heterogeneity in Single-Cell RNA Sequencing Data.” Genome Biology 16 (December): 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish Adam, Diekhans Mark, Ferreira Anne-Maud, Johnson Rory, Jungreis Irwin, Loveland Jane, Mudge Jonathan M., et al. 2019. “GENCODE Reference Annotation for the Human and Mouse Genomes.” Nucleic Acids Research 47 (D1): D766–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert Luke A., Larson Matthew H., Morsut Leonardo, Liu Zairan, Brar Gloria A., Torres Sandra E., Stern-Ginossar Noam, et al. 2013. “CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes.” Cell 154 (2): 442–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golonzhka Olga, Nord Alex, Tang Paul L. F., Lindtner Susan, Ypsilanti Athena R., Ferretti Elisabetta, Visel Axel, Selleri Licia, and Rubenstein John L. R.. 2015. “Pbx Regulates Patterning of the Cerebral Cortex in Progenitors and Postmitotic Neurons.” Neuron 88 (6): 1192–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Shiaoching, Zheng Chen, Doughty Martin L., Losos Kasia, Didkovsky Nicholas, Schambra Uta B., Nowak Norma J., et al. 2003. “A Gene Expression Atlas of the Central Nervous System Based on Bacterial Artificial Chromosomes.” Nature 425 (6961): 917–25. [DOI] [PubMed] [Google Scholar]
- Hawrylycz Michael J., Lein Ed S., Guillozet-Bongaarts Angela L., Shen Elaine H., Ng Lydia, Miller Jeremy A., van de Lagemaat Louie N., et al. 2012. “An Anatomically Comprehensive Atlas of the Adult Human Brain Transcriptome.” Nature 489 (7416): 391–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz Sven, Benner Christopher, Spann Nathanael, Bertolino Eric, Lin Yin C., Laslo Peter, Cheng Jason X., Murre Cornelis, Singh Harinder, and Glass Christopher K.. 2010. “Simple Combinations of Lineage-Determining Transcription Factors Prime Cis-Regulatory Elements Required for Macrophage and B Cell Identities.” Molecular Cell 38 (4): 576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyne Henrike O., Singh Tarjinder, Stamberger Hannah, Rami Abou Jamra Hande Caglayan, Craiu Dana Jonghe Peter De, et al. 2018. “De Novo Variants in Neurodevelopmental Disorders with Epilepsy.” Nature Genetics 50 (7): 1048–53. [DOI] [PubMed] [Google Scholar]
- Huber Wolfgang, Carey Vincent J., Gentleman Robert, Anders Simon, Carlson Marc, Carvalho Benilton S., Bravo Hector Corrada, et al. 2015. “Orchestrating High-Throughput Genomic Analysis with Bioconductor.” Nature Methods 12 (2): 115–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Fumitaka, Kreimer Anat, Ashuach Tal, Ahituv Nadav, and Yosef Nir. 2018. “Massively Parallel Characterization of Regulatory Dynamics during Neural Induction.” bioRxiv. 10.1101/370452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama Yuta, Nishiyama Masaaki, Shoji Hirotaka, Ohkawa Yasuyuki, Kawamura Atsuki, Sato Tetsuya, Suyama Mikita, Takumi Toru, Miyakawa Tsuyoshi, and Nakayama Keiichi I.. 2016. “CHD8 Haploinsufficiency Results in Autistic-like Phenotypes in Mice.” Nature 537 (7622): 675–79. [DOI] [PubMed] [Google Scholar]
- Korb Erica, Herre Margaret, Ilana Zucker-Scharff Jodi Gresack, Allis C. David, and Darnell Robert B.. 2017. “Excess Translation of Epigenetic Regulators Contributes to Fragile X Syndrome and Is Alleviated by Brd4 Inhibition.” Cell 170 (6): 1209–23.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labun Kornel, Montague Tessa G., Krause Maximilian, Torres Cleuren Yamila N., Tjeldnes Håkon, and Valen Eivind. 2019. “CHOPCHOP v3: Expanding the CRISPR Web Toolbox beyond Genome Editing.” Nucleic Acids Research 47 (W1): W171–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Jin, Christoforo Grey, Christoforo Grey, Foo CS, Probert Chris, Kundaje Anshul, Boley Nathan, kohpangwei, Kim Daniel, and Dacre Mike. 2016. Kundajelab/atac_dnase_pipelines: 0.3.0 10.5281/zenodo.156534. [DOI]
- Li Mingfeng, Santpere Gabriel, Yuka Imamura Kawasawa Oleg V. Evgrafov, Gulden Forrest O., Pochareddy Sirisha, Sunkin Susan M., et al. 2018. “Integrative Functional Genomic Analysis of Human Brain Development and Neuropsychiatric Risks.” Science 362 (6420). 10.1126/science.aat7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Qunhua. 2014. IDR: Irreproducible Discovery Rate (version 1.2). https://CRAN.R-project.org/package=idr.
- Li Qunhua, Brown James B., Huang Haiyan, and Bickel Peter J.. 2011. “Measuring Reproducibility of High-Throughput Experiments.” 10.1214/11-AOAS466. [DOI]
- Matharu Navneet, Rattanasopha Sawitree, Tamura Serena, Maliskova Lenka, Wang Yi, Bernard Adelaide, Hardin Aaron, Eckalbar Walter L., Vaisse Christian, and Ahituv Nadav. 2019. “CRISPR-Mediated Activation of a Promoter or Enhancer Rescues Obesity Caused by Haploinsufficiency.” Science 363 (6424). 10.1126/science.aau0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney Wes. 2012. Python for Data Analysis. Sebastopol, CA: O’Reilly. [Google Scholar]
- McLean Cory Y., Bristor Dave, Hiller Michael, Clarke Shoa L., Schaar Bruce T., Lowe Craig B., Wenger Aaron M., and Bejerano Gill. 2010. “GREAT Improves Functional Interpretation of Cis-Regulatory Regions.” Nature Biotechnology 28 (5): 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuleman Wouter, Muratov Alexander, Rynes Eric, Halow Jessica, Lee Kristen, Bates Daniel, Diegel Morgan, et al. 2019. “Index and Biological Spectrum of Accessible DNA Elements in the Human Genome.” bioRxiv. 10.1101/822510. [DOI] [Google Scholar]
- Mo Alisa, Mukamel Eran A., Davis Fred P., Luo Chongyuan, Henry Gilbert L., Picard Serge, Urich Mark A., et al. 2015. “Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain.” Neuron 86 (6): 1369–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux Bradley J., Arlotta Paola, Hirata Tustomu, Hibi Masahiko, and Macklis Jeffrey D.. 2005. “Fezl Is Required for the Birth and Specification of Corticospinal Motor Neurons.” Neuron 47 (6): 817–31. [DOI] [PubMed] [Google Scholar]
- Nowakowski Tomasz J., Bhaduri Aparna, Pollen Alex A., Alvarado Beatriz, Mostajo-Radji Mohammed A., Lullo Elizabeth Di, Haeussler Maximilian, et al. 2017. “Spatiotemporal Gene Expression Trajectories Reveal Developmental Hierarchies of the Human Cortex.” Science 358 (6368): 1318–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski Tomasz J., Pollen Alex A., Sandoval-Espinosa Carmen, and Kriegstein Arnold R.. 2016. “Transformation of the Radial Glia Scaffold Demarcates Two Stages of Human Cerebral Cortex Development.” Neuron 91 (6): 1219–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary Dennis D. M., Chou Shen-Ju, and Sahara Setsuko. 2007. “Area Patterning of the Mammalian Cortex.” Neuron 56 (2): 252–69. [DOI] [PubMed] [Google Scholar]
- O’Leary Nuala A., Wright Mathew W., Brister J. Rodney, Ciufo Stacy, Haddad Diana, McVeigh Rich, Rajput Bhanu, et al. 2016. “Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation.” Nucleic Acids Research 44 (D1): D733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak Neelroop N., Luo Rui, Zhang Alice, Won Hyejung, Lowe Jennifer K., Chandran Vijayendran, Horvath Steve, and Geschwind Daniel H.. 2013. “Integrative Functional Genomic Analyses Implicate Specific Molecular Pathways and Circuits in Autism.” Cell 155 (5): 1008–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabiraman Kartik, Golonzhka Olga, Lindtner Susan, Nord Alex S., Taher Leila, Hoch Renee, Silberberg Shanni N., et al. 2014. “Transcriptional Regulation of Enhancers Active in Protodomains of the Developing Cerebral Cortex.” Neuron 82 (5): 989–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa Fabian, Varoquaux Gaël, Gramfort Alexandre, Michel Vincent, Thirion Bertrand, Grisel Olivier, Blondel Mathieu, et al. 2011. “Scikit-Learn: Machine Learning in Python.” Journal of Machine Learning Research 12 (November): 2825–30. [Google Scholar]
- Pollard Katherine S., Hubisz Melissa J., Rosenbloom Kate R., and Siepel Adam. 2010. “Detection of Nonneutral Substitution Rates on Mammalian Phylogenies.” Genome Research 20 (1): 110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan Aaron R., and Hall Ira M.. 2010. “BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features.” Bioinformatics 26 (6): 841–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Suhas S. P., Huntley Miriam H., Durand Neva C., Stamenova Elena K., Bochkov Ivan D., Robinson James T., Sanborn Adrian L., et al. 2014. “A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping.” Cell 159 (7): 1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2018. “R: A Language and Environment for Statistical Computing.” Vienna, Austria: R Foundation for Statistical Computing; https://www.r-project.org. [Google Scholar]
- Roadmap Epigenomics Consortium Anshul Kundaje, Meuleman Wouter, Ernst Jason, Bilenky Misha, Yen Angela, Heravi-Moussavi Alireza, et al. 2015. “Integrative Analysis of 111 Reference Human Epigenomes.” Nature 518 (7539): 317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouillard Andrew D., Gundersen Gregory W., Fernandez Nicolas F., Wang Zichen, Monteiro Caroline D., McDermott Michael G., and Ma’ayan Avi. 2016. “The Harmonizome: A Collection of Processed Datasets Gathered to Serve and Mine Knowledge about Genes and Proteins.” Database: The Journal of Biological Databases and Curation 2016 (July). 10.1093/database/baw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JLR, and Campbell K. 2013. “Neurogenesis in the Basal Ganglia. Patterning and Cell Type Specification in the Developing CNS and PNS.” Elsevier. [Google Scholar]
- Rubenstein John, and Pasko Rakic. 2013. Neural Circuit Development and Function in the Healthy and Diseased Brain: Comprehensive Developmental Neuroscience. Academic Press. [Google Scholar]
- Sandberg Magnus, Taher Leila, Hu Jianxin, Black Brian L., Nord Alex S., and Rubenstein John L. R.. 2018. “Genomic Analysis of Transcriptional Networks Directing Progression of Cell States during MGE Development.” Neural Development 13 (1): 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders Stephan J., He Xin, Willsey A. Jeremy, Ercan-Sencicek A. Gulhan, Samocha Kaitlin E., Cicek A. Ercument, Murtha Michael T., et al. 2015. “Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci.” Neuron 87 (6): 1215–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterstrom F Kyle, Kosmicki Jack A., Wang Jiebiao, Breen Michael S., Rubeis Silvia De, An Joon-Yong, Peng Minshi, et al. 2020. “Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism.” Cell 180 (3): 568–84.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sestan Nenad, and State Matthew W.. 2018. “Lost in Translation: Traversing the Complex Path from Genomics to Therapeutics in Autism Spectrum Disorder.” Neuron 100 (2): 406–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seuntjens Eve, Nityanandam Anjana, Miquelajauregui Amaya, Debruyn Joke, Stryjewska Agata, Goebbels Sandra, Nave Klaus-Armin, Huylebroeck Danny, and Tarabykin Victor. 2009. “Sip1 Regulates Sequential Fate Decisions by Feedback Signaling from Postmitotic Neurons to Progenitors.” Nature Neuroscience 12 (11): 1373–80. [DOI] [PubMed] [Google Scholar]
- Shim Sungbo, Kwan Kenneth Y., Li Mingfeng, Lefebvre Veronique, and Sestan Nenad. 2012. “Cis-Regulatory Control of Corticospinal System Development and Evolution.” Nature 486 (7401): 74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel Adam, Bejerano Gill, Pedersen Jakob S., Hinrichs Angie S., Hou Minmei, Rosenbloom Kate, Clawson Hiram, et al. 2005. “Evolutionarily Conserved Elements in Vertebrate, Insect, Worm, and Yeast Genomes.” Genome Research 15 (8): 1034–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberberg Shanni N., Taher Leila, Lindtner Susan, Sandberg Magnus, Nord Alex S., Vogt Daniel, Mckinsey Gabriel L., et al. 2016. “Subpallial Enhancer Transgenic Lines: A Data and Tool Resource to Study Transcriptional Regulation of GABAergic Cell Fate.” Neuron 92 (1): 59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- State Matthew W., and Šestan Nenad. 2012. “Neuroscience. The Emerging Biology of Autism Spectrum Disorders.” Science 337 (6100): 1301–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian Aravind, Tamayo Pablo, Mootha Vamsi K., Mukherjee Sayan, Ebert Benjamin L., Gillette Michael A., Paulovich Amanda, et al. 2005. “Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles.” Proceedings of the National Academy of Sciences of the United States of America 102 (43): 15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkin Susan M., Ng Lydia, Lau Chris, Dolbeare Tim, Gilbert Terri L., Thompson Carol L., Hawrylycz Michael, and Dang Chinh. 2013. “Allen Brain Atlas: An Integrated Spatio-Temporal Portal for Exploring the Central Nervous System.” Nucleic Acids Research 41 (Database issue): D996–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov Artem, Vilella Albert J., Cuppen Edwin, Nijman Isaac J., and Prins Pjotr. 2015. “Sambamba: Fast Processing of NGS Alignment Formats.” Bioinformatics 31 (12): 2032–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre-Ubieta Luis de la, Jason L. Stein, Hyejung Won, Opland Carli K., Liang Dan, Lu Daning, and Geschwind Daniel H.. 2018. “The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis.” Cell 172 (1–2): 289–304.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker Eric S., Segall Samantha, Gopalakrishna Deepak, Wu Yongqin, Vernon Mike, Polleux Franck, and Lamantia Anthony-Samuel. 2008. “Molecular Specification and Patterning of Progenitor Cells in the Lateral and Medial Ganglionic Eminences.” The Journal of Neuroscience: The Official Journal of the Society for Neuroscience 28 (38): 9504–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velmeshev Dmitry, Schirmer Lucas, Jung Diane, Haeussler Maximilian, Perez Yonatan, Mayer Simone, Bhaduri Aparna, Goyal Nitasha, Rowitch David H., and Kriegstein Arnold R.. 2019. “Single-Cell Genomics Identifies Cell Type-Specific Molecular Changes in Autism.” Science 364 (6441): 685–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel Axel, Minovitsky Simon, Dubchak Inna, and Pennacchio Len A.. 2007. “VISTA Enhancer Browser--a Database of Tissue-Specific Human Enhancers.” Nucleic Acids Research 35 (Database issue): D88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel Axel, Taher Leila, Girgis Hani, May Dalit, Golonzhka Olga, Hoch Renee V., McKinsey Gabriel L., et al. 2013. “A High-Resolution Enhancer Atlas of the Developing Telencephalon.” Cell 152 (4): 895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers Lisenka E. L. M., van Ravenswaaij Conny M. A., Admiraal Ronald, Hurst Jane A., de Vries Bert B. A., Janssen Irene M., van der Vliet Walter A., et al. 2004. “Mutations in a New Member of the Chromodomain Gene Family Cause CHARGE Syndrome.” Nature Genetics 36 (9): 955–57. [DOI] [PubMed] [Google Scholar]
- Werling Donna M., Brand Harrison, An Joon-Yong, Stone Matthew R., Zhu Lingxue, Glessner Joseph T., Collins Ryan L., et al. 2018. “An Analytical Framework for Whole-Genome Sequence Association Studies and Its Implications for Autism Spectrum Disorder.” Nature Genetics 50 (5): 727–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeremy Willsey, A., Sanders Stephan J., Li Mingfeng, Dong Shan, Tebbenkamp Andrew T., Muhle Rebecca A., Reilly Steven K., et al. 2013. “Coexpression Networks Implicate Human Midfetal Deep Cortical Projection Neurons in the Pathogenesis of Autism.” Cell 155 (5): 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won Hyejung, Torre-Ubieta Luis de la, Stein Jason L., Parikshak Neelroop N., Huang Jerry, Opland Carli K., Gandal Michael J., et al. 2016. “Chromosome Conformation Elucidates Regulatory Relationships in Developing Human Brain.” Nature 538 (7626): 523–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Yong, Liu Tao, Meyer Clifford A., Eeckhoute Jérôme, Johnson David S., Bernstein Bradley E., Nusbaum Chad, et al. 2008. “Model-Based Analysis of ChIP-Seq (MACS).” Genome Biology 9 (9): R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2: pRE and OCR annotations, related to Figure 2 and Figure S2.
Table S2A: pRE genomic coordinates, conservation score, nearest genes, telencephalic regions which overlap an OCR, pRE prediction score.
Table S2B: OCR genomic coordinates, conservation score, nearest genes, telencephalic regions which overlap an OCR, pRE prediction score.
Table S2C: GREAT analysis of pREs.
Table S2D: pRE genomic coordinates, overlaps with histone ChIp-seq peaks from PFC.
Table S2E: OCR genomic coordinates, overlaps with histone ChIP-seq peaks from PFC.
Table S3: Region-specific pREs proximal to differentially expressed genes between PFC, V1, MGE. Related to Figure 3 and Figure S3.
Table S3A: MGE and V1 specific pREs proximal to differentially expressed genes between MGE and V1.
Table S3B: MGE and PFC specific pREs proximal to differentially expressed genes between MGE and PFC.
Table S3C: V1 and PFC specific pREs proximal to differentially expressed genes between V1 and PFC.
Table S4: Temporal differences in chromatin accessibility at cortical pREs, related to Figure 4 and Figure S4.
Table S4A: GREAT analysis of early (14gw) and late (19gw) frontal cortex pREs. Table S4B: 14gw and 19gw specific pREs proximal to differentially expressed genes between the two timepoints.
Table S5: Layer specific OCRs, related to Figure 5 and Figure S5.
Table S5A: OCRs annotated whether they overlap upper layer or deep layer OCRs.
Table S5B: Upper layer OCRs annotated by whether they contain motifs for TFs expressed in immature neurons, or mature superficial neurons, or both.
Table S6: OCRs proximal to genes related to neurodevelopmental disorders or markers of particular cell types in the developing telencephalon, related to Figure 6 and Figure S6.
Table S6A: Percentage of genes per gene set located within 50kb of a region-specific open chromatin region. Fragile-X (FMRP) has a high percentage of genes proximal to RS OCRs, and is enriched across all regions.
Table S6B: Region-specific OCRs proximal to disease genes (ASD, DD, FMRP targets, and CHD8 targets).
Table S6C: Region-specific OCRs proximal to marker genes of different cell types in the developing telencephalon (Nowakowski et al 2017).
Table S7: pREs containing de novo variants and luciferase assay cloning oligonucleotides.
Table S7A: pREs that contain more than one de novo ASD patient variant (An et al 2018), related to Figure 7 and Figure S7.
Table S7B: Oligonucleotide sequences of forward and reverse primers used to amplify pREs and OCRs from human genomic DNA, then cloned into the minimal promoter pGl4.23 luciferase vector (Promega) using SacI and XhoI restriction sites (underlined) in the vector’s multiple cloning site. Related to Figure 2 and Figure S1.
Figure S1: Predicting neurodevelopmental enhancers, related to Figure 1
S1A) Enrichment of ATAC-seq reads centered at RefSeq TSSs, calculated using the ENCODE standard (Methods). Coverage is normalized using the average raw coverage at regions flanking the TSS (offsets from −2000 to −1900 and +1900 to +2000, indicated by the blue vertical lines) as background, resulting in a median TSS signal to background ratio of 42.3 across samples.
S1B) The ten largest and smallest model coefficients (L2-penalized) from a logistic regression classifier trained using various epigenetic and sequence features (Methods) to distinguish between developmental brain enhancers versus non-brain enhancers (ex: heart, limb) and failed enhancer candidates validated by VISTA. These coefficients represent features most predictive of the positive (brain enhancer, blue) and negative (non-brain enhancer and failed enhancers, red) classes. Predictive positive features include open chromatin and H3K27ac in neural progenitors (N2), H3K27ac in hESC cells with neural markers, conserved TF binding sites, and other open chromatin and activating histone marks in neural cell types.
S1C) Mean firefly luciferase levels in human neuroblastoma cells, normalized to Renilla. Eight randomly selected OCRs (blue bars) that are not pREs were cloned upstream of a minimal promoter and firefly luciferase ORF in the pGL4.23 vector. The green bar indicates the only sequence tested, a pRE proximal to GSX2, which had increased luciferase activity compared to empty vector (red bar). Error bars indicate standard error across two replicate experiments.
S1D) Phastcons and PhyloP scores (100-way alignments, vertebrate conservation) of pREs, OCRs, and VISTA enhancers.
S1E) Percent of PFC pREs that overlap called peaks from 11 histone ChIP-seq datasets from midfetal PFC (ages 15gw-22gw). Histone marks are classified based on their association with activating, repressing, or bivalently regulating gene regulatory elements and transcription.
Figure S2: Tracking OCR activation and inactivation across midfetal development, related to Figure 1
S2A) The following sets of PFC OCRs are binned according to orientation and distance to gene TSS in the human genome: OCRs that do not change chromatin accessibility between 14gw and 18gw in the PFC, OCRs that gained accessibility from 14gw to 18gw, and OCRs that lost accessibility from 14gw to 18gw. Below, TF motif analysis of sets of PFC OCRs that change chromatin accessibility from 14gw to 18gw, and subsets of those OCRs where increase or decrease in chromatin accessibility is accompanied by gain or loss of H3K27ac enrichment, respectively. The top significant TF motifs are shown for OCRs where there was no change in accessibility, while all the significant (p<0.05) TF motifs are listed for all other sets of OCRs.
S2B) Example of an OCR (highlighted in yellow) proximal to the EOMES/TBR2 gene locus, which loses both chromatin accessibility and H3K27ac in the PFC between 14gw and 18gw.
S2C) Example of OCRs (highlighted in yellow) proximal to the SEMA3A gene locus, which gain both chromatin accessibility and H3K27ac in the PFC between 14gw and 18gw.
Figure S3: Regional differences in chromatin accessibility at pREs, related to Figure 3
S3A) In situ hybridization for Kcnq3, Pppp2r1b, and Trim2 mRNA (purple) in wildtype murine embryos at E14.5, from Eurexpress database (www.eurexpress.org) (Diez-Roux et al. 2011). Kcnq3 is expressed in the cortex (both rostral and caudal) and not the basal ganglia (bg), Pppp2r1b is expressed in rostral cortex, and Trim2 is expressed in caudal cortex.
S3B) Summary of region-specific OCRs and pREs identified in this study.
S3C) Annotation of genomic features at region-specific pREs compared to pREs shared across multiple brain regions.
Figure S4: Temporal differences in chromatin accessibility at pREs, related to Figure 4
S4A) In situ hybridization for Chd7 mRNA (purple) in wildtype murine embryos at E14.5, from Eurexpress database (www.eurexpress.org) (Diez-Roux et al. 2011). Expression in the ventricular zone (VZ) of cortex is indicated.
S4B) TF motif analysis of 422 PFC pREs that lose chromatin accessibility as well as H3K27ac peaks from 14gw to 18gw. All significant (p<0.05) TF motifs are listed.
Figure S5: Laminar differences in chromatin accessibility in the developing PFC, related to Figure 5.
S5A,B) ATAC-seq reads combined from multiple samples from upper layer and deep layer microdissections of PFC at 18gw and 19gw, mapped to A) upper and B) deep layer marker genes, respectively. Layer-specific pREs are indicated in yellow. Y-axis scale is 0 to 50.
S5C) ATAC-seq reads combined from multiple samples from upper layer and deep layer microdissections of PFC at 18gw and 19gw, nearby upper layer gene SATB2. Highlighted in yellow are two pREs that are upper-layer specific (not OCR in deep layer). One of these two is also a conserved OCR in purified populations of Layer 5 neurons in Rbp4-CRE transgenic mice, but not a conserved layer 6 neuron OCR in Ntsr1-CRE transgenic mice. Y-axis scale is 0 to 50.
Figure S6: Identifying pREs that regulate neurodevelopmental disorder genes, related to Figure 6.
S6A) Postnatal day 2 sections of BCL11A enhancer transgenic mouse, a separate founder line than the one shown in Figure 6B. GFP reporter expression in purple (RNA in situ hybridization for GFP). Cortical, striatal, and hippocampal expression is indicated. S6B) Enrichment analysis of genes with a TSS proximal (within 50 kb) to region-specific OCRs. ASD genes (Sanders et al. 2015), biological targets of Fragile X Mental Retardation protein (FMRP) (Darnell et al. 2011), biological targets of ASD gene CHD8 (Cotney et al. 2015), and developmental delay (DDD) genes (Deciphering Developmental Disorders Study 2015) were compared to liver (Subramanian et al. 2005) and olfactory receptor (Rouillard et al. 2016) gene sets.
S6C) Enrichment analysis of genes having a TSS within a set maximum distance of region-specific OCRs, using multiple distance thresholds from 5 kb to 100 kb in increments of 5kb. A general trend for higher enrichment is seen for more proximal distance thresholds, though these points are not always statistically significant (indicated by point shape) due to the reduced number of region-specific OCRs for testing.
Figure S7: CWAS approach for testing enrichment of ASD case vs. control de novo variants, related to Figure 7.
Category wide association study (CWAS) on de novo ASD mutations from 1,902 quartet families (An et al. 2018) using genome annotations such as conservation scores, gene proximity to neurodevelopmental disorder genes, OCRs and pREs. Although pRE-containing categories did not cross the threshold for statistical significance (p-value = 7.5*10−6 after Bonferroni correction for 6,711 effective tests), they had higher relative risks (labeled and highlighted in grey; ASD=ASD gene locus) than the same categories using OCRs rather than pREs (labeled in red x’s and highlighted in grey). This demonstrates the value of our machine learning approach for identifying a subset of OCRs related to ASD.
Data Availability Statement
Raw ATAC-seq and ChIP-seq sequencing data has been deposited to dbGAP (phs002033.v1.p1). Processed bigwigs, peaks, and OCR/pRE annotations have been deposited to GEO (GSE149268).