

Summary
CRISPR-Cas defense systems have been coopted multiple times in nature for guide RNA-directed transposition by Tn7-like elements. Prototypic Tn7 uses dedicated proteins for two targeting pathways, one targeting a neutral and conserved attachment site in the chromosome and a second directing transposition into mobile plasmids facilitating cell-to-cell transfer. We show that Tn7-CRISPR-Cas elements evolved a system of guide RNA categorization to accomplish the same two-pathway lifestyle. Multiple mechanisms allow functionally distinct guide RNAs for transposition, a conventional system capable of acquiring guide RNAs to new plasmid and phage targets and a second providing long-term memory for access to chromosomal sites upon entry into a new host. Guide RNAs are privatized to be recognized only by the transposon-adapted system via sequence-specialization, mismatch tolerance and selective regulation to avoid toxic self-targeting by endogenous CRISPR-Cas defense systems. This information reveals promising avenues to engineer guide RNAs for enhanced CRISPR-Cas functionality for genome modification.
Keywords: CRISPR regulation, Guide RNA categorization, RNA-directed transposition
In Brief…
Transposons have coopted CRISPR-Cas systems for RNA-guided transposition, using mechanisms that allow for target site choice, and that prevent guide RNA use by canonical CRISPR-Cas systems—features that could be useful for biotech applications.
Graphical Abstract
Introduction
CRISPR-Cas systems are widespread in bacteria and archaea providing an efficient defense system from predation from bacteriophages and the burden imposed by other mobile elements (Makarova et al., 2020). CRISPR-Cas systems have also been repurposed in nature for other functions that benefit from programmable guide RNA-based sequence recognition (Faure et al., 2019a). One compelling collection of systems involves the occurrence on multiple occasions where a specialized class of transposons called Tn7-like elements have coopted different CRISPR-Cas systems for guide RNA-targeted transposition (Faure et al., 2019b; Peters et al., 2017). Beyond providing an intriguing example of the propensity of evolution to mix and match useful functions, these systems have also been repurposed in the laboratory as promising tools for genome modification (Klompe et al., 2019; Strecker et al., 2019). Multiple questions remain with the basic functioning of Tn7-CRISPR-Cas elements, especially with how they interact with canonical CRISPR-Cas systems.
The largest group of Tn7-CRISPR-Cas elements adapted a specific subtype of CRISPR-Cas systems within the type I-F group for guide RNA-directed transposition (Peters, 2019). Canonical type I-F CRISPR-Cas systems, called type I-F1 systems, use four proteins (Cas8, Cas5, Cas7, and Cas6) as an effector complex called cascade to mature pre-crRNAs transcribed from a CRISPR array into functional guide RNA complexes. The repeats in the CRISPR array encode the guide RNA handles that are bound by Cas proteins, while target specificity is encoded in the spacers. Cas1 and Cas2 are proteins found across CRISPR-Cas systems for acquiring new spacers (also called adaptation) which are inserted into one end of the array adjacent to a special leader region. Type I CRISPR-Cas systems degrade targets recognized by the system using a helicase-nuclease protein Cas3. The Tn7-CRISPR-Cas elements that derived from the canonical I-F1 systems are called I-F3 systems (Makarova et al., 2020). The I-F3 Tn7-CRISPR-Cas systems lack Cas3 and the Cas8 and Cas5 proteins are naturally fused. Tn7-CRISPR-Cas elements lack a spacer acquisition system and must collect new targeting information in trans using Cas1-2 functions borrowed from canonical CRISPR-Cas systems.
Prototypic Tn7 is known for the control it has over target site selection using five element-encoded proteins to direct transposition into either a specific neutral chromosomal attachment (att) site or mobile genetic elements capable of cell-to-cell transfer (Waddell and Craig, 1988). A core machinery used for all transposition involves a heteromeric transposase, TnsA+TnsB, for the breaking and joining functions that underlie transposition that is controlled by a AAA+ regulator protein, TnsC (Bainton et al., 1991; Bainton et al., 1993; Stellwagen and Craig, 1998). TnsABC must function with one of two target site selection proteins, TnsD/TniQ or TnsE. Tn7 transposition mediated by the sequence specific DNA binding protein TnsD/TniQ allows high frequency transposition into its att site downstream of the glmS gene. TnsD/TniQ recognizes the coding region of the essential and highly conserved glmS gene, ensuring a place for the transposon to integrate in a new bacterial host (Mitra et al., 2010). Tn7 or diverged families of Tn7-like elements with homologs of TnsABCD have been identified in 10-20% of sequenced bacteria, but in the diverged families the TnsD/TniQ protein evolved new DNA binding specificities to recognize a wide variety of new att sites (Peters, 2019; Peters et al., 2017). TnsABC from prototypic Tn7 also functions with the element-encoded protein TnsE to recognize plasmids capable of cell-cell transfer to facilitate transfer of the element between bacteria (Parks et al., 2009; Peters and Craig, 2001; Shi et al., 2015).
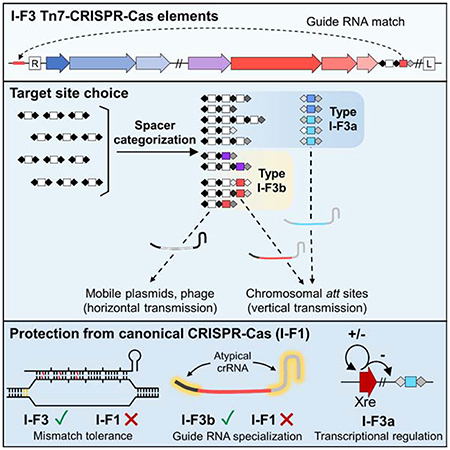
We present a comprehensive bioinformatic analysis of I-F3 Tn7-CRISPR-Cas elements that reveals mechanisms that allowed the evolution of guide RNA-directed transposition involving categorization of guide RNAs. This updated analysis indicates that all I-F3 Tn7-CRISPR-Cas insertion events are explained by guide RNAs encoded in CRISPR arrays within the element. A form of curation allows the I-F3 elements to maintain different classes of guide RNAs to mirror the two-pathway lifestyle found with prototypic Tn7, but with a guide-RNA-only system. Guide RNA-directed transposition into the chromosome occurs via CRISPR arrays that are under the control of a specialized transcriptional regulation system that directs pathway choice or using an atypical CRISPR repeat structure that allows the guide RNA to be private to the Tn7-CRISPR-Cas transposon. Guide RNAs encoded by the elements that recognize the chromosome also have mismatches that are tolerated for directing transposition, but not for interference by a canonical I-F1 system. The guide RNA attributes found in I-F3 Tn7-CRISPR-Cas elements help explain how they interact with related type I-F CRISPR-Cas systems, such as the ability to tolerate self-targeting guide RNAs that would otherwise cause canonical CRISPR-Cas systems to degrade the host chromosome.
Results
I-F3 Tn7-CRISPR-Cas element targeting is explained by spacers in atypical CRISPR array configurations
We conducted an updated bioinformatics analysis of the I-F3 family of Tn7-CRISPR-Cas elements (Peters, 2019; Peters et al., 2017). An analysis of over 53,000 genomes from gamma proteobacteria identified 802 Tn7-like elements that encode the type I-F3 CRISPR-Cas system found in two branches (Figure 1). One branch, I-F3a, primarily uses attachment sites adjacent to the yciA and guaC (IMPDH) genes. Elements in a second branch, I-F3b, are primarily found in an attachment site downstream of the ffs gene encoding the RNA component of the signal recognition particle and a minor branch with elements residing downstream of the rsmJ gene. As part of this analysis we reexamined CRISPR arrays and made a striking finding that altered our understanding of how transposition is targeted across all of the I-F3 elements. We can show that the insertion position of all elements can be explained by guide RNA-directed transposition; for essentially all of the I-F3 elements we can identify a spacer within element-encoded CRISPR arrays that matches a region ~48 bp from the right end of the element (Figures 1, 2a, and 2b)(Supplementary Table S1). In each of these cases the spacer in the array matches the same protospacer in the yciA, guaC, ffs, or rsmJ genes (Figure 2b). In addition to being at one of the ends of the gene to direct transposition just outside the reading frame, the spacers matching the yciA, guaC, and rsmJ genes are all found in the same reading frame register that aligns the variable wobble position of the codons with every sixth position in the guide RNA, a position known to flip out and not required to match the protospacer (Fineran et al., 2014; Jackson et al., 2014; Mulepati et al., 2014; Zhao et al., 2014). Around six percent of Tn7-CRISPR-Cas insertions identified in bacterial genomes are not located in one of the four major att sites (Supplemental Table S1). However, even with the insertions outside the major att sites we could still identify a spacer in the array that was specific to a protospacer ~48bp from the right end of the element (Figure 1).
Figure 1 -. Tn7-like elements with I-F3 CRISPR-Cas systems found in gamma proteobacteria.
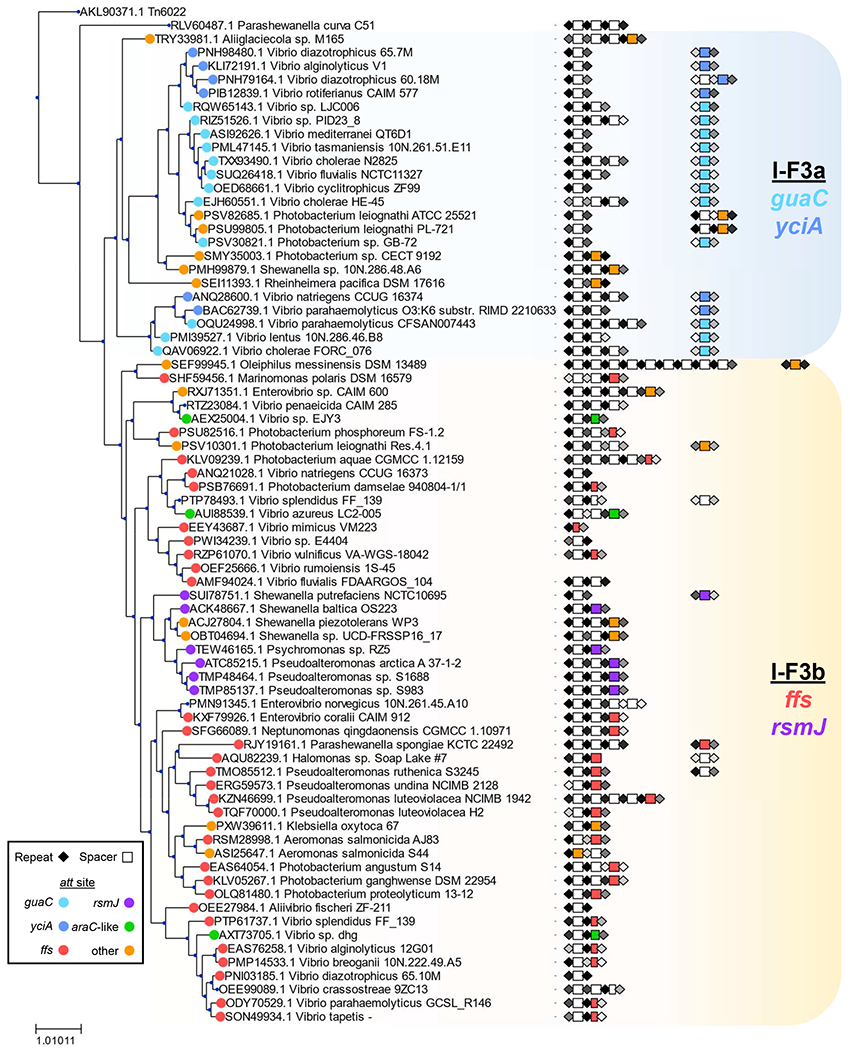
A TnsA protein similarity tree representing 802 elements indicated by host strain. Elements that were >90 percent identical are indicated with a single representative. A similarity score was calculated for repeats and indicated in shades of black (high) to grey (low). Spacers are indicated with rectangles using a color key (shorter rectangles indicate truncated spacers) (Supplementary Table S1). See text for details.
Figure 2 – Selected representatives from four att-site families of Tn7-like elements with I-F3 CRISPR-Cas systems.
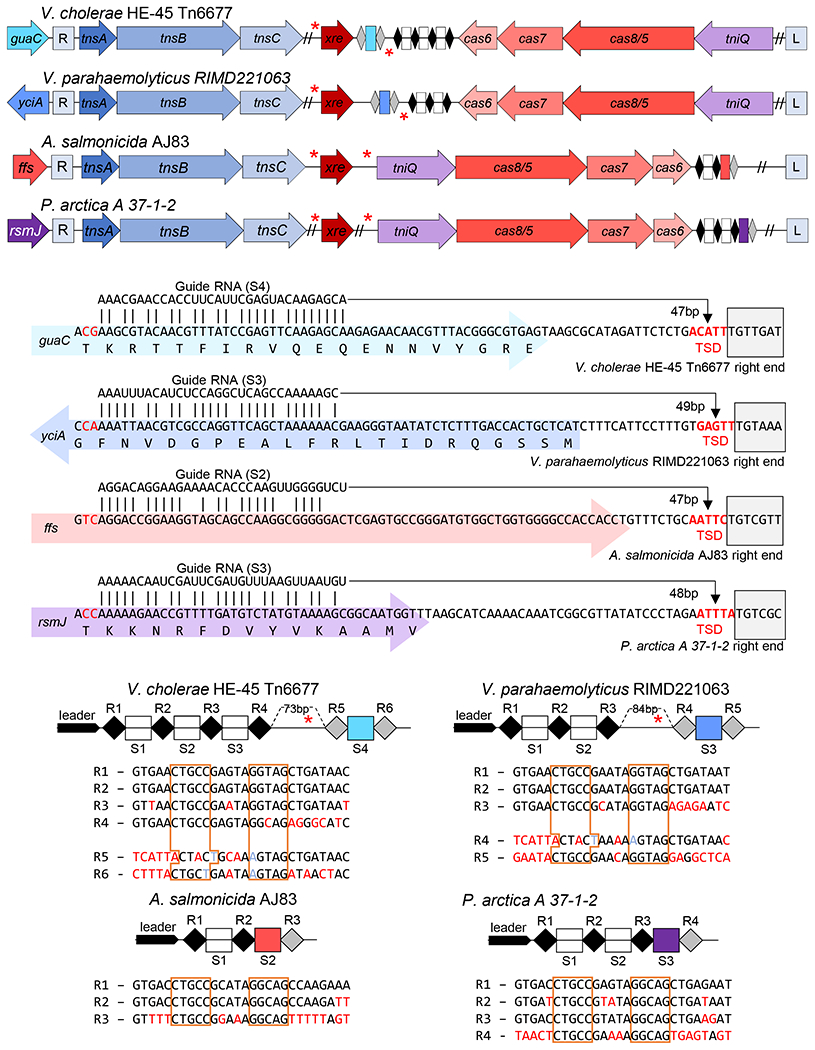
Representatives for three major families (att sites; yciA, guaC, and ffs) and one minor family (rsmJ) are indicated by host. (a) Transposition genes (tnsA, tnsB, tnsC, and tniQ/tnsD), Cas genes cas6, cas7, and cas8/5, and regulator xre are indicated. CRISPR arrays indicated as in Figure 1. The left (L) and Right (R) ends of the elements are indicated and putative Xre binding sites (red asterisks). (b) Matches between the guide RNA and protospacer are shown on each gene (colored block arrow) with the right end of the element (grey box) and host indicated. Distance from the protospacer to the target site duplication (TSD, bold red) is shown. (c) CRISPR array is indicated with the leader region and spacer (S#) and repeats (R#) indicated showing the sequence of the repeats. Sequence differences from the first repeat (red), noting changes maintaining the stem (light blue) and the inverted repeats that makes the stem (boxed orange) are indicated. The size of the gap in the array is indicated noting the putative Xre regulatory site.
Of additional interest, the spacers that recognize each one of the four major att sites are located in a specific position in the element-encoded CRISPR array(s). There are obvious trends with this position and the configuration of the CRISPR arrays that differed in the two major branches of I-F3 elements. In the branch of I-F3a elements, the spacer that matched the yciA or guaC att sites was located after a 70-90 bp gap in the array found immediately downstream of the tniQ, cas8/5, cas7, cas6 operon (Figures 2a and 2c)(see below). In these cases where the CRISPR array was not contiguous it was not clear if the array was transcribed as a single pre-crRNA and/or if all of the spacers were capable of being matured into functional guide RNA complexes (addressed below). In the I-F3b branch of elements that recognizes att sites associated with the ffs and rsmJ genes the att-site specific spacer tended to be found in the single CRISPR array located downstream of the tniQ-cas operon, but always as the last spacer in the array (Figures 1 and 2a).
Only one transposition event was identified in a plasmid rather than in the chromosome in our analysis. The Tn7-CRISPR-Cas element in Aeromonas salmonicida S44, Tn6900, was on a large plasmid (pS44-1) that is predicted to be mobile based on the presence of genes with known roles in conjugal DNA transfer (tra genes). Transposition into the site on the plasmid could still be explained by a guide RNA encoded in the array, however, in this case the spacer was at the leader-proximal position in the array (Supplemental Figure S1a–c). Interestingly, a near-identical Tn7-CRISPR-Cas element, Tn6899, found in the ffs att site in Aeromonas hydrophila AFG_SD03 (Boehmer et al., 2018) had a spacer that recognized the same plasmid-encoded gene, but at a different position (Supplemental Figure S1b–c), suggesting a possible plasmid vector important for the dispersal of these elements within Aeromonas.
In addition to their distinct position in the CRISPR array, the att spacers were flanked by repeats with novel sequences. New spacers are added to a CRISPR array at the leader-proximal end of the array in a process that duplicates the leader-proximal repeat (Xiao et al., 2017). Therefore, although repeats can diverge over time, the first and second repeats start out identical in CRISPR arrays. In I-F3 Tn7-CRISPR-Cas elements the terminal spacer that was used for guide RNA-directed transposition into the chromosome was invariably flanked by repeats that were highly diverged from the leader-proximal repeat (Figure 2c and Supplemental Table S1). We call the highly diverged repeats, “atypical” repeats, and a guide RNA formed from these sequences atypical guide RNAs.
Highly diverged atypical repeat-spacer units form functional guide RNA complexes
To help understand the unique nature of the CRISPR array structure found in I-F3 Tn7-CRISPR-Cas elements we established guide RNA-directed transposition in a heterologous and genetically tractable system, E. coli. Elements identified in a plasmid in Aeromonas salmonicida S44 and in the ffs attachment site in Aeromonas hydrophila AFG_SD03 were of particular interest because they were near-identical, but found in different species at distinct insertion points suggesting they were functional recently (Supplemental Figure S1a). For the transposition (Tns) and Cas proteins we utilized a coding sequence configuration we predicted to be active for transposition by looking for consensus across multiple elements found in Aeromonas.
Previous studies trying to establish Tn7-CRISPR-Cas transposition in a heterologous host relied heavily on indirect PCR-based techniques to assess transposition, techniques that are vulnerable to artifacts (Rice et al., 2020; Strecker et al., 2020). To get a complete picture of Tn7-CRISPR-Cas transposition, we used an assay that monitored full transposition events. A mini Tn7-CRISPR-Cas element was situated in the chromosome, the donor site for transposition in our assays, constructed with cis-acting transposon end-sequences predicted by putative TnsB-binding sites (Peters, 2014) flanking an antibiotic resistance determinant. In this assay, candidate transposition targets resided on a conjugal F plasmid. After inducing expression of the components of the guide RNA-directed transposition system, full transposition events were detected by mating the conjugal plasmid into a tester strain and screening for the antibiotic resistance gene in the mini transposon (Supplemental Figure S2a). The tnsABC, tniQ-cas8/5,7,6, and the CRISPR array were expressed from three separate expression vectors.
Initially we analyzed candidate guide RNAs produced from the wild-type configuration of the CRISPR array found in Tn6900 in A. salmonicida S44. In this configuration the leader-proximal spacer was a perfect match to a mobile plasmid-encoded gene from the native host and the second/terminal spacer had a degenerate match to the ffs protospacer with 10 mismatches (Figure 3a, Supplemental Figure S1c). Some of the mismatches between the spacer and the protospacer in the target were at every sixth position and therefore would not impact recognition of the ffs guide RNA target (Supplemental Figure S1c). Monitoring transposition following expression of the native array configuration confirmed that functional guide RNAs were produced both from the spacer with the canonical repeat structure at the leader-proximal position and the terminal spacer flanked by highly diverged atypical repeats (Figure 3b). Interestingly, guide RNA-mediated transposition occurred at a higher frequency with the ffs-specific spacer even though it contained mismatches and was flanked by atypical repeats (Figure 3b).
Figure 3 – I-F3b Tn6900 element derived from A. salmonicida S44 allows RNA-guided transposition with typical and atypical repeats.
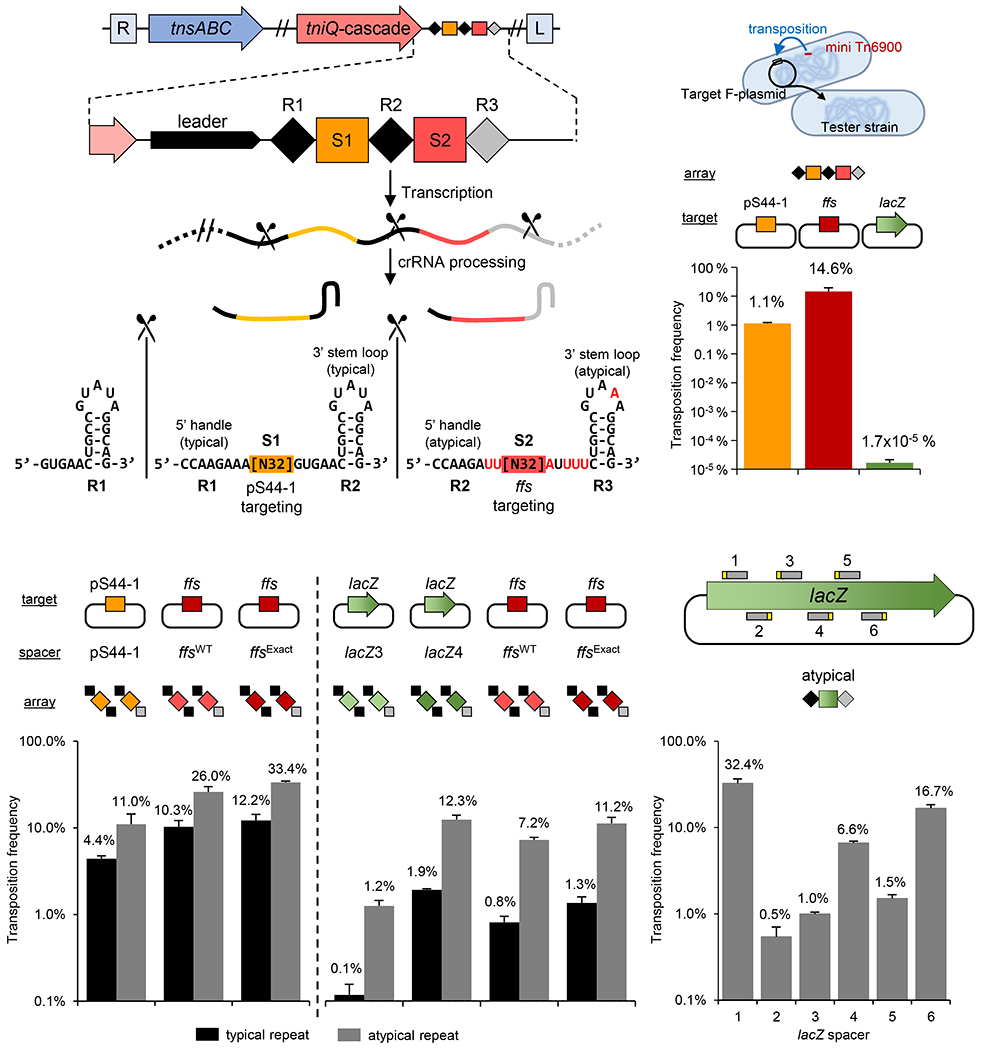
Various transposition targets were tested using the A. salmonicida S44 native array or as individual repeat-spacer-repeat units. (a) Simplified representation of transposition/CRISPR-associated genes, CRISPR array (marked as in Figure 1) and the resulting typical and atypical guide RNAs with the 5’ and 3’ handles indicated. Position of Cas6 processing is indicated (scissors). (b) Frequency of transposition found with the native A. salmonicida S44 array with targets constructed into an F plasmid; A. salmonicida S44 plasmid pS44-1 (pS44-1), chromosomal ffs att site (ffs) or a negative control, lacZ gene. (lacZ) (c-e) Transposition frequency found with a single repeat-spacer-repeat unit in various combinations of spacers with typical or atypical repeats from Tn6900 with the indicated targets constructed into an F plasmid. All data indicate mean +/− standard deviation (n=3).
To test the individual contributions of the spacer, protospacer, and repeat sequences, we designed CRISPR array constructs with leader-proximal (typical) or terminal (atypical) flanking repeats as a single guide RNA expression construct and tested various native and synthetic spacer sequences individually. Not only were the guide RNAs with the atypical repeats functional, but also consistently allowed a higher frequency of transposition when compared to the typical repeat when tested with three different spacers (Figure 3c). Additionally the ffs-specific spacer showed a higher frequency of transposition than the spacer directed at the plasmid target even though the plasmid spacer had a perfect match to its target and the ffs spacer had 10 mismatches to its target (several that were not at the sixth positions that are predicted to be flipped out) (Figure 3c). Altering the native ffs-specific spacer so that it was a perfect match to the ffs protospacer consistently allowed a modestly higher frequency of transposition (Figure 3c–d).
Guide RNA complexes were also designed using spacers matching different positions in lacZ (Figures 3d–e). We found that transposition frequency varied as much as 10-fold with different spacers, even though the sequences recognized all had the same candidate PAM sequence, a result that was not explained by the DNA strand that was targeted in the highly expressed lacZ gene (Figures 3d–e). However, regardless of the spacer tested, a modestly higher transposition frequency was consistently found with guide RNAs with the atypical repeats when compared with typical repeats in the Tn7-CRISPR-Cas system from A. salmonicida S44 (Figures 3c–d). These experiments confirmed that a functional guide RNA complex could be produced from atypical repeats and hinted that the functionality of these complexes may show important differences from typical repeats. We found that multiple different positions could also be targeted in the E. coli chromosome using guide RNA-directed transposition supporting a view that this was not a plasmid-specific process (Supplemental Figure S1d–e).
Atypical repeats form functionally distinct guide RNA complexes with the Tn7-CRISPR-Cas system from A. salmonicida S44
Our experiments suggested that the guide RNA produced from atypical repeats was functional and appeared to allow enhanced transposition activity with the system derived from the Tn6900 element in A. salmonicida S44. To get a better understanding of the relevance of differences in the repeat sequences, we compared the sequences of the leader-proximal typical repeats with the atypical repeats flanking the terminal spacers to look for common trends across the two branches (Figure 4a). In both branches there were common trends in the final repeat encoding the 3’ handle of the guide RNA with a tendency to lose the typical GTG (positions 1-3), a loss of conservation of the region cleaved from the final guide RNA (positions 21-28), and a general enrichment for adenines in the loop (Figure 4a). Functional differences from changes with the typical and atypical repeats were examined with Tn6900 by making changes to the repeat regions encoding the 5’ and 3’ handles of guide RNAs (Figure 4b). While not subject to an extensive analysis, changing the GUG region to an AUU in the 3’ handle of a typical repeat (typical*) or changing the AUU to a GUG in the atypical repeat (atypical*) resulted in only small changes to the frequency of guide RNA-directed transposition (Figure 4b), suggesting these conserved positions are not alone responsible for the atypical repeat frequency advantage and that a more complicated interdependency is at play.
Figure 4 – Analysis of atypical repeat sequences in Tn7-CRISPR-Cas elements, Tn6677 and Tn6900.
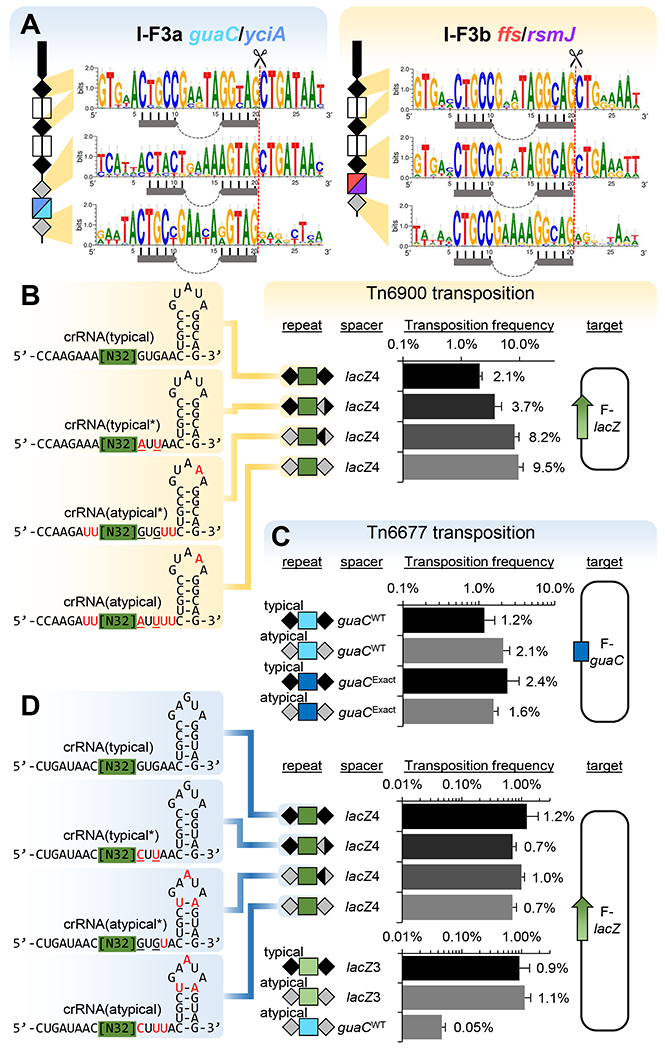
(a) Consensus sequence of the typical and atypical repeats as a function of position in the array. Symbols are as in previous figures with the stem-loop indicated (top, n=85 for I-F3a, n=74 for I-F3b), (middle, n=51 for I-F3a, n=41 for I-F3b) or (bottom, n=51 for I-F3a, n=41 for I-F3b). Frequency of transposition found with changes in typical and atypical guide RNAs for Tn6900 (b) or Tn6677 (c-d) with the indicated spacers and their associated targets. Typical guide RNA were tested, comparing with naturally-occurring changes from atypical repeats (red) or engineered mutations (underlined). All data indicate mean +/− standard deviation (n=3).
Previous work with a different I-F3 Tn7-CRISPR-Cas systems found with the Tn6677 element from Vibrio cholerae HE-45 indicated that guide RNA complexes could be directed to programed target sites in E. coli using guide RNAs (Klompe et al., 2019). The Tn6677 element is in the I-F3a branch of elements and provided a good point of comparison for understanding differences between the two branches of I-F3 Tn7-CRISPR-Cas elements (Figure 1). Tn6677 naturally resides in the att site downstream of guaC and consistent with the trends we identified above, this element carries the att site targeting spacer in a noncontiguous array with an atypical repeat structure (Figure 2). The Tns, Cas, and CRISPR array modules from Tn6677 were constructed under lactose and arabinose expression systems and tested in the transposition assay used for the Tn6900 derivative above (Supplemental Figure S2a). We found that transposition into the native guaC attachment site used by Tn6677 required the guaC-specific guide RNA encoded in the array and that the atypical repeats in Tn6677 were also functional (Figure 4c). However, unlike the Tn6900 derivative, a similar frequency of transposition was found with the typical and atypical arrays (Figure 4c) or with modest changes in the typical and atypical repeats with the Tn6677 element (Figure 4d). Naturally occurring Tn7-like and Tn7-CRISPR-Cas elements control the left to right orientation with which they insert. Consistent with previous work, Tn6677 seemed to be somewhat relaxed for orientation control (Supplemental Figure S2b) (Klompe et al., 2019). The Tn6900 derivative showed the expected bias for one orientation found with canonical Tn7 and found naturally when 24 independent insertions were analyzed (Supplemental Figure S2b). Tn6900 derivative insertions were ~48 bp from the protospacer and occurred with the target site duplication expected with full transposition (Supplemental Figure S2c).
Guide RNAs can be made private to I-F3 transposition by mismatch tolerance and a specialized function with atypical guide RNAs
A question not previously addressed with Tn7-CRISPR-Cas systems involves possible cross-talk between CRISPR arrays with other type I-F CRISPR-Cas systems. If the CRISPR array from a Tn7-CRISPR-Cas element with the guide RNAs specific to the chromosome could be used by a standard I-F1 system, the chromosome att site would be a target for degradation. This could limit the spread of I-F3 Tn7-CRISPR-Cas elements if it entered a new host that encoded a standard I-F1 CRISPR-Cas system. We investigated if the typical and atypical guide RNAs encoded in I-F3 CRISPR arrays could be accessed by a I-F1 system (Chowdhury et al., 2017). In our work testing the P. aerigunosa system we co-expressed the Cas proteins and a single spacer CRISPR array with a T7 expression system (Vorontsova et al., 2015). Repeats from the type I-F1 system from P. aerigunosa, the type I-F3a V. cholerae Tn6677 system or the type I-F3b system derived from A. salmonicida Tn6900 were examined using a transformation efficiency assay examining plasmids with and without a protospacer. In control experiments we observed robust interference using the I-F1 CRISPR-Cas system from P. aeruginosa PA14 (Figure 5). Transformation was decreased over three orders of magnitude with the plasmid encoding a protospacer compared to a plasmid that lacked the protospacer. Similarly, the typical repeats of the I-F3 systems from Tn6677 and Tn6900 also allowed robust interference with the plasmid transformation assay when they contained an exact match to the protospacer in the plasmid. This was not unexpected because the repeats from the canonical I-F1 and I-F3 Tn7-CRISPR-Cas systems are similar (Supplemental Figure S3) and it is likely that the I-F3 Tn7-CRISPR-Cas systems rely on standard I-F1 systems for spacer acquisition.
Figure 5 – P. aeruginosa type I-F1 Cascade can utilize heterologous I-F3 CRISPR arrays in a plasmid interference assay, but mismatches and I-F3b atypical guide RNAs allow privatization.
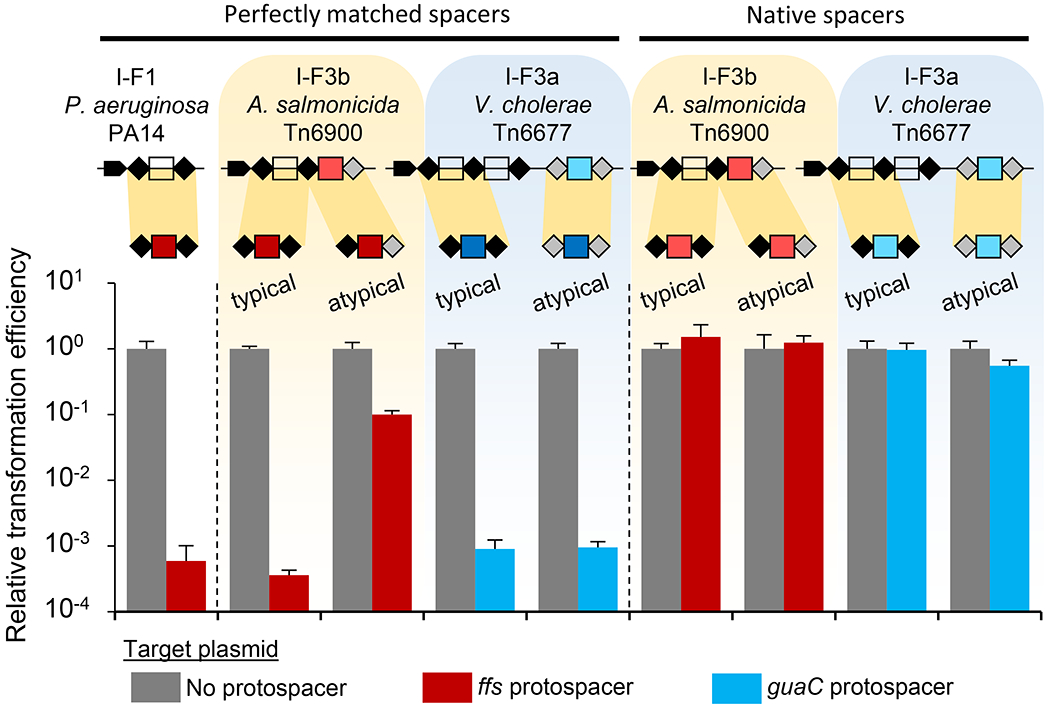
Expression of P. aeruginosa Cas proteins with various arrays reduces transformation efficiency for protospacer containing plasmid, but not control. Single unit arrays from P. aeruginosa PA14 and A. salmonicida S44 Tn6900 with ffs spacer, and V. cholerae HE-45 Tn6677 with guaC spacer were tested in typical and atypical repeat configurations as indicated. Spacers were either perfectly matched to protospacer or contained native mismatches. Repeat configuration sequences are presented in Supplemental Figure S3b. All data indicate mean +/− standard deviation (n=3).
We also tested the tolerance for mismatches in the I-F1 interference system, based on the observation that mismatches were common in the att site guide RNAs found in the Tn7-CRISPR-Cas systems (Supplementary Table S1). While the native mismatches had little or no effect on the ability to function for guide RNA-directed transposition with the Tn6900 derivative with 10 mismatches (Figure 3c) and Tn6677 element with 7 mismatches (Figure 4c), these same guide RNAs had a profound effect on interference with the I-F1 CRISPR-Cas system from P. aeruginosa PA14, allowing no observable interference in the transformation assay (Figure 5). This indicates a form of privatization, where mismatches with the guide RNA have minimal or no impact on guide RNA-directed transposition, they are rendered unusable by the I-F1 system tested in our work.
We also determined if the specialized atypical guide RNAs can be used by a canonical I-F1 system. When a spacer was situated with atypical repeats from the I-F3b system found with the Tn6900 derivative, guide RNA complexes formed with atypical repeats were drastically reduced in their ability to function for interference in the plasmid transformation assay, even with a perfect spacer-protospacer match (Figure 5). The compromised use with the atypical repeats for interference was in contrast to the enhanced use we found for guide RNA-directed transposition with the I-F3b system (Figures 3 and 4). This result indicates a second mechanism that would allow chromosomal-targeting spacers to be tolerated in hosts with standard I-F1 CRISPR-Cas systems by allowing them to remain private to the I-F3b system. This privatization was absent in the Tn6677 I-F3a system from V. cholerae. With the I-F3a Vc system, robust interference was found with either the typical repeats or the highly diverged atypical array from this element. However, the results below suggest that I-F3a Tn7-CRISPR-Cas elements can use a separate transcription network to help tolerate self-targeting spacers.
I-F3 elements utilize Xre-family transcriptional regulators to regulate CRISPR-Cas components
To better understand I-F3 Tn7-CRISPR-Cas element dissemination, we searched for genes conserved among diverse members of this group. One of the other genes found conserved across I-F3 Tn7-CRISPR-Cas elements were predicted Xre-family transcriptional regulators. The xre gene resides at a conserved position between the tnsABC and tniQ-cas8/5,7,6 operons in nearly all I-F3 elements (Figure 2a). While each of the two branches of I-F3 elements have xre genes, the predicted regulatory gene in each branch segregated with phylogenetically distinct families of controller (C) proteins associated with restriction-modification systems. I-F3a elements have a 68 amino acid Xre protein related to C.AhdI and I-F3b elements have a ~100 amino acid Xre protein related to C.Csp231I (Supplemental Figure S4a). Candidate regulatory features could also be identified with the tniQ-cas and CRISPR arrays based on homology with the previously established systems (Supplemental Figure S4b, see below)(Streeter et al., 2004).
We analyzed putative promoter regions in I-F3a elements and discovered candidate sites for Xre-mediated regulation upstream of xre as well as directly upstream of the att-targeting spacer in Tn6677 and other members of this branch of elements (Figure 6a). The regulatory regions were confirmed in vitro with two elements in the I-F3a branch, V. cholerae HE-45 Tn6677 (Vc) and V. parahaemolyticus RIMD221063 (Vp)(Figure 6c). A functional role for this interaction was shown by a LacZ reporter assay. Xre was found to autoregulate its own pXre promoter, which allowed minimal transcription without Xre, was activated by low amounts of Xre, and repressed as the expression of Xre increased (Figure 6e). Meanwhile the promoter identified for the att-targeting spacer (pAttGuide) was highly expressed when Xre was not present and increasingly repressed with increasing amounts of Xre induction (Figure 6e). As predicted and shown below, this system provides a burst of the atypical guide RNA that is specific to the guaC or yciA att sites with I-F3a elements upon entry into a new host via zygotic induction.
Figure 6 – Xre proteins regulate components of RNA-guided transposition.
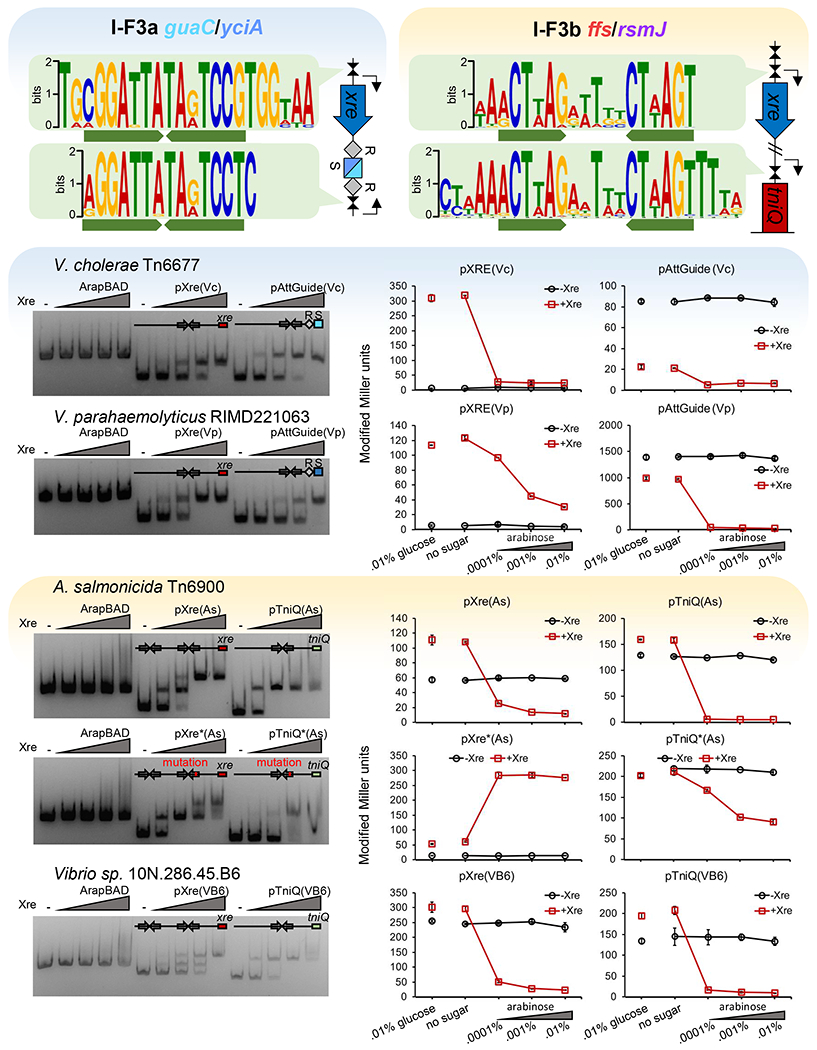
(a-b) Consensus sequence for putative Xre binding motifs in I-F3a and I-Fb elements.
(c-d) Xre-binding resolved by EMSA. DNA fragments with the transcription control regions were incubated with increasing amounts of Xre protein from the respective element before electrophoresis (100 nM DNA; protein:DNA ratios = 0,2,5,10,20:1). (e-f) Promoter function resolved by LacZ expression monitored by Miller units at various arabinose controlled Xre expression levels. Transcription control regions are indicated from V. cholerae HE-45 Tn6677 (Vc), V. parahaemolyticus RIMD221063 (Vp). A. salmonicida S44 Tn6900 (As), and Vibrio sp. 10N.286.45.B6 (VB6).
I-F3b elements were also surveyed for inverted repeat motifs to investigate the functional role of the conserved C.Csp231I-like Xre regulator. Like I-F3a elements, conserved motifs were found in the promoter region of xre that were nearly identical to those used by C.Csp231I (Figure 6b, Supplemental Figure S4b)(McGeehan et al., 2011). Unlike the I-F3a elements, the conserved motif could not be identified upstream of the CRISPR arrays with the I-F3b elements, and instead we found a single copy of this motif upstream of the tniQ-cas8/5,7,6 operon (Figures 2a and 6b). The regulatory regions were confirmed in vitro with two I-F3b elements from A. salmonicida S44 Tn6900 (As) and Vibrio sp. 10N.286.45.B6 (VB6). Binding to the two predicted motifs in the upstream region of Xre could be visualized as two separately migrating species (Figure 6d). Mutating the xre-proximal regulatory motif weakened interaction as demonstrated by the higher concentration of protein required to achieve a full mobility shift (Figure 6d). We additionally visualized interaction with the motif upstream of tniQ-cas8/5,7,6 and confirmed the sequence-specific nature of binding by utilizing a mutated motif which weakened interaction. LacZ reporter assays were again used to confirm a functional role in regulation. Xre regulator was shown to act as a repressor of its own pXre promoter (Figure 6f). Interestingly, mutation of the proximal binding site which impaired binding in vitro resulted in Xre regulator instead acting as an activator, suggesting interaction with the distal site activates transcription while interaction with the proximal site represses it (Figure 6f). Similar to the result with the I-F3a elements, the Xre regulator was able to repress tniQ-cas8/5,7,6 expression and this repression was impaired by mutation of the conserved binding motif (Figure 6f).
An additional assay was used to confirm zygotic induction following conjugal transfer of regulatory regions with examples from both the I-F3a and I-F3b systems. Consistent with the biochemistry and expression control data with the Xre proteins found in Tn7-CRISPR-Cas elements and previously literature with controller proteins, the Xre proteins allow tight repression in an established donor and a strong burst of expression when transmitted into a new recipient (Figure 7). Recipient strains expressing Xre regulators are immunized from this expression burst following conjugation. To signify the Xre-dependent control demonstrated with CRISPR-Cas promoters in Tn7-CRISPR-Cas elements we propose naming the xre genes rtaC and rtbC (RNA-guided transposon/transposition I-F3a or I-F3b csontroller).
Figure 7 -. Xre regulation program allows zygotic induction of transposition function promoters in a new host after conjugal transfer.
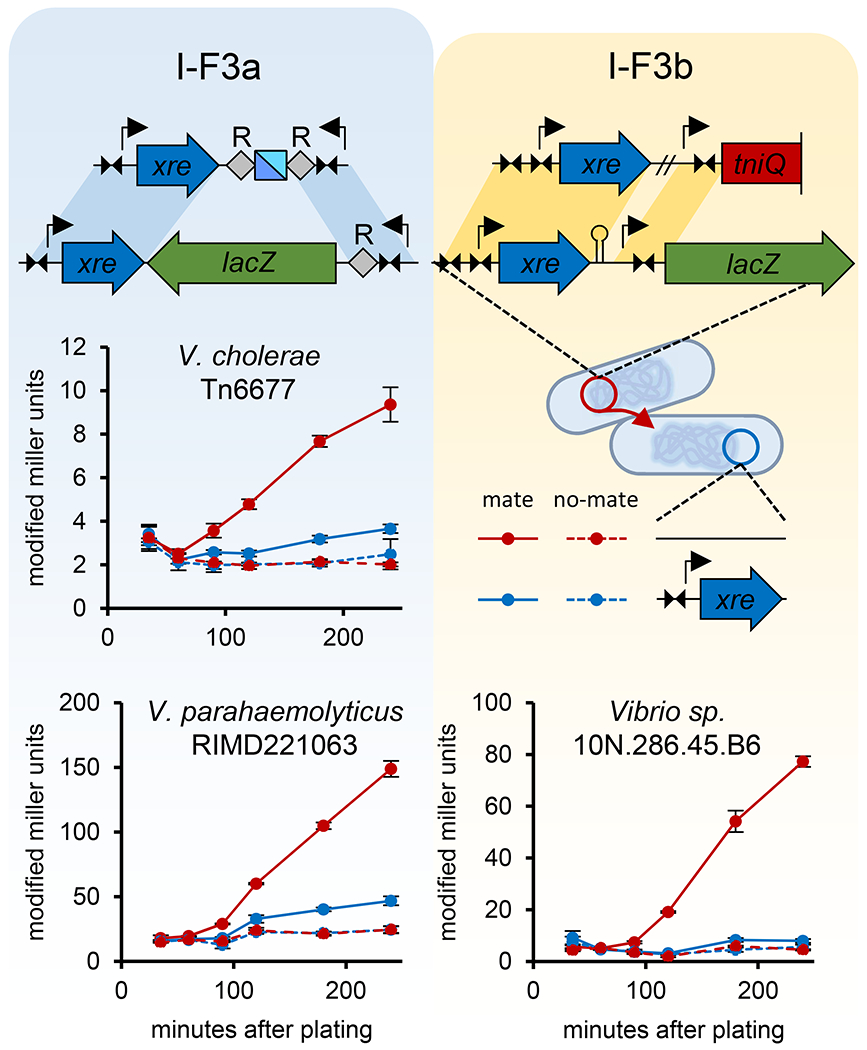
Transfer of lacZ fused to Xre transcriptional control regions results in a burst of expression in a naive recipient strain, but not recipients expressing the Xre regulator protein. Donor and recipient were plated together for mating (solid lines) or plated separately (dashed lines) as a control. Cells were harvested and controls mixed and LacZ expression monitored by Miller units as indicated. All data indicate mean +/− standard deviation (n=3).
Discussion
Our findings indicate that the major family of Tn7-CRISPR-Cas systems have evolved a categorization system using conventional and specialized guide RNAs. We find that all transposition can be accounted for by spacers in the element-encoded CRISPR array (Figure 1) and that transposition with members of both major branches of these elements require guide RNAs for att-targeted transposition (Figures 3 and 4). We also show that the spacers used to target chromosomal sites show certain characteristics; in addition to residing in the last position in the array (Figure 1), they are flanked by highly diverged repeats (Figure 2) and maintain mismatches that show little or no impact on guide RNA-directed transposition (Figures 3c and 4c), but that render them unusable for interreference with a conventional 1-F1 system (Figure 5). The repeat divergence appears to be specifically beneficial for a type I-F3b element from A. salmonicida S44, as these atypical guide RNA complexes are almost completely unusable for I-F1-mediated interference even when perfectly matched to a target (Figure 5), while allowing for a higher level of transposition than found with the typical guide RNAs (Figure 3). We show that each of the branches of I-F3 Tn7-CRISPR-Cas systems have separately coopted Xre family regulators (Supplementary Figure S4) that allow a burst of expression specifically for guide RNAs directing transposition into chromosomal att sites when they enter a new host (Figure 6 and 7). The transposon-encoded guide RNAs that allow long-term memory to direct transposition into chromosomal sites are therefore privatized to the transposon-adapted I-F3 system using mismatch tolerance, specialized atypical guide RNAs, and selective regulation to guard against toxic self-targeting by canonical CRISPR-Cas defense systems.
A significant consequence of I-F3 Tn7-CRISPR-Cas elements accessing the chromosome using an element-encoded guide RNA is the hazard of canonical I-F1 CRISPR-Cas systems using the self-targeting guide RNA. Self-targeting spacers that are accessed by other canonical CRISPR-Cas systems would be a liability whenever the element entered a host that already had this family of CRISPR-Cas systems. To this point, we find that a I-F1 system can use typical guide RNAs encoded in I-F3 CRISPR arrays in interference (Figure 5). Standard I-F1 systems are found in Vibrionales and Aeromonadales and with our analysis we found multiple specific examples where I-F3 Tn7-CRISPR-Cas elements reside in strains that have standard I-F1 systems (Supplemental Table S1). Self-targeting spacers have been identified in the work of others and have even been used as a tool to help identify anti-CRISPR systems (Borges et al., 2017). However, established members of anti-CRISPR systems are not widely conserved in elements with Tn7-CRISPR-Cas systems in our analysis (Wiegand et al., 2020).
Mismatch tolerance likely forms a major mechanism of privatization in I-F3 systems. We suspect that mismatches accumulate in I-F3 guide RNAs that recognize chromosomal positions due to strong negative selection when they are found in a host with a I-F1 system. It is notable that 94% of the diverse I-F3 elements reside in only four different chromosomal att sites indicating that acquiring new sites occurs very rarely. This is likely explained by the many factors that would theoretically limit evolving new chromosomal att site. For example, initial acquisition of spacers from the chromosome is rare and to become fixed in the population likely demands a spacer that recognizes a highly conserved gene at a position near one end of the gene to ensure that the gene itself is not inactivated by transposition. As expected, three of the four major att sites are associated with known essential genes (guaC, rsmJ, and ffs), while the function for the fourth is unknown (yciA). Guide RNAs that target protein coding genes show a concentration for mismatches at the 3rd positions coincident with the wobble positions (Supplemental Figure S5). Mismatches are known to be tolerated at the 6th positions in I-F systems, but the tolerance for mismatches at the 3rd positions appears to be a specific adaptation for cascade systems used for transposition (Figure 5). Mismatch tolerance is likely to be an insufficient protection from self-targeting spacers with diverse I-F1 systems as other mechanisms also contribute to this process (see below).
In the case of the I-F3b system from A. salmonicida S44, the atypical repeat appears to be a specific adaptation that allows a higher frequency of guide RNA targeted transposition (Figures 3 and 4) and privatization from a canonical I-F1 interference system (Figure 5). It is unresolved from our work how atypical guide RNAs in the I-F3b system can be private from the I-F1 system, but could result from poor processing by the canonical system. A better understanding of what allows atypical guide RNAs to function differently in the I-F3b element derived from A. salmonicida should offer avenues for optimizing guide RNA-directed transposition in future work by modifying the repeats. Interestingly, the type I-F3a Tn7-CRISPR-Cas system from Tn6677 did not show enhanced transposition with the atypical array found in this system; the frequency of transposition was the same with the typical and the highly diverged atypical repeat (Figures 4c and 4d). Mismatch tolerance and the regulator program revealed in our work are likely important protections from self-targeting spacers given that highly diverged repeats from Tn6677 were readily used by a canonical I-F1 interference system (Figure 5).
We suspect that there are multiple distinct ways that I-F3 Cas proteins have adapted to recognize specialized atypical repeats based on our bioinformatics. Interestingly, we notice that in one subbranch within the I-F3b elements the final spacer is truncated by 10 to 12 base pairs in length (Figure 1 and Supplemental Figure S6). These smaller spacers produce functional guide RNAs as predicted by the commensurate natural repositioning of the insertion closer to the protospacer (Supplemental Figure S6). Previous work in closely related CRISPR-Cas systems suggests that guide RNAs of this length will not be functional for targeting transposition nor robust interference (Klompe et al., 2019; Kuznedelov et al., 2016). However, the ability of the I-F3b systems to accommodate shorter guide RNAs could provide another mechanism of privatization from other I-F CRISPR-Cas systems. Intriguingly, naturally occurring minimal type I-F2 CRISPR-Cas systems tested in the laboratory are not functional for interference with similarly truncated guide RNAs, but can still form complexes capable of forming R-loops to matching protospacers (Gleditzsch et al., 2016).
Both branches of I-F3 elements use Xre (RtaC/RtbC) proteins for “zygotic” induction where the gene program is reset when they enter a new host that lacks the regulatory protein (Figures 6 and 7). This type of regulatory scheme is important for related controller (C) proteins with some restriction-modification systems to ensure that the new host is methylated before expression of the restriction component (Rodic et al., 2017). Using the I-F3a elements tested in our experiments as an example, the att-targeting spacer, guaC or yciA is tightly repressed in Xre (RtaC)-containing donor cells, but derepressed upon entering a new host (Figure 6 and 7). This expression profile would program the Tn7-CRISPR-Cas element to transpose into the neutral chromosomal att sites upon entry into the new host. The strong repression (as demonstrated in donor cells) then could help guard against potential self-targeting with this guide RNA in the presence of endogenous I-F1 systems. Presumably a basal expression level from the primary CRISPR array would allow the production of sufficient guide RNA complexes to recognize mobile elements to allow horizontal transfer of I-F3a elements into new hosts.
In the I-F3b elements, the TniQ-Cascade operon itself is subject to Xre(RtbC) regulation. Presumably this bolus of expression would facilitate transposition into the chromosomal ffs or rsmJ att site. In CRISPR-Cas systems, the total number of effective guide RNA complexes will be a function of the amount of cascade components as related to the number of different spacers and their expression levels (Martynov et al., 2017). Therefore, the I-F3b systems like the ones found with Tn6900 may draw additional benefit by swamping the cell with effector complexes to overwhelm any I-F1 systems potentially found in a new host. While I-F3b elements may not generally rely on transcriptional regulation to privatize the self-targeting spacer from any endogenous CRISPR-Cas system, they have nonetheless coopted a similar regulatory pathway to activate transposition function in a new host.
The outlier element found in Parashewanella curva C51 provides an interesting alternative hybrid two-pathway strategy from the other elements using the I-F3 CRISPR-Cas system (Figure 1). Unlike the other I-F3 family elements indicated in Figure 1, this element does not have a spacer that could form a guide RNA complex to recognize where it inserted in the chromosome. Interestingly, this element is from a small group of representatives that encode two TniQ proteins. One TniQ is associated with the Cascade operon and the second is similar to a TniQ protein found in other Tn7-like elements lacking CRISPR-Cas components that reside in an attachment site downstream of theparE gene encoding the essential and conserved toposiomerse IV, subunit B (Supplemental Figure S7). These elements are predicted to use guide RNA-directed transposition to recognize mobile elements, but presumably continue to rely on the mechanism used by prototypic Tn7 and most other Tn7-like elements (Peters, 2014), in this case with the TniQ protein recognizing the parE gene coding region. In two previously identified instances where type I-B CRISPR-Cas systems were captured by other Tn7-like members the elements have a similar configuration with two tniQ genes and likely function in a similar way to allow a two pathway life-style (Peters et al., 2017). It is unclear how Tn7-CRISPR-Cas elements with a type V-K CRISPR-Cas systems control the use of two pathways given these elements only encode a single tniQ gene (Faure et al., 2019b).
It will be interesting to learn how the adaptations found in I-F3 Tn7-CRISPR-Cas systems extend to canonical CRISPR-Cas systems and in other cases where CRISPR-Cas systems have been coopted for new functions. The examples shown here join numerous others where it is now clear that CRISPR-Cas activity is regulated to offset potential negative impacts of these potent systems (Hoyland-Kroghsbo et al., 2017; Patterson et al., 2016; Westra et al., 2010). Transposition levels with the I-F3b system developed in our work is robust, occurring in as much as one third of the cell population, suggesting that it should be a good candidate for future application-based work. The new information with atypical guide RNAs also suggests additional avenues for altering guide RNA activity for practical applications in Tn7-CRISPR-Cas systems or potentially CRISPR-Cas systems in general.
STAR Methods
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joseph E. Peters (joe.peters@cornell.edu).
Materials Availability
Select plasmids listed in the Key Resources Table generated in this study are being deposited in Addgene.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Escherichia coli DH5α | J. Peters Lab | N/A |
| Escherichia coli BW27783 | Coli Genetic Stock Center (CGSC), (Khlebnikov et al., 2002) | CGSC#12119 |
| Escherichia coli EMG2 | CGSC | CGSC#4401 |
| Escherichia coli BL21(DE3) | Novagen | Cat# 69450 |
| Escherichia coli BW27783 attTn7::miniTn7(miniTn6900(kanR)) | This paper | MTP997 |
| Escherichia coli BW27783 attTn7::miniTn7(miniTn6900(kanR)) ΔlacZ4787(::rrnB-3)::lacZ | This paper | MTP1191 |
| Escherichia coli BW27783 attTn7::miniTn7(miniTn6677(kanR)) | This paper | MTP1196 |
| Escherichia coli BL21-AI | Invitrogen | Cat# C607003 |
| Escherichia coli S17-1 lacZ::miniTn7(genR) | This paper | PO429 |
| Escherichia coli CW51 | J. Peters Lab, (Waddell and Craig, 1988) | JP606 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Purified proteins : Xre(Vc), Xre(Vp), Xre(As), Xre(VB6) | This paper | N/A |
| Critical Commercial Assays | ||
| Q5® High-Fidelity DNA polymerase | NEB | M0491L |
| NEBuilder® HiFi DNA Assembly Master Mix | NEB | E2621L |
| HiTrap Chelating HP | GE Healthcare | Cat# 17-0409-03 |
| Oligonucleotides | ||
| For cloning and other use in this study | This paper | See Table S3 |
| Recombinant DNA | ||
| pMS26_miniTn6900(kanR) | This paper | pMTP112, Addgene |
| pTA106_TnsABC(Tn6900) | This paper | pMTP130, Addgene |
| pBAD322G_TniQ-Cascade(Tn6900) | This paper | pMTP140, Addgene |
| pBAD33_CRISPR(Tn6900)-entry | This paper | pMTP170, Addgene |
| All other plasmids used and constructed in this study | This paper | See Table S2 |
| Synthesized gene fragments used in this study | This paper | See Table S4 |
| Software and Algorithms | ||
| HMMER3 | (Johnson et al., 2010) | http://hmmer.org/ |
| Biopython | (Cock et al., 2009) | https://biopython.org/ |
| Infernal | (Nawrocki et al., 2013) | http://infernal.janelia.org |
| Cd-hit | (Li et al., 2006) | https://github.com/weizhongli/cdhit |
| MUSCLE | (Edgar, 2004) | http://www.drive5.com/muscle |
| FastTree | (Price et al., 2009) | http://microbesonline.org/fasttree |
| ETEToolkit | (Huerta-Cepas et al., 2016) | http://etetoolkit.org |
| BBTools | BBMap, Bushnell B. | http://sourceforge.net/projects/bbmap/ |
| MEME | (Bailey et al., 2009) | http://meme.nbcr.net |
| WebLogo 3 | (Crooks et al., 2004) | http://weblogo.threeplusone.com/ |
Data and Code Availability
The dataset of I-F3 Tn7-CRISPR-Cas elements generated during this study is available as Table S1. This study did not generate any unique code.
Experimental Model and Subject Details
Escherichia coli strains were grown at 30 or 37°C in lysogeny broth (LB) or on LB agar (unless stated otherwise in the Method Details) supplemented with the following concentrations of antibiotics when appropriate: 100 μg/mL carbenicillin, 10 μg/mL gentamicin, 30 μg/mL chloramphenicol, 8 μg/mL tetracycline, 50 μg/mL kanamycin, 100 μg/mL spectinomycin.
Method Details
Identifying type I-F CRISPR-guided Tn7-like transposons
Annotated protein fasta files, genomic sequences and feature tables of Gammaproteobacteria were downloaded from National Center for Biotechnology Information (NCBI) FTP site on September 22, 2019. In total, there were 53,079 genomes for analysis.
Profile HMMs associated with TnsA (PF08722,PF08721), TnsB (PF00665), TnsC (PF11426,PF05621), TniQ(PF06527), Cas5f(PF09614), Cas6f(PF09618), Cas7f(PF09615) and Xre family proteins(PF01381), which can be downloaded from The European Bioinformatics Institute (EMBL-EBI) Pfam database, were used for detecting homologs with hmmsearch (HMMER3).
Candidate proteins were grouped into tnsABC operons and tniQ-cas operon based on their orientation and proximity. Then each tnsABC operon was grouped with its downstream tniQ-cas operon into one transposon functional unit. The Xre/HTH (helix turn helix) proteins situated between the two operons and are homologous to restriction controller proteins (blastp, identity >40%) were defined as candidate regulators.
CRISPR array detection
Manually curated CRISPR repeats of Tn7-CRISPR-Cas elements were used to create a DNA sequence profile, which was used as a query for nhmmscan searches (HMMER3) to find CRISPR repeats in the downstream 20-kb region of cas6. Putative repeats were grouped into arrays by their distances to each other. The distance between repeats was required to be >55 bp and <65 bp, the bit-score threshold is −1. The distance between last repeat and previous repeat was allowed to be between 43 bp and 55 bp, but in such cases its bit-score had to be >=0.3. The sum of bit-scores of repeats in an array cannot be lower than 6.0. The longest non-overlapping arrays are collected as putative CRISPR arrays. All repeats besides the final repeat from the first array downstream of cas6 were used to create an updated repeat profile, and the CRISPR detection procedure was repeated with the new profile twice.
Protospacer detection
To detect protospacers that match the transposon-associated CRISPR spacers, each spacer was converted into position-specific scoring matrix (PSSM) and used to search upstream 1-kb DNA of tnsA for matches with Biopython (threshold=11.0). Because every 6th base of spacers is flipped out in type I CRISPR Cascade complex, all 6th positions of the matrix are set to have equal weight on all four bases.
Except for ffs (SRP-RNA), the major attachment site genes that containing the candidate protospacers are classified with the annotations provided in NCBI. The attachment site SRP-RNA gene (ffs) is often poorly annotated, so it was reannotated using cmsearch (Infernal) and SRP-RNA profile (RF00169) available on RFAM (https://rfam.xfam.org/).
Constructing similarity trees
The TnsA, TniQ and Xre proteins were clustered using Cd-hit with identity threshold set to 90%. Multiple alignments of the representatives were done with MUSCLE. Similarity trees were made with FastTree using WAG evolutionary model and the discrete gamma model with 20 rate categories as previously described (Peters et al., 2017). The visualization of the trees, major attachment sites, CRISPR arrays and matched spacers was done with ETEToolkit.
Identifying shared promoter motifs of xre and CRISPR-Cas genes
The transposons were classified into two groups based on associated xre lengths (68 a.a. for I-F3a or ~100 a.a. for I-F3b) and similarities to C.AhdI and C.Csp231I. For each group, the 100bp upstream of xre, second CRISPR array, and tniQ-cas operon were collected and deduplicated with dedupe.sh (BBTools) with threshold of 70% identity or 30 edit distance. The sequences were then sent to MEME for motif detection and comparison.
Comparing consensus CRISPR repeat sequences of chromosome targeting spacers to those of other spacers
To make consensus sequences of CRISPR repeats, the transposon representatives with non-redundant TniQ were selected with Cd-hit and separated into two groups based on their attachment sites being ffs/rsmJ or guaC/yciA. The upstream and downstream CRISPR repeats of the chromosome targeting spacers, and repeats not flanking chromosome targeting spacers were collected and sequence logos created using WebLogo 3.
Transposition assays
All transposition assays were performed in MTP1191, or one of MTP997 or MTP1196 with an F plasmid derivative as indicated in Table S2.
For Tn6900 transposition, strains used to monitor transposition were made competent by growing cells in LB media to mid-log, washed, and resuspended in ice cold a 0.1M CaCl2 solution (Peters, 2007) and transformed with pMTP130, pMTP140, and a derivative of pMTP150, pMTP160, pMTP170, or pMTP190 as indicated in Table S2 onto LB agar supplemented with 100 μg/mL carbenicillin, 10 μg/mL gentamicin, 30 μg/mL chloramphenicol, and 0.2% w/v glucose. After 16 hours incubation at 37°C, several hundred transformants were washed up in M9 minimal media (Peters, 2007) supplemented with 0.2% w/v maltose and diluted to a calculated OD = 0.2 in M9 supplemented with 100 μg/mL carbenicillin, 10 μg/mL gentamicin, 30 μg/mL chloramphenicol, 0.2% w/v arabinose, and 100 μM IPTG to induce transposition.
For experiments monitoring transposition frequency through loss of sugar metabolism on MacConkey’s media, induction pools were incubated for 24 hours with shaking at 30°C before being serially diluted in LB and plated on MacConkeys 1% w/v lactose, sorbitol, or galactose. Plates were incubated at 37°C for 16 hours before colonies were counted.
For experiments monitoring transposition frequency by the mate out assay (Supplemental Figure 2a), after 24 hours incubation with shaking at 30°C, a portion of induced cultures were washed once and resuspended in LB supplemented with 0.2% w/v glucose. After 2 hours incubation at 37°C induced pools were mixed with prepared mid-log CW51 recipient strain at a ratio of 1:5 donor:recipient and incubated with gentle agitation for 90 minutes at 37°C to allow mating. After incubation cultures were vortexed, placed on ice, then serially diluted in LB 0.2% w/v glucose and plated on LB supplemented with 20 μg/mL nalidixic acid, 100 μg/mL rifampicin, 100 μg/mL spectinomycin, 50 μg/mL X-gal, with or without 50 μg/mL kanamycin to sample the entire transconjugant population or select for transposition respectively. Plates were incubated at 37°C for 36 hours before colonies were counted.
Tn6677 transposition assays were performed as above with function plasmids pMTP230, pMTP240, and a derivative of pMTP250, pMTP260, or pMTP270 as indicated in Table S2 with the exception of 8 μg/mL tetracycline replacing gentamicin when present.
In all experiments, non-target controls where the spacer did not match the target F plasmid were used, with transposition frequency similar to non-target rate in Figure 3B for A. salmonicida S44 transposition, or Figure 4D for Tn6677 transposition.
Screen orientation of transposition events
Individually isolated CW51 transconjugants with mini-element insertions from the mate out assay were purified on LB supplemented with 20 μg/mL nalidixic acid, 100 μg/mL rifampicin, 100 μg/mL spectinomycin, 50 μg/mL X-gal, and 50 μg/mL kanamycin. Colony PCR was performed using primer set A (JEP1386+JEP1958) or primer set B (JEP1387+JEP1958) to capture position and orientation of insertion events.
P. aeruginosa CRISPR interference assays
All interference assays were performed in BL21-AI. BL21-AI made electro competent by growing cells in LB media to mid-log, washed, and resuspended in ice cold water (Peters, 2007) and transformed with pOPO322, pCsy_complex, and a derivative of pCOLADuet-1 as indicated in Table S2 onto LB agar supplemented with 100 μg/mL carbenicillin, 100 μg/mL spectinomycin, 30 .μg/mL chloramphenicol, and 0.2% w/v glucose. Overnight cultures grown in LB agar supplemented with 100 μg/mL carbenicillin, 100 μg/mL spectinomycin, 30 μg/mL chloramphenicol were diluted 1:50 in LB supplemented with 100 μg/mL carbenicillin, 100 μg/mL spectinomycin, 30 μg/mL chloramphenicol, 100 μM IPTG and 1 mM arabinose. Cultures were grown to OD = 0.4 before electrocompetent cells were prepared by standard methods (Peters, 2007) and transformed with 1 ng pOPO275 or pOPO390. Cells were recovered in SOC at 37°C for one hour before being serially diluted and plated on LB supplemented with 100 μg/mL carbenicillin, 50 μg/mL kanamycin, 30 μg/mL chloramphenicol, and 100 μg/mL spectinomycin. Plates were incubated at 37°C for 16 hours before colonies were counted.
Xre protein purification
pOPO223, pOPO239, pOPO331 or pOPO360 were transformed into BL21 (DE3), which was cultured in Terrific Broth at 37°C and induced with 0.1mM IPTG during log-phase. Cells were cultured an additional 12-16 hours at 18°C before being collected with centrifugation and lysed by sonication in nickel buffer (20 mM HEPES–NaOH (pH 7.5), 500 mM NaCl, 30 mM imidazole, 5% (v/v) glycerol, 5 mM β-mercaptoethanol) supplemented with 0.15 mg/mL lysozyme. Lysate was cleared by centrifugation and loaded on Nickel-NTA column, washed with nickel buffer, and eluted over a 30 mM to 500 mM imidazole gradient in nickel buffer. Selected purified fractions were pooled, dialyzed and buffer exchanged into storage buffer (20 mM HEPES–NaOH (pH 7.5), 100 mM KCl, 5% (v/v) glycerol, 1mM DTT). The purified proteins were snap-frozen with liquid nitrogen and stored at −80°C.
Electrophoretic mobility shift assay (EMSA)
The promoter fragments of putative Xre regulated genes and their mutated variants were PCR amplified and purified. 100 nM DNA was incubated with different amounts of purified Xre proteins in equilibrium buffer (50 mM Tris–HCl (pH 8.0), 1 mM DTT, 10 mM MgCl2) at 25°C for 20 minutes then mixed with glycerol (final concentration 6%). EMSAs were performed in 6% non-denaturing TBE PAGE (Polyacrylamide gel) with 0.5x TBE as running buffer, running at 80V for one hour at room temperature. The gels were EtBr stained and visualized with UV imager.
DNA substrates were produced as follows: ArapBAD was amplified from pBAD24 (JEP175+JEP1364), pXre(Vp) and pAttguide(Vp) were amplified from V. parahaemolyticus RIMD221063 (JEP1956+JEP1957, pXre(Vp); JEP1954+JEP1955, pAttguide(Vp)), pXre(Vc) and pAttguide(Vc) were amplified from gBlock11 (JEP29+JEP30, pXre(Vc); JEP1553+JEP82, pAttguide(Vc)), pXre(As) was amplified from pOPO08 (JEP1321+JEP81), pTniQ(As) was amplified from pOPO09 (JEP1322+JEP81), pXre*(As) was amplified from pOPO10 (JEP1321+JEP81)., pTniQ*(As) was amplified from pOPO11 (JEP1322+JEP81)., pXre(VB6) was amplified from pOPO06 (JEP1553+JEP81), and pTniQ(VB6) was amplified from pOPO07 (JEP1554+JEP81).
In vivo promoter assay
pOPO256, pOPO258, pOPO364, or pOPO345 and a derivative of pOPO221 as indicated in Table S2 were transformed into BW27783 made competent by growing cells in LB media to mid-log, washed, and resuspended in ice cold a 0.1M CaCl2 solution (Peters, 2007) on LB agar supplemented with 100 μg/mL carbenicillin and 30 μg/mL chloramphenicol. Overnight cultures grown in LB supplemented with 100 μg/mL carbenicillin and 30 μg/mL chloramphenicol were diluted 1:100 into LB supplemented with 100 μg/mL carbenicillin, 30 μg/mL chloramphenicol and various concentrations of glucose or arabinose as indicated in Figure 6 and cultured for an additional 20 hours at 30°C. The LacZ activities were measured with standard Miller unit assay (Malke, 1993).
Zygotic induction assay
PO429 was made competent by growing cells in LB media to mid-log, washed, and resuspended in ice cold a 0.1M CaCl2 solution (Peters, 2007) and transformed with one of pOPO392, pOPO394, or pOPO435 on LB agar supplemented with 50 μg/mL kanamycin to produce donor strains. DH5α was made competent by growing cells in LB media to mid-log, washed, and resuspended in ice cold a 0.1M CaCl2 solution (Peters, 2007) and transformed with pETDuet-1, pOPO395, pOPO397 or pOPO438 on LB agar supplemented with 100 μg/mL carbenicillin to produce recipient strains. Overnight cultures of donors and recipients grown in LB supplemented with appropriate antibiotics were diluted 1:10 in the same media and grown for two hours, then washed with LB three times to remove antibiotics. Donors and recipient strains were mixed at a 1:2 ratio and spotted on LB agar for mating at 37°C. The LacZ activity of the mating cells at different time points were measured with standard Miller unit assay (Malke, 1993). The non-mating control was done by spotting donors and recipients separately on the same plate.
Strain construction
MTP997 and MTP1196 were constructed by transforming pMTP112 or pMTP113 into BW27783 made competent by growing cells in LB media to mid-log, washed, and resuspended in ice cold a 0.1M CaCl2 solution (Peters, 2007) on LB agar supplemented with 100 μg/mL carbenicillin grown at 30°C. Individual colonies were purified on LB agar supplemented with 50 μg/mL kanamycin grown at 42°C to select for miniTn7 insertion into the chromosome while curing pMS26 derivatives. Individual colonies were purified at 30°C on LB agar supplemented with carbenicillin or kanamycin to confirm loss of carbenicillin resistance.
MTP1191 was constructed by P1 transduction of MTP997 with bacteriophage grown on strain EMG2 to replace lacZ deletion with wild-type lac operon. Transductants were selected on M9 minimal media supplemented with 0.2% w/v lactose.
PO429 was constructed by using recombineering (Datsenko and Wanner, 2000) to replace wild-type lacZ with a lacZ::miniTn7(genR) allele PCR amplified from a miniTn7(genR) lacZ insertion library (Peters, unpublished).
Plasmid construction
Standard molecular cloning techniques were used to make the vectors described below using vendor instructions.
pMTP112 was constructed by ligating gBlock1 into the NotI site of pMS26 following digestion with NotI. The clone used has A. salmonicida left end proximal to the Tn7 right end. pMTP113 was constructed by assembling two PCR products amplified from pSL0527 (pDonor) (JEP1858+JEP1859 and JEP1860+JEP1861), one PCR product amplified from gBlock1 (JEP1862+JEP1863) and pMS26 digested with NotI using NEBuilder Hifi (NEB). pMTP114 was constructed by assembling two PCR products amplified from F plasmid (JEP1398+1340 and JEP1341+1399, GenBank: AP001918.1), one PCR product amplified from pMTP150 (JEP1343+JEP1344), one PCR product amplified from pBAD322S (JEP1345+JEP1346, GenBank: DQ131584.1) and pTSC29 digested with EcoRV using NEBuilder Hifi. pMTP115 was constructed by inserting a PCR product amplified from EMG2 (JEP1663+JEP1664, GenBank: U00096.3) into pMTP114 following digestion with BsaI using golden gate cloning (Engler et al., 2008). pMTP116 was constructed by inserting annealed oligos (JEP1485+JEP1486) into pMTP114 following digestion with BsaI using golden gate cloning. pMTP117 was constructed by inserting annealed and extended oligos (JEP1481+JEP1482) into pMP114 following digestion with BsaI using golden gate cloning. pMTP118 was constructed by inserting annealed and extended oligos (JEP1878+JEP1879) into pMTP114 following digestion with BsaI using golden gate cloning. pMTP130 was constructed by assembling gBlock2, gBlock3 and a PCR product amplified from pTA106 (JEP1146+JEP1467) digested by DraII with 3,800 bp fragment gel purified using NEBuilder Hifi. pMTP140 was constructed by assembling gBlock4, gBlock5, gBlock6 and pBAD322G digested with NcoI and HindIII using NEBuilder Hifi. pMTP150 was constructed by assembling two PCR products amplified from pBAD33 (JEP1766+JEP1767 and JEP1768+JEP1769) with gBlock7 and gBlock8 using NEBuilder Hifi.pMTP151 was constructed by inserting annealed oligos (JEP1477+JEP1478) into pMTP150 following digestion with BsaI using golden gate cloning. pMTP160 was constructed by assembling two PCR products amplified from pBAD33 (JEP1766+JEP1767 and JEP1768+JEP1769) with gBlock7 and one PCR product amplified from gBlock8 (JEP1475+JEP1773) using NEBuilder Hifi. pMTP161-165 were constructed by ligating annealed oligos (JEP1477+JEP1478, pMTP161; JEP1776+JEP1777, pMTP162; JEP1778+JEP1779, pMTP163; JEP1669+JEP1670, pMTP164; JEP1671+JEP1672, pMTP165). pMTP170 was constructed by assembling two PCR products amplified from pBAD33 (JEP1766+JEP1767 and JEP1770+JEP1769) with one PCR product amplified from gBlock7 (JEP1774+JEP1474) and one PCR product amplified from gBlock8 (JEP1475+JEP1775) using NEBuilder Hifi. pMTP171-183 were constructed by inserting annealed oligos (JEP1784+JEP1785, pMTP171; JEP1780+1781, pMTP172; JEP1782+JEP1783, pMTP173; JEP1794+JEP1795, pMTP174; JEP1796+JEP1797, pMTP175; JEP1786+JEP1787, pMTP176; JEP1788+JEP1789, pMTP177; JEP1798+JEP1799, pMTP178; JEP1800+JEP1801, pMTP179; JEP1808+JEP1809, pMTP180; JEP1810+JEP1811, pMTP181; JEP1816+JEP1817, pMTP182; JEP1818+JEP1819, pMTP183) into pMTP170 following digestion with BsaI using golden gate cloning. pMTP190 was constructed by assembling two PCR product amplified from pBAD33 (JEP1766+JEP1767 and JEP1771+JEP1769) using NEBuilder Hifi.pMTP191 and pMTP192 were constructed by annealing four oligos (JEP1928, JEP1929, JEP1930, JEP1931 : pMTP191; JEP1932, JEP1933, JEP1934, JEP1935 : pMTP192) and ligating with pMTP190 digested with XmaI and BsaI.
pMTP230 was constructed by assembling one PCR product amplified from pBAD33 (JEP1864+JEP1865), one PCR product amplified from pMTP130 (JEP1866+JEP1867) and pSL0284 digested with NcoI and PflFI with 3,707bp fragment gel purified using NEBuilder Hifi. pMTP240 was constructed by assembling a PCR product amplified from pBAD322 (JEP1868+JEP1869) with pSL0284 digested with NdeI and BglI with 5,152bp fragment gel purified using NEBuilder Hifi. pMTP250 was constructed by assembling a PCR product amplified from pCDFDuet-1 (JEP1838+JEP1839), a PCR product amplified from pBAD322 (JEP1834+JEP1835) and a PCR product amplified from pBBR1MCS-3 (JEP1836+JEP1837) using NEBuilder Hifi. pMTP260 and pMTP270 were constructed by annealing four oligos (JEP1870, JEP1871, JEP1872, JEP1873 : pMTP260; JEP1908, JEP1909, JEP1910, JEP1911 : pMTP270) and ligating with pMTP250 digested with XmaI and BsaI. pMTP261-264 were constructed by inserting annealed oligos (JEP1914+JEP1915, pMTP161; JEP1912+JEP1913, pMTP162; JEP1880+JEP1881, pMTP163; JEP1882+JEP1883, pMTP164) into pMTP260 following digestion with BsaI using golden gate cloning. pMTP271-274 were constructed by inserting annealed oligos (JEP1914+JEP1919, pMTP271; JEP1912+JEP1917, pMTP272; JEP1880+JEP1916, pMTP273; JEP1882+JEP1917, pMTP27) into pMTP270 following digestion with BsaI using golden gate cloning. pMTP275 and pMTP276 were constructed by annealing four oligos (JEP1920, JEP1921, JEP1922, JEP1923 : pMTP275; JEP1924, JEP1925, JEP1926, JEP1927 : pMTP276) and ligating with pMTP250 digested with XmaI and BsaI.
All F derivatives were made by using recombineering (Datsenko and Wanner, 2000) to replace a large region of plasmid F from strain EMG2 (GenBank: AP001918.1) with PCR fragments amplified from pMTP114 derivatives (JEP1376+1386. pMTP115, FΔ(finO-fxsA)::lacZ specR; pMTP116, FΔ(finO-fxsA)::cysHAs specR; pMTP117, FΔ(finO-fxsA)::ffsAs specR; pMTP118, FΔ(finO-fxsA)::guaCVc specR).
pOPO256 was constructed by ligating a PCR product amplified from gBlock9 (JEP1657+JEP1757) digested with NdeI and HindIII into pBAD33 digested with the same enzymes. The resulting construct was digested with NdeI and XbaI and ligated with phosphorylated annealed oligos (JEP1842+JEP1843). pOPO258 was constructed by assembling a PCR product amplified from gBlock10 (JEP1764+JEP1765) with pBAD33 digested with NdeI and HindIII using NEBuilder Hifi. The resulting construct was digested with NdeI and XbaI and ligated with phosphorylated annealed oligos (JEP1842+JEP1843). pOPO364 was constructed by ligating a PCR product amplified from gDNA of V. parahaemolyticus RIMD221063 (kindly provided by Tobias Doerr) (JEP1952+JEP1960) digested with NdeI and HindIII and phosphorylated annealed oligos (JEP1842+JEP1843) into pBAD33 digested with NdeI and HindIII. pOPO345 was constructed by ligating a PCR product amplified from gBlock11 (JEP1555+JEP1556) digested with SpeI and HindIII into pBAD33 digested with XbaI and HindIII. pOPO221 was constructed by ligating a PCR product amplified from pBAD24 (JEP1759+JEP1760) digested with BsaI and XhoI with a PCR product amplified from EMG2 (JEP1761+JEP1762) digested with the same enzymes. pOPO227-230, pOPO332, pOPO334, pOPO341, and pOPO337 were constructed by ligating fragments from gBlock10 or gBlock11 digested with XhoI and StuI (gBlock10 : pOPO227-230; gBlock11 : pOPO332, pOPO334, pOPO341, pOPO337) into pOPO221 digested with XhoI and SmaI. pOPO329 and pOPO330 were constructed by ligating PCR products amplified from gDNA of V. parahaemolyticus RIMD221063 (JEP1956+JEP1957, pOPO329; JEP1954+JEP1955, pOPO330) digested with XhoI and StuI into pOPO221 digested with XhoI and SmaI. pOPO223, pOPO239, pOPO331, pOPO360 were constructed by ligating PCR products amplified (from gBlock9, JEP1675+JEP1758, pMTP016; from gBlock10, JEP1556+JEP1764, pMTP017; from gDNA of V. parahaemolyticus RIMD221063, JEP1952+JEP1953, pMTP018; from gBlock11, JEP1950+1951, pMTP019) digested with NdeI and XhoI into pET22b(+) digested with the same enzymes. pOPO390 and pOPO275 was constructed by ligating annealed oligos (JEP2119, JEP2120, JEP1906+JEP1907 respectively) into a PCR product amplified from pCOLADuet-1 (JEP1902+JEP1903) digested with SapI. pOPO322 was constructed by assembling a PCR product amplified from pCas1_pCas2/3 (JEP1889+JEP1890) and pACYCDuet-1 digested with NcoI and AvrII using NEBuilder Hifi. pOPO392 was constructed by assembling PCR products amplified from gDNA of V. parahaemolyticus RIMD221063 (JEP2107+JEP2108) and pOPO330 (JEP2109+JEP2110) with pBBR1MCS-2 digested with NsiI and BamHI using NEBuilder Hifi. pOPO394 was constructed by assembling PCR products amplified from gBlock10 (JEP2111+JEP2112, JEP2113+JEP2114) and pOPO227 (JEP2115+JEP2116) with pBBR1MCS-2 digested with NsiI and BamHI using NEBuilder Hifi. pOPO435 was constructed by assembling PCR products amplified from gBlock11 (JEP2154+JEP2155, JEP2156+JEP2157) and pOPO337 (JEP2158+JEP2159) with pBBR1MCS-2 digested with NsiI and BamHI using NEBuilder Hifi. pOPO395 was constructed by assembling a PCR product amplified from gDNA of V. parahaemolyticus RIMD221063 (JEP2101+JEP2102) and pETDuet-1 digested with XbaI and AvrII using NEBuilder Hifi. pOPO397 was constructed by assembling PCR products amplified from gBlock10 (JEP2103+JEP2104, JEP2105+JEP2106) with pETDuet-1 digested with XbaI and AvrII using NEBuilder Hifi. pOPO438 was constructed by assembling PCR products amplified from gBlock11 (JEP2160+JEP2161, JEP2162+JEP2163) with pETDuet-1 digested with XbaI and AvrII using NEBuilder Hifi.
pOPO374 was constructed by ligating a PCR product amplified from pCDFDuet-1 (JEP1577+JEP1891) digested with BsaI and two pairs of phosphorylated annealed oligos (JEP1995+JEP1996, JEP1997+JEP1998). pOPO376 and pOPO378 were constructed with the same method, but with oligos (JEP2003+JEP2004, JEP2005+JEP2006) and (JEP2007+JEP2008, JEP2009+JEP2010).
pMTP281-286 were constructed by ligating a PCR product amplified from pCDFDuet-1 (JEP2032+JEP2033) digested with BsaI with four annealed oligos (JEP2063, JEP2064, JEP2065, JEP2066 : pMTP281; JEP2078, JEP2079, JEP2080, JEP2081 : pMTP282; JEP2035, JEP2036, JEP2037, JEP2038 : pMTP283; JEP2049, JEP2050, JEP2051, JEP2052 : pMTP284; JEP2067, JEP2068, JEP2069, JEP2066 : pMTP285; JEP2082, JEP2083, JEP2084, JEP2081 : pMTP286).
Quantification and Statistical Analysis
Statistical details are listed in the Figure Legends. When stated, experiments were performed with three biological replicates (n=3).
Supplementary Material
Supplemental Table S1 - Type I-F3 Tn7-CRISPR-Cas Elements in Gammaproteobacteria, Related to Figure 1.
List of 802 I-F3 Tn7-CRISPR-Cas elements indicated by the attachment site where located indicating the host genus, species, and strain with accession information (contig). Sequences were manually curated to identify the repeats and spacers. Repeats were scored by similarity to consensus sequence of non-terminal repeats of the first array. The spacer and spacer match (protospacer) is indicated with the size of the spacer and number of predicted base-pair contacts and where the matching spacer resided in the array. It is indicated if another I-F CRISPR-Cas system could be identified in the strain. See text for details.
Supplemental Table S2 – Plasmids Used in This Study, Related to STAR Methods.
Supplemental Table S3 – Oligos Used in This Study, Related to STAR Methods.
Supplemental Table S4 – Synthesized gene fragments (gBlocks) Used in This Study, Related to STAR Methods.
Supplemental Figure S1 – Aeromonas Element Features and Transposition, Related to Figure 2.
(a) Schematic representation of two nearly-identical I-F3b Tn7-CRISPR-Cas elements found in different bacterial species suggesting recent activity. Core features are indicated as in Figure 2a. Elements are located either in the chromosomal ffs site in A. hydrophila AFG_SD03 or inserted into a phosphoadenosine phosphosulfate reductase gene (cysH) found on a large conjugal plasmid (pS44-1) in A. salmonicida S44. The A. hydrophila element is split across several contigs interrupted by apparent IS element insertions.
(b) Spacers in the leader-proximal position of A. salmonicida S44 and A. hydrophila AFG_SD03 CRISPR arrays match protospacers in a plasmid encoded cysH. Relative position of the protospacers are indicated. Distance from the edge of the protospacer matching A.salmonicida S44 spacer to the central position in the target site duplication (TSD), the 5 bp TSD (underlined), as well as the terminal sequence of the transposon ends are shown.
(c) Repeats and spacers from Tn7-like CRISPR arrays in A. salmonicida S44 and A. hydrophila AFG_SD03. Repeats are annotated as in Figure 2c. Differences from the first repeat are indicated in red. Matches between the guide RNA and protospacer are indicated by a short vertical line. The putative I-F PAM is underlined.
(d) Protospacers on the chromosome or F plasmid are targeted at high efficiency with atypical guide RNA complexes. The same three lacZ guide RNA complexes were tested with either the F::lacZ plasmid or chromosome (lacZ in its native position) and insertion events were indicated by generating white verses red colonies on MacConkey’s lactose indicator media. Graph shows the mean +/− standard deviation of three biological replicates and number of white colonies and total colonies observed.
(e) Different genes on the chromosome can be targeted for atypical guide RNA-directed transposition in the E. coli chromosome. Two genes where tested with two spacers each (top and bottom strand) for galactose (galK) and sorbitol (srlD). Transposition frequency was assayed by monitoring gene inactivation leading to loss of sugar catabolism as visualized by white verses red colonies on the appropriate MacConkey’s indicator media. Graph shows the mean +/− standard deviation of three biological replicates.
Supplemental Figure S2 – Assaying Full Transposition Frequency and Position, Related to Figure 3.
(a) Mate out assay schematic. Target DNAs with the appropriate protospacer are recombined onto an F plasmid and transposition genes and arrays are supplied on expression vectors to mobilize a mini-Tn donor element located in the chromosome (Methods). After induction, transposition frequency is determined by mating out the population of F plasmids into a donor strain and quantifying antibiotic marker presence in transconjugants as shown.
(b) Transposition position and orientation in transconjugants are determined by PCR. An internal primer and two primers flanking the target site capture orientation of insertion. For Tn6900, pS44-1-targeted insertions were monitored; for Tn6677, guaCVc-targeted insertions were monitored. *For Tn6900 typical array insertion isolates 12, 13, and 14, the first PCR reaction failed and was repeated with the same template strain.
(c) Transposition position and target site duplication are confirmed by Sanger sequencing for Tn6900 transposition. Arrows indicate position of the central base of the target site duplication for isolated transposition events targeting pS44-1, with distance from protospacer to the central position of the target site duplication listed for eight transposition events, confirming previously described target site wobble. Graph shows one representative of the actual target site duplication (TSD).
Supplemental Figure S3 – Interference Assay Repeat Sequences, Related to Figure 5.
Repeat sequences used in interference assays. Differences from P. aeruginosa repeats are indicated in red. The orange box indicates the previously established conserved region that comprises the putative stem-loop in I-F repeats. N32 indicates the position encoded in the spacer.
Supplemental Figure S4 – I-F3 Xre Cluster Into Two Clades with Restriction-Modification C Proteins, Related to Figure 6.
(a) Similarity tree of Xre (midpoint rooted) with associated C proteins C.AhdI and C.Csp231I (marked in teal and fuchsia), indicating clustering in the two branches. Features are indicated as in Figure 1.
(b) Predicted regulator sequences for Xre and associated C proteins. Conserved inverted motif sequences are indicated by bold red text and black arrows. The start codon of the downstream gene is underlined, except for pAttGuide sequences, where the first three bases of the att-targeting spacer are underlined.
Supplemental Figure S5 – Comparing Spacers and Protospacers in Relation to Reading Frame, Relating to Figure 2.
The four major att-site targeting spacers are compared to the protospacer (target) in each host; ffs, guaC, yciA, and rsmJ. The percentage of mismatches is indicated by position in the spacer comparing unique spacer-protospacer combinations (related to a diagram of the guide RNA showing the predicted flipped-out 6th position in red). The amino acid sequence is indicated to relate to the coding sequence (Note that ffs is functional as an RNA and the yciA gene is encoded on the opposite strand of guaC and rsmJ). The consensus sequence for the unique spacers and protospacers are indicated as Weblogos. The total number of mismatches per spacer-protospacer is indicated excluding the 6th position which is flipped-out in the cascade complex in I-F systems. The number included in the tabulations is indicated (n).
Supplemental Figure S6 – Elements with Shortened Spacers and Their Insertion Positions, Related to Figure 2.
(a) ffs-integrated elements,
(b) araC-like integrated elements. Features are indicated as in Figure 2b. Arrays are shown for each element, with repeat sequences marked in blue, spacers marked in red, and conserved Cas6 binding motifs are underlined.
Supplemental Figure S7 – Schematic Representation of Elements Inserted Downstream of parE, Related to Figure 1.
Similarity tree of TniQ proteins indicates that Parashewanella curva C51 has representatives that group with elements that target the parE att site and elements that use the I-F3 CRISPR-Cas system. In cases where two TniQs are found in the element, the one used for the similarity tree is indicated with orange highlighting.
CRISPR-Cas systems have been naturally coopted for guide RNA-directed transposition
crRNAs are categorized to separately target the chromosome or plasmids and phage
crRNAs to neutral chromosome sites are privatized from canonical CRISPR-Cas systems
Specialized crRNAs reveal avenues to engineer enhanced transposition
Acknowledgements
This work was supported by NIH R01 GM129118. S-C.H. was supported by a fellowship from the Taiwan government. We thank Joshua Chu for assistance with experiments and Nancy Craig and Ailong Ke for manuscript comments. We thank John Cronan, Tobias Doerr, Elizabeth Raleigh, and Ailong Ke for reagents.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
Cornell University has filed patent applications related to this work with J.E.P. as inventor. The lab has corporate funding for research that is not directly related to the work in this publication.
References
- Bainton R, Gamas P, and Craig NL (1991). Tn7 transposition in vitro proceeds through an excised transposon intermediate generated by staggered breaks in DNA. Cell 65, 805–816. [DOI] [PubMed] [Google Scholar]
- Bainton RJ, Kubo KM, Feng J-N, and Craig NL (1993). Tn7 transposition: target DNA recognition is mediated by multiple Tn7-encoded proteins in a purified in vitro system. Cell 72, 931–943. [DOI] [PubMed] [Google Scholar]
- Boehmer T, Vogler AJ, Thomas A, Sauer S, Hergenroether M, Straubinger RK, Birdsell D, Keim P, Sahl JW, Williamson CHD, et al. (2018). Phenotypic characterization and whole genome analysis of extended-spectrum beta-lactamase-producing bacteria isolated from dogs in Germany. PLoS One 13, e0206252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges AL, Davidson AR, and Bondy-Denomy J (2017). The Discovery, Mechanisms, and Evolutionary Impact of Anti-CRISPRs. Annu Rev Virol 4, 37–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S, Carter J, Rollins MF, Golden SM, Jackson RN, Hoffmann C, Nosaka L, Bondy-Denomy J, Maxwell KL, Davidson AR, et al. (2017). Structure Reveals Mechanisms of Viral Suppressors that Intercept a CRISPR RNA-Guided Surveillance Complex. Cell 169, 47–57.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, and Wanner BL (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler C, Kandzia R, and Marillonnet S (2008). A one pot, one step, precision cloning method with high throughput capability. PloS one 3, e3647–e3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure G, Makarova KS, and Koonin EV (2019a). CRISPR-Cas: Complex Functional Networks and Multiple Roles beyond Adaptive Immunity. J Mol Biol 431, 3–20. [DOI] [PubMed] [Google Scholar]
- Faure G, Shmakov SA, Yan WX, Cheng DR, Scott DA, Peters JE, Makarova KS, and Koonin EV (2019b). CRISPR-Cas in mobile genetic elements: counter-defence and beyond. Nat Rev Microbiol 17, 513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineran PC, Gerritzen MJ, Suarez-Diez M, Kunne T, Boekhorst J, van Hijum SA, Staals RH, and Brouns SJ (2014). Degenerate target sites mediate rapid primed CRISPR adaptation. Proc Natl Acad Sci U S A 111, E1629–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleditzsch D, Muller-Esparza H, Pausch P, Sharma K, Dwarakanath S, Urlaub H, Bange G, and Randau L (2016). Modulating the Cascade architecture of a minimal Type I-F CRISPR-Cas system. Nucleic Acids Res 44, 5872–5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyland-Kroghsbo NM, Paczkowski J, Mukherjee S, Broniewski J, Westra E, Bondy-Denomy J, and Bassler BL (2017). Quorum sensing controls the Pseudomonas aeruginosa CRISPR-Cas adaptive immune system. Proc Natl Acad Sci U S A 114, 131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RN, Golden SM, van Erp PB, Carter J, Westra ER, Brouns SJ, van der Oost J, Terwilliger TC, Read RJ, and Wiedenheft B (2014). Structural biology. Crystal structure of the CRISPR RNA-guided surveillance complex from Escherichia coli. Science 345, 1473–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klompe SE, Vo PLH, Halpin-Healy TS, and Sternberg SH (2019). Transposon-encoded CRISPR–Cas systems direct RNA-guided DNA integration. Nature 571, 219–225. [DOI] [PubMed] [Google Scholar]
- Kuznedelov K, Mekler V, Lemak S, Tokmina-Lukaszewska M, Datsenko KA, Jain I, Savitskaya E, Mallon J, Shmakov S, Bothner B, et al. (2016). Altered stoichiometry Escherichia coli Cascade complexes with shortened CRISPR RNA spacers are capable of interference and primed adaptation. Nucleic Acids Res 44, 10849–10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, Charpentier E, Cheng D, Haft DH, Horvath P, et al. (2020). Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol 18, 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malke H (1993). Jeffrey H. Miller, A Short Course in Bacterial Genetics – A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor 1992. Cold Spring Harbor Laboratory Press. ISBN: 0–87969-349–5. Journal of Basic Microbiology 33, 278–278. [Google Scholar]
- Martynov A, Severinov K, and Ispolatov I (2017). Optimal number of spacers in CRISPR arrays. PLoS Comput Biol 13, e1005891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeehan JE, Streeter SD, Thresh SJ, Taylor JE, Shevtsov MB, and Kneale GG (2011). Structural analysis of a novel class of R-M controller proteins: C.Csp231I from Citrobacter sp. RFL231. J Mol Biol 409, 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra R, McKenzie GJ, Yi L, Lee CA, and Craig NL (2010). Characterization of the TnsD-attTn7 complex that promotes site-specific insertion of Tn7. Mobile DNA 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulepati S, Heroux A, and Bailey S (2014). Structural biology. Crystal structure of a CRISPR RNA-guided surveillance complex bound to a ssDNA target. Science 345, 1479–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks AR, Li Z, Shi Q, Owens RM, Jin MM, and Peters JE (2009). Transposition into replicating DNA occurs through interaction with the processivity factor. Cell 138, 685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson AG, Jackson SA, Taylor C, Evans GB, Salmond GPC, Przybilski R, Staals RHJ, and Fineran PC (2016). Quorum Sensing Controls Adaptive Immunity through the Regulation of Multiple CRISPR-Cas Systems. Mol Cell 64, 1102–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JE (2014). Tn7. Microbiology Spectrum 2, 1–20. [DOI] [PubMed] [Google Scholar]
- Peters JE (2019). Targeted transposition with Tn7 elements: safe sites, mobile plasmids, CRISPR/Cas and beyond. Mol Microbiol 112, 1635–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JE, and Craig NL (2001). Tn7 recognizes target structures associated with DNA replication using the DNA binding protein TnsE. Genes & Dev 15, 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JE, Makarova KS, Shmakov S, and Koonin EV (2017). Recruitment of CRISPR-Cas systems by Tn7-like transposons. Proceedings of the National Academy of Sciences 114, E7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice PA, Craig NL, and Dyda F (2020). Comment on “RNA-guided DNA insertion with CRISPR-associated transposases”. Science 368. [DOI] [PubMed] [Google Scholar]
- Rodic A, Blagojevic B, Zdobnov E, Djordjevic M, and Djordjevic M (2017). Understanding key features of bacterial restriction-modification systems through quantitative modeling. BMC Syst Biol 11, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Straus MR, Caron JJ, Wang H, Chung YS, Guarne A, and Peters JE (2015). Conformational toggling controls target site choice for the heteromeric transposase element Tn7. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen AE, and Craig NL (1998). Mobile DNA elements: controlling transposition with ATP-dependent molecular switches. Trends Biochem Sci 23, 486–490. [DOI] [PubMed] [Google Scholar]
- Strecker J, Ladha A, Gardner Z, Schmid-Burgk JL, Makarova KS, Koonin EV, and Zhang F (2019). RNA-guided DNA insertion with CRISPR-associated transposases. Science 365, 48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strecker J, Ladha A, Makarova KS, Koonin EV, and Zhang F (2020). Response to Comment on “RNA-guided DNA insertion with CRISPR-associated transposases”. Science 368. [DOI] [PubMed] [Google Scholar]
- Streeter SD, Papapanagiotou I, McGeehan JE, and Kneale GG (2004). DNA footprinting and biophysical characterization of the controller protein C.AhdI suggests the basis of a genetic switch. Nucleic Acids Res 32, 6445–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorontsova D, Datsenko KA, Medvedeva S, Bondy-Denomy J, Savitskaya EE, Pougach K, Logacheva M, Wiedenheft B, Davidson AR, Severinov K, et al. (2015). Foreign DNA acquisition by the I-F CRISPR-Cas system requires all components of the interference machinery. Nucleic Acids Res 43, 10848–10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell CS, and Craig NL (1988). Tn7 transposition: two transposition pathways directed by five Tn7-encoded genes. Genes Dev 2, 137–149. [DOI] [PubMed] [Google Scholar]
- Westra ER, Pul U, Heidrich N, Jore MM, Lundgren M, Stratmann T, Wurm R, Raine A, Mescher M, Van Heereveld L, et al. (2010). H-NS-mediated repression of CRISPR-based immunity in Escherichia coli K12 can be relieved by the transcription activator LeuO. Mol Microbiol 77, 1380–1393. [DOI] [PubMed] [Google Scholar]
- Wiegand T, Karambelkar S, Bondy-Denomy J, and Wiedenheft B (2020). Structures and Strategies of Anti-CRISPR-Mediated Immune Suppression. Annu Rev Microbiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Ng S, Nam KH, and Ke A (2017). How type II CRISPR-Cas establish immunity through Cas1-Cas2-mediated spacer integration. Nature 550, 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Sheng G, Wang J, Wang M, Bunkoczi G, Gong W, Wei Z, and Wang Y (2014). Crystal structure of the RNA-guided immune surveillance Cascade complex in Escherichia coli. Nature 515, 147–150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1 - Type I-F3 Tn7-CRISPR-Cas Elements in Gammaproteobacteria, Related to Figure 1.
List of 802 I-F3 Tn7-CRISPR-Cas elements indicated by the attachment site where located indicating the host genus, species, and strain with accession information (contig). Sequences were manually curated to identify the repeats and spacers. Repeats were scored by similarity to consensus sequence of non-terminal repeats of the first array. The spacer and spacer match (protospacer) is indicated with the size of the spacer and number of predicted base-pair contacts and where the matching spacer resided in the array. It is indicated if another I-F CRISPR-Cas system could be identified in the strain. See text for details.
Supplemental Table S2 – Plasmids Used in This Study, Related to STAR Methods.
Supplemental Table S3 – Oligos Used in This Study, Related to STAR Methods.
Supplemental Table S4 – Synthesized gene fragments (gBlocks) Used in This Study, Related to STAR Methods.
Supplemental Figure S1 – Aeromonas Element Features and Transposition, Related to Figure 2.
(a) Schematic representation of two nearly-identical I-F3b Tn7-CRISPR-Cas elements found in different bacterial species suggesting recent activity. Core features are indicated as in Figure 2a. Elements are located either in the chromosomal ffs site in A. hydrophila AFG_SD03 or inserted into a phosphoadenosine phosphosulfate reductase gene (cysH) found on a large conjugal plasmid (pS44-1) in A. salmonicida S44. The A. hydrophila element is split across several contigs interrupted by apparent IS element insertions.
(b) Spacers in the leader-proximal position of A. salmonicida S44 and A. hydrophila AFG_SD03 CRISPR arrays match protospacers in a plasmid encoded cysH. Relative position of the protospacers are indicated. Distance from the edge of the protospacer matching A.salmonicida S44 spacer to the central position in the target site duplication (TSD), the 5 bp TSD (underlined), as well as the terminal sequence of the transposon ends are shown.
(c) Repeats and spacers from Tn7-like CRISPR arrays in A. salmonicida S44 and A. hydrophila AFG_SD03. Repeats are annotated as in Figure 2c. Differences from the first repeat are indicated in red. Matches between the guide RNA and protospacer are indicated by a short vertical line. The putative I-F PAM is underlined.
(d) Protospacers on the chromosome or F plasmid are targeted at high efficiency with atypical guide RNA complexes. The same three lacZ guide RNA complexes were tested with either the F::lacZ plasmid or chromosome (lacZ in its native position) and insertion events were indicated by generating white verses red colonies on MacConkey’s lactose indicator media. Graph shows the mean +/− standard deviation of three biological replicates and number of white colonies and total colonies observed.
(e) Different genes on the chromosome can be targeted for atypical guide RNA-directed transposition in the E. coli chromosome. Two genes where tested with two spacers each (top and bottom strand) for galactose (galK) and sorbitol (srlD). Transposition frequency was assayed by monitoring gene inactivation leading to loss of sugar catabolism as visualized by white verses red colonies on the appropriate MacConkey’s indicator media. Graph shows the mean +/− standard deviation of three biological replicates.
Supplemental Figure S2 – Assaying Full Transposition Frequency and Position, Related to Figure 3.
(a) Mate out assay schematic. Target DNAs with the appropriate protospacer are recombined onto an F plasmid and transposition genes and arrays are supplied on expression vectors to mobilize a mini-Tn donor element located in the chromosome (Methods). After induction, transposition frequency is determined by mating out the population of F plasmids into a donor strain and quantifying antibiotic marker presence in transconjugants as shown.
(b) Transposition position and orientation in transconjugants are determined by PCR. An internal primer and two primers flanking the target site capture orientation of insertion. For Tn6900, pS44-1-targeted insertions were monitored; for Tn6677, guaCVc-targeted insertions were monitored. *For Tn6900 typical array insertion isolates 12, 13, and 14, the first PCR reaction failed and was repeated with the same template strain.
(c) Transposition position and target site duplication are confirmed by Sanger sequencing for Tn6900 transposition. Arrows indicate position of the central base of the target site duplication for isolated transposition events targeting pS44-1, with distance from protospacer to the central position of the target site duplication listed for eight transposition events, confirming previously described target site wobble. Graph shows one representative of the actual target site duplication (TSD).
Supplemental Figure S3 – Interference Assay Repeat Sequences, Related to Figure 5.
Repeat sequences used in interference assays. Differences from P. aeruginosa repeats are indicated in red. The orange box indicates the previously established conserved region that comprises the putative stem-loop in I-F repeats. N32 indicates the position encoded in the spacer.
Supplemental Figure S4 – I-F3 Xre Cluster Into Two Clades with Restriction-Modification C Proteins, Related to Figure 6.
(a) Similarity tree of Xre (midpoint rooted) with associated C proteins C.AhdI and C.Csp231I (marked in teal and fuchsia), indicating clustering in the two branches. Features are indicated as in Figure 1.
(b) Predicted regulator sequences for Xre and associated C proteins. Conserved inverted motif sequences are indicated by bold red text and black arrows. The start codon of the downstream gene is underlined, except for pAttGuide sequences, where the first three bases of the att-targeting spacer are underlined.
Supplemental Figure S5 – Comparing Spacers and Protospacers in Relation to Reading Frame, Relating to Figure 2.
The four major att-site targeting spacers are compared to the protospacer (target) in each host; ffs, guaC, yciA, and rsmJ. The percentage of mismatches is indicated by position in the spacer comparing unique spacer-protospacer combinations (related to a diagram of the guide RNA showing the predicted flipped-out 6th position in red). The amino acid sequence is indicated to relate to the coding sequence (Note that ffs is functional as an RNA and the yciA gene is encoded on the opposite strand of guaC and rsmJ). The consensus sequence for the unique spacers and protospacers are indicated as Weblogos. The total number of mismatches per spacer-protospacer is indicated excluding the 6th position which is flipped-out in the cascade complex in I-F systems. The number included in the tabulations is indicated (n).
Supplemental Figure S6 – Elements with Shortened Spacers and Their Insertion Positions, Related to Figure 2.
(a) ffs-integrated elements,
(b) araC-like integrated elements. Features are indicated as in Figure 2b. Arrays are shown for each element, with repeat sequences marked in blue, spacers marked in red, and conserved Cas6 binding motifs are underlined.
Supplemental Figure S7 – Schematic Representation of Elements Inserted Downstream of parE, Related to Figure 1.
Similarity tree of TniQ proteins indicates that Parashewanella curva C51 has representatives that group with elements that target the parE att site and elements that use the I-F3 CRISPR-Cas system. In cases where two TniQs are found in the element, the one used for the similarity tree is indicated with orange highlighting.
Data Availability Statement
The dataset of I-F3 Tn7-CRISPR-Cas elements generated during this study is available as Table S1. This study did not generate any unique code.