

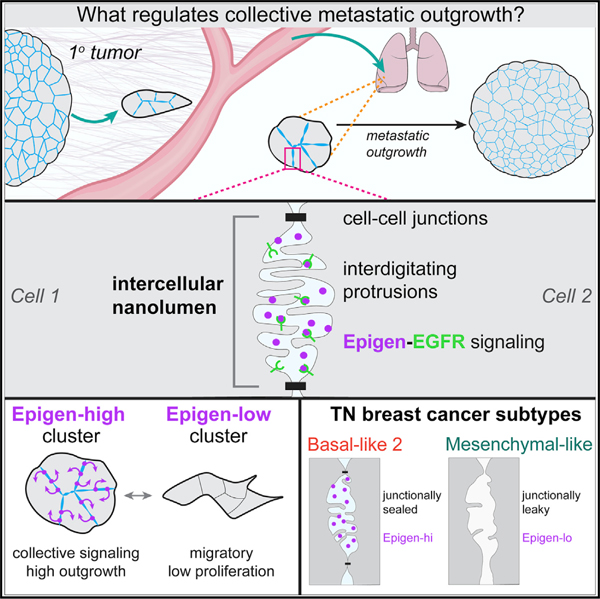
SUMMARY
Collective metastasis is defined as the cohesive migration and metastasis of multicellular tumor cell clusters. Disrupting various cell adhesion genes markedly reduces cluster formation and colonization efficiency, yet the downstream signals transmitted by clustering remain largely unknown. Here, we use mouse and human breast cancer models to identify a collective signal generated by tumor cell clusters supporting metastatic colonization. We show that tumor cell clusters produce the growth factor epigen and concentrate it within nanolumina—intercellular compartments sealed by cell-cell junctions and lined with microvilli-like protrusions. Epigen knockdown profoundly reduces metastatic outgrowth and switches clusters from a proliferative to a collective migratory state. Tumor cell clusters from basal-like 2, but not mesenchymal-like, triple-negative breast cancer cell lines have increased epigen expression, sealed nanolumina, and impaired outgrowth upon nanolumenal junction disruption. We propose that nanolumenal signaling could offer a therapeutic target for aggressive metastatic breast cancers.
Graphical Abstract
In Brief
Nanolumina encased within clusters of tumor cells harbor signaling molecules that promote metastasis.
INTRODUCTION
Although metastasis is often conceived of as a single cell pro-cess, multicellular tumor cell clusters have been directly observed at tumor invasive fronts, within the systemic circulation, and colonizing distant organs (Aceto et al., 2015; Cheung and Ewald, 2016; Friedl and Gilmour, 2009). Clinically, circulating tumor cell clusters are associated with poorer prognosis in many cancer types (Au et al., 2017; Giuliano et al., 2018). In different mouse models, multicellular clusters give rise to between 50%–97% of metastases, supporting a collective origin of metastasis (Aceto et al., 2014; Cheung et al., 2016). Yet, compared with single cell metastasis (Derynck and Weinberg, 2019; Lambert et al., 2017), the molecular mechanisms driving dissemination and outgrowth of multicellular clusters are far less understood.
The formation of clusters markedly enhances metastatic potential (Aceto et al., 2014; Cheung et al., 2016; Fidler, 1973; Maddipati and Stanger, 2015) and induces multiple molecular properties including increased stem cell-like features (Gkountela et al., 2019), evasion of natural killer cell targeting (Lo et al., 2020), radiotherapy resistance (Haeger et al., 2020), and resistance to metabolic stress (Labuschagne et al., 2019; Padmanaban et al., 2019), among others (Choi et al., 2015; Liu et al., 2019b). Although disrupting cell adhesion genes reduced cluster formation and metastatic potential in multiple studies, how cluster formation induces these molecular changes is less clear.
In developmental contexts, multicellularity and cell-cell adhesion are known to induce cooperative social-like signaling during tissue morphogenesis (Durdu et al., 2014; Friedl and Mayor, 2017; Gilmour et al., 2017; Gurdon, 1988; Mishra et al., 2019; Raff, 1992). In addition, cooperative interactions between tumor cells have been posited to promote metastasis (Archetti and Pienta, 2019; Ben-Jacob et al., 2012; Hickson et al., 2009; Janiszewska et al., 2019). Thus, clustering of tumor cells could give rise to complex signaling mechanisms distinct from those utilized by single cells.
In the present study, we used mouse and human models of breast cancer metastasis to identify collective signals generated by tumor cell clusters during metastatic outgrowth. We show that mouse and human tumor cell clusters harbor nanolumina—intercellular signaling compartments sealed by cell-cell junctions. Within nanolumina, we identify a diffusible growth factor, epigen, which controls switching between collective migratory and proliferative modes. Importantly, we find that epigen expression and nanolumen formation are enriched in a subtype of aggressive, triple-negative human breast cancers.
RESULTS
An Experimental System to Identify Metastasis-Promoting Signals Generated by Aggregation
To temporally control cluster formation and monitor downstream molecular events (Figure 1A), we used an aggressive mouse model of breast cancer, MMTV-PyMT, (Guy et al., 1992; Lin et al., 2003) in which collective metastasis has been reported (Cheung et al., 2013, 2016). When MMTV-PyMT tumor organoids were enzymatically dissociated and cultured in suspension, single tumor cells spontaneously assembled into loosely attached aggregates by 6 h and more compact clusters by 24 h (Figure S1A; Video S1). MMTV-PyMT membrane-Tomato-labeled tumor organoids were dissociated to single cells and then four input suspensions were generated: single cells aggregated for 0 h, for 6 h, for 24 h, and for 24 h then re-dissociated back to single cells. Equal cell numbers of each suspension were injected into immunocompromised NSG mice. 3 weeks later, loosely attached (6 h) clusters formed 141-fold and compact (24 h) clusters formed 532-fold more lung metastases than an equal number of single cells (Figures 1B and S1B). Well-organized (24 h) clusters re-dissociated back into single cells were negligibly metastatic (Figure 1B), indicating that metastatic potential in this system requires multicellular organization.
Figure 1. RNA-Seq Analysis of Tumor Cell Aggregation Identifies Epigen as a Cluster-Upregulated Gene Supporting Metastatic Outgrowth.
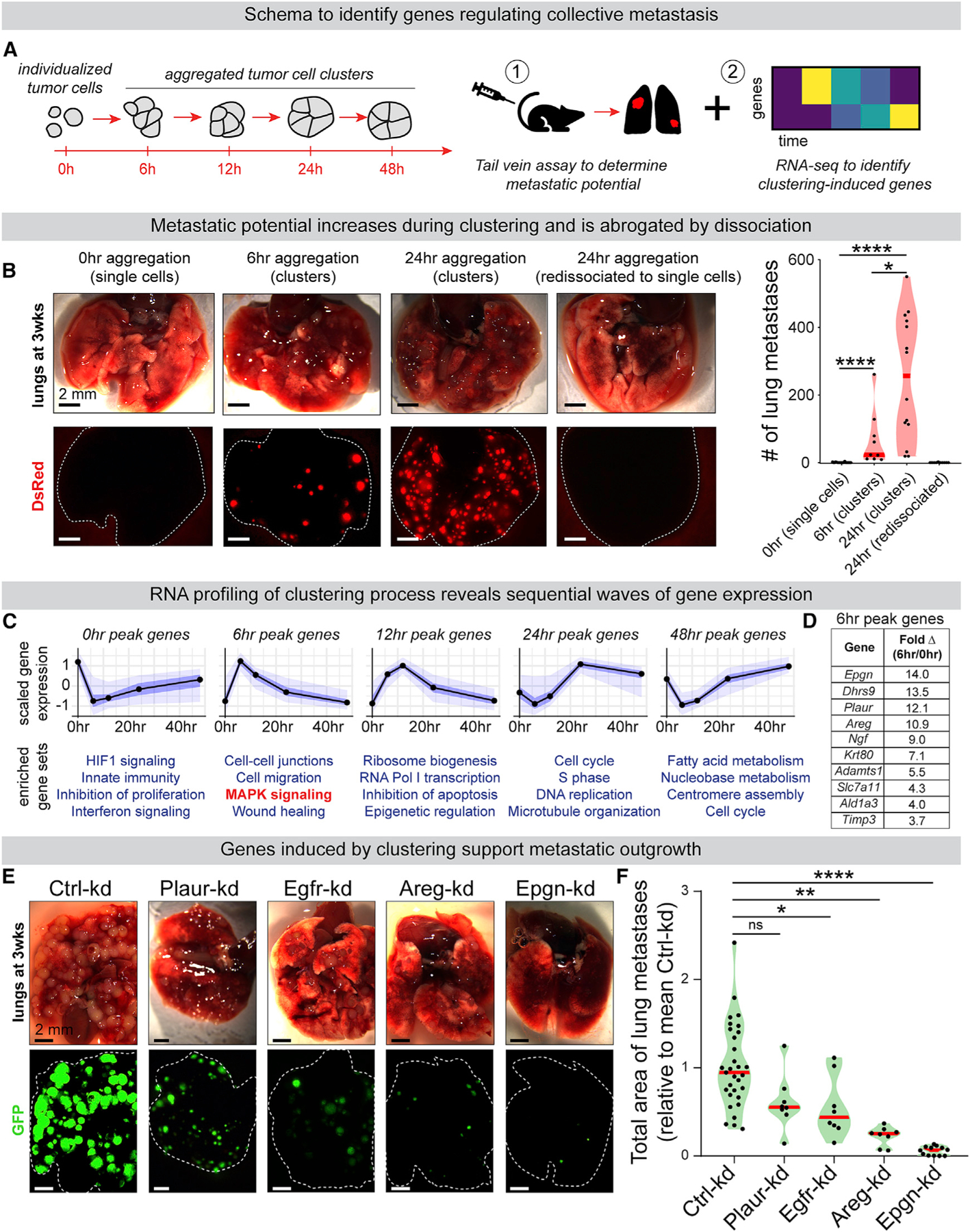
(A) MMTV-PyMT tumor organoids dissociated to single cells spontaneously aggregate into clusters in suspension. Single cells or aggregates were subjected to (1) tail vein metastasis assays and (2) RNA-seq to identify genes altered by aggregation.
(B) MMTV-PyMT mTomato+ tumor organoids were dissociated to single cells and aggregated for 0, 6, and 24 h. At 24 h, some clusters were re-dissociated back to single cells. At each time point, 200,000 cells were injected by tail vein into NSG mice. Left: lungs 3 weeks later. Right: # of mTomato+ metastases. n (# mice) = 19 (0 h), 9 (6 h), 14 (24 h), 10 (re-dissociated). p values = Mann-Whitney tests.
(C) MMTV-PyMT cells were collected for RNA-seq at 0, 6, 12, 24, and 48 h of aggregation. n = 455, 780, 367, 320, and 482 genes peaking at each time point, respectively (false discover rate [FDR] ≤0.05). n = 3. Top: mean-variance normalized gene expression. Black line, median; dark blue, 50% confidence interval (CI); light blue, 95% CI. Bottom: Gene ontology enrichment (Metascape, p value < 0.0001).
(D) The most differentially cluster-upregulated transcripts expressed at 0 h and 6 h, sorted by fold change. n = 3.
(E) Non-targeting control or shRNA transduced PyMT tumor cell clusters were injected by tail vein into NSG mice (200,000 clustered cells/mouse). Representative images of lungs 3 week later.
(F) Total GFP+ lung metastatic burden per mouse, relative to mean Ctrl-kd metastatic burden for each injection replicate. n (# of mice): Ctrl-kd = 31, Egfr-kd = 8, Areg-kd = 8, Epgn-kd = 12, Plaur-kd = 8. One-way ANOVA p < 0.0001. p values = Dunnett’s test.
See also Figures S1, S2, and S3, Tables S2 and S3, and Video S1.
Both single cells and clusters were trapped in the lungs of mice shortly after tail vein injection (Figure S1C), but clusters persisted while single cells were largely cleared by 3 weeks post-injection (Figures S1D and S1E). Likewise, intracardiac injection of clustered tumor cells generated significantly more metastases to multiple organs than injection of single cells (Figures S1F and S1G), indicating superior metastatic potential of clusters delivered by both venous and arterial routes.
Concordant with these in vivo observations, MMTV-PyMT tumor cell clusters had >4.7-fold higher outgrowth than single cells in 3D basement membrane-rich gels (Figure S2A). Proliferation was highest in clusters with tumor cells directly contacting multiple neighbors (Figures S2B and S2C). In addition, we used a caspase-3/7 fluorescent biosensor to measure apoptosis. MMTV-PyMT clusters had >2.2-fold higher survival than single cells (Figure S2D). Further, 8 of 10 individual human breast cancer tumor specimens, including those from metastases to brain and bone, had 2.5- to >70-fold higher survival as clusters (Figures S2E and S2F; Table S1). Together, these data establish a system to dissect the metastasis-promoting signals downstream of tumor cell cluster formation.
The Growth Factor Epigen Is the Most Induced Gene upon Clustering and Supports Efficient Metastatic Outgrowth in the Lung
We next performed an RNA sequencing (RNA-seq) time course of MMTV-PyMT cells throughout their aggregation from single cells to highly metastatic clusters (from 0 to 48 h after dissociation) (Figures 1A and 1C). This revealed sequential upregulation of different biological pathways: at 0 h, HIF1 and interferon stress signaling pathways; at 6 h, wound-healing and mitogen-activated protein kinase (MAPK)-associated genes; at 12 h, ribosome biogenesis; and at 24 and 48 h, genes involved in cell cycle and fatty acid metabolism (Figure 1C; Table S2). Analysis of early clusters (6 h) compared to single cells revealed that clustering rapidly induced expression of low-affinity EGFR ligands epigen and amphiregulin (Figure 1D). Epgn and was the most differentially expressed of all mRNAs (Table S3). Compared to high-affinity ligands like EGF, low-affinity EGFR ligands are associated with prolonged signaling and altered effector responses (Freed et al., 2017). uPar (Plaur) that can participate in ligand-independent EGFR activation (Liu et al., 2002; Smith and Marshall, 2010) was also highly induced in clusters. Therefore, we hypothesized that growth factor production and EGFR activation could support the superior metastatic colonization of clusters.
To test the role of EGFR-associated genes in collective metastatic outgrowth, we used lentiviral RNAi to deplete Egfr, Epgn, Areg, and Plaur in MMTV-PyMT tumor cell clusters (Figure S3A). Importantly, knockdowns suppressed outgrowth in 3D culture but did not disrupt the ability of tumor cells to aggregate, supporting the hypothesis that these factors act downstream of clustering (Figures S3B and S3C). Next, for each condition, equal numbers of clustered cells were injected by tail vein. 3 weeks later, we observed significant reductions in metastatic burden relative to control by Egfr-kd, Areg-kd, and Epgn-kd (Figures 1E and 1F). Strikingly, Epgn-kd reduced metastatic outgrowth in the lungs by >94% and strongly reduced the size, but not total number, of lung metastases (Figures S1H and S1I). Further, lung metastases had reduced reporter GFP expression in Epgn-kd but not non-targeting Ctrl-kd cluster injected mice, indicating selective pressure to escape epigen knockdown (Figures S1J and S1K). Taken together, these data indicate that efficient metastatic outgrowth depends on epigen expression.
Epigen Suppression Switches Tumor Cell Clusters from a Proliferative to a Migratory State
We next sought to determine the biological processes regulated by epigen expression. RNA-seq of Ctrl-kd and Epgn-kd MMTV-PyMT clusters revealed that Epgn-kd clusters downregulate cell cycle genes, upregulate migration-related genes (Figure 2A; Table S4), and their transcription most closely resembles nascent, non-proliferative clusters (Figure 1C). Gene set analysis revealed that Epgn-kd induces expression of genes in cadherin, desmosome, tight junction, and matrix metalloproteinase families (Figure 2C). Although Epgn-kd induced expression of some epithelial-mesenchymal transition (EMT)-associated genes, Epgn-kd organoids showed persistent expression of epithelial genes including Epcam and Cdh1 (Figure 2C), and expression of multiple cytokeratins including Krt5, Krt14, and Krt17 (Figures S3E and S3F) associated with basal/myoepithelial and invasive leader cells (Cheung et al., 2013, 2016).
Figure 2. Epigen Suppression Switches Tumor Cell Clusters from a Proliferative State to a Migratory State.
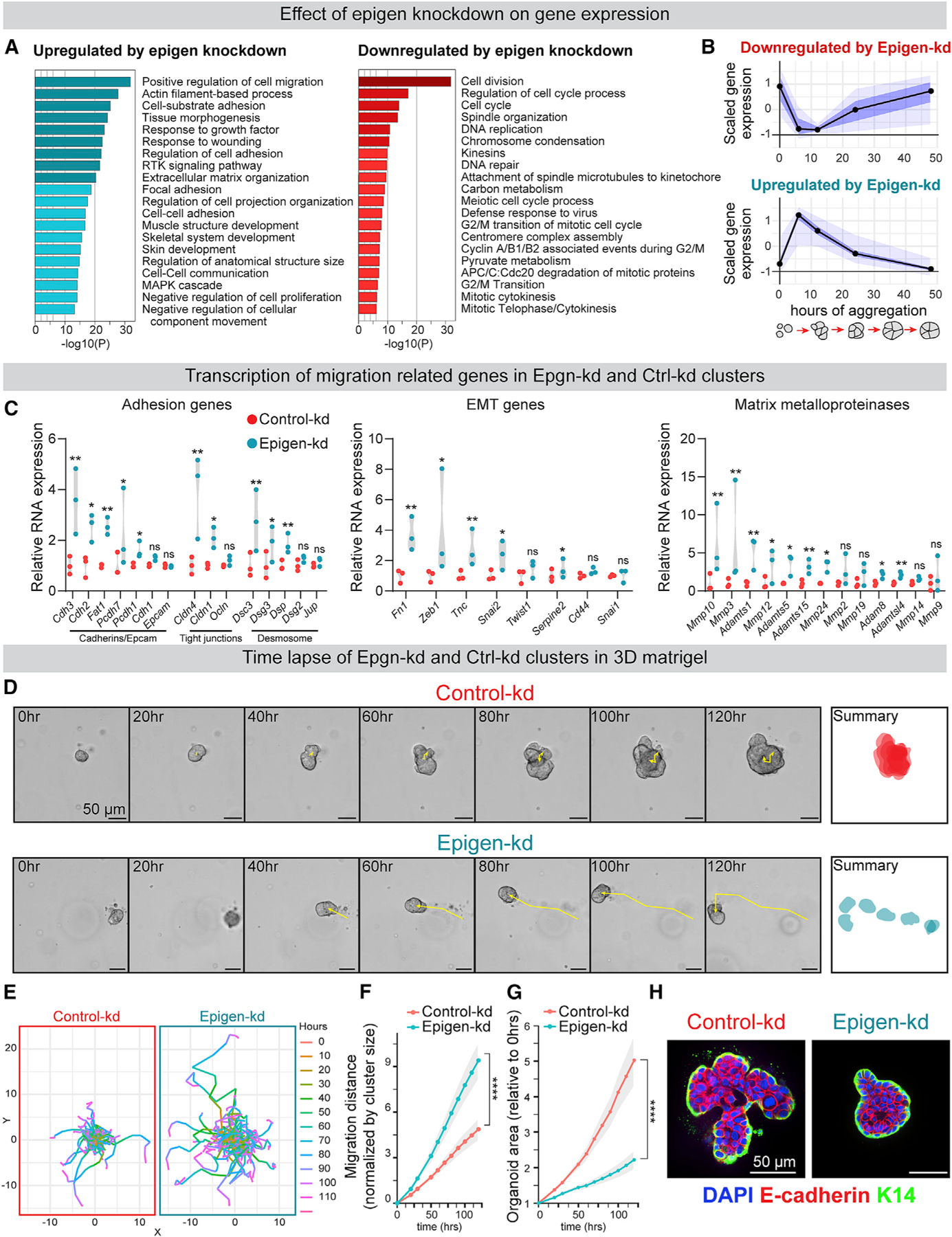
(A) Gene ontology analysis of differentially expressed genes in Epgn-kd clusters versus Ctrl-kd clusters (aggregated 24 h; Metascape, FDR ≤0.05).
(B) Comparison of aggregation induced genes (from Figure 1C) to genes differentially expressed in Epgn-kd versus Ctrl-kd clusters (FDR ≤0.01). n = 39 Epgn-kd downregulated genes, 107 upregulated genes. Black line, median; dark blue, 50% CI; light blue, 95% CI.
(C) RNA expression of selected gene families in Ctrl-kd and Epgn-kd MMTV-PyMT clusters. n = 3, adjusted p values.
(D) Time-lapse images of clusters in 3D basement membrane-rich gels. Right: summary of cluster size and location over 120 h.
(E) Cumulative migration distance of individual clusters in 3D culture, normalized to initial cluster size. n (# of clusters): Ctrl-kd = 106, Epgn-kd = 102. n = 4 biological replicates. p values = unpaired t tests.
(F) Cu mulative migration distance, normalized to initial cluster size, for Ctrl-kd and Epgn-kd clusters in 3D culture. n (# of clusters): Ctrl-kd = 106, Epgn-kd = 102. n = 4 biological replicates. p values = unpaired t tests.
(G) Area measurements, relative to starting cluster area, of Ctrl-kd and Epgn-kd clusters in 3D culture. Mean with 95% CI. n (# of clusters): Ctrl-kd = 106, Epgn-kd = 102. n = 4 biological replicates. p values = unpaired t tests.
(H) Keratin-14 and E-cadherin immunofluorescence of clusters in 3D culture.
To functionally test the effects of epigen suppression on collective outgrowth and migration, Ctrl-kd and Epgn-kd clusters were embedded in 3D basement membrane-rich gels and monitored by time-lapse microscopy (Figure 2D; Video S2). Compared with Ctrl-kd clusters, Epgn-kd clusters showed markedly increased migration but reduced outgrowth and proliferation (Figures 2E–2G and S3D). Consistent with retained mRNA expression of epithelial genes, Epgn-kd clusters migrated not as individual cells but as E-cad+/K14+/K17+ clusters (Figures 2H and S3F). We further confirmed that a second epigen short hairpin RNA (shRNA) reduced epigen expression and outgrowth (Figures S3G and S3H), and that outgrowth was rescued by an shRNA-resistant human epigen construct (Figure S3I). Taken together, these data reveal that epigen expression supports the transition of clustered tumor cells from a collective migratory to proliferative state.
Epigen Acts as a Collective Signaling Factor Shared Non-cell-Autonomously within Clusters
We reasoned that epigen could be secreted from clusters into their local microenvironment, facilitating long-range paracrine signaling (model 1 and Figure 3A). If this was the case, then co-culture with non-transduced clusters should partly rescue the outgrowth defect of nearby Epgn-kd clusters. However, time-lapse imaging indicated that Epgn-kd clusters co-cultured with non-transduced clusters had similar outgrowth as those cultured in 3D gels alone (Figures 3B and 3C). In addition, immunofluorescence revealed that epigen was largely restricted to intercellular areas in clusters from MMTV-PyMT, MMTV-Neu, and C3(1)-TAg mouse models of breast cancer that was depleted upon epigen knockdown (Figures 3D, 3E, and S4A–S4C). Likewise, mTomato-PyMT clusters transduced with an epigen-mGFP fusion construct had enriched GFP signal at intercellular regions (Figure 3F). These data indicate that epigen acts primarily as a short-range outgrowth signal.
Figure 3. Epigen Acts as a Collective Signaling Factor Shared Non-cell-Autonomously within Clusters.
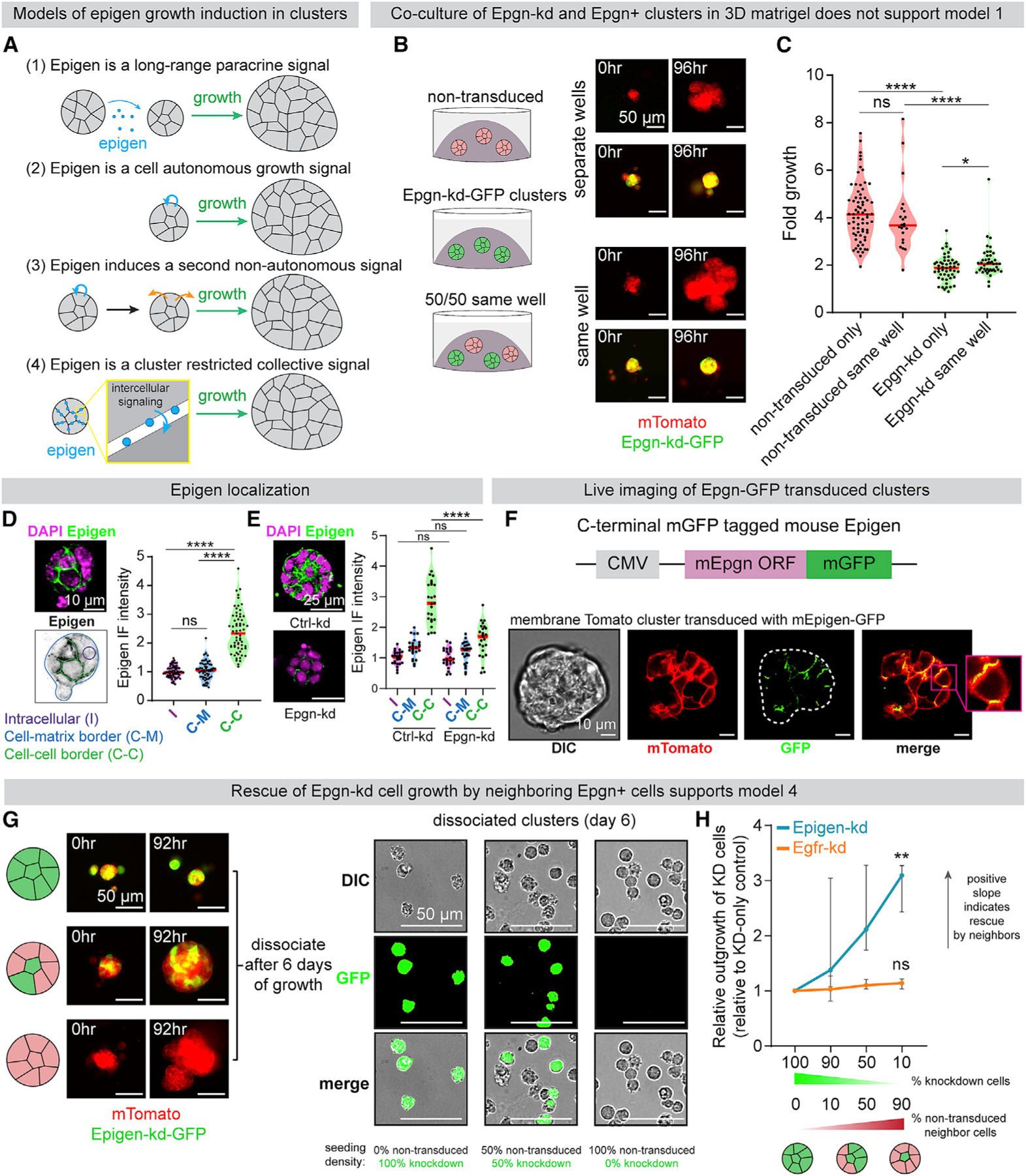
(A) Possible mechanisms by which epigen is a signal for cluster outgrowth.
(B) Epgn-kd and non-transduced clusters were cultured individually or co-cultured in the same 3D gel. Right: time lapse images of each condition.
(C) Time-lapse fold outgrowth measurements of (B). n = 71 non-transduced clusters alone, 22 in co-culture. n = 51 Epgn-kd clusters alone, 44 in co-culture. n = 2 biological replicates. p values = Mann-Whitney test.
(D) Immunofluorescence of epigen in a MMTV-PyMT cluster ex vivo. Bottom: schematic of IF quantification. Right: quantification of epigen signal. n = 3 biological replicates, n = 63 clusters. p values = Mann-Whitney tests.
(E) Immunofluorescence of epigen in Ctrl-kd and Epgn-kd clusters. Right: quantification of mean intensity in different regions. n = 2 biological replicates, 30 clusters per condition. p values = Mann-Whitney test.
(F) Live imaging of a mTomato PyMT cluster expressing epigen-mGFP.
(G) Left: time-lapse images of Epgn-kd-GFP and non-transduced mTomato-PyMT cells aggregated at different ratios to form pure or mosaic clusters. Right: dissociated cells after 6 days of culture.
(H) Relative outgrowth of Epgn-kd or Egfr-kd cells when mixed with different proportions of non-transduced cells. Epgn-kd n = 3 biological replicates (36,022 cells). Egfr-kd n = 4 biological replicates (19,911 cells). Median and 95% CI. p values = unpaired t tests versus 100% knockdown clusters.
See also Figure S4.
We next sought to test whether epigen acts non-cell-autonomously or in a cell autonomous manner (model 2). If epigen ligand is shared non-cell-autonomously among neighboring cells, adding epigen-expressing cells to a cluster should rescue the outgrowth of adjacent epigen deficient cells. Epgn-kd cells were mixed with non-transduced cells to form mosaic clusters (Figure 3G). After 6 days, clusters were dissociated and the number of Epgn-kd cells was counted to assess their outgrowth. Mixing non-transduced cells and Epgn-kd cells in a 9:1 ratio increased the outgrowth of Epgn-kd cells 3-fold higher than when cultured alone (Figure 3H). Further, Epgn-kd cells aggregated with Ctrl-kd cells migrated markedly less than pure Epgn-kd clusters (Figure S4E).
These data suggest that epigen induces outgrowth of neighboring cells non-cell-autonomously. Consistent with this hypothesis, recombinant epigen induced outgrowth in a dose-dependent manner (Figure S4D). However, an alternative explanation is that epigen does not directly promote outgrowth but rather induces expression of a second growth signal transmitted to neighboring cells (model 3). To test this, we performed mosaic mixing experiments with non-transduced cells and Egfr-kd cells. Egfr-kd cells in mosaic clusters did not grow significantly more than when cultured as pure Egfr-kd clusters (Figure 3H), demonstrating that non-transduced cells do not produce a second signal downstream of EGFR that can rescue Egfr-kd outgrowth. Thus, epigen’s intercellular enrichment, short-range restriction, and non-cell-autonomous activity all indicate that epigen acts within clusters as a shared, collective growth signal (model 4).
Secreted Epigen Is Concentrated within Intercellular Nanolumina
We next used transmission electron microscopy (TEM) to characterize the physical properties at the site of epigen accumulation in MMTV-PyMT clusters. To our surprise, we observed that intercellular membranes, rather than being closely apposed, were often separated by hollow cavities, bounded by electron-dense cell-cell junctions and lined by microvilli-like protrusions (Figures 4A and S4F). We refer to these intercellular cavities as “nanolumina.” Importantly, epigen knockdown did not alter nanolumina morphology, indicating that epigen expression is not required for nanolumina formation (Figure S4G).
Figure 4. Epigen Is Stored and Concentrated within Intercellular Nanolumina.
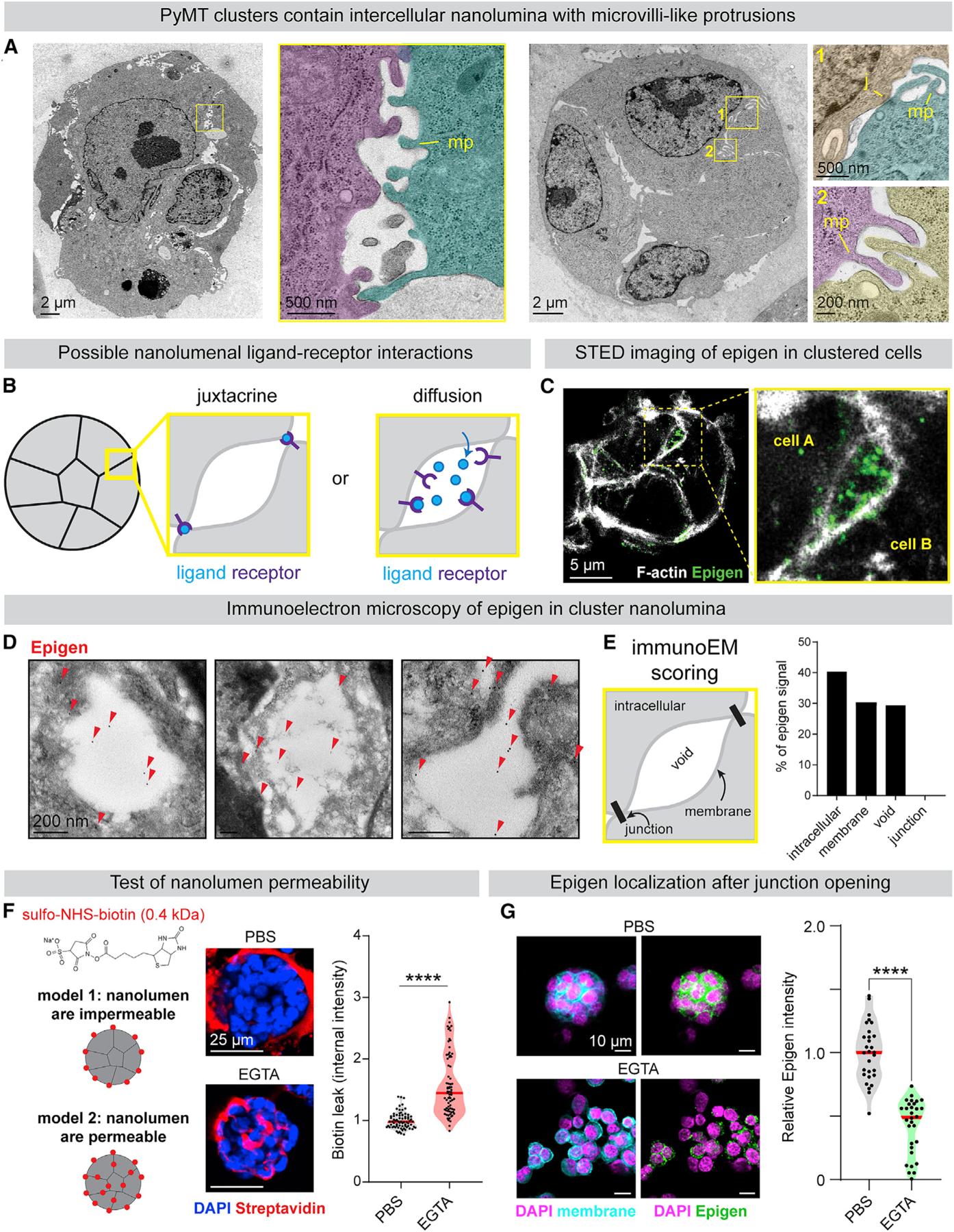
(A) TEM images of MMTV-PyMT clusters (pseudo-colored). MP, microvilli-like protrusions; J, cell-cell junction.
(B) Models of intercellular epigen signaling as a membrane-restricted juxtacrine ligand or a diffusible paracrine ligand.
(C) Epigen immunofluorescence stimulated emission depletion microscopy images in a MMTV-PyMT cluster.
(D) Immunogold labeling of epigen in MMTV-PyMT clusters. Arrows, gold labeling.
(E) Schematic of features scored for Immunogold signal. Right: quantification of epigen Immunogold signal. n = 268 gold particles.
(F) To assess junctional permeability, clusters were incubated with sulfo-NHS-biotin. EGTA was used as a positive control to disrupt cell-cell junctions. Right: quantification of streptavidin-fluorescein isothiocyanate (FITC) fluorescence. n = 3 biological replicates. n = 75 clusters per condition. p values = Mann-Whitney tests.
(G) Immunofluorescence of epigen in clusters ± 1 mM EGTA treatment. Right: median epigen intensity per cluster. n = 3 biological replicates, 59 clusters. p value = Mann-Whitney test.
See also Figures S4 and S5.
We hypothesized that the nanolumina observed in tumor cell clusters could concentrate soluble signaling molecules such as epigen (Figure 4B), not unlike the luminal signaling compartments observed in collectively migrating zebrafish lateral line primordium (Durdu et al., 2014). Alternatively, epigen could localize to areas of direct cell-cell contact and activate EGFR in a membrane-restricted juxtracrine manner. Consistent with the first model, stimulated emission depletion microscopy revealed concentration of epigen within intercellular spaces (Figure 4C). Immunoelectron microscopy also confirmed the presence of epigen within nanolumenal cavities and along nanolumenal membranes but not at direct cell-cell junctions (Figures 4D and 4E). Epigen was not restricted to areas expressing the apical marker Muc1, suggesting that these are not apically polarized lumina (Figure S4H). Together, these findings suggest that epigen diffuses within shared nanolumina.
The accumulation of epigen within nanolumina implied a mechanism for preventing ligand diffusion out of clusters. To characterize junctional permeability, we treated clusters with sulfo-NHS-biotin that labels membrane surface proteins but is excluded from the cytosol. At baseline, biotin was excluded from areas of cell-cell contact, predominantly labeling areas of cell-matrix contact (Figure 4F). Disruption of junctions by calcium chelation, Latrunculin A, or ochratoxin A allowed biotin to leak between clustered cells (Figures 4F and S4I). Likewise, calcium chelation reduced epigen immunofluorescence in clusters (Figure 4G). Although we could not measure the nanolumenal concentration of epigen directly, we estimated it to be >5,000-fold higher than the extra-cluster concentration (223 versus 0.04 ng/mL, Figure S4J). Thus, restricted junctional permeability facilitates a high local concentration of epigen between clustered cells.
To assess the dominant receptor tyrosine kinase pathway active in clusters in an unbiased manner, we performed protein array analysis of 39 phospho-RTKs in single-cell versus cluster lysates. ERBB family members pTyr-EGFR and pTyr-HER2, which can heterodimerize with EGFR, were the most cluster-up-regulated phospho-RTKs (Figure S5A). We observed induction of EGFR effectors pERK1/2 and pAKT in clustered tumor cells (Figures S5B–S5D). In addition, pEGFR and HER2 localized to intercellular areas between clustered tumor cells (Figures S5E and S5F). Using immunoelectron microscopy, we confirmed pEGFR and HER2 localized primarily along nanolumenal membranes, as would be expected of transmembrane receptors (Figures S5G and S5H). These data show that nanolumina form a productive compartment for epigen-EGFR signaling.
Epigen Suppression Reduces Outgrowth of Primary Tumors and Spontaneous Metastases
Having identified the site and mechanism of epigen signaling ex vivo, we next assessed epigen expression and nanolumina formation during metastasis. MMTV-PyMT mTomato+ clusters were transplanted into mammary fat pads of NSG mice. Intercellular epigen was highly expressed in primary tumors and frequently in locally disseminated tumor cell clusters (Figures 5A and 5B). Likewise, distant metastases consistently expressed intercellular epigen (Figures 5D–5F and S6D), pEGFR, and HER2 (Figures S6E and S6F). In contrast, epigen was absent or weakly expressed in locally disseminated single cells (Figure 5B) and in surrounding normal tissues (Figures 5A–5F, S6B, and S6D). In addition, locally disseminated tumor cell clusters adopted two morphologies: strand-like/protrusive and nest-like/non-protrusive. In primary tumors, protrusive clusters had lower epigen expression than their non-protrusive counterparts (Figure 5C) although they retained cytokeratin expression (Figure S6C). Likewise, protrusive clusters adjacent to brain metastases expressed epigen at lower levels than their non-protrusive counterparts (Figure 5F). These findings reveal that epigen expression is lowest in locally disseminated single cells and protrusive clusters and highest in non-protrusive clusters and in primary and metastatic tumors.
Figure 5. Epigen Suppression Reduces Both Primary and Metastatic Tumor Outgrowth.
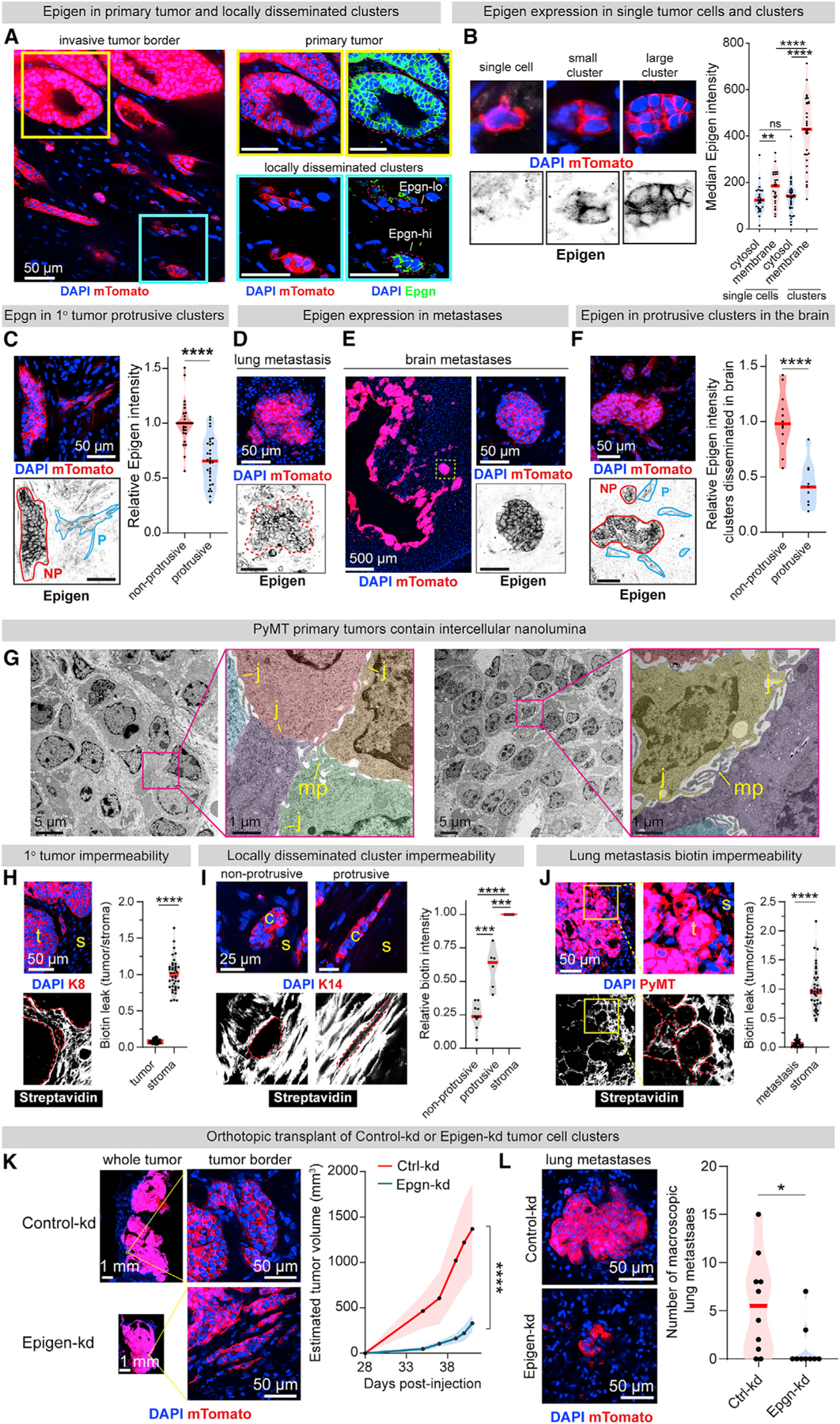
(A) Immunofluorescence of epigen in a mTomato-PyMT primary tumor and adjacent clusters 10 weeks after orthotopic transplant in non-fluorescent NSG mouse.
(B) Quantification of epigen expression in locally disseminated single tumor cells and non-protrusive tumor cell clusters. n = 27 single cells, 33 clusters. n = 2 tumors. p values = Mann-Whitney tests.
(C) Immunofluorescence of epigen in locally disseminated clusters adjacent to a primary tumor. NP, non-protrusive; p, protrusive. Right: relative epigen intensity. n = 3 tumors. n = 29 non-protrusive, 31 protrusive clusters (clusters with intermediate morphology were not scored). p value = Mann-Whitney test.
(D) Epigen immunofluorescence of a spontaneous lung metastasis 10 weeks after mTomato-PyMT organoid orthotopic transplant.
(E) Epigen immunofluorescence in brain metastases 6 weeks after mTomato-PyMT cluster intracardiac injection.
(F) Epigen immunofluorescence in the brain 6 weeks after intracardiac mTomato-PyMT cluster injection. n = 3 mice. n = 12 non-protrusive, 9 protrusive clusters. p value = Mann-Whitney test.
(G) TEM of an MMTV-PyMT tumor. Pseudo-color highlights different cells. MP, microvilli-like protrusions; J, cell-cell junctions.
(H) Immunofluorescence of Keratin-8 (K8) and FITC-streptavidin in MMTV-PyMT primary tumors treated with sulfo-NHS-biotin. Right, FITC-streptavidin quantification. n = 3 tumors, 50 tumor areas (“t”), 50 stromal (“s”) areas. p value = Mann-Whitney test.
(I) Immunofluorescence of Keratin-14 and FITC-streptavidin in locally disseminated clusters from tumors fixed after sulfo-NHS-biotin treatment. Right: for each cluster, internal FITC-streptavidin signal relative to adjacent stroma. n = 15 clusters (9 non-protrusive, 6 protrusive), n = 2 tumors. p values = Mann-Whitney tests.
(J) Immunofluorescence of PyMT and FITC-streptavidin in mouse lungs 2 weeks after tail vein injection of MMTV-PyMT clusters, lungs treated with sulfo-NHS-biotin before fixation. Right: FITC-streptavidin quantification. n = 3 mice, 50 metastasis areas (“t”), 32 stromal areas (“s”). p value = Mann-Whitney test.
(K) Primary tumors 6 weeks after orthotopic transplant of Ctrl-kd or Epgn-kd PyMT clusters. Right: estimated tumor volume. n = 10 Ctrl-kd mice, 9 Epgn-kd mice. p value = Mann-Whitney test.
(L) Left: lung metastases 6 weeks after orthotopic transplant. Right: # of visible GFP+ lung macrometastases per mouse. n = 10 Ctrl-kd mice, 9 Epgn-kd mice. p value = Mann-Whitney test.
See also Figure S6.
TEM of MMTV-PyMT primary tumors revealed extensive intercellular nanolumina similar to those observed ex-vivo (Figure 5G). When tumors were exposed to sulfo-NHS-biotin prior to fixation, biotin was found extensively in the tumor-adjacent stroma but was largely excluded from tumor cell nests (Figure 5H). We observed increased biotin permeability in protrusive compared with non-protrusive clusters (Figure 5I). Like primary tumors, lung metastases were largely biotin impermeable (Figure 5J). Thus, restricted nanolumenal permeability and epigen accumulation occur at phases of metastasis associated with outgrowth.
To assess the functional role of epigen in vivo, Ctrl-kd and Epgn-kd clusters were orthotopically transplanted into the mammary fat pads of NSG mice. We observed significantly slower growth of Epgn-kd tumors versus control tumors (Figure 5K) and a modest increase in locally disseminated clusters in the adjacent stroma (Figure S6G). Importantly, Epgn-kd lung metastases were significantly less proliferative (Figure S6H) and smaller (Figure S6I) than Ctrl-kd metastases. Although Epgn-kd mice harbored fewer macrometastases (Figure 7L), the number of micrometastases was similar between Ctrl-kd and Epgn-kd mouse lungs (Figure S6J), supporting a larger effect on metastatic outgrowth than initial seeding. Epgn-kd tumors lost expression of the shRNA-GFP construct more frequently than Ctrl-kd tumors (Figure S6K), indicating selective pressure to revert to Epgn-high phenotypes. Thus, epigen suppression reduces primary tumor and metastatic outgrowth, while increasing local dissemination of clusters, consistent with our ex vivo findings.
Figure 7. HCC70 Outgrowth Depends on Epigen Expression and Is Sensitive to IFNγ that Induces Nanolumen Permeability.
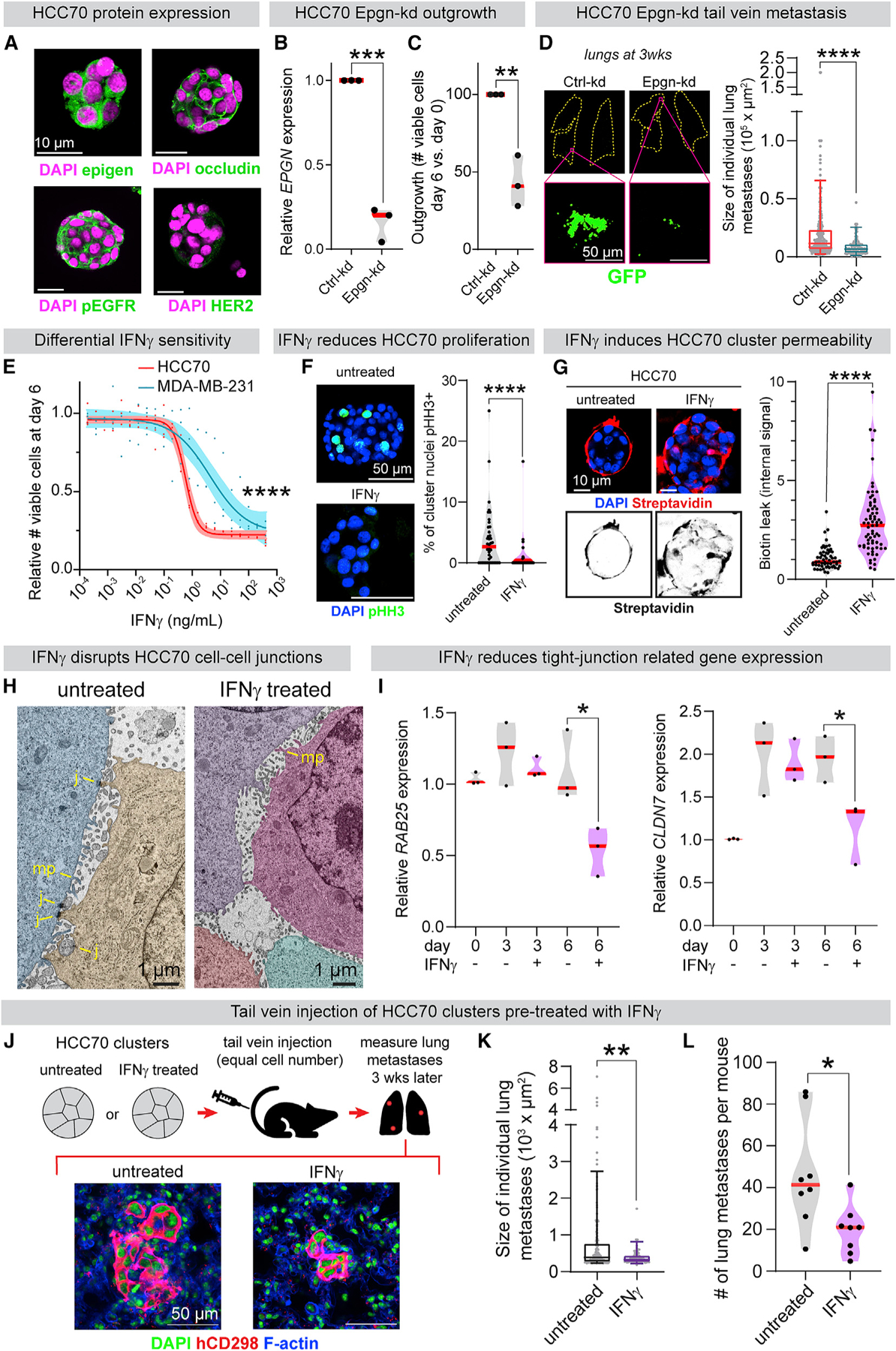
(A) Immunofluorescence of HCC70 (basal-like 2, epigen high cell line) clusters.
(B) qPCR of EPGN in Ctrl-kd and Epgn-kd HCC70 clusters. n = 3 biological replicates. p values = unpaired t tests.
(C) Ctrl-kd and Epgn-kd HCC70 viable cell count (day 6 versus day 0). n = 3 biological replicates. p values = unpaired t tests.
(D) Left: lungs 3 weeks after tail vein injection of Ctrl-kd or Epgn-kd HCC70 clusters. Right: individual metastasis areas. n (# of metastases): Ctrl-kd = 299, Epgn-kd = 87. n = 7 mice/condition. Boxplot, 5%–95%. p value = Mann-Whitney test.
(E) Viable cell quantification after 6 days of IFNγ treatment. Line, non-linear regression; band, 95% CI. n = 3 biological replicates. p value = extra sum-of-squares F test.
(F) pHH3 IF to mark mitotic cells in HCC70 clusters after 6 days ± 3 ng/mL IFNγ treatment. Right: % pHH3+ nuclei in clusters from each condition. n = 3 biological replicates. n = 40 untreated, 34 IFNγ-treated clusters. p value = Mann-Whitney test.
(G) Biotin permeability of HCC70 clusters after 6 days ± 3 ng/mL IFNγ. Right: internal FITC-streptavidin intensity per cluster. n = 60 untreated, 70 IFNγ-treated clusters. n = 3 biological replicates. p value = Mann-Whitney test.
(H) TEM of HCC70 clusters after 6 days ± IFNγ treatment (3 ng/mL). Cells pseudo-colored. MP, microvilli-like protrusions; J, cell-cell junctions.
(I) qPCR of (left) RAB25 and (right) CLDN7 in HCC70 clusters ± 3 ng/mL IFNγ treatment. n = 3 biological replicates. p value = unpaired t test.
(J) HCC70 clusters (24 h aggregation) were treated ± 2 ng/mL IFNγ. After 6 days, clusters from each condition were injected by tail vein into NSG mice (200,000 viable clustered cells per mouse). 3 weeks later, lung metastases were scored using a human-specific CD298 antibody.
(K) Sizes of individual HCC70 metastases 3 weeks after cluster tail vein injection. n = 8 mice/condition, 217 untreated metastases, 79 IFNγ pre-treated metastases. Boxplot, 5%–95%. p value = Mann-Whitney test.
(L) Number of lung metastases per mouse 3 weeks after HCC70 cluster tail vein injection ± IFNγ (2 ng/mL) pre-treatment. n = 8 mice/condition. p value = Mann-Whitney test.
See also Figure S7.
Epigen Expression and Nanolumina Formation Are Associated with Basal-like 2 Triple-Negative Breast Cancer
We next sought to determine the generality of our findings to human breast cancer. Tumor cell clusters were collected from a breast cancer patient recruited to an institutional review board (IRB)-approved study at Fred Hutchinson Cancer Research Center. At the time of enrollment, the patient had ER+/PR+/HER2− breast cancer that was metastatic to orbit, lung, liver, and bone. CTC clusters were detected at the second and third time points, coinciding with clinical evidence of disease progression (Figures 6A and 6B). Palliative paracentesis revealed malignant ascites containing cytokeratin-positive/CD45-negative tumor cell clusters (Figure 6C), phosphorylated EGFR at intercellular contacts (Figure 6C), and enriched epigen expression relative to adjacent CD45+ cells (Figure S7B). Further, TEM revealed nanolumina with microvilli-like protrusions in ascites-derived tumor cell clusters (Figure 6D). Malignant ascites were also collected from a second patient with metastatic ER+/PR+/HER2− breast cancer. At the time of paracentesis, she was treated with letrozole and abemaciclib and had metastatic disease to bone, liver, chest wall, stomach, and peritoneum. Similarly, tumor cell clusters collected from this patient demonstrated intercellular pEGFR and epigen expression as well as intercellular nanolumina (Figures S7A and S7B). These clinical observations support the disease relevance of nanolumenal signaling for in vivo metastasis.
Figure 6. Epigen Expression and Nanolumina Formation Are Associated with the Basal-like 2 Subgroup of Triple-Negative Breast Cancer.
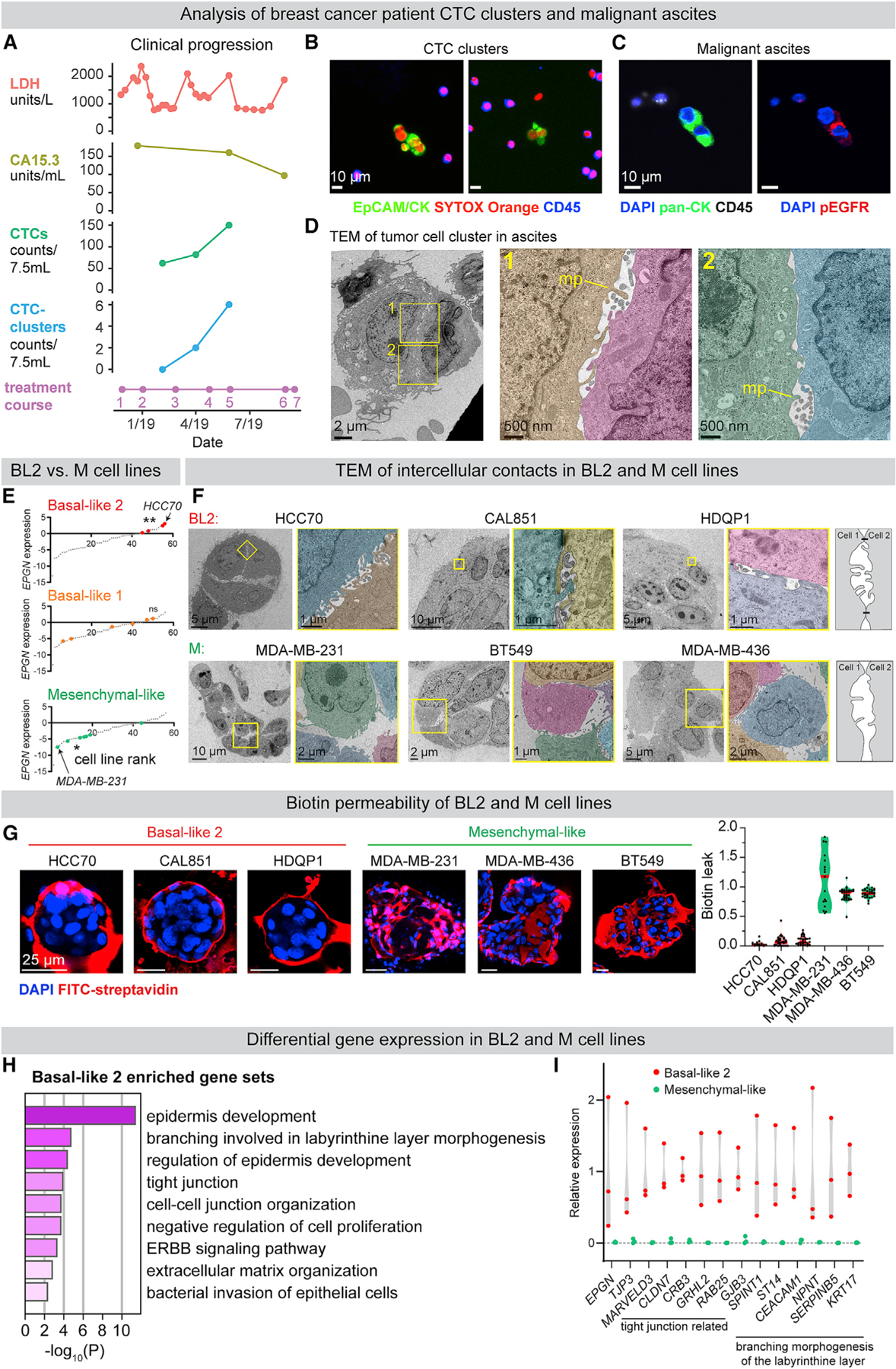
(A) Clinical progression of a patient with metastatic ER+/PR+/HER2− breast cancer treatment course: (1) vinorelbine, (2) cytoxan/methotrexate, (3) drug holiday, (4) cytoxan/methotrexate, (5) irinotecan, (6) low dose doxorubicin, and (7) hospice. LDH, serum lactate dehydrogenase; CA15.3, cancer antigen 15–3.
(B) CTC clusters in the blood. EpCAM/pan-CK, epithelial cells; CD45, immune cells; SYTOX Orange, nucleic acid stain.
(C) pEGFR (Y1068) IF of tumor cell clusters in malignant ascites (pan-CK+, epithelial cells; CD45+, immune cells).
(D) TEM of malignant ascites from a metastatic breast cancer patient. MP, microvilli-like protrusion.
(E) Epigen mRNA expression in human cell lines (Barretina et al., 2012) classified by triple-negative breast cancer sub-groups (Lehmann et al., 2011). p values = Kolmogorov-Smirnov tests.
(F) TEM of tumor cell clusters from basal-like 2 and mesenchymal-like breast cancer cell lines. Right: schematic of intercellular morphologies.
(G) Test of cluster permeability using sulfo-NHS-biotin. Right: quantification of biotin leak (internal biotin signal/membrane biotin signal) in clusters. n (# clusters/# biological replicates): HCC70 = 25/2, CAL851 = 30/3, HDQP1 = 30/3, MDA-MB-231 = 17/2, MDA-MB-436 = 30/3, and BT549 = 30/3.
(H) Gene sets (Metascape, FDR ≤0.01) enriched in BL2 (HCC70, CAL851, and HDQP1) versus M-like (MDA-MB-231, MDA-MB-436, and BT549) cell lines (CCLE).
(I) RNA expression of individual genes (CCLE) in BL2 (HCC70, CAL851, and HDQP1) versus M-like (MDA-MB-231, MDA-MB-436, and BT549) cell lines.
As a second approach, we examined epigen expression in two RNA-seq compendia of human breast cancer cell lines (Barretina et al., 2012; Klijn et al., 2015). Triple-negative cell lines have been previously classified based on their gene expression into subgroups: basal-like 1 (BL1), basal-like 2 (BL2), mesenchymal-like (M-like), and luminal androgen receptor (LAR) (Lehmann et al., 2011, 2016). We observed a strong association between EPGN expression and the basal-like 2 subgroup, whereas mesenchymal-like breast cancer cell lines had the lowest EPGN expression (Figures 6E and S7C). BL2 triple-negative breast cancers have increased growth factor signaling, basal/myoepithelial gene expression, and poor response rates to chemotherapy (Masuda et al., 2013; Ring et al., 2016).
TEM revealed considerable morphological differences between three BL2, epigen-high cell lines (HCC70, CAL851, and HDQP1) and three M-like, epigen-low cell lines (MDA-MB-231, MDA-MB-436, and BT549). BL2 cell clusters contained nanolumina with extensive microvilli-like protrusions, often sealed by electron-dense junctional complexes (Figure 6F). In contrast, M-like cell clusters were loosely organized and largely lacked junctionally sealed intercellular spaces. Further, BL2 cell clusters were highly biotin-impermeable, whereas biotin readily leaked into the intercellular spaces of M-like clusters (Figure 6G). Importantly, transcriptional analysis of these 6 cell lines revealed that BL2/epigen-high line gene expression was enriched for genes related to epithelial development and migration, branching morphogenesis of the placental labyrinthine layer (Rinkenberger and Werb, 2000), and tight junctions (Figures 6H and 6I; Table S5). M-like cell lines, on the other hand, were enriched for genes related to collagen metabolism and FGFR signaling relative to BL2 lines (Figure S7D; Table S5). These data show that epigen enrichment and nanolumina occur in aggressive metastatic breast cancers and reveal large differences in nanolumina formation, junctional permeability, and morphogenesis-related gene expression between BL2 and M-like cells.
HCC70 Outgrowth Depends on Epigen Expression and Is Sensitive to Interferon (IFN)γ that Induces Nanolumen Permeability
HCC70 was the highest EPGN expressing cell line in both RNA-seq datasets (Barretina et al., 2012; Klijn et al., 2015). Notably, when compared to a transcriptomic dataset of metastatic solid tumors (MET500), HCC70 cells modeled the gene expression of basal-like breast cancers more closely than other tested cell lines (Liu et al., 2019a; Robinson et al., 2017). We observed that HCC70 clusters expressed epigen, pEGFR, and the tight junction protein occludin, but not HER2 (Figure 7A). Importantly, EPGN knockdown in HCC70 clusters significantly reduced their outgrowth ex vivo (Figures 7B and 7C). No significant differences in migration or outgrowth were observed in 2D (Figures S7E and S7F). Transduction with an EPGN single guide RNA (sgRNA) likewise reduced HCC70 cluster outgrowth (Figure S7G). To assess epigen’s role in metastatic outgrowth, Epgn-kd HCC70 cell clusters were injected by tail vein into immunocompromised NSG mice. 3 weeks later, the Epgn-kd clusters had formed significantly smaller metastases than Ctrl-kd clusters (Figure 7D). Thus, epigen dependence and nanolumina formation occur in both mouse and human models of breast cancer.
The existence of nanolumina suggested that this structure could be targeted to reduce collective signaling during metastasis. The cytokine IFNγ can enhance paracellular diffusion (Bruewer et al., 2003; Madara and Stafford, 1989) and increase epithelial flux to amplify immune responses (Turner, 2009). We hypothesized that IFNγ could weaken the barriers sealing nanolumina and inhibit cluster outgrowth. Consistent with this hypothesis, IFNγ treatment strongly reduced HCC70 tumor cell cluster outgrowth (IC50 = 0.58 ng/mL) (Figure 7E). IFNγ reduced proliferation but did not increase cell death in HCC70 clusters (Figures 7F and S7H). Importantly, IFNγ treatment increased biotin permeability of HCC70 clusters (Figure 7G) and altered nanolumenal morphology (Figure 7H). Additionally, HCC70 clusters had reduced mRNA expression of tight junction related genes RAB25 and CLDN7 after IFNγ treatment (Figure 7I). Conversely, MDA-MB-231 clusters were far less sensitive to IFNγ treatment (Figure 7E), lowly expressed RAB25 and CLDN7 (Figure S7I), and were highly biotin-permeable with or without IFNγ treatment (Figure S7J).
Next, we tested the effects of IFNγ on metastatic outgrowth. After 6 days of IFNγ (2 ng/mL) treatment, equal viable cell numbers of HCC70 clustered cells were injected by tail vein into NSG mice (Figure 7J). 3 weeks later, HCC70 metastases in the lungs were identified using a human-specific antibody (Lawson et al., 2015). IFNγ pre-treated clusters formed significantly smaller and fewer metastases than untreated controls (Figures 7K and 7L). Although IFNγ is likely to have pleotropic effects, these data suggest that a nanolumen-opening agent could impair the collective metastatic outgrowth of tumor cell clusters.
DISCUSSION
Metastasis is regulated by factors both internal to the cancer cell as well as arising from interactions with its tumor microenvironment (Fidler and Kripke, 2015; Lambert et al., 2017; Massagué and Obenauf, 2016). Our study reveals an additional dimension: the shared intercellular exchange of growth factors between clustered tumor cells. Here, we find that multicellular clusters form nanolumina—intercellular compartments that accumulate a high local concentration of epigen (Figure S7K). In this way, tumor cell clusters construct their own internal microenvironment and collective signals to drive metastatic outgrowth.
Recent studies have demonstrated that tumor cells can cooperatively support polyclonal growth and metastasis through secreted paracrine signals between genetically distinct clones (Cleary et al., 2014; Janiszewska et al., 2019; Marusyk et al., 2014). Here, we uncover a distinct mechanism in which cooperative signaling is generated by the physical architecture of nanolumina. In this regard, our findings share parallels with the concentration of FGF within lumens in migrating lateral line primordia (Durdu et al., 2014), but the compartments we observe here are typically nanometer-scale in diameter and do not appear apically polarized. Recent findings have similarly emphasized the impact of 3D topology on signaling during development and in generating synthetic biological systems (Durdu et al., 2014; Gilmour et al., 2017; Shyer et al., 2015; Simian and Bissell, 2017; Toda et al., 2018; Xavier da Silveira Dos Santos and Liberali, 2019). Our study supports the parallel importance of 3D multicellular architecture in shaping signaling during metastasis.
Although single cell metastasis has been conceptualized as a series of switch-like transitions (Lambert et al., 2017; Massagué and Obenauf, 2016), the factors regulating transitions during collective metastasis are far less understood. Our findings reveal an epigen signaling axis regulating switching between proliferative and migratory modes. Importantly, loss of epigen does not result in dissolution of clusters to individualized tumor cells. Rather, our findings suggest that the nanolumen compartment and its epigen content are dynamically regulated. In this study, we also observed that locally disseminated clusters that are highly protrusive are more biotin permeable than rounded, non-protrusive clusters. Future studies are indicated to understand the plasticity, regulatory factors, and metastatic potential of clusters with differing permeability. In addition, interrogating nanolumen composition could reveal other signaling molecules regulating transitions during multicellular metastasis (Grigore et al., 2016; Gritsenko et al., 2020; Haeger et al., 2020; Labernadie et al., 2017; Silvestri et al., 2020; Zhang et al., 2019).
Nanolumenal signaling could also play an important role in normal development and homeostasis. Normal mammary epithelia contain interdigitating structures similar to the nanolumina characterized here, but whose function has remained unknown (Ewald et al., 2012). The regulation of epigen signaling during phases of metastatic outgrowth and migration suggest parallels with the proliferation and collective migration of developing mammary terminal end buds (Macias and Hinck, 2012; Paine and Lewis, 2017; Visvader and Stingl, 2014). Likewise, the enrichment of genes related to labyrinthine placental morphogenesis (Rinkenberger and Werb, 2000) in BL2 cancer cells may hint at a primordial developmental role for nanolumina that is being co-opted during metastasis, analogous to the co-option of EMT programs in mesenchymal cancer cells (Dongre and Weinberg, 2019; Nieto et al., 2016). Future studies are warranted to interrogate nanolumen signaling in normal and cancer contexts and to develop approaches to therapeutically target nanolumina in collective metastasis.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kevin Cheung (kcheung@fredhutch.org).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
RNA sequencing datasets have been deposited to the NCBI Sequence Read Archive (PRJNA648151, PRJNA648435).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal models
All mice were maintained under specific-pathogen-free conditions, and experiments conformed to the guidelines as approved by the Institutional Animal Care and Use Committee of Fred Hutchinson Cancer Research Center (FHCRC). FVB/N-Tg(MMTV-PyVT) 634Mul/J (MMTV-PyMT) were maintained and tumor growth was monitored every 2 days. MMTV-PyMT mice were crossed with ROSA mTomato/mGFP mice (Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J) to generate mTmG-PyMT mice with mTomato+ cell membranes. For injection experiments, immunocompromised NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) were used. 6–15 week old female mice were used for all procedures.
Human breast cancer patient samples
Deidentified human breast cancer primary or metastatic samples were received from (1) the Cooperative Human Tissues Network, a program funded by the National Cancer Institute, shipped overnight on wet ice or (2) received same day from the University of Washington/Seattle Cancer Care Alliance, with patients consented and samples obtained under a Fred Hutch IRB approved study (FH5306). Tumor samples were processed as previously described to generate organoids (Nguyen-Ngoc et al., 2012), detailed below. Blood and ascites fluid were collected from patients consented and samples obtained under a Fred Hutch IRB approved study (FH8649) for longitudinal monitoring of circulating tumor cells in metastatic breast cancer patients. CTCs and CTC-clusters were enumerated in these fluid samples using a RareCyte assay (Kaldjian et al., 2018). For details on the receptor status and pathology of each deidentified human sample used, see Table S1.
Cell lines
293FT (ThermoFisher Scientific R70007), MDA-MB-231-YFP (generated from ATCC HTB-26), CAL-85–1 (DSMZ ACC 440), HDQ-P1 (DSMZ ACC 494), and MDA-MB-436 (ATCC HTB-130) cell lines were grown at 37°C, 5% CO2 in DMEM high glucose (GIBCO 10569–010) supplemented with 10% fetal bovine serum (Sigma-Aldrich F0926–500ML) and 1X penicillin/streptomycin (Sigma-Aldrich P4333). BT549 (ATCC HTB-122) and HCC70 (ATCC CRL2315) cell lines were grown at 37°C, 5% CO2 in RPMI (GIBCO 61870–127) supplemented with 10% fetal bovine serum (Sigma-Aldrich F0926–500ML) and 1X penicillin/streptomycin (Sigma-Aldrich P4333). For non-adherent culture, cell lines were cultured in complete media + 2% (v/v) Matrigel. All cell lines used were from human females. MDA-MB-231-YFP cells were a gift from Cyrus Ghajar.
METHOD DETAILS
Tail vein and intracardiac injection of NSG mice
For single cell versus cluster experiments, mTomato-PyMT organoids were dissociated to single cells at day 0 using Accumax (20 minutes at 37°C). 200,000 cells in 200 uL DPBS were injected per mouse in the tail vein. To generate clusters, single cells were plated in non-adherent dishes at 150,000 cells/mL in media +2% basement membrane-rich gel (v/v) and then injected as above 6hr or 24 hr later into Nod scid gamma (NSG) immunocompromised mice. shRNA knockdown injections were all carried out with 24hr aggregated clusters. DIC images were taken before injection and assessed to ensure similar number and size of clusters injected between conditions. 3 weeks after tail vein injection mice were euthanized and lungs imaged under a dissecting microscope for quantification of fluorescent (metastatic) area. For intracardiac injections, 100,000 mTomato-PyMT clustered cells were injected into the left ventricle in 100 µL of PBS using a 26 g needle with ultrasound guidance with 2.5% isoflurane anesthesia. 6 weeks later the brain, liver, lung, femur, kidneys, and ovaries were collected. Collected organs were fixed in 4% PFA for 4 hr, then transferred to 25% sucrose in DPBS overnight at 4°C before embedding in OCT and storing at −80°C.
Mouse mammary tumor organoid culture
Organoids were isolated from MMTV-PyMT, C3(1)TAg, or MMTV-Neu mouse mammary tumors as previously described (Nguyen-Ngoc et al., 2015). Mice were harvested as the largest tumor neared 1.5 cm in diameter. MMTV-PyMT mammary tumors were dissected, mechanically disrupted with a scalpel, and then digested in a collagenase-trypsin solution for 30–60 minutes shaking at an angle at 100–150 rpm at 37°C. The digestion solution (in 20 mL of DMEM/F12) contained 2 mg/mL collagenase (Sigma C2139), 2mg/mL trypsin (GIBCO 27250–018), 5% fetal bovine serum, 5 µg/mL human insulin (Sigma-Aldrich I9278), 50 µg/mL gentamicin (GIBCO 15750–060). Tumor fragments were centrifuged for 10 minutes at 1500 rpm, resuspended in 4 mL DMEM/F12 and treated with 40 µL (2000 U/mL) of DNase (Sigma D4263) for 3 minutes then resuspended in 10mL DMEM/F12. The solution was centrifuged for 4 s at 1500 rpm (453 g) to isolate multicellular organoids, then resuspended in 10 mL DMEM/F12. This was repeated for a total of 4 washes. For 3D culture, 100–200 clusters were embedded in 100 uL of growth-factor reduced Matrigel (Corning), the Matrigel was allowed to polymerize for 30–60 minutes, then 1 mL of organoid media (DMEM-F12, 2.5 nM FGF2, insulin-transferrin selenium, & penicillin/streptomycin) was added. For suspension culture, clusters were cultured in non-adherent dishes in organoid media + 2% (v/v) Matrigel (which we term low basement membrane suspension culture). Tips and tubes used to handle organoids were first coated in 2.5% BSA in DPBS to prevent loss of material. To create single cell suspensions, organoids were centrifuged, resuspended in Accumax (Innovative Cell Technologies) for 20 minutes at 37°C, and counted on a hemocytometer to ensure a low number of residual clusters.
Human cell line and patient tumor organoid culture
Human cell lines were cultured in complete media (DMEM or RPMI with penicillin/streptomycin + 10% FBS) on tissue-culture treated plates, or in non-adherent plates supplemented with 2% Matrigel (v/v). To assess migration in 2D, 100,000 viable cells in 10 uL media were seeded as a droplet at the center of a Collagen I coated 24 well plate. After 30 minutes at 37°C, 1 mL of complete media was added. Tiled DIC and GFP images were taken 1 day and 6 days after plating. Deidentified human breast cancer primary or metastatic samples were received from the Cooperative Human Tissues Network and the Seattle Cancer Care Alliance. Tumor samples were processed as previously described to generate organoids (Nguyen-Ngoc et al., 2012). Samples were washed with DPBS containing 200 U/mL penicillin/200 µg/mL streptomycin (Sigma-Aldrich P4333) and 5 µg/mL Amphotericin B (ThermoFisher Scientific 15290–018). They were then minced with a scalpel and resuspended in digestion solution. The digestion solution (in 40 mL of DMEM/F12) contained 2 mg/mL collagenase (Sigma C2139), 5% fetal bovine serum, 1X penicillin/streptomycin (Sigma-Aldrich P4333), and 5 µg/mL human insulin (Sigma-Aldrich I9278). Samples were incubated at 37°C shaking at 200 rpm for 2–5 hours. Tumor fragments were centrifuged for 10 minutes at 1500 rpm, resuspended in 4 mL DMEM/F12 and treated with 40 µL (2000 U/mL) of DNase (Sigma D4263) for 3 minutes then resuspended in fresh 10mL DMEM/F12. The solution was centrifuged for 4 s at 1500 rpm (453 g) to isolate multicellular organoids. Human organoids were cultured with 2% (v/v) or 100% growth factor reduced Matrigel (Corning) with complete HuMEC Ready media (Fisher 12752010).
Scoring metastatic foci and outgrowth after tail vein injection
3 weeks after tail vein injection, lungs were removed and imaged under a dissecting microscope. Bright field and DsRed images were taken for all experiments using mTomato-PyMT cells, GFP images were collected for cells expressing GFP-shRNAs. To measure outgrowth in whole lungs or lung sections, fluorescence images were thresholded equally in FIJI software (Schindelin et al., 2012) and total fluorescent area was measured. For each tail vein injected mouse, the total area of lung metastases was measured as the cm2 GFP+ area in the lungs using FIJI software. For each animal, the total area of lung metastases was normalized by the mean total metastatic area of Ctrl-kd mice within each cohort of tail vein injections. This allowed the effect size of control versus gene knockdowns to be compared, accounting for intrinsic differences in organoid line growth rates between injections. Statistical analysis was performed using one-way ANOVA followed by Dunnett’s Test for multiple comparisons. To measure individual metastasis areas, the area of each visible metastasis was measured manually using FIJI software. To identify and measure micrometastases, 50 um thick lung sections were imaged at 10X magnification, tiled, and assembled. Metastasis counts from lung sections were normalized to the area of the lung imaged.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde (10 min), permeabilized 30 minutes with 0.5% Triton-X, and blocked 1–2 hr at room temperature with 10% FBS/1% BSA/0.1% Triton-X in DPBS. For cells in suspension culture or in malignant ascites, cells were centrifuged onto tissue pathology slides using a Cytospin 4 (A78300003), 800 rpm for 5 minutes. Slides were fixed for 10 minutes in 4% paraformaldehyde and then treated as above. Primary antibodies were added in block solution and incubated at 4°C overnight. Secondary antibodies (1:200) were incubated for 2–3 hr at room temperature with 5% host serum. Confocal mages were acquired using an Andor CSU-W confocal spinning disk on a Leica DMi8 inverted microscope. For Leica 3X STED imaging, cells were stained with Alexa Fluor 594 Phalloidin (ThermoFisher Scientific A12381) and imaged using a Leica TCS SP8. DAPI was not included for STED imaging due to high background.
Time-lapse imaging for apoptosis, migration, and outgrowth analysis
Single or clustered cells were plated in growth factor reduced basement membrane-rich gels. Differential interference contrast (DIC) and fluorescent images were captured hourly using a Leica SPE at 10X or 20X magnification. For growth assays, DIC images were acquired. For survival assays, 30 minutes before imaging NucView 488 (Biotium) in PBS was added to the media at 1:1000 to mark nuclei in cells with active executioner caspases. Exposure times were < 30ms for BF/DIC, and ~150ms for fluorescence. Temperature was maintained at 37°C and CO2 at 5%. Motility and NucView positivity were used to score apoptosis in single cells and clusters. Clusters were marked as dead when the large majority of cells had died i.e., if a few cells were still alive in the apoptotic debris after most the cluster died, the cluster was not scored as alive. FIJI software was used to score the area of cells or clusters to measure outgrowth (as final area/initial area). Objects smaller than 250 µm2 were scored as single cells (this cutoff may have occasionally included small MMTV-PyMT doublets). Images were acquired using a Leica DMi8 TCS SPE. For migration analysis, the centroid position of each individual organoid was tracked over time relative to static objects in the gel (e.g., debris). Each migration track was then normalized by the number of cells per organoid, determined by dividing the area of the organoid at time = 0 hr by the mean µm2 area per cell, which here we determined to be ~76 µm2 (n = 71 organoids, 8613 cells). Cumulative path was determined by summing over the path length of each migration track.
Mixed culture experiments measuring Epgn-kd outgrowth with non-transduced neighbors
On day zero, 22,500 cells were plated in non-adherent 96 well plate at 100% knockdown, 90% knockdown/10% non-transduced (e.g., 20250 knockdown cells, 2250 non-transduced cells), 50% knockdown/50% non-transduced, or 10% knockdown/90% non-transduced. On day 6, each well was centrifuged and resuspended in Accumax for 20 minutes at 37°C. Dissociated single cells were resuspended in organoid media and plated in glass-bottomed 8-well plates, allowed to settle, then imaged at 40X magnification (at least 10 fields per well). GFP positive and negative cells were manually counted in each field. For GFP+ cell growth calculations, this measurement was normalized to the starting number of GFP+ knockdown cells plated.
Transmission electron microscopy
Tumor cell clusters from cell lines, organoids, patient samples, or primary tumors (minced with a scalpel to < 1mm3) were centrifuged, washed in DPBS, then fixed in 1/2 strength Karnovsky’s buffer (2% PFA/2.5% glutaraldehyde in 0.1M cacodylate buffer) at 4°C at 16 hr. Samples were then processed by the Fred Hutchinson Cellular Imaging core. Samples were visualized using a JEOL-1400 transmission electron microscope operated at 120 kV. For ascites-derived samples, tumor cells were identified based on gross morphological differences from stromal and blood cells in the same sample, as well as by the presence of electron dense cell-cell junctions.
Immunoelectron microscopy
For pEGFR and epigen immunogold staining, MMTV-PyMT clusters were collected and fixed in 4% PFA. For HER2 immunogold staining, MMTV-PyMT clusters were collected in 4% PFA + 0.1% glutaraldehyde. Approximately 70nm ultra thin sections were picked up on nickel grids (from Ted Pella, Inc., performed by Fred Hutchinson Cellular Imaging core). Grids were blocked with 50 mM glycine for 20 minutes, washed in PBS 3 times, blocked in 5% BSA for 30 minutes, washed with incubation buffer (1% BSA-C, 0.16% Tween-20 in PBS) 6 × 4 minutes, incubated with primary antibody (R&D MAB11271, Abcam ab40815, or CST 2165) for 2 hr in incubation buffer, washed in PBS 4 times, washed in incubation buffer 6 × 4 minutes, incubated with secondary antibody (10nm gold goat-anti-rabbit or goat-anti-rat), washed in incubation buffer 10 × 4 minutes, post-fixed in 2% glutaraldehyde for 10 minutes, and washed in warm DI water. Samples were visualized using a JEOL-1400 transmission electron microscope with a Gatan Rio 4K camera. Images were compared against a negative control not incubated with primary antibody to ensure secondary specificity.
Biotin permeability testing and immunofluorescence
To test the permeability of tumor cell clusters, NHS-biotin was added to a final concentration of 0.8 mM in PBS for 30 minutes at 37°C. Treatment with 1mM EGTA, Latrunculin A, or Ochratoxin A was for 1hr, prior to biotin incubation. Treatment with interferon gamma was for 6 days prior to biotin permeability experiments. Cells were washed three times with cold PBS then centrifuged onto tissue pathology slides using a Cytospin 4 (800 rpm for 5 minutes) and fixed with 4% PFA for 10 minutes. Freshly dissected primary tumors or lungs were immersed in 0.8 mM sulfo-NHS-biotin in PBS for 30 minutes then washed 3 times with PBS prior to fixation and embedding in OCT. Immunofluorescence was conducted (as above) with FITC-conjugated streptavidin to localize biotin. Human breast cancer cell lines were cultured for 6 days as clusters in suspension before assessing biotin permeability.
Immunofluorescence quantification
Phalloidin staining and DAPI were used to define membrane and nuclear areas, respectively. Using the freehand line tool in FIJI software, lines along cell-matrix (outer membrane), cell-cell (intercellular) membranes, and intracellular (cytosolic) areas were drawn and median fluorescence intensity was measured for each channel. Values were normalized to cytosolic signal. For biotin leak-in assays, the freehand line tool was used to draw along the cell-matrix membranes of the cluster and measure FITC-streptavidin intensity. Then the polygon selection tool was used to measure FITC-streptavidin intensity in the cluster core (excluding the cell-matrix contacts) and in adjacent stromal regions. To assess biotin permeability in tumor versus stroma areas, mean FITC-streptavidin signal was measured in FIJI in equally sized squares in adjacent pure tumor or pure stroma areas. To score protrusive versus non-protrusive epigen immunofluorescence intensity, the polygon tool in FIJI software was used to measure epigen signal in locally disseminated mTomato+ PyMT clusters. Only protrusive and non-protrusive clusters in the same field of view were compared to one another, to account for differences in staining intensity between different regions of the tumor and different biological replicates. Protrusive clusters were identified based on the presence of strand-like organization and membrane protrusions. Non-protrusive clusters had more rounded organization and smooth borders. Clusters with intermediate or unclear morphology were not scored. Clusters with one type of morphology in one region, and another in a different region, were not scored. To count DAPI+ nuclei in clusters, a single confocal slice from the thickest part of the cluster was used. To assess cell death in clusters, propidium iodide (a fluorescent dye excluded from viable cells) was added to cell media at 0.5 ug/mL for 10 minutes prior to imaging.
Lentivirus production and transduction
Lentivirus was produced in HEK293FT cells using PsPax and MD2.G packaging plasmids for pLKO.1 (Moffat et al., 2006), LentiCRISPRv2 (Sanjana et al., 2014), or lentiviral shRNA vectors (transOMIC). Supernatants were concentrated using Lenti-X (Clontech), resuspended in 1/100th the supernatant volume of PBS, and frozen at −80°C. Viruses were titered using a p24 ELISA (Retrotek) Tumor organoids were dissociated to single cells in Accumax and resuspended at 150,000 viable cells/mL in organoid media + 2% (v/v) Matrigel. Protamine sulfate was added at 8ug/mL to enhance transduction efficiency. For plasmids with PuroR, 2 days after transduction puromycin was included in the media at 1–2 ug/mL for selection. Cells were maintained in puromycin for at least 5 days before experiments and puromycin was included in the media throughout ex vivo culture. CRISPR sgRNA transduced cells were kept in puromycin at least 10 days before conducting experiments.
RTK array and quantification
The R&D Proteome Profiler Mouse Phospho-RTK Array Kit (R&D ARY014) was performed according to manufacturer instructions using 125 ug of dissociated single cell or clustered (6 hr aggregation) MMTV-PyMT lysates. X-ray film was exposed to membranes for 1 to 10 minutes. To quantify signal, the pixel intensity of each coordinate was measured using FIJI software for film scans in which the signal was not over or under exposed. The signal of the negative control (PBS) was then subtracted from each measurement.
Western Blotting
Clusters were lysed in RIPA buffer with protease and phosphatase inhibitors for 30 minutes at 4°C and then centrifuged (15 minutes at 12000 g). Clusters were cultured in 2% basement membrane suspension prior to lysate collection. Protein concentration was quantified using a BCA assay (Pierce), lysates were loaded onto a 4%–12% Bis-Tris NuPage protein gel. Semi-dry transfers (iBlot2, P3 5–8min) were performed using PVDF membranes which were then blocked with 5% BSA in TBST for 1 hr. Primary antibodies were incubated overnight at 4°C in 1% BSA in TBST, diluted 1:1000. Species specific LI-COR 680 and 800 secondary antibodies were used in TBST plus 1% BSA. Primary antibodies used: ERK1/2 (CST 4695), pERK1/2 (CST 4370), pAkt (CST 4060), Akt (CST 4691), beta-tubulin (Abcam ab40815), EGFR (Millipore 06–847). Band intensity was measured using the “Analyze Gels” tool in FIJI software, relative to beta-tubulin as a loading control.
Mammary fat pad orthotopic transplantation
MMTV-PyMT-mTomato organoids were resuspended in 50% basement membrane-rich gel in DMEM/F12 (vol/vol) on ice. 9–15 week old NSG mice were anesthetized with 2.5% isoflurane and the surgical site was sterilized with ethanol and chlorhexidine. A 1 cm midline incision was made, allowing the #4 mammary fat pad to be exposed. 50,000 clustered cells per gland (aggregated at 250,000 viable cells/mL overnight) were injected into the left and right #4 mammary gland. The surgical area was locally infiltrated with 0.25% bupivacaine for pain relief. Surgical wounds were closed with 9mm autoclips and tissue glue. Triple antibiotic ointment was applied to the incision. Mice were monitored closely with autoclip removal two weeks after surgery. Tumor volume was estimated based on caliper measurements as: (4/3) x (π)x (width/2) x (width/2) x (length/2). At endpoint weeks, mice were euthanized, and primary tumors and lungs were fixed in 4% PFA for 4 hr, then transferred to 25% sucrose in DPBS overnight at 4°C before embedding in OCT and storing at −80°C. Lung metastases were quantified using a fluorescence dissecting microscope at endpoint. Ctrl-kd and Epgn-kd tumors were harvested at the same time, when the first Ctrl-kd tumors reached the maximum allowable size (15 mm diameter).
RNA sequencing and bioinformatic analysis
MMTV-PyMT cells or clusters in suspension culture (non-adherent plates in media +2% Corning Matrigel) were collected for RNA extraction. Cells were pelleted 5 min. at 300 g then resuspended in 1mL of Trizol. Samples were incubated at room temperature for 5 min. to ensure lysis, and then stored at −20°C. RNA samples were quantified using Qubit 2.0 Fluorometer and RNA integrity was checked with Agilent TapeStation (Agilent, RIN range 8.7 to 9.3). RNA sequencing libraries were prepared by GENEWIZ using the NEBNext Ultra RNA Library Prep Kit for Illumina following manufacturer’s instructions. The sequencing libraries were validated on the Agilent TapeStation and quantified by using Qubit 2.0 Fluorometer as well as by quantitative PCR (KAPA Biosystems, Wilmington, MA, USA). The sequencing libraries were clustered on 1 lane of a flowcell. After clustering, the flowcell was loaded on the Illumina HiSeq instrument (4000 or equivalent) according to manufacturer’s instructions. The samples were sequenced using a 2×150bp Paired End (PE) configuration. Image analysis and base calling were conducted by the HiSeq Control Software (HCS).
Raw sequence data (.bcl files) generated from Illumina HiSeq was converted into fastq files and de-multiplexed using Illumina’s bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification. Raw sequencing data was demultiplexed to generated two fastq files per sample with between 24.1 to 32.7 million reads per sample. Kallisto pseudoalignment v0.46.0 (Bray et al., 2016) was used for transcript abundance estimation, and differential transcript expression analysis was performed using Sleuth v0.30.0 (Pimentel et al., 2017). Kallisto was run with bootstrap-samples = 100, and the transcript target was Ensembl (release 97) Mus musculus transcriptome (Mus_musculus.GRCm38.cdna.all.fa). For gene-level analyses, aggregated transcripts per million (TPM) scaled using the average transcript length and averaged over samples and to library size were generated with tximport and were used in further downstream differential gene expression analysis using limma-voom.
To visualize expression dynamics, genes were sequentially ordered in the dataset by their time-point of maximum expression, mean-variance normalized, and clustered together by time-point of maximum expression. Gene cluster enriched biological processes and signaling pathways were identified using Metascape (Zhou et al., 2019).
RNA sequencing datasets
Human RNA sequencing data from the Genotype-Tissue Expression (GTEx) Project were accessed using the GTEx portal (https://www.gtexportal.org/home/gene/EPGN). Breast cancer cell line RNA sequencing datasets were accessed from the Broad Institute CCLE portal (https://portals.broadinstitute.org/ccle) (Barretina et al., 2012) or EMBL-EBI (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-2706/) (Klijn et al., 2015).
Epigen ELISA
Epigen ELISAs were performed according to manufacturer instructions (R&D DuoSet) using MMTV-PyMT single cell or day 1 cluster (18hrs aggregation) lysates in RIPA buffer diluted at least 5-fold in ELISA reagent diluent. At least 30 ug of total protein was loaded. Recombinant epigen (R&D 1127-EP) was used as a standard. Antibodies MAB11271 (R&D) and AF1127 (R&D) were used for coating and detection, respectively.
Real-time qPCR
Human or mouse tumor cell clusters were pelleted and snap frozen in liquid nitrogen. RNA was extracted using an RNEasy mini kit (QIAGEN). 250ng-1ug of RNA was used to generate cDNA with a SuperScript III First-Strand Synthesis kit (ThermoFisher) with oligo(dT) primers. RT-qPCR was performed using PowerUp SYBR Green master mix in 10–20 uL reactions on a QuantStudio 5 real-time PCR instrument. Pre-designed Millipore Sigma KiCqStart™ primers were used (https://www.kicqstart-primers-sigmaaldrich.com/KiCqStartPrimers.php), primer pair IDs can be found in the Key Resources Table. Data were analyzed using the ΔΔCt method.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat-anti-Epigen (for mouse cells) | R&D | MAB11271 |
| Goat-anti-Epigen (for ELISAs) | R&D | AF1127 |
| Rabbit-anti-Epigen (for human cells) | Bioss | bs-5767R |
| Rabbit-anti-Her2 | Cell Signaling Technology | 2165 |
| Rabbit-anti-pEGFR (Tyr1068/1092) | Abcam | ab40815 |
| Rabbit-anti-Keratin 17 (D73C7) | Cell Signaling Technology | 4543 |
| Rat-anti-E-Cadherin (DECMA-1) | EMD Millipore | MABT26 |
| Mouse-anti-human-CD298 | BioLegend | 341704 |
| Rabbit-anti-phospho-Histone H3 (Ser10) | Cell Signaling Technology | 9701 |
| Rat-anti-Keratin 8 | DSHB | TROMA-I |
| Alexa Fluor 647 anti-human CD45 | BioRad | MCA87A647T |
| Alexa Fluor 488 pan-cytokeratin | Cell Signaling Technology | 4523 |
| Rabbit-anti-keratin 14 | BioLegend | 905301 |
| Rat-anti-PyMT | Abcam | ab15085 |
| Rabbit-anti-EGFR | EMD Millipore | 06–847 |
| Rabbit-anti-pERK1/2 (Thr202/Tyr204) | Cell Signaling Technology | 4370 |
| Rabbit-anti-ERK1/2 | Cell Signaling Technology | 4695 |
| Rabbit-anti-pAkt (Ser473) | Cell Signaling Technology | 4060 |
| Rabbit-anti-Akt | Cell Signaling Technology | 4691 |
| Rabbit-anti-Beta tubulin | Abcam | ab6046 |
| Rabbit-anti-Muc1 | Abcam | ab45167 |
| Rabbit-anti-Occludin | Thermo Fisher Scientific | 71–1500 |
| Goat-anti-Rat IgG (H&L) EM grade 10nm | Electron Microscopy Sciences | 25189 |
| Aurion F(ab’)2 Fragment Goat-anti-Rabbit IgG (H&L) | Electron Microscopy Services | 25365 |
| Biological Samples | ||
| Human breast cancer, tumor samples | Cooperative Human Tissues Network | See Table S1 |
| Human breast cancer, tumor samples | Seattle Cancer Care Alliance | See Table S1 |
| Human breast cancer, blood samples | Seattle Cancer Care Alliance | See Table S1 |
| Human breast cancer, ascites samples | Seattle Cancer Care Alliance | See Table S1 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NucView 488 | Biotium | 10403 |
| EZ-Link Sulfo-NHS-Biotin No Weigh Format | Thermo Fisher Scientific | A39256 |
| Streptavidin-FITC | Thermo Fisher Scientific | SA10002 |
| Recombinant human IFN-γ Carrier Free | Biolegend | 570204 |
| Latrunculin A | Tocris | 3973 |
| Ochratoxin A | Fisher Scientific | 12–911 |
| Recombinant epigen | GenScript | Z03107–50 |
| Critical Commercial Assays | ||
| Proteome Profiler Mouse phospho-RTK Array | R&D | ARY014 |
| Collagen I, Coated Plate, 24 well | ThermoFisher Scientific | A1142802 |
| Deposited Data | ||
| RNA sequencing of MMTV-PyMT cells during aggregation | This paper | NCBI PRJNA648151 |
| RNA sequencing of Ctrl-kd versus Epgn-kd MMTV-PyMT organoids | This paper | NCBI PRJNA648435 |
| Broad Institute Cancer Cell Line Encyclopedia | Barretina et al., 2012 | NCBI PRJNA155793 |
| RNA-seq of 675 commonly used human cancer cell lines | Klijn et al., 2015 | E-MTAB-2706 |
| GTEx Analysis Release V8 | The Genotype-Tissue Expression project | dbGaP Accession phs000424.v8.p2 |
| Experimental Models: Cell Lines | ||
| Human: 293FT | ThermoFisher Scientific | R70007 |
| Human: HCC70 | ATCC | CRL-2315 |
| Human: CAL-85–1 | DSMZ | ACC 440 |
| Human: HDQ-P1 | DSMZ | ACC 494 |
| Human: BT549 | ATCC | HTB-122 |
| Human: MDA-MB-436 | ATCC | HTB-130 |
| Human: YFP-MDA-MB-231 | Cyrus Ghajar | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: FVB/NTg(MMTV-PyVT)634Mul/J(MMTV-PyMT) | The Jackson Laboratory | 002374 |
| Mouse: ROSA mTomato/mGFP | The Jackson Laboratory | 007576 |
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | 005557 |
| Mouse: FVB-Tg(C3–1-TAg)cJeg/JegJ | Andrew Ewald | N/A |
| Mouse: FVB/N-Tg(MMTVneu)202Mul/J | Sasha Stanton | N/A |
| Oligonucleotides | ||
| Mouse beta actin qPCR primer | Millipore Sigma | KiCqStart M_ActB_1 |
| Mouse Areg qPCR primer | Millipore Sigma | KiCqStart M_Areg_1 |
| Mouse Epgn qPCR primer | Millipore Sigma | KiCqStart M_Epgn_1 |
| Mouse Egfr qPCR primer | Millipore Sigma | KiCqStart M_Egfr_2 |
| Mouse Plaur qPCR primer | Millipore Sigma | KiCqStart M_Plaur_1 |
| Human beta actin qPCR primer | Millipore Sigma | KiCqStart M_ActB_1 |
| Human RPL32 qPCR primer | Millipore Sigma | KiCqStart M_RPL32_1 |
| Human EPGN qPCR primer | Millipore Sigma | KiCqStart M_EPGN_1 |
| Human RAB25 qPCR primer | Millipore Sigma | KiCqStart M_RAB25_1 |
| Human CLDN7 qPCR primer | Millipore Sigma | KiCqStart M_CLDN7_1 |
| CRISPR non-targeting control GTAGCGAACGTGTCCGGCGT | Patrick Paddison | N/A |
| EPGN CRISPR sgRNA #2 GAAGTTCTCACACCTTTGCC | GenScript | N/A |
| Recombinant DNA | ||
| PsPax2 | A gift from Didier Trono | Addgene #12260 |
| MD2.G | A gift from Didier Trono | Addgene #12259 |
| shCtrl Non-targeting #4 (Ctrl-kd) | transOMIC | TLMSU1400-NT4 |
| pZIP-mCMV-ZsGreen-Mouse-shEGFR | transOMIC | ULTRA-3213354 |
| pZIP-mCMV-ZsGreen-Mouse-Plaur | transOMIC | ULTRA-3232040 |
| pZIP-mCMV-ZsGreen-Mouse-shAreg | transOMIC | ULTRA-3205528 |
| pZIP-mCMV-ZsGreen-Mouse-shEpgn | transOMIC | ULTRA-3383713 |
| pLenti-C-mGFP-hEpgn | OriGene Technologies | RC214501L4 |
| pLenti-C-mGFP-mEpgn | OriGene Technologies | MR219218L4 |
| pLKO.1 TRC control | Moffat et al., 2006 | Addgene #10879 |
| pLKO.1 TRC shEpgn #175159 | Sigma Aldrich | SHCLNG-NM_053087 |
| LentiCRISPRv2 | Sanjana et al., 2014 | Addgene #52961 |
| Software and algorithms | ||
| GraphPad Prism 8 | Graphpad Software | https://www.graphpad.com/scientific-software/prism/ |
| FIJI v2.0.0-rc-69/1.52p | Schindelin et al., 2012 | https://fiji.sc/ |
| R v3.6.1 | R Core Team | https://www.r-project.org/ |
| Metascape | Zhou et al., 2019 | https://metascape.org/ |
| bcl2fastq v2.17 | Illumina, Inc. | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html |
| Kallisto v0.46.0 | Bray et al., 2016 | http://pachterlab.github.io/kallisto/download |
| Sleuth v0.30.0 | Pimentel et al., 2017 | https://www.rdocumentation.org/packages/sleuth/versions/0.30.0 |
| Tximport v1.12.3 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/tximport.html |
| Limma v3.40.6 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/limma.html |
QUANTIFICATION AND STATISTICAL ANALYSIS
Bars are presented as mean ± standard deviation. Red lines denote medians, unless otherwise noted. Graphs were created and statistical tests conducted in GraphPad Prism 8. Non-parametric tests were used when data were not normally distributed or when the median was a better representation of the sample than the mean. Experiments using cell lines on different days or using organoids generated from different mice were considered biological replicates. All statistical tests are two-sided. p ≤ 0.05 was considered significant. p values: “ns” p > 0.05, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Supplementary Material
(A) MMTV-PyMT mTomato+ single cells were aggregated for 0, 6, or 24 hr and stained for DAPI. Right: quantification of number of nuclei per entity (single confocal slice), with a mean number of cells per cluster of 4.4 (SD ± 4.2) and 5.4 (SD ± 5.2) at 6 and 24 hr, respectively. n = 3 biological replicates. n (# entities, single cells or clusters): 0hr = 307, 6hr = 359, 24hr = 326. P values = unpaired t tests.
(B) Number of metastases formed 3 weeks after mTomato-PyMT tail vein relative to the number of entities (single cells or clusters) injected based on measurements in (A). % success = (100 x number of metastases)/number of entities injected. n (# mice) = 19 (0hr), 9 (6hr), 14 (24hr), 10 (re-dissociated). P values = Mann-Whitney tests. Medians: 0hr = 0%, 6hr = 0.16%, 24hr clusters = 1.72%, 24hr redissociated = 0%.
(C) Experimental schematic: 200,000 mTomato-PyMT cells were injected by tail vein into NSG mice either as single cells or clusters. 0, 24, and 48 hr after injection the lungs were harvested. Non-consecutive sections were imaged and the number of mTomato+ clusters and single cells was quantified for each field of view.
(D) Quantification of the number of mTomato+ single cells or clusters detected in lung sections at 0, 24, and 48 hr after tail vein injection. For each mouse 3 sections were imaged (10 25x fields per section). n = 4 mice per time point.
(E) To determine whether single tumor cells form micro-metastases that are not visible at low magnification, lungs were sectioned 3 weeks after tail vein injection of single mTomato+ PyMT cells. Sections were imaged at 10x magnification and the number of mTomato+ single cells and clusters was counted. n = 6 mice, 1 section per mouse.
(F) Schematic of intracardiac injection of mTomato-PyMT clusters or single cells into the left ventricle of immunocompromised mice to generate systemic metastases. Right, organs 6 weeks after intracardiac injection.
(G) Quantification of systemic metastases 6 weeks after intracardiac injection of mTomato-PyMT clusters. n = 4 single cell injected mice, 7 cluster injected mice. p values = Mann-Whitney test.
(H) Lung sections of mice injected by tail vein with Ctrl-kd or Epgn-kd clusters. Red signal = mTomato+ metastases. Right, quantification of the number of metastases per lung section. n = 5 mice per condition, 1–2 sections per mouse. P value = Mann-Whitney test.
(I) Size of individual lung metastases 3 weeks after tail vein injection of Ctrl-kd or Epgn-kd clusters. Boxplot = 5%–95%. P value = Mann-Whitney test.
(J) Left: 24hr aggregated Ctrl-kd-GFP and Epgn-kd-GFP clusters shortly before tail vein injection. Right: lung sections 3 weeks after tail vein injection of mTomato-PyMT Ctrl-kd-GFP or Epgn-kd-GFP clusters.
(K) Quantification of double positive (GFP+ and mTomato+) and shRNA escape (mTomato+ but GFP−) areas in Ctrl-kd or Epgn-kd lung metastases 3 weeks after tail vein injection. n = 3 mice per condition. P values = unpaired t tests.
(A) Time lapse images of a MMTV-PyMT single cell and cluster cultured in 3D basement membrane-rich gels for 4 days. Right, outgrowth quantification as fold change in area. n = 8 mice, 23 single cells, 500 clusters. P value = Mann-Whitney test.
(B) Outgrowth measured by fold change in area over 4 days of time lapse imaging for MMTV-PyMT single cells and clusters of different sizes in 3D basement membrane-rich gels. P values = Mann-Whitney tests. n (# of single cells or clusters): 0–500 um2 = 39, 500–1000 um2 = 39, 1000–2000 um2 = 58, > 2000 um2 = 387. n = 8 biological replicates.
(C) % of nuclei in mitosis (scored by pHH3 staining) binned by number of neighboring cells with visible direct membrane-membrane contact. n = 4 biological replicates. n (# cells): 0 contacts (single cells) = 453, 1 contact = 698, 2 contacts = 704, 3 contacts = 667. P values = unpaired t tests.
(D) Time lapse DIC and GFP images of a MMTV-PyMT single cell and cluster in the presence of a caspase-3/7 activated green fluorescing DNA dye in 3D basement membrane-rich gels. Right, quantification of MMTV-PyMT single cell versus cluster survival over time. Clusters were marked as dead once a majority of cells in the clusters apoptosed. n = 4 mice, 79 single cells and 105 clusters. P value = Mantel-Cox.
(E) Time lapse DIC and GFP (Caspase 3/7 biosensor) images of a single cell and cluster derived from a human breast cancer brain metastasis. Right, summary of single cell versus cluster survival after 72hrs of time lapse in 3D basement membrane-rich gels from 9 human breast tumor specimens (HU1–9). n = 283 single cells and 197 clusters. P value = pairwise t test.
(F) Survival of single cells versus clusters from 10 individual human breast cancer patient samples (HU1–10). P values = Mantel-Cox.
(A) Validation of MMTV-PyMT shRNA knockdowns by qPCR. Red line = mean. # of biological replicates; Plaur-kd n = 3, Egfr-kd n = 4, Areg-kd n = 2, Epgn-kd n= 3.
(B) Left, Images of Ctrl and knockdown cells after 24 hr of aggregation, plated at equal cell densities. Middle, quantification of suspension object areas (MATLAB) prior to tail vein injection. n (# entities): Ctrl-kd = 6931, Egfr-kd = 3561, Areg-kd = 3158, Epgn-kd = 3367. Right, number of nuclei (by DAPI staining) of Ctrl-kd or Epgn-kd clusters aggregated for 24hrs. n = 3 biological replicates. n = 100 Ctrl-kd clusters, 66 Epgn-kd clusters. P value = unpaired t test.
(C) DIC time lapse images of Ctrl-kd, Plaur-kd, Egfr-kd, Epgn-kd, Areg-kd MMTV-PyMT clusters growing in 3D basement membrane-rich gels for 90 hr. Right, quantification of fold growth by change in area over 4 days of time lapse imaging. Ctrl-kd: n = 8 mice, 246 clusters. Plaur-kd: n = 2 mice, n = 31 clusters. Egfr-kd: n = 3 mice, 84 clusters. Areg-kd: n = 3 mice, 108 clusters. Epgn-kd: n = 3 mice, 90 clusters.
(D) % of Ctrl-kd and Epgn-kd nuclei in mitosis after 24 hr of aggregation, measured as % phospho-histone H3 positive. n = 4 biological replicates. n (# nuclei): Ctrl-kd = 2636, Epgn-kd = 1790. P value = unpaired t test.
(E) RNA expression (RNA-seq) of cytokeratin genes in Ctrl-kd and Epgn-kd MMTV-PyMT clusters, n = 3. Adjusted p values.
(F) Immunofluorescence of keratin-17 in Ctrl-kd and Epgn-kd clusters in 3D basement membrane-rich gels after 6 days of outgrowth. Right, quantification of % cells in each cluster keratin-17 positive by immunofluorescence. n = 2 biological replicates. n (# clusters): non-transduced = 34, Ctrl-kd = 39, Epgn-kd = 27. P values = unpaired t tests.
(G) Validation of MMTV-PyMT TRC-Epgn-kd by qPCR. n = 3 biological replicates. P value = unpaired t test.
(H) Relative viable cell number of TRC-Ctrl-kd and TRC-Epgn-kd after 6 days of outgrowth in low basement membrane suspension culture. n = 3 biological replicates. P value = unpaired t test.
(I) DIC images of Ctrl-kd and Epgn-kd clusters after 4 days growth with and without transduction with an shRNA resistant human epigen construct (MOI < 1). Right, quantification of viable cell number using CellTiter-Glo. n = 3 biological replicates. P value = unpaired t test.
(A–B) Immunofluorescence of epigen in tumor cell clusters ex vivo from MMTV-Neu tumors (A) and C3(1)Tag tumors (B).
(C) Quantification of epigen immunofluorescence signal along cytosolic, cell-cell, or cell-matrix areas in C3(1)-TAg clusters. n = 60 clusters, 3 biological replicates. P values = Mann-Whitney test.
(D) Relative number of viable cells after 6 days of treatment of PyMT cells in suspension with recombinant epigen. n = 3 biological replicates. P value = unpaired t test.
(E) Cumulative migration distance of individual clusters, normalized to initial cluster cell number, in 3D basement membrane-rich gels over 120 hr. Mixed clusters were generated by aggregating Ctrl-kd and Epgn-kd cells at a 1:1 ratio. Data are plotted as mean with 5%–95% CI. n (# of clusters): Non-transduced = 49, Ctrl-kd = 50, Epgn-kd = 47, Mixed 1:1 = 35. n (# of biological replicates) = 2. P values = unpaired t tests.
(F) Ultrastructure of tumor cell cluster using high pressure freezing electron microscopy. Pseudo-color highlights different cells. MP = microvilli-like protrusions. J = cell-cell junction.
(G) Transmission electron microscopy of Ctrl-kd and Epgn-kd PyMT clusters. Right, nanolumenal area and perimeter measurements. n = 40 nanolumina measured per condition (measured junction to junction). MP = microvilli-like protrusions. J = cell-cell junction.
(H) Immunofluorescence of epigen and apical marker Muc-1 in a MMTV-PyMT cluster.
(I) Biotin leak-in assay of MMTV-PyMT clusters without treatment, treated with ochratoxin A (200 µm, 1 hr) (McLaughlin et al., 2004), or treated with latrunculin A (0.2 µm, 1hr) (Shen and Turner, 2005) to disrupt cell-cell junctions.
(J) Estimated concentration of epigen in nanolumen, based on TEM estimates of nanolumenal volume per cell and ELISA measurements of epigen protein per cell. Secreted epigen was also measured by epigen ELISA of 18 hr tumor cell cluster supernatants (extra-cluster concentration). Supernatants: n = 3 biological replicates. Cell lysates: n = 2 biological replicates. P values = unpaired t tests.
(A) Quantification of receptor tyrosine kinase phospho-tyrosine array signal for MMTV-PyMT cluster (6hrs aggregation) versus PyMT single cell (0hr aggregation) lysates. n = 3 biological replicates. P values = unpaired t tests.
(B) Immunoblots of single cell (0 hr) versus cluster (6hrs aggregation) MMTV-PyMT lysates for phospho (Thr 202/Tyr204) and total ERK1/2. Right: quantification of n = 3 biological replicates. P values = unpaired t tests.
(C) Immunoblots of single cell (0hr) versus cluster (6hrs aggregation) MMTV-PyMT lysates for phospho (S473) and total Akt. Right: quantification of n = 3 biological replicates. P values = unpaired t tests.
(D) Immunoblot of EGFR and EGFR effectors pERK1/2 in non-transduced, Ctrl-kd, and Egfr-kd PyMT tumor cell clusters. Right: quantification of n = 3 biological replicates. P values = unpaired t tests.
(E) Immunofluorescence of pEGFR (Y1068) in a MMTV-PyMT cluster ex vivo. Right, quantification of pEGFR at intracellular (I), cell-matrix (C-M), and cell-cell (C-C) spaces in MMTV-PyMT clusters. n = 68 clusters, n = 3 biological replicates. P values = Mann-Whitney tests.
(F) Immunofluorescence of HER2 in a MMTV-PyMT cluster ex vivo. Right, quantification of HER2 immunofluorescence at intracellular (I), cell-matrix (C-M), and cell-cell (C-C) spaces in MMTV-PyMT clusters. n = 40 clusters, n = 2 biological replicates. P values = Mann-Whitney tests.
(G) Immunogold labeling of pEGFR along nanolumina in MMTV-PyMT clusters. Arrows = pEGFR immunogold signal. Right, quantification of labeling in intercellular areas, along nanolumenal membranes, along nanolumenal villi, at cell-cell junctions, and within nanolumenal voids. n = 93 gold particles.
(H) Immunogold labeling of HER2 along nanolumina in MMTV-PyMT clusters. Arrows = HER2 immunogold signal. Right, quantification labeling in intracellular areas, along nanolumenal membranes, along nanolumenal villi, at cell-cell junctions, and within nanolumenal voids. n = 45 gold particles.
(A) Immunofluorescence of epigen and keratin-14 in normal FVB mouse back skin as an antibody positive control, as epigen is known to be expressed in hair follicles (Kochupurakkal et al., 2005).
(B) Genotype-Tissue Expression (GTEx) RNA sequencing data of EPGN in normal human tissues including (left) the top 10 EPGN expressing tissues by median expression and (right) frequent sites of breast cancer metastasis.
(C) Pan-cytokeratin, Keratin-14, Keratin-17, and epigen immunofluorescence of protrusive tumor strands on the border of mTomato-PyMT orthotopic tumors 10 weeks after transplant of non-transduced (pan-CK, K17) or Ctrl-kd clusters (K14).
(D) Immunofluorescence of epigen in a liver metastasis 6 weeks after intracardiac injection of mTomato-PyMT clusters.
(E) pEGFR (Y1068) immunofluorescence of a primary tumor 10 weeks after mTomato-PyMT organoid orthotopic transplant into the mammary fat pad and of a liver metastasis 6 weeks after intracardiac injection of mTomato-PyMT clusters.
(F) HER2 immunofluorescence of a primary tumor 10 weeks after mTomato-PyMT organoid orthotopic transplant into the mammary fat pad and of a liver metastasis 6 weeks after intracardiac injection of mTomato-PyMT clusters.
(G) 20X tiled images were taken of sections from Ctrl-kd and Epgn-kd orthotopic transplant tumors. Right, quantification of the number of locally disseminated clusters (mTomato+) per mm2 stromal area (DAPI+/mTomato−). n = 6 Ctrl-kd tumors (148 clusters), 6 Epgn-kd tumors (218 clusters). P values = unpaired t tests.
(H) Left, images of lung metastases 6 weeks after orthotopic transplant of Ctrl-kd or Epgn-kd organoids, stained for mitotic marker pHH3. Right, quantification of % of lung metastasis cells pHH3+. n = 3 Ctrl-kd mice (925 cells), n = 3 Epgn-kd mice (157 cells). P value = unpaired t test.
(I) Area measurements of individual metastases (including disseminated single cells) in lungs 6 weeks after orthotopic transplant of Ctrl-kd or Epgn-kd organoids. n = 5 mice per condition. n = 47 Ctrl-kd metastases, 72 Epgn-kd metastases. P value = Mann-Whitney test.
(J) Number of metastases detected in each lung section from mice 6 weeks after orthotopic transplant of Ctrl-kd or Epgn-kd organoids. n = 5 mice per condition. P value = unpaired t test.
(K) mTomato+ and GFP+ areas were measured in Ctrl-kd and Epgn-kd orthotopic tumor sections using 20X tiled images. Right, quantification of the GFP+ area as a percent of the mTomato+ area. n = 4 Ctrl-kd tumors, 4 Epgn-kd tumors. P value = unpaired t test.
(A) Shown are representative tumor cell clusters obtained directly from the ascites fluid of a metastatic ER+/PR+/HER2-breast cancer patient with disease to bone, liver, chest wall, stomach and peritoneum. Left: image of ascites cluster stained for pan-cytokeratin, CD45, and pEGFR. Right: TEM images of ascites clusters. MP = microvilli-like protrusions. J = cell-cell junction.
(B) Epigen immunofluorescence of tumor cell clusters in malignant ascites from patients #1 and #2 using a pan-cytokeratin antibody to identify tumor cell clusters and CD45 to identify immune cells.
(C) Epigen mRNA expression in two panels of breast cancer cell lines (Barretina et al., 2012; Klijn et al., 2015). Triple-negative subgroups: BL2 = basal-like 2, BL1 = basal-like 1, M = mesenchymal, LAR = luminal androgen receptor. L-HER2+ = luminal like HER2+ cell lines. HER2E = basal like HER2+ cell lines (Cancer Genome Atlas Network, 2012; Watson et al., 2018).
(D) Gene sets (Metascape, FDR % 0.05) enriched in M-like (MDA-MB-231, MDA-MB-436, and BT549) versus BL2 (HCC70, CAL851, HDQP1) cell lines (CCLE).
(E) GFP images of Ctrl-kd and Epgn-kd cells grown on collagen I coated plates to assess 2D migration. Right, quantification of the colony area one day after plating (D1) and of the dense monolayer (“confluent”) and invasive outer layer (“subconfluent”) formed 6 days after plating (D6). n = 3 biological replicates. P values = unpaired t tests.
(F) Relative number of viable cells of non-transduced, Ctrl-kd, and Epgn-kd HCC70 cells after 4 days of growth (seeded at equal cell number) in 2D on tissue culture treated plates. n = 3 biological replicates. P value = unpaired t test.
(G) Left: qPCR validation of HCC70 clusters transduced with non-targeting or epigen-targeting CRISPR sgRNAs. Cells were not clonally selected i.e., these represent mixed populations of cells with < 100% knockout. n = 2 biological replicates. Right, relative number of viable cells after 4 days of growth as clusters in low-basement membrane suspension. n = 3 biological replicates. P values = unpaired t tests.
(H) Number of non-viable nuclei per 1000 um2 area in HCC70 clusters ex vivo, measured by confocal imaging of propidium iodide staining after 6 days with or without 3 ng/mL IFNγ treatment. n = 3 biological replicates. n = 76 untreated clusters, 72 IFNγ treated clusters.
(I) Fold change of CLDN7 mRNA (relative to RPL32 housekeeping control) for MDA-MB-231 and HCC70 clusters after 6 days with or without 3ng/mL IFNγ.n=3 biological replicates. P values = unpaired t tests. RAB25 was not detected by qPCR in MDA-MB-231 cells at any time point.
(J) Representative images of biotin leak-in assay of MDA-MB-231 clusters after 6 days of treatment with or without 3ng/mL IFNγ. Right, quantification of biotin leak-in (internal FITC-streptavidin intensity). n = 18 untreated clusters, 16 IFNγ treated clusters. n = 2 biological replicates. P value = Mann-Whitney test.
(K) Here, we find that tumor cell clusters generate sealed, intercellular nanolumina. During phases of outgrowth, these nanolumina have restricted permeability and contain high concentrations of the growth factor epigen. Collective signaling by epigen within nanolumina promotes primary and metastatic tumor outgrowth. In contrast, low epigen expression is found in protrusive locally disseminated clusters. High epigen expression and nanolumenal impermeability were found in basal-like 2 human breast cancer cells, whereas mesenchymal-like cells were highly permeable and lowly expressed epigen. Suppression of epigen, or targeting of nanolumenal junctions, in basal-like 2 breast cancer cells reduced collective metastatic outgrowth.
Highlights.
Breast tumor cell clusters contain nanolumina—sealed, intercellular cavities
Clusters produce the growth factor epigen and concentrate it in nanolumina
Epigen is regulated during metastasis, and its suppression reduces outgrowth
Basal-like 2 breast cancer clusters highly express epigen and form nanolumina
ACKNOWLEDGMENTS
We thank the following people: S. Beronja and P. Paddison for providing shRNA and sgRNA reagents; A. Ewald and S. Stanton for providing mouse tumors; C. Tran for research coordinator support at SCCA; A. Hsieh, J. Cooper, and V. Vasioukhin for critical feedback on this work. Most of all, we thank the patients participating in this study for their generosity. This study was supported by the Department of Defense Breast Cancer Research Program (BCRP; W81XWH-18–1–0098), the NIH/NCI (R37CA234488), the Burroughs Wellcome Fund Career Award for Medical Scientists, the Breast Cancer Research Foundation (BCRF-18–035), the V Foundation (V Foundation Scholar Award), the Phi Beta Psi Sorority, Seattle Translational Tumor Research, and the Shared Resources of the FHCRC/UW Cancer Consortium (P30 CA015704).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.08.045.
DECLARATION OF INTERESTS
E.D.W. and K.J.C. are inventors on a pending patent application related to this work.
REFERENCES
- Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, et al. (2014). Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158, 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aceto N, Toner M, Maheswaran S, and Haber DA (2015). En Route to Metastasis: Circulating Tumor Cell Clusters and Epithelial-to-Mesenchymal Transition. Trends Cancer 1, 44–52. [DOI] [PubMed] [Google Scholar]
- Archetti M, and Pienta KJ (2019). Cooperation among cancer cells: applying game theory to cancer. Nat. Rev. Cancer 19, 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au SH, Edd J, Haber DA, Maheswaran S, Stott SL, and Toner M (2017). Clusters of Circulating Tumor Cells: a Biophysical and Technological Perspective. Curr. Opin. Biomed. Eng 3, 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehér J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Jacob E, Coffey DS, and Levine H (2012). Bacterial survival strategies suggest rethinking cancer cooperativity. Trends Microbiol. 20, 403–410. [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, and Nusrat A (2003). Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J. Immunol 171, 6164–6172. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KJ, and Ewald AJ (2016). A collective route to metastasis: Seeding by tumor cell clusters. Science 352, 167–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KJ, Gabrielson E, Werb Z, and Ewald AJ (2013). Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 155, 1639–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KJ, Padmanaban V, Silvestri V, Schipper K, Cohen JD, Fairchild AN, Gorin MA, Verdone JE, Pienta KJ, Bader JS, and Ewald AJ (2016). Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 113, E854–E863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JW, Kim JK, Yang YJ, Kim P, Yoon KH, and Yun SH (2015). Urokinase exerts antimetastatic effects by dissociating clusters of circulating tumor cells. Cancer Res 75, 4474–4482. [DOI] [PubMed] [Google Scholar]
- Cleary AS, Leonard TL, Gestl SA, and Gunther EJ (2014). Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature 508, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, and Weinberg RA (2019). EMT and Cancer: More Than Meets the Eye. Dev. Cell 49, 313–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongre A, and Weinberg RA (2019). New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol 20, 69–84. [DOI] [PubMed] [Google Scholar]
- Durdu S, Iskar M, Revenu C, Schieber N, Kunze A, Bork P, Schwab Y, and Gilmour D (2014). Luminal signalling links cell communication to tissue architecture during organogenesis. Nature 515, 120–124. [DOI] [PubMed] [Google Scholar]
- Ewald AJ, Huebner RJ, Palsdottir H, Lee JK, Perez MJ, Jorgens DM, Tauscher AN, Cheung KJ, Werb Z, and Auer M (2012). Mammary collective cell migration involves transient loss of epithelial features and individual cell migration within the epithelium. J. Cell Sci 125, 2638–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidler IJ (1973). The relationship of embolic homogeneity, number, size and viability to the incidence of experimental metastasis. Eur. J. Cancer 9, 223–227. [DOI] [PubMed] [Google Scholar]
- Fidler IJ, and Kripke ML (2015). The challenge of targeting metastasis. Cancer Metastasis Rev 34, 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed DM, Bessman NJ, Kiyatkin A, Salazar-Cavazos E, Byrne PO, Moore JO, Valley CC, Ferguson KM, Leahy DJ, Lidke DS, and Lemmon MA (2017). EGFR Ligands Differentially Stabilize Receptor Dimers to Specify Signaling Kinetics. Cell 171, 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, and Gilmour D (2009). Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol 10, 445–457. [DOI] [PubMed] [Google Scholar]
- Friedl P, and Mayor R (2017). Tuning Collective Cell Migration by Cell-Cell Junction Regulation. Cold Spring Harb. Perspect. Biol 9, a029199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour D, Rembold M, and Leptin M (2017). From morphogen to morphogenesis and back. Nature 541, 311–320. [DOI] [PubMed] [Google Scholar]
- Giuliano M, Shaikh A, Lo HC, Arpino G, De Placido S, Zhang XH, Cristofanilli M, Schiff R, and Trivedi MV (2018). Perspective on Circulating Tumor Cell Clusters: Why It Takes a Village to Metastasize. Cancer Res 78, 845–852. [DOI] [PubMed] [Google Scholar]
- Gkountela S, Castro-Giner F, Szczerba BM, Vetter M, Landin J, Scherrer R, Krol I, Scheidmann MC, Beisel C, Stirnimann CU, et al. (2019). Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 176, 98–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigore AD, Jolly MK, Jia D, Farach-Carson MC, and Levine H (2016). Tumor Budding: The Name is EMT. Partial EMT. J. Clin. Med 5, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritsenko PG, Atlasy N, Dieteren CEJ, Navis AC, Venhuizen JH, Veelken C, Schubert D, Acker-Palmer A, Westerman BA, Wurdinger T, et al. (2020). p120-catenin-dependent collective brain infiltration by glioma cell networks. Nat. Cell Biol 22, 97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurdon JB (1988). A community effect in animal development. Nature 336, 772–774. [DOI] [PubMed] [Google Scholar]
- Guy CT, Cardiff RD, and Muller WJ (1992). Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol. Cell. Biol 12, 954–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeger A, Alexander S, Vullings M, Kaiser FMP, Veelken C, Flucke U, Koehl GE, Hirschberg M, Flentje M, Hoffman RM, et al. (2020). Collective cancer invasion forms an integrin-dependent radioresistant niche. J. Exp. Med 217, e20181184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson J, Diane Yamada S, Berger J, Alverdy J, O’Keefe J, Bassler B, and Rinker-Schaeffer C (2009). Societal interactions in ovarian cancer metastasis: a quorum-sensing hypothesis. Clin. Exp. Metastasis 26, 67–76. [DOI] [PubMed] [Google Scholar]
- Janiszewska M, Tabassum DP, Castaño Z, Cristea S, Yamamoto KN, Kingston NL, Murphy KC, Shu S, Harper NW, Del Alcazar CG, et al. (2019). Subclonal cooperation drives metastasis by modulating local and systemic immune microenvironments. Nat. Cell Biol 21, 879–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaldjian EP, Ramirez AB, Sun Y, Campton DE, Werbin JL, Varshavskaya P, Quarre S, George T, Madan A, Blau CA, and Seubert R (2018). The RareCyte® platform for next-generation analysis of circulating tumor cells. Cytometry A 93, 1220–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klijn C, Durinck S, Stawiski EW, Haverty PM, Jiang Z, Liu H, Degenhardt J, Mayba O, Gnad F, Liu J, et al. (2015). A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol 33, 306–312. [DOI] [PubMed] [Google Scholar]
- Kochupurakkal BS, Harari D, Di-Segni A, Maik-Rachline G, Lyass L, Gur G, Kerber G, Citri A, Lavi S, Eilam R, et al. (2005). Epigen, the last ligand of ErbB receptors, reveals intricate relationships between affinity and mitogenicity. J. Biol. Chem 280, 8503–8512. [DOI] [PubMed] [Google Scholar]
- Labernadie A, Kato T, Brugués A, Serra-Picamal X, Derzsi S, Arwert E, Weston A, González-Tarragó V, Elosegui-Artola A, Albertazzi L, et al. (2017). A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol 19, 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labuschagne CF, Cheung EC, Blagih J, Domart MC, and Vousden KH (2019). Cell Clustering Promotes a Metabolic Switch that Supports Metastatic Colonization. Cell Metab 30, 720–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AW, Pattabiraman DR, and Weinberg RA (2017). Emerging Biological Principles of Metastasis. Cell 168, 670–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, Zhou A, Eyob H, Balakrishnan S, Wang CY, et al. (2015). Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 526, 131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, and Pietenpol JA (2011). Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest 121, 2750–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann BD, Jovanović B, Chen X, Estrada MV, Johnson KN, Shyr Y, Moses HL, Sanders ME, and Pietenpol JA (2016). Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 11, e0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ, and Pollard JW (2003). Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am. J. Pathol 163, 2113–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Aguirre Ghiso J, Estrada Y, and Ossowski L (2002). EGFR is a transducer of the urokinase receptor initiated signal that is required for in vivo growth of a human carcinoma. Cancer Cell 1, 445–457. [DOI] [PubMed] [Google Scholar]
- Liu K, Newbury PA, Glicksberg BS, Zeng WZD, Paithankar S, Andrechek ER, and Chen B (2019a). Evaluating cell lines as models for metastatic breast cancer through integrative analysis of genomic data. Nat. Commun 10, 2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Taftaf R, Kawaguchi M, Chang YF, Chen W, Entenberg D, Zhang Y, Gerratana L, Huang S, Patel DB, et al. (2019b). Homophilic CD44 Interactions Mediate Tumor Cell Aggregation and Polyclonal Metastasis in Patient-Derived Breast Cancer Models. Cancer Discov 9, 96–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo HC, Xu Z, Kim IS, Pingel B, Aguirre S, Kodali S, Liu J, Zhang W, Muscarella AM, Hein SM, et al. (2020). Resistance to natural killer cell immunosurveillance confers a selective advantage to polyclonal metastasis. Nat. Cancer 1, 709–722. [DOI] [PubMed] [Google Scholar]
- Macias H, and Hinck L (2012). Mammary gland development. Wiley Interdiscip. Rev. Dev. Biol 1, 533–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madara JL, and Stafford J (1989). Interferon-gamma directly affects barrier function of cultured intestinal epithelial monolayers. J. Clin. Invest 83, 724–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddipati R, and Stanger BZ (2015). Pancreatic Cancer Metastases Harbor Evidence of Polyclonality. Cancer Discov 5, 1086–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, and Polyak K (2014). Non-cell-autonomous driving of tumour growth supports subclonal heterogeneity. Nature 514, 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J, and Obenauf AC (2016). Metastatic colonization by circulating tumour cells. Nature 529, 298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda H, Baggerly KA, Wang Y, Zhang Y, Gonzalez-Angulo AM, Meric-Bernstam F, Valero V, Lehmann BD, Pietenpol JA, Hortobagyi GN, et al. (2013). Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin. Cancer Res 19, 5533–5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin J, Padfield PJ, Burt JP, and O’Neill CA (2004). Ochratoxin A increases permeability through tight junctions by removal of specific claudin isoforms. Am. J. Physiol. Cell Physiol 287, C1412–C1417. [DOI] [PubMed] [Google Scholar]
- Mishra AK, Campanale JP, Mondo JA, and Montell DJ (2019). Cell interactions in collective cell migration. Development 146, dev172056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298. [DOI] [PubMed] [Google Scholar]
- Nguyen-Ngoc KV, Cheung KJ, Brenot A, Shamir ER, Gray RS, Hines WC, Yaswen P, Werb Z, and Ewald AJ (2012). ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc. Natl. Acad. Sci. USA 109, E2595–E2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen-Ngoc KV, Shamir ER, Huebner RJ, Beck JN, Cheung KJ, and Ewald AJ (2015). 3D culture assays of murine mammary branching morphogenesis and epithelial invasion. Methods Mol. Biol 1189, 135–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA, Huang RY, Jackson RA, and Thiery JP (2016). EMT: 2016. Cell 166, 21–45. [DOI] [PubMed] [Google Scholar]
- Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS, and Ewald AJ (2019). E-cadherin is required for metastasis in multiple models of breast cancer. Nature 573, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paine IS, and Lewis MT (2017). The Terminal End Bud: the Little Engine that Could. J. Mammary Gland Biol. Neoplasia 22, 93–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel H, Bray NL, Puente S, Melsted P, and Pachter L (2017). Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 14, 687–690. [DOI] [PubMed] [Google Scholar]
- Raff MC (1992). Social controls on cell survival and cell death. Nature 356, 397–400. [DOI] [PubMed] [Google Scholar]
- Ring BZ, Hout DR, Morris SW, Lawrence K, Schweitzer BL, Bailey DB, Lehmann BD, Pietenpol JA, and Seitz RS (2016). Generation of an algorithm based on minimal gene sets to clinically subtype triple negative breast cancer patients. BMC Cancer 16, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinkenberger J, and Werb Z (2000). The labyrinthine placenta. Nat. Genet 25, 248–250. [DOI] [PubMed] [Google Scholar]
- Robinson DR, Wu Y-M, Lonigro RJ, Vats P, Cobain E, Everett J, Cao X, Rabban E, Kumar-Sinha C, Raymond V, et al. (2017). Integrative clinical genomics of metastatic cancer. Nature 548, 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, and Turner JR (2005). Actin depolymerization disrupts tight junctions via caveolae-mediated endocytosis. Mol. Biol. Cell 16, 3919–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyer AE, Huycke TR, Lee C, Mahadevan L, and Tabin CJ (2015). Bending gradients: how the intestinal stem cell gets its home. Cell 161, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestri VL, Henriet E, Linville RM, Wong AD, Searson PC, and Ewald AJ (2020). A tissue-engineered 3D microvessel model reveals the dynamics of mosaic vessel formation in breast cancer. Cancer Res Published online July 14, 2020 10.1158/0008-5472. [DOI] [PMC free article] [PubMed]
- Simian M, and Bissell MJ (2017). Organoids: A historical perspective of thinking in three dimensions. J. Cell Biol 216, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HW, and Marshall CJ (2010). Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol 11, 23–36. [DOI] [PubMed] [Google Scholar]
- Toda S, Blauch LR, Tang SKY, Morsut L, and Lim WA (2018). Programming self-organizing multicellular structures with synthetic cell-cell signaling. Science 361, 156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JR (2009). Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol 9, 799–809. [DOI] [PubMed] [Google Scholar]
- Visvader JE, and Stingl J (2014). Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes Dev 28, 1143–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson SS, Dane M, Chin K, Tatarova Z, Liu M, Liby T, Thompson W, Smith R, Nederlof M, Bucher E, et al. (2018). Microenvironment-Mediated Mechanisms of Resistance to HER2 Inhibitors Differ between HER2+ Breast Cancer Subtypes. Cell Syst 6, 329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier da Silveira Dos Santos A, and Liberali P (2019). From single cells to tissue self-organization. FEBS J 286, 1495–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Goliwas KF, Wang W, Taufalele PV, Bordeleau F, and Reinhart-King CA (2019). Energetic regulation of coordinated leader-follower dynamics during collective invasion of breast cancer cells. Proc. Natl. Acad. Sci. USA 116, 7867–7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) MMTV-PyMT mTomato+ single cells were aggregated for 0, 6, or 24 hr and stained for DAPI. Right: quantification of number of nuclei per entity (single confocal slice), with a mean number of cells per cluster of 4.4 (SD ± 4.2) and 5.4 (SD ± 5.2) at 6 and 24 hr, respectively. n = 3 biological replicates. n (# entities, single cells or clusters): 0hr = 307, 6hr = 359, 24hr = 326. P values = unpaired t tests.
(B) Number of metastases formed 3 weeks after mTomato-PyMT tail vein relative to the number of entities (single cells or clusters) injected based on measurements in (A). % success = (100 x number of metastases)/number of entities injected. n (# mice) = 19 (0hr), 9 (6hr), 14 (24hr), 10 (re-dissociated). P values = Mann-Whitney tests. Medians: 0hr = 0%, 6hr = 0.16%, 24hr clusters = 1.72%, 24hr redissociated = 0%.
(C) Experimental schematic: 200,000 mTomato-PyMT cells were injected by tail vein into NSG mice either as single cells or clusters. 0, 24, and 48 hr after injection the lungs were harvested. Non-consecutive sections were imaged and the number of mTomato+ clusters and single cells was quantified for each field of view.
(D) Quantification of the number of mTomato+ single cells or clusters detected in lung sections at 0, 24, and 48 hr after tail vein injection. For each mouse 3 sections were imaged (10 25x fields per section). n = 4 mice per time point.
(E) To determine whether single tumor cells form micro-metastases that are not visible at low magnification, lungs were sectioned 3 weeks after tail vein injection of single mTomato+ PyMT cells. Sections were imaged at 10x magnification and the number of mTomato+ single cells and clusters was counted. n = 6 mice, 1 section per mouse.
(F) Schematic of intracardiac injection of mTomato-PyMT clusters or single cells into the left ventricle of immunocompromised mice to generate systemic metastases. Right, organs 6 weeks after intracardiac injection.
(G) Quantification of systemic metastases 6 weeks after intracardiac injection of mTomato-PyMT clusters. n = 4 single cell injected mice, 7 cluster injected mice. p values = Mann-Whitney test.
(H) Lung sections of mice injected by tail vein with Ctrl-kd or Epgn-kd clusters. Red signal = mTomato+ metastases. Right, quantification of the number of metastases per lung section. n = 5 mice per condition, 1–2 sections per mouse. P value = Mann-Whitney test.
(I) Size of individual lung metastases 3 weeks after tail vein injection of Ctrl-kd or Epgn-kd clusters. Boxplot = 5%–95%. P value = Mann-Whitney test.
(J) Left: 24hr aggregated Ctrl-kd-GFP and Epgn-kd-GFP clusters shortly before tail vein injection. Right: lung sections 3 weeks after tail vein injection of mTomato-PyMT Ctrl-kd-GFP or Epgn-kd-GFP clusters.
(K) Quantification of double positive (GFP+ and mTomato+) and shRNA escape (mTomato+ but GFP−) areas in Ctrl-kd or Epgn-kd lung metastases 3 weeks after tail vein injection. n = 3 mice per condition. P values = unpaired t tests.
(A) Time lapse images of a MMTV-PyMT single cell and cluster cultured in 3D basement membrane-rich gels for 4 days. Right, outgrowth quantification as fold change in area. n = 8 mice, 23 single cells, 500 clusters. P value = Mann-Whitney test.
(B) Outgrowth measured by fold change in area over 4 days of time lapse imaging for MMTV-PyMT single cells and clusters of different sizes in 3D basement membrane-rich gels. P values = Mann-Whitney tests. n (# of single cells or clusters): 0–500 um2 = 39, 500–1000 um2 = 39, 1000–2000 um2 = 58, > 2000 um2 = 387. n = 8 biological replicates.
(C) % of nuclei in mitosis (scored by pHH3 staining) binned by number of neighboring cells with visible direct membrane-membrane contact. n = 4 biological replicates. n (# cells): 0 contacts (single cells) = 453, 1 contact = 698, 2 contacts = 704, 3 contacts = 667. P values = unpaired t tests.
(D) Time lapse DIC and GFP images of a MMTV-PyMT single cell and cluster in the presence of a caspase-3/7 activated green fluorescing DNA dye in 3D basement membrane-rich gels. Right, quantification of MMTV-PyMT single cell versus cluster survival over time. Clusters were marked as dead once a majority of cells in the clusters apoptosed. n = 4 mice, 79 single cells and 105 clusters. P value = Mantel-Cox.
(E) Time lapse DIC and GFP (Caspase 3/7 biosensor) images of a single cell and cluster derived from a human breast cancer brain metastasis. Right, summary of single cell versus cluster survival after 72hrs of time lapse in 3D basement membrane-rich gels from 9 human breast tumor specimens (HU1–9). n = 283 single cells and 197 clusters. P value = pairwise t test.
(F) Survival of single cells versus clusters from 10 individual human breast cancer patient samples (HU1–10). P values = Mantel-Cox.
(A) Validation of MMTV-PyMT shRNA knockdowns by qPCR. Red line = mean. # of biological replicates; Plaur-kd n = 3, Egfr-kd n = 4, Areg-kd n = 2, Epgn-kd n= 3.
(B) Left, Images of Ctrl and knockdown cells after 24 hr of aggregation, plated at equal cell densities. Middle, quantification of suspension object areas (MATLAB) prior to tail vein injection. n (# entities): Ctrl-kd = 6931, Egfr-kd = 3561, Areg-kd = 3158, Epgn-kd = 3367. Right, number of nuclei (by DAPI staining) of Ctrl-kd or Epgn-kd clusters aggregated for 24hrs. n = 3 biological replicates. n = 100 Ctrl-kd clusters, 66 Epgn-kd clusters. P value = unpaired t test.
(C) DIC time lapse images of Ctrl-kd, Plaur-kd, Egfr-kd, Epgn-kd, Areg-kd MMTV-PyMT clusters growing in 3D basement membrane-rich gels for 90 hr. Right, quantification of fold growth by change in area over 4 days of time lapse imaging. Ctrl-kd: n = 8 mice, 246 clusters. Plaur-kd: n = 2 mice, n = 31 clusters. Egfr-kd: n = 3 mice, 84 clusters. Areg-kd: n = 3 mice, 108 clusters. Epgn-kd: n = 3 mice, 90 clusters.
(D) % of Ctrl-kd and Epgn-kd nuclei in mitosis after 24 hr of aggregation, measured as % phospho-histone H3 positive. n = 4 biological replicates. n (# nuclei): Ctrl-kd = 2636, Epgn-kd = 1790. P value = unpaired t test.
(E) RNA expression (RNA-seq) of cytokeratin genes in Ctrl-kd and Epgn-kd MMTV-PyMT clusters, n = 3. Adjusted p values.
(F) Immunofluorescence of keratin-17 in Ctrl-kd and Epgn-kd clusters in 3D basement membrane-rich gels after 6 days of outgrowth. Right, quantification of % cells in each cluster keratin-17 positive by immunofluorescence. n = 2 biological replicates. n (# clusters): non-transduced = 34, Ctrl-kd = 39, Epgn-kd = 27. P values = unpaired t tests.
(G) Validation of MMTV-PyMT TRC-Epgn-kd by qPCR. n = 3 biological replicates. P value = unpaired t test.
(H) Relative viable cell number of TRC-Ctrl-kd and TRC-Epgn-kd after 6 days of outgrowth in low basement membrane suspension culture. n = 3 biological replicates. P value = unpaired t test.
(I) DIC images of Ctrl-kd and Epgn-kd clusters after 4 days growth with and without transduction with an shRNA resistant human epigen construct (MOI < 1). Right, quantification of viable cell number using CellTiter-Glo. n = 3 biological replicates. P value = unpaired t test.
(A–B) Immunofluorescence of epigen in tumor cell clusters ex vivo from MMTV-Neu tumors (A) and C3(1)Tag tumors (B).
(C) Quantification of epigen immunofluorescence signal along cytosolic, cell-cell, or cell-matrix areas in C3(1)-TAg clusters. n = 60 clusters, 3 biological replicates. P values = Mann-Whitney test.
(D) Relative number of viable cells after 6 days of treatment of PyMT cells in suspension with recombinant epigen. n = 3 biological replicates. P value = unpaired t test.
(E) Cumulative migration distance of individual clusters, normalized to initial cluster cell number, in 3D basement membrane-rich gels over 120 hr. Mixed clusters were generated by aggregating Ctrl-kd and Epgn-kd cells at a 1:1 ratio. Data are plotted as mean with 5%–95% CI. n (# of clusters): Non-transduced = 49, Ctrl-kd = 50, Epgn-kd = 47, Mixed 1:1 = 35. n (# of biological replicates) = 2. P values = unpaired t tests.
(F) Ultrastructure of tumor cell cluster using high pressure freezing electron microscopy. Pseudo-color highlights different cells. MP = microvilli-like protrusions. J = cell-cell junction.
(G) Transmission electron microscopy of Ctrl-kd and Epgn-kd PyMT clusters. Right, nanolumenal area and perimeter measurements. n = 40 nanolumina measured per condition (measured junction to junction). MP = microvilli-like protrusions. J = cell-cell junction.
(H) Immunofluorescence of epigen and apical marker Muc-1 in a MMTV-PyMT cluster.
(I) Biotin leak-in assay of MMTV-PyMT clusters without treatment, treated with ochratoxin A (200 µm, 1 hr) (McLaughlin et al., 2004), or treated with latrunculin A (0.2 µm, 1hr) (Shen and Turner, 2005) to disrupt cell-cell junctions.
(J) Estimated concentration of epigen in nanolumen, based on TEM estimates of nanolumenal volume per cell and ELISA measurements of epigen protein per cell. Secreted epigen was also measured by epigen ELISA of 18 hr tumor cell cluster supernatants (extra-cluster concentration). Supernatants: n = 3 biological replicates. Cell lysates: n = 2 biological replicates. P values = unpaired t tests.
(A) Quantification of receptor tyrosine kinase phospho-tyrosine array signal for MMTV-PyMT cluster (6hrs aggregation) versus PyMT single cell (0hr aggregation) lysates. n = 3 biological replicates. P values = unpaired t tests.
(B) Immunoblots of single cell (0 hr) versus cluster (6hrs aggregation) MMTV-PyMT lysates for phospho (Thr 202/Tyr204) and total ERK1/2. Right: quantification of n = 3 biological replicates. P values = unpaired t tests.
(C) Immunoblots of single cell (0hr) versus cluster (6hrs aggregation) MMTV-PyMT lysates for phospho (S473) and total Akt. Right: quantification of n = 3 biological replicates. P values = unpaired t tests.
(D) Immunoblot of EGFR and EGFR effectors pERK1/2 in non-transduced, Ctrl-kd, and Egfr-kd PyMT tumor cell clusters. Right: quantification of n = 3 biological replicates. P values = unpaired t tests.
(E) Immunofluorescence of pEGFR (Y1068) in a MMTV-PyMT cluster ex vivo. Right, quantification of pEGFR at intracellular (I), cell-matrix (C-M), and cell-cell (C-C) spaces in MMTV-PyMT clusters. n = 68 clusters, n = 3 biological replicates. P values = Mann-Whitney tests.
(F) Immunofluorescence of HER2 in a MMTV-PyMT cluster ex vivo. Right, quantification of HER2 immunofluorescence at intracellular (I), cell-matrix (C-M), and cell-cell (C-C) spaces in MMTV-PyMT clusters. n = 40 clusters, n = 2 biological replicates. P values = Mann-Whitney tests.
(G) Immunogold labeling of pEGFR along nanolumina in MMTV-PyMT clusters. Arrows = pEGFR immunogold signal. Right, quantification of labeling in intercellular areas, along nanolumenal membranes, along nanolumenal villi, at cell-cell junctions, and within nanolumenal voids. n = 93 gold particles.
(H) Immunogold labeling of HER2 along nanolumina in MMTV-PyMT clusters. Arrows = HER2 immunogold signal. Right, quantification labeling in intracellular areas, along nanolumenal membranes, along nanolumenal villi, at cell-cell junctions, and within nanolumenal voids. n = 45 gold particles.
(A) Immunofluorescence of epigen and keratin-14 in normal FVB mouse back skin as an antibody positive control, as epigen is known to be expressed in hair follicles (Kochupurakkal et al., 2005).
(B) Genotype-Tissue Expression (GTEx) RNA sequencing data of EPGN in normal human tissues including (left) the top 10 EPGN expressing tissues by median expression and (right) frequent sites of breast cancer metastasis.
(C) Pan-cytokeratin, Keratin-14, Keratin-17, and epigen immunofluorescence of protrusive tumor strands on the border of mTomato-PyMT orthotopic tumors 10 weeks after transplant of non-transduced (pan-CK, K17) or Ctrl-kd clusters (K14).
(D) Immunofluorescence of epigen in a liver metastasis 6 weeks after intracardiac injection of mTomato-PyMT clusters.
(E) pEGFR (Y1068) immunofluorescence of a primary tumor 10 weeks after mTomato-PyMT organoid orthotopic transplant into the mammary fat pad and of a liver metastasis 6 weeks after intracardiac injection of mTomato-PyMT clusters.
(F) HER2 immunofluorescence of a primary tumor 10 weeks after mTomato-PyMT organoid orthotopic transplant into the mammary fat pad and of a liver metastasis 6 weeks after intracardiac injection of mTomato-PyMT clusters.
(G) 20X tiled images were taken of sections from Ctrl-kd and Epgn-kd orthotopic transplant tumors. Right, quantification of the number of locally disseminated clusters (mTomato+) per mm2 stromal area (DAPI+/mTomato−). n = 6 Ctrl-kd tumors (148 clusters), 6 Epgn-kd tumors (218 clusters). P values = unpaired t tests.
(H) Left, images of lung metastases 6 weeks after orthotopic transplant of Ctrl-kd or Epgn-kd organoids, stained for mitotic marker pHH3. Right, quantification of % of lung metastasis cells pHH3+. n = 3 Ctrl-kd mice (925 cells), n = 3 Epgn-kd mice (157 cells). P value = unpaired t test.
(I) Area measurements of individual metastases (including disseminated single cells) in lungs 6 weeks after orthotopic transplant of Ctrl-kd or Epgn-kd organoids. n = 5 mice per condition. n = 47 Ctrl-kd metastases, 72 Epgn-kd metastases. P value = Mann-Whitney test.
(J) Number of metastases detected in each lung section from mice 6 weeks after orthotopic transplant of Ctrl-kd or Epgn-kd organoids. n = 5 mice per condition. P value = unpaired t test.
(K) mTomato+ and GFP+ areas were measured in Ctrl-kd and Epgn-kd orthotopic tumor sections using 20X tiled images. Right, quantification of the GFP+ area as a percent of the mTomato+ area. n = 4 Ctrl-kd tumors, 4 Epgn-kd tumors. P value = unpaired t test.
(A) Shown are representative tumor cell clusters obtained directly from the ascites fluid of a metastatic ER+/PR+/HER2-breast cancer patient with disease to bone, liver, chest wall, stomach and peritoneum. Left: image of ascites cluster stained for pan-cytokeratin, CD45, and pEGFR. Right: TEM images of ascites clusters. MP = microvilli-like protrusions. J = cell-cell junction.
(B) Epigen immunofluorescence of tumor cell clusters in malignant ascites from patients #1 and #2 using a pan-cytokeratin antibody to identify tumor cell clusters and CD45 to identify immune cells.
(C) Epigen mRNA expression in two panels of breast cancer cell lines (Barretina et al., 2012; Klijn et al., 2015). Triple-negative subgroups: BL2 = basal-like 2, BL1 = basal-like 1, M = mesenchymal, LAR = luminal androgen receptor. L-HER2+ = luminal like HER2+ cell lines. HER2E = basal like HER2+ cell lines (Cancer Genome Atlas Network, 2012; Watson et al., 2018).
(D) Gene sets (Metascape, FDR % 0.05) enriched in M-like (MDA-MB-231, MDA-MB-436, and BT549) versus BL2 (HCC70, CAL851, HDQP1) cell lines (CCLE).
(E) GFP images of Ctrl-kd and Epgn-kd cells grown on collagen I coated plates to assess 2D migration. Right, quantification of the colony area one day after plating (D1) and of the dense monolayer (“confluent”) and invasive outer layer (“subconfluent”) formed 6 days after plating (D6). n = 3 biological replicates. P values = unpaired t tests.
(F) Relative number of viable cells of non-transduced, Ctrl-kd, and Epgn-kd HCC70 cells after 4 days of growth (seeded at equal cell number) in 2D on tissue culture treated plates. n = 3 biological replicates. P value = unpaired t test.
(G) Left: qPCR validation of HCC70 clusters transduced with non-targeting or epigen-targeting CRISPR sgRNAs. Cells were not clonally selected i.e., these represent mixed populations of cells with < 100% knockout. n = 2 biological replicates. Right, relative number of viable cells after 4 days of growth as clusters in low-basement membrane suspension. n = 3 biological replicates. P values = unpaired t tests.
(H) Number of non-viable nuclei per 1000 um2 area in HCC70 clusters ex vivo, measured by confocal imaging of propidium iodide staining after 6 days with or without 3 ng/mL IFNγ treatment. n = 3 biological replicates. n = 76 untreated clusters, 72 IFNγ treated clusters.
(I) Fold change of CLDN7 mRNA (relative to RPL32 housekeeping control) for MDA-MB-231 and HCC70 clusters after 6 days with or without 3ng/mL IFNγ.n=3 biological replicates. P values = unpaired t tests. RAB25 was not detected by qPCR in MDA-MB-231 cells at any time point.
(J) Representative images of biotin leak-in assay of MDA-MB-231 clusters after 6 days of treatment with or without 3ng/mL IFNγ. Right, quantification of biotin leak-in (internal FITC-streptavidin intensity). n = 18 untreated clusters, 16 IFNγ treated clusters. n = 2 biological replicates. P value = Mann-Whitney test.
(K) Here, we find that tumor cell clusters generate sealed, intercellular nanolumina. During phases of outgrowth, these nanolumina have restricted permeability and contain high concentrations of the growth factor epigen. Collective signaling by epigen within nanolumina promotes primary and metastatic tumor outgrowth. In contrast, low epigen expression is found in protrusive locally disseminated clusters. High epigen expression and nanolumenal impermeability were found in basal-like 2 human breast cancer cells, whereas mesenchymal-like cells were highly permeable and lowly expressed epigen. Suppression of epigen, or targeting of nanolumenal junctions, in basal-like 2 breast cancer cells reduced collective metastatic outgrowth.
Data Availability Statement
RNA sequencing datasets have been deposited to the NCBI Sequence Read Archive (PRJNA648151, PRJNA648435).