

ABSTRACT
In response to stress, the obligate intracellular pathogen Chlamydia trachomatis stops dividing and halts its biphasic developmental cycle. The infectious, extracellular form of this bacterium is highly susceptible to killing by the host immune response, and by pausing development, Chlamydia can survive in an intracellular, “aberrant” state for extended periods of time. The relevance of these aberrant forms has long been debated, and many questions remain concerning how they contribute to the persistence and pathogenesis of the organism. Using reporter cell lines, fluorescence microscopy, and a dipeptide labeling strategy, we measured the ability of C. trachomatis to synthesize, assemble, and degrade peptidoglycan under various aberrance-inducing conditions. We found that all aberrance-inducing conditions affect chlamydial peptidoglycan and that some actually halt the biosynthesis pathway early enough to prevent the release of an immunostimulatory peptidoglycan component, muramyl tripeptide. In addition, utilizing immunofluorescence and electron microscopy, we determined that the induction of aberrance can detrimentally affect the development of the microbe’s pathogenic vacuole (the inclusion). Taken together, our data indicate that aberrant forms of Chlamydia generated by different environmental stressors can be sorted into two broad categories based on their ability to continue releasing peptidoglycan-derived, immunostimulatory muropeptides and their ability to secrete effector proteins that are normally expressed at the mid- and late stages of the microbe’s developmental cycle. Our findings reveal a novel, immunoevasive feature inherent to a subset of aberrant chlamydial forms and provide clarity and context to the numerous persistence mechanisms employed by these ancient, genetically reduced microbes.
KEYWORDS: persistence, immune evasion, pathoadaptation, Chlamydia trachomatis, peptidoglycan, secretion systems
INTRODUCTION
Chlamydia trachomatis is the leading cause of infectious blindness and bacterial sexually transmitted infectious disease, with a hundred million individuals infected worldwide (1). Over 1.7 million new genital tract infections are reported to the CDC annually (2, 3). Due to the large number of asymptomatic infections that go unreported each year, estimates place the true number of annual infections in the United States at closer to 3 million (4). The disease burden falls disproportionately on young women between the ages of 15 and 24 years, and while infections are treatable, untreated infections can result in pelvic inflammatory disease, ectopic pregnancy, and infertility in women and epididymitis or urethritis in men (5).
Chlamydia species utilize a distinctive, biphasic developmental cycle (Fig. 1a), alternating between a small (≤0.5-μm), extracellular, infectious form (elementary body [EB]) and a larger (∼1-μm), intracellular, replicative form (reticulate body [RB]). Because each developmental form is highly specialized and can carry out only either the infection or replication process, preventing the development of either form effectively halts the infection cycle. EBs are rapidly internalized upon attachment to a host cell and utilize a type III secretion (T3S) system (T3SS) to inject over 150 different effector proteins into the cell’s cytosol. These effectors have a number of diverse activities. They permit the Chlamydia-containing vacuole (inclusion) to escape the cell’s endosomal maturation pathway and lysosomal fusion, inhibit the host cell from triggering apoptosis, alter the dynamics of the infected cell’s cytoskeletal proteins, and redirect intracellular vesicle trafficking (6). The expression of these effectors is highly regulated, and the timing of their expression and secretion largely aligns with their function and role in the microbe’s developmental process. Inclusion membrane proteins (Incs) are one class of effectors that are inserted into the outer leaflet of the inclusion membrane and permit Chlamydia to control the vast majority of vesicle fusion events that occur within the infected cell as the inclusion enlarges. This allows the microbe to control the overall composition of the inclusion and direct its localization, as well as that of several organelles, within the host cell. Within ∼8 to 12 h of invading a cell, EBs begin differentiating into RBs, initiate replication, and begin to divide by a process that is dependent on the synthesis and turnover of septal peptidoglycan (PG) (7, 8). At roughly 24 h postinfection (hpi), RBs begin asynchronously converting back into EBs. Chlamydia EBs eventually exit their host cell by one of two pathways: either (i) the inclusion expands enough to lyse the host cell, expelling the newly formed EBs, or (ii) portions of the inclusion begin to bleb off into the extracellular environment in a process termed extrusion (9).
FIG 1.
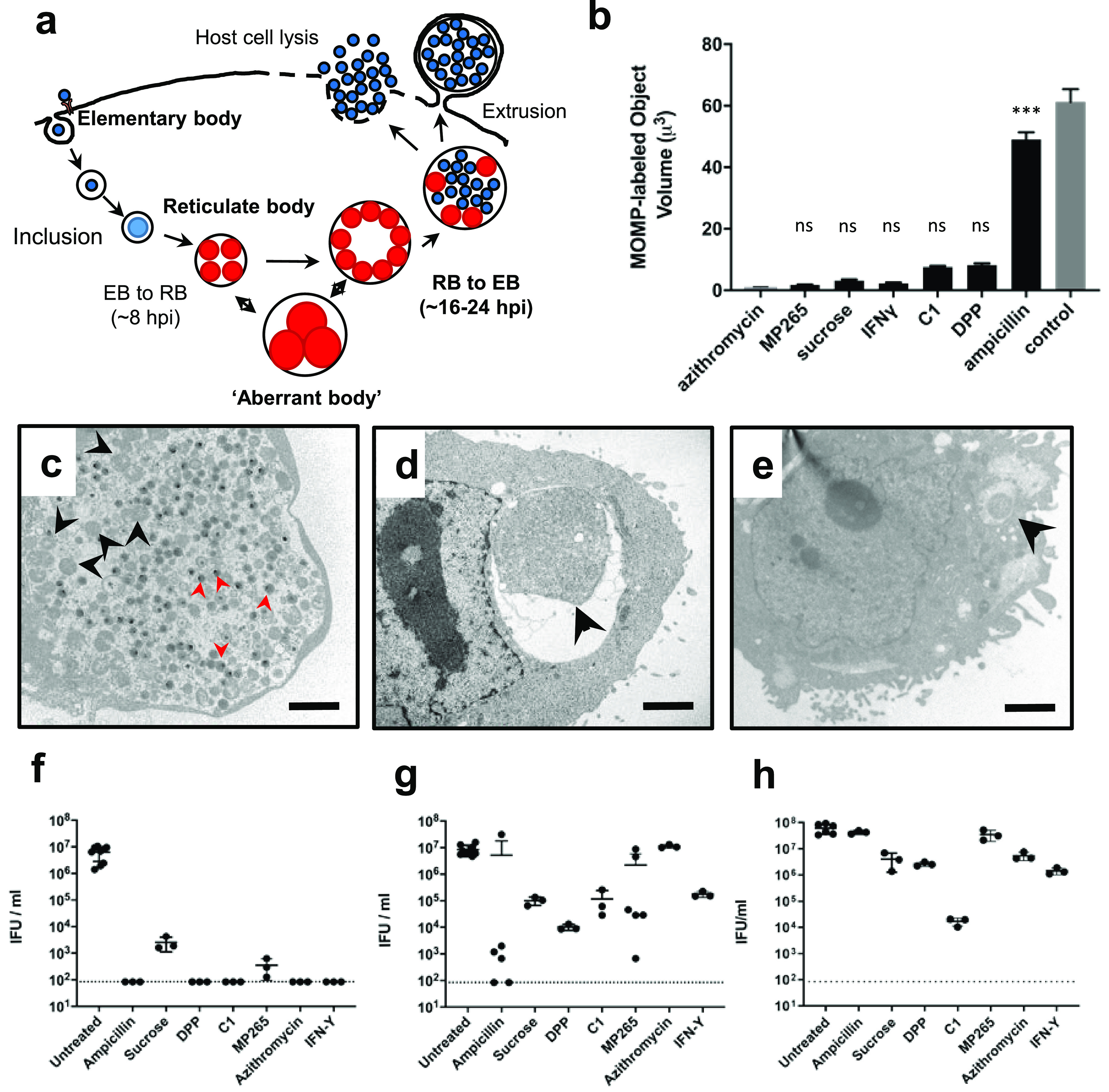
C. trachomatis aberrant body formation results in cell division arrest and pausing of the developmental cycle. (a) Diagram of the Chlamydia developmental cycle. (b) Volume measurements of C. trachomatis present within inclusions at 24 hpi under untreated and aberrance-inducing conditions. Data were collected by fluorescence microscopy over three independent experiments. Each data point represents the collective total of contiguous, MOMP-labeled material (∼100 inclusion measurements taken per group). (c to e) Electron micrographs of HeLa cells infected with C. trachomatis at 24 hpi that were either untreated (c) or treated with AMP (d) or the iron chelator DPP (e). Black arrowheads indicate RBs and ABs, and red arrowheads indicate the much smaller, electron-dense EBs. Bars, ∼2 μm. (f to h) Inclusion-forming unit (IFU) counts were conducted at 44 h postinfection/posttreatment (f) and after recovery in the absence of treatment for an additional 24 h (g) and 48 h (h). The dotted lines represent the assay’s limit of detection. C1, T3SS inhibitor compound C1; MP265, MreB polymerization inhibitor. Each data point plotted represents data from an independent experiment, and error bars represent the standard deviations from the means.
Researchers have long known of the existence of an additional, nonreplicative form of Chlamydia that is capable of remaining viable within the inclusion vacuole for extended periods of time. These forms are referred to as “aberrant bodies” (ABs) and have been classically distinguished by their enlarged size (∼2 to 10 μm). Chlamydia AB formation is the result of cell division inhibition while cell growth continues unabated (Fig. 1a). Because cell division is required for the bacterium to eventually return to its infectious form, halting the process also inhibits the production of infectious EBs. This pause in development is reversible; in most cases, upon removal of the stressor, cell division resumes, and, eventually, infectious progeny are once again observed. This unusual morphology was noted in early studies investigating the effects of the antibiotic penicillin on intracellular Chlamydia species (10). It was originally proposed that this unique morphology resulted from the inhibition of the Chlamydia penicillin-binding proteins (PBPs) (the target of all β-lactam-based antibiotics) that are essential for bacterial cell division (11). Subsequent studies identified additional stressors that induce Chlamydia AB formation in tissue culture cells. Examples include the addition of the cytokine interferon gamma (IFN-γ) (12), tryptophan (13) and iron (14) starvation, heat shock (15), amino acid starvation (16, 17), infection of monocytes (18) and macrophages (19, 20), coinfection with viruses (21, 22), and a number of other pressures (23). Aberrant forms have also been observed in vivo (24–26), indicating that they can develop during infection. This phenotype is not unique to members of the Chlamydiaceae, as prominent members of the Chlamydiales have recently been shown to exhibit the aberrance phenotype under a number of experimental conditions in vitro (27).
Given their appearance under a wide array of culture conditions, it has been proposed that AB formation results from an underlying stress response pathway that provides Chlamydia species a level of protection during infection (28). In the context of pathogenic Chlamydia species, “persistence” has generally been defined as (i) the reversible inhibition of cell division that results in an interruption of the pathogen’s developmental cycle and (ii) a corresponding increase in bacterial cell size. While the mechanisms of cell division arrest have been previously investigated in great detail for a number of aberrance-inducing conditions (29), considerably less effort has been spent on investigating the degree to which these persistent forms differ in their basic physiologies and their interactions with host cells. While a few studies have characterized the structural (30), transcriptional (31–33), and translational (34, 35) regulatory mechanisms underlying AB formation, there continues to be much speculation and debate concerning the relevance of Chlamydia ABs in vivo and their contribution to the pathogenesis and persistence of these organisms in human and animal infections (23, 36–38).
Currently, the defining features of Chlamydia’s “persistent state” are (i) the inhibition of cell division, (ii) cell size enlargement, (iii) the inhibition of EB production, and (iv) the reversibility of the process. In this study, we set out to explore the physiological underpinnings that define these aberrant forms of Chlamydia and delineate how their structural and functional differences impact the microbe’s interaction with its host cell. We found that two prominent factors differ between Chlamydia’s aberrant forms: (i) the synthesis and turnover of immunostimulatory peptidoglycan and (ii) the presentation of effector proteins on the inclusion surface that are associated with the mid- and late stages of the pathogen’s developmental cycle.
RESULTS
C. trachomatis aberrant body formation results in cell division arrest and temporarily pauses the developmental cycle.
We began by testing a panel of aberrance-inducing conditions to determine if the ABs produced met the classical criteria for chlamydial persistence. Seven aberrance-inducing conditions were compared: β-lactam antibiotics (ampicillin [AMP]), nonionic osmotic stress (induced by sucrose supplementation), MreB inhibition (induced by the polymerization inhibitor MP265), T3SS inhibition (induced by the T3S inhibitor compound C1), iron depletion (induced by 2,2′-bipyridyl [DPP]), and tryptophan starvation (induced by interferon gamma [IFN-γ]). At 24 hpi, we fixed and labeled the outer surface of Chlamydia trachomatis L2 strain 434/Bu under different treatment conditions and conducted an imaging analysis to assess the average volume of microbial material present within individual inclusions. By 24 h, C. trachomatis inclusions are filled with hundreds of RBs and EBs and therefore do not provide sufficient resolution for delineating individual RB cell boundaries by fluorescence microscopy (Fig. 1b, control). Azithromycin inhibits protein synthesis in Chlamydia spp., preventing RB growth and division. The RB volume and diameter in infected cells treated with azithromycin at 8 hpi were ∼0.9 μm3 and ∼1.2 μm, respectively. These measurements align well with what has been reported previously regarding the average RB size early in chlamydial development (39), and we used this condition to serve as a positive control representing the normal RB size in our subsequent analysis. All aberrance-inducing conditions tested halted or severely limited cell division in C. trachomatis within 24 h; however, we found that the resultant ABs differed dramatically in size (Fig. 1b to e). AMP induction resulted in the largest AB volumes and average diameters (∼50 μm3 and ∼5 μm, respectively), with the rest of the conditions resulting in ABs with much smaller volumes and average diameters (∼5 to 10 μm3 and ∼1.5 to 3 μm, respectively). Examination of inclusions under different aberrance-inducing conditions by electron microscopy revealed that small ABs were generally found within small inclusions compared to both untreated and AMP-treated controls (Fig. 1c to e).
Because cell division and differentiation are linked in Chlamydia species, the inhibition of cell division results in the halting of the developmental cycle and prevents the formation of Chlamydia’s infectious form. In order to validate that our AB induction conditions successfully halted EB formation, we lysed infected cells at 44 hpi and infected new cell monolayers with these lysates. After 24 h, we counted the resultant inclusions and used them as a readout for the number of infectious EBs resulting from the previous infection. The majority of aberrance-inducing conditions tested effectively halted EB production, and few, if any, infectious forms were recoverable after 44 h (Fig. 1f).
While “aberrance” is defined by the absence of cell division and a corresponding increase in chlamydial cell size, for this morphotype to be defined as “persistence,” the phenotype must also be reversible. To assess this, we removed the aberrance-inducing medium from each treatment group and allowed the cells to recover (as described in Materials and Methods). Under most conditions, we observed a partial recovery of infectious progeny production after 24 h (Fig. 1g) and nearly full recovery after 48 h (Fig. 1h). Only C. trachomatis treated with compound C1 failed to recover to within one log of the untreated control group within 48 h. Previous studies have found that compounds similar to C1 used as T3SS inhibitors may also function as iron chelators (40, 41), and we hypothesized that this dual functionality might explain why C. trachomatis was unable to recover as efficiently under this treatment condition. When exogenous iron sulfate was added to the infection medium containing compound C1, we observed a partial recovery of infectious progeny, indicating that C1 may also act as an iron chelator (see Fig. S1 in the supplemental material). Overall, our observations were largely consistent with those of previous studies (8, 10, 14, 31, 42), and we concluded that our testing conditions were suitable for inducing persistence in C. trachomatis L2 strain 434/Bu.
Persistent forms of Chlamydia differ in their capacity to assemble septal peptidoglycan.
Peptidoglycan (PG) is a principal component of the bacterial cell wall and is essential for maintaining cell shape, regulating osmotic pressure, and constricting the septum during the last stage of the bacterial division process (43). Recent technical advances have allowed us to label PG in Chlamydia spp. utilizing a novel dipeptide incorporation strategy (7, 44). This labeling is specific for newly synthesized PG (Fig. 2a) as the incorporation of the probes occurs in living microbes during the initial synthesis of muramyl-pentapeptide in the bacterial cell cytoplasm (45). Previously, we determined that members of the Chlamydiaceae (pathogenic Chlamydiae) possess PG (7) (Fig. 2b, row 1, column 1). Whereas other bacterial species, including other members of the Chlamydiales (46), synthesize PG as a major component of their cell wall, pathogenic Chlamydia species limit PG to their septum (8), representing a significant reduction in PG abundance in these microbes and highlighting its importance for chlamydial cell division. As aberrant forms of Chlamydia no longer divide, we hypothesized that this deficiency was likely due to the inhibition of either PG synthesis or assembly.
FIG 2.
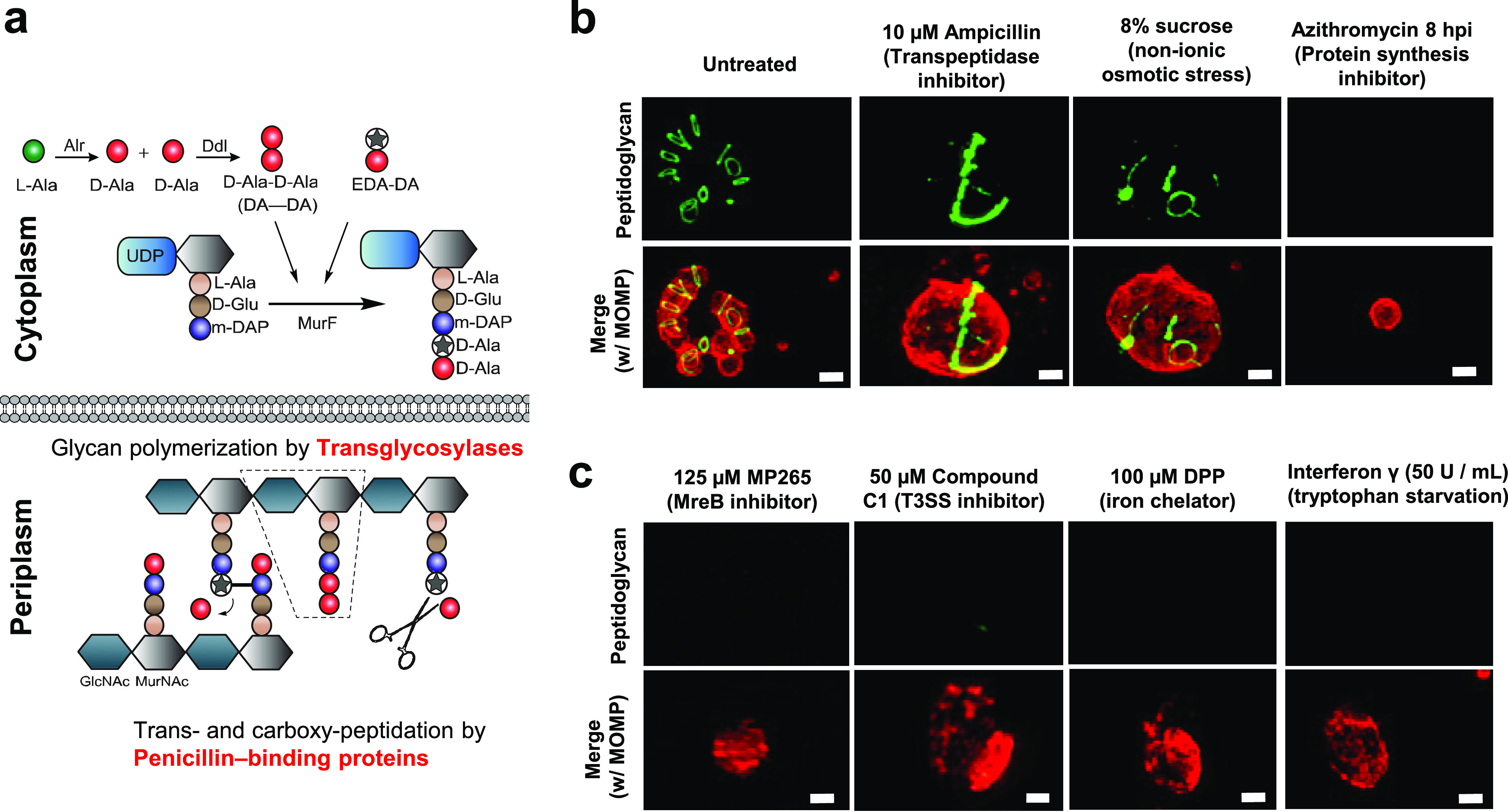
Aberrant Chlamydia forms differ in their abilities to synthesize peptidoglycan. (a) The Park nucleotide biosynthesis pathway in Gram-negative bacteria. Peptidoglycan can be labeled utilizing modified ethynyl-D-alanyl-D-alanine (EDA-DA) (green), which is subsequently incorporated into newly synthesized stem peptides (7). Alr, alanine racemase; Ddl, d-alanine-d-alanine ligase; MurE, UDP-N-acetylmuramoyl-l-alanyl-d-glutamate-diaminopimelate ligase; MurF, UDP-N-acetylmuramoyl-tripeptide-d-alanyl-d-alanine ligase. (b and c) Chlamydia-infected cells were grown under a variety of aberrance-inducing conditions. Peptidoglycan was labeled via click chemistry, and Chlamydia RBs and ABs were visualized utilizing a monoclonal antibody to the major outer membrane protein (MOMP) (red). Images are representative of 20 to 30 viewing planes over three independent experiments. ***, P < 0.001; ns, not significant. Bars, ∼1 μm.
Our labeling studies revealed that C. trachomatis is still capable of synthesizing PG glycan chains when subjected to β-lactam antibiotics that induce AB formation; however, PG localization is no longer restricted to a single division septum (Fig. 2b, row 1, column 2) (7, 8). In contrast, when PG assembly is completely prevented by inhibiting MreB, the major organizer of the microbe’s division complex (47), no PG labeling is discernible (Fig. 2c, row 1, column 1) (8). We hypothesized that different inducers of persistence may similarly differ in whether they cause cell division arrest as a result of deficiencies in either PG assembly or PG synthesis. To distinguish between these two possibilities, we tested our panel of aberrance-inducing conditions in conjunction with our PG labeling assay. We found that sucrose-induced aberrance produced ABs that contained labeled PG in an irregular arrangement (Fig. 2b, row 1, column 3; Fig. S5), similar to that observed in β-lactam-induced aberrance. Conversely, inhibition of the microbe’s T3SS, iron depletion, and tryptophan depletion all resulted in ABs that lacked detectable PG (Fig. 2c, row 1, columns 2 to 4). As the mechanism of action by which this second category of stressors induces aberrance involves the downregulation of transcription or posttranscriptional regulation (31–35), we also examined the effect of azithromycin on the chlamydial PG ring when added at 8 hpi, a time point immediately following to the EB-to-RB transition (7, 48). This treatment resulted in normal-sized RBs (∼1 to 1.5 μm) that completely lacked discernible PG (Fig. 2b, row 1, column 4). Taken together, these results indicate that all chlamydial ABs examined exhibited alterations in their PG but that they diverged in terms of whether those changes were in PG localization or abundance.
Aberrant Chlamydia forms differ in their ability to release PG-derived, immunostimulatory muropeptides.
While PG is continuously synthesized during bacterial cell growth and division, changes in cell size and shape and the constriction of the division plane also require localized PG degradation. This process results in replicating microbes shedding PG-derived muropeptides into the environment. In the case of invading pathogens, these muropeptides are recognizable by a host’s innate immune system, specifically the nucleotide oligomerization domain (NOD) receptors (43). NOD1 recognizes peptides containing a meso-diaminopimelic acid (mDAP), which is normally present at the third position of the PG stem peptide in Gram-negative bacteria. NOD1 signaling triggers the production of a number of host cell cytokines via an NF-κB-dependent pathway, and both NOD1 and NOD2 recognize intracellular Chlamydia pneumoniae and play an essential role in bacterial clearance in vivo (49).
As a number of chlamydial ABs appeared to lack detectable PG labeling, we wanted to eliminate the possibility that PG biosynthesis was still occurring but was simply being degraded too rapidly to be visualized. We utilized a human NOD1 reporter cell line to determine the abundance of PG-derived muropeptides released by C. trachomatis under three aberrance-inducing conditions thought to be most physiologically relevant to human infections: β-lactam antibiotics, iron restriction, and IFN-γ. As we failed to detect PG labeling under our iron restriction and IFN-γ-induced conditions, we hypothesized that the block of PG biosynthesis must be earlier in the synthesis pathway than the addition of the dipeptide probes to the PG stem peptide by the UDP-N-acetylmuramoyl-tripeptide-d-alanyl-d-alanine ligase (MurF) (Fig. 2a). We found that under AMP-induced aberrance, significant amounts of NOD1 ligand continued to be released by C. trachomatis at 24 hpi (Fig. 3a), whereas under iron-depleted conditions (Fig. 3b) and in the presence of IFN-γ (Fig. 3c), NOD1 signaling was significantly reduced. This reduction was specific for Chlamydia-infected cells, as interferon and iron depletion had no discernible effect on NOD1 signaling induced by the NOD-stimulatory ligand l-Ala-γ-d-Glu-mDAP (TriDAP). No significant signal was observed in the Null1 cell line (which contains an empty expression vector) under any of the conditions tested (Fig. 3a to c), indicating that NF-κB expression was NOD1 specific. When combined with PG-targeting antibiotics (AMP), iron depletion continued to exhibit a complete loss of NOD1 signaling (Fig. 3a), indicating that the PG synthesis inhibition phenotype is dominant under the conditions tested. This result indicates that the block on PG biosynthesis under iron/tryptophan depletion conditions occurs prior to the synthesis of muramyl tripeptide. Taken together, these results indicate that chlamydial ABs induced by penicillin-derived antibiotics and iron/tryptophan starvation differ in their capacities to synthesize, assemble, and subsequently shed immunostimulatory, PG-derived muropeptides. The inability to detect muramyl tripeptide in infected cells under iron/tryptophan depletion conditions also indicates that PG biosynthesis is inhibited prior to the addition of mDAP to the PG stem peptide by the chlamydial UDP-N-acetylmuramoyl-l-alanyl-d-glutamate-diaminopimelate ligase, MurE (50).
FIG 3.

Aberrant Chlamydia forms differ in their abilities to synthesize immunostimulatory peptidoglycan. A HEK hNOD1 SEAP reporter cell line was utilized to quantify the relative abundances of PG shed when cells were infected with C. trachomatis in the presence of ampicillin (AMP) (a), the iron chelator DPP (a and b), and interferon gamma (IFN) (c). TriDAP was used in each experiment as a positive control for NOD1 signaling, and the Null1 cell line was used as a negative control. Data presented are the means from three independent biological replicates, and error bars represent standard deviations from the means. **, P < 0.005; ns, not significant.
The timing of persistence induction directly affects Chlamydia morphology and PG-derived muropeptide release.
During active infections, C. trachomatis develops asynchronously, with infected cells in close proximity often containing bacteria in completely different stages of the pathogen’s developmental cycle. The ability of C. trachomatis to form enlarged, aberrant bodies is largely dependent on when induction conditions are introduced during the chlamydial developmental cycle. Previous studies have used fast-acting, aberrance-inducing compounds to investigate how persistence induction affects C. trachomatis at different stages of its development. For example, when MreB is inhibited prior to the EB-to-RB transition, development completely halts, and very few EBs successfully transition to RBs (8). Similar results have been reported when protein synthesis is inhibited prior to the EB-to-RB transition, and protein synthesis is known to be globally affected by aberrance induction under iron restriction (35). To determine whether this is the case for other aberrance-inducing conditions, we examined the effect that individual aberrance inducers had on chlamydial morphology when added at different time points in the developmental cycle. We added the fast-acting T3SS inhibitor C1 at key points throughout the developmental cycle of C. trachomatis: 2 hpi (immediately after invasion), 8 hpi (EB-to-RB transition), and 22 hpi (RBs undergoing active cell division). We found that compound C1 exhibited a phenotype similar to those of the MreB-inhibiting compounds A22 and MP265: when it was added prior to the EB-to-RB transition (at 2 hpi), only PG-negative, EB-sized particles were observable in infected cell monolayers at 24 hpi (Fig. 4b), and when it was added just after the EB-to-RB transition (8 hpi), enlarged ABs that lacked discernible PG were observed (Fig. 4c).
FIG 4.
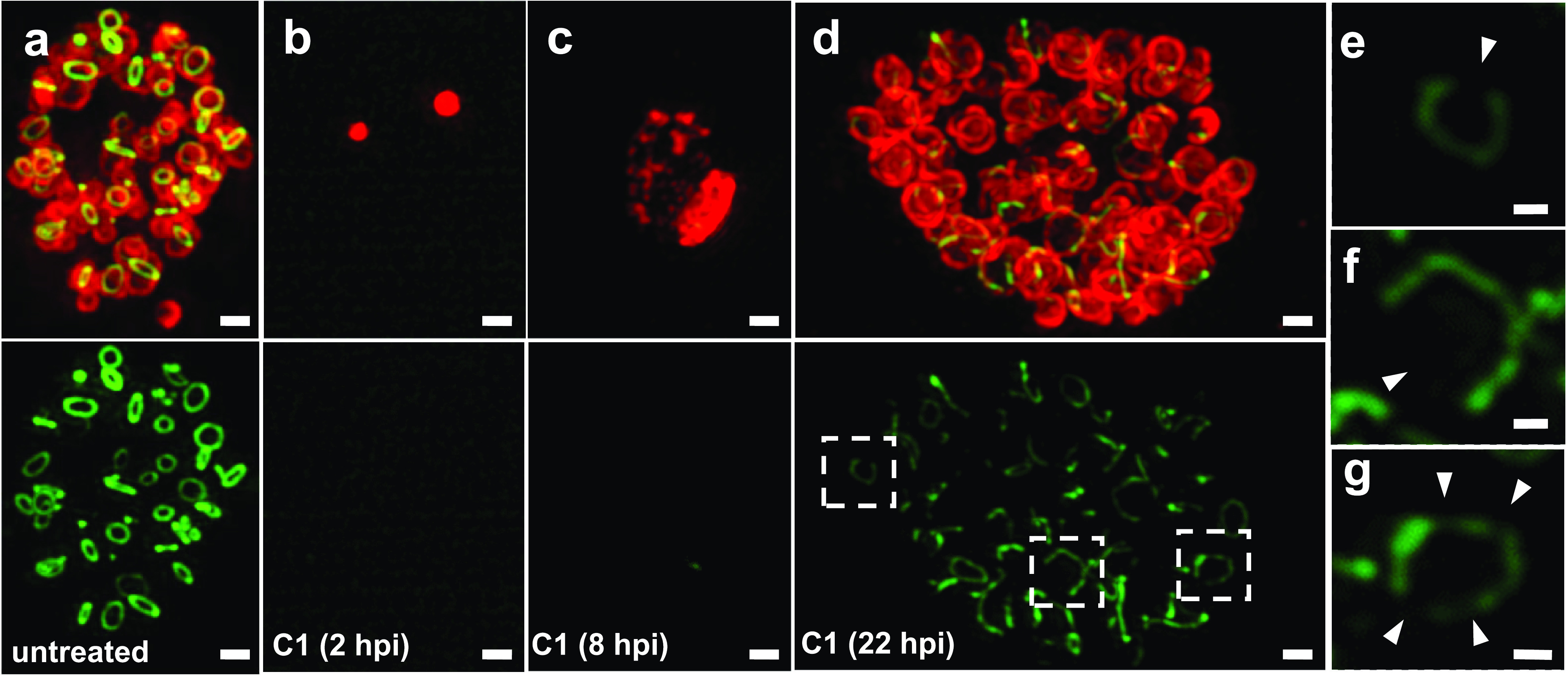
The presence of PG and differences in cell/inclusion size are dependent on the timing of persistence induction. (a) Chlamydia-infected HeLa cells were grown in the presence of the PG-labeling reagent EDA-DA for 24 h. (b to d) Persistence was induced with the T3SS inhibitor compound C1 at 2 hpi (b), 8 hpi (c), and 22 hpi (d). (e to g) Enhanced/magnified regions of interest from panel d. Arrowheads indicate discernible breaks in the RB PG rings. Images are representative of 20 to 30 viewing planes over three separate and independent experiments. Bars, ∼1 μm.
Unlike a PG sacculus that largely retains its form when PG synthesis is inhibited, septal PG in Chlamydia species is more transient and begins to degrade within minutes of inhibiting PG synthesis (8). Given the absence of PG in the majority of ABs examined (Fig. 2) and the lack of immunostimulatory muropeptides released into host cells (Fig. 3), we postulated that induction of aberrance in Chlamydia likely affects PG biosynthesis. We sought to test this by inducing aberrance with compound C1 for a short, 2-h duration later in the developmental cycle (at 22 hpi) and subsequently examined the effects on PG localization. We found that 2-h-treated RBs appeared large and exhibited a swollen morphology compared to untreated controls (Fig. 4d). PG was discernible, but the labeling intensity appeared to be significantly reduced. A closer examination of individual ABs revealed the presence of incomplete PG rings that appeared to be undergoing degradation (Fig. 4e to g), matching previous observations with the MreB inhibitors (8). These data largely support our previous conclusion that PG synthesis and degradation pathways are decoupled in C. trachomatis and also indicate that during persistence induced by T3S inhibition, PG degradation continues despite a halt in PG biosynthesis.
We hypothesized that if chlamydial persistence halts new PG biosynthesis but does not stop PG degradation, then RBs that transition to aberrant forms later in development will actively degrade and shed their PG in a time-dependent manner. Inhibition of protein synthesis has been previously shown to prevent PG biosynthesis in C. trachomatis (51), and iron sequestration has previously been linked to a general reduction of translation in C. trachomatis (35). Therefore, we hypothesized that iron restriction likely prevents PG biosynthesis in C. trachomatis. To examine the temporal kinetics of PG muropeptide release under aberrance-inducing conditions, we compared NOD1 signaling in Chlamydia-infected cells in which persistence via iron depletion was induced at different time points throughout the developmental cycle. We found that the earlier in the developmental cycle that DPP was added to the infected cells, the greater the decrease in Chlamydia-specific NOD1 signaling (Fig. S2). No discernible difference was observed between untreated cells and those treated at 12 and 16 hpi, indicating that the abundance of immunostimulatory peptides present at 12 hpi was sufficient to effectively saturate the assay’s upper detection limit. In comparison, the timing of exposure with the immunostimulatory ligand for cells treated with exogenous TriDAP appeared to have no effect on NOD1 signaling (Fig. S2). Taken together, these results indicate that some persistence induction conditions inhibit the synthesis of chlamydial PG in a time-dependent manner but do not inhibit PG degradation.
Persistent forms of Chlamydia differ in the presentation of mid- and late-stage effector proteins.
Chlamydia maturation is temporally regulated, and Inc proteins are secreted and integrate into the inclusion membrane at different time points during the pathogen’s development. Secreted effectors are categorized into three general groups that fit into different phases of the developmental cycle: early, mid, and late (31, 52). As an inclusion “matures,” it gradually accumulates newly secreted Inc proteins, which results in a change of the overall composition of the inclusion membrane. Many of these early- and mid-stage Inc proteins are involved in redirecting exocytic vesicles to the inclusion, enabling it to steadily enlarge over time (6). We reasoned that an interruption in the inclusion maturation pathway could explain the reduction in the inclusion size that we previously observed under various persistence-inducing conditions (Fig. 1b to e). To test this hypothesis, we selected four Inc proteins that are expressed and secreted at different times during inclusion development (52). ct229 and incG are expressed early in development, with peak expression at 2 and 12 hpi, respectively. incA is expressed closer to the midway point in the inclusion maturation pathway, with peak expression at ∼20 hpi, and ct813 is a late-stage effector whose expression peaks at ∼30 hpi. Using polyclonal antibodies to each of these four Inc proteins, we examined whether they were present or absent on the inclusion membranes in Chlamydia-infected cells under different aberrance-inducing conditions. We found that CT229 was present under all conditions tested (Fig. S3a), whereas IncG was present on AMP- and sucrose-treated cells and, to a lesser degree, on cells treated with the iron chelator DPP (Fig. S3b). IncA and CT813 were present on inclusions only in the control and AMP- and sucrose-treated groups, and all other conditions resulted in no discernible labeling (Fig. S4a and b). When combined with our inclusion size (Fig. 1b) and PG labeling (Fig. 2b and c) data, imaging analysis suggests that a strong correlation exists between inclusion maturation and size and PG biosynthesis in persistent forms of C. trachomatis.
Persistent forms of Chlamydia exhibit differences in their abilities to undergo homotypic fusion and induce actin cage formation.
Given the differences in Inc presentation that we observed on the inclusion membranes of different persistent forms of C. trachomatis, we hypothesized that persistent forms also differ in their interactions with host cells, particularly interactions that occur at later stages of development. A Chlamydia inclusion will traffic to the microtubule-organizing center (MTOC) of a host cell relatively early in development (within 4 to 5 hpi) in a process dependent on dynein and chlamydial protein synthesis (53). As this process occurs early in development, we predicted that it would not be adversely affected by the induction of aberrance because the relevant effector proteins would have already been generated and secreted. Supporting this premise, we found that under all of the aberrance-inducing conditions tested, all inclusions were capable of successfully trafficking to the cell nucleus (Fig. S5a).
In contrast to trafficking to the MTOC, homotypic vesicle fusion of C. trachomatis inclusions within a single cell occurs later in the developmental cycle and is mediated by the secreted effector protein IncA (54). Therefore, we hypothesized that in cells infected at an increased multiplicity of infection (MOI), inclusions exposed to aberrance-inducing conditions with discernible labeling of IncA (Fig. 5a) should successfully undergo homotypic fusion, resulting in fewer inclusions per cell, while those lacking discernible labeling of IncA should exhibit higher numbers of inclusions per cell. We found that in cells infected at an MOI of 10:1, DPP-treated cells contained higher numbers of inclusions per cell than untreated and AMP-treated cells (Fig. 5b). We also noted that despite containing more inclusions per cell, these inclusions still appeared to congregate in the same area of the cell (Fig. 5c), further supporting the premise that early developmental processes such as inclusion trafficking to the MTOC are retained by these persistent forms. Despite being in proximity, inclusions in DPP-treated cells appeared to be incapable of fusion, in line with the observed absence of IncA (Fig. 5a).
FIG 5.
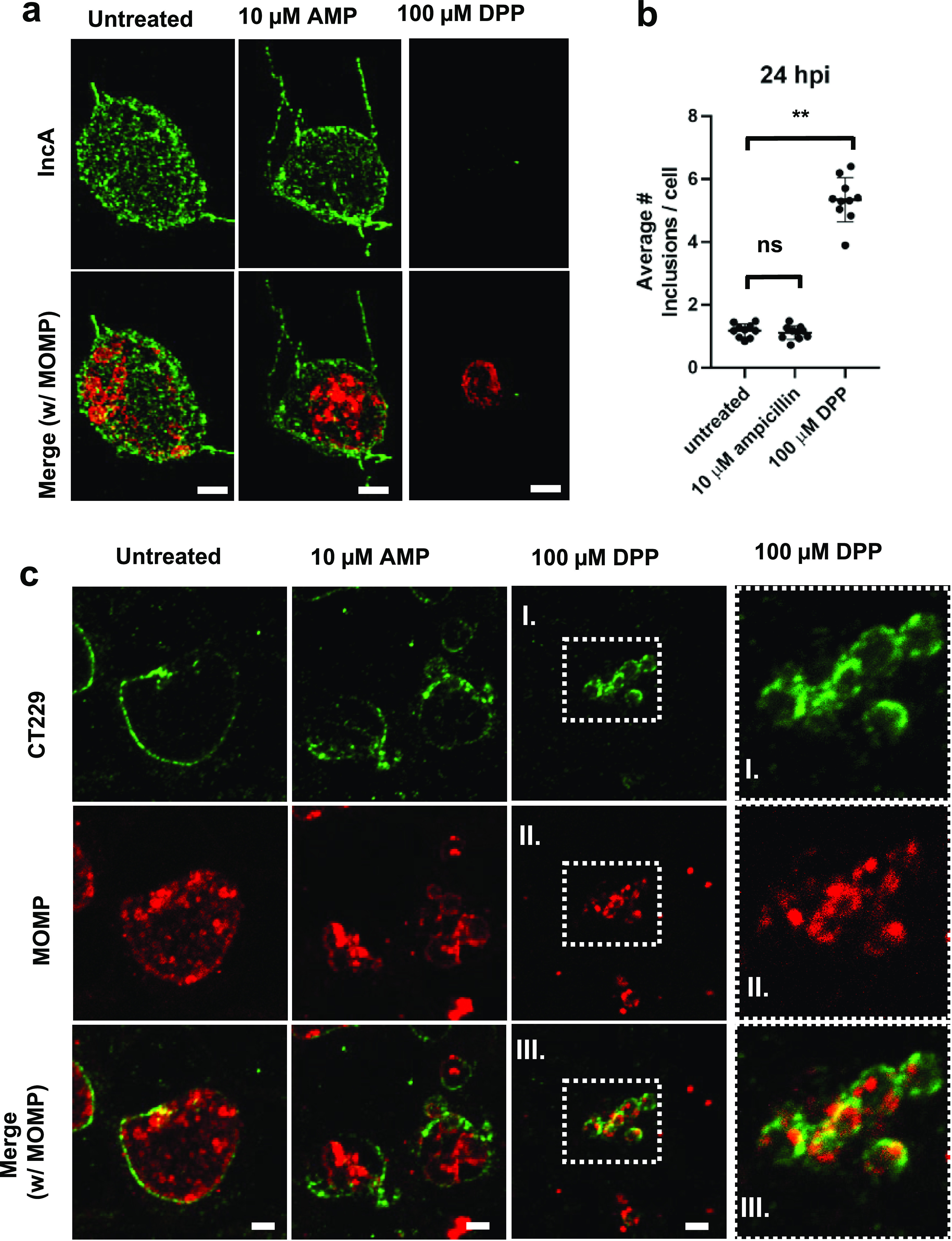
Chlamydia ABs differ in their presentation of the mid-stage inclusion membrane protein IncA. HeLa cells were infected at an MOI of 1. Cells were either left untreated or treated with various aberrance inducers, and cells under all conditions were fixed at 24 hpi. Immunolabeling was conducted for IncA as well as MOMP. (a) Ampicillin (AMP) and DPP treatments. See Fig. S4 in the supplemental material for the rest of the conditions. Images are representative of between 10 and 20 inclusions observed under each condition, and the experiment was carried out three times. (b) To assess the fusogenic potential of inclusions under aberrance induction, HeLa cells were infected at an MOI of 10, fixed at 24 hpi, and labeled for CT229. Inclusions present per cell were measured by counting cell and inclusion numbers across 10 random imaging planes under each condition (untreated, AMP treatment, and DPP treatment). Data points represent average values of the numbers of inclusions per cell per imaging plane examined with 10 imaging planes under each condition. The assay was carried out at least twice under each condition tested. Lines represent mean values for all data points under each condition, and error bars represent standard deviations. **, P < 0.005; ns, not significant. (c) Representative images from the analysis of fusogenic and nonfusogenic inclusions. The rightmost panels are magnifications of the boxed areas. Bars, ∼2 μm.
Actin rearrangement is important for maintaining the integrity of the chlamydial inclusion (55) as well as enabling Chlamydia to eventually exit the host cell (56). During normal chlamydial development, actin recruitment around the chlamydial inclusion is directly mediated by the late-stage effector protein CT813 (57). The recruitment of actin to the inclusion begins at around 20 hpi and continues to increase as the chlamydial inclusion matures (56). As we observed an absence of CT813 labeling in a number of chlamydial ABs, we hypothesized that these inclusions should be defective in their ability to recruit actin. We found that actin cage formation roughly corresponded to that in our CT813 imaging study (Fig. 6a; Fig. S5b) in that actin cages were observed only in untreated and AMP-treated cells (Fig. 6b; Fig. S5b). Taken together, these results indicate that while all Chlamydia ABs are effectively paused in the developmental cycle, only a subset of ABs also paused the maturation of the chlamydial inclusion.
FIG 6.
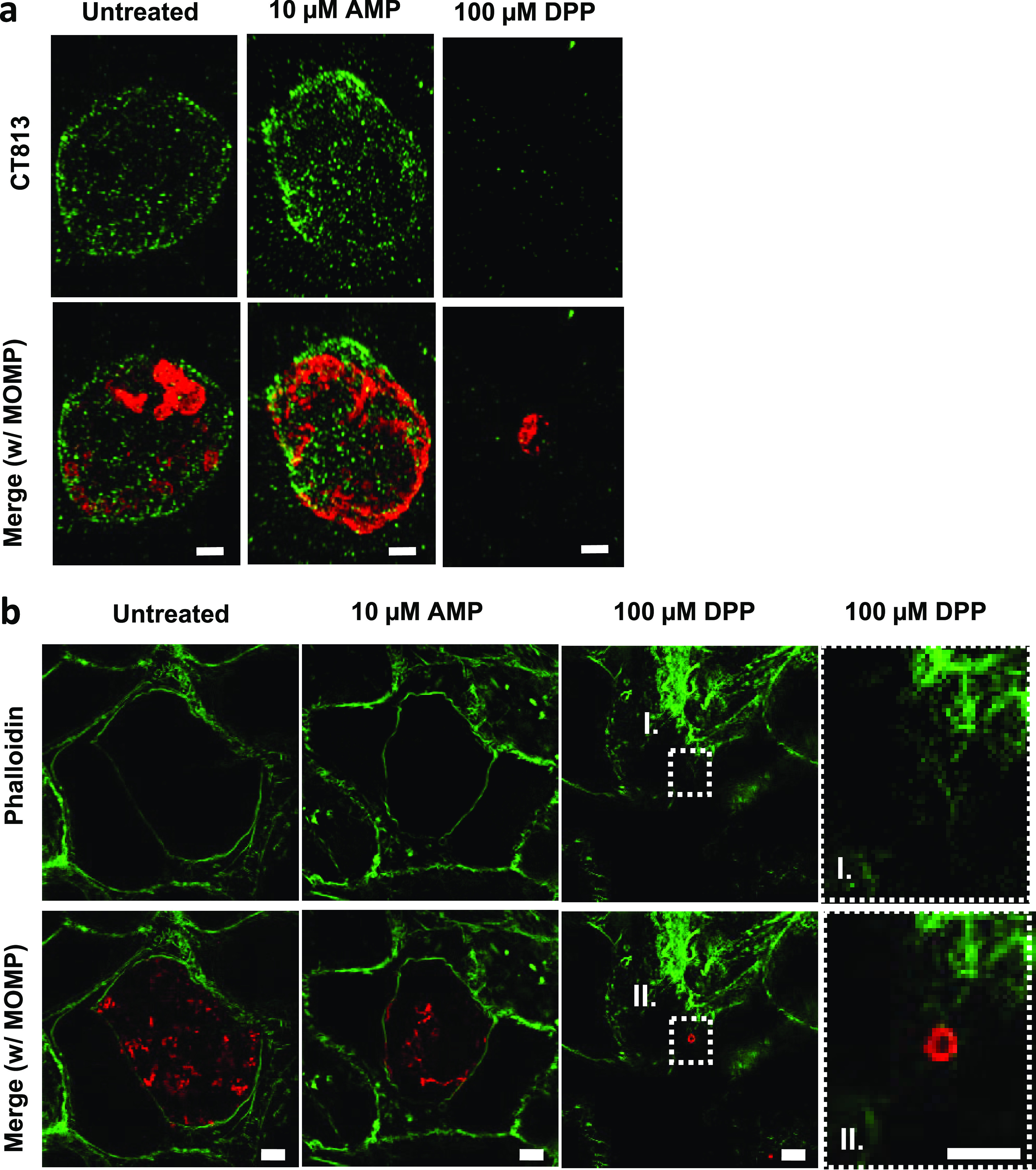
Chlamydia ABs differ in their presentations of the late-stage inclusion membrane protein CT813. HeLa cells were infected at an MOI of 1. At 2 hpi, cells were either left untreated or treated with various aberrance inducers, and cells under all conditions were fixed at 24 hpi. Immunolabeling was conducted for the inclusion membrane protein CT813 as well as MOMP. (a) Ampicillin (AMP) and DPP treatments. See Fig. S4 in the supplemental material for the rest of the conditions. Bars, ∼2 μm. Images are representative of between 10 and 20 inclusions observed under each condition over the course of three separate experiments. To assess actin cage formation, HeLa cells were infected at an MOI of 1, fixed at 46 hpi, and labeled for MOMP and phalloidin. (b) AMP and DPP treatments. The rightmost panels are magnifications of the boxed areas. The remaining aberrance-inducing conditions are presented in Fig. S5 in the supplemental material. Images are representative of 20 fields of view under each condition examined over three separate experiments. Bars, ∼5 μm.
Iron-depletion-induced persistence enhances the susceptibility of C. trachomatis to azithromycin treatment.
“Persistence” is often used to describe a general survival mechanism by which pathogens avoid clearance by a host, but it is also frequently used to describe a mechanism to avoid killing by antibiotic treatment. We sought to determine whether C. trachomatis ABs are protected against treatment with azithromycin, a frontline antibiotic commonly used to treat human chlamydial infections (58). We found that viable EBs were recoverable from the subinhibitory azithromycin treatment group, while treatment with AMP or DPP induced aberrance and prevented EB formation (Fig. 7). Interestingly, we successfully recovered EBs in the AMP/azithromycin-treated group after recovery but were unable to detect viable EBs in the DPP/azithromycin-treated group after recovery (Fig. 7). These results indicate that iron-depletion-induced persistence enhances the susceptibility of C. trachomatis to killing by azithromycin, while AMP-induced persistence does not.
FIG 7.

Iron-depletion-induced persistence enhances the susceptibility of C. trachomatis to azithromycin treatment. Subinhibitory doses of azithromycin (AZM) were added to Chlamydia-infected cells (18 hpi) either alone or following the addition of 10 μg/ml ampicillin (AMP) or 100 μM the iron chelator DPP at the time of infection. EBs were harvested at 44 hpi from treated cells, or cells were washed and Chlamydia cells were allowed to recover for an additional 48 h. Inclusion-forming units (IFU) were then calculated by conducting serial dilutions, infecting fresh monolayers, and counting new inclusions at 24 hpi. The dotted line represents the limit of detection for the assay. Data were gathered over three separate experiments, the data plotted represent the results of three independent biological replicates, and error bars represent the standard deviations from the means. **, P < 0.01.
DISCUSSION
For decades, researchers have debated whether aberrant forms of Chlamydia play any role during active infections. Because these forms have been defined solely by their enlarged size and inability to divide and produce infectious progeny, discussion concerning how they could potentially benefit the pathogen in vivo has been largely restricted to these phenotypes. Recent studies have demonstrated that aberrant Chlamydia bacteria differ in their transcriptional (31, 32) and translational (34) profiles. The investigation of aberrance in Chlamydia-related bacteria has also provided evidence that this phenotype transcends the human and zoonotic pathogens (the Chlamydiales) and likely is important for many other members of the Chlamydiae phylum (27). However, very little is known about the underlying physiology of chlamydial ABs and the degree to which they differ in their interactions with the microbe’s intracellular niche. Our results indicate that persistent forms of C. trachomatis differ significantly in their interactions with host cells as a result of disparities in PG biosynthesis and the secretion of mid- and late-stage effector proteins (Fig. 8). In addition, our data also challenge the notion that bacterial cell enlargement is a prerequisite for persistence in C. trachomatis, as many of our treatment conditions did not result in a statistically significant increase in the average size of intracellular C. trachomatis.
FIG 8.
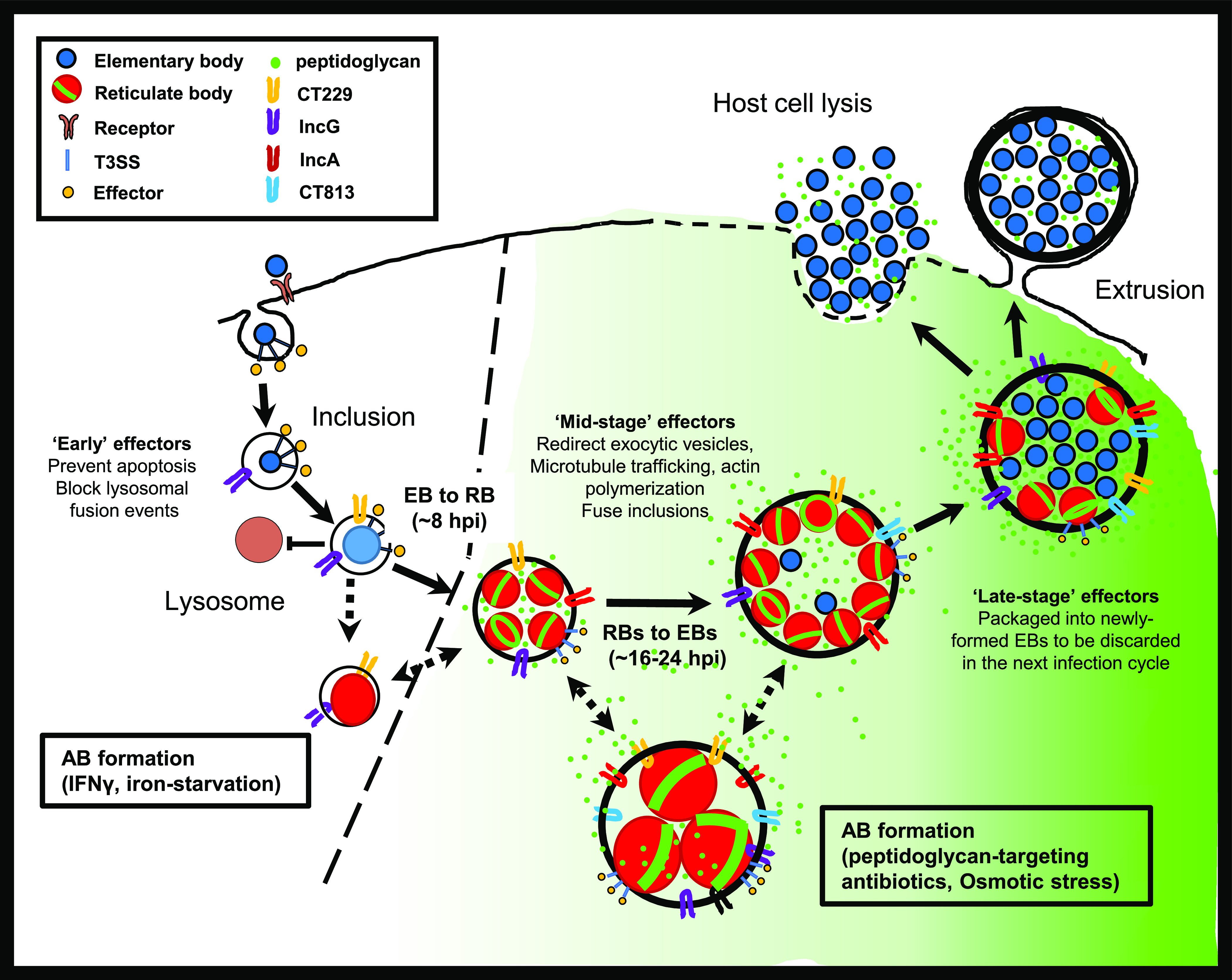
Persistence halts the Chlamydia developmental cycle while simultaneously altering bacteria-host cell interactions. The induction of persistence in response to a cell’s innate response to infection results in C. trachomatis significantly reducing the synthesis and shedding of immunostimulatory peptidoglycan (green) while concurrently minimizing the deleterious reorganization of host cell processes. This is not the case when C. trachomatis-infected cells are treated with peptidoglycan-targeting antibiotics or exposed to osmotic shock. We propose that inducing persistence prior to mid-stage development (dotted line) allows C. trachomatis to remain intracellular and escape clearance by the cell-mediated immune response for extended periods of time.
In the presence of β-lactam antibiotics, actively growing bacterial cells are highly susceptible to cell lysis as a result of the weakening of their cell walls and the inherent osmotic pressure differential across their cell membranes (59). Chlamydia species reside within an osmotically stable intracellular environment that protects them from the bactericidal effects of PG-targeting antibiotics. Inhibitors of MreB, a cytoskeletal protein that is the major organizer of the chlamydial division complex and directs septal PG synthesis, also induces aberrance in C. trachomatis (47). Iron depletion via chelators such as DPP induced aberrance in Chlamydia species through the downregulation of translation and T3SS function (35). In primate-derived cell lines, IFN-γ induction in response to intracellular pathogens results in the expression of the enzyme indoleamine deoxygenase (IDO), which effectively depletes cellular pools of tryptophan (60). Numerous studies have demonstrated the importance of IFN-γ in fighting/clearing Chlamydia infections (61–64). Tryptophan depletion significantly alters protein expression in a number of Chlamydia spp. and also results in the induction of aberrant bodies (31). It has also been suggested that these aberrant forms may also lower Chlamydia’s sensitivity to frontline antibiotics. There are numerous reports of Chlamydia-infected patients exhibiting antibiotic treatment failures despite the demonstrated absence of any known resistance genes being carried by the pathogen (65, 66).
When Chlamydia species are exposed to PG-targeting antibiotics such as β-lactams, d-cycloserine, and fosmidomycin, the synthesis of PG precursors continues, but the completion of PG assembly is inhibited (7, 8, 51, 67). This counterintuitive result was clarified when researchers determined that bacteria respond to PG-induced stress by continuing to degrade and resynthesize peptidoglycan (68). We now report that nonionic osmotic stress also appears to inhibit chlamydial cell division in a similar fashion. However, the greater novelty inherent to this work is that for all of the other aberrance-inducing conditions that we examined, both PG assembly and synthesis appeared to be inhibited, while PG degradation continued unabated. PG degradation is largely carried out by two major enzymatic activities, amidase activity and lytic transglycosylase activity, and both have been successfully characterized in Chlamydia species (69, 70). Changes in the transcription of PG-processing enzymes, such as the chlamydial amidase (encoded by amiA) and the PG synthase (encoded by ftsW), have been shown to occur during IFN-γ-induced persistence (31), hinting at one potential regulatory pathway underlying this phenotype. However, given previous studies linking persistence to a global downregulation of genes essential for protein synthesis (35) and our data indicating that (i) persistence enhances susceptibility to protein synthesis inhibitors and (ii) protein synthesis inhibition directly results in the disappearance of the PG ring, PG degradation in aberrant C. trachomatis may occur as a result of a reduction in protein synthesis. Future studies comparing the relative half-lives and activities of PG biosynthesis and degradation enzymes in Chlamydia species will bring some clarity on this point. Another possibility is that differences in the ability to replicate DNA between aberrance-inducing conditions could account for the observed differences in PG synthesis and release. A recent study established that a breakdown in genome replication in C. trachomatis results in the disruption of PG biosynthesis (71). While genome replication has been shown to proceed under some persistence-inducing conditions (72, 73), it is frequently delayed compared to untreated controls (32, 74). PG-dependent septum formation is regulated by nucleoid occlusion in a number of bacterial species (75–78), and the inability of C. trachomatis to properly complete replication or the segregation of its chromosomes during persistence could potentially explain the absence of a PG septum during persistence.
We previously hypothesized that reduced utilization of PG in Chlamydia spp. also reduces the generation of immunostimulatory, PG-derived muropeptides, conferring a pathoadaptive advantage (7). In support of this theory, a rudimentary PG-recycling pathway has recently been characterized in C. trachomatis, which the microbe utilizes to limit the shedding of PG-derived immunostimulatory peptides (79). Our finding that persistence induction results in the inhibition of PG synthesis also supports this basic premise, as limiting the production and release of immunostimulatory muropeptides during active infection would likely be beneficial to these organisms. PG signaling through NOD1 is a large contributor to the overall interleukin-8 (IL-8) response to C. trachomatis (80). IL-8 is the most highly induced inflammatory cytokine and a major driver of inflammation during C. trachomatis, C. muridarum, and C. pneumoniae infections (81, 82). IL-8 transcript levels are significantly upregulated at ∼15 hpi, and this upregulation is dependent on chlamydial protein synthesis (83). Interestingly, this timing roughly correlates with the time points at which we see PG-induced NOD1 signaling peak in our in vitro experiments, suggesting that PG production by C. trachomatis is likely driving this signaling cascade. Although DPP-induced iron starvation can affect host cell processes (35), it does not appear to significantly alter NOD1 signaling activity (as indicated by our positive control in Fig. 3b). It is also possible that additional alterations in cell-microbe interactions are affected, which may indirectly affect Chlamydia growth and, thereby, peptidoglycan biosynthesis.
We found that a number of aberrance-inducing conditions resulted in inclusions lacking mid- and late-stage Inc proteins on their membranes and that this appeared to correlate with the above-described PG synthesis phenotype. PG-positive ABs were present in larger, mature inclusions containing all four Inc proteins, and PG-negative ABs were housed in smaller, immature inclusions that lacked mid- and late-stage Incs in their inclusion membranes. These data strongly suggest that these mid- and late-stage T3S effectors are either not being produced or are not being secreted, under these aberrance-inducing conditions. Previous studies have reported that inhibitors of the chlamydial T3SS prevent the presentation of various inclusion membrane proteins (such as IncA and CT813) on the inclusion surface and result in Chlamydia exhibiting a swollen, aberrant-like morphology (42, 84). These inclusions were also significantly smaller than those of untreated controls, indicating that they fail to undergo the normal maturation process associated with chlamydial development (8).
Our observations take on additional significance when the functions of the Inc proteins examined in this study are fully considered. CT229 is expressed early in chlamydial development and aids in the formation and maintenance of the inclusion by recruiting and binding several Rab GTPases that alter host vesicular trafficking, thus enabling C. trachomatis to avoid the host cell surveillance and suicide programs (85–88). Chlamydia inclusions that lack CT229 lyse prematurely during development, resulting in host cell death (89). IncG functions by binding with the 14‐3‐3β protein (90) and is thought to interact directly with lipid droplets and recruit them to the growing chlamydial inclusion (6). IncA enables homotypic vesicle fusion of adjoining Chlamydia inclusions, an event that significantly enhances chlamydial development (54). Actin recruitment to the inclusion is mediated directly by CT813; CT813 recruits ARF1 and ARF4 to the inclusion membrane, which are essential for repositioning Golgi complex fragments around the inclusion (91, 92). As CT813 is not present on inclusions whose maturation processes have been paused early in development, it is likely that host cell actin and Golgi complexes remain in their original, unaltered states. It is currently unclear whether the absence of CT813 on inclusions is beneficial to infected host cells or provides additional immunoevasive benefit to persistent chlamydial forms. While only four inclusion membrane proteins were examined in this study, we suspect that our results are representative of T3S effectors as a whole and that aberrance induction halts inclusion maturation.
Assuming that Chlamydia’s aberrant forms enhance persistence in vivo by lowering its immunogenic profile, it follows that the timing of persistence induction is likely critical. Chlamydia begins synthesizing PG upon transitioning to the replicative form. The longer the microbe’s developmental cycle proceeds, the greater the number of RBs produced and the higher the concentration of muropeptides that will accumulate in infected cells. As chlamydial development is not synchronized between cells during active infections, aberrance induction likely occurs at various stages of the developmental cycle (Fig. 9). We postulate that the time at which C. trachomatis pauses its developmental cycle is likely what determines an individual bacterium’s fate. Because persistence induction pauses T3S (either directly or indirectly), halting the developmental cycle too early would prevent the secretion of early effector proteins essential for host cell invasion, resulting in either apoptosis or targeting of immature inclusions to lysosomes, where they are subsequently destroyed. Conversely, if persistence is induced late in infection, RB replication and shedding of PG prior to AB formation will have already resulted in NOD1 signaling, IL-8 secretion, and targeting of infected cells by the cell-mediated immune response. Under this principle, if development is instead halted at a midway point in development, between 8 and 12 hpi, theoretically, the inclusion would be relatively stable within a host cell and sufficiently removed from the endocytic pathway while also limiting the release of immunostimulatory PG muropeptides and the subsequent targeting of the host cell for cell-mediated killing.
FIG 9.
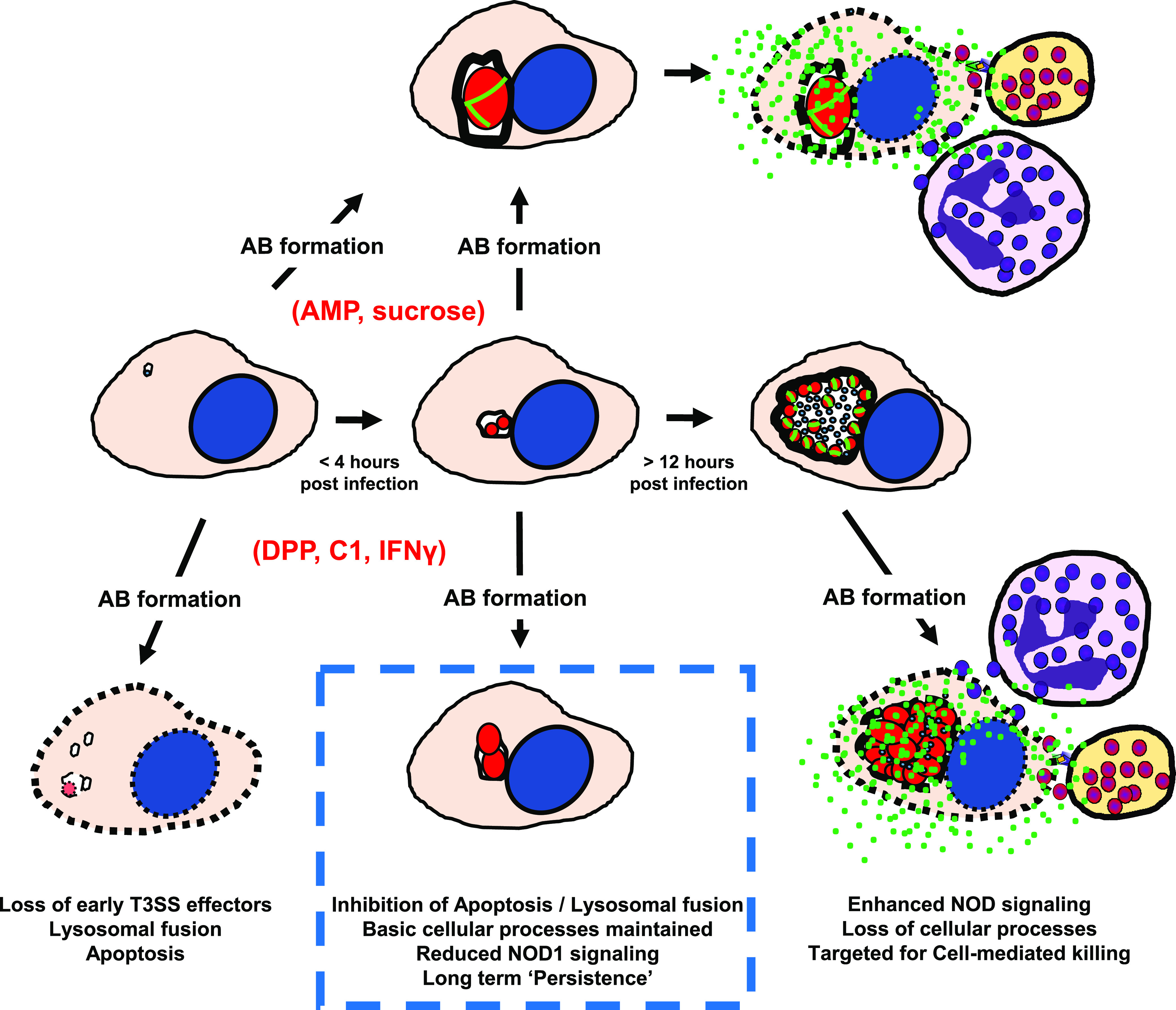
Modeling chlamydial persistence in the context of infection. For stress conditions that do not affect PG synthesis and inclusion maturation (AMP and sucrose), C. trachomatis benefits from remaining intracellular but eventually will be targeted by cell-mediated killing mechanisms. Under stress conditions that halt PG synthesis and inclusion maturation (DPP, C1, and IFN-γ), the timing of induction directly affects the pathogen’s survival. If aberrance is induced early, prior to the establishment of the inclusion and the secretion of antiapoptotic factors, the microbe will be eradicated by either lysosomal fusion or host cell apoptosis. If aberrance is induced late in development, host cell processes such as chromosomal segregation and actin remodeling will be irreversibly affected, and immunostimulatory PG release will result in cytokine production, leading to cell-mediated killing. In this persistence model, Chlamydia survival is maximized when aberrance is induced subsequent to the host cell’s intracellular defenses being overcome but prior to the initiation of a cytokine-induced, innate immune response.
There has been much debate as to whether Chlamydia’s aberrant state provides a level of protection against antibiotic treatment. Treatment failures have continued to increase over the last 2 decades (93), and genetic analysis of the genomes from clinical samples indicates that the acquisition of canonical antibiotic resistance mechanisms is not occurring (94). We found that the susceptibility of C. trachomatis to azithromycin was enhanced by iron depletion, indicating that this aberrance mechanism does not appear to confer protection. A major mechanism for persistence induction is thought to be the downregulation of protein synthesis (35), and it logically follows that the effect of a subinhibitory dose of a protein synthesis inhibitor (azithromycin) under these conditions would be additive, resulting in a lethal phenotype. Our findings are consistent with those of a previous study that examined the effects of subinhibitory concentrations of azithromycin on chlamydial development. Researchers found that C. trachomatis is more resistant to killing by low doses of azithromycin if it is allowed to first enter the mid-stage of its developmental cycle (95). These data largely fit our time-dependent model, which illustrates how mid-stage persistence may be selectively advantageous in vivo, with C. trachomatis exhibiting the highest tolerance to aberrance-inducing conditions at a midway point in its development.
As only a few observational studies have reported the presence of ABs in vivo, these reports have been viewed with a healthy degree of skepticism. The consensus within the field has been that chlamydial ABs represent rare, dead-end morphotypes that are likely cleared rapidly by the cell’s natural defenses or by the host’s innate immune response. However, our results offer alternative propositions: (i) that ABs exist in much smaller inclusions, making them more difficult to identify by conventional screening approaches in vivo; (ii) that ABs resist killing by host cell apoptosis and the endosomal maturation pathway because they retain early Inc proteins in their inclusion membrane; and (iii) that ABs inhibit PG biosynthesis prior to incorporating mDAP into the PG stem peptide and subsequently fail to release NOD1-stimulating muropeptides into the host cell, thus limiting the recruitment of innate and cellular defenses. As NOD1/NF-κB-mediated IL-8 signaling is a hallmark of the immune response to chlamydial infections, the prevention of this signaling pathway by persistent forms of Chlamydia inhibiting PG synthesis likely enables the pathogen to mask itself from the host’s response to infection. We propose that these findings offer a significant, biologically relevant purpose for ABs during Chlamydia infections and link the persistent state with enhanced evasion of both extracellular and intracellular host defenses. Future examinations of PG and Inc protein abundance and localization in tissue specimens will be needed to definitively demonstrate the importance of persistent forms in the pathogenesis of Chlamydia species.
MATERIALS AND METHODS
Bacterial strains and cell lines.
C. trachomatis serovar L2 strain 434/Bu was provided by Anthony Maurelli (University of Florida). Chlamydial infections were carried out in HeLa cells (also provided by Anthony Maurelli) unless otherwise noted. HEK-Blue-hNOD1 and -Null1 cells were purchased from InvivoGen and propagated according to the manufacturer’s instructions. Cell lines were passaged in high-glucose Dulbecco’s modified Eagle medium (DMEM; Gibco) and 10% fetal bovine serum (FBS; HyClone).
Chlamydial infections.
HeLa cells were plated on a glass coverslip in 24-well tissue culture-treated plate wells (Costar) at a confluence of ∼70 to 80%. Cells were infected with C. trachomatis L2 434/Bu in cold DMEM (Gibco) (250 μl per well) and placed on a rocker in a 37°C tissue culture for 2 h. Subsequently, DMEM was removed and replaced with DMEM supplemented with 10% FBS (HyClone) and 1× minimal essential medium (MEM) nonessential amino acid solution (Sigma) (250 μl per well). The following aberrance-inducing conditions were tested in this study: 50 U IFN-γ, 100 μM DPP (iron chelator), 10 μM ampicillin, 6 to 8% sucrose, 50 μM C1 (T3SS inhibitor), and 125 μM MP265 (polymerization inhibitor of the bacterial cytoskeletal protein MreB). The concentration of azithromycin used was 3 μg/ml. IFN-γ and DPP were added at the time of infection. Ampicillin and sucrose were added at 2 hpi, while C1, azithromycin, or MP265 was added at 8 hpi, following the EB-to-RB conversion.
Analysis of the total bacterial volume within individual inclusions.
Volumetric measurements were performed by utilizing an immunolabeling/fluorescence approach. Infected cells were either left untreated or subjected to aberrance-inducing conditions for 24 h. Cells were fixed and permeabilized with methanol for 5 min, gently washed three times with 1× phosphate-buffered saline (PBS), further permeabilized with 0.5% Triton X-100 for an additional 5 min, and subsequently washed three times with 1× PBS. To prevent nonspecific binding, cells were blocked for 1 h with 3% bovine serum albumin (BSA) (Sigma), subsequently labeled with goat anti-major outer membrane protein (MOMP) (1:500 dilution in 3% BSA) for 1 h, washed with 3% BSA, and incubated with donkey anti-goat Alexa Fluor 594 (1:2,000 in 3% BSA). Cells were washed with a solution containing 3% BSA and 1× PBS, and coverslips were subsequently mounted on slides with ProLong gold antifade mounting medium and stored in the dark at 4°C until imaged via confocal microscopy. All confocal imaging was conducted with a Zeiss 710 laser scanning microscope, confocal z-stacks were taken using the 40× objective, and all processing was conducted using Zen 2012 software (Carl Zeiss). Settings were fixed at the beginning of image acquisition, and the same parameters were applied for collection and postprocessing of all images taken. ImageJ was used for all subsequent image analyses utilizing the Fiji add-on “3D Objects Counter” to identify the 3-dimensional spatial constraints of individual Chlamydia inclusions based on positive MOMP labeling and calculate their corresponding volumes in cubic micrometers. Statistical significance values were calculated utilizing one-way analysis of variance (ANOVA) coupled with a multiple-comparison test, with each condition being compared with the azithromycin treatment group. RB diameters were calculated using radius values derived from the RB volumes, with the assumption that C. trachomatis RBs and ABs are spherical: d = 2r = 2[3(V/4π)(1/3)].
Quantification of inclusion-forming units (IFU).
Chlamydia EBs were mechanically harvested at 44 hpi with glass beads at the indicated time points, suspended in 200 μl of 1× sucrose-phosphate-glutamate (SPG), and stored at −80°C. For recovery assays, at 44 hpi, cells were washed with cold DMEM and replaced with DMEM supplemented with 10% FBS (HyClone) and 1× MEM nonessential amino acid solution (Sigma) for an additional 24 or 48 h prior to EB harvests. To ascertain infectious titers, EBs were serially diluted and used to infect HeLa cells that had been plated 24 h prior (200 μl of 200,000 cells/ml per well seeded in a 96-well plate) in technical duplicates. Plates were centrifuged for 1 h at 2,000 × g at 35°C to enhance synchronous infection. At 24 hpi, cells were fixed and permeabilized with methanol for 10 min, incubated with a Pathfinder direct flourescent antigen (DFA) kit (Bio-Rad) for 30 min, and washed twice with 1× PBS. Ninety-five-percent glycerol was placed in each well, and inclusions were subsequently counted via epifluorescence microscopy. Twenty fields of view were counted in duplicate under each treatment condition.
Fluorescence labeling of PG, cell nuclei, actin filaments, and inclusion membrane proteins.
PG labeling was carried out as previously described (7, 8). Briefly, HeLa cells were infected with C. trachomatis L2 434/Bu as described above, with 1 mM EDA-DA probe added to the infection medium at 2 hpi. At 24 hpi, infection medium was removed, and cells were washed three times with 1× PBS. Cells were fixed and permeabilized with methanol for 5 min and gently washed three times with 1× PBS. Cells were then further permeabilized and blocked as described above. A click chemistry reaction was then performed utilizing the Click-iT cell reaction buffer kit (Invitrogen) and Alexa Fluor 488-azide (10 μM). Subsequent MOMP labeling was then conducted, as described above. For labeling inclusion membrane proteins, fixed/permeabilized cells were incubated for 1 h with polyclonal mouse or rabbit serum raised against the inclusion membrane protein IncA, CT229, CT813, or IncG. Cells were then washed with 3% BSA and incubated with donkey anti-mouse or donkey anti-rabbit Alexa Fluor 488 (1:1,000 in 3% BSA). For visualizing cell nuclei, methanol-fixed and permeabilized cells were incubated with Hoechst stain for 3 min and then washed with 3% BSA and 1× PBS. For labeling host cell actin, at 46 hpi, cells were fixed with 3.7% paraformaldehyde for 15 min at room temperature and subsequently washed with 1× PBS. Cells were then permeabilized with 0.01% Triton X-100 for 25 min and subsequently washed twice with 1× PBS. Cells were then blocked for 1 h with 1% BSA and incubated with Alexa Fluor 488 phalloidin (catalog number A12379; Thermo Fisher Scientific) that was prepared in methanol as described by the manufacturer to a final concentration of 1× for 1 h, before washing labeled cells with 1% BSA and PBS. All coverslips were mounted on slides with ProLong gold antifade mounting medium and stored in the dark at 4°C prior to imaging via confocal (Zeiss 710) or structured-illumination (Elyra PS.1) microscopy.
Homotypic fusion assay.
HeLa cells were infected with C. trachomatis L2 434/Bu at an MOI of 10, and infections were allowed to proceed normally or in the presence of either AMP or DPP. At 24 hpi, infection medium was removed, and cells were washed, fixed, and labeled, as described above. Cells were incubated with goat anti-MOMP and polyclonal rabbit anti-CT229 for 1 h, washed with 3% BSA, and coincubated with chicken anti-goat Alexa Fluor 594 and donkey anti-rabbit Alexa Fluor 488 before being washed, and coverslips were mounted on slides with ProLong gold antifade mounting medium. An analysis of inclusions present per cell was conducted by counting cell and inclusion numbers across 10 random imaging planes under each condition examined (untreated, AMP treated, and DPP treated). Data points represent average values of the number of inclusions per cell per imaging plane examined (10 under each condition). Lines represent mean values of all data points under each condition, and error bars represent standard deviations. Statistical significance values were calculated utilizing one-way ANOVA coupled with a multiple-comparison test (**, P < 0.005; ns, not significant).
Infection of HEK-Blue-hNOD1 and -Null1 cells with C. trachomatis and NF-κB reporter assay.
HEK-Blue cells expressing the hNOD1 receptor and carrying the NF-κB SEAP (secreted embryonic alkaline phosphatase) reporter gene (InvivoGen) were used according to the manufacturer’s instructions and adapted to assess NOD1-specific NF-κB activity during infection with C. trachomatis. Briefly, 3 × 105 cells/ml of HEK-Blue-hNOD1 or -Null1 cells were plated in 96-well plates (total reaction volume of 200 μl per well [∼6.0 × 104 cells per well]) and allowed to settle/adhere for 6 h at 37°C. These cells were infected with C. trachomatis L2 434/Bu at an MOI of 1. To enhance the synchronization of the infections, plates were centrifuged for 1 h at 2,000 × g. Plates were then incubated in a CO2 incubator at 37°C, and supernatants were collected for subsequent analysis of SEAP activity at the indicated time points. A colorimetric reporter assay was then utilized in order to quantify the abundance of SEAP in cell supernatants, providing an estimate of the relative abundance of residual stimulatory ligand (MurNAc-l-Ala-d-Glu-mDAP) in experimental and control cells. Twenty microliters of the supernatants collected from infected cells was added to 180 μl of the SEAP detection solution (InvivoGen), followed by incubation at 37°C for ∼8 h. SEAP enzymatic activity was then quantified using a plate reader set to 650 nm. Infected cells were compared to uninfected controls as well as infected cells that had been treated with either the iron chelator DPP or IFN-γ. Uninfected cells treated with the known NOD1 signaling ligand TriDAP (1 μg/ml) were used as a positive control. Additionally, to ensure that changes in alkaline phosphatase activity were NOD1 dependent under each of the experimental conditions tested, all experiments were carried out in parallel in the HEK-Blue-Null1 cell line, which contains the empty expression vector but lacks hNOD1. HEK-Blue NOD1 SEAP reporter assays were carried out in three separate experiments, statistical analysis was conducted by 2-way ANOVA, and significance values were analyzed by utilizing Sidak’s multiple-comparison test. Values plotted are means of the raw optical density at 650 nm (OD650) measurements.
Azithromycin study.
HeLa cells were plated at a confluence of ∼70 to 80% in 24-well tissue culture-treated plate wells (Costar). Cells were infected with C. trachomatis L2 434/Bu in cold DMEM and placed on a rocker in a 37°C tissue culture for 2 h. Subsequently, DMEM was removed and replaced with DMEM supplemented with 10% FBS (HyClone) and 1× MEM nonessential amino acid solution (Sigma). DPP was added at the time of infection, while AMP was added at 2 hpi. At 18 hpi, 3 μg/ml azithromycin was added to infected cells. Infections were allowed to proceed for an additional 24 h, and the monolayers were then gently washed with 1× PBS. Half of the wells were then mechanically harvested with glass beads and 200 μl of 1× sucrose-phosphate-glutamate buffer and stored at −80°C. In the other half of the wells, the medium was replaced with DMEM supplemented with 10% FBS (HyClone) and 1× MEM nonessential amino acid solution (Sigma) for an additional 48 h, at which point they were also lysed, and EBs were harvested. IFU were then quantified under both treatment and recovery conditions, as described above. The assay was conducted three separate times. Statistical significance values were calculated by utilizing Student t tests comparing the groups indicated.
Investigation of compound C1 effects under high-iron conditions.
HeLa cells were plated at a confluence of ∼70 to 80% in 24-well tissue culture-treated plate wells (Costar). Cells were infected with C. trachomatis L2 434/Bu in cold DMEM and placed on a rocker in a 37°C tissue culture for 2 h. Subsequently, DMEM was removed and replaced with DMEM supplemented with 10% FBS (HyClone) and 1× MEM nonessential amino acid solution (Sigma). DPP was added at the time of infection, while C1 was added at 8 hpi. A total of 0 or 250 μM FeSO4 was added at the time of infection. At 44 hpi, monolayers were washed with 1× PBS, and Chlamydia EBs were mechanically harvested with glass beads at the indicated time points, suspended in 200 μl of 1× SPG, and stored at −80°C. Inclusion-forming units were quantified as described above. The assay was conducted three separate times. Statistical significance values were calculated by utilizing one-way ANOVA coupled with a multiple-comparison test.
ACKNOWLEDGMENTS
We thank Anthony Maurelli (University of Florida) for providing us Chlamydia strains. We thank our long-time collaborator Michael VanNieuwenhze (Indiana University) and his laboratory for providing us with the “clickable” d-alanine dipeptides (EDA-DA) used in our peptidoglycan-labeling experiments. We also thank Ted Hackstadt (NIAID), Daniel Rockey (Oregon State University), Ken Fields (University of Kentucky), and Raphael Valdivia (Duke University) for graciously sharing their inclusion membrane protein and T3SS-specific polyclonal antibodies for use in these studies. We thank D. Scott Merrell (Uniformed Services University) for providing us with DPP and Dennis McDaniel (Uniformed Services University) for preparing samples for transmission electron microscopic analysis and conducting electron microscopy. We thank Kieran Broder for his assistance with conducting inclusion-forming unit counts. Finally, we thank both Anthony Maurelli and Ann Jerse (Uniformed Services University) for their helpful comments in preparing the manuscript.
This work was supported by a MIRA ESI award (R35 GM138202) and a USU faculty start-up award (HP73LIEC18) to G.W.L. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. The opinions and assertions expressed herein are those of the author(s) and do not necessarily reflect the official policy or position of the Uniformed Services University or the Department of Defense.
Conceptualization and Design, M.R.B. and G.W.L.; Data Curation and Formal Analysis, M.R.B. and G.W.L.; Investigation, Methodology, Validation, Visualization, and Writing – Original Draft, M.R.B. and G.W.L.; Writing – Review & Editing, M.R.B. and G.W.L.; Funding Acquisition, Project Administration, and Supervision, G.W.L. Both authors read and approved the final manuscript.
We declare that no competing interests exist.
Footnotes
Supplemental material is available online only.
Contributor Information
George W. Liechti, Email: george.liechti@usuhs.edu.
Craig R. Roy, Yale University School of Medicine
REFERENCES
- 1.World Health Organization. 2007. Global strategy for prevention and control of sexually transmitted infections: 2006-2015. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.CDC. 2016. Sexually transmitted disease surveillance 2015. CDC, Atlanta, GA. [Google Scholar]
- 3.CDC. 2019. Sexually transmitted disease surveillance 2018. CDC, Atlanta, GA. [Google Scholar]
- 4.Satterwhite CL, Torrone E, Meites E, Dunne EF, Mahajan R, Ocfemia MC, Su J, Xu F, Weinstock H. 2013. Sexually transmitted infections among US women and men: prevalence and incidence estimates, 2008. Sex Transm Dis 40:187–193. 10.1097/OLQ.0b013e318286bb53. [DOI] [PubMed] [Google Scholar]
- 5.Brunham RC, Rey-Ladino J. 2005. Immunology of Chlamydia infection: implications for a Chlamydia trachomatis vaccine. Nat Rev Immunol 5:149–161. 10.1038/nri1551. [DOI] [PubMed] [Google Scholar]
- 6.Elwell C, Mirrashidi K, Engel J. 2016. Chlamydia cell biology and pathogenesis. Nat Rev Microbiol 14:385–400. 10.1038/nrmicro.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liechti GW, Kuru E, Hall E, Kalinda A, Brun YV, VanNieuwenhze M, Maurelli AT. 2014. A new metabolic cell-wall labelling method reveals peptidoglycan in Chlamydia trachomatis. Nature 506:507–510. 10.1038/nature12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liechti G, Kuru E, Packiam M, Hsu YP, Tekkam S, Hall E, Rittichier JT, VanNieuwenhze M, Brun YV, Maurelli AT. 2016. Pathogenic Chlamydia lack a classical sacculus but synthesize a narrow, mid-cell peptidoglycan ring, regulated by MreB, for cell division. PLoS Pathog 12:e1005590. 10.1371/journal.ppat.1005590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hybiske K, Stephens RS. 2007. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc Natl Acad Sci U S A 104:11430–11435. 10.1073/pnas.0703218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsumoto A, Manire GP. 1970. Electron microscopic observations on the effects of penicillin on the morphology of Chlamydia psittaci. J Bacteriol 101:278–285. 10.1128/JB.101.1.278-285.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haeusser DP, Margolin W. 2016. Splitsville: structural and functional insights into the dynamic bacterial Z ring. Nat Rev Microbiol 14:305–319. 10.1038/nrmicro.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beatty WL, Byrne GI, Morrison RP. 1993. Morphologic and antigenic characterization of interferon gamma-mediated persistent Chlamydia trachomatis infection in vitro. Proc Natl Acad Sci U S A 90:3998–4002. 10.1073/pnas.90.9.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beatty WL, Belanger TA, Desai AA, Morrison RP, Byrne GI. 1994. Tryptophan depletion as a mechanism of gamma interferon-mediated chlamydial persistence. Infect Immun 62:3705–3711. 10.1128/IAI.62.9.3705-3711.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raulston JE. 1997. Response of Chlamydia trachomatis serovar E to iron restriction in vitro and evidence for iron-regulated chlamydial proteins. Infect Immun 65:4539–4547. 10.1128/IAI.65.11.4539-4547.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kahane S, Friedman MG. 1992. Reversibility of heat shock in Chlamydia trachomatis. FEMS Microbiol Lett 76:25–30. 10.1016/0378-1097(92)90358-u. [DOI] [PubMed] [Google Scholar]
- 16.Coles AM, Reynolds DJ, Harper A, Devitt A, Pearce JH. 1993. Low-nutrient induction of abnormal chlamydial development: a novel component of chlamydial pathogenesis? FEMS Microbiol Lett 106:193–200. 10.1111/j.1574-6968.1993.tb05958.x. [DOI] [PubMed] [Google Scholar]
- 17.Pearce J, Gaston H, Deane K, Devitt A, Harper A, Jecock R. 1994. ‘Persistent’ forms and persistence of Chlamydia. Trends Microbiol 2:257–259. 10.1016/0966-842x(94)90632-7. [DOI] [PubMed] [Google Scholar]
- 18.Koehler L, Nettelnbreker E, Hudson AP, Ott N, Gerard HC, Branigan PJ, Schumacher HR, Drommer W, Zeidler H. 1997. Ultrastructural and molecular analyses of the persistence of Chlamydia trachomatis (serovar K) in human monocytes. Microb Pathog 22:133–142. 10.1006/mpat.1996.0103. [DOI] [PubMed] [Google Scholar]
- 19.Nettelnbreker E, Zeidler H, Bartels H, Dreses-Werringloer U, Daubener W, Holtmann H, Kohler L. 1998. Studies of persistent infection by Chlamydia trachomatis serovar K in TPA-differentiated U937 cells and the role of IFN-gamma. J Med Microbiol 47:141–149. 10.1099/00222615-47-2-141. [DOI] [PubMed] [Google Scholar]
- 20.Gracey E, Lin A, Akram A, Chiu B, Inman RD. 2013. Intracellular survival and persistence of Chlamydia muridarum is determined by macrophage polarization. PLoS One 8:e69421. 10.1371/journal.pone.0069421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deka S, Vanover J, Dessus-Babus S, Whittimore J, Howett MK, Wyrick PB, Schoborg RV. 2006. Chlamydia trachomatis enters a viable but non-cultivable (persistent) state within herpes simplex virus type 2 (HSV-2) co-infected host cells. Cell Microbiol 8:149–162. 10.1111/j.1462-5822.2005.00608.x. [DOI] [PubMed] [Google Scholar]
- 22.Borel N, Dumrese C, Ziegler U, Schifferli A, Kaiser C, Pospischil A. 2010. Mixed infections with Chlamydia and porcine epidemic diarrhea virus—a new in vitro model of chlamydial persistence. BMC Microbiol 10:201. 10.1186/1471-2180-10-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bavoil PM. 2014. What’s in a word: the use, misuse, and abuse of the word “persistence” in Chlamydia biology. Front Cell Infect Microbiol 4:27. 10.3389/fcimb.2014.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borel N, Summersgill JT, Mukhopadhyay S, Miller RD, Ramirez JA, Pospischil A. 2008. Evidence for persistent Chlamydia pneumoniae infection of human coronary atheromas. Atherosclerosis 199:154–161. 10.1016/j.atherosclerosis.2007.09.026. [DOI] [PubMed] [Google Scholar]
- 25.Pospischil A, Borel N, Chowdhury EH, Guscetti F. 2009. Aberrant chlamydial developmental forms in the gastrointestinal tract of pigs spontaneously and experimentally infected with Chlamydia suis. Vet Microbiol 135:147–156. 10.1016/j.vetmic.2008.09.035. [DOI] [PubMed] [Google Scholar]
- 26.Rank RG, Whittimore J, Bowlin AK, Wyrick PB. 2011. In vivo ultrastructural analysis of the intimate relationship between polymorphonuclear leukocytes and the chlamydial developmental cycle. Infect Immun 79:3291–3301. 10.1128/IAI.00200-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scherler A, Jacquier N, Kebbi-Beghdadi C, Greub G. 2020. Diverse stress-inducing treatments cause distinct aberrant body morphologies in the Chlamydia-related bacterium, Waddlia chondrophila. Microorganisms 8:89. 10.3390/microorganisms8010089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fisher RA, Gollan B, Helaine S. 2017. Persistent bacterial infections and persister cells. Nat Rev Microbiol 15:453–464. 10.1038/nrmicro.2017.42. [DOI] [PubMed] [Google Scholar]
- 29.Panzetta ME, Valdivia RH, Saka HA. 2018. Chlamydia persistence: a survival strategy to evade antimicrobial effects in-vitro and in-vivo. Front Microbiol 9:3101. 10.3389/fmicb.2018.03101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moulder JW. 1993. Why is Chlamydia sensitive to penicillin in the absence of peptidoglycan? Infect Agents Dis 2:87–99. [PubMed] [Google Scholar]
- 31.Belland RJ, Nelson DE, Virok D, Crane DD, Hogan D, Sturdevant D, Beatty WL, Caldwell HD. 2003. Transcriptome analysis of chlamydial growth during IFN-gamma-mediated persistence and reactivation. Proc Natl Acad Sci U S A 100:15971–15976. 10.1073/pnas.2535394100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muramatsu MK, Brothwell JA, Stein BD, Putman TE, Rockey DD, Nelson DE. 2016. Beyond tryptophan synthase: identification of genes that contribute to Chlamydia trachomatis survival during gamma interferon-induced persistence and reactivation. Infect Immun 84:2791–2801. 10.1128/IAI.00356-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ouellette SP, Rueden KJ, Rucks EA. 2016. Tryptophan codon-dependent transcription in Chlamydia pneumoniae during gamma interferon-mediated tryptophan limitation. Infect Immun 84:2703–2713. 10.1128/IAI.00377-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonner CA, Byrne GI, Jensen RA. 2014. Chlamydia exploit the mammalian tryptophan-depletion defense strategy as a counter-defensive cue to trigger a survival state of persistence. Front Cell Infect Microbiol 4:17. 10.3389/fcimb.2014.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brinkworth AJ, Wildung MR, Carabeo RA. 2018. Genomewide transcriptional responses of iron-starved Chlamydia trachomatis reveal prioritization of metabolic precursor synthesis over protein translation. mSystems 3:e00184-17. 10.1128/mSystems.00184-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Borel N, Pospischil A, Hudson AP, Rupp J, Schoborg RV. 2014. The role of viable but non-infectious developmental forms in chlamydial biology. Front Cell Infect Microbiol 4:97. 10.3389/fcimb.2014.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wyrick PB. 2010. Chlamydia trachomatis persistence in vitro: an overview. J Infect Dis 201(Suppl 2):S88–S95. 10.1086/652394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Workowski KA, Lampe MF, Wong KG, Watts MB, Stamm WE. 1993. Long-term eradication of Chlamydia trachomatis genital infection after antimicrobial therapy. Evidence against persistent infection. JAMA 270:2071–2075. 10.1001/jama.1993.03510170061031. [DOI] [PubMed] [Google Scholar]
- 39.Lee JK, Enciso GA, Boassa D, Chander CN, Lou TH, Pairawan SS, Guo MC, Wan FYM, Ellisman MH, Sutterlin C, Tan M. 2018. Replication-dependent size reduction precedes differentiation in Chlamydia trachomatis. Nat Commun 9:45. 10.1038/s41467-017-02432-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Slepenkin A, Enquist PA, Hägglund U, de la Maza LM, Elofsson M, Peterson EM. 2007. Reversal of the antichlamydial activity of putative type III secretion inhibitors by iron. Infect Immun 75:3478–3489. 10.1128/IAI.00023-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He Q-Z, Zeng H-C, Huang Y, Hu Y-Q, Wu Y-M. 2015. The type III secretion system (T3SS) of Chlamydophila psittaci is involved in the host inflammatory response by activating the JNK/ERK signaling pathway. Biomed Res Int 2015:652416. 10.1155/2015/652416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolf K, Betts HJ, Chellas-Gery B, Hower S, Linton CN, Fields KA. 2006. Treatment of Chlamydia trachomatis with a small molecule inhibitor of the Yersinia type III secretion system disrupts progression of the chlamydial developmental cycle. Mol Microbiol 61:1543–1555. 10.1111/j.1365-2958.2006.05347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wolf AJ, Underhill DM. 2018. Peptidoglycan recognition by the innate immune system. Nat Rev Immunol 18:243–254. 10.1038/nri.2017.136. [DOI] [PubMed] [Google Scholar]
- 44.Hsu Y-P, Booher G, Egan A, Vollmer W, VanNieuwenhze MS. 2019. d-Amino acid derivatives as in situ probes for visualizing bacterial peptidoglycan biosynthesis. Acc Chem Res 52:2713–2722. 10.1021/acs.accounts.9b00311. [DOI] [PubMed] [Google Scholar]
- 45.Kuru E, Radkov A, Meng X, Egan A, Alvarez L, Dowson A, Booher G, Breukink E, Roper DI, Cava F, Vollmer W, Brun Y, VanNieuwenhze MS. 2019. Mechanisms of incorporation for D-amino acid probes that target peptidoglycan biosynthesis. ACS Chem Biol 14:2745–2756. 10.1021/acschembio.9b00664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pilhofer M, Aistleitner K, Biboy J, Gray J, Kuru E, Hall E, Brun YV, VanNieuwenhze MS, Vollmer W, Horn M, Jensen GJ. 2013. Discovery of chlamydial peptidoglycan reveals bacteria with murein sacculi but without FtsZ. Nat Commun 4:2856. 10.1038/ncomms3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ouellette SP, Karimova G, Subtil A, Ladant D. 2012. Chlamydia co-opts the rod shape-determining proteins MreB and Pbp2 for cell division. Mol Microbiol 85:164–178. 10.1111/j.1365-2958.2012.08100.x. [DOI] [PubMed] [Google Scholar]
- 48.Belland RJ, Zhong G, Crane DD, Hogan D, Sturdevant D, Sharma J, Beatty WL, Caldwell HD. 2003. Genomic transcriptional profiling of the developmental cycle of Chlamydia trachomatis. Proc Natl Acad Sci U S A 100:8478–8483. 10.1073/pnas.1331135100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shimada K, Chen S, Dempsey PW, Sorrentino R, Alsabeh R, Slepenkin AV, Peterson E, Doherty TM, Underhill D, Crother TR, Arditi M. 2009. The NOD/RIP2 pathway is essential for host defenses against Chlamydophila pneumoniae lung infection. PLoS Pathog 5:e1000379. 10.1371/journal.ppat.1000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patin D, Bostock J, Blanot D, Mengin-Lecreulx D, Chopra I. 2009. Functional and biochemical analysis of the Chlamydia trachomatis ligase MurE. J Bacteriol 191:7430–7435. 10.1128/JB.01029-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Packiam M, Weinrick B, Jacobs WR, Jr, Maurelli AT. 2015. Structural characterization of muropeptides from Chlamydia trachomatis peptidoglycan by mass spectrometry resolves “chlamydial anomaly.” Proc Natl Acad Sci U S A 112:11660–11665. 10.1073/pnas.1514026112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Almeida F, Borges V, Ferreira R, Borrego MJ, Gomes JP, Mota LJ. 2012. Polymorphisms in Inc proteins and differential expression of inc genes among Chlamydia trachomatis strains correlate with invasiveness and tropism of lymphogranuloma venereum isolates. J Bacteriol 194:6574–6585. 10.1128/JB.01428-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grieshaber SS, Grieshaber NA, Hackstadt T. 2003. Chlamydia trachomatis uses host cell dynein to traffic to the microtubule-organizing center in a p50 dynamitin-independent process. J Cell Sci 116:3793–3802. 10.1242/jcs.00695. [DOI] [PubMed] [Google Scholar]
- 54.Hackstadt T, Scidmore-Carlson MA, Shaw EI, Fischer ER. 1999. The Chlamydia trachomatis IncA protein is required for homotypic vesicle fusion. Cell Microbiol 1:119–130. 10.1046/j.1462-5822.1999.00012.x. [DOI] [PubMed] [Google Scholar]
- 55.Kumar Y, Valdivia RH. 2008. Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell Host Microbe 4:159–169. 10.1016/j.chom.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chin E, Kirker K, Zuck M, James G, Hybiske K. 2012. Actin recruitment to the Chlamydia inclusion is spatiotemporally regulated by a mechanism that requires host and bacterial factors. PLoS One 7:e46949. 10.1371/journal.pone.0046949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kokes M, Dunn JD, Granek JA, Nguyen BD, Barker JR, Valdivia RH, Bastidas RJ. 2015. Integrating chemical mutagenesis and whole-genome sequencing as a platform for forward and reverse genetic analysis of Chlamydia. Cell Host Microbe 17:716–725. 10.1016/j.chom.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lau CY, Qureshi AK. 2002. Azithromycin versus doxycycline for genital chlamydial infections: a meta-analysis of randomized clinical trials. Sex Transm Dis 29:497–502. 10.1097/00007435-200209000-00001. [DOI] [PubMed] [Google Scholar]
- 59.Tomasz A. 1979. The mechanism of the irreversible antimicrobial effects of penicillins: how the beta-lactam antibiotics kill and lyse bacteria. Annu Rev Microbiol 33:113–137. 10.1146/annurev.mi.33.100179.000553. [DOI] [PubMed] [Google Scholar]
- 60.Takikawa O, Kuroiwa T, Yamazaki F, Kido R. 1988. Mechanism of interferon-gamma action. Characterization of indoleamine 2,3-dioxygenase in cultured human cells induced by interferon-gamma and evaluation of the enzyme-mediated tryptophan degradation in its anticellular activity. J Biol Chem 263:2041–2048. 10.1016/S0021-9258(19)77982-4. [DOI] [PubMed] [Google Scholar]
- 61.Cotter TW, Ramsey KH, Miranpuri GS, Poulsen CE, Byrne GI. 1997. Dissemination of Chlamydia trachomatis chronic genital tract infection in gamma interferon gene knockout mice. Infect Immun 65:2145–2152. 10.1128/IAI.65.6.2145-2152.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perry LL, Feilzer K, Caldwell HD. 1997. Immunity to Chlamydia trachomatis is mediated by T helper 1 cells through IFN-gamma-dependent and -independent pathways. J Immunol 158:3344–3352. [PubMed] [Google Scholar]
- 63.Johansson M, Schon K, Ward M, Lycke N. 1997. Genital tract infection with Chlamydia trachomatis fails to induce protective immunity in gamma interferon receptor-deficient mice despite a strong local immunoglobulin A response. Infect Immun 65:1032–1044. 10.1128/IAI.65.3.1032-1044.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rottenberg ME, Gigliotti Rothfuchs AC, Gigliotti D, Svanholm C, Bandholtz L, Wigzell H. 1999. Role of innate and adaptive immunity in the outcome of primary infection with Chlamydia pneumoniae, as analyzed in genetically modified mice. J Immunol 162:2829–2836. [PubMed] [Google Scholar]
- 65.Sandoz KM, Rockey DD. 2010. Antibiotic resistance in Chlamydiae. Future Microbiol 5:1427–1442. 10.2217/fmb.10.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Borel N, Leonard C, Slade J, Schoborg RV. 2016. Chlamydial antibiotic resistance and treatment failure in veterinary and human medicine. Curr Clin Microbiol Rep 3:10–18. 10.1007/s40588-016-0028-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Slade JA, Brockett M, Singh R, Liechti GW, Maurelli AT. 2019. Fosmidomycin, an inhibitor of isoprenoid synthesis, induces persistence in Chlamydia by inhibiting peptidoglycan assembly. PLoS Pathog 15:e1008078. 10.1371/journal.ppat.1008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cho H, Uehara T, Bernhardt TG. 2014. Beta-lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell 159:1300–1311. 10.1016/j.cell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Klockner A, Otten C, Derouaux A, Vollmer W, Buhl H, De Benedetti S, Munch D, Josten M, Molleken K, Sahl HG, Henrichfreise B. 2014. AmiA is a penicillin target enzyme with dual activity in the intracellular pathogen Chlamydia pneumoniae. Nat Commun 5:4201. 10.1038/ncomms5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jacquier N, Yadav AK, Pillonel T, Viollier PH, Cava F, Greub G. 2019. A SpoIID homolog cleaves glycan strands at the chlamydial division septum. mBio 10:e01128-19. 10.1128/mBio.01128-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brothwell JA, Brockett M, Banerjee A, Stein BD, Nelson DE, Liechti GW. 2021. Genome copy number regulates inclusion expansion, septation, and infectious developmental form conversion in Chlamydia trachomatis. J Bacteriol 203:e00630-20. 10.1128/JB.00630-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lambden PR, Pickett MA, Clarke IN. 2006. The effect of penicillin on Chlamydia trachomatis DNA replication. Microbiology 152:2573–2578. 10.1099/mic.0.29032-0. [DOI] [PubMed] [Google Scholar]
- 73.Gerard HC, Krausse-Opatz B, Wang Z, Rudy D, Rao JP, Zeidler H, Schumacher HR, Whittum-Hudson JA, Kohler L, Hudson AP. 2001. Expression of Chlamydia trachomatis genes encoding products required for DNA synthesis and cell division during active versus persistent infection. Mol Microbiol 41:731–741. 10.1046/j.1365-2958.2001.02550.x. [DOI] [PubMed] [Google Scholar]
- 74.Pokorzynski ND, Brinkworth AJ, Carabeo R. 2019. A bipartite iron-dependent transcriptional regulation of the tryptophan salvage pathway in Chlamydia trachomatis. Elife 8:e42295. 10.7554/eLife.42295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bailey MW, Bisicchia P, Warren BT, Sherratt DJ, Mannik J. 2014. Evidence for divisome localization mechanisms independent of the Min system and SlmA in Escherichia coli. PLoS Genet 10:e1004504. 10.1371/journal.pgen.1004504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bernhardt TG, de Boer PA. 2005. SlmA, a nucleoid-associated, FtsZ binding protein required for blocking septal ring assembly over chromosomes in E. coli. Mol Cell 18:555–564. 10.1016/j.molcel.2005.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu LJ, Errington J. 2004. Coordination of cell division and chromosome segregation by a nucleoid occlusion protein in Bacillus subtilis. Cell 117:915–925. 10.1016/j.cell.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 78.Adams DW, Wu LJ, Errington J. 2015. Nucleoid occlusion protein Noc recruits DNA to the bacterial cell membrane. EMBO J 34:491–501. 10.15252/embj.201490177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Singh R, Liechti G, Slade JA, Maurelli AT. 2020. Chlamydia trachomatis oligopeptide transporter performs dual functions of oligopeptide transport and peptidoglycan recycling. Infect Immun 88:e00086-20. 10.1128/IAI.00086-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Buchholz KR, Stephens RS. 2008. The cytosolic pattern recognition receptor NOD1 induces inflammatory interleukin-8 during Chlamydia trachomatis infection. Infect Immun 76:3150–3155. 10.1128/IAI.00104-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Opitz B, Forster S, Hocke AC, Maass M, Schmeck B, Hippenstiel S, Suttorp N, Krull M. 2005. Nod1-mediated endothelial cell activation by Chlamydophila pneumoniae. Circ Res 96:319–326. 10.1161/01.RES.0000155721.83594.2c. [DOI] [PubMed] [Google Scholar]
- 82.Welter-Stahl L, Ojcius DM, Viala J, Girardin S, Liu W, Delarbre C, Philpott D, Kelly KA, Darville T. 2006. Stimulation of the cytosolic receptor for peptidoglycan, Nod1, by infection with Chlamydia trachomatis or Chlamydia muridarum. Cell Microbiol 8:1047–1057. 10.1111/j.1462-5822.2006.00686.x. [DOI] [PubMed] [Google Scholar]
- 83.Buchholz KR, Stephens RS. 2006. Activation of the host cell proinflammatory interleukin-8 response by Chlamydia trachomatis. Cell Microbiol 8:1768–1779. 10.1111/j.1462-5822.2006.00747.x. [DOI] [PubMed] [Google Scholar]
- 84.Muschiol S, Bailey L, Gylfe A, Sundin C, Hultenby K, Bergstrom S, Elofsson M, Wolf-Watz H, Normark S, Henriques-Normark B. 2006. A small-molecule inhibitor of type III secretion inhibits different stages of the infectious cycle of Chlamydia trachomatis. Proc Natl Acad Sci U S A 103:14566–14571. 10.1073/pnas.0606412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rzomp KA, Moorhead AR, Scidmore MA. 2006. The GTPase Rab4 interacts with Chlamydia trachomatis inclusion membrane protein CT229. Infect Immun 74:5362–5373. 10.1128/IAI.00539-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mirrashidi KM, Elwell CA, Verschueren E, Johnson JR, Frando A, Von Dollen J, Rosenberg O, Gulbahce N, Jang G, Johnson T, Jager S, Gopalakrishnan AM, Sherry J, Dunn JD, Olive A, Penn B, Shales M, Cox JS, Starnbach MN, Derre I, Valdivia R, Krogan NJ, Engel J. 2015. Global mapping of the Inc-human interactome reveals that retromer restricts Chlamydia infection. Cell Host Microbe 18:109–121. 10.1016/j.chom.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sixt BS, Bastidas RJ, Finethy R, Baxter RM, Carpenter VK, Kroemer G, Coers J, Valdivia RH. 2017. The Chlamydia trachomatis inclusion membrane protein CpoS counteracts STING-mediated cellular surveillance and suicide programs. Cell Host Microbe 21:113–121. 10.1016/j.chom.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Faris R, Merling M, Andersen SE, Dooley CA, Hackstadt T, Weber MM. 2019. Chlamydia trachomatis CT229 subverts Rab GTPase-dependent CCV trafficking pathways to promote chlamydial infection. Cell Rep 26:3380–3390.e5. 10.1016/j.celrep.2019.02.079. [DOI] [PubMed] [Google Scholar]
- 89.Weber MM, Lam JL, Dooley CA, Noriea NF, Hansen BT, Hoyt FH, Carmody AB, Sturdevant GL, Hackstadt T. 2017. Absence of specific Chlamydia trachomatis inclusion membrane proteins triggers premature inclusion membrane lysis and host cell death. Cell Rep 19:1406–1417. 10.1016/j.celrep.2017.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Scidmore MA, Hackstadt T. 2001. Mammalian 14-3-3beta associates with the Chlamydia trachomatis inclusion membrane via its interaction with IncG. Mol Microbiol 39:1638–1650. 10.1046/j.1365-2958.2001.02355.x. [DOI] [PubMed] [Google Scholar]
- 91.Wesolowski J, Weber MM, Nawrotek A, Dooley CA, Calderon M, St Croix CM, Hackstadt T, Cherfils J, Paumet F. 2017. Chlamydia hijacks ARF GTPases to coordinate microtubule posttranslational modifications and Golgi complex positioning. mBio 8:e02280-16. 10.1128/mBio.02280-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wesolowski J, Paumet F. 2017. Taking control: reorganization of the host cytoskeleton by Chlamydia. F1000Res 6:2058. 10.12688/f1000research.12316.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Horner PJ. 2012. Azithromycin antimicrobial resistance and genital Chlamydia trachomatis infection: duration of therapy may be the key to improving efficacy. Sex Transm Infect 88:154–156. 10.1136/sextrans-2011-050385. [DOI] [PubMed] [Google Scholar]
- 94.Suchland RJ, Dimond ZE, Putman TE, Rockey DD. 2017. Demonstration of persistent infections and genome stability by whole-genome sequencing of repeat-positive, same-serovar Chlamydia trachomatis collected from the female genital tract. J Infect Dis 215:1657–1665. 10.1093/infdis/jix155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xue Y, Zheng H, Mai Z, Qin X, Chen W, Huang T, Chen D, Zheng L. 2017. An in vitro model of azithromycin-induced persistent Chlamydia trachomatis infection. FEMS Microbiol Lett 364:fnx145. 10.1093/femsle/fnx145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download IAI00685-20_Supp_1_seq1.pdf, PDF file, 0.08 MB (81.3KB, pdf)
Supplemental material. Download IAI00685-20_Supp_2_seq2.pdf, PDF file, 0.8 MB (771.4KB, pdf)
Supplemental material. Download IAI00685-20_Supp_3_seq3.pdf, PDF file, 1.8 MB (1.8MB, pdf)
Supplemental material. Download IAI00685-20_Supp_4_seq4.pdf, PDF file, 1.5 MB (1.5MB, pdf)
Supplemental material. Download IAI00685-20_Supp_5_seq5.pdf, PDF file, 6.5 MB (6.5MB, pdf)
Supplemental material. Download IAI00685-20_Supp_6_seq6.pdf, PDF file, 0.04 MB (45.3KB, pdf)